Co-reporter:Norman L. Allinger
Journal of Computer-Aided Molecular Design 2011 Volume 25( Issue 4) pp:295-316
Publication Date(Web):2011 April
DOI:10.1007/s10822-011-9422-4
Molecular mechanics gives us a well known model of molecular structure. It is less widely recognized that valence bond theory gives us structures which offer a direct interpretation of molecular mechanics formulations and parameters. The electronic effects well-known in physical organic chemistry can be directly interpreted in terms of valence bond structures, and hence quantitatively calculated and understood. The basic theory is outlined in this paper, and examples of the effects, and their interpretation in illustrative examples is presented.
Co-reporter:Jenn-Huei Lii and Norman L. Allinger
The Journal of Physical Chemistry A 2008 Volume 112(Issue 46) pp:11903-11913
Publication Date(Web):October 22, 2008
DOI:10.1021/jp804581h
An expanded treatment of hydrogen bonding has been developed for MM4 force field calculations, which is an extension from the traditional van der Waals-electrostatic model. It adds explicit hydrogen-bond angularity by the inclusion of lone-pair directionality. The vectors that account for this directionality are placed along the hydrogen acceptor and its chemically intuitive electron pairs. No physical lone-pairs are used in the calculations. Instead, an H-bond angularity function, and a lone-pair directionality function, are incorporated into the hydrogen-bond term. The inclusion of the lone-pair directionality results in improved accuracy in hydrogen-bonded geometries and interaction energies. In this work is described hydrogen bonding in alcohols, and also in water and hydrogen fluoride dimer. The extension to other compounds such as aldehydes, ketones, amides, and so on is straightforward and will be discussed in future work. The conformational energies of ethylene glycol are discussed.
Co-reporter:Jenn-Huei Lii, Kuo-Hsiang Chen, Glenn P. Johnson, Alfred D. French, Norman L. Allinger
Carbohydrate Research 2005 Volume 340(Issue 5) pp:853-862
Publication Date(Web):11 April 2005
DOI:10.1016/j.carres.2005.01.032
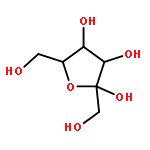
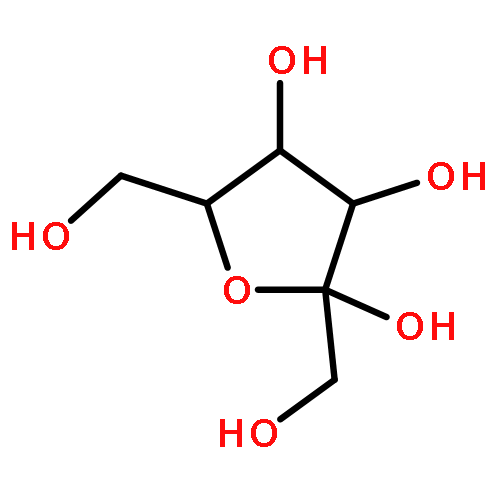
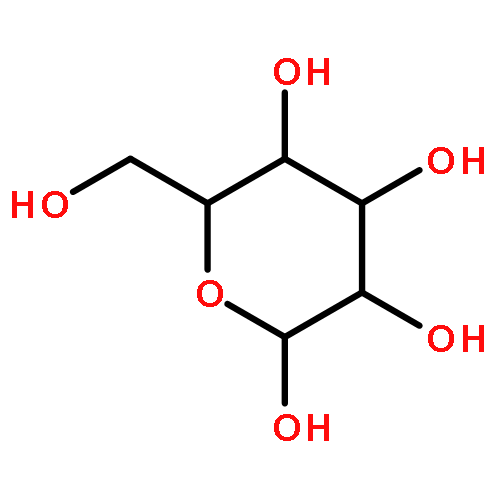
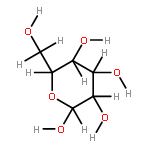
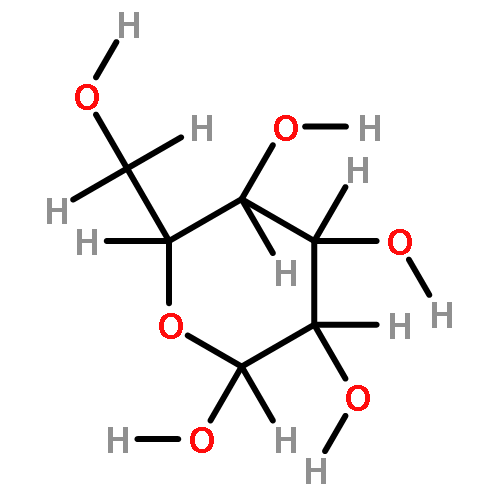
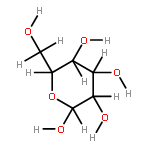
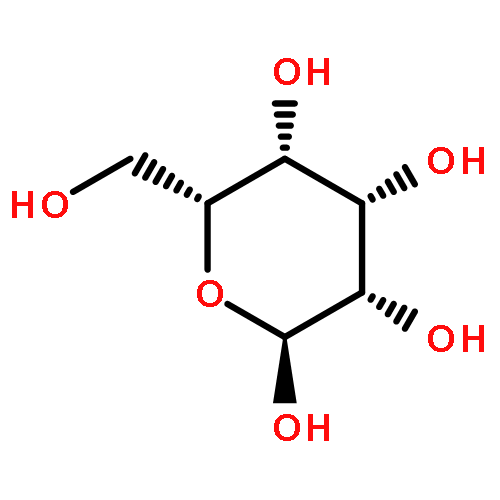
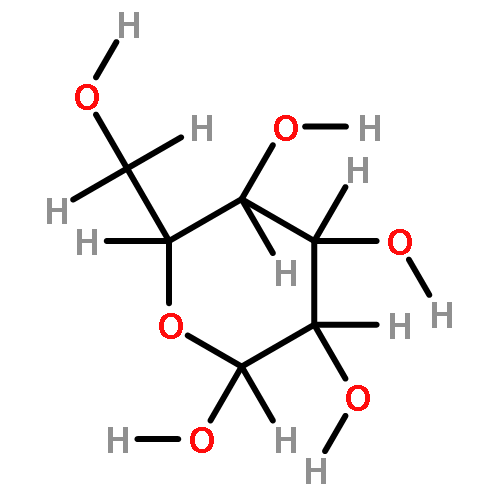
The rotational barrier for a methyl group at the end of an anomeric system is sometimes lower than we might have anticipated. Thus, in the trans–trans conformation of dimethoxymethane, the barrier to methyl rotation is calculated (B3LYP/6-311++G(2d,2p)) to be 2.22 kcal/mol, just slightly smaller than the corresponding barrier to rotation of the methyl group in methyl propyl ether of 2.32 kcal/mol. However, if the methyl being rotated in dimethoxymethane is placed into a gauche conformation, that rotational barrier is reduced to 1.52 kcal/mol. This substantial (0.80 kcal/mol relative to methyl propyl ether) reduction in barrier height in the latter case is attributed mainly to the change in the bond order of the C–O bond to which the methyl is attached, as a function of conformation, which in turn is a result of the anomeric effect. We have called this barrier lowering the external-anomeric torsional effect. This effect is apparently widespread in carbohydrates, and it results in the changing of conformational energies by up to about 2 kcal/mol. If polysaccharide potential surfaces are to be accurately mapped by molecular mechanics, this effect clearly needs to be accounted for.Torsional energies for rotation of an aglycon depend on the exo-anomeric conformation.
.jpg)














