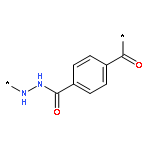
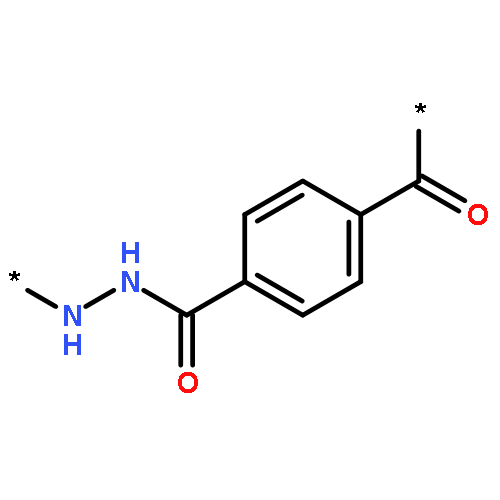
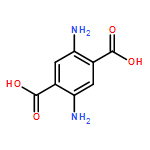
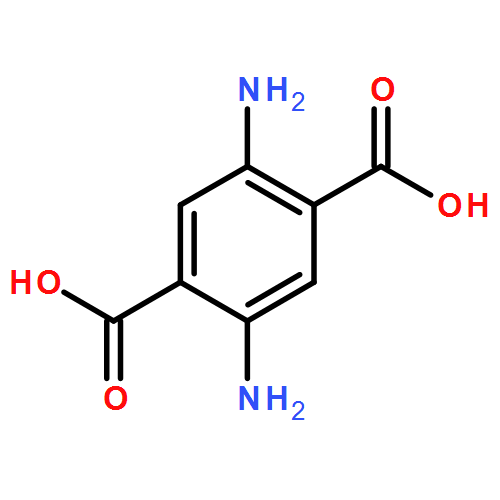
Co-reporter:Junfeng Huang;Hongqiang Qin;Jing Dong;Chunxia Song;Yangyang Bian;Mingming Dong;Kai Cheng;Fangjun Wang;Deguang Sun;Liming Wang;Mingliang Ye;Hanfa Zou
Journal of Proteome Research September 5, 2014 Volume 13(Issue 9) pp:3896-3904
Publication Date(Web):2017-2-22
DOI:10.1021/pr500454g
Current sample preparation protocols for quantitative phosphoproteome analysis are tedious and time-consuming. Here, a facile in situ sample processing approach (iSPA) is developed by using macroporous Ti(IV)-IMAC microspheres as the preparation “beds”, where all sample preparation procedures including the enrichment of phosphoproteins, tryptic digestion of proteins, enrichment, and isotope labeling of phosphopeptides are performed in situ sequentially. As a result of the in situ processing design and the seamless procedures, extra steps for desalting and buffer exchanging, which are always required in conventional approaches, are avoided, and the sample loss and contamination could be greatly reduced. Thus, better sensitivity and accuracy for the quantitative phosphoproteome analysis were obtained. This strategy was further applied to differential phosphoproteome analysis of human liver tissues with or without hepatocellular carcinoma (HCC). In total, 8548 phosphorylation sites were confidently quantified from three replicate analyses of 0.5 mg of human liver protein extracts.Keywords: dimethyl labeling; hepatocellular carcinoma; human liver; in situ sample processing approach; on-beads digestion; phosphoprotein enrichment; phosphoproteome quantification; solid phase labeling;
Co-reporter:Xinning Jiang;Mingliang Ye;Hanfa Zou;Kai Cheng
Journal of Proteome Research May 7, 2010 Volume 9(Issue 5) pp:2743-2751
Publication Date(Web):Publication Date (Web): March 24, 2010
DOI:10.1021/pr9009904
The development of new phosphoproteomic technologies has led to a rapid increase in the number of phosphoprotein identifications. Managing and extracting valuable information from the phosphoproteome data sets and generating output information in user-friendly formats require special data management and process platform. Even though a few proteome pipelines have been developed, they are mainly designed for processing data set of unmodified peptide/protein identifications. Because of the different characteristics of phosphorylated peptides/proteins, these pipelines are inconvenient, sometimes inappropriate, to process the phosphoproteome data sets. In this study, a software suite named ArMone was specially designed for the management and analysis of phosphoproteome data. It can readily identify phosphopeptides with high reliability and high sensitivity, and can effectively pinpoint the most probable phosphorylation site. A few well-designed postvalidation process tools are also available to extract and export valuable information. ArMone is a stand-alone application with friendly graphic user interface. It can run on different operating systems and can process data sets obtained by most of the commonly used database search engines.Keywords: automatic validation; bioinformatics; phosphoproteome analysis; software;
Co-reporter:Deguang Sun;Fangjun Wang;Rui Chen;Xinning Jiang;Guanghui Han;Liming Wang;Mingliang Ye;Hanfa Zou
Journal of Proteome Research February 6, 2009 Volume 8(Issue 2) pp:651-661
Publication Date(Web):Publication Date (Web): January 21, 2009
DOI:10.1021/pr8008012
The study of protein glycosylation has lagged far behind the progress of current proteomics because of the enormous complexity, wide dynamic range distribution and low stoichiometric modification of glycoprotein. Solid phase extraction of tryptic N-glycopeptides by hydrazide chemistry is becoming a popular protocol for the analysis of N-glycoproteome. However, in silico digestion of proteins in human proteome database by trypsin indicates that a significant percentage of tryptic N-glycopeptides is not in the preferred detection mass range of shotgun proteomics approach, that is, from 800 to 3500 Da. And the quite big size of glycan groups may block trypsin to access the K, R residues near N-glycosites for digestion, which will result in generation of big glycopeptides. Thus many N-glycosites could not be localized if only trypsin was used to digest proteins. Herein, we describe a comprehensive way to analyze the N-glycoproteome of human liver tissue by combination of hydrazide chemistry method and multiple enzyme digestion. The lysate of human liver tissue was digested with three proteases, that is, trypsin, pepsin and thermolysin, with different specificities, separately. Use of trypsin alone resulted in identification of 622 N-glycosites, while using pepsin and thermolysin resulted in identification of 317 additional N-glycosites. Among the 317 additional N-glycosites, 98 (30.9%) could not be identified by trypsin in theory because the corresponding in silico tryptic peptides are either too small or too big to detect in mass spectrometer. This study clearly demonstrated that the coverage of N-glycosites could be significantly increased due to the adoption of multiple enzyme digestion. A total number of 939 N-glycosites were identified confidently, covering 523 noredundant glycoproteins from human liver tissue, which leads to the establishment of the largest data set of glycoproteome from human liver up to now.Keywords: Glycoproteomics; Human liver proteome project; Hydrazide chemistry; Multiple enzyme digestion;
Co-reporter:Qi Wang;Keyun Wang;Mingliang Ye
Analyst (1876-Present) 2017 vol. 142(Issue 19) pp:3536-3548
Publication Date(Web):2017/09/25
DOI:10.1039/C7AN00954B
Protein methylation is an important post-translational modification (PTM) that plays crucial roles in the regulation of diverse biological processes. Though many efforts have been devoted to the investigation of protein methylation, the analysis of non-histone methylation at the proteome level is still a great challenge. The alteration of the protein/peptide physicochemical properties caused by methylation is very small, thus it is difficult to develop highly efficient enrichment approaches to separate methylated peptides from a pool of diverse background peptides. The mass shifts caused by methylations are identical to the substitutions of some amino acids, thus it is difficult to confidently identify methylated peptides. In this review, we report on recent advances in the development of methods for large-scale analysis of non-histone protein methylation. Especially the methods for efficient enrichment and the approaches for controlling identification confidence have been covered.
Co-reporter:Jingyao Bai, Zhongshan Liu, Hongwei Wang, Xin You, Junjie Ou, Yehua Shen, Mingliang Ye
Journal of Chromatography A 2017 Volume 1498(Volume 1498) pp:
Publication Date(Web):19 May 2017
DOI:10.1016/j.chroma.2016.12.031
•A hydrophilic hybrid monolith was synthesized via thiol-ene click reaction.•The obtained columns exhibited good separation ability for neutral polar compounds.•The matrix was modified into hydrazide material for glycopeptides enrichment.A macroporous hydrophilic organic-silica hybrid monolithic column was synthesized via photoinitiated thiol-ene click polymerization reaction of 1-thioglycerol-modified polyhedral oligomeric vinylsilsesquioxane (vinylPOSS) and dithiothreitol (DTT) in a binary porogenic system consisting of tetrahydrofuran (THF) and dodecanol. 1-Thioglycerol was used to modify vinylPOSS in order to form a precursor with good solubility in the binary porogenic system. The influences of both the ratio of 1-thioglycerol/vinylPOSS and the porogenic solvents on the morphology and permeability of hybrid monoliths were studied in detail. The physical properties of hybrid monolith were characterized by scanning electron microscopy (SEM), Fourier transform infrared (FT-IR) spectroscopy and nitrogen adsorption/desorption measurement. The chromatographic performance was evaluated by separation of neutral polar compounds in capillary liquid chromatography (cLC). The resulting column possessed homogeneous macroporous structure and showed hydrophilic interaction liquid chromatography (HILIC) separation mechanism with high efficiency of 65,000 N m−1 for formamide. Ultimately, the hybrid matrix was grafted with hydrazine groups and then exhibited the ability of glycopeptides enrichment.
