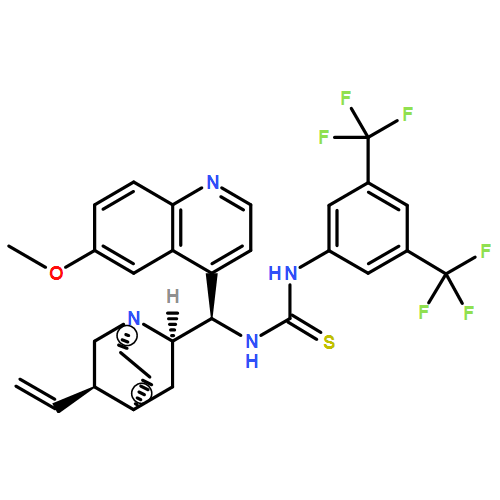
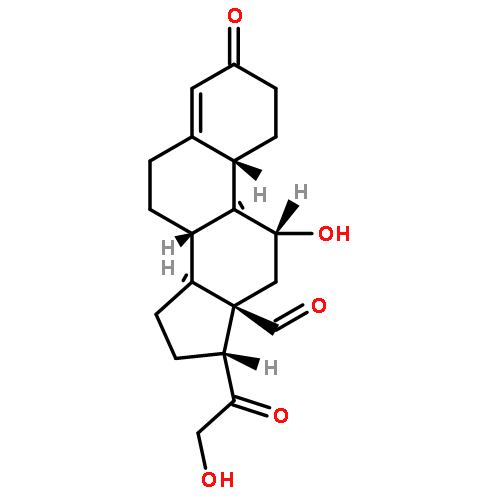
Co-reporter:Li-Qiang Han, Xia Yuan, Xing-Yu Wu, Ri-Dong Li, Bo Xu, Qing Cheng, Zhen-Ming Liu, Tian-Yan Zhou, Hao-Yun An, Xin Wang, Tie-Ming Cheng, Ze-Mei Ge, Jing-Rong Cui, Run-Tao Li
European Journal of Medicinal Chemistry 2017 Volume 125() pp:925-939
Publication Date(Web):5 January 2017
DOI:10.1016/j.ejmech.2016.10.023
•Two series of novel urea-containing peptide boronic acids were designed and synthesized as proteasome inhibitors.•The SAR was investigated and one of the two series showed impressive anti-proteasome and anti-tumor activities.•More human tumor cell lines were tested and compound I-14 was selected to evaluate its anticancer activity in vivo.•Compound I-14 showed obvious in vivo anticancer activity with much lower toxicity comparing to Bortezomib.A novel class of urea-containing peptide boronic acids as proteasome inhibitors was designed by introducing a urea scaffold to replace an amido bond. Compounds were synthesized and their antitumor activities were evaluated. After two rounds of optimizations, the compound I-14 was found to be a potent proteasome inhibitor. Compared with Bortezomib, I-14 showed higher potency against the chymotrypsin-like activity of human 20S proteasome (IC50 < 1 pM), similar potency against four different cancer cell lines (IC50 < 10 nM), and better pharmacokinetic profile. Furthermore, I-14 significantly inhibited tumor growth in Bel7404 mouse xenograft model. The excellent proteasome inhibition by I-14 was rationalized through docking and molecular dynamics studies.
Co-reporter:Xiaolei Du;Dawei Yin;Zemei Ge;Xin Wang
RSC Advances (2011-Present) 2017 vol. 7(Issue 39) pp:24547-24550
Publication Date(Web):2017/05/03
DOI:10.1039/C7RA03069J
The efficient asymmetric Michael addition reactions of pyrrolones with chalcones catalyzed by a simple and commercially available chiral 1,2-diaminocyclohexane-2-(N-Boc-amino)benzoic acid have been developed to provide the corresponding Michael adducts in good yields (up to 90%) and high enantioselectivities (up to 95% ee).
Co-reporter:Liqiang Han, Yanzhao Wen, Ridong Li, Bo Xu, Zemei Ge, Xin Wang, Tieming Cheng, Jingrong Cui, Runtao Li
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 15(Issue 15) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.bmc.2017.05.049
•Two series of peptide-boronic acids as proteasome were designed and synthesized.•Most compounds showed significant antiproliferative activity against MGC803.•Compound II-7 exhibited potent activity against 5 cancer cell lines.•Compound II-7 showed excellent subunit selectivity over proteasome comparing with Bortezomib.On the basis of the application of proline-boronic acid as pharmacophore in the kinase inhibitors and our previous research results, using proline-boronic acid as warhead, two series of peptide proline-boronic acids, dipeptide proline-boronic acids (I) and tripeptide proline-boronic acids (II), were designed and synthesized. All the synthesized compounds were first evaluated for their biological activity against MGC803 cell, and then, the best compound II-7 was selected to test its anti-tumor spectrum on six human tumor cell lines and proteasome inhibition against three subunits. The results indicated that series II have much better biological activities than series I. The compound II-7 exhibited not only excellent biological activities with IC50 values of nM level in both cell and proteasome models, but also much better subunit selectivity. Thus, proline-boronic acid as warhead is reasonable in the design of proteasome inhibitors.Download high-res image (107KB)Download full-size image
Co-reporter:Yong Hua, Wenjie Zhang, Xin Wang, Zemei Ge, Runtao Li
Tetrahedron 2017 Volume 73, Issue 30(Issue 30) pp:
Publication Date(Web):27 July 2017
DOI:10.1016/j.tet.2017.05.098
A novel kind of five-membered endocyclic sulfoximines, 1-aryl-3-(arylimino)-3,5-dihydro-1,4,2- dithiazole 1-oxides, were synthesized through addition/cyclization of chloromethyl aryl sulfoximines with aryl isothiocyanates under mild reaction conditions in moderate to good yields.Download high-res image (102KB)Download full-size image
Co-reporter:Songwen Lin, Yingbo Li, Yufen Zheng, Laichun Luo, Qi Sun, Zemei Ge, Tieming Cheng, Runtao Li
European Journal of Medicinal Chemistry 2017 Volume 127(Volume 127) pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.ejmech.2016.12.055
•Phosphoramide mustard functionality incorporated into the quinazoline scaffold as EGFR/HER2 inhibitors were proposed.•The mechanism studies were supported on DNA damage.•Compound 10d is a potential candidate for treatment of lung cancer.•MTD study indicated that compound 10d had no obvious acute toxicity.A series of novel compounds with phosphoramide mustard functionality incorporated into the quinazoline scaffold of EGFR/HER2 inhibitors were designed and synthesized as multi-target-directed ligands against tumor cells. In vitro assays showed that tumor cell lines with high HER2 level were more sensitive to the compounds than tumor cells with low HER2 level. Compound 10d (EMB-3) was one of the most potent inhibitors with IC50 of 7.4 nM and 82 nM against EGFR and HER2, respectively. The mechanism studies were also supported by the effect of 10d-induced DNA damage in MDA-MB-468 cells. In vivo efficacy study showed that 10d could significantly inhibit H522 tumor xenograft model with a TGI of 68% at dose of 100 mg/kg (QDx28, p.o.) and no significant body weight loss was observed. MTD study indicated that compound 10d had no acute toxicity to mice at doses up to 900 mg/kg (single dose).Download high-res image (156KB)Download full-size image
