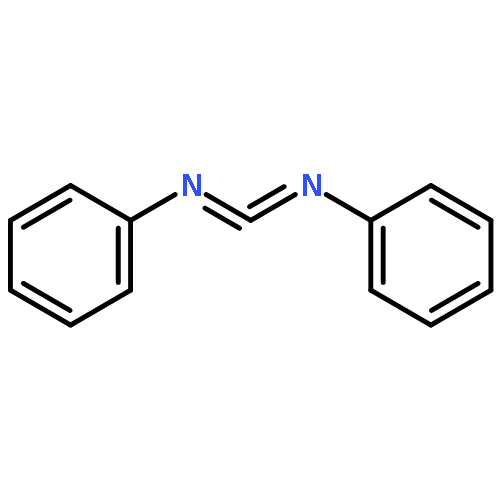
Co-reporter:Chao Wang and Hui Chen
Journal of the American Chemical Society September 20, 2017 Volume 139(Issue 37) pp:13038-13038
Publication Date(Web):August 28, 2017
DOI:10.1021/jacs.7b06343
AurF and CmlI are currently the only two known diiron arylamine oxygenases. On the basis of extensive quantum mechanical/molecular mechanical (QM/MM) spectroscopic and mechanistic modelings, here we predict that the key oxygenated intermediates in AurF and CmlI, so-called P, are uniformly hydroperoxo species having similar structures. As a basis for mechanistic unification in AurF and CmlI, the proposed diferric-hydroperoxo P is calculated to be able to promote the arylamine N-oxygenation with highly accessible kinetics. This convergent μ-η0:η2 structural assignment of P’s in AurF and CmlI can rationalize many conundrums for P, including the different Mössbauer spectroscopic parameters, low O–O vibrational frequency, ambiphilic reactivity, and inertness toward C–H activation. In view of the very limited knowledge about hydroperoxo species in diiron enzymes, the novel diferric-hydroperoxo-mediated N-oxygenation mechanism revealed in this work opens up a new avenue for understanding the O2 activation mode in nature. For elucidating the structures of transient oxidants for diiron enzymes, the promising approach of QM/MM Mössbauer spectroscopic modeling is highlighted as a key problem solver in mechanistic enzymatic research.
Co-reporter:Xuefeng Cong, Fei Fan, Pengchen Ma, Meiming Luo, Hui Chen, and Xiaoming Zeng
Journal of the American Chemical Society October 25, 2017 Volume 139(Issue 42) pp:15182-15182
Publication Date(Web):October 3, 2017
DOI:10.1021/jacs.7b08579
The cleavage of aromatic carbon–nitrogen bonds catalyzed by transition metals is of high synthetic interest because such bonds are common in organic chemistry. However, few metal catalysts can be used to selectively break C(aryl)–N bonds in electronically neutral molecules. We report here the first low-valent, high-spin chromium-catalyzed cleavage of C(aryl)–N bonds in electronically neutral aniline derivatives at room temperature. By using simple and inexpensive chromium(II) chloride as precatalyst, accompanied by an imino auxiliary, the selective arylative and alkylative C–C coupling of C(aryl)–N bonds can be achieved. Crossover experiments indicate that a low-valent chromium species, formed in situ by reduction of CrCl2 with Grignard reagent, is responsible for the catalytic cleavage of C(aryl)–N bonds. DFT calculations show that facile insertion of the C(aryl)–N bond by chromium(0) can take place in a high-spin quintet (S = 2) ground state, whereas the lower-spin singlet (S = 0) and triplet (S = 1) states are inaccessible in energy. It was found that both donation of the sole paired d electrons in the d6 shell of high-spin chromium(0) to the antibonding orbital of the C(aryl)–N bond and the nitrogen ligating interaction to the metal center with its lone pair play important roles in the cleavage of the C(aryl)–N bond by the zerovalent chromium species.
Co-reporter:Lianrui Hu, Kejuan Chen, and Hui Chen
Journal of Chemical Theory and Computation October 10, 2017 Volume 13(Issue 10) pp:4841-4841
Publication Date(Web):September 7, 2017
DOI:10.1021/acs.jctc.7b00708
Accurate modelings of reactions involving 3d transition metals (TMs) are very challenging to both ab initio and DFT approaches. To gain more knowledge in this field, we herein explored typical σ-bond activations of H–H, C–H, C–Cl, and C–C bonds promoted by nickel(0), a low-valent late 3d TM. For the key parameters of activation energy (ΔE‡) and reaction energy (ΔER) for these reactions, various issues related to the computational accuracy were systematically investigated. From the scrutiny of convergence issue with one-electron basis set, augmented (A) basis functions are found to be important, and the CCSD(T)/CBS level with complete basis set (CBS) limit extrapolation based on augmented double-ζ and triple-ζ basis pair (ADZ and ATZ), which produces deviations below 1 kcal/mol from the reference, is recommended for larger systems. As an alternative, the explicitly correlated F12 method can accelerate the basis set convergence further, especially after its CBS extrapolations. Thus, the CCSD(T)-F12/CBS(ADZ-ATZ) level with computational cost comparable to the conventional CCSD(T)/CBS(ADZ-ATZ) level, is found to reach the accuracy of the conventional CCSD(T)/A5Z level, which produces deviations below 0.5 kcal/mol from the reference, and is also highly recommendable. Scalar relativistic effects and 3s3p core–valence correlation are non-negligible for achieving chemical accuracy of around 1 kcal/mol. From the scrutiny of convergence issue with the N-electron basis set, in comparison with the reference CCSDTQ result, CCSD(T) is found to be able to calculate ΔE‡ quite accurately, which is not true for the ΔER calculations. Using highest-level CCSD(T) results of ΔE‡ in this work as references, we tested 18 DFT methods and found that PBE0 and CAM-B3LYP are among the three best performing functionals, irrespective of DFT empirical dispersion correction. With empirical dispersion correction included, ωB97XD is also recommendable due to its improved performance.
Co-reporter:Lianrui Hu and Hui Chen
Journal of the American Chemical Society November 8, 2017 Volume 139(Issue 44) pp:15564-15564
Publication Date(Web):October 24, 2017
DOI:10.1021/jacs.7b06086
Iron-catalyzed alkene [2+2] cycloaddition reactions represent a promising stepwise pathway to effect the kinetically hindered concerted [2+2] cycloaddition. However, the fundamental reactivity paradigm of these reactions remains unclear. Based on high level combined CASPT2/DFT modelings, herein we reveal an unprecedented substrate-dependent two-state reactivity scenario for the key C—C coupling in this iron catalysis, in which the representative substrates of mono-olefins only and mono-olefin plus 1,3-diene exhibit different reactivity paradigms. The role of the redox-active ligand is found to generate a ferric oxidation state for the metallacyclic intermediate of C—C coupling, thereby rendering a thermodynamically more accessible FeIII/FeI reductive elimination process compared with the otherwise FeII/Fe0 one. The enhancement of the spin state transition efficiency between the singlet and triplet states is predicted as an alternative way to increase the C—C coupling reactivity in the cross [2+2] cycloaddition reactions between mono-olefins and dienes. This work highlights the ab initio multi-reference method in describing very complicated open-shell iron catalysis.
Co-reporter:Jian Liu, Lianrui Hu, Lei Wang, Hui Chen, and Liang Deng
Journal of the American Chemical Society March 15, 2017 Volume 139(Issue 10) pp:3876-3876
Publication Date(Web):February 21, 2017
DOI:10.1021/jacs.7b00484
Transition-metal alkylidenes are important reactive organometallic intermediates, and our current knowledge on them has been mainly restricted to those with closed-shell electronic configurations. In this study, we present an exploration on open-shell iron alkylidenes with a weak-field tripodal amido-phosphine-amido ligand. We found that a high-spin (amido-phosphine-amido)iron(II) complex can react with (p-tolyl)2CN2 to afford a high-spin (amido-ylide-amido)iron(II) complex, 2, which could transfer its alkylidene moiety to a variety of alkenes, either the electron-rich or electron-deficient ones, to form cyclopropane derivatives. The reaction of 2 with cis-β-deuterio-styrene gave deuterated cyclopropane derivatives with partial loss of the stereochemical integrity with respect to the cis-styrene. Kinetic study on the cyclopropanation reaction of 2 with 4-fluoro-styrene disclosed the activation parameters of ΔH⧧ = 23 ± 1 kcal/mol and ΔS⧧ = −20 ± 3 cal/mol/K, which are comparable to those of the cyclopropanation reactions involving transition-metal alkylidenes. However, the cyclopropanation of para-substituted styrenes by 2 shows a nonlinear Hammett plot of log(kX/kH) vs σp. By introduction of a radical parameter, a linear plot of log(kX/kH) vs 0.59σp + 0.55σc• was obtained, which suggests the “nucleophilic” radical nature of the transition state of the cyclopropanation step. In corroboration with the experimental observations, density functional theory calculation on the reaction of 2 with styrene suggests the involvement of an open-shell (amido-phosphine-amido)iron alkylidene intermediate that is higher in energy than its (amido-ylide-amido)iron(II) precursor and an “outer-sphere” radical-type mechanism for the cyclopropanation step. The negative charge distribution on the alkylidene carbon atoms of the open-shell states (S = 2 and 1) explains the high activity of the cyclopropanation reaction toward electron-deficient alkenes. The study demonstrates the unique activity of open-shell iron alkylidene species beyond its closed-shell analogues, thus pointing out their potential synthetic usage in catalysis.
Co-reporter:Lianrui Hu and Hui Chen
ACS Catalysis January 6, 2017 Volume 7(Issue 1) pp:285-285
Publication Date(Web):November 23, 2016
DOI:10.1021/acscatal.6b02694
Metal–imido complexes are critical intermediates in transition metal-catalyzed C–H amination reactions. Discerning the factors that control their reactivity, however, remains largely open for exploration, particularly for the territory of cobalt–imido’s. Herein we describe a systematic computational exploration of this new frontier via the C–H activation mechanisms of typical well-defined cobalt–imido complexes, whose formal oxidation states cover an extremely wide range from Co(II) to Co(V). Hydrogen atom abstraction (HAA) is found to be the rate-limiting step in all these systems, with the open-shell electronic states of radical character consistently bearing kinetic advantage over the closed-shell ones. Surprisingly, there is no correlation found between the cobalt oxidation state and the HAA reactivity. To render a more accessible HAA channel, the dichotomous EER/anti-EER electron-shift scenarios for the open-shell electronic structure are dependent on the cobalt oxidation states [Co(III), different from others], implying a paradigm shift from an EER to an anti-EER scenario in the periodic table from Fe to Co. In contrast to the kinetic factor that determines the HAA reactivity, the reaction outcomes of C–H activation (amination or cyclometalation product) in cobalt–imido complexes are found to be controlled by the thermodynamic stabilities of the products. Our results for the cobalt–imido complexes imply that, in addition to HAA chemistry of metal–oxo’s, the HAA promoted by metal–imido species could also be subject to the radical-facilitated reactivity. From this work, it is predictable that the stabilization of the less reactive closed-shell singlet state relative to other more reactive open-shell states is generally not beneficial to the HAA reactivity of cobalt–imido species.Keywords: amination; cobalt-imido; cyclometalation; C−H activation; DFT calculation; exchange-enhanced reactivity; hydrogen atom abstraction; radical-facilitated reactivity;
Co-reporter:Pankaj Kumar; Yong-Min Lee; Lianrui Hu; Jianwei Chen; Young Jun Park; Jiannian Yao; Hui Chen; Kenneth D. Karlin;Wonwoo Nam
Journal of the American Chemical Society 2016 Volume 138(Issue 24) pp:7753-7762
Publication Date(Web):May 24, 2016
DOI:10.1021/jacs.6b04040
Metal–nitrosyl complexes are key intermediates involved in many biological and physiological processes of nitric oxide (NO) activation by metalloproteins. In this study, we report the reactivities of mononuclear cobalt(III)–nitrosyl complexes bearing N-tetramethylated cyclam (TMC) ligands, [(14-TMC)CoIII(NO)]2+ and [(12-TMC)CoIII(NO)]2+, in NO-transfer and dioxygenation reactions. The Co(III)–nitrosyl complex bearing 14-TMC ligand, [(14-TMC)CoIII(NO)]2+, transfers the bound nitrosyl ligand to [(12-TMC)CoII]2+ via a dissociative pathway, {[(14-TMC)CoIII(NO)]2+ → {(14-TMC)Co···NO}2+}, thus affording [(12-TMC)CoIII(NO)]2+ and [(14-TMC)CoII]2+ as products. The dissociation of NO from the [(14-TMC)CoIII(NO)]2+ complex prior to NO-transfer is supported experimentally and theoretically. In contrast, the reverse reaction, which is the NO-transfer from [(12-TMC)CoIII(NO)]2+ to [(14-TMC)CoII]2+, does not occur. In addition to the NO-transfer reaction, dioxygenation of [(14-TMC)CoIII(NO)]2+ by O2 produces [(14-TMC)CoII(NO3)]+, which possesses an O,O-chelated nitrato ligand and where, based on an experiment using 18O-labeled O2, two of the three O-atoms in the [(14-TMC)CoII(NO3)]+ product derive from O2. The dioxygenation reaction is proposed to occur via a dissociative pathway, as proposed in the NO-transfer reaction, and via the formation of a Co(II)–peroxynitrite intermediate, based on the observation of phenol ring nitration. In contrast, [(12-TMC)CoIII(NO)]2+ does not react with O2. Thus, the present results demonstrate unambiguously that the NO-transfer/dioxygenation reactivity of the cobalt(III)–nitrosyl complexes bearing TMC ligands is significantly influenced by the ring size of the TMC ligands and/or the spin state of the cobalt ion.
Co-reporter:Yihua Sun; Hao Tang; Kejuan Chen; Lianrui Hu; Jiannian Yao; Sason Shaik
Journal of the American Chemical Society 2016 Volume 138(Issue 11) pp:3715-3730
Publication Date(Web):February 23, 2016
DOI:10.1021/jacs.5b12150
C–H bond activation/functionalization promoted by low-valent iron complexes has recently emerged as a promising approach for the utilization of earth-abundant first-row transition metals to carry out this difficult transformation. Herein we use extensive density functional theory and high-level ab initio coupled cluster calculations to shed light on the mechanism of these intriguing reactions. Our key mechanistic discovery for C–H arylation reactions reveals a two-state reactivity (TSR) scenario in which the low-spin Fe(II) singlet state, which is initially an excited state, crosses over the high-spin ground state and promotes C–H bond cleavage. Subsequently, aryl transmetalation occurs, followed by oxidation of Fe(II) to Fe(III) in a single-electron transfer (SET) step in which dichloroalkane serves as an oxidant, thus promoting the final C–C coupling and finalizing the C–H functionalization. Regeneration of the Fe(II) catalyst for the next round of C–H activation involves SET oxidation of the Fe(I) species generated after the C–C bond coupling. The ligand sphere of iron is found to play a crucial role in the TSR mechanism by stabilization of the reactive low-spin state that mediates the C–H activation. This is the first time that the successful TSR concept conceived for high-valent iron chemistry is shown to successfully rationalize the reactivity for a reaction promoted by low-valent iron complexes. A comparative study involving other divalent middle and late first-row transition metals implicates iron as the optimum metal in this TSR mechanism for C–H activation. It is predicted that stabilization of low-spin Mn(II) using an appropriate ligand sphere should produce another promising candidate for efficient C–H bond activation. This new TSR scenario therefore emerges as a new strategy for using low-valent first-row transition metals for C–H activation reactions.
Co-reporter:Ya-Ke Li, Zhen Yuan, Yan-Xia Zhao, Chongyang Zhao, Qing-Yu Liu, Hui Chen, and Sheng-Gui He
Journal of the American Chemical Society 2016 Volume 138(Issue 39) pp:12854-12860
Publication Date(Web):September 8, 2016
DOI:10.1021/jacs.6b05454
Laser ablation generated RhAl3O4+ heteronuclear metal oxide cluster cations have been mass-selected using a quadrupole mass filter and reacted with CH4 or CD4 in a linear ion trap reactor under thermal collision conditions. The reactions have been characterized by state-of-the-art mass spectrometry and quantum chemistry calculations. The RhAl3O4+ cluster can activate four C–H bonds of a methane molecule and convert methane to syngas, an important intermediate product in methane conversion to value-added chemicals. The Rh atom is the active site for activation of the C–H bonds of methane. The high electron-withdrawing capability of Rh atom is the driving force to promote the conversion of methane to syngas. The polarity of Rh oxidation state is changed from positive to negative after the reaction. This study has provided the first example of methane conversion to syngas by heteronuclear metal oxide clusters under thermal collision conditions. Furthermore, the molecular level origin has been revealed for the condensed-phase experimental observation that trace amounts of Rh can promote the participation of lattice oxygen of chemically very inert support (Al2O3) to oxidize methane to carbon monoxide.
Co-reporter:Han-Xiao Wang, Zheng Meng, Jun-Feng Xiang, Yu-Xiang Xia, Yihua Sun, Shu-Zhen Hu, Hui Chen, Jiannian Yao and Chuan-Feng Chen
Chemical Science 2016 vol. 7(Issue 1) pp:469-474
Publication Date(Web):08 Oct 2015
DOI:10.1039/C5SC03511B
The manipulation of supramolecular devices to carry out sophisticated and programmed tasks is bound up with the spatial allocation of their components, especially the threading direction of the guest, which controls the host–guest orientation in the device. However, insights are needed to probe more possibilities for steering the threading direction. We have developed a new system consisting of a three-dimensional nonsymmetric oxacalixarene (H) with a fixed comformation and (bi)pyridinium salts (G1–G3), in which we found that based on the intrinsic discrepancies between the two semi-cavities of H, the electron densities of the axles greatly affect the threading direction. This was unequivocally demonstrated by NMR spectra and single crystal structures. With elaborate design, unidirectional threading was achieved, resulting in an oriented rotaxane. Therefore, we describe a new approach in which the threading direction and final orientation may be finely controlled by adjustment of the structure of the guest.