Co-reporter:Jingyao Bai, Junjie Ou, Haiyang Zhang, Shujuan Ma, Yehua Shen, Mingliang Ye
Journal of Chromatography A 2017 Volume 1514(Volume 1514) pp:
Publication Date(Web):8 September 2017
DOI:10.1016/j.chroma.2017.07.070
•Thiol-maleimide click reaction was firstly adopted to prepare capillary monoliths.•Ultra-high column efficiency (180,500 N/m) was acquired on poly(BMI-co-3SH).•The tryptic digests of BSA and HeLa were positively identified in cLC–MS.One-step thiol-maleimide polymerization reaction was firstly adopted for direct preparation of polymeric monoliths via alkaline-catalyzed reaction of 4,4′-bis(maleimidophenyl)methane (BMI) and trimethylolpropane tris(3-mercaptopropionate) (3SH)/pentaerythriol tetra(3-mercaptopropionate) (4SH) in the presence of a small amount of triethylamine (TEA). The polymerization could be performed within 3 h, which was faster than thermal-initiated free radical polymerization. Two kinds of monoliths, poly(BMI-co-3SH) (marked as I) and poly(BMI-co-4SH) (marked as II), were characterized with scanning electron microscopy (SEM), attenuated total reflection Fourier-transformed infrared spectroscopy (ATR-FTIR), thermal gravimetric analysis (TGA) and mercury intrusion porosimetry (MIP). Satisfactory chromatographic separation ability and column efficiency were gained for analysis of small molecular compounds such as alkylbenzenes, polynuclear aromatic hydrocarbons (EPA 610) and phenols in reversed-phase capillary liquid chromatography (cLC). High column efficiency (180,500 N/m) for butylbenzene was acquired on poly(BMI-co-3SH) column I-2, which was higher than those on most reported polymeric monoliths. A retention-independent efficient performance of small molecules was obtained by plotting of plate height (H) of alkylbenzenes versus the linear velocity (u). A term values in van Deemter equation of I-2 (1.72-0.24 μm) and poly(BMI-co-4SH) column II-2 (5.28-4.14 μm) were smaller than those of traditional organic/hybrid monoliths. Finally, as a practical application, 53 and 2184 unique peptides from the tryptic digests of bovine serum albumin (BSA) and HeLa cell proteins were positively identified with poly(BMI-co-3SH) monolith in cLC–MS.
Co-reporter:Guowang Xu, Mingliang Ye
Journal of Chromatography A 2017 Volume 1498(Volume 1498) pp:
Publication Date(Web):19 May 2017
DOI:10.1016/j.chroma.2017.03.082
Co-reporter:Zhongshan Liu, Jing Liu, Zheyi Liu, Hongwei Wang, Junjie Ou, Mingliang Ye, Hanfa Zou
Journal of Chromatography A 2017 Volume 1498(Volume 1498) pp:
Publication Date(Web):19 May 2017
DOI:10.1016/j.chroma.2017.01.029
•Vinyl-functionalized monolithic columns with 75 and 150 μm i.d. were prepared.•The monolithic columns were facilely modified via thiol-ene click reaction.•The effect of flow rate on cLC–MS/MS performance was investigated.•The SCX and RP monolithic columns were applied in two-dimensional separation.The vinyl-functionalized hybrid monolithic columns (75 and 150 μm i.d.) were prepared via sol-gel chemistry of tetramethoxysilane (TMOS) and vinyltrimethoxysilane (VTMS). The content of accessible vinyl groups was further improved after the monolithic column was post-treated with vinyldimethylethoxysilane (VDMES). The surface properties of monolithic columns were tailored via thiol-ene click reaction by using 1-octadecanethiol, sodium 3-mercapto-1-propanesulfonate and 2,2′-(ethylenedioxy)diethanethiol/vinylphosphonic acid, respectively. The preparing octadecyl-functionalized monolithic columns were adopted for proteomics analysis in cLC–MS/MS. A 37-cm-long × 75-μm-i.d. monolithic column could identify 3918 unique peptides and 1067 unique proteins in the tryptic digest of proteins from HeLa cells. When a 90-cm-long × 75-μm-i.d. monolithic column was used, the numbers of unique peptides and proteins were increased by 82% and 32%, respectively. Furthermore, strong cation exchange (SCX) monolithic columns (4 cm in length × 150 μm i.d.) were also prepared and coupled with the 37-cm-long × 75-μm-i.d. octadecyl-functionalized monolithic column for two-dimensional SCX-RPLC–MS/MS analysis, which could identify 17114 unique peptides and 3211 unique proteins.
Co-reporter:Yating Yao, Jing Dong, Mingming Dong, Fangjie Liu, Yan Wang, Jiawei Mao, Mingliang Ye, Hanfa Zou
Journal of Chromatography A 2017 Volume 1498(Volume 1498) pp:
Publication Date(Web):19 May 2017
DOI:10.1016/j.chroma.2017.03.026
•A new Ti4+-IMAC adsorbent was synthesized and applied for phosphopeptide enrichment.•It exhibited excellent specificity of over 99% for the enrichment of phosphopeptides.•The spin tip was fitted for the analysis of minute amount of complex samples.In this study, we developed a centrifugation assisted solid phase extraction (SPE) method for the selective enrichment of phosphopeptides using a new Ti4+-IMAC material synthesized in-house. This new material has the feature of big size and large specific surface area which makes it more suitable to enrich phosphopeptides in a SPE way. The spin tips loaded with the Ti4+-IMAC material were applied to enrich phosphopeptides from the complex protein digests. It was found that phosphopeptides can be specifically enriched from tryptic digest of bovine serum albumin and β-casein at a molar ratio up to 1000:1. And about 4700 unique phosphorylated peptides can be identified with the specificity as high as 99% from the tryptic digest of HeLa cell proteins. This tip was demonstrated to have good column-to-column reproducibility. Furthermore, it is fitted to analyze minute amount of sample. Compared with the conventional solution method, the SPE method facilitated the rapid and complete separation of the material with solution, which making it a time-saving and convenient method for phosphopeptide enrichment. Compared with the commercial TiO2 material, this new materials yielded much more phosphopeptide identifications and much higher enrichment specificity.
Co-reporter:Hongwei Wang, Yating Yao, Ya Li, Shujuan Ma, Xiaojun Peng, Junjie Ou, Mingliang Ye
Analytica Chimica Acta 2017 Volume 979(Volume 979) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.aca.2017.05.004
•It was the first time to prepare OT column by ring-opening polymerization.•The ratio of ethanol/H2O at 13/1 (v/v) was used in the synthesis of the OT phases.•The OT column was successfully applied in cLC-MS/MS analysis of tryptic digest.An open tubular (OT) column (25 μm i.d.) was prepared by in situ ring-opening polymerization of octaglycidyldimethylsilyl polyhedral oligomeric silsesquioxanes (POSS-epoxy) with 4-aminophenyl disulfide (APDS) in a binary porogenic system of ethanol/H2O. It was found that porogenic composition played an important role in the formation of OT stationary phases. The ratio of ethanol/H2O at 6/1 (v/v) would lead to the fabrication of hybrid monoliths, while the ratio of ethanol/H2O at 13/1 (v/v) would result in the synthesis of OT phases. In addition, the effects of precursor content and reaction duration on the thickness of OT stationary phases were investigated. Either lower precursor content or shorter reaction duration would produce thinner layer of OT column. The repeatability of OT columns was evaluated through relative standard deviation (RSD%) with benzene as the analyte. The run-to-run, column-to-column and batch-to-batch repeatabilities were 1.7%, 4.8% and 5.6%, respectively, exhibiting satisfactory repeatability of the OT column. Then tryptic digest of mouse liver proteins was used to evaluate the performance of the resulting OT columns (25 μm i.d. × 2.5 m in length) by cLC-MS/MS analysis, demonstrating their potential in proteome analysis.Download high-res image (147KB)Download full-size image
Co-reporter:Yanan Li, Yan Wang, Mingming Dong, Hanfa Zou, and Mingliang Ye
Analytical Chemistry 2017 Volume 89(Issue 4) pp:
Publication Date(Web):January 24, 2017
DOI:10.1021/acs.analchem.6b03812
Temporal tyrosine phosphorylation (pTyr) plays a crucial role in numerous cellular functions. The characterization of the tyrosine phosphorylation states of cells is of great interest for understanding the underlying mechanisms. In this study, we developed sensitive and cost-effective methods for the assay of the global protein tyrosine phosphorylation in complex samples by using a novel engineered pTyr binding protein, Src SH2 domain triple-point mutant (Trm-SH2). Taking the advantage of the pan-specific interaction of Trm-SH2 to pTyr, a high throughput approach was developed to determine the total protein tyrosine phosphorylation level in a sample. This method allowed the detection of 0.025 ng of tyrosine phosphorylated proteins. The Trm-SH2 was further exploited to develop a method to profile the global tyrosine phosphorylation state. When this approach was applied to analyze the tyrosine phosphoproteome upon stimulation, distinct patterns were observed. This approach is readily used in many research and clinical fields for the analysis of tyrosine phosphorylated proteins in complex samples, including classifying aberrant phosphotyrosine-dependent signaling in cancer.
