Co-reporter:Qi-Zheng Sun, Gui-Feng Lin, Lin-Li Li, Xi-Ting Jin, Lu-Yi Huang, Guo Zhang, Wei Yang, Kai Chen, Rong Xiang, Chong Chen, Yu-Quan Wei, Guang-Wen Lu, and Sheng-Yong Yang
Journal of Medicinal Chemistry July 27, 2017 Volume 60(Issue 14) pp:6337-6337
Publication Date(Web):July 10, 2017
DOI:10.1021/acs.jmedchem.7b00665
Autophagy inducers represent new promising agents for the treatment of a wide range of medical illnesses. However, safe autophagy inducers for clinical applications are lacking. Inhibition of cdc2-like kinase 1 (CLK1) was recently found to efficiently induce autophagy. Unfortunately, most of the known CLK1 inhibitors have unsatisfactory selectivity. Herein, we report the discovery of a series of new CLK1 inhibitors containing the 1H-[1,2,3]triazolo[4,5-c]quinoline scaffold. Among them, compound 25 was the most potent and selective, with an IC50 value of 2 nM against CLK1. The crystal structure of CLK1 complexed with compound 25 was solved, and the potency and kinase selectivity of compound 25 were interpreted. Compound 25 was able to induce autophagy in in vitro assays and displayed significant hepatoprotective effects in the acetaminophen (APAP)-induced liver injury mouse model. Collectively, due to its potency and selectivity, compound 25 could be used as a chemical probe or agent in future mechanism-of-action or autophagy-related disease therapy studies.
Co-reporter:Zhen Fang, Tian-qi Wang, Hui Li, Guo Zhang, Xiao-ai Wu, Li Yang, Yu-lan Peng, Jun Zou, Lin-li Li, Rong Xiang, Sheng-yong Yang
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 14(Issue 14) pp:
Publication Date(Web):15 July 2017
DOI:10.1016/j.bmcl.2017.05.002
Herein we report the discovery of a series of new small molecule inhibitors of histone lysine demethylase 4D (KDM4D). Molecular docking was first performed to screen for new KDM4D inhibitors from various chemical databases. Two hit compounds were retrieved. Further structural optimization and structure-activity relationship (SAR) analysis were carried out to the more selective one, compound 2, which led to the discovery of several new KDM4D inhibitors. Among them, compound 10r is the most potent one with an IC50 value of 0.41 ± 0.03 μM against KDM4D. Overall, compound 10r could be taken as a good lead compound for further studies.Download high-res image (94KB)Download full-size image
Co-reporter:Guo-Bo Li, Shuang Ma, Ling-Ling Yang, Sen Ji, Zhen Fang, Guo Zhang, Li-Jiao Wang, Jie-Min Zhong, Yu Xiong, Jiang-Hong Wang, Shen-Zhen Huang, Lin-Li Li, Rong Xiang, Dawen Niu, Ying-Chun Chen, and Sheng-Yong Yang
Journal of Medicinal Chemistry 2016 Volume 59(Issue 18) pp:8293-8305
Publication Date(Web):August 18, 2016
DOI:10.1021/acs.jmedchem.6b00604
Psoriasis is a chronic T-cell-mediated autoimmune disease, and FMS-like tyrosine kinase 3 (FLT3) has been considered as a potential molecular target for the treatment of psoriasis. In this investigation, structural optimization was performed on a lead compound, 1-(4-(1H-pyrazolo[3,4-d]pyrimidin-4-yloxy)phenyl)-3-(4-chloro-3-(trifluoromethyl)phenyl)urea (1), which showed a moderate inhibitory activity againt FLT3. A series of pyrazolo[3,4-d]pyrimidine derivatives were synthesized, and structure–activity relationship analysis led to the discovery of a number of potent FLT3 inhibitors. One of the most active compounds, 1-(4-(1H-pyrazolo[3,4-d]pyrimidin-4-yloxy)-3-fluorophenyl)-3-(5-tert-butylisoxazol-3-yl)urea (18b), was then chosen for in-depth antipsoriasis studies because this compound displayed the highest potency in a preliminary antipsoriasis test. Compound 18b exhibited significant antipsoriatic effects in the K14-VEGF transgenic mouse model of psoriasis, and no recurrence was found 15 days later after the last administration. Detailed mechanisms of action of compound 18b were also investigated. Collectively, compound 18b could be a potential drug candidate for psoriasis treatment.
Co-reporter:Chun-Hui Zhang, Kai Chen, Yan Jiao, Lin-Li Li, Ya-Ping Li, Rong-Jie Zhang, Ming-Wu Zheng, Lei Zhong, Shen-Zhen Huang, Chun-Li Song, Wan-Ting Lin, Jiao Yang, Rong Xiang, Bing Peng, Jun-Hong Han, Guang-Wen Lu, Yu-Quan Wei, and Sheng-Yong Yang
Journal of Medicinal Chemistry 2016 Volume 59(Issue 21) pp:9788-9805
Publication Date(Web):October 14, 2016
DOI:10.1021/acs.jmedchem.6b00943
Co-reporter:Guo-Bo Li, Lu-Yi Huang, Hui Li, Sen Ji, Lin-Li Li and Sheng-Yong Yang
RSC Advances 2016 vol. 6(Issue 66) pp:61137-61140
Publication Date(Web):20 Jun 2016
DOI:10.1039/C6RA11240D
Herein, we sought to discover new p300 HAT inhibitors from natural products by a customized structure-based virtual screening method. The natural compounds NP-2 (spinosine), NP-3 (palmatine), NP-9 (venenatine), and NP-15 (taxodione) were found to be potent p300 HAT inhibitors, of which IC50 values are 0.69 μM, 1.05 μM, 0.58 μM, 4.85 μM, respectively.
Co-reporter:Chan Zou;Yu Xiong;Lu-Yi Huang;Chun-Li Song;Xiao-Ai Wu;Lin-Li Li
Chemical Biology & Drug Design 2016 Volume 87( Issue 4) pp:569-574
Publication Date(Web):
DOI:10.1111/cbdd.12689
Receptor interacting protein 1 (RIP1) kinase plays an important role in necroptosis, and inhibitors of the RIP1 kinase are thought to have a potential therapeutic value in the treatment of diseases related to necrosis. Herein, we report the structural optimization of a RIP1 kinase inhibitor, 1-(2,4-dichlorobenzyl)-3-nitro-1H-pyrazole (1a). A number of 1-benzyl-1H-pyrazole derivatives were synthesized and structure-activity relationship (SAR) analysis led to the discovery of a potent compound, 4b, which showed a Kd value of 0.078 μm against the RIP1 kinase and an EC50 value of 0.160 μm in a cell necroptosis inhibitory assay. Compound 4b also displayed considerable ability to protect the pancreas in an l-arginine-induced pancreatitis mouse model.
Co-reporter:Xiaoai Wu, Zhen Fang, Bo Yang, Lei Zhong, Qiuyuan Yang, Chunhui Zhang, Shenzhen Huang, Rong Xiang, Takayoshi Suzuki, Lin-Li Li, Sheng-Yong Yang
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 9) pp:2284-2288
Publication Date(Web):1 May 2016
DOI:10.1016/j.bmcl.2016.03.048
Herein we report the discovery of a series of new KDM5A inhibitors. A three-dimensional (3D) structure model of KDM5A jumonji domain was firstly established based on homology modeling. Molecular docking-based virtual screening was then performed against commercial chemical databases. A number of hit compounds were retrieved. Further structural optimization and structure–activity relationship (SAR) analysis were carried out to the most active hit compound, 9 (IC50: 2.3 μM), which led to the discovery of several new KDM5A inhibitors. Among them, compound 15e is the most potent one with an IC50 value of 0.22 μM against KDM5A. This compound showed good selectivity for KDM5A and considerable ability to suppress the demethylation of H3K4me3 in intact cells. Compound 15e could be taken as a good lead compound for further studies.
Co-reporter:Chun-Hui Zhang; Ming-Wu Zheng; Ya-Ping Li; Xing-Dong Lin; Mei Huang; Lei Zhong; Guo-Bo Li; Rong-Jie Zhang; Wan-Ting Lin; Yan Jiao; Xiao-Ai Wu; Jiao Yang; Rong Xiang; Li-Juan Chen; Ying-Lan Zhao; Wei Cheng; Yu-Quan Wei
Journal of Medicinal Chemistry 2015 Volume 58(Issue 9) pp:3957-3974
Publication Date(Web):April 2, 2015
DOI:10.1021/acs.jmedchem.5b00270
A series of 3-(phenylethynyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine derivatives were designed and synthesized. Structure–activity relationship (SAR) analysis of these compounds led to the discovery of compound 1j, which showed the highest inhibitory potency against the Src kinase and the most potent antiviability activity against the typical TNBC cell line MDA-MB-231 among all the synthesized compounds. Further kinase inhibition assays showed that compound 1j was a multikinase inhibitor and potently inhibited Src (IC50 = 0.0009 μM) and MAPK signaling protein kinases B-RAF and C-RAF. In an MDA-MB-231 xenograft mouse model, a once-daily dose of compound 1j at 30 mg/kg for 18 days completely suppressed the tumor growth with a tumor inhibition rate larger than 100% without obvious toxicity. It also displayed good pharmacokinetic properties in a preliminary pharmacokinetic assay. Western blot and immunohistochemical assays revealed that compound 1j significantly inhibited Src and MAPK signaling and markedly induced apoptosis in tumor tissues.
Co-reporter:Guo-Bo Li, Sen Ji, Ling-Ling Yang, Rong-Jie Zhang, Kai Chen, Lei Zhong, Shuang Ma, Sheng-Yong Yang
European Journal of Medicinal Chemistry 2015 Volume 93() pp:523-538
Publication Date(Web):26 March 2015
DOI:10.1016/j.ejmech.2015.02.019
•We built a new automatic tool for structure-based lead optimization called LEADOPT.•Ligand efficiency is used as a measure to sort the generated molecules in LEADOPT.•Twelve ADMET properties are evaluated in LEADOPT.•We obtained some new potent VEGFR2 and SYK inhibitors using LEADOPT.Lead optimization is one of the key steps in drug discovery, and currently it is carried out mostly based on experiences of medicinal chemists, which often suffers from low efficiency. In silico methods are thought to be useful in improving the efficiency of lead optimization. Here we describe a new in silico automatic tool for structure-based lead optimization, termed LEADOPT. The structural modifications in LEADOPT mainly include two operations: fragment growing and fragment replacing, which are restricted to carry out in the active pocket of target protein with the core scaffold structure of ligand kept unchanged. The bioactivity of the newly generated molecules is estimated by ligand efficiency rather than a commonly used scoring function. Twelve important pharmacokinetic and toxic properties are evaluated using SCADMET, a program for the prediction of pharmacokinetic and toxic properties. LEADOPT was first evaluated using two retrospective cases, in which it showed a very good performance. LEADOPT was then applied to the structural optimizations of the VEGFR2 inhibitor, sorafenib, and the SYK inhibitor, R406. Though just several compounds were synthesized, we have obtained some compounds that are more potent than sorafenib and R406 in enzymatic and functional assays. All of these have validated, at least to some extent, the effectiveness of LEADOPT.A new in silico automatic tool for structure-based lead optimization termed LEADOPT was developed. It was successfully applied to the structural optimizations of the VEGFR2 inhibitor, sorafenib, and the SYK inhibitor, R406.
Co-reporter:Wen-Jing Wang;Qi Huang;Jun Zou;Lin-Li Li
Chemical Biology & Drug Design 2015 Volume 86( Issue 1) pp:1-8
Publication Date(Web):
DOI:10.1111/cbdd.12470
Most of the scoring functions currently used in structure-based drug design belong to ‘universal’ scoring functions, which often give a poor correlation between the calculated scores and experimental binding affinities. In this investigation, we proposed a simple strategy to construct target-specific scoring functions based on known ‘universal’ scoring functions. This strategy was applied to Chemscore, a widely used empirical scoring function, which led to a new scoring function, termed TS-Chemscore. TS-Chemscore was validated on 14 protein targets, which cover a wide range of biological target categories. The results showed that TS-Chemscore significantly improved the correlation between the calculated scores and experimental binding affinities compared with the original Chemscore. TS-Chemscore was then applied in virtual screening to retrieve novel JAK3 and YopH inhibitors. Top 30 compounds for each target were selected for experimental validation. Six active compounds for JAK3 and four for YopH were obtained. These compounds were out of the lists of top 30 compounds sorted by Chemscore. Collectively, TS-Chemscore established in this study showed a better performance in virtual screening than its counterpart Chemscore.
Co-reporter:Xing-Dong Lin, Hui-Wen Yang, Shuang Ma, Wei-Wei Li, Chun-Hui Zhang, Wen-Jing Wang, Rong Xiang, Lin-Li Li, Sheng-Yong Yang
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 20) pp:4534-4538
Publication Date(Web):15 October 2015
DOI:10.1016/j.bmcl.2015.08.068
In this investigation, a series of 6-phenylimidazo[2,1-b]thiazole derivatives were synthesized. Structure–activity relationship (SAR) analysis of these compounds based on cellular assays led to the discovery of a number of compounds that showed potent activity against FLT3-dependent human acute myeloid leukemia (AML) cell line MV4-11, but very weak or no activity against FLT3-independent human cervical cancer cell line Hela. FLT3 kinase inhibition assays were then performed on the three most active compounds. Among these compounds, 6-(4-(3-(5-(tert-butyl)isoxazol- 3-yl)ureido)phenyl)-N-(3-(dimethylamino)propyl)imidazo[2,1-b]thiazole-3-carboxamide (19) exhibited the highest potency in both cellular (MV4-11, IC50: 0.002 μM) and enzymatic (FLT3, IC50: 0.022 μM) assays. Further in-depth in vitro anti-AML activity and mechanism of action studies were carried out on compound 19.
Co-reporter:Xu-Ri Yu, Yun Tang, Wen-Jing Wang, Sen Ji, Shuang Ma, Lei Zhong, Chun-Hui Zhang, Jiao Yang, Xiao-Ai Wu, Zheng-Yan Fu, Lin-Li Li, Sheng-Yong Yang
Bioorganic & Medicinal Chemistry Letters 2015 25(22) pp: 5449-5453
Publication Date(Web):
DOI:10.1016/j.bmcl.2015.06.095
Co-reporter:Qi-Zheng Sun;Yong Xu;Jing-Jing Liu;Chun-Hui Zhang;Ze-Rong Wang
Molecular Diversity 2014 Volume 18( Issue 2) pp:403-409
Publication Date(Web):2014 May
DOI:10.1007/s11030-014-9508-8
Here, we describe the structural optimization of a known EGFR inhibitor (compound 1) that showed weak off-target activity against RET. Twenty-six analogs of 1 were synthesized. SAR analysis led to the discovery of several compounds that showed considerable potency against the RET-dependent thyroid cancer cell line TT. Kinase inhibitory potency was then measured for the most active compound (2u) in the cellular assay. The results showed that 2u is a potent RET inhibitor with an \(\hbox {IC}_{50}\) value of 7 nM.
Co-reporter:Heng-Xiu Yan;Wei-Wei Li;Yan Zhang;Xia-Wei Wei;Li-Xin Fu
Immunologic Research 2014 Volume 60( Issue 1) pp:112-126
Publication Date(Web):2014 October
DOI:10.1007/s12026-014-8521-4
Psoriasis is a common chronic T-cell-mediated autoimmune skin disease, and traditional immunotherapies for psoriasis have focused on the direct inhibition of T cells, which often causes toxicity and lacks long-term effectiveness. Safe and effective therapeutic strategies are strongly needed for psoriasis. In this study, we show for the first time a significant accumulation of FLT3+ CD11c+ dendritic cells (DCs) in human psoriatic lesions and in the skin of experimental preclinical K14-VEGF transgenic homozygous mice, our animal model, although not an exact match for human psoriasis, displays many characteristics of inflammatory skin inflammation. SKLB4771, a potent and selective FLT3 inhibitor that we designed and synthesised, was used to treat cutaneous inflammation and psoriasis-like symptoms of disease in mice and almost completely cured the psoriasis-like disease without obvious toxicity. Mechanistic studies indicated that SKLB4771 treatment significantly decreased the number and activation of pDCs and mDCs in vitro and in vivo, and subsequent T-cell cascade reactions mediated by Th1/Th17 pathways. These findings show that targeted inhibition of FLT3, and hence direct interference with DCs, may be a novel therapeutic approach for the treatment of psoriasis.