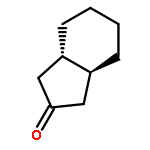
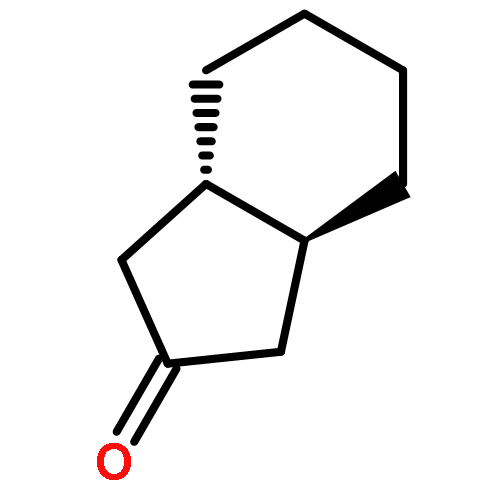
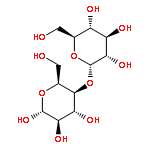
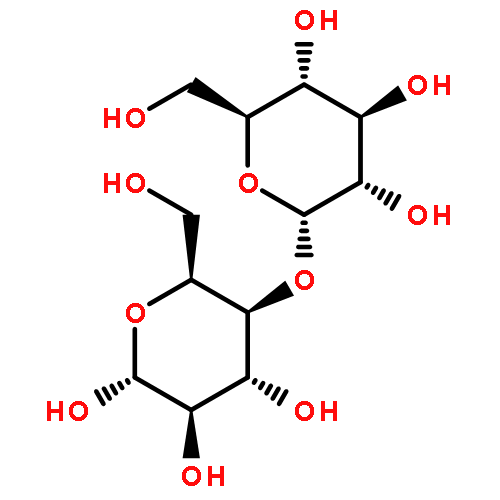
![Benzene,1-methyl-4-[(2-methyl-1-propen-1-yl)sulfonyl]-](http://img.cochemist.com/ccimg/16200/16192-03-3.png)
![Benzene,1-methyl-4-[(2-methyl-1-propen-1-yl)sulfonyl]-](http://img.cochemist.com/ccimg/16200/16192-03-3_b.png)








![Bicyclo[3,2,1]octane-3-one](http://img.cochemist.com/ccimg/14300/14252-05-2.png)
![Bicyclo[3,2,1]octane-3-one](http://img.cochemist.com/ccimg/14300/14252-05-2_b.png)


![Cyclopenta[def]fluorene](http://img.cochemist.com/ccimg/13400/13357-45-4.png)
![Cyclopenta[def]fluorene](http://img.cochemist.com/ccimg/13400/13357-45-4_b.png)



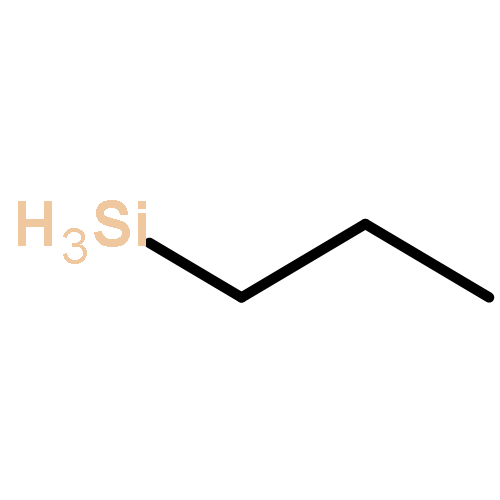




![Tricyclo[4.2.0.02,5]octane,(1R,2R,5S,6S)-rel-](http://img.cochemist.com/ccimg/13100/13027-75-3.png)
![Tricyclo[4.2.0.02,5]octane,(1R,2R,5S,6S)-rel-](http://img.cochemist.com/ccimg/13100/13027-75-3_b.png)
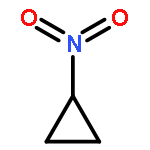
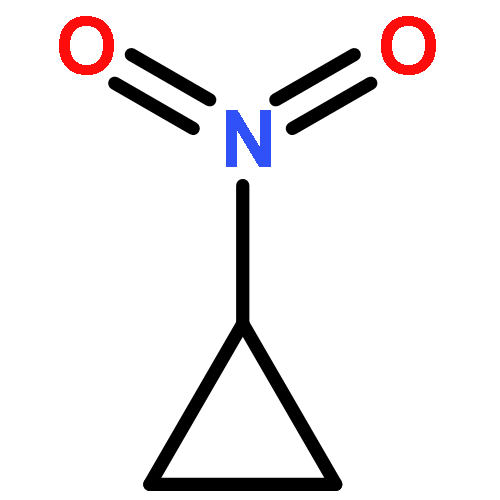
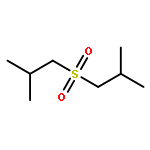
![Bicyclo[2.2.1]heptan-2-one, 1-methyl-](http://img.cochemist.com/ccimg/10300/10218-04-9.png)
![Bicyclo[2.2.1]heptan-2-one, 1-methyl-](http://img.cochemist.com/ccimg/10300/10218-04-9_b.png)
![Bicyclo[3.3.1]nonan-3-one](http://img.cochemist.com/ccimg/10100/10036-09-6.png)
![Bicyclo[3.3.1]nonan-3-one](http://img.cochemist.com/ccimg/10100/10036-09-6_b.png)
![N,N-DIMETHYL-2-{[2-PHENYL-8-(TRIFLUOROMETHYL)-4-QUINAZOLINYL]SULF<WBR />ANYL}ACETAMIDE](http://img.cochemist.com/ccimg/7800/7783-29-1.png)
![N,N-DIMETHYL-2-{[2-PHENYL-8-(TRIFLUOROMETHYL)-4-QUINAZOLINYL]SULF<WBR />ANYL}ACETAMIDE](http://img.cochemist.com/ccimg/7800/7783-29-1_b.png)


















![Bicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/6300/6221-55-2.png)
![Bicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/6300/6221-55-2_b.png)










![pentacyclo[4.4.0.0~2,5~.0~3,8~.0~4,7~]decane](http://img.cochemist.com/ccimg/5700/5603-27-0.png)
![pentacyclo[4.4.0.0~2,5~.0~3,8~.0~4,7~]decane](http://img.cochemist.com/ccimg/5700/5603-27-0_b.png)
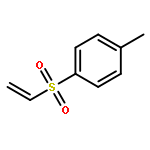
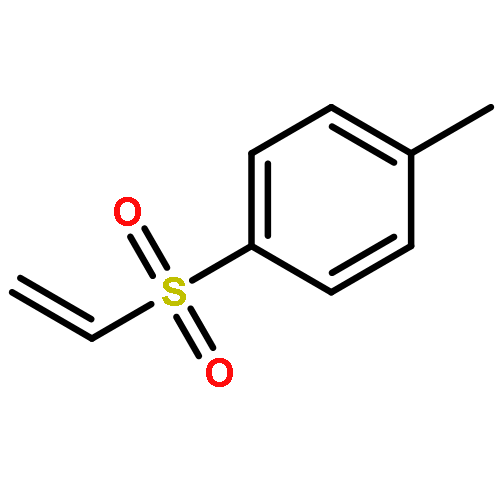




![Bicyclo[3.2.1]octan-2-one](http://img.cochemist.com/ccimg/5100/5019-82-9.png)
![Bicyclo[3.2.1]octan-2-one](http://img.cochemist.com/ccimg/5100/5019-82-9_b.png)