Co-reporter:Bin Liu, Xin Wang, Zemei Ge and Runtao Li
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 10) pp:2944-2949
Publication Date(Web):09 Feb 2016
DOI:10.1039/C6OB00179C
Herein we report a novel iridium(III)-catalyzed ortho-mono-alkynylation of 7-azaindoles under mild conditions. This approach provides a general and straightforward access to form novel 7-azaindole derivatives with ample substrate scope and broad group tolerance.
Co-reporter:Yihuan Lu, Yuqiong Zhao, Shucheng Wang, Xin Wang, Zemei Ge and Runtao Li
RSC Advances 2016 vol. 6(Issue 14) pp:11378-11381
Publication Date(Web):22 Jan 2016
DOI:10.1039/C5RA26524J
A simple and efficient synthesis of 2-thio-5-amino substituted benzoquinones via KI catalyzed cascade oxidation/S-Michael addition/oxidation/N-Michael addition/oxidation starting from hydroquinone, amines and S-alkylisothiouronium salts was described. Various 2-thio-5-amino substituted benzoquinones were obtained by this method in moderate to good yields. The use of S-alkylisothiouronium salts as thiol equivalents is more convenient and environmental friendly.
Co-reporter:Shucheng Wang, Xuhu Huang, Zemei Ge, Xin Wang and Runtao Li
RSC Advances 2016 vol. 6(Issue 68) pp:63532-63535
Publication Date(Web):28 Jun 2016
DOI:10.1039/C6RA09046J
A metal-free process for the C-3 alkylation of imidazopyridines has been developed. Various alkylation products including alkyl ester-, cyano-, ketone- and amide-substituted imidazopyridines were prepared in good yields. With this method, a highly efficient and concise formal synthesis of alpidem (A) and zolpidem (B) has been completed.
Co-reporter:Shucheng Wang, Xuhu Huang, Qi Wang, Zemei Ge, Xin Wang and Runtao Li
RSC Advances 2016 vol. 6(Issue 14) pp:11754-11757
Publication Date(Web):22 Jan 2016
DOI:10.1039/C5RA27878C
A novel and highly efficient cascade synthesis of sulfonated quinoline dione derivatives from N-(2-cyanoaryl) methylacrylamides and sulfonylhydrazides in good yields is described. The reaction proceeds through radical addition, cyclization, and imine hydrolysis, in which one new C–C bond and one CO bond were formed.
Co-reporter:Ridong Li, Penglin Leng, Bin Liu, Xin Wang, Zemei Ge, Runtao Li
Tetrahedron 2016 Volume 72(Issue 37) pp:5707-5712
Publication Date(Web):15 September 2016
DOI:10.1016/j.tet.2016.07.075
A novel and efficient one-pot procedure for the synthesis of S-vinyl dithiocarbamates from electron-deficient allenes, amines and CS2 was presented. The reactions proceed at room temperature for 10–30 min without any catalyst to afford the products in high yields, excellent regioselectivity and stereoselectivity.
Co-reporter:Bowen Li, Shuo Zhou, Shucheng Wang, Xingyi Sun, Zemei Ge, Runtao Li
Tetrahedron 2016 Volume 72(27–28) pp:3885-3889
Publication Date(Web):7 July 2016
DOI:10.1016/j.tet.2016.05.011
An efficient method for the synthesis of organic sulfonic acid potassiums containing dithiocarbamate side chains was developed through the reaction of amines, carbon disulfide and sultones in the presence of K3PO4 in water at room temperature. The organic sulfonic acid potassium derivatives are easily transformed into the corresponding organic sulfonic acids, which were further reacted with amines to afford the important organic sulfonamides containing dithiocarbamate side chains.
Co-reporter:Ri-Dong Li, Hui-Ling Wang, Ying-Bo Li, Zhong-Qing Wang, Xin Wang, Yi-Tao Wang, Ze-Mei Ge, Run-Tao Li
European Journal of Medicinal Chemistry 2015 Volume 93() pp:381-391
Publication Date(Web):26 March 2015
DOI:10.1016/j.ejmech.2015.02.030
•A series of dual dithiocarbamates were synthesized.•Most compounds showed significant antiproliferative activities against H460.•Compound 14m exhibited potent activity against nine types of tumor cells.•Compound 14m showed excellent antitumor potency in vitro against HepG2 and MCF-7.A series of dual dithiocarbamates were synthesized and evaluated for their in-vitro anticancer activities on human non-small cell lung cancer cell line H460. Nine compounds exhibited significant antiproliferative activities with IC50 less than 1 μM. Among them, compound 14m showed the highest inhibitory activity against H460 cell and inhibited the growth of nine types of tumor cells with IC50 values less than 1 μM. It also achieved IC50 of 54 nM and 23 nM against HepG2 and MCF-7 cell lines, respectively. Preliminary structure–activity relationship study indicated that: a) when the methyl group (region A) is substituted with benzene rings, ortho substitution on the benzene ring is favored for activity; b) substitution with heterocyclic structures at region A exhibited greater impact on the anti-tumor activity of compounds, in which pyridine ring, thiazole ring, coumarin and benzo[b]thiophene are favored and quinoline ring is the most favored; c) substitution with different amines (region B) also showed marked effect on the activity of compounds and dimethylamine and morpholine are preferred to other tested amines.
Co-reporter:Zhushuang Bai, Ling Ji, Zemei Ge, Xin Wang and Runtao Li
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 19) pp:5363-5366
Publication Date(Web):13 Apr 2015
DOI:10.1039/C5OB00708A
The first bifunctional thiourea catalyzed asymmetric Michael addition reactions of nitroalkanes to 2-furanones are described. The highly functionalized γ-lactones with two or three consecutive stereogenic carbons were obtained in high yields (up to 99%), high diastereoselectivities (up to >20:1 dr) and enantioselectivities (up to >99% ee).
Co-reporter:Dawei Yin, Wei Wang, Yangqiu Peng, Zemei Ge, Tieming Cheng, Xin Wang and Runtao Li
RSC Advances 2015 vol. 5(Issue 22) pp:17296-17299