Co-reporter:Xiang Su, Yihua Sun, Jiannian Yao, Hui Chen and Chao Chen
Chemical Communications 2016 vol. 52(Issue 24) pp:4537-4540
Publication Date(Web):26 Feb 2016
DOI:10.1039/C6CC00452K
Acid-promoted efficient, site- and stereo-selective bicyclization of alkynes to polycyclic compounds (benzobicyclo[3.2.1]octanes) was realized with atom- and step-economy. The reaction proceeded through two C–C bonds formed on remote alkyl C–H bonds via twice long-distance cationic rearrangement.
Co-reporter:Qin Jiang, Ming Wang, Lifen Yang, Hui Chen, and Lanqun Mao
Analytical Chemistry 2016 Volume 88(Issue 20) pp:10322
Publication Date(Web):September 30, 2016
DOI:10.1021/acs.analchem.6b03383
Highly selective detection of intracellular glutamine (Gln) is very essential to understand the roles of Gln in some biological processes. Here, we report a new fluorescent method for selective imaging of Gln in live cells with an aldehyde-containing iridium complex, [Ir(pba)2(DMSO)2]PF6 (Hpba = 4-(2-pyridiyl)benzaldehyde) (Ir1), as the probe. Density functional theory (DFT) calculation and experimental results suggest that the coordination and hydrogen bonding interaction between Ir1 and Gln synergistically stabilize the Ir1–Gln complex, modulate charge-transfer characteristics and emission of Ir1, and as a consequence, enable Ir1 as the probe for the fluorescent sensing of Gln. The sensing strategy is well-responsive to Gln without interference from other amino acids or Gln-containing peptides and is demonstrated to be useful for in situ Gln imaging in live cells. The study provides a new method for fluorescent imaging of Gln in live cells, which is envisioned to find interesting applications in understanding the roles of Gln in some physiological processes.
Co-reporter:Rui Zhang, Chongyang Zhao, Xiumin Li, Zongyao Zhang, Xicheng Ai, Hui Chen and Rui Cao
Dalton Transactions 2016 vol. 45(Issue 32) pp:12772-12778
Publication Date(Web):19 Jul 2016
DOI:10.1039/C6DT02187E
A homoleptic, all-alkynyl-stabilized [Au8Ag8(ArCC)16] (1, Ar = 3,5-di-tert-butylphenyl) cluster was synthesized and characterized with a single crystal X-ray structure. Reactions of 3,5-di-tert-butyl-phenylacetylene with Ag(I) and Au(I) gave [Ag(ArCC)]n and Au(PPh3)(ArCC), respectively, where both have unusually high solubility in nonpolar organic solvents. In addition to drastically increased solubility, the two bulky tert-butyl substituents on the phenyl ring can confine the metal core to a certain size by preventing infinite aggregation of d10 metals. This feature makes the isolation of an all-alkynyl-stabilized Au–Ag cluster possible. Complex 1 is intensely luminescent with a very high quantum yield of 0.67 in solution at room temperature. Theoretical studies offered valuable insights into the intriguing photophysical properties, and revealed the significant role of metal–alkynyl bond interactions and enhanced molecular rigidity provided by tert-butyl groups.
Co-reporter:Teng Jia;Chongyang Zhao;Dr. Ruoyu He;Dr. Hui Chen;Dr. Congyang Wang
Angewandte Chemie 2016 Volume 128( Issue 17) pp:5354-5357
Publication Date(Web):
DOI:10.1002/ange.201600365
Abstract
Stoichiometric C−H bond activation of arenes mediated by iron carbonyls was reported by Pauson as early as in 1965, yet the catalytic C−H transformations have not been developed. Herein, an iron-catalyzed annulation of N−H imines and internal alkynes to furnish cis-3,4-dihydroisoquinolines is described, and represents the first iron-carbonyl-catalyzed C−H activation reaction of arenes. Remarkablely, this is also the first redox-neutral [4+2] annulation of imines and alkynes proceeding by C−H activation. The reaction also features only cis stereoselectivity and excellent atom economy as neither base, nor external ligand, nor additive is required. Experimental and theoretical studies reveal an oxidative addition mechanism for C−H bond activation to afford a dinuclear ferracycle and a synergetic diiron-promoted H-transfer to the alkyne as the turnover-determining step.
Co-reporter:Yihua Sun ;Dr. Hui Chen
ChemPhysChem 2016 Volume 17( Issue 1) pp:119-127
Publication Date(Web):
DOI:10.1002/cphc.201500817
Abstract
To enable the selection of more accurate computational methods for the future theoretical exploration of the reaction mechanism of Ir-catalyzed olefin hydrogenation, we compared high-level ab initio coupled cluster and DFT calculations with a simplified model of Pfaltz's Ir/P,N-type catalyst for all four previously proposed IrI/IrIII and IrIII/IrV mechanisms. Through the systematic assessment of the DFT performances, the DFT empirical dispersion correction (DFT-D3) is found to be indispensable for improving the accuracy of relative energies between the IrI/IrIII and IrIII/IrV mechanisms. After including the DFT-D3 correction, the three best performing density functionals (DFs) are B2-PLYP, BP86, and TPSSh. In these recommended DFs, the computationally more expensive double-hybrid functional B2-PLYP-D3 has a balanced and outstanding performance for calculations of the reaction barriers, reaction energies, and energy gaps between different mechanisms, whereas the less costly BP86-D3 and TPSSh-D3 methods have outstanding, but relatively less uniform performances.
Co-reporter:Teng Jia;Chongyang Zhao;Dr. Ruoyu He;Dr. Hui Chen;Dr. Congyang Wang
Angewandte Chemie International Edition 2016 Volume 55( Issue 17) pp:5268-5271
Publication Date(Web):
DOI:10.1002/anie.201600365
Abstract
Stoichiometric C−H bond activation of arenes mediated by iron carbonyls was reported by Pauson as early as in 1965, yet the catalytic C−H transformations have not been developed. Herein, an iron-catalyzed annulation of N−H imines and internal alkynes to furnish cis-3,4-dihydroisoquinolines is described, and represents the first iron-carbonyl-catalyzed C−H activation reaction of arenes. Remarkablely, this is also the first redox-neutral [4+2] annulation of imines and alkynes proceeding by C−H activation. The reaction also features only cis stereoselectivity and excellent atom economy as neither base, nor external ligand, nor additive is required. Experimental and theoretical studies reveal an oxidative addition mechanism for C−H bond activation to afford a dinuclear ferracycle and a synergetic diiron-promoted H-transfer to the alkyne as the turnover-determining step.
Co-reporter:Chao Wang, Chongyang Zhao, Lianrui Hu, and Hui Chen
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 21) pp:4427-4432
Publication Date(Web):October 24, 2016
DOI:10.1021/acs.jpclett.6b02061
Cyanobacterial aldehyde-deformylating oxygenase (cADO) is a nonheme diiron enzyme that catalyzes the conversion of aldehyde to alk(a/e)ne, an important transformation in biofuel research. In this work, we report a highly desired computational study for probing the mechanism of cADO. By combining our QM/MM results with the available 57Fe Mössbauer spectroscopic data, the gained detailed structural information suggests construction of asymmetry from the symmetric diiron cofactor in an aldehyde substrate and O2 activation. His160, one of the two iron-coordinate histidine residues in cADO, plays a pivotal role in this asymmetric aldehyde activation process by unprecedented reversible dissociation from the diiron cofactor, a behavior unknown in any other nonheme dinuclear or mononuclear enzymes. The revealed intrinsically asymmetric interactions of the substrate/O2 with the symmetric cofactor in cADO are inspirational for exploring diiron subsite resolution in other nonheme diiron enzymes.
Co-reporter:Lei Wang; Lianrui Hu; Hezhong Zhang; Hui Chen;Liang Deng
Journal of the American Chemical Society 2015 Volume 137(Issue 44) pp:14196-14207
Publication Date(Web):October 27, 2015
DOI:10.1021/jacs.5b09579
High-valent iron imido species are implicated as reactive intermediates in many iron-catalyzed transformations. However, isolable complexes of this type are rare, and their reactivity is poorly understood. Herein, we report the synthesis, characterization, and reactivity studies on novel three-coordinate iron(IV) bisimido complexes with aminocarbene ligation. Using our recently reported synthetic method for [LFe(NDipp)2] (L = IMes, 1; Me2-cAAC, 2), four new iron(IV) imido complexes, [(IPr)Fe(NDipp)2] (3) and [(Me2-cAAC)Fe(NR)2] (R = Mes, 4; Ad, 5; CMe2CH2Ph, 6), were prepared from the reactions of three-coordinate iron(0) compounds with organic azides. Characterization data acquired from 1H and 13C NMR spectroscopy, 57Fe Mössbauer spectroscopy, and X-ray diffraction studies suggest a low-spin singlet ground state for these iron(IV) complexes and the multiple-bond character of their Fe–N bonds. A reactivity study taking the reactions of 1 as representative revealed an intramolecular alkane dehydrogenation of 1 to produce the iron(II) complex [(IMes)Fe(NHDipp)(NHC6H3-2-Pri-6-CMe═CH2)] (7), a Si–H bond activation reaction of 1 with PhSiH3 to produce the iron(II) complex [(IMes)Fe(NHDipp)(NDippSiPhH2)] (8), and a [2+2]-addition reaction of 1 with PhNCNPh and p-PriC6H4NCO to form the corresponding open-shell formal iron(IV) monoimido complexes [(IMes)Fe(NDipp)(N(Dipp)C(NPh)(═NPh))] (9) and [(IMes)Fe(NDipp)(N(Dipp)C(O)N(p-PriC6H4))] (10), as well as [NDipp]-group-transfer reactions with CO and ButNC. Density functional theory calculations suggested that the alkane chain dehydrogenation reaction starts with a hydrogen atom abstraction mechanism, whereas the Si–H activation reaction proceeds in a [2π+2σ]-addition manner. Both reactions have the pathways at the triplet potential energy surfaces being energetically preferred, and have formal iron(IV) hydride and iron(IV) silyl species as intermediates, respectively. The low-coordinate nature and low d-electron count (d4) of iron(IV) imido complexes are thought to be the key features endowing their unique reactivity.
Co-reporter:Jiantao Hu, Tianlong Lan, Yihua Sun, Hui Chen, Jiannian Yao and Yu Rao
Chemical Communications 2015 vol. 51(Issue 80) pp:14929-14932
Publication Date(Web):12 Aug 2015
DOI:10.1039/C5CC04952K
A novel palladium catalyzed hydroxylation of unactivated aliphatic C(sp3)–H bonds was successfully developed. Different from conventional methods, water serves as the hydroxyl group source in the reaction. This new reaction demonstrates good reactivity and broad functional group tolerance. The C–H hydroxylated products can be readily transformed into various highly valuable chemicals via known transformations. Based on experimental and theoretical studies, a mechanism involving the Pd(II)/(IV) pathway is proposed for this hydroxylation reaction.
Co-reporter:Lianrui Hu and Hui Chen
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 10) pp:4601-4614
Publication Date(Web):August 20, 2015
DOI:10.1021/acs.jctc.5b00373
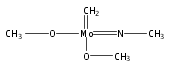
Using high level ab initio coupled cluster calculations as reference, the performances of 15 commonly used density functionals (DFs) on activation energy calculations for typical Mo/W-mediated reactions have been systematically assessed for the first time in this work. The selected representative Mo/W-mediated reactions cover a wide range from enzymatic reactions to organometallic reactions, which include Mo-catalyzed aldehyde oxidation (aldehyde oxidoreductase), Mo-catalyzed dimethyl sulfoxide (DMSO) reduction (DMSO reductase), W-catalyzed acetylene hydration (acetylene hydratase), Mo/W-mediated olefin metathesis, Mo/W-mediated olefin epoxidation, W-mediated alkyne metathesis, and W-mediated C–H bond activation. Covering both Mo- and W-mediated reactions, four DFs of B2GP-PLYP, M06, B2-PLYP, and B3LYP are uniformly recommended with and without DFT empirical dispersion correction. Among these four DFs, B3LYP is notably improved in performance by DFT empirical dispersion correction. In addition to the absolute value of calculation error, if the trend of DFT results is also a consideration, B2GP-PLYP, B2-PLYP, and M06 keep better performance than other functionals tested and constitute our final recommendation of DFs for both Mo- and W-mediated reactions.
Co-reporter:Yuanyuan Sun, Lianrui Hu, and Hui Chen
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 4) pp:1428-1438
Publication Date(Web):February 20, 2015
DOI:10.1021/ct5009119
In this work, the performances of 19 density functional theory (DFT) methods are calibrated comparatively on Ru- and Rh-promoted σ-bond (C–H, O–H, and H–H) activations. DFT calibration reference is generated from explicitly correlated coupled cluster CCSD(T)-F12 calculations, and the 4s4p core–valence correlation effect of the two 4d platinum group transition metals is also included. Generally, the errors of DFT methods for calculating energetics of Ru-/Rh-mediated reactions appear to correlate more with the magnitude of energetics itself than other factors such as metal identity. For activation energy calculations, the best performing functionals for both Ru and Rh systems are MN12SX < CAM-B3LYP < M06-L < MN12L < M06 < ωB97X < B3LYP < LC-ωPBE (in the order of increasing mean unsigned deviations, MUDs, of less than 2 kcal/mol). For reaction energy calculations, best functionals with MUDs less than 2 kcal/mol are PBE0 < CAM-B3LYP ≈ N12SX. The effect of the DFT empirical dispersion correction on the performance of the DFT methods is beneficial for most density functionals tested in this work, reducing their MUDs to different extents. After including empirical dispersion correction, ωB97XD, B3LYP-D3, and CAM-B3LYP-D3 (PBE0-D3, B3LYP-D3, and ωB97XD) are the three best performing DFs for activation energy (reaction energy) calculations, from which B3LYP-D3 and ωB97XD can notably be recommended uniformly for both the reaction energy and reaction barrier calculations. The good performance of B3LYP-D3 in quantitative description of the energetic trends further adds value to B3LYP-D3 and singles this functional out as a reasonable choice in the Ru/Rh-promoted σ-bond activation processes.
Co-reporter:Jian Zhang
The Journal of Physical Chemistry C 2015 Volume 119(Issue 5) pp:2761-2769
Publication Date(Web):January 12, 2015
DOI:10.1021/jp511264r
An anthranol derivative (DTDDP) and an anthraquinone derivative diastereomer (DDDP) without typical donor–acceptor (D–A) structural features were synthesized from 1,4-diaminoanthraquinone via a one-step reaction. The absorption and fluorescence emission properties of DTDDP and DDDP were investigated in common organic solvents with different polarity. In spite of a lack of apparent D–A structure, DTDDP exhibited apparent solvatochromic fluorescence emission with shift >80 nm in aprotic media. The linear correlation of the Stokes shift of DTDDP in a Lippert–Mataga plot was observed, and the change in the dipole moment of DTDDP upon excitation, 10.7 D, was determined. Time-dependent density functional theory (TDDFT) calculation confirmed the marked hole–electron separation of the excited DTDDP molecule as compared to that of the excited DDDP molecule. The experimental results and the theoretical calculation revealed the excited-state intramolecular charge-transfer nature of the emitting state of DTDDP molecules, which is responsible for the observed solvatochromic fluorescence emission features. The photooxidation-mediated facile conversion of DTDDP to its anthraquinone form counterpart DDDP was also confirmed.
Co-reporter:Dr. Yongbing Liu;Lianrui Hu;Dr. Hui Chen;Dr. Haifeng Du
Chemistry - A European Journal 2015 Volume 21( Issue 8) pp:3495-3501
Publication Date(Web):
DOI:10.1002/chem.201405388
Abstract
The stereoselective hydrogenation of alkynes to alkenes is an extremely useful transformation in synthetic chemistry. Despite numerous reports for the synthesis of Z-alkenes, the hydrogenation of alkynes to give E-alkenes is still not well resolved. In particular, selective preparation of both Z- and E-alkenes by the same catalytic hydrogenation system using molecular H2 has rarely been reported. In this paper, a novel strategy of using simple alkenes as promoters for the HB(C6F5)2-catalyzed metal-free hydrogenation of alkynes was adopted. Significantly, both Z- and E-alkenes can be furnished by hydrogenation with molecular H2 in high yields with excellent stereoselectivities. Further experimental and theoretical mechanistic studies suggest that interactions between H and F atoms of the alkene promoter, borane intermediate, and H2 play an essential role in promoting the hydrogenolysis reaction.
Co-reporter:Hao Tang, Xu-Ri Huang, Jiannian Yao, and Hui Chen
The Journal of Organic Chemistry 2015 Volume 80(Issue 9) pp:4672-4682
Publication Date(Web):April 2, 2015
DOI:10.1021/acs.joc.5b00580
