Co-reporter:María C. Gélvez-Rueda, Eline M. Hutter, Duyen H. Cao, Nicolas Renaud, Constantinos C. Stoumpos, Joseph T. Hupp, Tom J. Savenije, Mercouri G. Kanatzidis, and Ferdinand C. Grozema
The Journal of Physical Chemistry C November 30, 2017 Volume 121(Issue 47) pp:26566-26566
Publication Date(Web):November 3, 2017
DOI:10.1021/acs.jpcc.7b10705
The optoelectronic properties of hybrid perovskites can be easily tailored by varying their components. Specifically, mixing the common short organic cation (methylammonium (MA)) with a larger one (e.g., butyl ammonium (BA)) results in 2-dimensional perovskites with varying thicknesses of inorganic layers separated by the large organic cation. In both of these applications, a detailed understanding of the dissociation and recombination of electron–hole pairs is of prime importance. In this work, we give a clear experimental demonstration of the interconversion between bound excitons and free charges as a function of temperature by combining microwave conductivity techniques with photoluminescence measurements. We demonstrate that the exciton binding energy varies strongly (between 80 and 370 meV) with the thickness of the inorganic layers. Additionally, we show that the mobility of charges increases with the layer thickness, in agreement with calculated effective masses from electronic structure calculations.
Co-reporter:Damla Inan, Rajeev K. Dubey, Nick Westerveld, Jorrit Bleeker, Wolter F. Jager, and Ferdinand C. Grozema
The Journal of Physical Chemistry A June 22, 2017 Volume 121(Issue 24) pp:4633-4633
Publication Date(Web):May 30, 2017
DOI:10.1021/acs.jpca.7b03806
We report here the synthesis and photophysical study of a series of electron donor–acceptor molecules, in which electron-donating 4-methoxyphenoxy groups are attached to the 1,7-bay positions of four different perylene tetracarboxylic acid derivatives, namely, perylene tetraesters 1, perylene monoimide diesters 2, perylene bisimides 3, and perylene monobenzimidazole monoimides 4. These perylene derivatives are used because of their increasing order of electron-accepting capability upon moving from 1 to 4. Two additional donor–acceptor molecules are synthesized by linking electron-donating 4-methoxyphenyl groups to the imide position of perylene monoimide diester 2 and perylene bisimide 3. The motivation for this study is to achieve a good control over the photoinduced charge-transfer (CT) process in perylene-based systems by altering the position of electron donors and tuning the electron deficiency of perylene core. A comprehensive study of the photophysical properties of these molecules has shown a highly systematic trend in the magnitude of CT as a function of increased electron deficiency of the perylene core and solvent polarity. Importantly, just by changing the attachment of electron-donating group from “bay” to “imide” position, we are able to block the CT process. This implies that the positioning of the electron donor at the perylene core strongly influences the kinetics of the photoinduced CT process. In these compounds, the CT process is characterized by the quenching of fluorescence and singlet excited-state lifetimes as compared to model compounds bearing non-electron-donating 4-tert-butylphenoxy groups. Transient absorption spectroscopy did not reveal spectra of CT states. This most likely implies that the CT state is not accumulated, because of the faster charge recombination.
Co-reporter:Simge Tarkuç, Rienk Eelkema, Ferdinand C. Grozema
Tetrahedron 2017 Volume 73, Issue 33(Issue 33) pp:
Publication Date(Web):17 August 2017
DOI:10.1016/j.tet.2017.04.037
In this contribution we describe a combined experimental and theoretical study of the relation between the molecular structure and the electronic properties of conjugated donor-acceptor type chromophores for light-harvesting applications. A series of model systems was synthesized where a central anthracene (electron donor) is connected to dicyanovinyl units (electron acceptor) through a π-conjugated spacer. The study of the redox and optical properties of these chromophores and of reference compounds without dicyanovinyl units allows us correlate the electronic properties to the presence of the electron withdrawing groups and the molecular conformation. Comparison with calculated electronic structure shows that the construction of chromophores that consist of electron donating and accepting units does not always follow the simple rules that are generally used in the design of such molecules. The results show a subtle relation between the charge transfer character and the geometry of the molecules. In some cases this leads to significant contribution of charge transfer excitation to the absorption spectra of some chromophores while such contributions are completely absent in others.A systematic study of the relation between the molecular structure and the electronic properties of new dicyanovinyl (DCV) substituted acceptor-donor-acceptor chromophores is described. A combination of theoretical and experimental methods shows a subtle relation between the charge transfer character and the geometry of the molecules.Download high-res image (136KB)Download full-size image
Co-reporter:Natalie Gorczak, Nicolas Renaud, Elena Galan, Rienk Eelkema, Laurens D. A. Siebbeles and Ferdinand C. Grozema
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 9) pp:6773-6779
Publication Date(Web):05 Feb 2016
DOI:10.1039/C5CP06728F
Quantum interference is a well-known phenomenon that dictates charge transport properties of single molecule junctions. However, reports on quantum interference in donor-bridge-acceptor molecules are scarce. This might be due to the difficulties in meeting the conditions for the presence of quantum interference in a donor-bridge-acceptor system. The electronic coupling between the donor, bridge, and acceptor moieties must be weak in order to ensure localised initial and final states for charge transfer. Yet, it must be strong enough to allow all bridge orbitals to mediate charge transfer. We present the computational route to the design of a donor-bridge-acceptor molecule that features the right balance between these contradicting requirements and exhibits pronounced interference effects.