Publication Date(Web):15 Dec 2014
DOI:10.1039/C4RA13796E
The first TMG catalyzed intramolecular aza-MBH reaction based on furanone derivatives was developed and successfully applied in the synthesis of a novel fused heterocyclic scaffold, furo[3,4-c]quinolin-3(1H)-ones in moderate to good yields.
Co-reporter:Yu Zhang, Bin Liu, Xingyu Wu, Ridong Li, Xianling Ning, Yu Liu, Zhenming Liu, Zemei Ge, Runtao Li, Yuxin Yin
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 15) pp:4815-4823
Publication Date(Web):1 August 2015
DOI:10.1016/j.bmc.2015.05.041
Pyruvate kinase M2 (PKM2) is a key protein responsible for cancer’s Warburg effect. Activation of PKM2 may alter aberrant metabolism in cancer cells, which suggests PKM2 as a tumor selective therapeutic target. In this paper, the lead compound 8 was first discovered as a new kind of PKM2 activator from a random screening of an in-house compound library. Then, a series of lead compound 8 analogs were designed, synthesized and evaluated for their activation of PKM2 and anticancer activities. 7-Azaindole analog 32 was identified as the most potent PKM2 activator. Compounds with potent enzyme activity also exhibited selective anti-proliferation activity on cancer cell lines HCT116, Hela and H1299 compared with non-tumor cell line BEAS-2B. The structure–activity relationships of these compounds were supported by molecular docking results. Preliminary pharmacological studies also showed that compound 32 arrests the cell cycle at the G2/M phase in HCT116 cell line.
Co-reporter:Laichun Luo;Lanlan Meng;Yangqiu Peng;Yongning Xing;Qi Sun;Zemei Ge
European Journal of Organic Chemistry 2015 Volume 2015( Issue 3) pp:631-637
Publication Date(Web):
DOI:10.1002/ejoc.201403156
Abstract
A ZnCl2-promoted one-pot synthesis of multisubstituted thiazolo[4,5-b]pyridines and thieno[2,3-b:4,5-b′]dipyridines from mercaptonitrile salts, α-bromo ketones and ketones has been developed. This approach involves a tandem SN2 alkylation/Thorpe–Ziegler cyclization/Friedländer reaction, which allows the creation of two hetero rings and four new bonds in a single operation. The reaction proceeds smoothly under solvent-free conditions to afford fused pyridines in moderate to good yields.
Co-reporter:Xuhu Huang, Shucheng Wang, Bowen Li, Xin Wang, Zemei Ge and Runtao Li
RSC Advances 2015 vol. 5(Issue 29) pp:22654-22657
Publication Date(Web):23 Feb 2015
DOI:10.1039/C4RA17237J
An efficient approach to sulfenyl imidazoheterocycles has been developed via iodine–triphenylphosphine mediated direct sulfenylation of imidazoheterocycles with sodium sulfinates. The reactions proceed smoothly under transition-metal-free conditions with a broad range of substrate scope, giving the desired products in moderate to excellent yields.
Co-reporter:Jie Wang, Hailiang Tan, Qi Sun, Zemei Ge, Xin Wang, Yinye Wang, Runtao Li
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 11) pp:2340-2344
Publication Date(Web):1 June 2015
DOI:10.1016/j.bmcl.2015.04.013
A series of pyridazino[3,4,5-de]quinazolin-3(2H)-one derivatives were designed and synthesized as PARP-1 inhibitors. Most of the synthesized compounds showed good inhibitory activities of PARP-1 and four of them achieved at the IC50 values ranging from 0.0914 μM to 0.244 μM. Two compounds, 1a and 1b, were further tested for their neuroprotective effect in the PC12 cell model injured by H2O2 and both of them exhibited excellent activities.
Co-reporter:Shucheng Wang, Xuhu Huang, Yanzhao Wen, Zemei Ge, Xin Wang, Runtao Li
Tetrahedron 2015 Volume 71(Issue 42) pp:8117-8122
Publication Date(Web):21 October 2015
DOI:10.1016/j.tet.2015.08.039
A novel and highly efficient cascade synthesis of spirooxindole δ-lactone derivatives from N-aryl hydroxymethylacrylamides and xanthates in good yields is described. The reaction proceeds through a radical addition/cyclization and ester exchange, in which two new C–C bonds and one C–O bond were formed.
Co-reporter:Shucheng Wang, Xuhu Huang, Bowen Li, Zemei Ge, Xin Wang, Runtao Li
Tetrahedron 2015 Volume 71(Issue 12) pp:1869-1875
Publication Date(Web):25 March 2015
DOI:10.1016/j.tet.2015.01.049
A convenient, high yielding synthesis of oxindoles by metal-free di-functionalization of alkenes with xanthates has been developed. This transformation involves a radical addition/cyclization process. Various arylalkylation products including alkyl ester-, benzyl-, cyano-, ketone-, amine-, and amide-substituted oxindoles were prepared in good to excellent yields.
Co-reporter:Laichun Luo, Lanlan Meng, Qi Sun, Zemei Ge and Runtao Li
RSC Advances 2014 vol. 4(Issue 13) pp:6845-6849
Publication Date(Web):06 Jan 2014
DOI:10.1039/C3RA46606J
An efficient approach to thiazolo[4,5-b]azepine-5,8-diones and thieno[3,2-b]azepine-5,8-diones has been developed via a domino synthesis of multifunctionalized thiazoles/thiophenes and further intramolecular cyclization. This transformation proceeded rapidly under mild conditions without use of metal catalyst.
Co-reporter:Laichun Luo, Lanlan Meng, Qi Sun, Zemei Ge, Runtao Li
Tetrahedron Letters 2014 Volume 55(Issue 1) pp:259-263
Publication Date(Web):1 January 2014
DOI:10.1016/j.tetlet.2013.11.014
A NBS-mediated sequential one-pot synthesis of multifunctionalized thiazoles and thiophenes from 1,3-dicarbonyl compounds and mercaptonitrile salts has been developed under mild conditions. This transformation involves sequential bromination/SN2 alkylation/Thorpe–Ziegler cyclization/regio-selective elimination of a –COR group, affording the desired products in moderate to good yields. The sequence of the leaving reactivity of –COR groups was determined and a possible mechanism was proposed.