Co-reporter:Zhenzhen Deng, Mingming Dong, Yan Wang, Jing Dong, Shawn S.-C. Li, Hanfa Zou, and Mingliang Ye
Analytical Chemistry 2017 Volume 89(Issue 4) pp:
Publication Date(Web):January 18, 2017
DOI:10.1021/acs.analchem.6b04288
Tyrosine phosphorylation (pTyr) is important for normal physiology and implicated in many human diseases, particularly cancer. Identification of pTyr sites is critical to dissecting signaling pathways and understanding disease pathologies. However, compared with serine/threonine phosphorylation (pSer/pThr), the analysis of pTyr at the proteome level is more challenging due to its low abundance. Here, we developed a biphasic affinity chromatographic approach where Src SH2 superbinder was coupled with NeutrAvidin affinity chromatography, for tyrosine phosphoproteome analysis. With the use of competitive elution agent biotin-pYEEI, this strategy can distinguish high-affinity phosphotyrosyl peptides from low-affinity ones, while the excess competitive agent is readily removed by using NeutrAvidin agarose resin in an integrated tip system. The excellent performance of this system was demonstrated by analyzing tyrosine phosphoproteome of Jurkat cells from which 3,480 unique pTyr sites were identified. The biphasic affinity chromatography method for deep Tyr phosphoproteome analysis is rapid, sensitive, robust, and cost-effective. It is widely applicable to the global analysis of the tyrosine phosphoproteome associated with tyrosine kinase signal transduction.
Co-reporter:Hongqiang QinKai Cheng, Jun Zhu, Jiawei Mao, Fangjun Wang, Mingming DongRui Chen, Zhimou Guo, Xinmiao Liang, Mingliang Ye, Hanfa Zou
Analytical Chemistry 2017 Volume 89(Issue 3) pp:
Publication Date(Web):December 28, 2016
DOI:10.1021/acs.analchem.6b02887
The diversity of O-linked glycan structures has drawn increasing attention due to its vital biological roles. However, intact O-glycopeptides with different glycans are typically not well elucidated using the current methods. In this work, an integrated strategy was developed for comprehensive analysis of O-GalNAc glycosylation by combining hydrophilic interaction chromatography (HILIC) tip enrichment, beam-type collision induced decomposition (beam-CID) detection, and in silico deglycosylation method for spectra interpretation. In this strategy, the intact O-GalNAc glycopeptides were selectively enriched and the original spectra obtained by time-of-flight (TOF)-CID were preprocessed using an in silico deglycosylation method, enabling direct searching without setting multiple glycosylation modifications, which could significantly decrease the search space. This strategy was applied to analyze the O-GalNAc glycoproteome of human serum, leading to identification of 407 intact O-GalNAc glycopeptides from 93 glycoproteins. About 81% of the glycopeptides contained at least one sialic acid, which could reveal the microheterogeneity of O-GalNAc glycosylation. Up until now, this is the largest data set of intact O-GalNAc glycoforms from complex biological samples at the proteome level. Furthermore, this method is readily applicable to study O-glycoform heterogeneity in other complex biological systems.
Co-reporter:Yuting Cong, Lianghai Hu, Zhang Zhang, Yin Gao, Mingming Dong, Hongqiang Qin, Mingliang Ye, Jingkai Gu, Hanfa Zou
Talanta 2017 Volume 165() pp:664-670
Publication Date(Web):1 April 2017
DOI:10.1016/j.talanta.2017.01.023
•PRM enabled the qualitative and quantitative analysis of mAbs and its glycosylation.•Absolute quantitation of mAbs and their glycoforms was achieved in serum and tissues.•HILIC enrichment enabled the in-depth analysis of low abundant glycopeptides.Monoclonal antibodies (mAbs), are one of the most important protein drugs have attracted increasing attention. However, the pharmacokinetics of mAbs has not been fully investigated due to the complexity of protein drugs. Traditonal immuno-based approaches can not recognize the proteoforms of mAbs because of the long development cycles, prohibitive cost, and interactions between different proteins. Therefore, reliable qualitative and quantitative analysis of the proteoforms of mAbs in biological samples is of crucial importance. Herein, a novel method was developed for absolute quantitation of mAbs and their glycoforms in complex biological samples such as serum and tissues. With the combination of HILIC enrichment and parallel reaction monitoring by high resolution mass spectrometry, most of the glycoforms can be accurately quantified at the fmol level through the use of the model mAb of bevacizumab. More importantly, the structural confirmation can be achieved simultaneously without the need for additional experiments. This strategy can be readily applied to the pharmacokinetic study of glycosylation modification and biomarker discovery for clinical applications.
Co-reporter:Zhongshan Liu, Junjie Ou, Hongwei Wang, Xin You, and Mingliang Ye
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 46) pp:32060
Publication Date(Web):November 3, 2016
DOI:10.1021/acsami.6b11572
Four kinds of either hydrazide-linked or amide-linked polymers were facilely synthesized by using hydrazine, tetrakis(4-aminophenyl)methane (TAPM), terephthaloyl chloride (TPC), and trimesoyl chloride (TMC) as building blocks. The morphology, porosity, composition, and surface property of polymers were characterized by scanning electron microscopy, transmission electron microscopy, nitrogen adsorption–desorption measurement, 13C/CP-MAS NMR, X-ray photoelectron spectroscopy, etc. The results indicated that building blocks had important effects on morphology and porosity. Poly(TMC–TAPM) synthesized with TMC and TAPM showed the highest surface area of 241.9 m2 g–1. In addition, note that a hollow structure with ∼20 nm wall thickness was formed in poly(TMC–hydrazine) prepared with TMC and hydrazine. Further study indicated that both carboxyl groups (−COOH) and hydrazide groups (−CONH–NH2) existed on the surface of poly(TMC–hydrazine), besides the mainly hydrazide linkage (−CONH–NHOC−). Taking advantages of good hydrophilicity and special functional groups on the surface, we finally adopted poly(TMC–hydrazine) to enrich glycopeptides from tryptic digest via both hydrophilic interaction chromatography method with identification of 369 unique N-glycosylation sites and hydrazide chemistry method with identification of 88 unique N-glycosylation sites, respectively.Keywords: amide; enrichment of glycopeptide; hollow structure; hydrazide; porous organic polymer
Co-reporter:Lianfang Chen, Junjie Ou, Hongwei Wang, Zhongshan Liu, Mingliang Ye, and Hanfa Zou
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 31) pp:20292
Publication Date(Web):July 18, 2016
DOI:10.1021/acsami.6b06225
Although thousands of metal–organic frameworks (MOFs) have been fabricated and widely applied in gas storage/separations, adsorption, catalysis, and so on, few kinds of MOFs have been used as adsorption materials while simultaneously serving as matrixes to analyze small molecules for laser desorption/ionization mass spectrometry (LDI-MS). Herein, a new concept is introduced to design and synthesize MOFs as both adsorption materials and matrixes according to the structure of ligands and common matrixes. The proof of concept design was demonstrated by selection of 2,5-pyridinedicarboxylic acid (PDC) and 2,5-dihydroxyterephthalic acid (DHT) as ligands for synthesis of MOFs. Two Zr(IV)-based MOFs of UiO-66-PDC and UiO-66-(OH)2 were synthesized and applied for the first time as new matrixes for analysis of small molecules by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS). Both of them showed low matrix interferences, high ionization efficiency, and good reproducibility when used as matrixes. A variety of small molecules, including saccharides, amino acids, nucleosides, peptides, alkaline drugs, and natural products, were analyzed. In addition, UiO-66-(OH)2 exhibited potential for application in the quantitative determination of glucose and pyridoxal 5′-phosphate. Furthermore, thanks to its intrinsically large surface area and highly ordered pores, UiO-66-(OH)2 also showed sensitive and specific enrichment of phosphopeptides prior to MS analysis. These results demonstrated that this strategy can be used to efficiently screen tailor-made MOFs as matrixes to analyze small molecules by MALDI-TOF-MS.Keywords: LDI-MS; metal−organic frameworks; small molecules; UiO-66-(OH)2; UiO-66-PDC
Co-reporter:Fangjie Liu, Hao Wan, Zhongshan Liu, Hongwei Wang, Jiawei Mao, Mingliang Ye, and Hanfa Zou
Analytical Chemistry 2016 Volume 88(Issue 10) pp:5058
Publication Date(Web):April 21, 2016
DOI:10.1021/acs.analchem.6b00701
In this study, we developed a Ti(IV) monolithic spin tip for phosphoproteome analysis of a minute amount of biological sample for the first time. The surface of polypropylene pipet tip was activated by the photoinitiator benzophenone under UV light radiation followed by polymerization of ethylene glycol methacrylate phosphate and bis-acrylamide in the tip to form a porous monolith with reactive phosphate groups. The as-prepared tips grafted with monolithic adsorbent were then chelated with titanium(IV) ion for phosphopeptide enrichment. It was found that the tips enabled fast and efficient capture of phosphopeptides from microscale complex samples. The monolithic tip was demonstrated to have a detection limit as low as 5 fmol β-casein tryptic digest, along with an exceptionally high specificity to capture phosphopeptides from complex tryptic digest mixed with an unphosphorylated protein and a phosphorylated protein at a molar ratio up to 1000:1. When the tip was applied to enrich phosphopeptides from 5 μg of tryptic digest of complex HeLa cell proteins, 1185 high confidence of phosphorylated sites were successfully identified with the specificity as high as 92.5%. So far, this is the most sensitive phosphoproteomics analysis using a standard liquid chromatography–tandem mass spectrometry (LC–MS/MS) system for proteome-wide phosphorylation analysis in mammalian cells.