Co-reporter:Ling-Ling Yang ; Guo-Bo Li ; Shuang Ma ; Chan Zou ; Shu Zhou ; Qi-Zheng Sun ; Chuan Cheng ; Xin Chen ; Li-Jiao Wang ; Shan Feng ; Lin-Li Li
Journal of Medicinal Chemistry 2013 Volume 56(Issue 4) pp:1641-1655
Publication Date(Web):January 30, 2013
DOI:10.1021/jm301537p
We describe the structural optimization of a hit compound, 1-(4-(1H-pyrazolo[3,4-d]pyrimidin-4-ylamino)phenyl)-3-(3-methoxyphenyl)urea (1), which exhibits inhibitory activity but low potency against FLT3 and VEGFR2. A series of pyrazolo[3,4-d]pyrimidine derivatives were synthesized, and structure–activity relationship analysis using cell- and transgenic-zebrafish-based assays led to the discovery of a number of compounds that exhibited both high potency against FLT3-driven human acute myeloid leukemia (AML) MV4-11 cells and a considerable antiangiogenic effect in transgenic-zebrafish-based assays. The compound 1-(4-(1H-pyrazolo[3,4-d]pyrimidin −4-yloxy)phenyl)-3-(4-chloro-3-(trifluoromethyl)phenyl)urea (33), which exhibited the highest activity in preliminary in vivo anti-AML assays, was chosen for further anti-AML studies. The results demonstrated that compound 33 is a multikinase inhibitor that potently inhibits FLT3 and VEGFR2. In an MV4-11 xenograft mouse model, a once-daily dose of compound 33 at 10 mg/kg for 18 days led to complete tumor regression without obvious toxicity. Western blot and immunohistochemical analyses were performed to determine the mechanism of action of compound 33.
Co-reporter:Guo-Bo Li, Ling-Ling Yang, Wen-Jing Wang, Lin-Li Li, and Sheng-Yong Yang
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 3) pp:592-600
Publication Date(Web):February 9, 2013
DOI:10.1021/ci300493w
Scoring functions have been widely used to assess protein–ligand binding affinity in structure-based drug discovery. However, currently commonly used scoring functions face some challenges including poor correlation between calculated scores and experimental binding affinities, target-dependent performance, and low sensitivity to analogues. In this account, we propose a new empirical scoring function termed ID-Score. ID-Score was established based on a comprehensive set of descriptors related to protein–ligand interactions; these descriptors cover nine categories: van der Waals interaction, hydrogen-bonding interaction, electrostatic interaction, π-system interaction, metal–ligand bonding interaction, desolvation effect, entropic loss effect, shape matching, and surface property matching. A total of 2278 complexes were used as the training set, and a modified support vector regression (SVR) algorithm was used to fit the experimental binding affinities. Evaluation results showed that ID-Score outperformed other selected commonly used scoring functions on a benchmark test set and showed considerable performance on a large independent test set. ID-Score also showed a consistent higher performance across different biological targets. Besides, it could correctly differentiate structurally similar ligands, indicating higher sensitivity to analogues. Collectively, the better performance of ID-Score enables it as a useful tool in assessing protein–ligand binding affinity in structure-based drug discovery as well as in lead optimization.
Co-reporter:Huan-Zhang Xie;Hai Lan;You-Li Pan;Jun Zou;Ze-Rong Wang;Lin-Li Li;Qi Huang;Hui Zhang
Chemical Biology & Drug Design 2013 Volume 81( Issue 2) pp:175-184
Publication Date(Web):
DOI:10.1111/cbdd.12084
In this investigation, a common feature pharmacophore model of anaplastic lymphoma kinase inhibitors was developed based on several known anaplastic lymphoma kinase inhibitors that were co-crystallized with anaplastic lymphoma kinase. The established pharmacophore model Hypo1 was carefully validated and then adopted to screen two in silico chemical databases, Specs (202 408 compounds) and Enamine (1 105 894 compounds), for retrieving novel anaplastic lymphoma kinase inhibitors. The hit compounds were further filtered using a fast bumping-check tool and molecular docking. Finally, 25 compounds were selected and purchased from market. The bioactivity of these compounds was firstly measured at the cellular level against a typical anaplastic lymphoma kinase mutant-driven cancer cell line, Karpas299. And six of them showed a good anti-viability activity. The kinase inhibitory potency against the recombinant human anaplastic lymphoma kinase kinase was tested to the most active compound at the cellular level, T0508-5181 (from Specs), which gave a half maximal inhibitory concentration (IC50) of 5.3 μm.
Co-reporter:Guo-Bo Li, Ling-Ling Yang, Yong Xu, Wen-Jing Wang, Lin-Li Li, Sheng-Yong Yang
Journal of Molecular Graphics and Modelling 2013 Volume 44() pp:278-285
Publication Date(Web):July 2013
DOI:10.1016/j.jmgm.2013.07.005
•A target database termed TargetDB was compiled.•Docking- and pharmacophore-based target prediction protocols were established.•A combined target prediction strategy was developed.•A probabilistic fusion method was presented for target ranking.Herein, a combined molecular docking-based and pharmacophore-based target prediction strategy is presented, in which a probabilistic fusion method is suggested for target ranking. Establishment and validation of the combined strategy are described. A target database, termed TargetDB, was firstly constructed, which contains 1105 drug targets. Based on TargetDB, the molecular docking-based target prediction and pharmacophore-based target prediction protocols were established. A probabilistic fusion method was then developed by constructing probability assignment curves (PACs) against a set of selected targets. Finally the workflow for the combined molecular docking-based and pharmacophore-based target prediction strategy was established. Evaluations of the performance of the combined strategy were carried out against a set of structurally different single-target compounds and a well-known multi-target drug, 4H-tamoxifen, which results showed that the combined strategy consistently outperformed the sole use of docking-based and pharmacophore-based methods. Overall, this investigation provides a possible way for improving the accuracy of in silico target prediction and a method for target ranking.
Co-reporter:Hui Zhang;Ming-Li Xiang;Jun-Yu Liang;Tao Zeng;Xiao-Nuo Zhang
Molecular Diversity 2013 Volume 17( Issue 4) pp:767-772
Publication Date(Web):2013 November
DOI:10.1007/s11030-013-9473-7
S6K1 has emerged as a potential target for the treatment for obesity, type II diabetes and cancer diseases. Discovery of S6K1 inhibitors has thus attracted much attention in recent years. In this investigation, a hybrid virtual screening method that involves pharmacophore hypothesis, genetic function approximation (GFA) model, and molecular docking technology has been used to discover S6K1 inhibitors especially with novel scaffolds. The common feature pharmacophore hypothesis and GFA regression model of S6K1 inhibitors were first developed and applied in a virtual screen of the Specs database for retrieving S6K1 inhibitors. Then, the molecular docking method was carried out to re-filter these screened compounds. Finally, 60 compounds with promising S6K1 inhibitory activity were carefully selected and have been handed over to the other group to complete the follow-up compound synthesis (or purchase) and activity test.
Co-reporter:Wei-Wei Li ; Xiao-Yan Wang ; Ren-Lin Zheng ; Heng-Xiu Yan ; Zhi-Xing Cao ; Lei Zhong ; Ze-Rong Wang ; Pan Ji ; Ling-Ling Yang ; Li-Jiao Wang ; Yong Xu ; Jing-Jing Liu ; Jiao Yang ; Chun-Hui Zhang ; Shuang Ma ; Shan Feng ; Qi-Zheng Sun ; Yu-Quan Wei
Journal of Medicinal Chemistry 2012 Volume 55(Issue 8) pp:3852-3866
Publication Date(Web):March 27, 2012
DOI:10.1021/jm300042x
Structure–activity relationship (SAR) studies of 2-(quinazolin-4-ylthio)thiazole derivatives, which are for optimizing the in vitro and in vivo antiacute myeloid leukemia (AML) activity of a previously identified FLT3 inhibitor 2-(6,7-dimethoxyquinazolin-4-ylthio)thiazole (1), are described. SAR studies centering around the head (thiazole) and tails (6- and 7-positions) of the quinazoline moiety of 1 led to the discovery of a series of compounds that exhibited significantly increased potency against FLT3-driven AML MV4-11 cells. Preliminary in vivo assays were carried out on three highly active compounds, whose results showed that 1-{5-[7-(3-morpholinopropoxy)quinazolin-4-ylthio]-[1,3,4]thiadiazol-2-yl}-3-p-tolylurea (20c) had the highest in vivo activity. Further in vitro and in vivo anti-AML studies were then performed on 20c; in an MV4-11 xenograft mouse model, a once-daily dose of 20c at 100 mg/kg for 18 days led to complete tumor regression without obvious toxicity. Western blot and immunohistochemical analysis were carried out to illustrate the mechanism of action of 20c.
Co-reporter:Jiao Yang ; Li-Jiao Wang ; Jing-Jing Liu ; Lei Zhong ; Ren-Lin Zheng ; Yong Xu ; Pan Ji ; Chun-Hui Zhang ; Wen-Jing Wang ; Xing-Dong Lin ; Lin-Li Li ; Yu-Quan Wei
Journal of Medicinal Chemistry 2012 Volume 55(Issue 23) pp:10685-10699
Publication Date(Web):November 1, 2012
DOI:10.1021/jm301365e
This paper describe the structural optimization of a hit compound, N2-(4-(4-methylpiperazin-1-yl)phenyl)-N8-phenyl-9H-purine-2,8-diamine (1), which is a reversible kinase inhibitor targeting both EGFR-activating and drug-resistance (T790M) mutations but has poor binding affinity. Structure–activity relationship studies led to the identification of 9-cyclopentyl-N2-(4-(4-methylpiperazin-1-yl)phenyl)-N8-phenyl-9H-purine-2,8-diamine (9e) that exhibits significant in vitro antitumor potency against the non-small-cell lung cancer (NSCLC) cell lines HCC827 and H1975, which harbor EGFR-activating and drug-resistance mutations, respectively. Compound 9e was further assessed for potency and selectivity in enzymatic assays and in vivo anti-NSCLC studies. The results indicated that compound 9e is a highly potent kinase inhibitor against both EGFR-activating and resistance mutations and has good kinase spectrum selectivity across the kinome. In vivo, oral administration of compound 9e at a dose of 5 mg/kg caused rapid and complete tumor regression in a HCC827 xenograft model, and an oral dose of 50 mg/kg initiated a considerable antitumor effect in an H1975 xenograft model.
Co-reporter:Ling-Ling Yang, Guo-Bo Li, Heng-Xiu Yan, Qi-Zheng Sun, Shuang Ma, Pan Ji, Ze-Rong Wang, Shan Feng, Jun Zou, Sheng-Yong Yang
European Journal of Medicinal Chemistry 2012 Volume 56() pp:30-38
Publication Date(Web):October 2012
DOI:10.1016/j.ejmech.2012.08.007
Aberrant activation of casein kinase 1 (CK1) has been demonstrated to be implicated in the pathogenesis of cancer and various central nervous system disorders. Discovery of CK1 inhibitors has thus attracted much attention in recent years. In this account, we describe the discovery of N6-phenyl-1H-pyrazolo[3,4-d]pyrimidine-3,6-diamine derivatives as novel CK1 inhibitors. An optimal common-feature pharmacophore hypothesis, termed Hypo2, was firstly generated, followed by virtual screening using Hypo2 against several chemical databases. One of the best hit compounds, N6-(4-chlorophenyl)-1H-pyrazolo[3,4-d]pyrimidine-3,6-diamine, was chosen for the subsequent hit-to-lead optimization under the guide of Hypo2, which led to the discovery of a new lead compound (1-(3-(3-amino-1H-pyrazolo[3,4-d]pyrimidin-6-ylamino)phenyl)-3-(3-chloro-4-fluorophenyl)urea) that potently inhibits CK1 with an IC50 value of 78 nM.Graphical abstractPharmacophore-based virtual screening together with subsequent hit-to-lead optimization led to the discovery of a new lead compound that potently inhibits CK1 with an IC50 value of 78 nM.Highlights► We described a newly proposed pharmacophore modeling protocol. ► We developed an optimal pharmacophore model of CK1, termed Hypo2. ► Hypo2 was used to retrieve potential CK1 inhibitors from several chemical databases. ► Further structural optimization was carried out on one of the best hit compounds. ► We discovered a new lead compound that inhibits CK1 with an IC50 value of 78 nM.
Co-reporter:Hua-Lin Wan;Ze-Rong Wang;Lin-Li Li;Chuan Cheng;Pan Ji;Jing-Jing Liu;Hui Zhang;Jun Zou
Chemical Biology & Drug Design 2012 Volume 80( Issue 3) pp:366-373
Publication Date(Web):
DOI:10.1111/j.1747-0285.2012.01415.x
Bruton’s tyrosine kinase has emerged as a potential target for the treatment for B-cell malignancies and autoimmune diseases. Discovery of Bruton’s tyrosine kinase inhibitors has thus attracted much attention recently. In this investigation, we introduced a hybrid protocol of virtual screening methods including support vector machine model-based virtual screening, pharmacophore model-based virtual screening and docking-based virtual screening for retrieving new Bruton’s tyrosine kinase inhibitors from commercially available chemical databases. Performances of the hybrid virtual screening approach were evaluated against a test set, which results showed that the hybrid virtual screening approach significantly shortened the overall screening time, and considerably increased the hit rate and enrichment factor compared with the individual method (SB-VS, PB-VS and DB-VS) or their combinations by twos. This hybrid virtual screening approach was then applied to screen several chemical databases including Specs (202 408 compounds) and Enamine (980 000 compounds) databases. Thirty-nine compounds were selected from the final hits and have been shifted to experimental studies.
Co-reporter:Lei Di-wu, Lin-Li Li, Wen-Jing Wang, Huan-Zhang Xie, Jiao Yang, Chun-Hui Zhang, Qi Huang, Lei Zhong, Shan Feng, Sheng-Yong Yang
Journal of Molecular Graphics and Modelling 2012 Volume 36() pp:42-47
Publication Date(Web):June 2012
DOI:10.1016/j.jmgm.2012.03.004
Protein kinase casein kinase 2 (CK2), a member of the serine/threonine kinase family, has been established as one of the most attractive targets for molecularly targeted cancer therapy. The discovery of CK2 inhibitors has thus attracted much attention in recent years. In this investigation, a hybrid virtual screening approach based on Bayesian classification model, pharmacophore hypothesis and molecular docking was proposed and employed to identify CK2 inhibitors. We first established a naïve Bayes classification model of CK2 inhibitors/non-inhibitors and pharmacophore hypotheses of CK2 inhibitors. The docking parameters and scoring functions were also optimized in advance. The three virtual screening methods were sequentially used to screen two large chemical libraries, Specs and Enamine, for retrieving new CK2 inhibitors. Finally 30 compounds were selected from the final hits for in vitro CK2 kinase inhibitory assays. Five compounds with completely novel scaffolds showed a good inhibitory potency against CK2, which have good potentials for a future hit-to-lead optimization.Graphical abstractA hybrid virtual screening protocol based on Bayesian model, pharmacophore hypothesis and molecular docking was employed to identify novel CK2 inhibitors and five compounds with new scaffolds were discovered.Highlights► A naïve Bayes classification model of CK2 inhibitors/non-inhibitors was developed. ► A hybrid protocol was proposed and applied to identify novel CK2 inhibitors. ► Five new CK2 inhibitors with novel scaffolds were identified.
Co-reporter:Z-X Cao, J-J Liu, R-L Zheng, J Yang, L Zhong, Y Xu, L-J Wang, C-H Zhang, B-L Wang, S Ma, Z-R Wang, H-Z Xie, Y-Q Wei and S-Y Yang
Leukemia 2012 26(8) pp:1892-1895
Publication Date(Web):April 3, 2012
DOI:10.1038/leu.2012.67