Co-reporter:Peng Liu;Ao-Ze Su;Yuan-Qiang Wang;Ze-Mei Ge;Tie-Ming Cheng
Molecular Diversity 2014 Volume 18( Issue 4) pp:737-743
Publication Date(Web):2014 November
DOI:10.1007/s11030-014-9526-6
A novel four-component one-pot approach for the synthesis of 2-amino-1,3,4-thiadiazoles from primary amines, carbon disulfide, hydrazine, and acyl chlorides has been developed. A series of 5-substituted-2-amino-1,3,4-thiadiazoles were synthesized in medium-to-good yields utilizing this newly developed method.
Co-reporter:Wei Wang, Junfeng Wang, Shuo Zhou, Qi Sun, Zemei Ge, Xin Wang and Runtao Li
Chemical Communications 2013 vol. 49(Issue 13) pp:1333-1335
Publication Date(Web):19 Dec 2012
DOI:10.1039/C2CC35488H
The efficient asymmetric Michael addition reactions of aryl methyl ketones with 2-furanones were catalyzed by a simple and commercially available chiral 1,2-diphenyl-1,2-ethanediamine and p-TSA·H2O as cocatalyst with good yields (up to 95%) and excellent enantioselectivities (up to >99% ee). A bi-functional catalytic mechanism for the reaction was proposed.
Co-reporter:Pengchao Gao, Penglin Leng, Qi Sun, Xin Wang, Zemei Ge and Runtao Li
RSC Advances 2013 vol. 3(Issue 38) pp:17150-17155
Publication Date(Web):17 Jul 2013
DOI:10.1039/C3RA42503G
A novel atom-economic three-component one-pot reaction of a primary amine, an S-alkylisothiouronium salt and a Michael receptor is reported, which affords a guanidinium salt and thioether simultaneously. The guanidine moiety is involved in catalyzing the conjugated Michael addition of the mercaptan. The reaction proceeds under ambient conditions using a non-toxic EtOH–H2O mixture as the solvent, and the two products can be very easily purified. Complete atom economy is achieved by fully utilizing the S-alkylisothiouronium salt and converting the previously wasted mercaptan by-product into the valuable thioether.
Co-reporter:Hailiang Tan, Jie Wang, Yu Zhang, Yongning Xing, Qi Sun, Runtao Li
Tetrahedron 2013 69(38) pp: 8299-8304
Publication Date(Web):
DOI:10.1016/j.tet.2013.05.105
Co-reporter:Yu-peng Liu, Jin-ming Liu, Xin Wang, Tie-ming Cheng, Run-tao Li
Tetrahedron 2013 69(25) pp: 5242-5247
Publication Date(Web):
DOI:10.1016/j.tet.2013.04.006
Co-reporter:Qiao-Yan Li;Ze-Mei Ge;Tie-Ming Cheng;Run-Tao Li
Molecular Diversity 2012 Volume 16( Issue 3) pp:431-439
Publication Date(Web):2012 August
DOI:10.1007/s11030-012-9376-z
A convenient and practical method for the synthesis of 2-alkylthio-4-amino-5-cyano-6-aryl(alkyl)pyrimidines has been developed via a three-component, one-pot reaction from aldehydes, malononitrile and S-alkylisothiouronium salts in water at room temperature. A series of polysubstituted pyrimidines were prepared by this method in moderate to excellent yields. In addition, two kinds of pyrimidine-fused heterocyclic derivatives with potential pharmacological activity were constructed from our 2-alkylthio-4-amino-5-cyano-6-arylpyrimidines.
Co-reporter:Xin Ai, Xin Wang, Jin-ming Liu, Ze-mei Ge, Tie-ming Cheng, Run-tao Li
Tetrahedron 2010 66(29) pp: 5373-5377
Publication Date(Web):
DOI:10.1016/j.tet.2010.05.054
Co-reporter:Yongqiang Zhu, Shuyang Yao, Bo Xu, Zemei Ge, Jingrong Cui, Tieming Cheng, Runtao Li
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 19) pp:6851-6861
Publication Date(Web):1 October 2009
DOI:10.1016/j.bmc.2009.08.023
A series of tripeptide boronate proteasome inhibitors were designed and synthesized on the basis of our previously built tripeptide aldehyde 3D-QSAR models. All the synthesized compounds were evaluated for their proteasome-inhibitory activities in an isolated 20S rabbit proteasome, and selected compounds were evaluated for their antitumor activities in vitro against four human cancer cell lines. Biological results showed bulky and negative substituents at P2 position improved the proteasome-inhibitory potency obviously, which completely conformed to the theoretical models, while those at P3 position thoroughly deviated from the 3D-QSAR model. Most of the screened compounds showed less than 1 nM inhibitory potency and high selectivity against 20S proteasome, of which 7f is the most potent (IC50 = 0.079 nM) and twofold more active than bortezomib (IC50 = 0.161 nM). Cell viability indicated hydrophilic 4-hydroxyphenyl substituent at P2 or P3 position was not favorable to the cellular activities. Especially for the two hematologic cancer cell lines, HL-60 and U266, 7f inhibited them at the level of less than 10 nM and was more potent than the control bortezomib. It is being considered a promising new lead to be developed for the treatment of various cancers.Series of tripeptide boronic acid proteasome inhibitors were designed and synthesized. The most active compound inhibited 20S proteasome with IC50 less than 0.1 nM and inhibited two hematologic cell lines with IC50 less than 10 nM.
Co-reporter:Shuai Xia, Xin Wang, Ze-mei Ge, Tie-ming Cheng, Run-tao Li
Tetrahedron 2009 65(5) pp: 1005-1009
Publication Date(Web):
DOI:10.1016/j.tet.2008.11.084
Co-reporter:Qi Sun, Cai-Qin Yue, Jia Ye, Chang-Ling Li, Tie-Ming Cheng, Run-Tao Li
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 22) pp:6245-6249
Publication Date(Web):15 November 2007
DOI:10.1016/j.bmcl.2007.09.026
Two novel classes of monospirocyclopiperazinium salts were designed, synthesized, and evaluated for their in vivo analgesic activities. Some interesting structure–activity relationships are revealed: (1) Spirocyclopiperazinium moiety is favorable to improve the analgesic activity; (2) The size and conformation of spirocyclopiperazinium moiety significantly affects the analgesic activity; (3) Phenylethyl group of 3d is a crucial pharmacophore. Among the compounds synthesized, 3d exhibited the most potent activity with low toxicity. Further antinociceptive mechanism studies of 3d showed that these compounds will be a new kind of analgesics.