Bidentate directing group (DG) strategy is a promising way to achieve sp2 and more inert sp3 C–H bond activations in transition metal (TM) catalysis. In this work, we systematically explored the assisting effects exerted by bidentate DGs in the C–H bond activations. Through DFT calculations and well-defined comparative analysis, we for the first time unified the rationale of the reactivity promoted by bidentate DG in sp2 and sp3 C–H activations, which are generally consistent with available experimental discoveries about the C–H activation reactivity up to date. In addition to the general rationale of the reactivity, the assisting effects of several typical bidentate DGs were also quantitatively evaluated and compared to reveal their relative promoting ability for C–H activation reactivity. Finally, the effect of the ligating group charge and the position of the ligating group charge in bidentate DGs were also investigated, based on which new types of DGs were designed and proposed to be potentially effective in C–H activation. The deeper understanding and new insight about the bidentate DG strategy gained in this work would help to enhance its further experimental development in sp2 and sp3 C–H bond activations.
Co-reporter:Zi-Yu Li;Lianrui Hu;Qing-Yu Liu;Dr. Chuan-Gang Ning;Dr. Hui Chen;Dr. Sheng-Gui He;Dr. Jiannian Yao
Chemistry - A European Journal 2015 Volume 21( Issue 49) pp:17748-17756
Publication Date(Web):
DOI:10.1002/chem.201503060
Abstract
Although early transition metal (ETM) carbides can activate CH bonds in condensed-phase systems, the electronic-level mechanism is unclear. Atomic clusters are ideal model systems for understanding the mechanisms of bond activation. For the first time, CH activation of a simple alkane (ethane) by an ETM carbide cluster anion (MoC3−) under thermal-collision conditions has been identified by using high-resolution mass spectrometry, photoelectron imaging spectroscopy, and high-level quantum chemical calculations. Dehydrogenation and ethene elimination were observed in the reaction of MoC3− with C2H6. The CH activation follows a mechanism of oxidative addition that is much more favorable in the carbon-stabilized low-spin ground electronic state than in the high-spin excited state. The reaction efficiency between the MoC3− anion and C2H6 is low (0.23±0.05) %. A comparison between the anionic and a highly efficient cationic reaction system (Pt++C2H6) was made. It turned out that the potential-energy surfaces for the entrance channels of the anionic and cationic reaction systems can be very different.
Co-reporter:Chunhua Dong, Xinzheng Yang, Jiannian Yao, and Hui Chen
Organometallics 2015 Volume 34(Issue 1) pp:121-126
Publication Date(Web):December 18, 2014
DOI:10.1021/om500985q
Density functional theory (DFT) study of the reactions of mononuclear phenolate diamine zinc hydride complexes and CO2 reveals a direct insertion mechanism with a rate-determining C–H bond formation step. A total of 16 zinc hydride complexes with various functional groups, including 3 experimental structures and 13 newly proposed complexes, have been optimized. The influences of various substituents at different positions, the ring size of nitrogen bidentate ligands, and the ortho groups of nitrogen on the reaction rate are investigated. Computational results indicate that the ortho effect of nitrogen is the most effective factor in reducing the reaction energy barrier, and the complex with isopropyls as the ortho groups of the nitrogen atom has the lowest barrier of 10.9 kcal/mol.
Co-reporter:Ruoyu He ; Xiqing Jin ; Hui Chen ; Zhi-Tang Huang ; Qi-Yu Zheng ;Congyang Wang
Journal of the American Chemical Society 2014 Volume 136(Issue 18) pp:6558-6561
Publication Date(Web):April 22, 2014
DOI:10.1021/ja503520t
An expedient Mn-catalyzed three-component synthesis of 1,5-amino/keto alcohols from Grignard reagents, imines/nitriles, and tetrahydrofuran (THF) is described, which deviates from the classic Grignard addition to imines/nitriles in THF solvent. THF is split and “sewn” in an unprecedented manner in the reaction, leading to the formation of two geminal C–C bonds via C–H and C–O cleavage. Mechanistic experiments and DFT calculations reveal radical and organo-Mn intermediates in the catalytic cycle and the α-arylative ring-opening of THF as the key reaction step.
Co-reporter:Hao Tang, Bingwei Zhou, Xu-Ri Huang, Congyang Wang, Jiannian Yao, and Hui Chen
ACS Catalysis 2014 Volume 4(Issue 2) pp:649
Publication Date(Web):January 13, 2014
DOI:10.1021/cs401141k
The strategy using N,N-bidentate directing groups is a promising way to achieve selective C(sp2)–H activation inaccessible by that of monodentate directing groups. Herein, through theoretical calculations, we present a rationale behind this strategy, which deciphers its key roles in C–H activation promoted by Ni, Pd, Ru, and Cu. The calculations reveal two key points: (a) Between the two coordination sites of the N,N-bidentate directing group, the proximal one influences more the C–H activation barrier ΔG‡, whereas the distal site affects more the free energy change ΔG relevant to the substrate coordination. (b) Enlarging/shrinking the chelation ring can exert different effects on the reactivity, depending on the metal identity and the ring size. Importantly, our computational results are in full agreement with previous experimental findings concerning reactivity. Furthermore, a prediction about the unprecedented reactivity from our theory is confirmed by our experiments, lending more credence to the rationale and insights gained in this study.Keywords: C(sp2)−H activation; coordination free energy; density functional theory; N,N-bidentate directing group; reaction barrier; transition metal
Co-reporter:Bingwei Zhou, Pengchen Ma, Hui Chen and Congyang Wang
Chemical Communications 2014 vol. 50(Issue 93) pp:14558-14561
Publication Date(Web):06 Oct 2014
DOI:10.1039/C4CC07598F
Since 1987, the stoichiometric two-step C–H conjugate addition reactions have been developed. Herein we describe the first manganese-catalyzed one-step direct aromatic C–H conjugate addition to α,β-unsaturated carbonyls, which is accelerated by a catalytic amount of dicyclohexylamine. Experimental and computational studies substantiated the validity of the proposed catalytic cycle.
Co-reporter:Jing Peng, Chao Chen, Junjie Chen, Xiang Su, Chanjuan Xi, and Hui Chen
Organic Letters 2014 Volume 16(Issue 14) pp:3776-3779
Publication Date(Web):July 10, 2014
DOI:10.1021/ol501655g
Copper-catalyzed arylcarbocyclization reaction of alkynes was realized with diaryliodonium salts through C–C bond formation on an inert C(sp3)–H bond. This method provides an efficient cyclization of alkyl alkynes to generate carbocycles with good step-economy. Theoretical study revealed an interesting Cu-catalyzed concerted pathway of the C–C bond formation.
Co-reporter:Runhua Kang, Kejuan Chen, Jiannian Yao, Sason Shaik, and Hui Chen
Inorganic Chemistry 2014 Volume 53(Issue 14) pp:7130-7136
Publication Date(Web):June 26, 2014
DOI:10.1021/ic500008c
Ligand effects of some representative monomeric Ru-based water oxidation catalysts on the key O–O formation step are revealed in this work. Three effects, namely, cis-effect, net charge effect, and steric hindrance effect, are identified, which can exert sizable modulation on the O–O formation barriers for the two widely accepted O–O formation mechanisms of WNA (water nucleophilic attack) and I2M (direct coupling of two high-valent metal oxo units). The study demonstrates that, through the way of ligand design, there remains a large space for improving O–O bond formation reactivity.
Co-reporter:Yihua Sun and Hui Chen
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 2) pp:579-588
Publication Date(Web):December 23, 2013
DOI:10.1021/ct4010855
By employing high-level coupled cluster CCSD(T)-F12 calculations as reference, we herein systematically assessed the performance of 16 popular density functional theory (DFT) approximations for typical rhenium-catalyzed reactions. The reactions under study cover those catalyzed by low-valent rhenium(I)/(III) carbonyl complexes as well as high-valent organorhenium(VII) bisperoxo complex. Without DFT dispersion correction, the four best-performing functionals for the barrier heights are B2GP-PLYP, TPSSh, B3LYP, and PBE0 with the mean unsigned deviations (MUDs) under 1.6 kcal/mol. Among these four functionals, B2GP-PLYP generates more accurate barrier heights, while B3LYP and TPSSh behave more reliably in the barrier trend description for these Re-catalyzed reactions. In general, herein the hybrid functionals are better choices than pure GGA or pure meta-GGA functionals. DFT empirical dispersion corrections were found to have beneficial effects on MUDs only for four tested functionals of BMK, CAM-B3LYP, LC-ωPBE, and ωB97X. Often associated with very large errors up to about 15 kcal/mol in barrier height for many tested functionals, the reaction catalyzed by high-valent rhenium(VII) bisperoxo is apparently different from the ones catalyzed by low-valent rhenium(I)/(III) carbonyl complexes. For reactions catalyzed by Re(I)/(III) carbonyl complexes, ωB97XD with dispersion correction performs excellently (MUD = 0.63 kcal/mol) and hence is highly recommended for these Re(I)/Re(III)-mediated reactions.
Co-reporter:Dr. Baode Ma;Tingting Miao;Yihua Sun;Yanmei He;Dr. Ji Liu;Dr. Yu Feng;Dr. Hui Chen;Dr. Qing-Hua Fan
Chemistry - A European Journal 2014 Volume 20( Issue 32) pp:9969-9978
Publication Date(Web):
DOI:10.1002/chem.201402709
Abstract
A series of tunable G0–G3 dendritic 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP) ligands was prepared by attaching polyaryl ether dendrons onto the four phenyl rings on the P atoms. Their ruthenium complexes were employed in the asymmetric hydrogenation of β-ketoesters, α-ketoesters, and α-ketoamides to reveal the effects of dendron size on the catalytic properties. The second- and third-generation catalysts exhibited excellent enantioselectivities, which are remarkably higher than those obtained from the small molecular catalysts and the first-generation catalyst. Molecular modeling indicates that the incorporation of bulky dendritic wedges can influence the steric environments around the metal center. In addition, the ruthenium catalyst bearing a second-generation dendritic ligand could be recycled and reused seven times without any obvious decrease in enantioselectivity.
Co-reporter:Shengjie Xu;Kejuan Chen;Dr. Hui Chen;Dr. Jiannian Yao;Dr. Xiaozhang Zhu
Chemistry - A European Journal 2014 Volume 20( Issue 50) pp:16442-16447
Publication Date(Web):
DOI:10.1002/chem.201405145
Abstract
Herein, a RhIII-catalyzed stereocontrolled synthesis of benzo[k]fluoranthenes is reported. It was found that the unexpected E/Z isomerization was highly sensitive to the electronic effects of the substituents on the aryl groups. Theoretical calculations revealed that this controllable stereochemistry originates from the mediation of rhodacyclopentadiene intermediates during the isomerization. The fact that similar stereochemistry was observed when using an IrIII catalyst further suggests a certain generality of this discovery toward some other transition metals.
Co-reporter:Shengjie Xu;Kejuan Chen;Dr. Hui Chen;Dr. Jiannian Yao;Dr. Xiaozhang Zhu
Chemistry - A European Journal 2014 Volume 20( Issue 50) pp:
Publication Date(Web):
DOI:10.1002/chem.201485061
Co-reporter:Bingwei Zhou ; Hui Chen ;Congyang Wang
Journal of the American Chemical Society 2013 Volume 135(Issue 4) pp:1264-1267
Publication Date(Web):January 3, 2013
DOI:10.1021/ja311689k
The first manganese-catalyzed aromatic C–H alkenylation with terminal alkynes is described. The procedure features an operationally simple catalyst system containing commercially available MnBr(CO)5 and dicyclohexylamine (Cy2NH). The reaction occurs readily in a highly chemo-, regio-, and stereoselective manner delivering anti-Markovnikov E-configured olefins in high yields. Experimental study and DFT calculations reveal that (1) the reaction is initiated by a C–H activation step via the cooperation of manganese and base; (2) manganacycle and alkynylmanganese species are the key reaction intermediates; and (3) the ligand-to-ligand H-transfer and alkynyl-assisted C–H activation are the key steps rendering the reaction catalytic in manganese.
Co-reporter:Guiling Zhang, Kejuan Chen, Hui Chen, Jiannian Yao, and Sason Shaik
Inorganic Chemistry 2013 Volume 52(Issue 9) pp:5088-5096
Publication Date(Web):April 5, 2013
DOI:10.1021/ic3028644
Mononuclear Ru-based water oxidation catalysts (WOCs) constitute an important class of WOCs for water splitting. This work constitutes a systematic study of Ru–O2 complexes of mononuclear ruthenium WOCs, with a focus on the thermodynamics of water-assisted O2 release in various electronic states and conformations. Our extensive DFT study reveals several factors that affect the O2 release thermodynamics: (1) steric effect from the ligand sphere of Ru; (2) trans effect of ligands trans to O2; (3) oxygen cis coordinating effect; (4) carbon coordinating effect; and (5) Ru coordination strength. Some of these effects could selectively stabilize/destabilize some states/conformations of the Ru–O2 complexes relative to Ru–OH2 complexes, and affect thereby the O2 release thermodynamics. The identification and rationalization of factors for O2 release thermodynamics, as in this work, could be helpful toward a better understanding of this final step of the ruthenium-catalyzed water oxidation.
Co-reporter:Runhua Kang, Jiannian Yao, and Hui Chen
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 4) pp:1872-1879
Publication Date(Web):March 20, 2013
DOI:10.1021/ct400004j
Mononuclear Ru-based water oxidation catalysts (WOCs) are an important class of WOCs for water splitting. In this work, through high-level coupled cluster calculations (CCSD(T)/CBS), we have examined a variety of density functionals for their performances in the whole catalytic cycle of water oxidation catalyzed by mononuclear Ru-based WOCs. The tested functionals cover a wide range from pure GGA and meta-GGA to hybrids and double hybrids (TPSS, OLYP, BP86, M06-L, B3LYP, PBE0, M06, M06-2X, TPSSh, CAM-B3LYP, wB97X, B2-PLYP, B2GP-PLYP). Depending on different reaction types and species in the catalytic cycle, the performances of different DFTs vary severely, whose trends are summarized in the paper. Our results indicate that using a single approximate functional to accurately model all reactions involved in the whole Ru-based WOC catalytic cycle is still a very challenging task. In the current status, PBE0 and M06 may be recommended for the whole catalytic cycle. Generally, this study provides a guide for selecting an appropriate DFT method in modeling each of the various steps in water oxidation catalyzed by Ru-based WOCs. The sensitivity of DFT and ab initio results upon the degree of basis set completeness found in this work is also worthy of attention in the future theoretical study of mononuclear Ru-based WOCs.
Co-reporter:Yuanyuan Sun and Hui Chen
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 11) pp:4735-4743
Publication Date(Web):September 23, 2013
DOI:10.1021/ct400432x
Coupled cluster CCSD(T) calculations with core–valence correlation and complete basis set (CBS) limit extrapolation are used to benchmark the performance of commonly used density functionals in computing energy barriers for Zr-mediated reactions involving zirconocene species. These reactions include (a) insertions of the Zr–H bond of Cp2Zr(H)Cl into C═C, C≡C, and C═O bonds and (b) C–H activations by Zr═N bond in Cp2Zr═NH. The best performing functionals are M06-L, M06, and M06-2X in the M06 series, all having mean unsigned deviations (MUD) less than 2 kcal/mol. The worst performing functional is OLYP, with a distinctly large MUD of more than 10 kcal/mol. Considering also the trends in barrier heights and the systematic barrier height deviation, our best recommended functional is M06-2X. In this work, DFT empirical dispersion correction (DFT-D3) is found to improve the performance of barrier height values for most functionals (especially of OLYP and B3LYP). With DFT empirical dispersion correction, we also recommend M06-2X for reaction barrier calculations of Zr-mediated reactions.
Co-reporter: Dr. Hui Chen;Xiang-Yu Kong; Dr. Weijun Zheng; Dr. Jiannian Yao; Dr. Anil K. Kalam; Dr. Puru Jena
ChemPhysChem 2013 Volume 14( Issue 14) pp:3303-3308
Publication Date(Web):
DOI:10.1002/cphc.201300677
Abstract
Hyperhalogens were recently identified as a new class of highly electronagative species which are composed of metals and superhalogens. In this work, high-level theoretical calculations and photoelectron spectroscopy experiments are systematically conducted to investigate a series of coinage-metal-containing hyperhalogen anions, Cu(BO2)2−, Ag(BO2)2−, and Au(BO2)2−. The vertical electron detachment energy (VDE) of Ag(BO2)2− is anomalously higher than those of Au(BO2)2− and Cu(BO2)2−. In quantitative agreement with the experiment, high-level ab initio calculations reveal that spin–orbit coupling (SOC) lowers the VDE of Au(BO2)2− significantly. The sizable magnitude of about 0.5 eV of SOC effect on the VDE of Au(BO2)2− demonstrates that SOC plays an important role in the electronic structure of gold hyperhalogens. This study represents a new paradigm for relativistic electronic structure calculations for the one-electron-removal process of ionic AuIL2 complexes, which is characterized by a substantial SOC effect.
Co-reporter:Jing Peng;Dr. Chao Chen;Yong Wang;Zhenbang Lou;Dr. Ming Li;Dr. Chanjuan Xi;Dr. Hui Chen
Angewandte Chemie International Edition 2013 Volume 52( Issue 29) pp:7574-7578
Publication Date(Web):
DOI:10.1002/anie.201303347
Co-reporter:Wenzhen Lai, Jiannian Yao, Sason Shaik, and Hui Chen
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 9) pp:2991-2996
Publication Date(Web):August 22, 2012
DOI:10.1021/ct3005936