Co-reporter:Jorge Follana-Berná, Damla Inan, Vicente M. Blas-Ferrando, Natalie Gorczak, Javier Ortiz, Félix Manjón, Fernando Fernández-Lázaro, Ferdinand C. Grozema, and Ángela Sastre-Santos
The Journal of Physical Chemistry C 2016 Volume 120(Issue 46) pp:26508-26513
Publication Date(Web):October 4, 2016
DOI:10.1021/acs.jpcc.6b07160
The synthesis and characterization of different conjugated phthalocyanine–perylenemonoimidebenzimidazole [ZnPc-PBIm(OR)4] and nonconjugated phthalocyanine–perylenediimide [ZnPc-PDI(OR)4] dyads are carried out. UV–vis, 1H NMR, and electrochemistry measurements reveal the interaction between perylene and phthalocyanine moieties in the ground state in the conjugated hybrid and the lack of interaction in the nonconjugated one. Ultrafast transient absorption measurements show that a state with substantial charge-transfer character is formed in both compounds, but the rates for the formation and recombination from this state are much faster for the conjugated compound.
Co-reporter:María C. Gélvez-Rueda
The Journal of Physical Chemistry C 2016 Volume 120(Issue 30) pp:16577-16585
Publication Date(Web):July 11, 2016
DOI:10.1021/acs.jpcc.6b06722
Organic–inorganic hybrid halide perovskites are a promising class of materials for photovoltaic application with reported power efficiencies over ∼22%. However, not much is known about the influence of the organic dipole rotation and phase transitions on charge carrier dynamics. Here, we report substantial changes in mobility and lifetime of charge carriers in CH3NH3PbI3 after the low-temperature tetragonal (β) to orthorhombic (γ) phase transition. By using microwave conductivity measurements, we observed that the mobility and lifetime of ionized charge carriers increase as the temperature decreases and a sudden increment is seen after the β–γ phase transition. For CH3NH3PbI3, the mobility and the half-lifetime increase by a factor of 3–6 compared with the values before the β–γ phase transition. We attribute the considerable change in the dynamics at low temperature to the decrease of the inherent dynamic disorder of the organic cation (CH3NH3+) inside the perovskite crystal structure.
Co-reporter:Natalie Gorczak, Nicolas Renaud, Simge Tarkuç, Arjan J. Houtepen, Rienk Eelkema, Laurens D. A. Siebbeles and Ferdinand C. Grozema
Chemical Science 2015 vol. 6(Issue 7) pp:4196-4206
Publication Date(Web):11 May 2015
DOI:10.1039/C5SC01104C
Destructive quantum interference has been shown to strongly reduce charge tunneling rates across molecular bridges. The current consensus is that destructive quantum interference occurs in cross-conjugated molecules, while linearly conjugated molecules exhibit constructive interference. Our experimental results on photoinduced charge transfer in donor-bridge-acceptor systems, however, show that hole transfer is ten times faster through a cross-conjugated biphenyl bridge than through a linearly conjugated biphenyl bridge. Electronic structure calculations reveal that the surprisingly low hole transfer rate across the linearly conjugated biphenyl bridge is caused by the presence of destructive instead of constructive interference. We find that the specific molecular orbital symmetry of the involved donor and acceptor states leads to interference conditions that are different from those valid in single molecule conduction experiments. Furthermore, the results indicate that by utilizing molecular orbital symmetry in a smart way new opportunities of engineering charge transfer emerge.
Co-reporter:Yaroslav V. Aulin, Martijn van Sebille, Michiel Moes and Ferdinand C. Grozema
RSC Advances 2015 vol. 5(Issue 130) pp:107896-107903
Publication Date(Web):07 Dec 2015
DOI:10.1039/C5RA20602B
This paper studies photochemical upconversion in solutions of octaethyl porphyrin (OEP) and diphenyl anthracene (DPA). The system has been widely used as a standard model system in the field of photochemical upconversion. Although, the kinetics of elementary processes contributing to photochemical upconversion in it have been extensively studied, there has been no research on the efficiency of upconversion in the system, despite of the fact that this parameter is detrimental for potential applications of photochemical upconversion process. We determine the yield of photochemical upconversion in a number of metal based OEP/DPA systems. Additionally, we studied the dependence of kinetic, and efficiency parameters of the process on the core metal of the porphyrin. We showed that the overall efficiency of photochemical upconversion depends significantly on the core metal of the triplet sensitizer porphyrin molecule. We attribute this effect to the differences in efficiency of triplet energy transfer from metal based OEP to DPA depending on the core metal.