Co-reporter:Yong-Qiang Zhu, Jian-Feng Pei, Zhen-Ming Liu, Lu-Hua Lai, Jing-Rong Cui, Run-Tao Li
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 5) pp:1483-1496
Publication Date(Web):1 March 2006
DOI:10.1016/j.bmc.2005.10.003
The ubiquitin–proteasome pathway plays a crucial role in the regulation of many physiological processes and in the development of a number of major human diseases, such as cancer, Alzheimer’s, Parkinson’s, diabetes, etc. As a new target, the study on the proteasome inhibitors has received much attention recently. Three-dimensional quantitative structure–activity relationship (3D-QSAR) studies using comparative molecule field analysis (CoMFA) and comparative molecule similarity indices analysis (CoMSIA) techniques were applied to analyze the binding affinity of a set of tripeptide aldehyde inhibitors of 20S proteasome. The optimal CoMFA and CoMSIA models obtained for the training set were all statistically significant with cross-validated coefficients (q2) of 0.615, 0.591 and conventional coefficients (r2) of 0.901, 0.894, respectively. These models were validated by a test set of eight molecules that were not included in the training set. The predicted correlation coefficients (r2) of CoMFA and CoMSIA are 0.944 and 0.861, respectively. The CoMFA and CoMSIA field contour maps agree well with the structural characteristics of the binding pocket of β5 subunit of 20S proteasome, which suggests that the 3D-QSAR models built in this paper can be used to guide the development of novel inhibitors of 20S proteasome.
Co-reporter:Ying-Bo Li, Zhong-Qing Wang, Xu Yan, Mei-Wan Chen, Jiao-Lin Bao, Guo-Sheng Wu, Ze-Mei Ge, De-Min Zhou, Yi-Tao Wang, Run-Tao Li
Cancer Letters (28 October 2013) Volume 340(Issue 1) pp:88-96
Publication Date(Web):28 October 2013
DOI:10.1016/j.canlet.2013.07.005
Highlights•We reported IC-4 as a novel irreversible TKI for EGFR.•IC-4 inhibited proliferation, induced apoptosis and a G2/M arrest in breast cancer cells.•IC-4 exhibited anti-angiogenesis effects in vitro and in vivo.Accumulating evidence suggested that the irreversible tyrosine kinase inhibitors (TKIs) have potential to override the acquired resistance to target-based therapies. Herein, we reported IC-4 as a novel irreversible TKI for epidermal growth factor receptor (EGFR). IC-4 potentially suppressed proliferation, induced apoptosis and a G2/M cell cycle arrest in breast cancer cells, correlating with inhibition of EGF-induced EGFR activation, but independent of DNA damage. In addition, IC-4 exhibited anti-angiogenetic activities both in vitro and in vivo. It suppressed cell viability and proliferation induced by various growth factors in human umbilical vein endothelial cells (HUVECs). IC-4 also inhibited HUVECs migration and tube formation. In transgenic zebrafish embryo model, IC-4 was shown to suppress formation of intersegmental vessel and development of subintestinal vessels. Taken together, these results demonstrated that IC-4 is a new irreversible EGFR-TKI, exhibiting potent anti-breast cancer and anti-angiogenetic effects.
Co-reporter:Bin Liu, Xuhu Huang, Xin Wang, Zemei Ge and Runtao Li
Inorganic Chemistry Frontiers 2015 - vol. 2(Issue 7) pp:NaN800-800
Publication Date(Web):2015/04/29
DOI:10.1039/C5QO00104H
Herein we reported a Pd-catalyzed ortho-acetoxylation of arenes through a six-membered system using an amide-oxazoline as the directing group. A wide range of aryls and heteroaryls are tolerated. The approach provides general and straightforward access to phenol esters without the need for extra oxidants.
Co-reporter:Bin Liu, Xuhu Huang, Xin Wang, Zemei Ge and Runtao Li
Inorganic Chemistry Frontiers 2015 - vol. 2(Issue 7) pp:NaN860-860
Publication Date(Web):2015/05/27
DOI:10.1039/C5QO90028J
Correction for ‘Palladium catalyzed amide-oxazoline directed C–H acetoxylation of arenes’ by Bin Liu et al., Org. Chem. Front., 2015, DOI: 10.1039/c5qo00104h.
Co-reporter:Zhushuang Bai, Ling Ji, Zemei Ge, Xin Wang and Runtao Li
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 19) pp:NaN5366-5366
Publication Date(Web):2015/04/13
DOI:10.1039/C5OB00708A
The first bifunctional thiourea catalyzed asymmetric Michael addition reactions of nitroalkanes to 2-furanones are described. The highly functionalized γ-lactones with two or three consecutive stereogenic carbons were obtained in high yields (up to 99%), high diastereoselectivities (up to >20:1 dr) and enantioselectivities (up to >99% ee).
Co-reporter:Wei Wang, Junfeng Wang, Shuo Zhou, Qi Sun, Zemei Ge, Xin Wang and Runtao Li
Chemical Communications 2013 - vol. 49(Issue 13) pp:NaN1335-1335
Publication Date(Web):2012/12/19
DOI:10.1039/C2CC35488H
The efficient asymmetric Michael addition reactions of aryl methyl ketones with 2-furanones were catalyzed by a simple and commercially available chiral 1,2-diphenyl-1,2-ethanediamine and p-TSA·H2O as cocatalyst with good yields (up to 95%) and excellent enantioselectivities (up to >99% ee). A bi-functional catalytic mechanism for the reaction was proposed.
Co-reporter:Bin Liu, Xin Wang, Zemei Ge and Runtao Li
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 10) pp:NaN2949-2949
Publication Date(Web):2016/02/09
DOI:10.1039/C6OB00179C
Herein we report a novel iridium(III)-catalyzed ortho-mono-alkynylation of 7-azaindoles under mild conditions. This approach provides a general and straightforward access to form novel 7-azaindole derivatives with ample substrate scope and broad group tolerance.