![bicyclo[8.2.2]tetradeca-1(12),10,13-triene](http://img.cochemist.com/ccimg/4700/4685-74-9.png)
![bicyclo[8.2.2]tetradeca-1(12),10,13-triene](http://img.cochemist.com/ccimg/4700/4685-74-9_b.png)
![2-chloro-N-{3-methyl-1-[5-({2-[(5-methyl-4-phenyl-1,3-thiazol-2-yl)amino]-2-oxoethyl}sulfanyl)-4-prop-2-en-1-yl-4H-1,2,4-triazol-3-yl]butyl}benzamide](http://img.cochemist.com/ccimg/4500/4493-27-0.png)
![2-chloro-N-{3-methyl-1-[5-({2-[(5-methyl-4-phenyl-1,3-thiazol-2-yl)amino]-2-oxoethyl}sulfanyl)-4-prop-2-en-1-yl-4H-1,2,4-triazol-3-yl]butyl}benzamide](http://img.cochemist.com/ccimg/4500/4493-27-0_b.png)

![undecacyclo[9.9.0.0~2,9~.0~3,7~.0~4,20~.0~5,18~.0~6,16~.0~8,15~.0~10,14~.0~12,19~.0~13,17~]icosane (non-preferred name)](/data/chemimg/51900/4493-23-6.png)
![undecacyclo[9.9.0.0~2,9~.0~3,7~.0~4,20~.0~5,18~.0~6,16~.0~8,15~.0~10,14~.0~12,19~.0~13,17~]icosane (non-preferred name)](/data/chemimg/51900/4493-23-6_b.png)






![bicyclo[7.2.2]trideca-1(11),9,12-triene](http://img.cochemist.com/ccimg/3800/3761-63-5.png)
![bicyclo[7.2.2]trideca-1(11),9,12-triene](http://img.cochemist.com/ccimg/3800/3761-63-5_b.png)



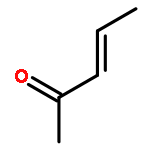
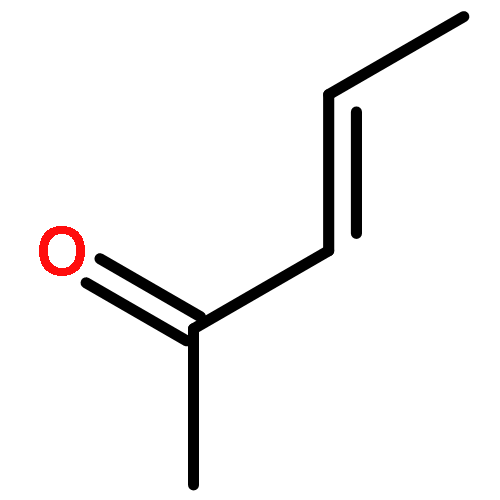



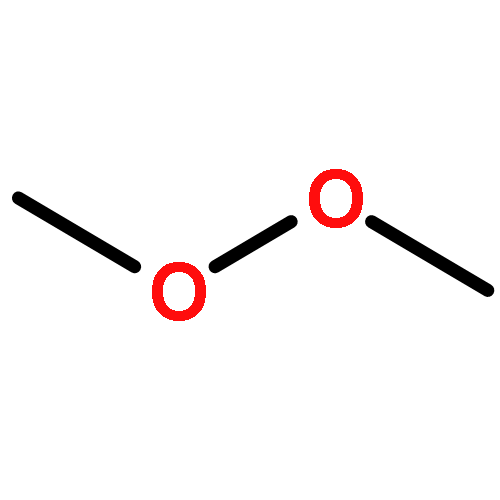

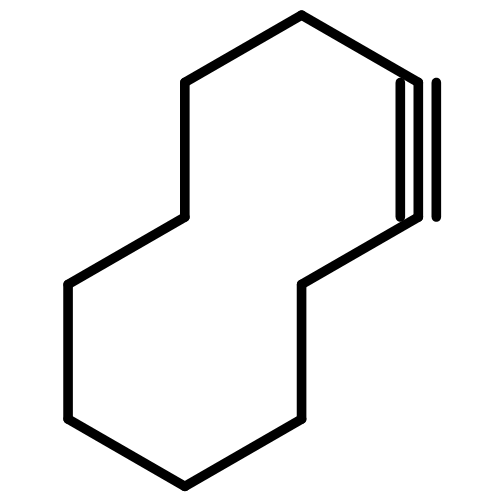










![Bicyclo[4.4.1]undeca-1,3,5,7,9-pentaene](http://img.cochemist.com/ccimg/2500/2443-46-1.png)
![Bicyclo[4.4.1]undeca-1,3,5,7,9-pentaene](http://img.cochemist.com/ccimg/2500/2443-46-1_b.png)
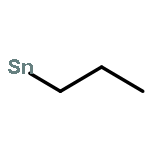
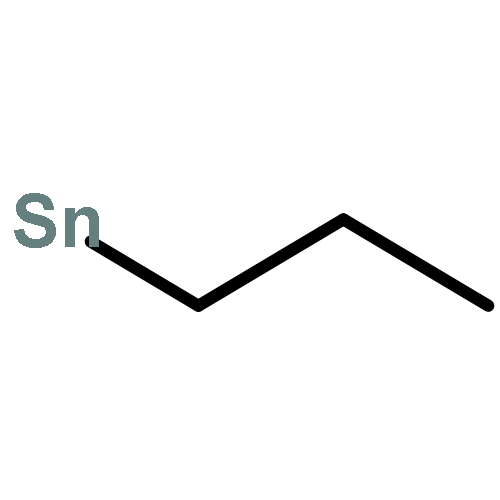
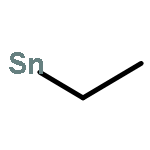
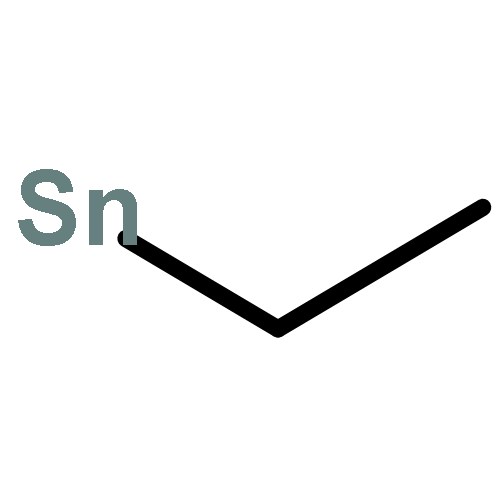
![Tricyclo[9.3.1.14,8]hexadeca-1(15),4,6,8(16),11,13-hexaene](http://img.cochemist.com/ccimg/2400/2319-97-3.png)
![Tricyclo[9.3.1.14,8]hexadeca-1(15),4,6,8(16),11,13-hexaene](http://img.cochemist.com/ccimg/2400/2319-97-3_b.png)
![Propane,2-[(1-methylethyl)sulfinyl]-](http://img.cochemist.com/ccimg/2300/2211-89-4.png)
![Propane,2-[(1-methylethyl)sulfinyl]-](http://img.cochemist.com/ccimg/2300/2211-89-4_b.png)
