Leukemia is a biologically heterogeneous hematological disease and has become one of the leading causes of cancer-related mortality worldwide.1 Recent studies of the pathogenesis of leukemia have revealed that mutations and/or aberrant expression of specific protein tyrosine kinases, such as FMS-like tyrosine kinase 3 (FLT3) and Abl, are responsible for the development of several common types of leukemia.2, 3, 4 For example, activating mutations in FLT3 are found in approximately 1/3 of acute myeloid leukemia (AML) patients, with the most prevalent activating mutations being ‘internal tandem duplications’ (ITD) in the juxtamembrane domain.5, 6 Several large-scale studies have demonstrated that FLT3/ITD is strongly associated with leukocytosis and a poor prognosis.7, 8, 9 Thus, FLT3 is considered to be an important molecular target in the treatment of AML.Despite many efforts in the past few years to develop FLT3 inhibitors for the treatment of AML, no FLT3 inhibitors have been approved for clinical use. Furthermore, except for a small number of inhibitors (such as AC220), most of the known FLT3 inhibitors that are currently in clinical trials do not show impressive clinical efficacy.10, 11, 12 These facts imply a requirement to develop new-generation FLT3 inhibitors with improved clinical efficacy, especially novel multikinase inhibitors because they are thought to enhance the therapeutic effect of FLT3 inhibitors in AML.13 Under these circumstances, we recently discovered a novel multikinase inhibitor termed SKLB1028 (Figure 1a). In this letter, we report the preclinical efficacy of SKLB1028 in AML; a brief description regarding the chemical characterization of SKLB1028 is provided in the Supplementary Material. SKLB1028 also showed considerable potency in models of chronic myeloid leukemia (CML) that harbor Abl mutants, which will also be reported here.The kinase-inhibition profile of SKLB1028 against a panel of recombinant human protein kinases was measured by the ‘gold standard’ radiometric kinase assay approach, and the results are shown in Supplementary Table S1. SKLB1028 potently inhibited the wild-type and L858R-mutant EGFR with half-maximal inhibitory concentration (IC50) values of 0.031 and 0.004 μM, respectively. The IC50 values of SKLB1028 against FLT3 (Figure 1b) and Abl were 0.055 and 0.081 μM, respectively. Of note, this compound also inhibited the Abl-T315I mutant with considerable potency (IC50: 0.071 μM); this AblT315I mutant is associated with imatinib resistance. Furthermore, SKLB1028 inhibited FYN, HCK, KDR, PDGFRβ, CSF1R, FGFR1 and FGFR2 with moderate activity (the IC50 values were 0.214, 0.487, 0.330, 0.360, 1.040, 1.20 and 0.980 μM, respectively). SKLB1028 displayed negligible inhibitory activity against the 24 additional protein kinases that were tested. These data demonstrate that SKLB1028 is a multikinase inhibitor with high potency against EGFR, FLT3 and Abl and has good kinase-spectrum selectivity.The growth-inhibitory potencies of SKLB1028 against various leukemia cell lines as well as several other cell lines were examined using the MTT assay method, and the results are presented in Supplementary Table S2. SKLB1028 potently inhibited the growth of MV4-11 cells that express FLT3-ITD, with an IC50 value of 0.002 μM (Figure 1c), but moderately inhibited the proliferation of RS4-11 cells that express wt-FLT3, with an observed IC50 of 0.790 μM. SKLB1028 also potently inhibited the growth of Ba/F3 cells that stably express human FLT3ITD (Ba/F3-FLT3-ITD), with an IC50 of 0.01 μM, but was nontoxic toward parental Ba/F3 cells at concentrations up to 5 μM. SKLB1028 inhibited the cell growth of K562 cells that express the Bcr-Abl mutant with an IC50 of 0.190 μM. SKLB1028 exhibited only a weak inhibitory potency in the human leukemia cell lines SU-DHL-6, TF-1, Karpas299 and Jurkat. Finally, SKLB1028 at a concentration of 10 μM did not inhibit the growth of the human myeloma cell line RPMI8226, human normal cell line LO2 and Chinese hamster ovary cell line CHO (Supplementary Table S2).The ability of SKLB1028 to inhibit the activation of FLT3 and downstream signaling proteins in intact cells was assessed using western blot analysis. As shown in Figure 1d, SKLB1028 inhibited FLT3 phosphorylation in a dose-dependent manner. Consistent with the downregulation of the phosphorylation of FLT3, the phosphorylation of the downstream signaling proteins STAT5 and ERK1/2 was also significantly inhibited at concentrations >0.003 μM (Figure 1e). We further examined the influence of SKLB1028 treatment on the cell cycle of MV4-11 cells using flow cytometry, and the results indicated that SKLB1028 could induce cell cycle arrest in G1 phase (Supplementary Figure S1). By observing the morphological changes in the nuclei using Hoechst staining, we found that the nuclei of SKLB1028-treated MV4-11 cells shrank, became rounded up, and eventually divided into several apoptotic bodies (Supplementary Figure S2), indicating cell apoptosis. The apoptosis induced by SKLB1028 was further confirmed using western blot analysis, in which we observed a dose-dependent decrease in the level of pro-caspase-3 and a dose-dependent increase in the level of the cleaved caspase-3 fragment in SKLB1028-treated MV4-11 cells at concentrations of 10–100 nM (Figure 2f).The in vivo anti-leukemia activities of SKLB1028 were evaluated using the FLT3-ITD-positive MV4-11 and the Bcr-Abl-positive K562 xenograft models. In the MV4-11 model, when the tumor grew to a volume of 300–500 mm3, the mice were grouped and treated orally once daily with 5, 10 or 20 mg/kg SKLB1028 for 18 days. The tumor volumes were measured every 3 days. Treatment with SKLB1028 at 10 or 20 mg/kg/day resulted in rapid and complete tumor regression in all mice in both groups (Figure 2a). SKLB1028 treatment at 5 mg/kg/day stopped the tumor growth and resulted in slight tumor regression. After 7 days of additional treatment, the mice treated with 20 mg/kg/day SKLB1028 were monitored (but not treated) for the subsequent 20 days, and no tumor re-growth was observed in any of the mice in this group. Histological and immunohistochemical analyses were performed to evaluate the anti-tumor mechanism of action of SKLB1028 in the MV4-11 model. As shown in Figure 2c, SKLB1028 significantly inhibited the tumor-cell proliferation (the percentage of Ki67-expressing cells in viable tumor tissue was significantly lower following SKLB1028 treatment) and induced apoptosis in the tumor tissue (the percentage of TUNEL-positive apoptotic cells was considerably higher in SKLB1028-treated tumors than in vehicle-treated tumors).In the K562 model, a series of doses of SKLB1028 was administered orally once daily when the tumors grew to 150–300 mm3. SKLB1028 treatment at a dose of 70 mg/kg/day significantly inhibited tumor growth compared with the vehicle group (Figure 2b). The tumor-inhibition rate of SKLB1028 at a dose of 70 mg/kg/day was 71.3%, whereas imatinib mesylate displayed a weak tumor-suppressing effect at the same dose. Again, histological and immunohistochemical analyses were performed to evaluate the mechanism of action of SKLB1028 in the K562 model. In this model, SKLB1028 considerably inhibited the tumor-cell proliferation and induced apoptosis in the tumor tissue in a manner similar to MV4-11 (Figure 2d).We further examined the influence of SKLB1028 on survival time using a bone marrow engraftment model of MV4-11 (Supplementary Material). Supplementary Figure S3 shows that compared with vehicle and positive control (sunitinib, 10 mg/kg/day) groups, SKLB1028-treated mice demonstrated prolonged survival in a dose-dependent manner with mean survival times (MSTs) of 65, 71 and 80 days for the 5, 10 and 20 mg/kg/day groups, respectively. The MSTs for the vehicle and positive control groups were 48.5 and 61 days, respectively. An initial sub-acute toxicity test was also performed, which indicated that SKLB1028 could be continuously administered at the dose of 100 mg/kg/day in rats with no observed adverse effects (Supplementary Figure S4 and Tables S4–S6). Finally, the pharmacokinetic characteristics of SKLB1028 following per os administration to male rats were analyzed. As shown in Supplementary Figure S5, the compound was well absorbed, achieving a maximum plasma level (Cmax) of 607 μg/l within 4 h after oral administration at a dose of 60 mg/kg. Furthermore, the apparent plasma half-life was approximately 7.2 h, and the AUC(0−24h) was approximately 6.969 mg/l/h.In summary, SKLB1028 is a novel multikinase inhibitor that displays exceptional activity in models of FLT3-driven AML and considerable potency in CML that harbors Abl mutations. SKLB1028 has the convenience of oral administration, favorable pharmacokinetic properties and low toxicity. Collectively, preclinical assessment of the pharmacodynamics, pharmacokinetics and initial toxicity of SKLB1028 supports the use of SKLB1028 as a promising candidate for clinical studies in patients with leukemia. Finally, we must mention that SKLB1028 differs from other known FLT3 inhibitors in that it is also a typical EGFR inhibitor. This characteristic of SKLB1028 is noteworthy because of the recent unexpected findings that EGFR inhibitors, such as 4,5-dianilinophthalimide (DAPH1), gefitinib and erlotinib, are able to promote differentiation and induce apoptosis in AML blasts in vitro.14, 15 Because AML cells do not express EGFR, the observed activity of EGFR inhibitors on AML cells cannot be due to inhibition of EGFR, but must have resulted from inhibition of an off-target kinase that is as yet uncharacterized. Whether SKLB1028 has the same effect on AML or acts on the same off-target kinases as other EGFR inhibitors remains to be determined.The authors declare no conflict of interest.This work was supported by the National Natural Science Foundation of China (81172987), the National S&T Major Project (2012ZX09102-101-002) and SRFDP (20100181110025).Supplementary Information accompanies the paper on the Leukemia website
Co-reporter:Qing-Qing Xie, Lei Zhong, You-Li Pan, Xiao-Yan Wang, Jian-Ping Zhou, Lei Di-wu, Qi Huang, Yu-Lan Wang, Ling-Ling Yang, Huan-Zhang Xie, Sheng-Yong Yang
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 9) pp:3675-3680
Publication Date(Web):September 2011
DOI:10.1016/j.ejmech.2011.05.031
Aberrant c-Met activation has been demonstrated to be implicated in tumorigenesis and anti-cancer drug resistance. Discovery of c-Met inhibitors has attracted much attention in recent years. In this study, a support vector machine (SVM) classification model that discriminates c-Met inhibitors and non-inhibitors was first developed. Evaluation through screening a test set indicates that combined SVM-based and docking-based virtual screening (SB/DB-VS) considerably increases hit rate and enrichment factor compared with the individual methods. Thus the combined SB/DB-VS approach was adopted to screen PubChem, Specs, and Enamine for c-Met inhibitors. 75 compounds were selected for in vitro assays. Eight compounds display a good inhibitory potency against c-Met. Five of them are found to have novel scaffolds, implying a good potential for further chemical modification.Combined SVM-based and molecular docking-based virtual screening approach was used to screening several large compounds libraries to retrieve c-Met inhibitors. Some active compounds with novel scaffolds were found.Highlights► Combined SVM/docking-based virtual screening was evaluated to be effective. ► This combined approach was used to screen large databases for c-Met inhibitors. ► Eight compounds display a good inhibitory potency against c-Met. ► Five of them are found to have novel scaffolds.
Co-reporter:Ji-Xia Ren, Lin-Li Li, Ren-Lin Zheng, Huan-Zhang Xie, Zhi-Xing Cao, Shan Feng, You-Li Pan, Xin Chen, Yu-Quan Wei, and Sheng-Yong Yang
Journal of Chemical Information and Modeling 2011 Volume 51(Issue 6) pp:1364-1375
Publication Date(Web):May 27, 2011
DOI:10.1021/ci100464b
In this investigation, we describe the discovery of novel potent Pim-1 inhibitors by employing a proposed hierarchical multistage virtual screening (VS) approach, which is based on support vector machine-based (SVM-based VS or SB-VS), pharmacophore-based VS (PB-VS), and docking-based VS (DB-VS) methods. In this approach, the three VS methods are applied in an increasing order of complexity so that the first filter (SB-VS) is fast and simple, while successive ones (PB-VS and DB-VS) are more time-consuming but are applied only to a small subset of the entire database. Evaluation of this approach indicates that it can be used to screen a large chemical library rapidly with a high hit rate and a high enrichment factor. This approach was then applied to screen several large chemical libraries, including PubChem, Specs, and Enamine as well as an in-house database. From the final hits, 47 compounds were selected for further in vitro Pim-1 inhibitory assay, and 15 compounds show nanomolar level or low micromolar inhibition potency against Pim-1. In particular, four of them were found to have new scaffolds which have potential for the chemical development of Pim-1 inhibitors.
Co-reporter:Jun Zou, Shi-Dong Luo, Yu-Quan Wei and Sheng-Yong Yang
Molecular BioSystems 2011 vol. 7(Issue 1) pp:169-179
Publication Date(Web):26 Oct 2010
DOI:10.1039/C0MB00004C
Understanding the regulation of mitotic entry is one of the most important goals of modern cell biology, and computational modeling of mitotic entry has been a subject of several recent studies. However, there are still many regulation mechanisms that remain poorly characterized. Two crucial aspects are how mitotic entry is controlled by its upstream regulators Aurora-A and Plk1, and how mitotic entry is coordinated with other biological events, especially G2/M checkpoint. In this context, we reconstructed a comprehensive computational model that integrates the mitotic entry network and the G2/M checkpoint system. Computational simulation of this model and subsequent experimental verification revealed that Aurora-A and Plk1 are redundant to the activation of cyclin B/Cdk1 during normal mitotic entry, but become especially important for cyclin B/Cdk1 activation during G2/M checkpoint recovery. Further analysis indicated that, in response to DNA damage, Chk1-mediated network rewiring makes cyclin B/Cdk1 more sensitive to the down-regulation of Aurora-A and Plk1. In addition, we demonstrated that concurrently targeting Aurora-A and Plk1 during G2/M checkpoint recovery achieves a synergistic effect, which suggests the combinational use of Aurora-A and Plk1 inhibitors after chemotherapy or radiotherapy. Thus, the results presented here provide novel insights into the regulation mechanism of mitotic entry and have potential value in cancer therapy.
Co-reporter:Guo-Bo Li, Ling-Ling Yang, Shan Feng, Jian-Ping Zhou, Qi Huang, Huan-Zhang Xie, Lin-Li Li, Sheng-Yong Yang
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 6) pp:1736-1740
Publication Date(Web):15 March 2011
DOI:10.1016/j.bmcl.2011.01.087