![6-Quinazolinol, 4-[(3-bromophenyl)amino]-7-methoxy-](http://img.cochemist.com/ccimg/295400/295330-61-9.png)
![6-Quinazolinol, 4-[(3-bromophenyl)amino]-7-methoxy-](http://img.cochemist.com/ccimg/295400/295330-61-9_b.png)































![2-(2-phenylimidazo[1,2-a]pyridin-3-yl)acetonitrile](http://img.cochemist.com/ccimg/885300/885272-84-4.png)
![2-(2-phenylimidazo[1,2-a]pyridin-3-yl)acetonitrile](http://img.cochemist.com/ccimg/885300/885272-84-4_b.png)
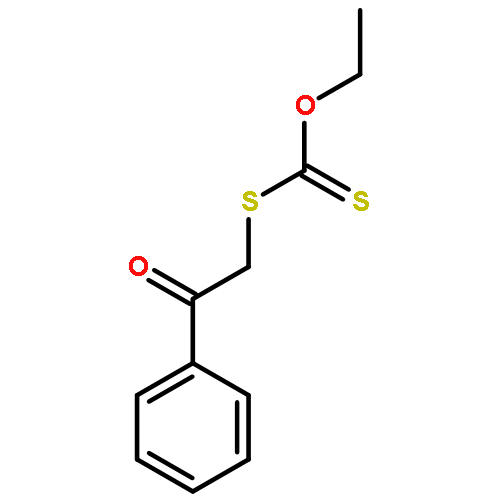
![CARBONODITHIOIC ACID, O-ETHYL S-[2-(4-MORPHOLINYL)-2-OXOETHYL] ESTER](http://img.cochemist.com/ccimg/682200/682153-32-8.png)
![CARBONODITHIOIC ACID, O-ETHYL S-[2-(4-MORPHOLINYL)-2-OXOETHYL] ESTER](http://img.cochemist.com/ccimg/682200/682153-32-8_b.png)