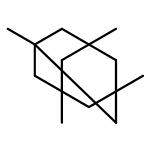







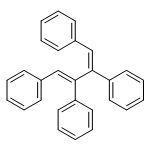




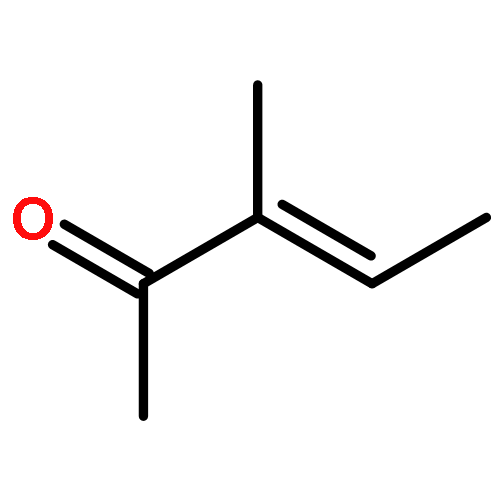
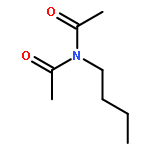
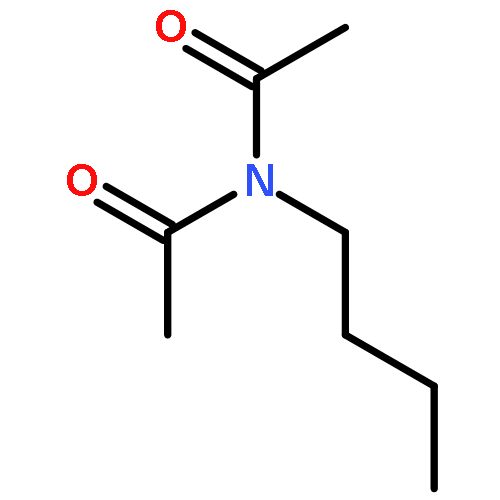


![N,N-dimethyltricyclo[3.3.1.1~3,7~]decane-1-carboxamide](http://img.cochemist.com/ccimg/1600/1502-00-7.png)
![N,N-dimethyltricyclo[3.3.1.1~3,7~]decane-1-carboxamide](http://img.cochemist.com/ccimg/1600/1502-00-7_b.png)



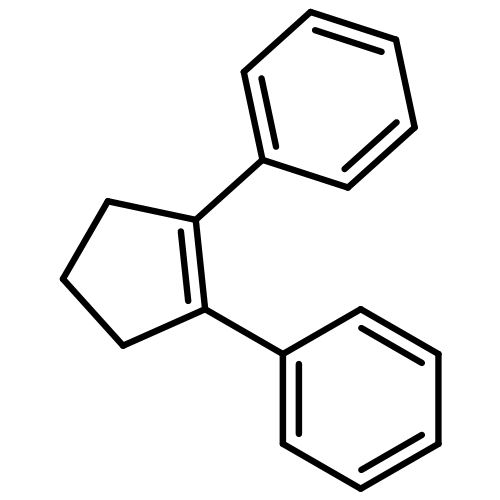




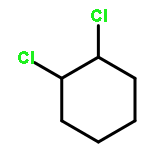
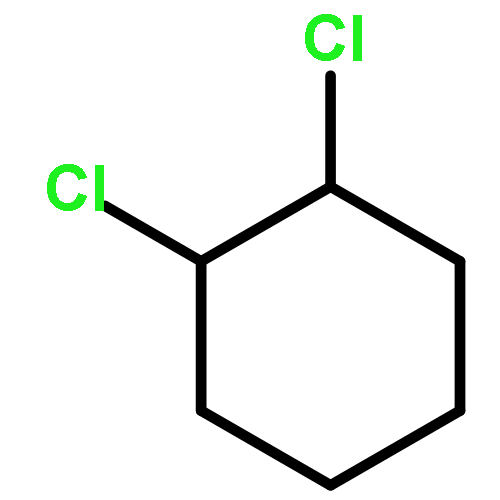
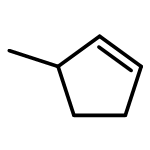














![Bicyclo[2.2.2]oct-2-ene](http://img.cochemist.com/ccimg/1000/931-64-6.png)
![Bicyclo[2.2.2]oct-2-ene](http://img.cochemist.com/ccimg/1000/931-64-6_b.png)

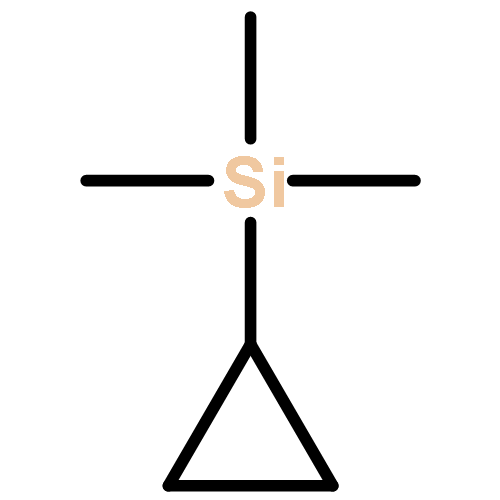



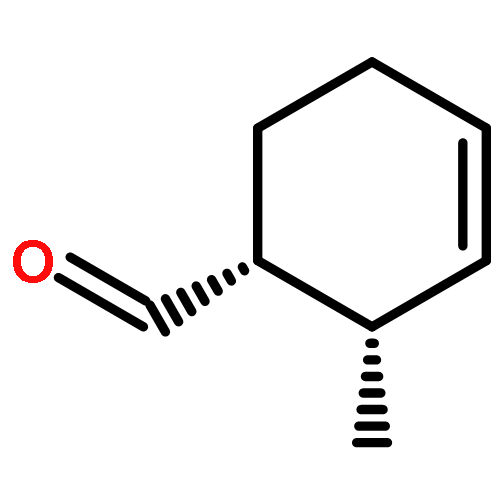
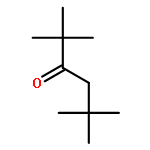
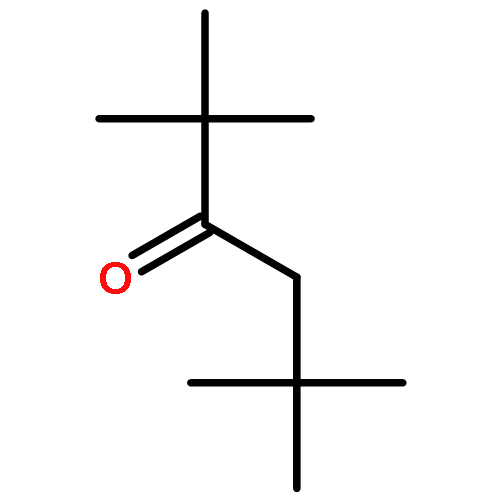




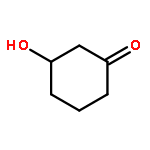
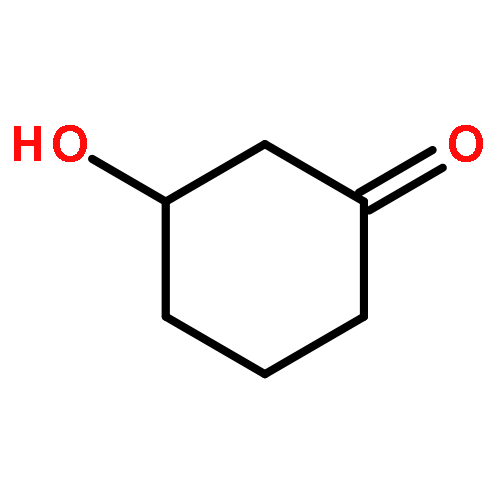






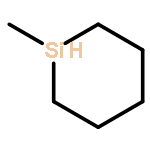
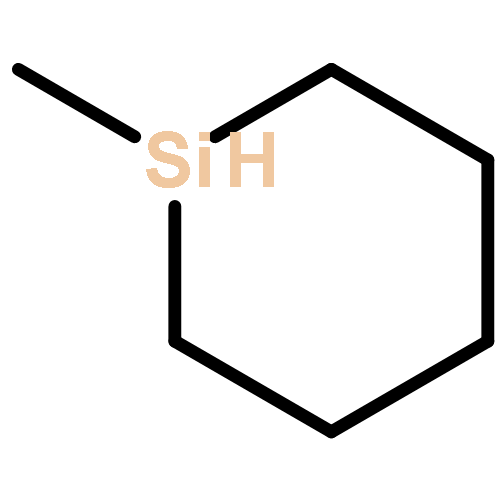




