Using the recently proposed corrective LCCSD(T) method as a reference, we systematically assess the widely used approximate density functionals to reproduce C–H bond activation barriers by pincer complexes of the late platinum group transition metals (TMs) (TM = Rh, Pd, Ir, Pt). The pincer ligands explored here cover a wide range of PNP, PCP, POCOP, NCN, and SCS types. Interestingly, B3LYP is found to be the most accurate functional, followed by several others previously identified as well-performing functionals, like B2GP-PLYP, B2-PLYP, and PBE0. However, all tested functionals were found to exhibit the following uniform trends: (1) the DFT barriers for reactions of group 9 TM (Rh and Ir) pincer complexes show higher accuracy compared with those for group 10 TM (Pd and Pt) reactions; (2) within the same group, 5d TM pincer complexes have higher accuracy than 4d TM ones. Consequently, the barriers for C–H activation by Pd(II) pincer complexes were found to be the least accurate among the four TMs in almost all functionals tested here. The DFT empirical dispersion correction (DFT-D3) is shown to have a very small effect on barrier height. This study has some implications for other σ-bond activations like H–H, C–C, and C–halogen bonds by late platinum group pincer complexes.
Co-reporter:Runhua Kang, Wenzhen Lai, Jiannian Yao, Sason Shaik, and Hui Chen
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 9) pp:3119-3127
Publication Date(Web):July 19, 2012
DOI:10.1021/ct3003942
To improve the accuracy of local coupled cluster (LCC) methods in computing activation energies, we propose herein a new computational scheme. Its applications to various types of late-transition-metal-catalyzed reactions involving Au, Pt, and Ir indicate that the new corrective approach for LCC methods can downsize the mean unsigned deviation and maximum deviation, from the CCSD(T)/CBS reference, to about 0.3 and 0.9 kcal/mol. Using this method, we also calibrated the performance of popular density functionals, with respect to the same test set of reactions. It is concluded that the best functional is the general-purpose double hybrid functional B2GP-PLYP. Other well-performing functionals include the “kinetic” functionals M06-2X and BMK, which have a large percentage of HF exchange, and general-purpose functionals like PBE0 and wB97X. Comparatively, general-purpose functionals like PBE0 and TPSSh perform much better than the tested “kinetic” functionals for Pt-/Ir-catalyzed reactions, while the opposite is true for Au-catalyzed reactions. In contrast, wB97X performs more uniformly in these two classes of reactions. These findings hint that even within the scope of late transition metals, different types of reactions may require different types of optimal DFT methods. Empirical dispersion correction of DFT was found to have a small or no effect on the studied reactions barriers.
Co-reporter:Kejuan Chen, Guiling Zhang, Hui Chen, Jiannian Yao, David Danovich, and Sason Shaik
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 5) pp:1641-1645
Publication Date(Web):March 21, 2012
DOI:10.1021/ct3000537
The transition metal-dependent spin–orbit coupling (SOC) and outer-core (5s5p) correlation effects in Ir- and Pt-catalyzed C–H activation processes are studied here using high level ab initio computations. The catalysts involve complexes with oxidation states: Ir(I), Ir(III), Pt(0), and Pt(II). It is demonstrated that for these heavy 5d transition metal-containing systems, the SOC effect and outer-core correlation effect on C–H activation are up to the order of ∼1 kcal/mol, and should be included if chemical accuracy is aimed. The interesting trends in our studied systems are: (1) the SOC effect consistently increases the C–H activation barriers and is apparently larger in higher oxidation states (Pt(II) and Ir(III)) than in low-oxidation states (Pt(0) and Ir(I)); and (2) the magnitude of outer-core (5s5p) correlation effects is larger in less coordinate-saturated system. The effect of basis set on the outer-core correlation correction is significant; larger basis sets tend to increase the C–H activation barriers.
Co-reporter:Hui Chen, Kyung-Bin Cho, Wenzhen Lai, Wonwoo Nam, and Sason Shaik
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 3) pp:915-926
Publication Date(Web):February 3, 2012
DOI:10.1021/ct300015y
We present a systematic study using density functional theory (DFT) and coupled cluster (CCSD(T)) computations with an aim of characterizing a non-heme ferric–superoxo complex [(TMC)Fe(O2)]2+ (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) that was proposed to perform allylic C–H activation of cyclohexene (Lee, Y.-M. et al. J. Am. Chem. Soc.2010, 132, 10668). As such, we investigated a series of iron–O2 species without and with a sixth ligand bound to the iron ion in different O2 coordination modes (end-on and side-on) and different spin states. Most of the iron–O2 complexes were found to be iron(III)–superoxo species, Fe(III)(O2–), with high-spin (S = 5/2) or intermediate-spin (S = 3/2) ferric centers coupled ferromagnetically or antiferromagnetically to the superoxide anion radical. One iron(IV)–peroxo state, Fe(IV)(O22–), was also examined. The preference for ferromagnetic or antiferromagnetic coupling modes between the superoxo and ferric radicals was found to depend on the FeOO angle, where a side-on tilt favors ferromagnetic coupling whereas the end-on tilt favors antiferromagnetic states. Experimental findings, e.g., the effects of solvent, spin state, and redox potential of non-heme Fe(II) complexes on O2 activation, were corroborated in this work. Solvent effects were found to disfavor O2 binding, relative to the unbound ferrous ion and O2. The potential H-abstraction reactivity of the iron(III)–superoxo species was considered in light of the recently proposed exchange-enhanced reactivity principle (Shaik, S.; Chen, H.; Janardanan, D. Nat. Chem.2011, 3, 19). It is concluded that localization and/or decoupling of an unpaired electron in the d-block of high-spin Fe(III) center in the S = 2 and 3 ferric–superoxo complexes during H abstractions enhances exchange stabilization and may be the root cause of the observed reactivity of [(TMC)Fe(O2)]2+.
Co-reporter:Wenzhen Lai, Rui Cao, Geng Dong, Sason Shaik, Jiannian Yao, and Hui Chen
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 17) pp:2315-2319
Publication Date(Web):August 6, 2012
DOI:10.1021/jz3008535
O–O bond formation catalyzed by a variety of β-octafluoro hangman corrole metal complexes was investigated using density functional theory methods. Five transition metal elements, Co, Fe, Mn, Ru, and Ir, that are known to lead to water oxidation were examined. Our calculations clearly show that the formal CoV catalyst has a CoIV–corrole•+ character and is the most efficient water oxidant among all eight transition-metal complexes. The O–O bond formation barriers were found to change in the following order: Co(V) ≪ Fe(V) < Mn(V) < Ir(V) < Co(IV) < Ru(V) < Ir(IV) < Mn(IV). The efficiency of water oxidation is discussed by analysis of the O–O bond formation step. Thus, the global trend is determined by the ability of the ligand d-block to accept two electrons from the nascent OH–, as well as by the OH• affinity of the TM(IV)═O species of the corresponding TM(V)═O·H2O complex. Exchange-enhanced reactivity (EER) is responsible for the high catalytic activity of the Co(V) species in its S = 1 state.Keywords: density functional theory; hangman corrole; nucleophilic attack; O−O bond formation; transition metal; water oxidation;
Co-reporter:Runhua Kang, Hui Chen, Sason Shaik, and Jiannian Yao
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 12) pp:4002-4011
Publication Date(Web):November 11, 2011
DOI:10.1021/ct200656p
Gold–substrate interaction is essential in gold-catalyzed organic transformations. This study uses high-level coupled cluster calculations with core–valence correlation and complete basis set (CBS) limit extrapolation as a reference, for assessing the performance of popular density functional theory (DFT) approximations for a variety of Au(I)/Au(III) complexes with unsaturated aliphatic hydrocarbon CnHm substrates (ethene, ethyne, and allene). The tested functionals cover from LDA to GGA and meta-GGA, and to hybrids and double hybrids (LSDA, PBE, M06-L, TPSS, B3LYP, PBE0, M06, M06-2X, TPSSh, B2-PLYP, B2GP-PLYP). Both the geometry and bond dissociation energy (De) of the Au–CnHm complexes are studied. Our findings show that B2GP-PLYP, PBE0, and B2-PLYP are the best performing functionals for this set of Au–CnHm complexes. DFT dispersion correction (DFT-D3), though very helpful for some functionals (e.g., B3LYP and B2-PLYP), does not uniformly improve the results of all functionals. Ab initio methods like MP2 and SCSMP2 are also tested. MP2 is found to be the worst performing method, and while SCSMP2 greatly improves the results, still its accuracy is lower than that of the best functionals, B2GP-PLYP, PBE0, and B2-PLYP.
Co-reporter:Hui Chen, Wenzhen Lai, Jiannian Yao, and Sason Shaik
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 10) pp:3049-3053
Publication Date(Web):September 9, 2011
DOI:10.1021/ct200614g
Nonheme perferryl FeV═O species are studied herein by means of coupled cluster (CCSD(T)) calculations with a complete basis set limit estimate and density functional B3LYP computations. It is shown that the high-spin/low-spin (HS/LS) energy order in these FeV═O species is highly dependent on the electronic nature of the ligand sphere and the geometric position of ligands relative to the FeV═O moiety. When only σ-donor amines ligate FeV═O, the LS state is slightly lower than the HS states. However, when a strong π-donor ligand such as hydroxyl is cis to FeV═O, the HS state becomes highly favored. And on the contrary, if the π-donor ligand is trans to FeV═O, the LS state is predicted here to be highly favored. This last type of perferryl complex has not yet been made by experimental means. Generally, our findings are consistent with the available experimental data.(4a, 6, 7) Some implications of these findings on the behavior of experimental systems are discussed.
Co-reporter:Chunhua Dong, Mingsong Ji, Xinzheng Yang, Jiannian Yao, Hui Chen
Journal of Organometallic Chemistry (15 March 2017) Volume 833() pp:
Publication Date(Web):15 March 2017
DOI:10.1016/j.jorganchem.2017.01.021
•Mechanistic insights of Rh catalyzed transfer hydroformylation.•Newly proposed cobalt complex is a promising low-cost catalyst.•Electronic structure analysis for key transition states and intermediates.The mechanisms of the transfer hydroformylation reactions catalyzed by rhodium, cobalt, and iridium complexes were studied by using the density functional theory. There are nine stages in each catalytic cycle: oxidation addition and C−H activation, hydrogen transfer and benzoic acid dissociation, anti-insertion reaction (decarbonylation), β-H elimination, nucleophilic substitution 1 (SN1) of ligand, C=C insertion, C=O insertion, coordination of benzoic acid and hydrogen transfer, and reductive elimination and aldehyde dissociation for catalyst regeneration. The total free energy barriers of the reactions catalyzed by Rh, Co and Ir complexes are 25.1 (3Rh → TS11,12-Rh), 27.3 (1Co → TS5,6-Co) and 41.5 (14Ir → TS14,1-Ir) kcal/mol, respectively. Such barriers indicate that the newly proposed cobalt complex could be a potential low-cost catalyst for the transfer hydroformylation reaction under mild conditions. The electronic structures of key intermediates and transition states in the reactions were analyzed by using the natural bond orbital theory and the Multiwfn program.Download high-res image (149KB)Download full-size image
Co-reporter:Chuanling Song, Yihua Sun, Jianwu Wang, Hui Chen, Jiannian Yao, Chen-Ho Tung and Zhenghu Xu
Inorganic Chemistry Frontiers 2015 - vol. 2(Issue 10) pp:NaN1373-1373
Publication Date(Web):2015/08/06
DOI:10.1039/C5QO00205B
A new and efficient strategy for the synthesis of tetraaryl-substituted olefins has been developed. With a Cu/Pd-catalyzed isomerization/insertion/oxidative coupling cascade reaction of cyclopropene with internal alkynes, a wide variety of cis-tetrasubstituted olefins were synthesized in good yields as single stereoisomers. The photophysical properties of these novel tetraarylethenes were fully characterized and proved to be good AIE (aggregation-induced emission) luminogens. Experimental studies and theoretical calculation indicated that Cu(I) and Pd(II) were the actual catalysts. A novel deprotonative Cu-catalyzed cyclopropene cycloisomerization and subsequent successive Cu/Pd transmetalation relay mechanism was proposed for the discovered reaction.
Co-reporter:Xiang Su, Yihua Sun, Jiannian Yao, Hui Chen and Chao Chen
Chemical Communications 2016 - vol. 52(Issue 24) pp:NaN4540-4540
Publication Date(Web):2016/02/26
DOI:10.1039/C6CC00452K
Acid-promoted efficient, site- and stereo-selective bicyclization of alkynes to polycyclic compounds (benzobicyclo[3.2.1]octanes) was realized with atom- and step-economy. The reaction proceeded through two C–C bonds formed on remote alkyl C–H bonds via twice long-distance cationic rearrangement.
Co-reporter:Jiantao Hu, Tianlong Lan, Yihua Sun, Hui Chen, Jiannian Yao and Yu Rao
Chemical Communications 2015 - vol. 51(Issue 80) pp:NaN14932-14932
Publication Date(Web):2015/08/12
DOI:10.1039/C5CC04952K
A novel palladium catalyzed hydroxylation of unactivated aliphatic C(sp3)–H bonds was successfully developed. Different from conventional methods, water serves as the hydroxyl group source in the reaction. This new reaction demonstrates good reactivity and broad functional group tolerance. The C–H hydroxylated products can be readily transformed into various highly valuable chemicals via known transformations. Based on experimental and theoretical studies, a mechanism involving the Pd(II)/(IV) pathway is proposed for this hydroxylation reaction.
Co-reporter:Rui Zhang, Chongyang Zhao, Xiumin Li, Zongyao Zhang, Xicheng Ai, Hui Chen and Rui Cao
Dalton Transactions 2016 - vol. 45(Issue 32) pp:NaN12778-12778
Publication Date(Web):2016/07/19
DOI:10.1039/C6DT02187E
A homoleptic, all-alkynyl-stabilized [Au8Ag8(ArCC)16] (1, Ar = 3,5-di-tert-butylphenyl) cluster was synthesized and characterized with a single crystal X-ray structure. Reactions of 3,5-di-tert-butyl-phenylacetylene with Ag(I) and Au(I) gave [Ag(ArCC)]n and Au(PPh3)(ArCC), respectively, where both have unusually high solubility in nonpolar organic solvents. In addition to drastically increased solubility, the two bulky tert-butyl substituents on the phenyl ring can confine the metal core to a certain size by preventing infinite aggregation of d10 metals. This feature makes the isolation of an all-alkynyl-stabilized Au–Ag cluster possible. Complex 1 is intensely luminescent with a very high quantum yield of 0.67 in solution at room temperature. Theoretical studies offered valuable insights into the intriguing photophysical properties, and revealed the significant role of metal–alkynyl bond interactions and enhanced molecular rigidity provided by tert-butyl groups.
Co-reporter:Han-Xiao Wang, Zheng Meng, Jun-Feng Xiang, Yu-Xiang Xia, Yihua Sun, Shu-Zhen Hu, Hui Chen, Jiannian Yao and Chuan-Feng Chen
Chemical Science (2010-Present) 2016 - vol. 7(Issue 1) pp:NaN474-474
Publication Date(Web):2015/10/08
DOI:10.1039/C5SC03511B
The manipulation of supramolecular devices to carry out sophisticated and programmed tasks is bound up with the spatial allocation of their components, especially the threading direction of the guest, which controls the host–guest orientation in the device. However, insights are needed to probe more possibilities for steering the threading direction. We have developed a new system consisting of a three-dimensional nonsymmetric oxacalixarene (H) with a fixed comformation and (bi)pyridinium salts (G1–G3), in which we found that based on the intrinsic discrepancies between the two semi-cavities of H, the electron densities of the axles greatly affect the threading direction. This was unequivocally demonstrated by NMR spectra and single crystal structures. With elaborate design, unidirectional threading was achieved, resulting in an oriented rotaxane. Therefore, we describe a new approach in which the threading direction and final orientation may be finely controlled by adjustment of the structure of the guest.
Co-reporter:Bingwei Zhou, Pengchen Ma, Hui Chen and Congyang Wang
Chemical Communications 2014 - vol. 50(Issue 93) pp:NaN14561-14561
Publication Date(Web):2014/10/06
DOI:10.1039/C4CC07598F
Since 1987, the stoichiometric two-step C–H conjugate addition reactions have been developed. Herein we describe the first manganese-catalyzed one-step direct aromatic C–H conjugate addition to α,β-unsaturated carbonyls, which is accelerated by a catalytic amount of dicyclohexylamine. Experimental and computational studies substantiated the validity of the proposed catalytic cycle.
.jpg)
![N,N-bis(2-chloroethyl)-N-methyl-N-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanaminium bromide](http://img.cochemist.com/ccimg/1347000/1346936-27-3.png)
![N,N-bis(2-chloroethyl)-N-methyl-N-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanaminium bromide](http://img.cochemist.com/ccimg/1347000/1346936-27-3_b.png)
![1,3,2-Dioxaborolane, 2-[9,9-bis(6-bromohexyl)-9H-fluoren-2-yl]-4,4,5,5-tetramethyl-](/data/chemimg/148200/1030020-61-1.png)
![1,3,2-Dioxaborolane, 2-[9,9-bis(6-bromohexyl)-9H-fluoren-2-yl]-4,4,5,5-tetramethyl-](/data/chemimg/148200/1030020-61-1_b.png)