![Propanoic acid, 2-[(ethoxythioxomethyl)thio]-, methyl ester](/data/chemimg/106200/351491-23-1.png)
![Propanoic acid, 2-[(ethoxythioxomethyl)thio]-, methyl ester](/data/chemimg/106200/351491-23-1_b.png)








![Propanoic acid, 2-[(ethoxythioxomethyl)thio]-2-methyl-, ethyl ester](/data/chemimg/1798300/227205-62-1.png)
![Propanoic acid, 2-[(ethoxythioxomethyl)thio]-2-methyl-, ethyl ester](/data/chemimg/1798300/227205-62-1_b.png)








![Imidazo[1,2-a]pyridine-3-acetic acid, 6-chloro-2-phenyl-, ethyl ester](http://img.cochemist.com/ccimg/194000/193979-43-0.png)
![Imidazo[1,2-a]pyridine-3-acetic acid, 6-chloro-2-phenyl-, ethyl ester](http://img.cochemist.com/ccimg/194000/193979-43-0_b.png)




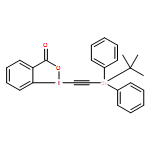
![1-[2-tri(propan-2-yl)silylethynyl]-1λ3,2-benziodoxol-3-one](/data/chemimg/1772700/181934-30-5.png)
![1-[2-tri(propan-2-yl)silylethynyl]-1λ3,2-benziodoxol-3-one](/data/chemimg/1772700/181934-30-5_b.png)







![Acetic acid, [(ethoxythioxomethyl)thio]-, methyl ester](http://img.cochemist.com/ccimg/124000/123972-89-4.png)
![Acetic acid, [(ethoxythioxomethyl)thio]-, methyl ester](http://img.cochemist.com/ccimg/124000/123972-89-4_b.png)





















![Cyclopenta[b]thiophene-4,6-dione](http://img.cochemist.com/ccimg/101000/100925-76-6.png)
![Cyclopenta[b]thiophene-4,6-dione](http://img.cochemist.com/ccimg/101000/100925-76-6_b.png)





![O-Ethyl S-[2-(4-methoxyphenyl)-2-oxoethyl] carbonodithioate](http://img.cochemist.com/ccimg/93700/93624-01-2.png)
![O-Ethyl S-[2-(4-methoxyphenyl)-2-oxoethyl] carbonodithioate](http://img.cochemist.com/ccimg/93700/93624-01-2_b.png)




![Imidazo[1,2-a]pyridine,6-chloro-2-(4-chlorophenyl)-](http://img.cochemist.com/ccimg/89000/88964-99-2.png)
![Imidazo[1,2-a]pyridine,6-chloro-2-(4-chlorophenyl)-](http://img.cochemist.com/ccimg/89000/88964-99-2_b.png)










![2-(6-Chloro-2-(4-chlorophenyl)imidazo[1,2-a]pyridin-3-yl)-N,N-dipropylacetamide](http://img.cochemist.com/ccimg/82700/82626-01-5.png)
![2-(6-Chloro-2-(4-chlorophenyl)imidazo[1,2-a]pyridin-3-yl)-N,N-dipropylacetamide](http://img.cochemist.com/ccimg/82700/82626-01-5_b.png)




























![Methanone, [4-amino-2-(methylthio)-5-thiazolyl]phenyl-](http://img.cochemist.com/ccimg/39800/39736-27-1.png)
![Methanone, [4-amino-2-(methylthio)-5-thiazolyl]phenyl-](http://img.cochemist.com/ccimg/39800/39736-27-1_b.png)
![1-[4-AMINO-2-(METHYLSULFANYL)-1,3-THIAZOL-5-YL]ETHANONE](http://img.cochemist.com/ccimg/39800/39736-26-0.png)
![1-[4-AMINO-2-(METHYLSULFANYL)-1,3-THIAZOL-5-YL]ETHANONE](http://img.cochemist.com/ccimg/39800/39736-26-0_b.png)












![Acetic acid, [(ethoxythioxomethyl)thio]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/27300/27240-57-9.png)
![Acetic acid, [(ethoxythioxomethyl)thio]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/27300/27240-57-9_b.png)











![2-Phenylbenzo[d]imidazo[2,1-b]thiazole](http://img.cochemist.com/ccimg/17900/17833-07-7.png)
![2-Phenylbenzo[d]imidazo[2,1-b]thiazole](http://img.cochemist.com/ccimg/17900/17833-07-7_b.png)








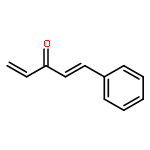
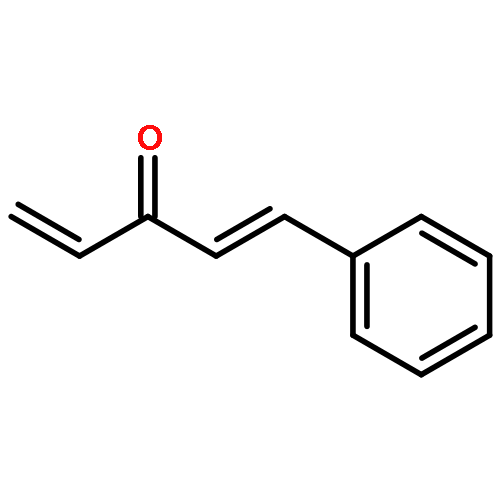
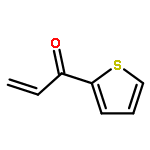