Development of glutamate non-competitive antagonists of mGluR1 (Metabotropic glutamate receptor subtype 1) has increasingly attracted much attention in recent years due to their potential therapeutic application for various nervous disorders. Since there is no crystal structure reported for mGluR1, ligand-based virtual screening (VS) methods, typically pharmacophore-based VS (PB-VS), are often used for the discovery of mGluR1 antagonists. Nevertheless, PB-VS usually suffers a lower hit rate and enrichment factor. In this investigation, we established a multistep ligand-based VS approach that is based on a support vector machine (SVM) classification model and a pharmacophore model. Performance evaluation of these methods in virtual screening against a large independent test set, M-MDDR, show that the multistep VS approach significantly increases the hit rate and enrichment factor compared with the individual SB-VS and PB-VS methods. The multistep VS approach was then used to screen several large chemical libraries including PubChem, Specs, and Enamine. Finally a total of 20 compounds were selected from the top ranking compounds, and shifted to the subsequent in vitro and in vivo studies, which results will be reported in the near future.
Co-reporter:Xiao-Fei Zeng, Wei-Wei Li, Hui-Jin Fan, Xiao-Yan Wang, Pan Ji, Ze-Rong Wang, Shuang Ma, Lin-Li Li, Xiao-Feng Ma, Sheng-Yong Yang
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 16) pp:4742-4744
Publication Date(Web):15 August 2011
DOI:10.1016/j.bmcl.2011.06.075
Development of fatty acid synthase (FAS) inhibitors has increasingly attracted much attention in recent years due to their potential therapeutic use in obesity and cancers. In this investigation, pharmacophore modeling based on the first crystal structure of human KS domain of FAS was carried out. The established pharmacophore model was taken as a 3D query for retrieving potent FAS inhibitors from the chemical database Specs. Docking study was further carried out to refine the obtained hit compounds. Finally, a total of 28 compounds were selected based on the ranking order and visual examination, which were first evaluated by a cell line-based assay. Seven compounds that have good inhibition activity against two FAS overexpressing cancer cell lines were further evaluated by an enzyme-based assay. One compound with a new chemical scaffold was found to have low micromolar inhibition potency against FAS, which has been subjected to further chemical structural modification.
Co-reporter:Wei-Wei Li;Jin-Juan Chen;Ren-Lin Zheng;Wen-Qin Zhang;Zhi-Xing Cao;Ling-Ling Yang;Xiao-Yu Qing;Liang-Xue Zhou;Li Yang;Luo-Ding Yu ;Li-Juan Chen ;Yu-Quan Wei
ChemMedChem 2010 Volume 5( Issue 4) pp:513-516
Publication Date(Web):
DOI:10.1002/cmdc.200900537
Co-reporter:Qi Huang, Lin-Li Li, Sheng-Yong Yang
Journal of Molecular Graphics and Modelling 2010 Volume 28(Issue 8) pp:775-787
Publication Date(Web):June 2010
DOI:10.1016/j.jmgm.2010.02.002
This account describes a new pharmacophore-based de novo design method of drug-like molecules (PhDD). The method PhDD first generates a set of new molecules that completely conform to the requirements of a given pharmacophore model, followed by a series of assessments to the generated molecules, including assessments of drug-likeness, bioactivity, and synthetic accessibility. PhDD is tested on three typical examples, namely, pharmacophore hypotheses of histone deacetylase (HDAC), cyclin-dependent kinase 2 (CDK2) and HIV-1 integrase (IN) inhibitors. The test results demonstrate that PhDD is able to generate molecules with novel structures but having similar biological functions with existing inhibitors. The validity of PhDD together with its ability of assessing synthetic accessibility makes it a useful tool in rational drug design.
Co-reporter:Ji-Xia Ren, Lin-Li Li, Jun Zou, Li Yang, Jin-Liang Yang, Sheng-Yong Yang
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 11) pp:4259-4265
Publication Date(Web):November 2009
DOI:10.1016/j.ejmech.2009.07.008
Inhibitors of transforming growth factor-β Type I Receptor (ALK5) have been thought as potential drugs for the treatment of fibrosis and cancer and a considerable number of ALK5 inhibitors have been reported recently. In order to clarify the essential structure–activity relationship for the known ALK5 inhibitors as well as identify new lead compounds against ALK5, 3D pharmacophore models have been established based on the known ALK5 inhibitors. The best pharmacophore model, Hypo1, was used as a 3D search query to perform a virtual screening. The hit compounds were subsequently subjected to filtering by Lipinski's rule of five and docking studies to refine the retrieved hits. Finally a total of 100 compounds were obtained and some of them were selected for further in vitro and in vivo assay studies. A quantitative pharmacophore model of ALK5 inhibitors has been established and evaluated. Based on this model, a virtual screening and docking study was carried out to identify new ALK5 inhibitors.
Co-reporter:Hui-Yuan Wang;Lin-Li Li;Zhi-Xing Cao;Shi-Dong Luo;Yu-Quan Wei
Chemical Biology & Drug Design 2009 Volume 73( Issue 1) pp:115-126
Publication Date(Web):
DOI:10.1111/j.1747-0285.2008.00751.x
In this study, 3D-pharmacophore models of Aurora B kinase inhibitors have been developed by using HipHop and HypoGen modules in Catalyst software package. The best pharmacophore model, Hypo1, which has the highest correlation coefficient (0.9911), consists of one hydrogen-bond acceptor, one hydrogen-bond donor, one hydrophobic aliphatic moiety and one ring aromatic feature. Hypo1 was validated by test set and cross-validation methods. And the specificity of Hypo1 to Aurora B inhibitors was examined with the use of selective inhibitors against Aurora B and its paralogue Aurora A. The results clearly indicate that Hypo1 can differentiate selective inhibitors of Aurora B from those of Aurora A, and the ring aromatic feature likely plays some important roles for the specificity of Hypo1. Then Hypo1 was used as a 3D query to screen several databases including Specs, NCI, Maybridge and Chinese Nature Product Database (CNPD) for identifying new inhibitors of Aurora B. The hit compounds were subsequently subjected to filtering by Lipinski’s rule of five and docking studies to refine the retrieved hits, and some compounds selected from the top ranked hits have been suggested for further experimental assay studies.
Co-reporter:Huan-Zhang Xie, Lin-Li Li, Ji-Xia Ren, Jun Zou, Li Yang, Yu-Quan Wei, Sheng-Yong Yang
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 7) pp:1944-1949
Publication Date(Web):1 April 2009
DOI:10.1016/j.bmcl.2009.02.049
In this investigation, chemical features based 3D pharmacophore models were developed based on the known inhibitors of Spleen tyrosine kinase (Syk) with the aid of hiphop and hyporefine modules within catalyst. The best quantitative pharmacophore model, Hypo1, was used as a 3D structural query for retrieving potential inhibitors from chemical databases including Specs, NCI, MayBridge, and Chinese Nature Product Database (CNPD). The hit compounds were subsequently subjected to filtering by Lipinski’s rule of five and docking studies to refine the retrieved hits. Finally 30 compounds were selected from the top ranked hit compounds and conducted an in vitro kinase inhibitory assay. Six compounds showed a good inhibitory potency against Syk, which have been selected for further investigation.
Co-reporter:Hui Zhang;Ming-Li Xiang;Chang-Ying Ma;Qi Huang;Wei Li;Yang Xie
Molecular Diversity 2009 Volume 13( Issue 2) pp:
Publication Date(Web):2009 May
DOI:10.1007/s11030-009-9108-1
In this investigation, three-class classification models of aqueous solubility (logS) and lipophilicity (logP) have been developed by using a support vector machine (SVM) method combined with a genetic algorithm (GA) for feature selection and a conjugate gradient method (CG) for parameter optimization. A 5-fold cross-validation and an independent test set method were used to evaluate the SVM classification models. For logS, the overall prediction accuracy is 87.1% for training set and 90.0% for test set. For logP, the overall prediction accuracy is 81.0% for training set and 82.0% for test set. In general, for both logS and logP, the prediction accuracies of three-class models are slightly lower by several percent than those of two-class models. A comparison between the performance of GA–CG–SVM models and that of GA–SVM models shows that the SVM parameter optimization has a significant impact on the quality of SVM classification model.
Co-reporter:Qing-Qing Xie, Huan-Zhang Xie, Ji-Xia Ren, Lin-Li Li, Sheng-Yong Yang
Journal of Molecular Graphics and Modelling 2009 Volume 27(Issue 6) pp:751-758
Publication Date(Web):February 2009
DOI:10.1016/j.jmgm.2008.11.008
In this study, chemical feature based pharmacophore models of type I and type II kinase inhibitors of Tie2 have been developed with the aid of HipHop and HypoRefine modules within Catalyst program package. The best HipHop pharmacophore model Hypo1_I for type I kinase inhibitors contains one hydrogen-bond acceptor, one hydrogen-bond donor, one general hydrophobic, one hydrophobic aromatic, and one ring aromatic feature. And the best HypoRefine model Hypo1_II for type II kinase inhibitors, which was characterized by the best correlation coefficient (0.976032) and the lowest RMSD (0.74204), consists of two hydrogen-bond donors, one hydrophobic aromatic, and two general hydrophobic features, as well as two excluded volumes. These pharmacophore models have been validated by using either or both test set and cross validation methods, which shows that both the Hypo1_I and Hypo1_II have a good predictive ability. The space arrangements of the pharmacophore features in Hypo1_II are consistent with the locations of the three portions making up a typical type II kinase inhibitor, namely, the portion occupying the ATP binding region (ATP-binding-region portion, AP), that occupying the hydrophobic region (hydrophobic-region portion, HP), and that linking AP and HP (bridge portion, BP). Our study also reveals that the ATP-binding-region portion of the type II kinase inhibitors plays an important role to the bioactivity of the type II kinase inhibitors. Structural modifications on this portion should be helpful to further improve the inhibitory potency of type II kinase inhibitors.
Co-reporter:Xiao-Qiang Deng;Hui-Yuan Wang;Ying-Lan Zhao;Ming-Li Xiang;Pei-Du Jiang;Zhi-Xing Cao;Yu-Zhu Zheng;Shi-Dong Luo;Luo-Ting Yu;Yu-Quan Wei
Chemical Biology & Drug Design 2008 Volume 71( Issue 6) pp:533-539
Publication Date(Web):
DOI:10.1111/j.1747-0285.2008.00663.x
Aurora-A has been identified as one of the most attractive targets for cancer therapy and a considerable number of Aurora-A inhibitors have been reported recently. In order to clarify the essential structure–activity relationship for the known Aurora-A inhibitors as well as identify new lead compounds against Aurora-A, 3D pharmacophore models were developed based on the known inhibitors. The best hypothesis, Hypo1, was used to screen molecular structural databases, including Specs and China Natural Products Database for potential lead compounds. The hit compounds were subsequently subjected to filtering by Lipinski’s rules and docking study to refine the retrieved hits and as a result to reduce the rate of false positive. Finally, 39 compounds were purchased for further in vitro assay against several human tumour cell lines including A549, MCF-7, HepG2 and PC-3, in which Aurora-A is overexpressed. Two compounds show very low micromolar inhibition potency against some of these tumour cells. And they have been selected for further investigation.
Co-reporter:Hui-Yuan Wang, Zhi-Xing Cao, Lin-Li Li, Pei-Du Jiang, Ying-Lan Zhao, Shi-Dong Luo, Li Yang, Yu-Quan Wei, Sheng-Yong Yang
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 18) pp:4972-4977
Publication Date(Web):15 September 2008
DOI:10.1016/j.bmcl.2008.08.033
Pharmacophore models of Polo-like kinase-1 (PLK1) inhibitors have been established by using the HipHop and HypoGen algorithms implemented in the Catalyst software package. The best quantitative pharmacophore model, Hypo1, which has the highest correlation coefficient (0.9895), consists of one hydrogen bond acceptor, one hydrogen bond donor, one hydrophobic feature, and one hydrophobic aliphatic feature. Hypo1 was further validated by test set and cross validation method. Then Hypo1 was used as a 3D query to screen several databases including Specs, NCI, Maybridge, and Chinese Nature Product Database (CNPD). The hit compounds were subsequently subjected to filtering by Lipinski’s rule of five and docking study to refine the retrieved hits and as a result to reduce the rate of false positive. Finally, a total of 20 compounds were selected and have been shifted to in vitro and in vivo studies. As far as we know, this is the first report on the pharmacophore modeling even the first publicly reported virtual screening study of PLK1 inhibitors.Pharmacophore model of PLK1 inhibitors was established. This model was then used as a 3D query to screen several chemical databases including Specs, NCI, Maybridge, and CNPD.
Co-reporter:Jun Zou, Huan-Zhang Xie, Sheng-Yong Yang, Jin-Juan Chen, Ji-Xia Ren, Yu-Quan Wei
Journal of Molecular Graphics and Modelling 2008 Volume 27(Issue 4) pp:430-438
Publication Date(Web):November 2008
DOI:10.1016/j.jmgm.2008.07.004
Pharmacophore modeling, including ligand- and structure-based approaches, has become an important tool in drug discovery. However, the ligand-based method often strongly depends on the training set selection, and the structure-based pharmacophore model is usually created based on apo structures or a single protein–ligand complex, which might miss some important information. In this study, multicomplex-based method has been suggested to generate a comprehensive pharmacophore map of cyclin-dependent kinase 2 (CDK2) based on a collection of 124 crystal structures of human CDK2–inhibitor complex. Our multicomplex-based comprehensive pharmacophore map contains almost all the chemical features important for CDK2–inhibitor interactions. A comparison with previously reported ligand-based pharmacophores has revealed that the ligand-based models are just a subset of our comprehensive map. Furthermore, one most-frequent-feature pharmacophore model consisting of the most frequent pharmacophore features was constructed based on the statistical frequency information provided by the comprehensive map. Validations to the most-frequent-feature model show that it can not only successfully discriminate between known CDK2 inhibitors and the molecules of focused inactive dataset, but also is capable of correctly predicting the activities of a wide variety of CDK2 inhibitors in an external active dataset. Obviously, this investigation provides some new ideas about how to develop a multicomplex-based pharmacophore model that can be used in virtual screening to discover novel potential lead compounds.
Co-reporter:Sheng-Yong Yang
Drug Discovery Today (June 2010) Volume 15(Issues 11–12) pp:444-450
Publication Date(Web):1 June 2010
DOI:10.1016/j.drudis.2010.03.013
Pharmacophore approaches have become one of the major tools in drug discovery after the past century's development. Various ligand-based and structure-based methods have been developed for improved pharmacophore modeling and have been successfully and extensively applied in virtual screening, de novo design and lead optimization. Despite these successes, pharmacophore approaches have not reached their expected full capacity, particularly in facing the demand for reducing the current expensive overall cost associated with drug discovery and development. Here, the challenges of pharmacophore modeling and applications in drug discovery are discussed and recent advances and latest developments are described, which provide useful clues to the further development and application of pharmacophore approaches.