Co-reporter:Keyun Wang, Mingming Dong, Jiawei Mao, Yan Wang, Yan Jin, Mingliang Ye, and Hanfa Zou
Analytical Chemistry 2016 Volume 88(Issue 23) pp:
Publication Date(Web):November 1, 2016
DOI:10.1021/acs.analchem.6b02872
Protein methylation is receiving more and more attention for its important regulating role in diverse biological processes including epigenetic regulation of gene transcription, RNA processing, DNA damage repair, and signal transduction. Global analysis of protein methylation at the proteome level requires the enrichment of methylated peptides with various forms; unfortunately, the immunoaffinity purification method can only enrich a subset of them due to lacking of pan specific antibody. Because methylation does not significantly alter the physicochemical properties of arginine or lysine residues, chemical approach for global methylome analysis is still at infancy. In this study, by exploiting the fact that the methylation on Arg and Lys prohibiting the cleavage by proteases for these sites, we developed an antibody-free method to enrich methylated peptides, which enabled the identification of 887 methylation forms on 768 sites from HepG2 cells. This technique allows the simultaneous analysis of both Lys and Arg methylation while it has better performance for the identification of Arg methylation. It should find broad applications in studying methylation regulated biological processes.
Co-reporter:Junfeng Huang, Hao Wan, Yating Yao, Jinan Li, Kai Cheng, Jiawei Mao, Jin Chen, Yan Wang, Hongqiang Qin, Weibing Zhang, Mingliang Ye, and Hanfa Zou
Analytical Chemistry 2015 Volume 87(Issue 20) pp:10199
Publication Date(Web):September 23, 2015
DOI:10.1021/acs.analchem.5b02669
Selective enrichment of glycopeptides from complex sample followed by cleavage of N-glycans by PNGase F to expose an easily detectable mark on the former glycosylation sites has become the popular protocol for comprehensive glycoproteome analysis. On account of the high enrichment specificity, hydrazide chemistry based solid-phase extraction of N-linked glycopeptides technique has sparked numerous interests. However, the enzymatic release of glycopeptides captured by hydrazide beads through direct incubation of the beads with PNGase F is not efficient due to the inherent steric hindrance effect. In this study, we developed a hydroxylamine assisted PNGase F deglycosylation (HAPD) method using the hydroxylamine to release glycopeptides captured on the hydrazide beads through the cleavage of hydrazone bonds by transamination followed with the PNGase F deglycosylation of the released glycopeptides in the free solution. Because of the homogeneous condition for the deglycosylation, the recovery of deglycosylated peptides (deglycopeptides) was improved significantly. It was found that 27% more N-glycosylation sites were identified by the HAPD strategy compared with the conventional method. Moreover, the ratio of identified N-terminal glycosylated peptides was improved over 5-fold.
Co-reporter:Yating Yao, Junfeng Huang, Kai Cheng, Yanbo Pan, Hongqiang Qin, Mingliang Ye, and Hanfa Zou
Analytical Chemistry 2015 Volume 87(Issue 22) pp:11353
Publication Date(Web):October 15, 2015
DOI:10.1021/acs.analchem.5b02711
A problem for “shot-gun” proteomics is that the peptides generated in the proteolysis step overwhelm the analytical capacity of current LC–MS/MS systems. A straightforward approach to overcome this problem is to reduce the sample complexity by isolating the representative peptides of each protein. In this study, we presented a facile solid-phase capture-release approach to selectively enrich the peptides with N-terminal serine/threonine from protein digests. This method exploited the highly efficient reaction between an aldehyde group and a hydrazine group. The excellent performance of this approach was validated using synthetic peptides as well as complex protein digests. It was found that high enrichment specificity could be obtained and the identifications for complex samples with and without enrichment were highly complementary. Besides, the enrichment of peptides with serine/threonine adjacent to different protease cleavage sites demonstrated that our method was able to enrich peptides from protein digests in a sequence specific way. As a result, this new approach provides a simple way to reduce sample complexity and facilitates the identification of low-abundance proteins.
Co-reporter:Zhang Zhang, Deguang Sun, Yuting Cong, Jiawei Mao, Junfeng Huang, Hongqiang Qin, Jing Liu, Guang Huang, Liming Wang, Mingliang Ye, and Hanfa Zou
Journal of Proteome Research 2015 Volume 14(Issue 9) pp:3892-3899
Publication Date(Web):August 10, 2015
DOI:10.1021/acs.jproteome.5b00306
An amine chemistry method was developed for the extraction of N-glycopeptides using amine-functionalized beads for glycoproteomics analysis. Two reductive amination reactions between primary amine and aldehyde were employed in this approach. The first one was to block the primary amines in the peptides by addition of formaldehyde and sodium cyanoborohydride into the peptide sample, and the second one was to couple the glycopeptides onto solid phase beads by incubating the glycopeptides containing aldehyde groups (oxidized by periodate) with the amine-functionalized beads in the presence of sodium cyanoborohydride. It was demonstrated that the blocking of primary amines in the peptides by the first reductive amination reaction prior to the periodate oxidation made the amine chemistry method very efficient and sensitive. This new method was validated by analysis of glycoprotein standards as well as proteome samples. It was found that this new method led to significant increase in the identification of N-glycosites compared with the conventional hydrazide chemistry method.
Co-reporter:Mingming Dong; Yangyang Bian; Jing Dong; Keyun Wang; Zheyi Liu; Hongqiang Qin; Mingliang Ye;Hanfa Zou
Journal of Proteome Research 2015 Volume 14(Issue 12) pp:5341-5347
Publication Date(Web):November 10, 2015
DOI:10.1021/acs.jproteome.5b00830
Among the natural amino acids, cysteine is unique since it can form a disulfide bond through oxidation and reduction of sulfhydryl and thus plays a pervasive role in modulation of proteins activities and structures. Crosstalk between phosphorylation and other post-translational modifications has become a recurrent theme in cell signaling regulation. However, the crosstalk between the phosphorylation and the formation and reductive cleavage of disulfide bond has not been investigated so far. To facilitate the study of this crosstalk, it is important to explore the subset of phosphoproteome where phosphorylations are occurred near to cysteine in the protein sequences. In this study, we developed a straightforward sequential enrichment method by combining the thiol affinity chromatography with the immobilized titanium ion affinity chromatography to selectively enrich cysteine-containing phosphopeptides. The high specificity and high sensitivity of this method were demonstrated by analyzing the samples of Jurkat cells. This “divide and conquer” strategy by specific analysis of a subphosphoproteome enables identification of more low abundant phosphosites than the conventional global phosphoproteome approach. Interestingly, amino acid residues surrounding the identified phosphosites were enriched with buried residues (L, V, A, C) while depleted with exposed residues (D, E, R, K). Also, the phosphosites identified by this approach showed a dramatic decrease in locating in disorder regions compared to that identified by conventional global phosphoproteome. Further analysis showed that more proline directed kinases and fewer acidophilic kinases were responsible for the phosphorylation sites of this subphosphoproteome.
Co-reporter:Fang-Jie LIU, Ming-Liang YE, Yan-Bo PAN, Han-Fa ZOU
Chinese Journal of Analytical Chemistry 2015 Volume 43(Issue 10) pp:1452-1458
Publication Date(Web):October 2015
DOI:10.1016/S1872-2040(15)60864-7
Polyacrylamide gel electrophoresis (PAGE) is a powerful protein separation technology. Combined with mass spectrometry, it could identify thousands of proteins in proteomics analysis. However, the time-consuming procedure restricts its broad applications in proteomics study. In this work, it was found that high concentration of trypsin did not compromise subsequent phosphopeptide enrichment after in-gel digestion, but could promote the in-gel digestion. Hence, a new, fast and robust digestion method was established. Firstly, 50 μg of HeLa cell protein was separated into 5 fractions by SDS-PAGE, and then the proteins were digested with high concentration trypsin for 30 min. Finally, the phosphopeptides were enriched by Ti-IMAC Tip. After liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis, about 2000 phosphorylation sites were identified in the experimental group, while less than 1500 phosphorylation sites were identified in control group. With the aid of high concentration of trypsin, in-gel digestion could be completed within only 30 min, and more phosphorylation site identifications and lower percentage of missed cleavages were acquired than those in control experiments. The experiment results demonstrated that high concentration of trypsin could not only accelerate the in-gel digestion, but also improve the phosphoproteome coverage.Figure A was the process of polyacrylamide gel electrophoresis. The proteins were separated by their molecular weights. B was the process of in gel digestion assisted by high concentration of trypsin. C were the acquired peptides extracted from the gel slices. D were the purified phosphopeptides through Ti-IMAC enrichment.