![bicyclo[2.2.2]octa-2,5,7-triene](http://img.cochemist.com/ccimg/600/500-24-3.png)
![bicyclo[2.2.2]octa-2,5,7-triene](http://img.cochemist.com/ccimg/600/500-24-3_b.png)
![bicyclo[2.2.2]octa-2,5-diene](http://img.cochemist.com/ccimg/600/500-23-2.png)
![bicyclo[2.2.2]octa-2,5-diene](http://img.cochemist.com/ccimg/600/500-23-2_b.png)
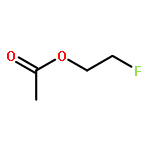
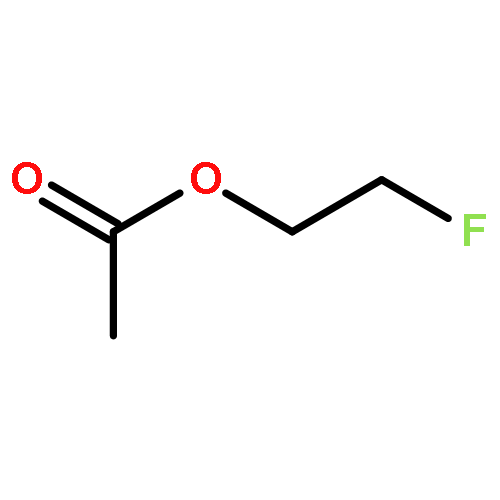
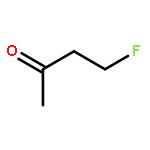
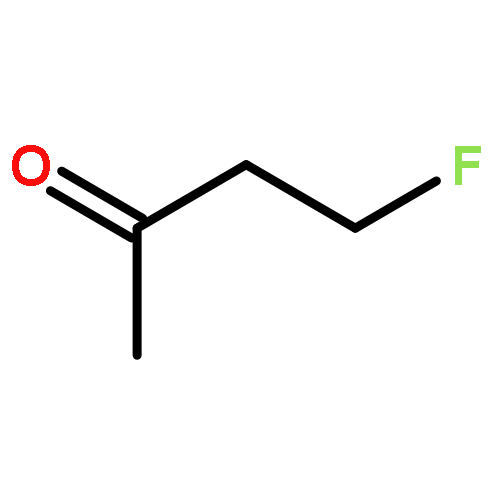
![1,12-DIMETHYLBENZO[A]ANTHRACENE](http://img.cochemist.com/ccimg/400/313-74-6.png)
![1,12-DIMETHYLBENZO[A]ANTHRACENE](http://img.cochemist.com/ccimg/400/313-74-6_b.png)





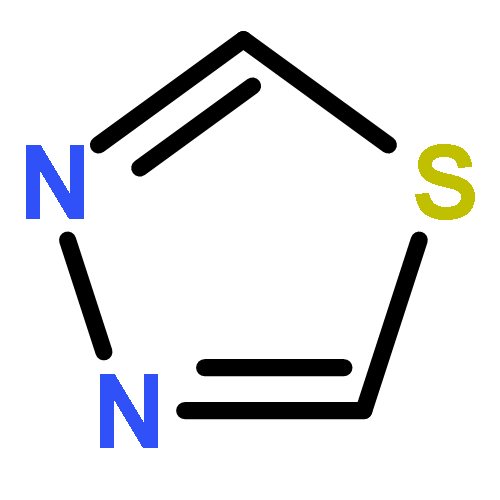
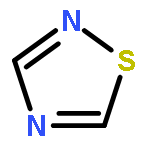
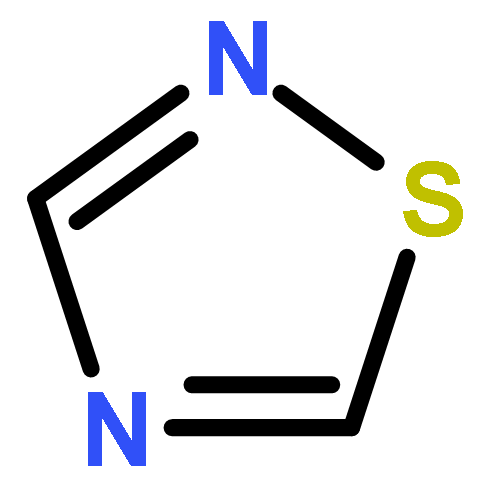








![Tricyclo[3.1.0.02,6]hexane](http://img.cochemist.com/ccimg/300/287-12-7.png)
![Tricyclo[3.1.0.02,6]hexane](http://img.cochemist.com/ccimg/300/287-12-7_b.png)
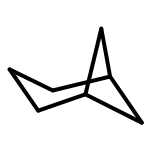
![Tricyclo[4.3.1.1(3,8)]undecane](http://img.cochemist.com/ccimg/300/281-46-9.png)
![Tricyclo[4.3.1.1(3,8)]undecane](http://img.cochemist.com/ccimg/300/281-46-9_b.png)
![9-Thiabicyclo[3.3.1]nonane](http://img.cochemist.com/ccimg/300/281-15-2.png)
![9-Thiabicyclo[3.3.1]nonane](http://img.cochemist.com/ccimg/300/281-15-2_b.png)
![2-Thiabicyclo[3.3.1]nonane](http://img.cochemist.com/ccimg/300/280-68-2.png)
![2-Thiabicyclo[3.3.1]nonane](http://img.cochemist.com/ccimg/300/280-68-2_b.png)
![Bicyclo[3.3.1]nonane](http://img.cochemist.com/ccimg/300/280-65-9.png)
![Bicyclo[3.3.1]nonane](http://img.cochemist.com/ccimg/300/280-65-9_b.png)
![6,7,8-Trioxabicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/300/280-21-7.png)
![6,7,8-Trioxabicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/300/280-21-7_b.png)
![7-Thiabicyclo[2.2.1]heptane](http://img.cochemist.com/ccimg/300/279-59-4.png)
![7-Thiabicyclo[2.2.1]heptane](http://img.cochemist.com/ccimg/300/279-59-4_b.png)
![1-Silabicyclo[2.2.1]heptane](http://img.cochemist.com/ccimg/300/279-30-1.png)
![1-Silabicyclo[2.2.1]heptane](http://img.cochemist.com/ccimg/300/279-30-1_b.png)
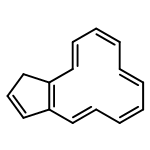
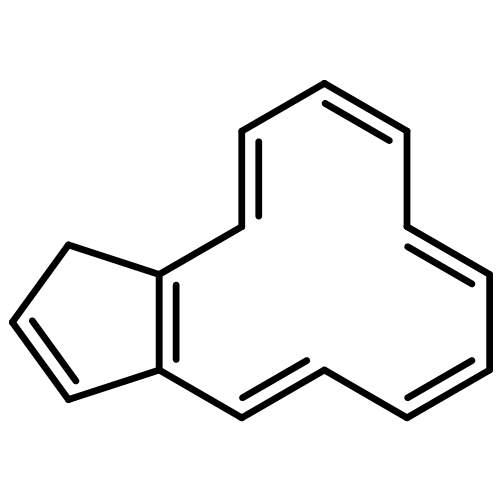
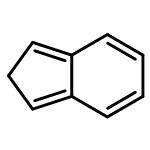
![5H-Dibenzo[a,d][7]annulene](http://img.cochemist.com/ccimg/300/256-81-5.png)
![5H-Dibenzo[a,d][7]annulene](http://img.cochemist.com/ccimg/300/256-81-5_b.png)
![Tricyclo[3.3.0.02,6]octane](http://img.cochemist.com/ccimg/300/250-21-5.png)
![Tricyclo[3.3.0.02,6]octane](http://img.cochemist.com/ccimg/300/250-21-5_b.png)
![Indeno[1,2-a]indene](http://img.cochemist.com/ccimg/300/206-71-3.png)
![Indeno[1,2-a]indene](http://img.cochemist.com/ccimg/300/206-71-3_b.png)
![Phenanthro[3,4-c]phenanthrene](http://img.cochemist.com/ccimg/200/187-83-7.png)
![Phenanthro[3,4-c]phenanthrene](http://img.cochemist.com/ccimg/200/187-83-7_b.png)
![bicyclo[2.2.0]hexane](http://img.cochemist.com/ccimg/200/186-04-9.png)
![bicyclo[2.2.0]hexane](http://img.cochemist.com/ccimg/200/186-04-9_b.png)
![tricyclo[1.1.0.0~2,4~]butane](http://img.cochemist.com/ccimg/200/157-39-1.png)
![tricyclo[1.1.0.0~2,4~]butane](http://img.cochemist.com/ccimg/200/157-39-1_b.png)