![(1R,4R)-2-Oxa-5-azabicyclo[2.2.1]heptane](http://img.cochemist.com/ccimg/661500/661470-56-0.png)
![(1R,4R)-2-Oxa-5-azabicyclo[2.2.1]heptane](http://img.cochemist.com/ccimg/661500/661470-56-0_b.png)


![2,5-Bis(trimethylstannyl)thieno[3,2-b]thiophene](http://img.cochemist.com/ccimg/470000/469912-82-1.png)
![2,5-Bis(trimethylstannyl)thieno[3,2-b]thiophene](http://img.cochemist.com/ccimg/470000/469912-82-1_b.png)



![2-Cyclohexen-1-one,2-[1-[[[(2E)-3-chloro-2-propen-1-yl]oxy]imino]propyl]-3-hydroxy-5-(tetrahydro-2H-pyran-4-yl)-](http://img.cochemist.com/ccimg/150000/149979-41-9.png)
![2-Cyclohexen-1-one,2-[1-[[[(2E)-3-chloro-2-propen-1-yl]oxy]imino]propyl]-3-hydroxy-5-(tetrahydro-2H-pyran-4-yl)-](http://img.cochemist.com/ccimg/150000/149979-41-9_b.png)
![2-Pyridinecarboxamide,N-(4-fluorophenyl)-6-[3-(trifluoromethyl)phenoxy]-](http://img.cochemist.com/ccimg/137700/137641-05-5.png)
![2-Pyridinecarboxamide,N-(4-fluorophenyl)-6-[3-(trifluoromethyl)phenoxy]-](http://img.cochemist.com/ccimg/137700/137641-05-5_b.png)
![Benzene, 1-bromo-4-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/125700/125662-16-0.png)
![Benzene, 1-bromo-4-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/125700/125662-16-0_b.png)