Co-reporter:Natalie Gorczak, Taiga Fujii, Ashutosh Kumar Mishra, Arjan J. Houtepen, Ferdinand C. Grozema, and Frederick D. Lewis
The Journal of Physical Chemistry B 2015 Volume 119(Issue 24) pp:7673-7680
Publication Date(Web):February 10, 2015
DOI:10.1021/jp512113w
The mechanism and dynamics of photoinduced electron injection and charge recombination have been investigated for several series of DNA hairpins. The hairpins possess a stilbenediether linker, which serves as an electron donor and base pair stems that possess different pyrimidine bases adjacent to the linker. Hairpins with adjacent thymine-adenine (T-A) base pairs undergo fast electron injection and relatively slow charge recombination with rate constants that are not strongly dependent upon the following base pair. Hairpins with adjacent cytosine-guanine (C-G) base pairs undergo reversible electron injection and much faster charge recombination than those with adjacent T-A base pairs. Hairpins with 5-fluorouracil or other halogenated pyrimidines in their first and second base pair undergo fast electron injection and multiexponential charge recombination. The difference in kinetic behavior for the different series of hairpins and its implications for the formation of long-lived charge-separated states are discussed and compared to results reported previously for other electron-donor chromophores.
Co-reporter:Natalie Gorczak, Marcel Swart and Ferdinand C. Grozema
Journal of Materials Chemistry A 2014 vol. 2(Issue 17) pp:3467-3475
Publication Date(Web):26 Feb 2014
DOI:10.1039/C3TC32475C
We calculated the energy landscape of charged molecules that is determined by electrostatic and induction interaction using the fully polarizable force field DRF90 in the bulk and at interfaces of the electron accepting material C60, and two exemplary electron donating materials pentacene and phthalocyanine. In particular, we compared the energy of a non-interacting electron–hole pair (NI-EH) without mutual electrostatic interactions to the energy of a Coulomb-bound interfacial charge-transfer state (CT). Our calculations show that due to electrostatic interactions with the environment a NI-EH state is destabilized on the phthalocyanine–C60 interface, whereas it is stabilized on the interface between pentacene and C60, even without the interaction with the counter charge. Upon adding the mutual electrostatic interaction between the opposite charges the electrostatic term overall stabilizes the CT state in both systems. This stabilization is not compensated by the reduced induction term. The resulting binding energy of the CT state amounts to several tenths of an eV, which contradicts the evidence of working solar cells based on these systems. The overestimated CT state binding energy for charges localized on a single molecule suggests that charge delocalization over multiple molecules might play an important role. Nevertheless, our results indicate clear opportunities to engineer electrostatic interactions at the interface that might lead to destabilization of NI-EH and hence to a lower binding energy of CT.
Co-reporter:D. Deniz Günbaş, Chenming Xue, Sameer Patwardhan, Maria C. Fravventura, Hao Zhang, Wolter F. Jager, Ernst J. R. Sudhölter, Laurens D. A. Siebbeles, Tom J. Savenije, Shi Jin and Ferdinand C. Grozema
Chemical Communications 2014 vol. 50(Issue 38) pp:4955-4958
Publication Date(Web):24 Mar 2014
DOI:10.1039/C4CC00330F
In this communication we report on the synthesis and charge mobility of highly soluble perylenebisimid derivatives. We show that introduction of alkylester side chains results in compounds combining a high solubility with charge mobilities up to 0.22 cm2 V−1 s−1. These materials are therefore interesting as an electron acceptor for solution-processed organic photovoltaics.
Co-reporter:Fatemeh Mirjani ; Nicolas Renaud ; Natalie Gorczak
The Journal of Physical Chemistry C 2014 Volume 118(Issue 26) pp:14192-14199
Publication Date(Web):June 6, 2014
DOI:10.1021/jp503398a
Singlet fission (SF) is a spin-allowed process by which a singlet excited state splits into a pair of triplet states. This process can potentially increase the efficiency of organic solar cells by a factor of 1.5. In this article, we study the dynamics of SF in different molecular aggregates of perylenediimide (PDI) derivatives, pentacene, and 1,3-diphenylisobenzofuran (DPB). To compute the SF rate, we have adopted a Markovian density matrix propagation approach to model SF in a molecular dimer. This approach allows accounting for both the coherent and incoherent processes that mediate the triplet formation. Our calculations show that SF can be much faster in PDI derivatives than in pentacene and DPB. Our analysis also indicates that SF is principally mediated by a superexchange mechanism that involves charge transfer states as virtual intermediates. In addition, because of the existence of different pathways for the formation of the triplet states, signatures of quantum interference are clearly observed.
Co-reporter:Natalie Gorczak, Simge Tarkuç, Nicolas Renaud, Arjan J. Houtepen, Rienk Eelkema, Laurens D. A. Siebbeles, and Ferdinand C. Grozema
The Journal of Physical Chemistry A 2014 Volume 118(Issue 22) pp:3891-3898
Publication Date(Web):May 14, 2014
DOI:10.1021/jp500839t
We report measurements of hole and electron transfer along identical oligo-p-phenylene molecular bridges of increasing length. Although the injection barriers for hole and electron transfer are similar, we observed striking differences in the distance dependence and absolute magnitude of the rates of these two processes. Electron transfer is characterized by an almost distance-independent, fast charge-transfer rate. Hole transfer presents a much slower rate that decreases significantly with the length of the bridge. Time-dependent density functional calculations show that the observed differences can be explained by the delocalization of the respective initial excitation. The evaluation of the initial state is therefore essential when comparing charge-transfer rates between different donor–bridge–acceptor systems.