Co-reporter:Zhenzhen Deng, Mingliang Ye, Yangyang Bian, Zheyi Liu, Fangjie Liu, Chunli Wang, Hongqiang Qin and Hanfa Zou
Chemical Communications 2014 vol. 50(Issue 90) pp:13960-13962
Publication Date(Web):11 Sep 2014
DOI:10.1039/C4CC04906C
A simple, cost-effective and high throughput method was developed for multiplexed kinase activity assay based on the multiplex isotope labeling of designed substrate peptides. This strategy was successfully applied to monitor the time-dependent consumption of substrates and generation of products in the single and multiple substrate systems.
Co-reporter:Yanbo Pan, Mingliang Ye, Hao Zheng, Kai Cheng, Zhen Sun, Fangjie Liu, Jing Liu, Keyun Wang, Hongqiang Qin, and Hanfa Zou
Analytical Chemistry 2014 Volume 86(Issue 2) pp:1170
Publication Date(Web):December 19, 2013
DOI:10.1021/ac403060d
An enzymatic approach to label peptide N-termini with isotope-coded affinity tags is presented. This method exploits the high activity of trypsin for peptide synthesis in organic solvents. A cosubstrate containing a stable isotope-coded Arg residue and a biotin tag was synthesized. When the cosubstrate was incubated with tryptic peptides and trypsin in ethanol solution, the stable isotope-coded affinity tag was specifically coupled onto the N-termini of peptides via the formation of new peptide bonds. The labeled peptides were specifically enriched by avidin affinity chromatography and then were submitted to liquid chromatography–tandem mass spectrometry (LC/MS/MS) for quantification. This enrichment step effectively reduced the interference by unlabeled peptides. The excellent performance of this approach was demonstrated by labeling standard peptides as well as a mouse liver digest. In addition to one amino acid residue, a few dipeptide tags were also introduced to the N-termini of peptides successfully by this enzymatic approach. It was found that the identifications for samples labeled with these tags were highly complementary. Coupling a short sequence tag onto peptides could be an effective approach to improve the coverage for proteome analysis.
Co-reporter:Fangjie Liu, Mingliang Ye, Yanbo Pan, Yi Zhang, Yangyang Bian, Zhen Sun, Jun Zhu, Kai Cheng, and Hanfa Zou
Analytical Chemistry 2014 Volume 86(Issue 14) pp:6786
Publication Date(Web):June 23, 2014
DOI:10.1021/ac5002146
Conventional sample preparation protocols for phosphoproteome analysis require multiple time-consuming and labor-intensive steps, including cell lysis, protein extraction, protein digestion, and phosphopeptide enrichment. In this study, we found that the presence of a large amount of trypsin in the sample did not interfere with phosphopeptide enrichment and subsequent LC-MS/MS analysis. Taking advantage of fast digestion achieved with high trypsin-to-protein ratio, we developed a novel concurrent lysis-digestion method for phosphoproteome analysis. In this method, the harvested cells were first placed in a lysis buffer containing a huge amount of trypsin. After ultrasonication, the cells were lysed and the proteins were efficiently digested into peptides within one step. Thereafter, tryptic digest was subjected to phosphopeptide enrichment, in which unphosphorylated peptides, trypsin, and other components incompatible with LC-MS/MS analysis were removed. Compared with conventional methods, better phosphoproteome coverage was achieved in this new one-step method. Because protein solubilization and cell lysis were facilitated by fast protein digestion, the complete transformation of cell pellets into the peptide mixture could be finished within 25 min, while it would take at least 16 h for conventional methods. Hence, our method, which integrated cell lysis, protein extraction, and protein digestion into one step, is rapid and convenient. It is expected to have broad applications in phosphoproteomics analysis.
Co-reporter:Zhang Zhang, Zhen Sun, Jun Zhu, Jing Liu, Guang Huang, Mingliang Ye, and Hanfa Zou
Analytical Chemistry 2014 Volume 86(Issue 19) pp:9830
Publication Date(Web):September 10, 2014
DOI:10.1021/ac5024638
Sialylated glycoproteins, which play important roles in tumor progression, have been extensively analyzed for the discovery of potential biomarkers for cancer diagnosis and prognosis. The site-specific N-sialoglycan occupancy rates of glycoproteins reflect the activities of glycosyltransferases and glycosidases in vivo and could be novel disease biomarkers. However, a high-throughput method to determine the N-sialoglycan occupancy rates is not available. On the basis of the fact that dihydroxy of sialic acid of glycan chains in glycoproteins can be specifically oxidized to aldehyde in mild periodate concentration while all types of glycan chains can be oxidized in high periodate concentration, we developed a modified protein-level hydrazide chemistry method for the determination of the N-sialoglycan occupancy rates. This method was first applied to determine the N-sialoglycan occupancy rates of two glycosites on human transferrin. These two sites were found to be fully sialylated and the N-sialoglycan occupancy rates were found to under significant decrease after the neuraminidase treatment. This method was then applied to analyze N-sialoglycan occupancy rates in proteome samples. We determined 496 and 632 site-specific N-sialoglycan occupancy rates on 334 and 394 proteins from hepatocellular carcinoma (HCC) and normal human liver tissues, respectively. By comparing the N-sialoglycan occupancy rates between the above two samples, we determined 76 N-sialoglycosites with more than a 2-fold change. This method was demonstrated to be an effective and high-throughput method for the analysis of the N-sialoglycan occupancy rates.
Co-reporter:Junfeng Huang, Fangjun Wang, Mingliang Ye, Hanfa Zou
Journal of Chromatography A 2014 Volume 1372() pp:1-17
Publication Date(Web):12 December 2014
DOI:10.1016/j.chroma.2014.10.107
•Major protein post-translational modifications (PTMs) and their enrichment strategies.•The challenges encountered in the current PTMs enrichment methods.•Separation strategies of PTM peptides for large scale PTMs analysis.Comprehensive analysis of the post-translational modifications (PTMs) on proteins at proteome level is crucial to elucidate the regulatory mechanisms of various biological processes. In the past decades, thanks to the development of specific PTM enrichment techniques and efficient multidimensional liquid chromatography (LC) separation strategy, the identification of protein PTMs have made tremendous progress. A huge number of modification sites for some major protein PTMs have been identified by proteomics analysis. In this review, we first introduced the recent progresses of PTM enrichment methods for the analysis of several major PTMs including phosphorylation, glycosylation, ubiquitination, acetylation, methylation, and oxidation/reduction status. We then briefly summarized the challenges for PTM enrichment. Finally, we introduced the fractionation and separation techniques for efficient separation of PTM peptides in large-scale PTM analysis.
Co-reporter:Yangyang Bian, Chunxia Song, Kai Cheng, Mingming Dong, Fangjun Wang, Junfeng Huang, Deguang Sun, Liming Wang, Mingliang Ye, Hanfa Zou
Journal of Proteomics 2014 Volume 96() pp:253-262
Publication Date(Web):16 January 2014
DOI:10.1016/j.jprot.2013.11.014
•An enzyme assisted RP-RPLC approach was developed with high orthogonality.•Identifications from TripleTOF 5600 and Orbitrap Velos were highly complementary.•22446 sites corresponding to 6526 phosphoproteins were identified from human liver.•The largest phosphoproteome dataset of human liver were constructed.•Many novel phosphorylation sites in the metabolic enzymes were identified.Protein phosphorylation is one of the most common post-translational modifications. It plays key roles in regulating diverse biological processes of liver tissues. To better understand the role of protein phosphorylation in liver functions, it is essential to perform in-depth phosphoproteome analysis of human liver. Here, an enzyme assisted reversed-phase-reversed-phase liquid chromatography (RP-RPLC) approach with both RPLC separations operated with optimized acidic mobile phase was developed. High orthogonal separation was achieved by trypsin digestion of the Glu-C generated peptides in the fractions collected from the first RPLC separation. The phosphoproteome coverage was further improved by using two types of instruments, i.e. TripleTOF 5600 and LTQ Orbitrap Velos. A total of 22,446 phosphorylation sites, corresponding to 6526 nonredundant phosphoproteins were finally identified from normal human liver tissues. Of these sites, 15,229 sites were confidently localized with Ascore ≥ 13. This dataset was the largest phosphoproteome dataset of human liver. It can be a public resource for the liver research community and holds promise for further biology studies.Biological significanceThe enzyme assisted approach enabled the two RPLC separations operated both with optimized acidic mobile phases. The identifications from TripleTOF 5600 and Orbitrap Velos are highly complementary. The largest phosphoproteome dataset of human liver was generated.