![Benzene, 1-(1,1-dimethylethyl)-4-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/114700/114606-26-7.png)
![Benzene, 1-(1,1-dimethylethyl)-4-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/114700/114606-26-7_b.png)
![Benzene, 1-methyl-3-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/107800/107770-92-3.png)
![Benzene, 1-methyl-3-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/107800/107770-92-3_b.png)


![Ethanone, 1-[4-[(4-methylphenyl)thio]phenyl]-](http://img.cochemist.com/ccimg/99500/99433-27-9.png)
![Ethanone, 1-[4-[(4-methylphenyl)thio]phenyl]-](http://img.cochemist.com/ccimg/99500/99433-27-9_b.png)






![1H-BENZ[DE]ISOQUINOLINE-1,3(2H)-DIONE, 2-(4-AMINOBUTYL)-](http://img.cochemist.com/ccimg/70100/70034-78-5.png)
![1H-BENZ[DE]ISOQUINOLINE-1,3(2H)-DIONE, 2-(4-AMINOBUTYL)-](http://img.cochemist.com/ccimg/70100/70034-78-5_b.png)
![2-[2,4-dichloro-5-(prop-2-yn-1-yloxy)phenyl]-5,6,7,8-tetrahydro[1,2,4]triazolo[4,3-a]pyridin-3(2H)-one](http://img.cochemist.com/ccimg/68100/68049-83-2.png)
![2-[2,4-dichloro-5-(prop-2-yn-1-yloxy)phenyl]-5,6,7,8-tetrahydro[1,2,4]triazolo[4,3-a]pyridin-3(2H)-one](http://img.cochemist.com/ccimg/68100/68049-83-2_b.png)




![Benzene, 1-methoxy-2-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/59400/59345-33-4.png)
![Benzene, 1-methoxy-2-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/59400/59345-33-4_b.png)








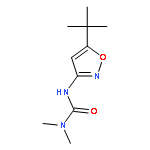
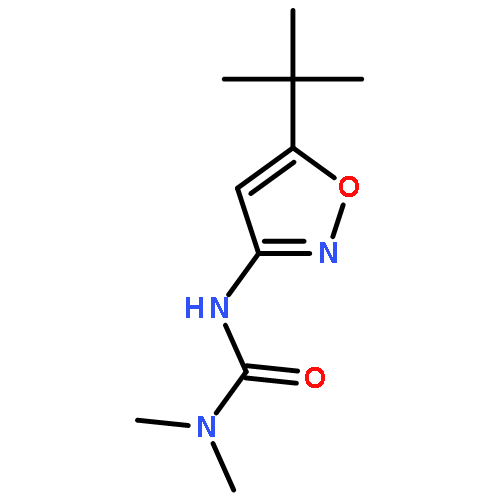
![methyl 5-[1-[(allyloxy)amino]butylidene]-2,2-dimethyl-4,6-dioxocyclohexanecarboxylate](http://img.cochemist.com/ccimg/55700/55634-91-8.png)
![methyl 5-[1-[(allyloxy)amino]butylidene]-2,2-dimethyl-4,6-dioxocyclohexanecarboxylate](http://img.cochemist.com/ccimg/55700/55634-91-8_b.png)
![Benzonitrile, 4-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/53300/53279-52-0.png)
![Benzonitrile, 4-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/53300/53279-52-0_b.png)


![4-Benzoxazolecarboxylicacid,5-(methylamino)-2-[[(2R,3R,6S,8S,9R,11R)-3,9,11-trimethyl-8-[(1S)-1-methyl-2-oxo-2-(1H-pyrrol-2-yl)ethyl]-1,7-dioxaspiro[5.5]undec-2-yl]methyl]-](http://img.cochemist.com/ccimg/52700/52665-69-7.png)
![4-Benzoxazolecarboxylicacid,5-(methylamino)-2-[[(2R,3R,6S,8S,9R,11R)-3,9,11-trimethyl-8-[(1S)-1-methyl-2-oxo-2-(1H-pyrrol-2-yl)ethyl]-1,7-dioxaspiro[5.5]undec-2-yl]methyl]-](http://img.cochemist.com/ccimg/52700/52665-69-7_b.png)


![NAPHTHALENE, 2-[(4-METHOXYPHENYL)THIO]-](http://img.cochemist.com/ccimg/51800/51739-39-0.png)
![NAPHTHALENE, 2-[(4-METHOXYPHENYL)THIO]-](http://img.cochemist.com/ccimg/51800/51739-39-0_b.png)
![Naphthalene, 1-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/51800/51739-38-9.png)
![Naphthalene, 1-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/51800/51739-38-9_b.png)
![BENZENE, 4-[(4-METHOXYPHENYL)ETHYNYL]-4-(TRIFLUOROMETHYL)-](http://img.cochemist.com/ccimg/40500/40474-01-9.png)
![BENZENE, 4-[(4-METHOXYPHENYL)ETHYNYL]-4-(TRIFLUOROMETHYL)-](http://img.cochemist.com/ccimg/40500/40474-01-9_b.png)

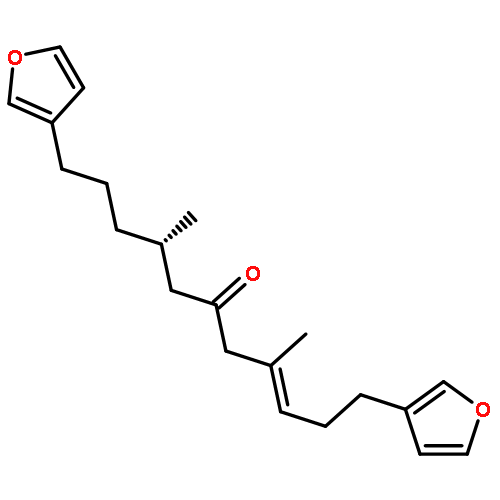
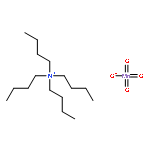

![2-[(1E,3Z)-3-(3-BUTYL-1,1-DIMETHYL-1,3-DIHYDRO-2H-BENZO[E]INDOL-2<WBR />-YLIDENE)-1-PROPEN-1-YL]-1,1,3-TRIMETHYL-1H-BENZO[E]INDOLIUM HEXA<WBR />FLUOROPHOSPHATE](http://img.cochemist.com/ccimg/35200/35122-96-4.png)
![2-[(1E,3Z)-3-(3-BUTYL-1,1-DIMETHYL-1,3-DIHYDRO-2H-BENZO[E]INDOL-2<WBR />-YLIDENE)-1-PROPEN-1-YL]-1,1,3-TRIMETHYL-1H-BENZO[E]INDOLIUM HEXA<WBR />FLUOROPHOSPHATE](http://img.cochemist.com/ccimg/35200/35122-96-4_b.png)

![BENZYL 6-OXA-3-AZABICYCLO[3.1.0]HEXANE-3-CARBOXYLATE](http://img.cochemist.com/ccimg/31900/31865-25-5.png)
![BENZYL 6-OXA-3-AZABICYCLO[3.1.0]HEXANE-3-CARBOXYLATE](http://img.cochemist.com/ccimg/31900/31865-25-5_b.png)


![Benzene, 1-[(4-methoxyphenyl)thio]-2-methyl-](http://img.cochemist.com/ccimg/22900/22894-90-2.png)
![Benzene, 1-[(4-methoxyphenyl)thio]-2-methyl-](http://img.cochemist.com/ccimg/22900/22894-90-2_b.png)
![Benzene, 1-chloro-4-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/22900/22865-55-0.png)
![Benzene, 1-chloro-4-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/22900/22865-55-0_b.png)



![Benzene,1-chloro-4-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/21000/20912-69-0.png)
![Benzene,1-chloro-4-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/21000/20912-69-0_b.png)






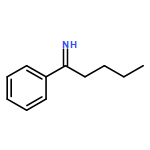
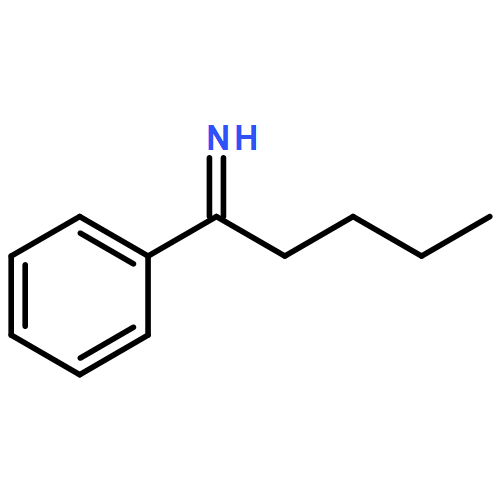