Co-reporter:Dr. Carlos R. Arroyo;Dr. Simge Tarkuc;Riccardo Frisenda;Dr. Johannes S. Seldenthuis;Charlotte H. M. Woerde;Dr. Rienk Eelkema;Dr. Ferdin C. Grozema;Dr. Herre S. J. vanderZant
Angewandte Chemie International Edition 2013 Volume 52( Issue 11) pp:3152-3155
Publication Date(Web):
DOI:10.1002/anie.201207667
Co-reporter:Sameer Patwardhan ; Sanchita Sengupta ; Laurens D. A. Siebbeles ; Frank Würthner
Journal of the American Chemical Society 2012 Volume 134(Issue 39) pp:16147-16150
Publication Date(Web):September 18, 2012
DOI:10.1021/ja3075192
We have studied the charge transport properties of self-assembled structures of semisynthetic zinc chlorins (ZnChls) in the solid state by pulsed radiolysis time-resolved microwave conductivity measurements. These materials can form either a two-dimensional (2D) brickwork-type slipped stack arrangement or a one-dimensional (1D) tubular assemblies, depending on the exact molecular structure of the ZnChls. We have observed efficient charge transport with mobilities as high as 0.07 cm2 V–1 s–1 for tubular assemblies of 31-hydroxy ZnChls and up to 0.28 cm2 V–1 s–1 for 2D stacked assemblies of 31-methoxy ZnChls at room temperature. The efficient charge transporting capabilities of these organized assemblies opens the way to supramolecular electronics based on biological systems.
Co-reporter:Sameer Patwardhan, Stefano Tonzani, Frederick D. Lewis, Laurens D. A. Siebbeles, George C. Schatz, and Ferdinand C. Grozema
The Journal of Physical Chemistry B 2012 Volume 116(Issue 37) pp:11447-11458
Publication Date(Web):August 13, 2012
DOI:10.1021/jp307146u
In this article, a theoretical study of the electronic and spectroscopic properties of well-defined DNA hairpins is presented. The excited states in the hairpins are described in terms of an exciton Hamiltonan model, and the structural dynamics of the DNA model systems is explicitly taken into account by molecular dynamics simulations. The results show that the model reproduces the experimentally observed absorption and circular dichroism spectra accurately in most cases. It is shown that structural disorder leads to excited states that are largely localized on a single base pair, even for regular DNA sequences consisting only of AT base pairs. Variations in the base pair sequence have a significant effect on the appearance of the spectra but also on the degree of delocalization of the excited state.
Co-reporter:Aleksey A. Kocherzhenko, Laurens D. A. Siebbeles, and Ferdinand C. Grozema
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 14) pp:1753-1756
Publication Date(Web):June 30, 2011
DOI:10.1021/jz200535j
This Letter proposes a realistic design of a single-molecule quantum-interference-based transistor. The transistor consists of a cross-conjugated donor–bridge–acceptor molecule and is chemically gated by a functional group that can be charged. Numerical simulations indicate that the device properties can be tuned to desired specifications by the choice of its constituting functional groups. The transistor does not require external contacts to control its operation. However, it can be chemically functionalized for easy integration into molecular electonic circuits, especially because its operation does not involve any conformational changes in the molecule. The upper operational frequency limit of the proposed device is found to be in the terahertz range.Keywords: charge transfer; donor-bridge-acceptor molecules; Hückel Hamiltonian; molecular electronics; quantum interference;
Co-reporter:Andrey Demenev and S. Holger Eichhorn, Tyler Taerum and Dmitrii F. Perepichka, Sameer Patwardhan, Ferdinand C. Grozema and Laurens D. A. Siebbeles, Richard Klenkler
Chemistry of Materials 2010 Volume 22(Issue 4) pp:1420
Publication Date(Web):January 8, 2010
DOI:10.1021/cm902453z
Reported here are the unique properties of N,N′,N′′-(3,4,5-tridodecyloxyphenyl)benzo[b,b′,b′′]tristhiophene-2,2′,2′′-tricarboxamide 3 as a new H-bonded discotic liquid crystal. Polarized optical microscopy and thermal analysis as well as variable temperature IR spectroscopy and X-ray diffraction confirm the presence of two thermotropic H-bonded hexagonal columnar mesophases that cover a temperature range from <−50 to 280 °C. Intermediate lyotropic mesophases of the highly viscous material aid the alignment of the hexagonal columnar mesophases, which is essential for a detailed structural characterization and applications. Solutions of 3 in heptane at concentrations as low as 1 wt % display isotropic organo-gel phases that consist of H-bonded networks of 3 but do not contain columnar stacks. 2D-X-ray diffraction studies on aligned samples of the thermotropic hexagonal columnar mesophases and DFT calculations on a tetramer of 3 reveal a helical columnar stacking of the individual benzotristhiophene units. Charge carrier mobility measured by time-resolved microwave conductivity is about 0.02 cm2 V−1 s−1 in both hexagonal columnar mesophases and quasi temperature independent even across the phase transition between the two mesophases. The temperature independence is explained by the interrelation between stacking distance and mutual rotation because of the persistent intracolumnar H-bonds between amide groups. Half-life of the charge carriers, on the other hand, drastically increases in the low temperature hexagonal columnar mesophase, which is most likely a result of changing molecular dynamic and conformational states of the side chains. DFT calculations of the frontier orbitals show that the benzotristhiophene core is the sole contributor to the LUMO but does not contribute to the HOMO, whereas the trialkoxyaniline groups are the sole contributors to the HOMO. This suggests that the observed combined mobility is that of electrons alone because no hole transport is expected to occur between trialkoxyaniline groups that are spaced apart by more than 4 Å. Indeed, an electron mobility of 2 × 10−3 cm2 V−1 s−1 but no transient signal for hole transport is obtained by time-of-flight charge carrier mobility measurements on a multi domain sample of 3.