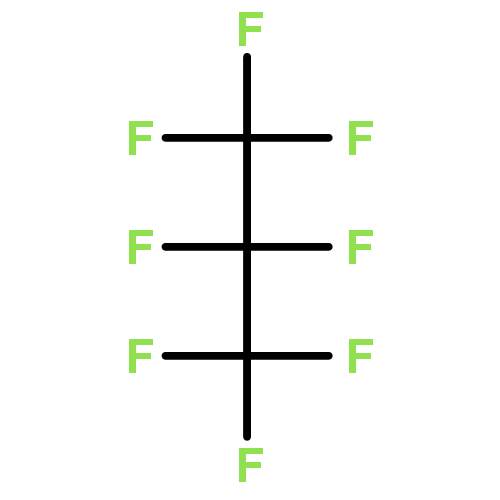






![1,1'-Bitricyclo[1.1.0.02,4]butane](http://img.cochemist.com/ccimg/119800/119703-39-8.png)
![1,1'-Bitricyclo[1.1.0.02,4]butane](http://img.cochemist.com/ccimg/119800/119703-39-8_b.png)
![1,1'-Bipentacyclo[4.2.0.02,5.03,8.04,7]octane](http://img.cochemist.com/ccimg/116600/116503-50-5.png)
![1,1'-Bipentacyclo[4.2.0.02,5.03,8.04,7]octane](http://img.cochemist.com/ccimg/116600/116503-50-5_b.png)
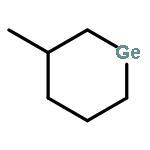
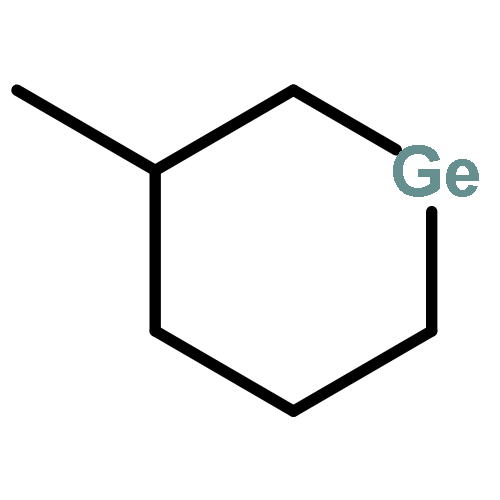
![1-SILABICYCLO[2.2.2]OCTA-2,5,7-TRIENE, 1-METHYL-](http://img.cochemist.com/ccimg/67800/67754-92-1.png)
![1-SILABICYCLO[2.2.2]OCTA-2,5,7-TRIENE, 1-METHYL-](http://img.cochemist.com/ccimg/67800/67754-92-1_b.png)
![1-Silatricyclo[3.3.1.13,7]decane, 1-methyl-](http://img.cochemist.com/ccimg/65700/65672-54-0.png)
![1-Silatricyclo[3.3.1.13,7]decane, 1-methyl-](http://img.cochemist.com/ccimg/65700/65672-54-0_b.png)
![1-Silabicyclo[2.2.1]heptane, 1-methyl-](http://img.cochemist.com/ccimg/61200/61192-29-8.png)
![1-Silabicyclo[2.2.1]heptane, 1-methyl-](http://img.cochemist.com/ccimg/61200/61192-29-8_b.png)

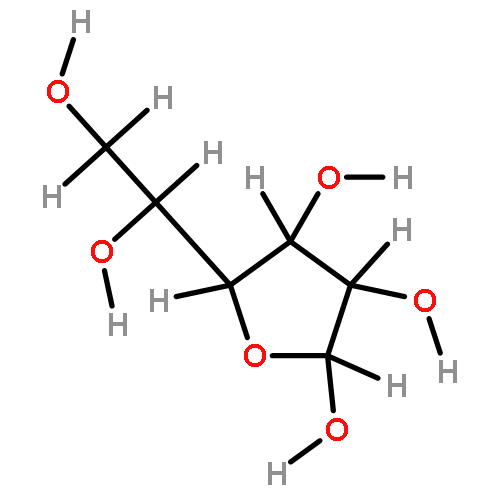


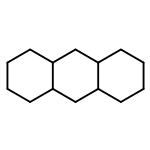
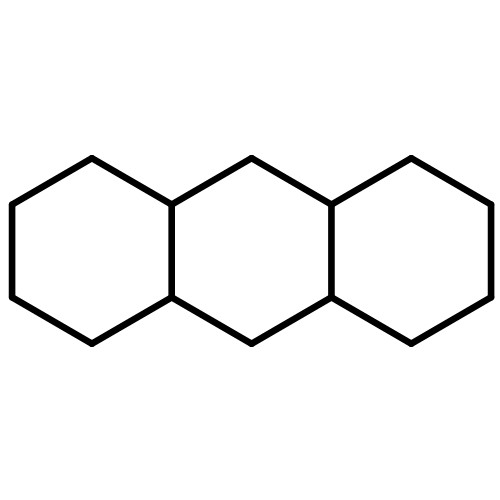
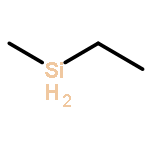
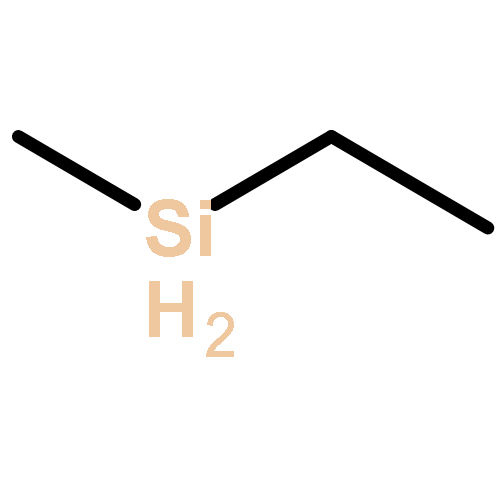
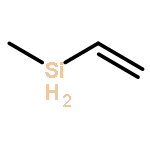
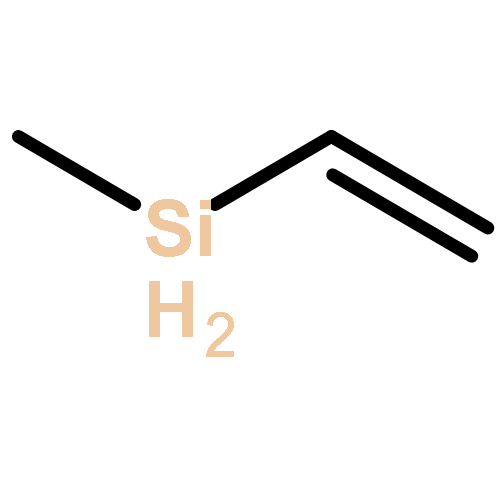