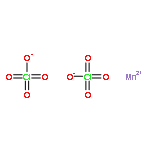
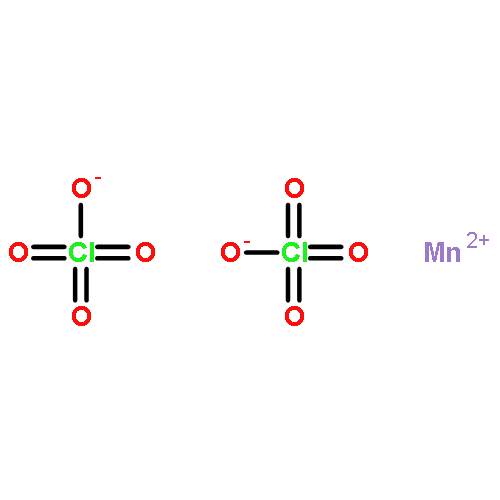

![Benzene,1-methoxy-4-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/6100/6013-47-4.png)
![Benzene,1-methoxy-4-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/6100/6013-47-4_b.png)


![4-tert-butyl-1-[(2,2-dichlorocyclopropyl)methyl]-2-nitrobenzene](http://img.cochemist.com/ccimg/5300/5216-30-8.png)
![4-tert-butyl-1-[(2,2-dichlorocyclopropyl)methyl]-2-nitrobenzene](http://img.cochemist.com/ccimg/5300/5216-30-8_b.png)


![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)


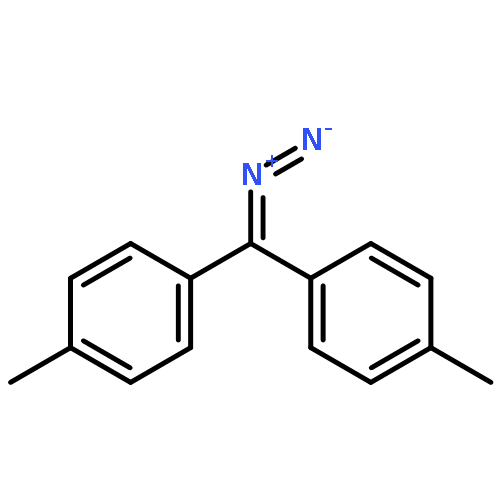


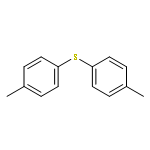
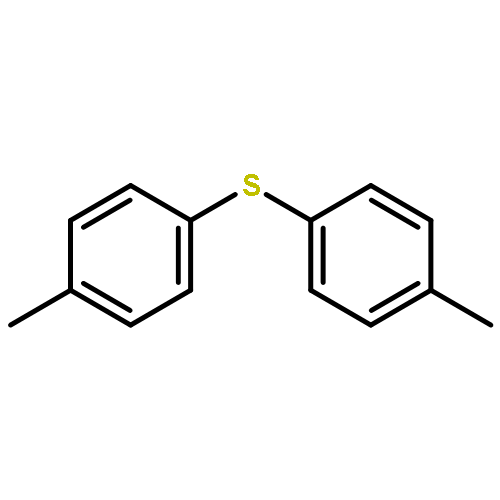









![Benzo[1,2-b:4,5-b']dithiophene, 4,8-bis(4,5-dioctyl-2-thienyl)-](/data/chemimg/3730500/1510594-39-4.png)
![Benzo[1,2-b:4,5-b']dithiophene, 4,8-bis(4,5-dioctyl-2-thienyl)-](/data/chemimg/3730500/1510594-39-4_b.png)


![Stannane, 1,1'-[4,8-bis[5-(2-ethylhexyl)-2-thienyl]benzo[1,2-b:4,5-b']dithiophene-2,6-diyl]bis[1,1,1-trimethyl-](/data/chemimg/139400/1352642-37-5.png)
![Stannane, 1,1'-[4,8-bis[5-(2-ethylhexyl)-2-thienyl]benzo[1,2-b:4,5-b']dithiophene-2,6-diyl]bis[1,1,1-trimethyl-](/data/chemimg/139400/1352642-37-5_b.png)
![Benzo[1,2-b:4,5-b']dithiophene, 4,8-bis[5-(2-ethylhexyl)-2-thienyl]-](/data/chemimg/139500/1352642-35-3.png)
![Benzo[1,2-b:4,5-b']dithiophene, 4,8-bis[5-(2-ethylhexyl)-2-thienyl]-](/data/chemimg/139500/1352642-35-3_b.png)





![Propanamide, N-[2-(methylthio)phenyl]-](/data/chemimg/137000/944775-06-8.png)
![Propanamide, N-[2-(methylthio)phenyl]-](/data/chemimg/137000/944775-06-8_b.png)


![1,3-Benzenedimethanol, 5,5'-[[5-[[3,5-bis[[3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]phenyl]methoxy]phenyl]methoxy]-1,3-phenylene]bis(methyleneoxy)]bis-](/data/chemimg/137000/943966-30-1.png)
![1,3-Benzenedimethanol, 5,5'-[[5-[[3,5-bis[[3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]phenyl]methoxy]phenyl]methoxy]-1,3-phenylene]bis(methyleneoxy)]bis-](/data/chemimg/137000/943966-30-1_b.png)
![1,3-Benzenedicarboxylic acid, 5,5'-[[5-[[3,5-bis[[3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]phenyl]methoxy]phenyl]methoxy]-1,3-phenylene]bis(methyleneoxy)]bis-, 1,1',3,3'-tetramethyl ester](/data/chemimg/137000/943966-29-8.png)
![1,3-Benzenedicarboxylic acid, 5,5'-[[5-[[3,5-bis[[3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]phenyl]methoxy]phenyl]methoxy]-1,3-phenylene]bis(methyleneoxy)]bis-, 1,1',3,3'-tetramethyl ester](/data/chemimg/137000/943966-29-8_b.png)
![1,3-Benzenedimethanol, 5-[[3,5-bis[[3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]phenyl]methoxy]phenyl]methoxy]-](/data/chemimg/137000/943966-28-7.png)
![1,3-Benzenedimethanol, 5-[[3,5-bis[[3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]phenyl]methoxy]phenyl]methoxy]-](/data/chemimg/137000/943966-28-7_b.png)
![1,3-Benzenedicarboxylic acid, 5-[[3,5-bis[[3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]phenyl]methoxy]phenyl]methoxy]-, 1,3-dimethyl ester](/data/chemimg/137000/943966-27-6.png)
![1,3-Benzenedicarboxylic acid, 5-[[3,5-bis[[3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]phenyl]methoxy]phenyl]methoxy]-, 1,3-dimethyl ester](/data/chemimg/137000/943966-27-6_b.png)



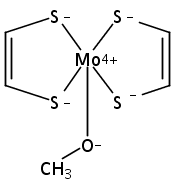
![Benzenepropanal, β-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4-methoxy-α-methyl-, (αS,βS)-](/data/chemimg/137000/915961-77-2.png)
![Benzenepropanal, β-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4-methoxy-α-methyl-, (αS,βS)-](/data/chemimg/137000/915961-77-2_b.png)
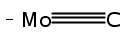

![1,3-Benzenedimethanol, 5,5'-[[5-(phenylmethoxy)-1,3-phenylene]bis(methyleneoxy)]bis-](/data/chemimg/137000/894094-41-8.png)
![1,3-Benzenedimethanol, 5,5'-[[5-(phenylmethoxy)-1,3-phenylene]bis(methyleneoxy)]bis-](/data/chemimg/137000/894094-41-8_b.png)
![1,3-Benzenedicarboxylic acid, 5,5'-[[5-(phenylmethoxy)-1,3-phenylene]bis(methyleneoxy)]bis-, tetraethyl ester (9CI)](/data/chemimg/137000/894094-40-7.png)
![1,3-Benzenedicarboxylic acid, 5,5'-[[5-(phenylmethoxy)-1,3-phenylene]bis(methyleneoxy)]bis-, tetraethyl ester (9CI)](/data/chemimg/137000/894094-40-7_b.png)
![1,3-Benzenedicarboxylic acid, 5-[[3,5-bis(phenylmethoxy)phenyl]methoxy]-, 1,3-dimethyl ester](/data/chemimg/137000/880345-03-9.png)
![1,3-Benzenedicarboxylic acid, 5-[[3,5-bis(phenylmethoxy)phenyl]methoxy]-, 1,3-dimethyl ester](/data/chemimg/137000/880345-03-9_b.png)


![Benzene,1,3-bis[[3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]phenyl]methoxy]-5-bromo-](http://img.cochemist.com/ccimg/863700/863676-42-0.png)
![Benzene,1,3-bis[[3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]phenyl]methoxy]-5-bromo-](http://img.cochemist.com/ccimg/863700/863676-42-0_b.png)
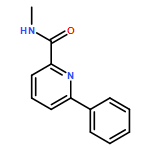
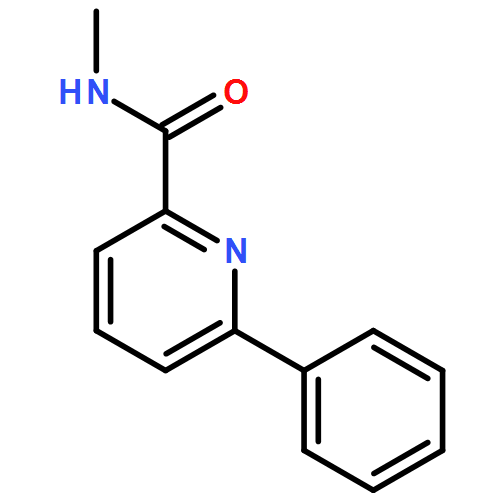














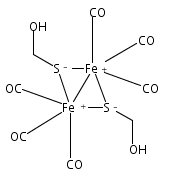


![2H-Naphtho[8a,1-b]furan-2-one, decahydro-9,10-dihydroxy-3,6,9-trimethyl-, (3R,3aS,6R,6aS,9S,10R,10aS)-](/data/chemimg/136900/382600-19-3.png)
![2H-Naphtho[8a,1-b]furan-2-one, decahydro-9,10-dihydroxy-3,6,9-trimethyl-, (3R,3aS,6R,6aS,9S,10R,10aS)-](/data/chemimg/136900/382600-19-3_b.png)





![2-Furanpropanal, β-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-α-methyl-, (αR,βR)-](/data/chemimg/136900/334988-61-3.png)
![2-Furanpropanal, β-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-α-methyl-, (αR,βR)-](/data/chemimg/136900/334988-61-3_b.png)
![1-Propanone, 1-[(4R)-4-(phenylmethyl)-2-thioxo-3-oxazolidinyl]-](/data/chemimg/136900/334968-78-4.png)
![1-Propanone, 1-[(4R)-4-(phenylmethyl)-2-thioxo-3-oxazolidinyl]-](/data/chemimg/136900/334968-78-4_b.png)










![Benzo[k]fluoranthene, 9-methoxy-7,12-diphenyl-](/data/chemimg/136900/253453-62-2.png)
![Benzo[k]fluoranthene, 9-methoxy-7,12-diphenyl-](/data/chemimg/136900/253453-62-2_b.png)
![Benzo[k]fluoranthene, 9-methyl-7,12-diphenyl-](/data/chemimg/136900/253453-55-3.png)
![Benzo[k]fluoranthene, 9-methyl-7,12-diphenyl-](/data/chemimg/136900/253453-55-3_b.png)
![Benzo[k]fluoranthene, 7,12-bis(4-fluorophenyl)-](/data/chemimg/136900/253436-26-9.png)
![Benzo[k]fluoranthene, 7,12-bis(4-fluorophenyl)-](/data/chemimg/136900/253436-26-9_b.png)




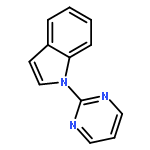
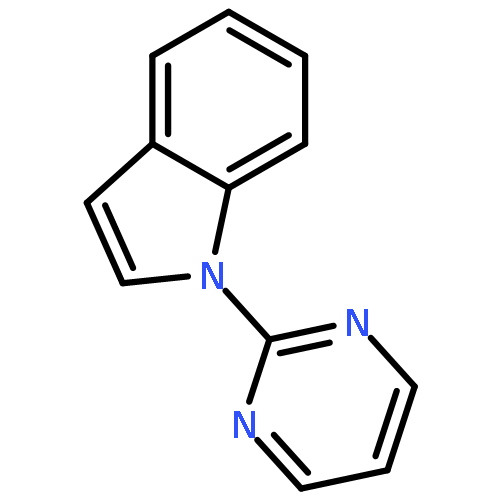
![Borane, [(1Z)-1,2-diphenylethenyl]bis(pentafluorophenyl)- (9CI)](/data/chemimg/136900/217966-69-3.png)
![Borane, [(1Z)-1,2-diphenylethenyl]bis(pentafluorophenyl)- (9CI)](/data/chemimg/136900/217966-69-3_b.png)