Co-reporter:Sameer Patwardhan ; Sanchita Sengupta ; Frank Würthner ; Laurens D. A. Siebbeles ;Ferdinand Grozema
The Journal of Physical Chemistry C 2010 Volume 114(Issue 48) pp:20834-20842
Publication Date(Web):October 8, 2010
DOI:10.1021/jp107184y
The optical properties of the supramolecular aggregates formed by semisynthetic 31-methoxy zinc chlorins were studied theoretically using an exciton theory description. The exciton coupling between the chromophores was calculated using a transition density formalism, making it possible to distinguish stereochemical differences that arise from the facial orientations of the chiral zinc chlorin molecules in the aggregate. It is shown that the precise stereochemical orientation of the molecules inside the stack strongly influences the observed optical properties. These calculations point to a structure with alternating twist angles along the stack, suggesting a different orientation of neighboring molecules. This study is the first step to systematically investigate the optical properties of chlorophyll aggregates, to unveil the supramolecular organization, and to correctly describe the energy transport processes.
Co-reporter:Ferdinand C. Grozema, Yuri A. Berlin, Laurens D. A. Siebbeles, and Mark A. Ratner
The Journal of Physical Chemistry B 2010 Volume 114(Issue 45) pp:14564-14571
Publication Date(Web):June 24, 2010
DOI:10.1021/jp1023422
Using a tight-binding model of charge transport in systems with static and dynamic disorder, we present a theoretical study of the positive charge transfer in molecular assemblies that involve a hole donor and an acceptor connected by fluorene and phenyl bridges. Two parameters that determine the rate of charge transfer within the proposed model are the charge transfer integral between neighboring units and the site energies. Fluctuations in the values of the charge transfer integral and the energy landscape for hole transport were calculated by taking into account variations of the dihedral angle between neighboring units and electrostatic interaction of positive charge moving along the bridge and the negative charge that remains on the hole donor. Analysis of the dynamics of hole transfer and the distribution of the positive charge during this process allows the conclusion that the rapid fall of the hole transfer rate coefficient observed in experiments with short bridges (three and four structural units for systems with fluorene and phenyl bridges, respectively) can be attributed to the electrostatic interaction. This interaction is responsible for the formation of the effective barrier between donor and acceptor with the height that increases as the number of structural bridge units remains less than 3 (fluorene bridge) or 4 (phenyl bridge). For longer bridges, however, the effective barrier changes only weakly and now the charge transport is mostly dominated by the fluctuation-assisted incoherent hole migration along the bridge. The latter mechanism exhibits much weaker dependence of the rate coefficient on the bridge length in agreement with the available experimental results.
Co-reporter:Aleksey A. Kocherzhenko, Ferdinand C. Grozema and Laurens D. A. Siebbeles
The Journal of Physical Chemistry C 2010 Volume 114(Issue 17) pp:7973-7979
Publication Date(Web):April 12, 2010
DOI:10.1021/jp9117216
Quantum interference effects occurring in molecules through which a charge can travel via multiple pathways can be the basis for new unconventional design principles in molecular scale electronics. However, these quantum interference effects can be reduced by interaction between the charge and molecular vibrations. In this work dephasing (decoherence) effects have been studied using a model that combines a (classical) molecular mechanics description of molecular vibrations with a quantum mechanical propagation of the charge. It is found that despite the clear effect of dephasing on the charge propagation, interference effects are largely retained at room temperature if vibrations are accounted for. Additionally, it is shown that taking electronic interactions between non-nearest neighbor atoms into account also diminishes interference effects but not sufficiently to destroy them completely. It is concluded that interference effects are strong enough to use them in a functional manner in molecular electronics. This opens up new ways to design molecular electronic components that exploit quantum interference.
Co-reporter:Ferdinand C. Grozema ; Stefano Tonzani ; Yuri A. Berlin ; George C. Schatz ; Laurens D. A. Siebbeles ;Mark A. Ratner
Journal of the American Chemical Society 2009 Volume 131(Issue 40) pp:14204-14205
Publication Date(Web):September 18, 2009
DOI:10.1021/ja906863k
DNA hairpins in which an electron donor and an electron acceptor are attached to the ends are excellent model systems for the study of charge transfer in weakly coupled π-stacked systems. In this communication we report on a computational study of the effect of the base pair sequence in these DNA hairpins on the kinetics of charge transfer. We show that the rate of charge transfer strongly depends on the actual position of a GC base pair in a sequence that otherwise only contains AT base pairs. This can be explained by evaluating the energy landscape through which the charge travels. It is shown that including the electrostatic interaction between electron and hole can explain the experimentally observed dependence on the position of the GC in the DNA. We conclude that electrostatic interactions are important to consider when explaining the charge transfer kinetics in GC containing DNA sequences.