![Bicyclo[3.3.1]nonan-9-one](http://img.cochemist.com/ccimg/18000/17931-55-4.png)
![Bicyclo[3.3.1]nonan-9-one](http://img.cochemist.com/ccimg/18000/17931-55-4_b.png)




![Bicyclo[2.2.1]heptan-7-one](http://img.cochemist.com/ccimg/10300/10218-02-7.png)
![Bicyclo[2.2.1]heptan-7-one](http://img.cochemist.com/ccimg/10300/10218-02-7_b.png)
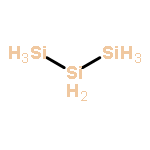



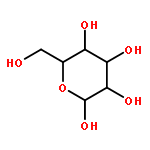






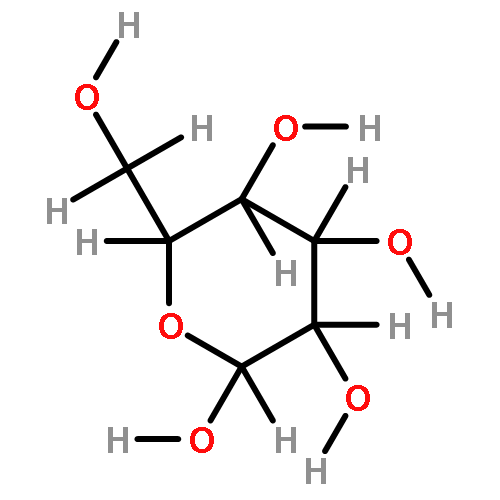
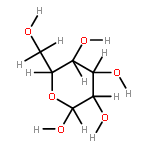
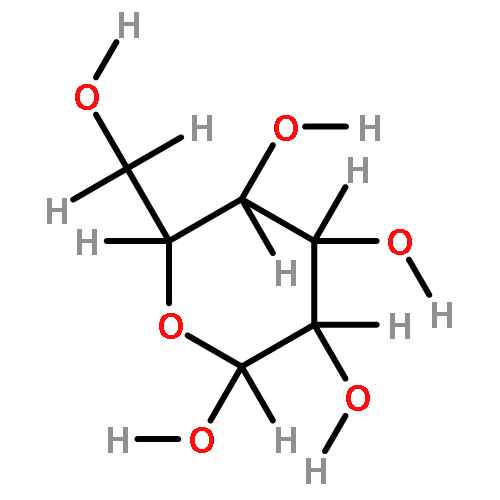
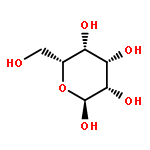

![dibenzo[ghi,mno]fluoranthene](http://img.cochemist.com/ccimg/5900/5821-51-2.png)
![dibenzo[ghi,mno]fluoranthene](http://img.cochemist.com/ccimg/5900/5821-51-2_b.png)

![hexacyclo[4.4.0.0~2,5~.0~3,9~.0~4,8~.0~7,10~]decane](http://img.cochemist.com/ccimg/4600/4572-17-2.png)
![hexacyclo[4.4.0.0~2,5~.0~3,9~.0~4,8~.0~7,10~]decane](http://img.cochemist.com/ccimg/4600/4572-17-2_b.png)




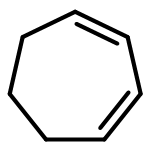
![Heptacyclo[6.4.0.02,7.03,6.04,11.05,10.09,12]dodecane](http://img.cochemist.com/ccimg/4500/4493-26-9.png)
![Heptacyclo[6.4.0.02,7.03,6.04,11.05,10.09,12]dodecane](http://img.cochemist.com/ccimg/4500/4493-26-9_b.png)
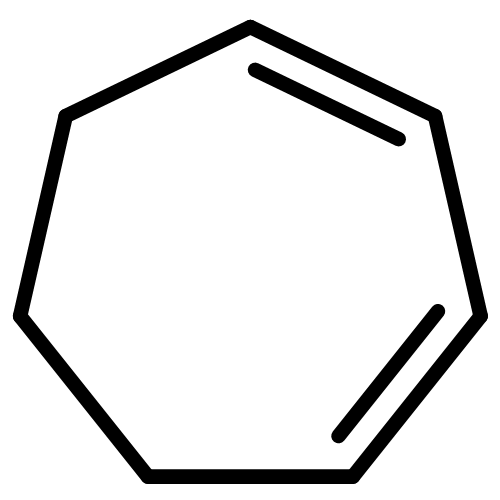





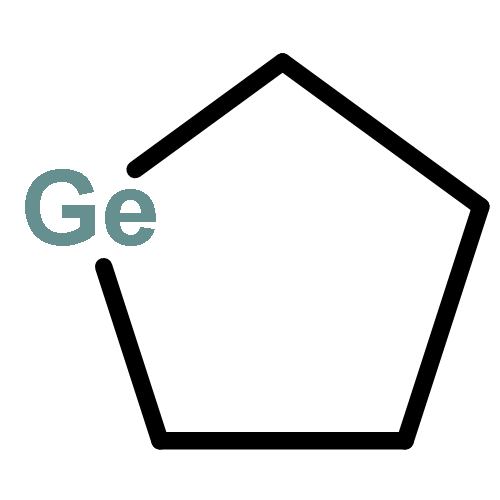


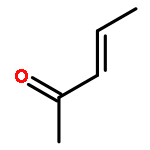
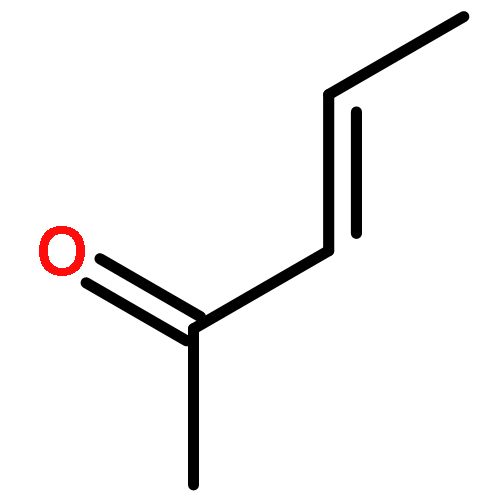
![Bicyclo[2.2.2]octan-2-one](http://img.cochemist.com/ccimg/2800/2716-23-6.png)
![Bicyclo[2.2.2]octan-2-one](http://img.cochemist.com/ccimg/2800/2716-23-6_b.png)
![Bicyclo[2.2.1]hept-1-yl](http://img.cochemist.com/ccimg/2700/2697-23-6.png)
![Bicyclo[2.2.1]hept-1-yl](http://img.cochemist.com/ccimg/2700/2697-23-6_b.png)