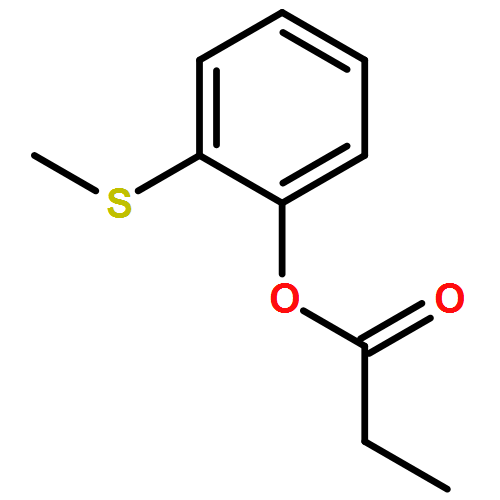
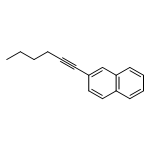
![Benzo[k]fluoranthene, 7,12-bis(4-methoxyphenyl)-](/data/chemimg/136900/211430-17-0.png)
![Benzo[k]fluoranthene, 7,12-bis(4-methoxyphenyl)-](/data/chemimg/136900/211430-17-0_b.png)
![Benzo[k]fluoranthene, 7,12-bis(4-methylphenyl)-](/data/chemimg/136900/211430-13-6.png)
![Benzo[k]fluoranthene, 7,12-bis(4-methylphenyl)-](/data/chemimg/136900/211430-13-6_b.png)
![1,3-Benzenedicarboxylicacid, 5,5'-[(5-hydroxy-1,3-phenylene)bis(methyleneoxy)]bis-,1,1',3,3'-tetramethyl ester](http://img.cochemist.com/ccimg/186700/186605-76-5.png)
![1,3-Benzenedicarboxylicacid, 5,5'-[(5-hydroxy-1,3-phenylene)bis(methyleneoxy)]bis-,1,1',3,3'-tetramethyl ester](http://img.cochemist.com/ccimg/186700/186605-76-5_b.png)



![2-Propenoic acid, 3-[4-[(3R)-4-hydroxy-3-methylbutoxy]phenyl]-, methyl ester, (E)-](/data/chemimg/136900/171674-98-9.png)
![2-Propenoic acid, 3-[4-[(3R)-4-hydroxy-3-methylbutoxy]phenyl]-, methyl ester, (E)-](/data/chemimg/136900/171674-98-9_b.png)


![Benzene, 1,3-dimethyl-5-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/165000/164989-61-1.png)
![Benzene, 1,3-dimethyl-5-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/165000/164989-61-1_b.png)





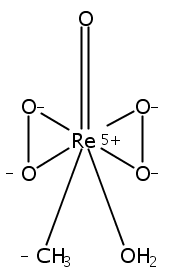





![Benzene, 1-[(1E)-3,3-dimethyl-1-buten-1-yl]-4-methyl-](/data/chemimg/136900/142039-30-3.png)
![Benzene, 1-[(1E)-3,3-dimethyl-1-buten-1-yl]-4-methyl-](/data/chemimg/136900/142039-30-3_b.png)


![Benzenemethanol,3,5-bis[[3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]phenyl]methoxy]-](http://img.cochemist.com/ccimg/137500/137472-16-3.png)
![Benzenemethanol,3,5-bis[[3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]phenyl]methoxy]-](http://img.cochemist.com/ccimg/137500/137472-16-3_b.png)



![Benzenemethanol, 3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]-](http://img.cochemist.com/ccimg/129600/129536-40-9.png)
![Benzenemethanol, 3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]-](http://img.cochemist.com/ccimg/129600/129536-40-9_b.png)





![3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin,decahydro-3,6,9-trimethyl-, (3R,5aS,6R,8aS,9R,12R,12aR)-](http://img.cochemist.com/ccimg/126200/126189-95-5.png)
![3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin,decahydro-3,6,9-trimethyl-, (3R,5aS,6R,8aS,9R,12R,12aR)-](http://img.cochemist.com/ccimg/126200/126189-95-5_b.png)
![Phosphine oxide, [2'-[bis(4-methoxyphenyl)phosphinyl][1,1'-binaphthalen]-2-yl]bis(4-methoxyphenyl)-](/data/chemimg/136900/123362-63-0.png)
![Phosphine oxide, [2'-[bis(4-methoxyphenyl)phosphinyl][1,1'-binaphthalen]-2-yl]bis(4-methoxyphenyl)-](/data/chemimg/136900/123362-63-0_b.png)






![5-Hexen-3-ol, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,4-dimethyl-,(2S,3R,4S)-](http://img.cochemist.com/ccimg/106400/106357-22-6.png)
![5-Hexen-3-ol, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,4-dimethyl-,(2S,3R,4S)-](http://img.cochemist.com/ccimg/106400/106357-22-6_b.png)


![Benzenamine,N-[(1,1-dimethylethyl)carbonimidoyl]-2,6-bis(1-methylethyl)-](http://img.cochemist.com/ccimg/105000/104961-67-3.png)
![Benzenamine,N-[(1,1-dimethylethyl)carbonimidoyl]-2,6-bis(1-methylethyl)-](http://img.cochemist.com/ccimg/105000/104961-67-3_b.png)






















![[3,5-BIS(METHOXYMETHOXY)PHENYL]METHANOL](http://img.cochemist.com/ccimg/76300/76280-60-9.png)
![[3,5-BIS(METHOXYMETHOXY)PHENYL]METHANOL](http://img.cochemist.com/ccimg/76300/76280-60-9_b.png)
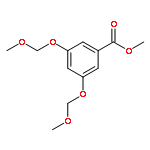

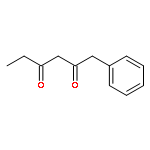
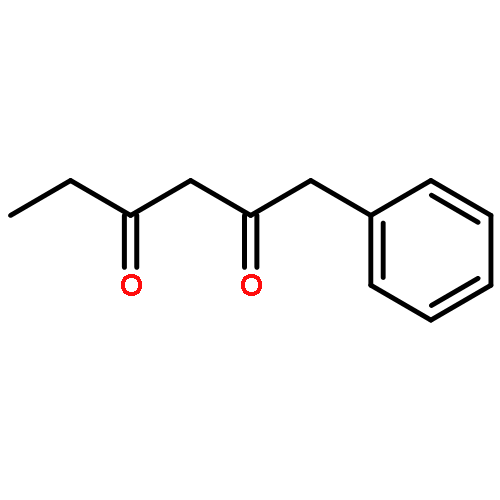
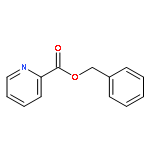













![1-METHYL-2-[(Z)-2-(2-METHYLPHENYL)ETHENYL]BENZENE](http://img.cochemist.com/ccimg/36900/36888-18-3.png)
![1-METHYL-2-[(Z)-2-(2-METHYLPHENYL)ETHENYL]BENZENE](http://img.cochemist.com/ccimg/36900/36888-18-3_b.png)








![Tricyclo[3.3.1.13,7]decane, 1-azido-](/data/chemimg/409500/24886-73-5.png)
![Tricyclo[3.3.1.13,7]decane, 1-azido-](/data/chemimg/409500/24886-73-5_b.png)
![1-phenyl-4-[(e)-2-phenylethenyl]benzene](http://img.cochemist.com/ccimg/21200/21175-18-8.png)
![1-phenyl-4-[(e)-2-phenylethenyl]benzene](http://img.cochemist.com/ccimg/21200/21175-18-8_b.png)




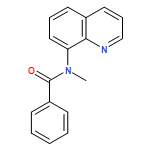
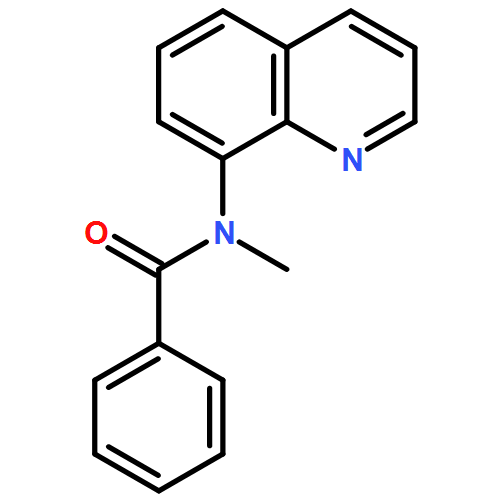


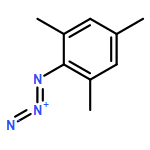
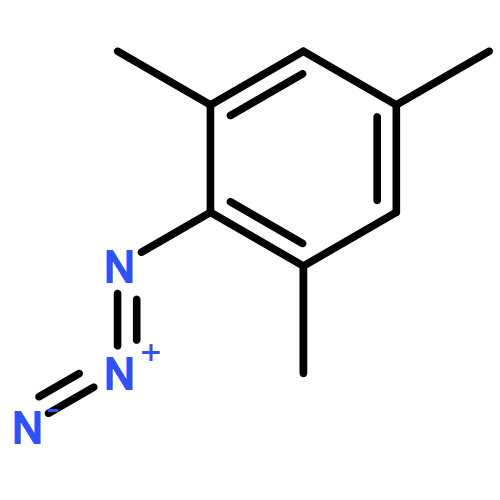





![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)





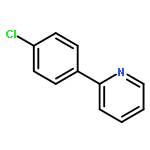
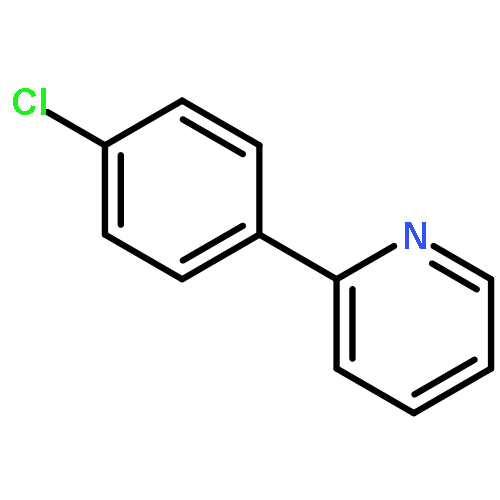
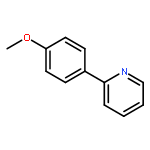
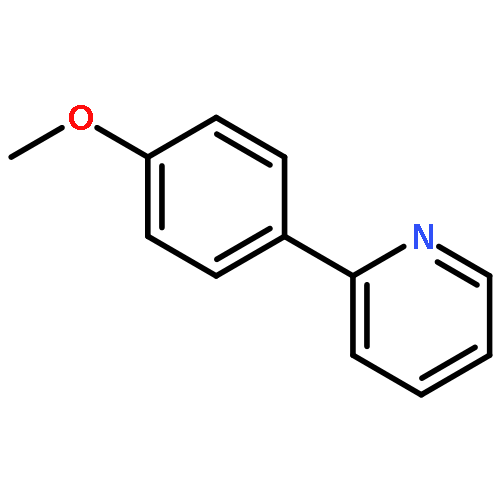





![1-chloro-4-[2-(4-chlorophenyl)ethyl]benzene](http://img.cochemist.com/ccimg/5300/5216-35-3.png)
![1-chloro-4-[2-(4-chlorophenyl)ethyl]benzene](http://img.cochemist.com/ccimg/5300/5216-35-3_b.png)

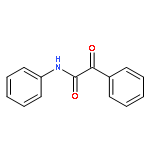
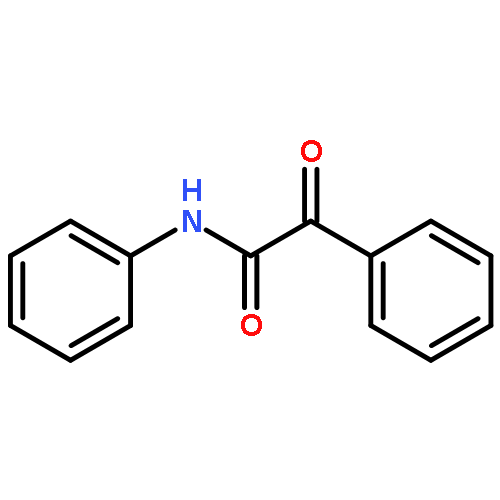


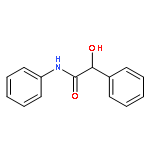
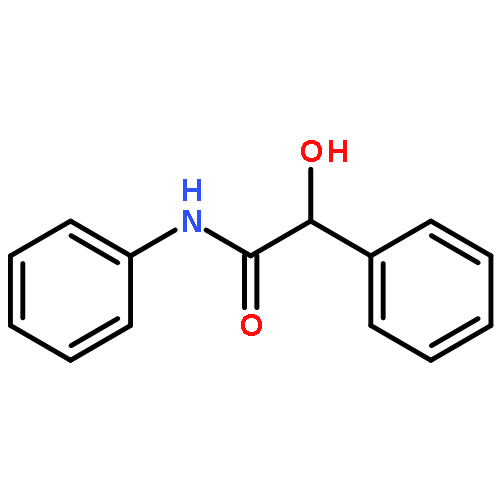

![Naphthalene, 2-[(1Z)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/3200/3132-88-5.png)
![Naphthalene, 2-[(1Z)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/3200/3132-88-5_b.png)
![Naphthalene, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/2900/2840-89-3.png)
![Naphthalene, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/2900/2840-89-3_b.png)




![2-Propenamide, N-[4-(2-phenyldiazenyl)phenyl]-](/data/chemimg/486300/2615-07-8.png)
![2-Propenamide, N-[4-(2-phenyldiazenyl)phenyl]-](/data/chemimg/486300/2615-07-8_b.png)