Co-reporter:Chunxia Song, Fangjun Wang, Kai Cheng, Xiaoluan Wei, Yangyang Bian, Keyun Wang, Yexiong Tan, Hongyang Wang, Mingliang Ye, and Hanfa Zou
Journal of Proteome Research 2014 Volume 13(Issue 1) pp:241-248
Publication Date(Web):2017-2-22
DOI:10.1021/pr400544j
Global quantification of the single amino-acid variations (SAAVs) is essential to investigate the roles of SAAVs in disease progression. However, few efforts have been made on this issue due to the lack of high -throughput approach. Here we presented a strategy by integration of the stable isotope dimethyl labeling with variation-associated database search to globally quantify the SAAVs at the first time. A protein database containing 87 745 amino acid variant sequences and 73 910 UniProtKB/Swiss-Prot canonical protein entries was constructed for database search, and higher energy collisional dissociation combined with collision-induced dissociation fragmentation modes were applied to improve the quantification coverage of SAAVs. Compared with target proteomics in which only a few sites could be quantified, as many as 282 unique SAAVs sites were quantified between hepatocellular carcinoma (HCC) and normal human liver tissues by our strategy. The variation rates in different samples were evaluated, and some interesting SAAVs with significant increase normalized quantification ratios, such as T1406N in CPS1 and S197R in HTATIP2, were observed to highly associate with HCC progression. Therefore, the newly developed strategy enables the large-scale comparative analysis of variations at the protein level and holds a promising future in the research related to variations.
Co-reporter:Yanbo Pan;Kai Cheng;Jiawei Mao;Fangjie Liu
Analytical and Bioanalytical Chemistry 2014 Volume 406( Issue 25) pp:6247-6256
Publication Date(Web):2014 October
DOI:10.1007/s00216-014-8071-6
Trypsin is the popular protease to digest proteins into peptides in shotgun proteomics, but few studies have attempted to systematically investigate the kinetics of trypsin-catalyzed protein digestion in proteome samples. In this study, we applied quantitative proteomics via triplex stable isotope dimethyl labeling to investigate the kinetics of trypsin-catalyzed cleavage. It was found that trypsin cleaves the C-terminal to lysine (K) and arginine (R) residues with higher rates for R. And the cleavage sites surrounded by neutral residues could be quickly cut, while those with neighboring charged residues (D/E/K/R) or proline residue (P) could be slowly cut. In a proteome sample, a huge number of proteins with different physical chemical properties coexists. If any type of protein could be preferably digested, then limited digestion could be applied to reduce the sample complexity. However, we found that protein abundance and other physicochemical properties, such as molecular weight (Mw), grand average of hydropathicity (GRAVY), aliphatic index, and isoelectric point (pI) have no notable correlation with digestion priority of proteins.
Co-reporter:Zhen Sun;Deguang Sun;Fangjun Wang;Kai Cheng;Zhang Zhang;Bo Xu
Clinical Proteomics 2014 Volume 11( Issue 1) pp:
Publication Date(Web):2014 December
DOI:10.1186/1559-0275-11-26
Dysregulation of glycoproteins is closely related with many diseases. Quantitative proteomics methods are powerful tools for the detection of glycoprotein alterations. However, in almost all quantitative glycoproteomics studies, trypsin is used as the only protease to digest proteins. This conventional method is unable to quantify N-glycosites in very short or long tryptic peptides and so comprehensive glycoproteomics analysis cannot be achieved.In this study, a comprehensive analysis of the difference of N-glycoproteome between hepatocellular carcinoma (HCC) and normal human liver tissues was performed by an integrated workflow combining the multiple protease digestion and solid phase based labeling. The quantified N-glycoproteins were analyzed by GoMiner to obtain a comparative view of cellular component, biological process and molecular function.An integrated workflow was developed which enabled the processes of glycoprotein coupling, protease digestion and stable isotope labeling to be performed in one reaction vessel. This workflow was firstly evaluated by analyzing two aliquots of the same protein extract from normal human liver tissue. It was demonstrated that the multiple protease digestion improved the glycoproteome coverage and the quantification accuracy. This workflow was further applied to the differential analysis of N-glycoproteome of normal human liver tissue and that with hepatocellular carcinoma. A total of 2,329 N-glycosites on 1,052 N-glycoproteins were quantified. Among them, 858 N-glycosites were quantified from more than one digestion strategy with over 99% confidence and 1,104 N-glycosites were quantified from only one digestion strategy with over 95% confidence. By comparing the GoMiner results of the N-glycoproteins with and without significant changes, the percentage of membrane and secreted proteins and their featured biological processes were found to be significant different revealing that protein glycosylation may play the vital role in the development of HCC.
Co-reporter:Fangjie Liu, Mingliang Ye, Chunli Wang, Zhengyan Hu, Yi Zhang, Hongqiang Qin, Kai Cheng, and Hanfa Zou
Analytical Chemistry 2013 Volume 85(Issue 15) pp:7024
Publication Date(Web):July 16, 2013
DOI:10.1021/ac4017693
Trypsin was immobilized on a variety of materials to improve digestion efficiency. However, because the immobilized trypsin will digest proteins during electrophoresis, direct immobilization of active trypsin in polyacrylamide gel will compromise the protein separation. To overcome this problem, here we report a novel polyacrylamide gel with switchable trypsin activity. It was prepared by copolymerization of the PEG–trypsin–aprotinin complex during the gel-casting step. Because the inhibitor aprotinin binds strongly with trypsin at alkaline pH, this novel gel does not display hydrolytic activity during electrophoresis. After electrophoresis, the activity of trypsin embedded in gel could be recovered by simply washing away the bound inhibitor at a low pH. It was demonstrated that this unique switchable activity design allowed high resolution of the complex protein mixture during electrophoresis and highly efficient digestion of the separated proteins in situ in the gel after electrophoresis.
Co-reporter:Jun Zhu, Fangjun Wang, Kai Cheng, Chunxia Song, Hongqiang Qin, Lianghai Hu, Daniel Figeys, Mingliang Ye, Hanfa Zou
Journal of Proteomics 2013 Volume 78() pp:389-397
Publication Date(Web):14 January 2013
DOI:10.1016/j.jprot.2012.10.006
As human serum is an important source for early diagnosis of many serious diseases, analysis of serum proteome and peptidome has been extensively performed. However, the serum phosphopeptidome was less explored probably because the effective method for database searching is lacking. Conventional database searching strategy always uses the whole proteome database, which is very time-consuming for phosphopeptidome search due to the huge searching space resulted from the high redundancy of the database and the setting of dynamic modifications during searching. In this work, a focused database searching strategy using an in-house collected human serum pro-peptidome target/decoy database (HuSPep) was established. It was found that the searching time was significantly decreased without compromising the identification sensitivity. By combining size-selective Ti (IV)-MCM-41 enrichment, RP–RP off-line separation, and complementary CID and ETD fragmentation with the new searching strategy, 143 unique endogenous phosphopeptides and 133 phosphorylation sites (109 novel sites) were identified from human serum with high reliability.Highlights► Searching space was greatly reduced by a focused database searching strategy. ► Human serum phosphopeptidome was extensively analyzed. ► Largest number of serum endogenous phosphopeptides were confidently identified.
Co-reporter:Chunli Wang, Mingliang Ye, Yangyang Bian, Fangjie Liu, Kai Cheng, Mingming Dong, Jing Dong, and Hanfa Zou
Journal of Proteome Research 2013 Volume 12(Issue 8) pp:3813-3821
Publication Date(Web):2017-2-22
DOI:10.1021/pr4002965
Understanding the specificity of kinases enables prediction of their substrates and uncovering kinase functions in signaling pathways. Traditionally synthesized peptide libraries are used to determine the kinase specificity. In this study, a proteomics-based method was developed to determine the specificity of kinase by taking the advantages of proteome-derived peptide libraries and quantitative proteomics. Proteome-derived peptide libraries were constructed by digesting proteins in total cell lysate followed with dephosphorylation of the resulting peptides. After incubating the peptide libraries with/without CK2 for in vitro kinase assay, stable isotopic labeling based quantitative phosphoproteomics was applied to distinguish the in vitro phosphosites generated by CK2. By using the above approach, 404 CK2 in vitro phosphosites were identified by 1D LC–MS/MS. Those sites allowed the statistic determination of the CK2 specificity. In addition to the easy construction of the proteome-derived peptide library, another significant advantage of this method over the method with synthesized peptide libraries is that the identified phosphosites could be directly mapped to proteins for the screening of putative kinase substrates. It was found that the confidence for substrate identification could be significantly improved by comparing the in vitro CK2 sites with the in vivo sites identified by phosphoproteomics analysis of the same cell lines. By applying this integrated strategy, 138 phosphosites from 105 putative CK2 substrates of high confidence were determined.