![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)


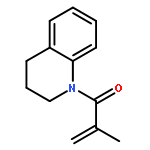









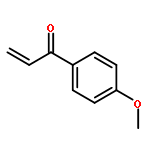
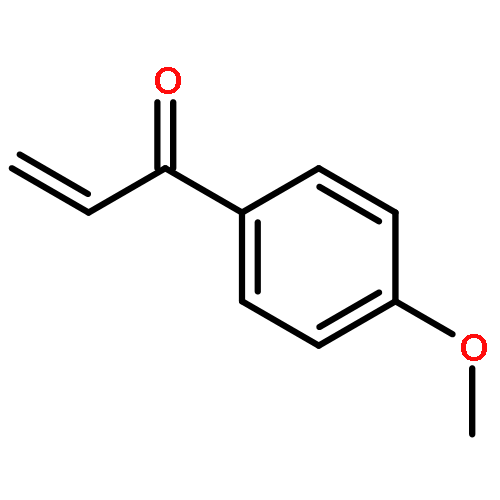
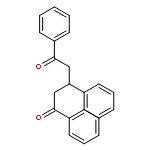



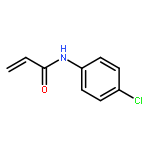
















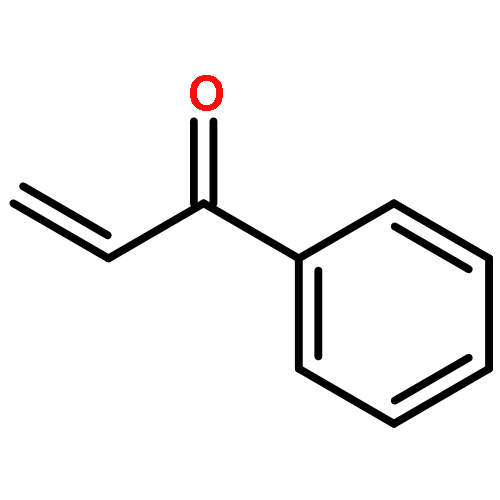




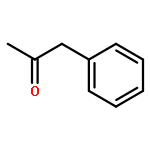
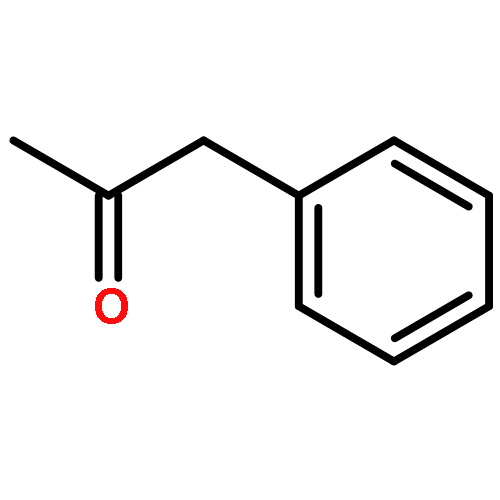




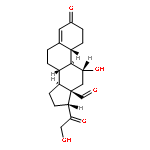
![1H-PYRROLO[2,3-B]PYRIDINE, 1-PHENYL-](http://img.cochemist.com/ccimg/343300/343238-82-4.png)
![1H-PYRROLO[2,3-B]PYRIDINE, 1-PHENYL-](http://img.cochemist.com/ccimg/343300/343238-82-4_b.png)
![Imidazo[1,2-a]pyridine, 2-phenyl-8-(phenylmethoxy)-](http://img.cochemist.com/ccimg/100600/100592-06-1.png)
![Imidazo[1,2-a]pyridine, 2-phenyl-8-(phenylmethoxy)-](http://img.cochemist.com/ccimg/100600/100592-06-1_b.png)
![2-(p-Tolyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/66000/65964-60-5.png)
![2-(p-Tolyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/66000/65964-60-5_b.png)







![Propanedinitrile, 2-[mercapto(methylthio)methylene]-, potassium salt (1:1)](/data/chemimg/1785300/20602-68-0.png)
![Propanedinitrile, 2-[mercapto(methylthio)methylene]-, potassium salt (1:1)](/data/chemimg/1785300/20602-68-0_b.png)
![Imidazo[1,2-a]pyrimidine, 2-phenyl-](http://img.cochemist.com/ccimg/15800/15764-47-3.png)
![Imidazo[1,2-a]pyrimidine, 2-phenyl-](http://img.cochemist.com/ccimg/15800/15764-47-3_b.png)
![6-PHENYLIMIDAZO[2,1-B]THIAZOLE](http://img.cochemist.com/ccimg/7100/7008-63-1.png)
![6-PHENYLIMIDAZO[2,1-B]THIAZOLE](http://img.cochemist.com/ccimg/7100/7008-63-1_b.png)

![2-Phenylimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/4200/4105-21-9.png)
![2-Phenylimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/4200/4105-21-9_b.png)
![Imidazo[1,2-a]pyridine, 2-(2-thienyl)-](http://img.cochemist.com/ccimg/4100/4045-03-8.png)
![Imidazo[1,2-a]pyridine, 2-(2-thienyl)-](http://img.cochemist.com/ccimg/4100/4045-03-8_b.png)
![6-methyl-2-phenyl-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/1100/1019-89-2.png)
![6-methyl-2-phenyl-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/1100/1019-89-2_b.png)
![7-Methyl-2-phenyl-1H-imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/900/885-91-6.png)
![7-Methyl-2-phenyl-1H-imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/900/885-91-6_b.png)
![8-methyl-2-phenyl-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/900/885-89-2.png)
![8-methyl-2-phenyl-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/900/885-89-2_b.png)