![Benzamide, 3-iodo-4-methyl-N-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/884700/884600-93-5.png)
![Benzamide, 3-iodo-4-methyl-N-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/884700/884600-93-5_b.png)
![Benzoic acid, 3-[(phenylamino)sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/866400/866324-02-9.png)
![Benzoic acid, 3-[(phenylamino)sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/866400/866324-02-9_b.png)


![BENZOIC ACID, 3-[(DIETHYLAMINO)SULFONYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/445300/445298-39-5.png)
![BENZOIC ACID, 3-[(DIETHYLAMINO)SULFONYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/445300/445298-39-5_b.png)
![1H-Pyrazolo[3,4-d]pyrimidin-4-amine, 3-iodo-1-(4-piperidinyl)-](http://img.cochemist.com/ccimg/330800/330793-49-2.png)
![1H-Pyrazolo[3,4-d]pyrimidin-4-amine, 3-iodo-1-(4-piperidinyl)-](http://img.cochemist.com/ccimg/330800/330793-49-2_b.png)
![4-(4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexanone](http://img.cochemist.com/ccimg/330800/330792-72-8.png)
![4-(4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexanone](http://img.cochemist.com/ccimg/330800/330792-72-8_b.png)
![Hydrazinecarboxamide, 2-[[5-(3-chlorophenyl)-2-furanyl]methylene]-](http://img.cochemist.com/ccimg/301200/301176-94-3.png)
![Hydrazinecarboxamide, 2-[[5-(3-chlorophenyl)-2-furanyl]methylene]-](http://img.cochemist.com/ccimg/301200/301176-94-3_b.png)