Co-reporter:Yanbo Pan;Dr. Mingliang Ye;Dr. Liang Zhao;Kai Cheng;Mingming Dong;Chunxia Song;Hongqiang Qin;Dr. Fangjun Wang;Dr. Hanfa Zou
Angewandte Chemie International Edition 2013 Volume 52( Issue 35) pp:9205-9209
Publication Date(Web):
DOI:10.1002/anie.201303429
Co-reporter:Yanbo Pan;Dr. Mingliang Ye;Dr. Liang Zhao;Kai Cheng;Mingming Dong;Chunxia Song;Hongqiang Qin;Dr. Fangjun Wang;Dr. Hanfa Zou
Angewandte Chemie 2013 Volume 125( Issue 35) pp:9375-9379
Publication Date(Web):
DOI:10.1002/ange.201303429
Co-reporter:Jun Zhu, Fangjun Wang, Rui Chen, Kai Cheng, Bo Xu, Zhimou Guo, Xinmiao Liang, Mingliang Ye, and Hanfa Zou
Analytical Chemistry 2012 Volume 84(Issue 11) pp:5146
Publication Date(Web):May 9, 2012
DOI:10.1021/ac3000732
Sample handling procedures including protein digestion, glycopeptide enrichment, and deglycosylation have significant impact on the performance of glycoproteome analysis. Several glycoproteomic analysis systems were developed to integrate some of these sample preparation procedures. However, no microsystem integrates all of above three procedures together. In this work, we developed a glycoproteomic microreactor enabling seamless integration of all these procedures. In this reactor, trypsin digestion was accelerated by adding acetonitrile to 80%, and after acidification of protein digest by trifluoroacetic acid (TFA), the following hydrophilic interaction chromatography (HILIC) enrichment and deglycosylation were sequentially performed without any desalting, lyophilization, or buffer exchange steps. The total processing time could be as short as 1.5 h. The detection limit of human IgG as low as 30 fmol was also achieved. When applied to human serum glycoproteome analysis, a total number of 92, 178, and 221 unique N-glycosylation sites were identified from three replicate analyses of 10 nL, 100 nL, and 1 μL of human serum, respectively. It was demonstrated that the glycoproteomic microreactor based method had very high sensitivity and was well suited for glycoproteome analysis of minute protein samples.
Co-reporter:Yangyang Bian, Mingliang Ye, Chunxia Song, Kai Cheng, Chunli Wang, Xiaoluan Wei, Jun Zhu, Rui Chen, Fangjun Wang, and Hanfa Zou
Journal of Proteome Research 2012 Volume 11(Issue 5) pp:2828-2837
Publication Date(Web):2017-2-22
DOI:10.1021/pr300242w
Complete coverage of all phosphorylation sites in a proteome is the ultimate goal for large-scale phosphoproteome analysis. However, only making use of one protease trypsin for protein digestion cannot cover all phosphorylation sites, because not all tryptic phosphopeptides are detectable in MS. To further increase the phosphoproteomics coverage of HeLa cells, we proposed a tandem digestion approach by using two different proteases. By combining the data set of the first Glu-C digestion and the second trypsin digestion, the tandem digestion approach resulted in the identification of 8062 unique phosphopeptides and 8507 phosphorylation sites in HeLa cells. The conventional trypsin digestion approach resulted in the identification of 3891 unique phosphopeptides and 4647 phosphorylation sites. It was found that the phosphorylation sites identified from the above two approaches were highly complementary. By combining above two data sets, in total we identified 10899 unique phosphopeptides and 11262 phosphorylation sites, corresponding to 3437 unique phosphoproteins with FDR < 1% at peptide level. We also compared the kinase motifs extracted from trypsin, Glu-C, or a second trypsin digestion data sets. It was observed that basophilic motifs were more frequently found in the trypsin and the second trypsin digestion data sets, and the acidic motifs were more frequently found in the Glu-C digestion data set. These results demonstrated that our tandem digestion approach is a good complement to the conventional trypsin digestion approach for improving the phosphoproteomics analysis coverage of HeLa cells.
Co-reporter:Mingming Dong, Mingliang Ye, Kai Cheng, Chunxia Song, Yanbo Pan, Chunli Wang, Yangyang Bian, and Hanfa Zou
Journal of Proteome Research 2012 Volume 11(Issue 9) pp:4673-4681
Publication Date(Web):2017-2-22
DOI:10.1021/pr300503z
The Ser/Thr protein kinases fall into three major subgroups, pro-directed, basophilic, and acidophilic, on the basis of the types of substrate sequences that they preferred. Despite many phosphoproteomics efforts that have been taken for global profiling of phosphopeptides, methodologies focusing on analyzing a particular type of kinase substrates have seldom been reported. Selective enrichment of phosphopeptides from basophilic kinase substrates is difficult because basophilic motifs are cleaved by trypsin during digestion. In this study, we develop a negative enrichment strategy to enhance the identification of basophilic kinase substrates. This method is based on an observation that high pH strong anion exchange (SAX) chromatography can separate tryptic phosphopeptides according to the number of acidic amino acidic residues that they have. Thus, SAX was applied to deplete acidic phosphopeptides from the phosphopeptide mixture, which improved the coverage for the detection of basophilic kinase substrates. The SAX depletion approach was further combined with online SCX-RP separation for large-scale analysis of mouse liver phosphoproteome, which resulted in the identification of 6944 phosphorylated sites. It was found that motifs associated with basophilic kinases prevail for these identified phosphorylated sites.
Co-reporter:Chunxia Song, Mingliang Ye, Guanghui Han, Xinning Jiang, Fangjun Wang, Zhiyuan Yu, Rui Chen and Hanfa Zou
Analytical Chemistry 2010 Volume 82(Issue 1) pp:53
Publication Date(Web):December 1, 2009
DOI:10.1021/ac9023044
Protein phosphorylation regulates a series of important biological processes in eukaryotes. However, the phosphorylation sites found up to now are far below than that actually exists in proteins due to the extreme complexity of the proteome sample. Here a new reversed-phase-reversed-phase liquid chromatography (RP-RPLC) approach was developed for multidimensional separation of phosphopeptides. In this approach, a large number of fractions were collected from the first dimensional RPLC separation at high pH. And then these fractions were pooled every two fractions with equal time interval, one from the early eluted section and another one from the later eluted section. The pooled fractions were finally submitted to RPLC−tandem mass spectrometry (MS/MS) analysis at low pH. It was found the resulting 2D separation was highly orthogonal and yielded more than 30% phosphopeptide identifications over the conventional RP-RPLC approach. This study provides a powerful approach for efficient separation of phosphopeptides and global phosphorylation analysis, where the orthogonality of 2D separation is greatly improved and the first dimensional separation is of high resolution.
Co-reporter:Mingming Dong, Minghuo Wu, Fangjun Wang, Hongqiang Qin, Guanghui Han, Jing Dong, Ren’an Wu, Mingliang Ye, Zhen Liu and Hanfa Zou
Analytical Chemistry 2010 Volume 82(Issue 7) pp:2907
Publication Date(Web):March 3, 2010
DOI:10.1021/ac902907w
Protein phosphorylation is one of the most biologically relevant and ubiquitous post-translational modifications. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) is a powerful tool for the analysis of protein phosphorylation by detection of phosphopeptides in phosphoprotein digest. Enrichment of phosphopeptides by immobilized metal ion affinity chromatography (IMAC) or metal oxide affinity chromatography (MOAC) followed with MALDI analysis is the common approach. However, the pH for loading and elution of phosphopeptides is incompatible with protein digestion as well as the preparation of the MALDI matrix solution. Therefore, some pretreatment steps, such as pH adjustment and desalting, are required, which make the approach tedious and insensitive. In this study, a strong anion-exchange (SAX) capillary monolith was prepared to enrich phosphopeptides from protein digest for MALDI-TOF MS analysis. It was found that phosphopeptides could be specifically retained on the SAX column at high pH around 8 and could be eluted by 5% formic acid. Thus, the protein digests without any pretreatment could be loaded onto the SAX column under basic pH condition; after removing nonphosphopeptides by washing, the bound phosphopeptides could be eluted directly onto a MALDI target and analyzed by MALDI-TOF MS. This approach significantly simplified the analytical procedures and reduced the sample loss. Because of the excellent MALDI MS compatible procedure and the microscale SAX column, a detection limit as low as 50 amol for the analysis of phosphopeptides from β-casein digest was achieved. To circumvent the inconvenience of the sample loading, a new simple sample introducing method based on capillary action was proposed, which further reduced the detection limit to 10 amol.
Co-reporter:Xinning Jiang, Mingliang Ye, Guanghui Han, Xiaoli Dong and Hanfa Zou
Analytical Chemistry 2010 Volume 82(Issue 14) pp:6168
Publication Date(Web):June 22, 2010
DOI:10.1021/ac100975t
Data dependent neutral loss triggered MS3 methodology (NLMS3) is often applied to acquire MS data for the analysis of phosphopeptides. Some phosphopeptides tend to seriously lose the phosphate and result in MS2 spectra with poor fragments and fragment-rich MS3 spectra, while some phosphopeptides do not lose phosphate and result in nice MS2 spectra. Since different phosphopeptides have fragment spectra with different characteristics, filtering all of the phosphopeptide identifications by setting a global filter criteria may be inappropriate and result in low sensitivity. In this study, we developed a classification filtering strategy to improve the phosphopeptide identification and phosphorylation site localization. Phosphopeptide identifications were classified into four classes according to their different characteristics, and then, the identifications from each class of mass spectra were processed and filtered separately using different filtering strategies. It was found that the overlap of phosphopeptide identifications from different classes was low and the classification strategy significantly improved the coverage of the phosphoproteome analysis. Compared with MS2 strategy and multiple stage activation (MSA) strategy, NLMS3 with the classification filtering strategy was demonstrated to have higher sensitivity and higher performance in localizing the phosphorylation to specific sites.