Co-reporter:Natalie Gorczak, Nicolas Renaud, Simge Tarkuç, Arjan J. Houtepen, Rienk Eelkema, Laurens D. A. Siebbeles and Ferdinand C. Grozema
Chemical Science (2010-Present) 2015 - vol. 6(Issue 7) pp:NaN4206-4206
Publication Date(Web):2015/05/11
DOI:10.1039/C5SC01104C
Destructive quantum interference has been shown to strongly reduce charge tunneling rates across molecular bridges. The current consensus is that destructive quantum interference occurs in cross-conjugated molecules, while linearly conjugated molecules exhibit constructive interference. Our experimental results on photoinduced charge transfer in donor-bridge-acceptor systems, however, show that hole transfer is ten times faster through a cross-conjugated biphenyl bridge than through a linearly conjugated biphenyl bridge. Electronic structure calculations reveal that the surprisingly low hole transfer rate across the linearly conjugated biphenyl bridge is caused by the presence of destructive instead of constructive interference. We find that the specific molecular orbital symmetry of the involved donor and acceptor states leads to interference conditions that are different from those valid in single molecule conduction experiments. Furthermore, the results indicate that by utilizing molecular orbital symmetry in a smart way new opportunities of engineering charge transfer emerge.
Co-reporter:Natalie Gorczak, Nicolas Renaud, Elena Galan, Rienk Eelkema, Laurens D. A. Siebbeles and Ferdinand C. Grozema
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 9) pp:NaN6779-6779
Publication Date(Web):2016/02/05
DOI:10.1039/C5CP06728F
Quantum interference is a well-known phenomenon that dictates charge transport properties of single molecule junctions. However, reports on quantum interference in donor-bridge-acceptor molecules are scarce. This might be due to the difficulties in meeting the conditions for the presence of quantum interference in a donor-bridge-acceptor system. The electronic coupling between the donor, bridge, and acceptor moieties must be weak in order to ensure localised initial and final states for charge transfer. Yet, it must be strong enough to allow all bridge orbitals to mediate charge transfer. We present the computational route to the design of a donor-bridge-acceptor molecule that features the right balance between these contradicting requirements and exhibits pronounced interference effects.
Co-reporter:Natalie Gorczak, Marcel Swart and Ferdinand C. Grozema
Journal of Materials Chemistry A 2014 - vol. 2(Issue 17) pp:NaN3475-3475
Publication Date(Web):2014/02/26
DOI:10.1039/C3TC32475C
We calculated the energy landscape of charged molecules that is determined by electrostatic and induction interaction using the fully polarizable force field DRF90 in the bulk and at interfaces of the electron accepting material C60, and two exemplary electron donating materials pentacene and phthalocyanine. In particular, we compared the energy of a non-interacting electron–hole pair (NI-EH) without mutual electrostatic interactions to the energy of a Coulomb-bound interfacial charge-transfer state (CT). Our calculations show that due to electrostatic interactions with the environment a NI-EH state is destabilized on the phthalocyanine–C60 interface, whereas it is stabilized on the interface between pentacene and C60, even without the interaction with the counter charge. Upon adding the mutual electrostatic interaction between the opposite charges the electrostatic term overall stabilizes the CT state in both systems. This stabilization is not compensated by the reduced induction term. The resulting binding energy of the CT state amounts to several tenths of an eV, which contradicts the evidence of working solar cells based on these systems. The overestimated CT state binding energy for charges localized on a single molecule suggests that charge delocalization over multiple molecules might play an important role. Nevertheless, our results indicate clear opportunities to engineer electrostatic interactions at the interface that might lead to destabilization of NI-EH and hence to a lower binding energy of CT.
Co-reporter:D. Deniz Günbaş, Chenming Xue, Sameer Patwardhan, Maria C. Fravventura, Hao Zhang, Wolter F. Jager, Ernst J. R. Sudhölter, Laurens D. A. Siebbeles, Tom J. Savenije, Shi Jin and Ferdinand C. Grozema
Chemical Communications 2014 - vol. 50(Issue 38) pp:NaN4958-4958
Publication Date(Web):2014/03/24
DOI:10.1039/C4CC00330F
In this communication we report on the synthesis and charge mobility of highly soluble perylenebisimid derivatives. We show that introduction of alkylester side chains results in compounds combining a high solubility with charge mobilities up to 0.22 cm2 V−1 s−1. These materials are therefore interesting as an electron acceptor for solution-processed organic photovoltaics.