![Benzoic acid,3-[(diethylamino)sulfonyl]-, hydrazide](http://img.cochemist.com/ccimg/96200/96134-80-4.png)
![Benzoic acid,3-[(diethylamino)sulfonyl]-, hydrazide](http://img.cochemist.com/ccimg/96200/96134-80-4_b.png)
![Benzoic acid,3-[(dimethylamino)sulfonyl]-, hydrazide](http://img.cochemist.com/ccimg/96200/96134-79-1.png)
![Benzoic acid,3-[(dimethylamino)sulfonyl]-, hydrazide](http://img.cochemist.com/ccimg/96200/96134-79-1_b.png)
![Hydrazinecarboxamide,2-[[3-methoxy-4-(phenylmethoxy)phenyl]methylene]-](http://img.cochemist.com/ccimg/81300/81263-72-1.png)
![Hydrazinecarboxamide,2-[[3-methoxy-4-(phenylmethoxy)phenyl]methylene]-](http://img.cochemist.com/ccimg/81300/81263-72-1_b.png)
![Hydrazinecarbothioamide, 2-[[5-(4-chlorophenyl)-2-furanyl]methylene]-](http://img.cochemist.com/ccimg/60700/60698-40-0.png)
![Hydrazinecarbothioamide, 2-[[5-(4-chlorophenyl)-2-furanyl]methylene]-](http://img.cochemist.com/ccimg/60700/60698-40-0_b.png)
![HYDRAZINECARBOXAMIDE, 2-[[5-(4-CHLOROPHENYL)-2-FURANYL]METHYLENE]-](http://img.cochemist.com/ccimg/58200/58136-77-9.png)
![HYDRAZINECARBOXAMIDE, 2-[[5-(4-CHLOROPHENYL)-2-FURANYL]METHYLENE]-](http://img.cochemist.com/ccimg/58200/58136-77-9_b.png)
![Hydrazinecarboxamide, 2-[[5-(4-nitrophenyl)-2-furanyl]methylene]-](http://img.cochemist.com/ccimg/58200/58136-76-8.png)
![Hydrazinecarboxamide, 2-[[5-(4-nitrophenyl)-2-furanyl]methylene]-](http://img.cochemist.com/ccimg/58200/58136-76-8_b.png)