Co-reporter:Guanghui Han, Mingliang Ye, Xinning Jiang, Rui Chen, Jian Ren, Yu Xue, Fangjun Wang, Chunxia Song, Xuebiao Yao and Hanfa Zou
Analytical Chemistry 2009 Volume 81(Issue 14) pp:5794
Publication Date(Web):June 12, 2009
DOI:10.1021/ac900702g
Since the emergence of proteomics, much attention has been paid to the development of new technologies for phosphoproteomcis analysis. Compared with large scale phosphorylation analysis at the proteome level, comprehensive and reliable phosphorylation site mapping of individual phosphoprotein is equally important. Here, we present a modified target-decoy database search strategy for confident phosphorylation site analysis of individual phosphoproteins without manual interpretation of spectra. Instead of using all protein sequences in a proteome database of an organism for the construction of a target-decoy database for phosphoproteome analysis, the composite database constructed for phosphorylation site analysis of individual phosphoproteins only included the sequences of the individual target proteins and a decoy version of a small inhomogeneous protein database. It was found that the confidence of phosphopeptide identifications could be effectively controlled when the acquired MS2 and MS3 spectra were searched against the above composite database followed with data processing. Because of the small size of the composite database, the computation time for the database search is very short, which allows the adoption of low-specificity proteases for protein digestion to increase the coverage of phosphorylation site mapping. The sensitivity and comprehensive phosphorylation site mapping of this approach was demonstrated by using two standard phosphoprotein samples of α-casein and β-casein, and this approach was further applied to analyze the phosphorylation of the cyclic AMP-dependent protein kinase (PKA), which resulted in the identification of 17 phosphorylation sites, including five novel sites on four PKA subunits.
Co-reporter:Zhiyuan Yu, Guanghui Han, Shutao Sun, Xinning Jiang, Rui Chen, Fangjun Wang, Ren’an Wu, Mingliang Ye, Hanfa Zou
Analytica Chimica Acta 2009 Volume 636(Issue 1) pp:34-41
Publication Date(Web):16 March 2009
DOI:10.1016/j.aca.2009.01.033
This study presented an approach to prepare monodisperse immobilized Ti4+ affinity chromatography (Ti4+-IMAC) microspheres for specific enrichment of phosphopeptides in phosphoproteome analysis. Monodisperse polystyrene seed microspheres with a diameter of ca. 4.8 μm were first prepared by a dispersion polymerization method. Monodisperse microspheres with a diameter of ca. 13 μm were prepared using the seed microspheres by a single-step swelling and polymerization method. Ti4+ ion was immobilized after chemical modification of the microspheres with phosphonate groups. The specificity of the Ti4+-IMAC microspheres to phosphopeptides was demonstrated by selective enrichment of phosphopeptides from mixture of tryptic digests of α-casein and bovine serum albumin (BSA) at molar ratio of 1 to 500 by MALDI-TOF MS analysis. The sensitivity of detection for phosphopeptides determined by MALDI-TOF MS was as low as 5 fmol for standard tryptic digest of β-casein. The Ti4+-IMAC microspheres were compared with commercial Fe3+-IMAC adsorbent and homemade Zr4+-IMAC microspheres for enrichment of phosphopeptides. The phosphopeptides and non-phosphopeptides identified by Fe3+-IMAC, Zr4+-IMAC and Ti4+-IMAC methods were 26, 114, 127 and 181, 11, 11 respectively for the same tryptic digest samples. The results indicated that the Ti4+-IMAC had the best performance for enrichment of phosphopeptides.
Co-reporter:Xinning Jiang, Xiaoli Dong, Mingliang Ye and Hanfa Zou
Analytical Chemistry 2008 Volume 80(Issue 23) pp:9326
Publication Date(Web):November 4, 2008
DOI:10.1021/ac8017229
The target−decoy database search strategy is often applied to determine the global false-discovery rate (FDR) of peptide identifications in proteome research. However, the confidence of individual peptide identification is typically not determined. In this study, we introduced an approach for the calculation of posterior probability of individual peptide identification from the “local false-discovery rate” (local FDR), which is also determined based on a target−decoy database search. The peptide identification scores output by the database search algorithm were weighted by their discriminating power using a Shannon information entropy based strategy. Then the local FDR of a peptide identification was calculated based on the fraction of decoy identifications among its nearest neighbors within a small space defined by these weighted scores. It was demonstrated that the calculated probability matched the actual probability precisely, and it provided powerful discriminating performance between true positive and false positive identifications. Hence, the sensitivity for peptide identification as well as protein identification was significantly improved when the calculated probability was used to process different proteome data sets. As an instance based strategy, this algorithm provides a safe way for the posterior probability calculation and should work well for data sets with different characteristics.
Co-reporter:Guanghui Han, Mingliang Ye and Hanfa Zou
Analyst 2008 vol. 133(Issue 9) pp:1128-1138
Publication Date(Web):01 Aug 2008
DOI:10.1039/B806775A
Protein phosphorylation is one of the most biologically relevant and ubiquitous post-translational modifications. The analysis of protein phosphorylation is very challenging due to its highly dynamic nature and low stoichiometry. In this article, recent techniques developed for phosphoproteome analysis are reviewed with an emphasis on the new developments in this field in China. To improve the performance of phosphoproteome analysis, many novel methods, either by application of new separation mechanisms or by adoption of new separation materials, were developed to specifically enrich phosphopeptides from complex protein digests. A series of new materials, including nanostructure materials, magnetic materials, and monolithic materials, were applied to prepare immobilized affinity chromatography or metal oxide affinity chromatography to improve the performance of phosphopeptide enrichment. Besides, new software tools were also developed to validate phosphopeptide identification and predict kinase specific phosphorylation sites.
Co-reporter:Xinning Jiang, Guanghui Han, Shun Feng, Xiaogang Jiang, Mingliang Ye, Xuebiao Yao and Hanfa Zou
Journal of Proteome Research 2008 Volume 7(Issue 4) pp:1640-1649
Publication Date(Web):2017-2-22
DOI:10.1021/pr700675j
Manual checking is commonly employed to validate the phosphopeptide identifications from database searching of tandem mass spectra. It is very time-consuming and labor intensive as the number of phosphopeptide identifications increases greatly. In this study, a simple automatic validation approach was developed for phosphopeptide identification by combining consecutive stage mass spectrometry data and the target-decoy database searching strategy. Only phosphopeptides identified from both MS2 and its corresponding MS3 were accepted for further filtering, which greatly improved the reliability in phosphopeptide identification. Before database searching, the spectra were validated for charge state and neutral loss peak intensity, and then the invalid MS2/MS3 spectra were removed, which greatly reduced the database searching time. It was found that the sensitivity was significantly improved in MS2/MS3 strategy as the number of identified phosphopeptides was 2.5 times that obtained by the conventional filter-based MS2 approach. Because of the use of the target-decoy database, the false-discovery rate (FDR) of the identified phosphopeptides could be easily determined, and it was demonstrated that the determined FDR can precisely reflect the actual FDR without any manual validation stage.
Co-reporter:Yang Yu, Yan Jin, Fangjun Wang, Jiaze Yan, Yanxia Qi, Mingliang Ye
Food Research International (June 2017) Volume 96() pp:182-190
Publication Date(Web):1 June 2017
DOI:10.1016/j.foodres.2017.04.002
•Protein digestomics of antler velvet was investigated by LC-MS/MS and bioactivity.•Antler velvet digests presented DPP-IV and PEP inhibitory activities.•The released bioactive peptides contributed to the bioactivity of antler velvet.Proteins are the most prominent bioactive component in deer antler velvet. The aim of the present study was to track the fate of protein of antler velvet by protein digestomics. The peptide profile identified by LC-MS/MS and the in vitro bioactivity of antler velvet aqueous extract (AAE) were investigated in simulated gastrointestinal digestion. A total of 23, 387 and 417 peptides in AAE, gastric and pancreatic digests were identified using LC-MS/MS, respectively. Collagens, the predominant proteins, released 34 peptides in gastric digests and 146 peptides in pancreatic digests. The gastric and pancreatic digests presented dipeptidyl peptidase IV (DPP-IV) and prolyl endopeptidase (PEP) inhibition activities. Four peptides from digests were proved to be DPP-IV and PEP inhibitory peptides. The results showed that the peptides released from antler velvet protein contributed to the bioactivity of antler velvet during digestion.Download high-res image (101KB)Download full-size image
Co-reporter:Zhenzhen Deng, Mingliang Ye, Yangyang Bian, Zheyi Liu, Fangjie Liu, Chunli Wang, Hongqiang Qin and Hanfa Zou
Chemical Communications 2014 - vol. 50(Issue 90) pp:NaN13962-13962
Publication Date(Web):2014/09/11
DOI:10.1039/C4CC04906C
A simple, cost-effective and high throughput method was developed for multiplexed kinase activity assay based on the multiplex isotope labeling of designed substrate peptides. This strategy was successfully applied to monitor the time-dependent consumption of substrates and generation of products in the single and multiple substrate systems.
.jpg)