![Poly[1,3-phenylene-(1Z)-1,2-ethenediyl[2,5-bis(octyloxy)-1,4-phenylene]
-(1Z)-1,2-ethenediyl]](http://img.cochemist.com/ccimg/850700/850636-06-5.png)
![Poly[1,3-phenylene-(1Z)-1,2-ethenediyl[2,5-bis(octyloxy)-1,4-phenylene]
-(1Z)-1,2-ethenediyl]](http://img.cochemist.com/ccimg/850700/850636-06-5_b.png)





![Poly[[[(3,7-dimethyloctyl)oxy]methoxy-1,4-phenylene]-1,2-ethenediyl]](http://img.cochemist.com/ccimg/250200/250164-11-5.png)
![Poly[[[(3,7-dimethyloctyl)oxy]methoxy-1,4-phenylene]-1,2-ethenediyl]](http://img.cochemist.com/ccimg/250200/250164-11-5_b.png)
![Poly[2,5-thiophenediyl[9,9-bis(2-ethylhexyl)-9H-fluorene-2,7-diyl]]](http://img.cochemist.com/ccimg/352100/352004-06-9.png)
![Poly[2,5-thiophenediyl[9,9-bis(2-ethylhexyl)-9H-fluorene-2,7-diyl]]](http://img.cochemist.com/ccimg/352100/352004-06-9_b.png)
![QUINOLINIUM, 7-[(2-AMINOETHYL)AMINO]-1-METHYL-, IODIDE](http://img.cochemist.com/ccimg/884600/884502-64-1.png)
![QUINOLINIUM, 7-[(2-AMINOETHYL)AMINO]-1-METHYL-, IODIDE](http://img.cochemist.com/ccimg/884600/884502-64-1_b.png)
![Ethanol, 2,2'-[(1E)-1,2-ethenediylbis(4,1-phenyleneoxy)]bis-](http://img.cochemist.com/ccimg/164000/163935-25-9.png)
![Ethanol, 2,2'-[(1E)-1,2-ethenediylbis(4,1-phenyleneoxy)]bis-](http://img.cochemist.com/ccimg/164000/163935-25-9_b.png)
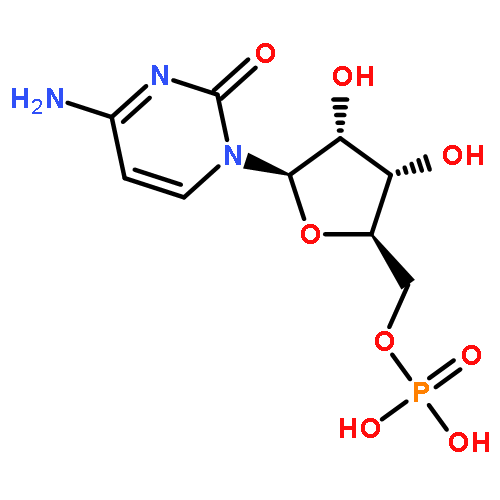
![[(2R,3S,5R)-2-[2-[[(2R,3S,5R)-5-(4-amino-2-oxo-pyrimidin-1-yl)-3-phosphonooxy-tetrahydrofuran-2-yl]methoxy]-2-[(2S,3S,5R)-5-(6-aminopurin-9-yl)-3-hydroxy-tetrahydrofuran-2-yl]ethyl]-5-(6-aminopurin-9-yl)tetrahydrofuran-3-yl] dihydrogen phosphate](http://img.cochemist.com/ccimg/70500/70419-16-8.png)
![[(2R,3S,5R)-2-[2-[[(2R,3S,5R)-5-(4-amino-2-oxo-pyrimidin-1-yl)-3-phosphonooxy-tetrahydrofuran-2-yl]methoxy]-2-[(2S,3S,5R)-5-(6-aminopurin-9-yl)-3-hydroxy-tetrahydrofuran-2-yl]ethyl]-5-(6-aminopurin-9-yl)tetrahydrofuran-3-yl] dihydrogen phosphate](http://img.cochemist.com/ccimg/70500/70419-16-8_b.png)



![1-bromo-4-[2-(4-bromophenyl)ethyl]benzene](http://img.cochemist.com/ccimg/19900/19829-56-2.png)
![1-bromo-4-[2-(4-bromophenyl)ethyl]benzene](http://img.cochemist.com/ccimg/19900/19829-56-2_b.png)
![Benzene,1,4-bis(octyloxy)-2,5-bis[2-[4-(2-phenylethenyl)phenyl]ethenyl]-](http://img.cochemist.com/ccimg/220500/220446-31-1.png)
![Benzene,1,4-bis(octyloxy)-2,5-bis[2-[4-(2-phenylethenyl)phenyl]ethenyl]-](http://img.cochemist.com/ccimg/220500/220446-31-1_b.png)



![2,9-bis(ethoxypropyl)anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone](http://img.cochemist.com/ccimg/59700/59642-64-7.png)
![2,9-bis(ethoxypropyl)anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone](http://img.cochemist.com/ccimg/59700/59642-64-7_b.png)


![Hexabenzo[bc,ef,hi,kl,no,qr]coronene](/data/chemimg/1472400/190-24-9.png)
![Hexabenzo[bc,ef,hi,kl,no,qr]coronene](/data/chemimg/1472400/190-24-9_b.png)






![Benzene,1,4-bis[2-[4-[2-[4-[2-[2,5-bis(octyloxy)-4-(2-phenylethenyl)phenyl]ethenyl]phenyl]ethenyl]-2,5-bis(octyloxy)phenyl]ethenyl]-](http://img.cochemist.com/ccimg/291600/291508-90-2.png)
![Benzene,1,4-bis[2-[4-[2-[4-[2-[2,5-bis(octyloxy)-4-(2-phenylethenyl)phenyl]ethenyl]phenyl]ethenyl]-2,5-bis(octyloxy)phenyl]ethenyl]-](http://img.cochemist.com/ccimg/291600/291508-90-2_b.png)
![Benzene,1,4-bis[2-[2,5-bis(octyloxy)-4-(2-phenylethenyl)phenyl]ethenyl]-](http://img.cochemist.com/ccimg/291600/291508-88-8.png)
![Benzene,1,4-bis[2-[2,5-bis(octyloxy)-4-(2-phenylethenyl)phenyl]ethenyl]-](http://img.cochemist.com/ccimg/291600/291508-88-8_b.png)