![(2E)-2-[(5-phenylfuran-2-yl)methylidene]hydrazinecarboxamide](http://img.cochemist.com/ccimg/13900/13803-15-1.png)
![(2E)-2-[(5-phenylfuran-2-yl)methylidene]hydrazinecarboxamide](http://img.cochemist.com/ccimg/13900/13803-15-1_b.png)

![Benzoic acid, 3-[(phenylamino)sulfonyl]-](http://img.cochemist.com/ccimg/1600/1576-45-0.png)
![Benzoic acid, 3-[(phenylamino)sulfonyl]-](http://img.cochemist.com/ccimg/1600/1576-45-0_b.png)
![ETHYL 6-(4-AMINOPHENYL)IMIDAZO[2,1-B][1,3]THIAZOLE-3-CARBOXYLATE](http://img.cochemist.com/ccimg/1397200/1397198-76-3.png)
![ETHYL 6-(4-AMINOPHENYL)IMIDAZO[2,1-B][1,3]THIAZOLE-3-CARBOXYLATE](http://img.cochemist.com/ccimg/1397200/1397198-76-3_b.png)
![ETHYL 6-(3-AMINOPHENYL)IMIDAZO[2,1-B][1,3]THIAZOLE-3-CARBOXYLATE](http://img.cochemist.com/ccimg/1397200/1397194-35-2.png)
![ETHYL 6-(3-AMINOPHENYL)IMIDAZO[2,1-B][1,3]THIAZOLE-3-CARBOXYLATE](http://img.cochemist.com/ccimg/1397200/1397194-35-2_b.png)