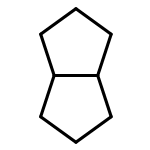


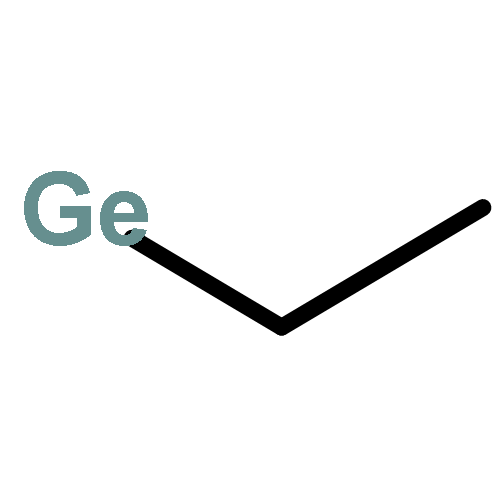














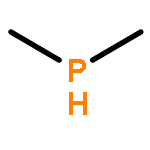
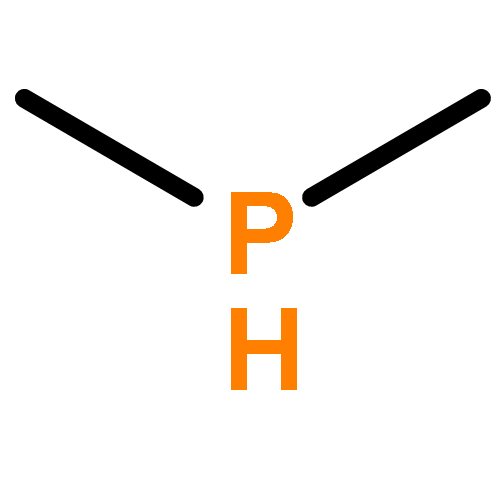
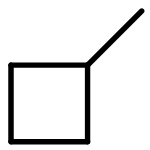
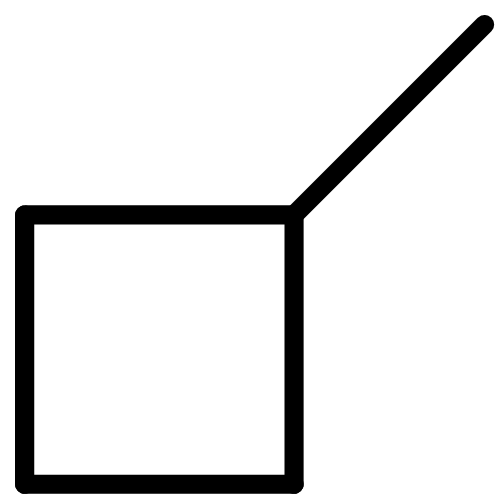


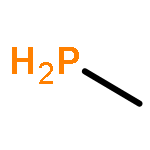
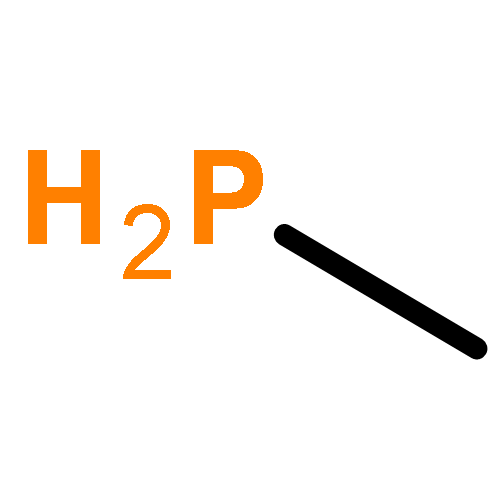
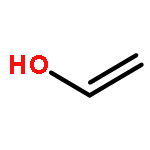
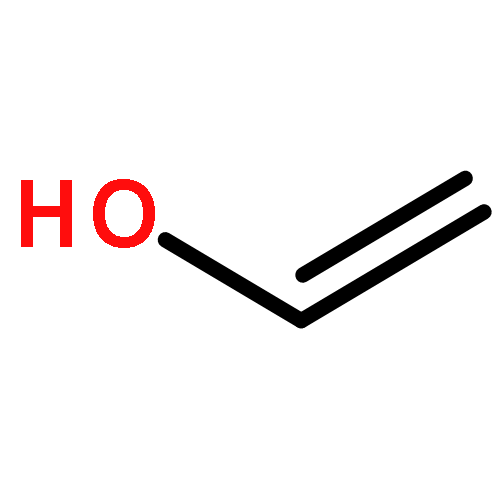



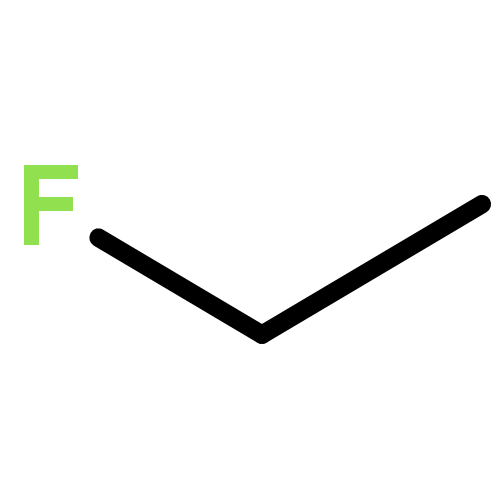
![BICYCLO[1.1.1]PENTANE](http://img.cochemist.com/ccimg/400/311-75-1.png)
![BICYCLO[1.1.1]PENTANE](http://img.cochemist.com/ccimg/400/311-75-1_b.png)
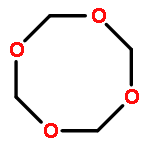
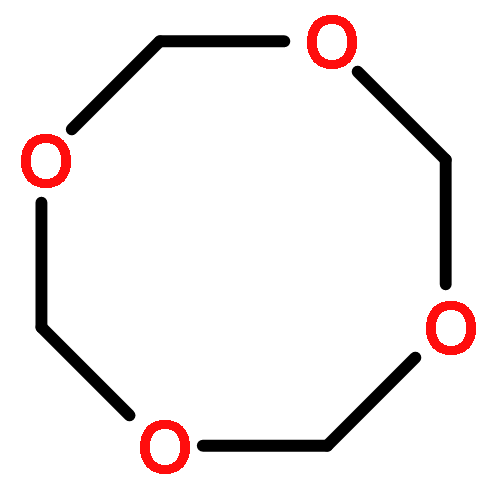
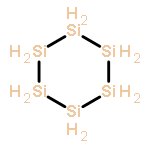




![Bicyclo[2.1.1]hexane(7CI,8CI,9CI)](http://img.cochemist.com/ccimg/300/285-86-9.png)
![Bicyclo[2.1.1]hexane(7CI,8CI,9CI)](http://img.cochemist.com/ccimg/300/285-86-9_b.png)
![Bicyclo[4.2.0]octane](http://img.cochemist.com/ccimg/300/278-30-8.png)
![Bicyclo[4.2.0]octane](http://img.cochemist.com/ccimg/300/278-30-8_b.png)




















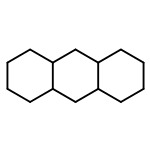
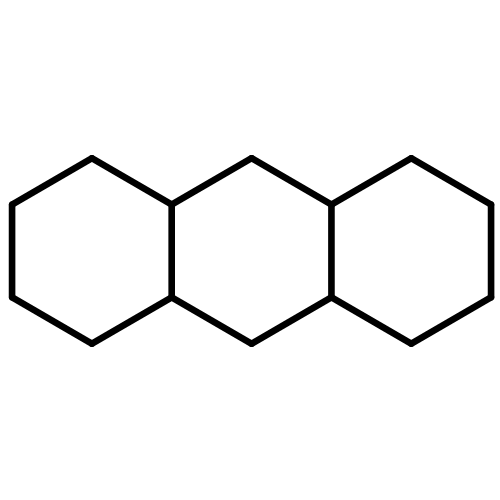



![Bicyclo[2.2.2]octane](http://img.cochemist.com/ccimg/300/280-33-1.png)
![Bicyclo[2.2.2]octane](http://img.cochemist.com/ccimg/300/280-33-1_b.png)




![Pentacyclo[4.2.0.02,5.03,8.04,7]octane](http://img.cochemist.com/ccimg/300/277-10-1.png)
![Pentacyclo[4.2.0.02,5.03,8.04,7]octane](http://img.cochemist.com/ccimg/300/277-10-1_b.png)