![Benzene, 1,1'-(1,2-ethenediyl)bis[4-[2-(4-methylphenyl)ethenyl]-](http://img.cochemist.com/ccimg/103400/103398-70-5.png)
![Benzene, 1,1'-(1,2-ethenediyl)bis[4-[2-(4-methylphenyl)ethenyl]-](http://img.cochemist.com/ccimg/103400/103398-70-5_b.png)
![2,5-BIS[5-[2-(5-HEXYLTHIOPHEN-2-YL)ETHYNYL]THIOPHEN-2-YL]THIOPHENE](http://img.cochemist.com/ccimg/710400/710338-93-5.png)
![2,5-BIS[5-[2-(5-HEXYLTHIOPHEN-2-YL)ETHYNYL]THIOPHEN-2-YL]THIOPHENE](http://img.cochemist.com/ccimg/710400/710338-93-5_b.png)



![1,4-BIS[2-[4-(2-PHENYLETHENYL)PHENYL]ETHENYL]BENZENE](/data/chemimg/487000/26260-75-3.png)
![1,4-BIS[2-[4-(2-PHENYLETHENYL)PHENYL]ETHENYL]BENZENE](/data/chemimg/487000/26260-75-3_b.png)
![6-FLUOROIMIDAZO[1,2-A]PYRIDINE-3-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/356600/356590-07-3.png)
![6-FLUOROIMIDAZO[1,2-A]PYRIDINE-3-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/356600/356590-07-3_b.png)
![Platinum, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/540800/31248-39-2.png)
![Platinum, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/540800/31248-39-2_b.png)



![Hexabenzo[a,d,g,j,m,p]coronene](/data/chemimg/350900/1065-80-1.png)
![Hexabenzo[a,d,g,j,m,p]coronene](/data/chemimg/350900/1065-80-1_b.png)
![2,9-bis(2,6-diisopropylphenyl)-5,6,12,13-tetrakis[4-(2-methyl-2-p Ropanyl)phenoxy]isoquinolino[4',5',6':6,5,10]anthra[2,1,9-def]iso Quinoline-1,3,8,10(2h,9h)-tetrone](http://img.cochemist.com/ccimg/112100/112078-08-7.png)
![2,9-bis(2,6-diisopropylphenyl)-5,6,12,13-tetrakis[4-(2-methyl-2-p Ropanyl)phenoxy]isoquinolino[4',5',6':6,5,10]anthra[2,1,9-def]iso Quinoline-1,3,8,10(2h,9h)-tetrone](http://img.cochemist.com/ccimg/112100/112078-08-7_b.png)


![5,12-Dibromo-2,9-bis(2,6-diisopropylphenyl)anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetraone](http://img.cochemist.com/ccimg/331900/331861-94-0.png)
![5,12-Dibromo-2,9-bis(2,6-diisopropylphenyl)anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetraone](http://img.cochemist.com/ccimg/331900/331861-94-0_b.png)





![2,9-Di(tridecan-7-yl)anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetraone](/data/chemimg/339200/110590-84-6.png)
![2,9-Di(tridecan-7-yl)anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetraone](/data/chemimg/339200/110590-84-6_b.png)
![Poly[(9,9-dioctylfluorenyl-2,7-diyl)-alt-(benzo[2,1,3]thiadiazol-4,7-diyl)]](/data/chemimg/107700/210347-52-7.png)
![Poly[(9,9-dioctylfluorenyl-2,7-diyl)-alt-(benzo[2,1,3]thiadiazol-4,7-diyl)]](/data/chemimg/107700/210347-52-7_b.png)

![Benzene,1,4-bis[2-[4-[2-[2,5-bis(octyloxy)-4-(2-phenylethenyl)phenyl]ethenyl]phenyl]ethenyl]-2,5-bis(octyloxy)-](http://img.cochemist.com/ccimg/291600/291508-89-9.png)
![Benzene,1,4-bis[2-[4-[2-[2,5-bis(octyloxy)-4-(2-phenylethenyl)phenyl]ethenyl]phenyl]ethenyl]-2,5-bis(octyloxy)-](http://img.cochemist.com/ccimg/291600/291508-89-9_b.png)





![Poly[[2-[(3,7-dimethyloctyl)oxy]-5-methoxy-1,4-phenylene]-1,2-ethenediyl]](http://img.cochemist.com/ccimg/177800/177716-59-5.png)
![Poly[[2-[(3,7-dimethyloctyl)oxy]-5-methoxy-1,4-phenylene]-1,2-ethenediyl]](http://img.cochemist.com/ccimg/177800/177716-59-5_b.png)
![Poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene]](http://img.cochemist.com/ccimg/138200/138184-36-8.png)
![Poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene]](http://img.cochemist.com/ccimg/138200/138184-36-8_b.png)






![Palladium, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/21200/24804-00-0.png)
![Palladium, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/21200/24804-00-0_b.png)