![PROPAN-2-YL (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-DIOXOPYRIMIDIN-1-YL)-4-FLUORO-3-HYDROXY-4-METHYLOXOLAN-2-YL]METHOXY-PHENOXYPHOSPHORYL]AMINO]PROPANOATE](http://img.cochemist.com/ccimg/1190400/1190307-88-0.png)
![PROPAN-2-YL (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-DIOXOPYRIMIDIN-1-YL)-4-FLUORO-3-HYDROXY-4-METHYLOXOLAN-2-YL]METHOXY-PHENOXYPHOSPHORYL]AMINO]PROPANOATE](http://img.cochemist.com/ccimg/1190400/1190307-88-0_b.png)


![3-chloro-4-[(4-methyl-1-piperazinyl)methyl]-Benzenamine](http://img.cochemist.com/ccimg/943400/943320-67-0.png)
![3-chloro-4-[(4-methyl-1-piperazinyl)methyl]-Benzenamine](http://img.cochemist.com/ccimg/943400/943320-67-0_b.png)


![1H-Pyrazolo[3,4-d]pyrimidin-4-amine, 3-iodo-1-methyl-](http://img.cochemist.com/ccimg/862800/862729-12-2.png)
![1H-Pyrazolo[3,4-d]pyrimidin-4-amine, 3-iodo-1-methyl-](http://img.cochemist.com/ccimg/862800/862729-12-2_b.png)
![IMIDAZO[2,1-B]THIAZOLE-3-CARBOXYLIC ACID, 6-(4-NITROPHENYL)-, ETHYL ESTER](http://img.cochemist.com/ccimg/752300/752244-21-6.png)
![IMIDAZO[2,1-B]THIAZOLE-3-CARBOXYLIC ACID, 6-(4-NITROPHENYL)-, ETHYL ESTER](http://img.cochemist.com/ccimg/752300/752244-21-6_b.png)
![4-[(4-ethyl-1-piperazinyl)methyl]-3-(trifluoromethyl)aniline](http://img.cochemist.com/ccimg/630200/630125-91-6.png)
![4-[(4-ethyl-1-piperazinyl)methyl]-3-(trifluoromethyl)aniline](http://img.cochemist.com/ccimg/630200/630125-91-6_b.png)
![3-METHYL-N-{[2-(PENTAFLUOROPHENYL)-1,3-BENZOXAZOL-5-YL]CARBAMOTHIOYL}BUTANAMIDE](/data/chemimg/631900/625370-80-1.png)
![3-METHYL-N-{[2-(PENTAFLUOROPHENYL)-1,3-BENZOXAZOL-5-YL]CARBAMOTHIOYL}BUTANAMIDE](/data/chemimg/631900/625370-80-1_b.png)




![<br>(3-Amino-6-(thiophen-2-yl)-4-(trifluoromethyl)thieno[2,3-b]pyridin-2-yl)(mo rpholino)methanone](http://img.cochemist.com/ccimg/400900/400863-57-2.png)
![<br>(3-Amino-6-(thiophen-2-yl)-4-(trifluoromethyl)thieno[2,3-b]pyridin-2-yl)(mo rpholino)methanone](http://img.cochemist.com/ccimg/400900/400863-57-2_b.png)
![<br>3-Amino-N,N-diethyl-6-phenyl-4-(trifluoromethyl)thieno[2,3-b]pyridine-2-car boxamide](http://img.cochemist.com/ccimg/400900/400863-50-5.png)
![<br>3-Amino-N,N-diethyl-6-phenyl-4-(trifluoromethyl)thieno[2,3-b]pyridine-2-car boxamide](http://img.cochemist.com/ccimg/400900/400863-50-5_b.png)










![2-Pyridinecarboxamide,4-[4-[[[[5-(1,1-dimethylethyl)-3-isoxazolyl]amino]carbonyl]amino]phenoxy]-N-methyl-](http://img.cochemist.com/ccimg/229000/228999-89-1.png)
![2-Pyridinecarboxamide,4-[4-[[[[5-(1,1-dimethylethyl)-3-isoxazolyl]amino]carbonyl]amino]phenoxy]-N-methyl-](http://img.cochemist.com/ccimg/229000/228999-89-1_b.png)
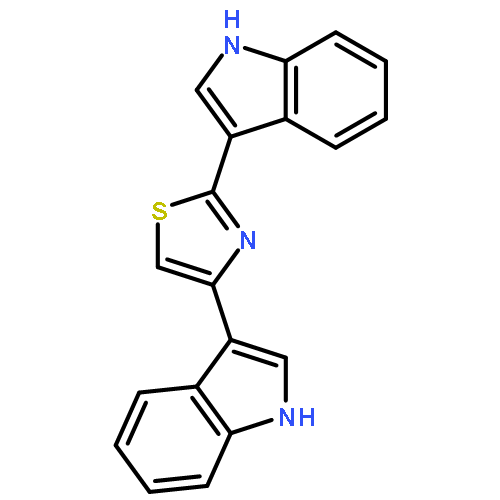
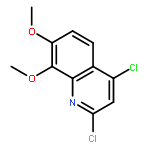
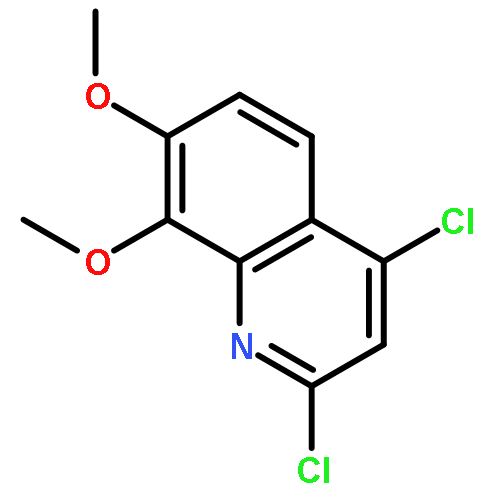








![4H-Pyrazolo[3,4-d]pyrimidin-4-one, 3,5-dihydro- (9CI)](http://img.cochemist.com/ccimg/161800/161746-79-8.png)
![4H-Pyrazolo[3,4-d]pyrimidin-4-one, 3,5-dihydro- (9CI)](http://img.cochemist.com/ccimg/161800/161746-79-8_b.png)
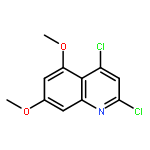
![Ethyl 3-amino-4-(trifluoromethyl)-6-phenylfuro[2,3-b]pyridine-2-carboxylate](http://img.cochemist.com/ccimg/160500/160436-48-6.png)
![Ethyl 3-amino-4-(trifluoromethyl)-6-phenylfuro[2,3-b]pyridine-2-carboxylate](http://img.cochemist.com/ccimg/160500/160436-48-6_b.png)
























![Imidazo[2,1-b]thiazole-6-carboxylic acid, 2-methyl-, ethyl ester](http://img.cochemist.com/ccimg/120200/120107-64-4.png)
![Imidazo[2,1-b]thiazole-6-carboxylic acid, 2-methyl-, ethyl ester](http://img.cochemist.com/ccimg/120200/120107-64-4_b.png)








![3-Amino-6-phenyl-4-(trifluoromethyl)thieno[2,3-b]pyridine-2-carboxylic acid](http://img.cochemist.com/ccimg/105000/104960-56-7.png)
![3-Amino-6-phenyl-4-(trifluoromethyl)thieno[2,3-b]pyridine-2-carboxylic acid](http://img.cochemist.com/ccimg/105000/104960-56-7_b.png)
![Propanedinitrile, 2-[2-[(1E)-2-[4-(dimethylamino)phenyl]ethenyl]-6-methyl-4H-pyran-4-ylidene]-](/data/chemimg/2248100/96042-30-7.png)
![Propanedinitrile, 2-[2-[(1E)-2-[4-(dimethylamino)phenyl]ethenyl]-6-methyl-4H-pyran-4-ylidene]-](/data/chemimg/2248100/96042-30-7_b.png)
![Disulfide, bis[4-(methylsulfonyl)phenyl]](http://img.cochemist.com/ccimg/92500/92424-06-1.png)
![Disulfide, bis[4-(methylsulfonyl)phenyl]](http://img.cochemist.com/ccimg/92500/92424-06-1_b.png)
![Benzoic acid,2-hydroxy-5-[[(4-methoxyphenyl)amino]sulfonyl]-](http://img.cochemist.com/ccimg/92300/92200-76-5.png)
![Benzoic acid,2-hydroxy-5-[[(4-methoxyphenyl)amino]sulfonyl]-](http://img.cochemist.com/ccimg/92300/92200-76-5_b.png)






![THIENO[2,3-B]PYRIDINE-2-CARBOXYLIC ACID, 3-AMINO-4-METHYL-6-PHENYL-](http://img.cochemist.com/ccimg/58400/58327-78-9.png)
![THIENO[2,3-B]PYRIDINE-2-CARBOXYLIC ACID, 3-AMINO-4-METHYL-6-PHENYL-](http://img.cochemist.com/ccimg/58400/58327-78-9_b.png)


![(2E)-1-[2,4-dihydroxy-3-(3-methylbut-2-en-1-yl)phenyl]-3-phenylprop-2-en-1-one](http://img.cochemist.com/ccimg/52700/52601-05-5.png)
![(2E)-1-[2,4-dihydroxy-3-(3-methylbut-2-en-1-yl)phenyl]-3-phenylprop-2-en-1-one](http://img.cochemist.com/ccimg/52700/52601-05-5_b.png)














![2,4-Pentadienal, 5-[4-(dimethylamino)phenyl]-, (2E,4E)-](http://img.cochemist.com/ccimg/20500/20432-36-4.png)
![2,4-Pentadienal, 5-[4-(dimethylamino)phenyl]-, (2E,4E)-](http://img.cochemist.com/ccimg/20500/20432-36-4_b.png)
![2-Propenal, 3-[4-(dimethylamino)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/20500/20432-35-3.png)
![2-Propenal, 3-[4-(dimethylamino)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/20500/20432-35-3_b.png)


![Benzaldehyde, 3-hydroxy-4-methoxy-, 2-[(3-hydroxy-4-methoxyphenyl)methylene]hydrazone](/data/chemimg/1744600/15332-12-4.png)
![Benzaldehyde, 3-hydroxy-4-methoxy-, 2-[(3-hydroxy-4-methoxyphenyl)methylene]hydrazone](/data/chemimg/1744600/15332-12-4_b.png)

![N-[(4-Nitrophenyl)sulfonyl]glycine](http://img.cochemist.com/ccimg/15100/15054-44-1.png)
![N-[(4-Nitrophenyl)sulfonyl]glycine](http://img.cochemist.com/ccimg/15100/15054-44-1_b.png)
![5,6,7,8-Tetrahydro-4H-cyclohepta[d]thiazol-2-amine](http://img.cochemist.com/ccimg/14300/14292-44-5.png)
![5,6,7,8-Tetrahydro-4H-cyclohepta[d]thiazol-2-amine](http://img.cochemist.com/ccimg/14300/14292-44-5_b.png)
![6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ylidenepropanedinitrile](http://img.cochemist.com/ccimg/14100/14003-22-6.png)
![6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ylidenepropanedinitrile](http://img.cochemist.com/ccimg/14100/14003-22-6_b.png)







![8-[5-(5,7-dimethoxy-4-oxo-4H-chromen-2-yl)-2-methoxyphenyl]-5,7-dimethoxy-2-(4-methoxyphenyl)-4H-chromen-4-one](http://img.cochemist.com/ccimg/3800/3778-26-5.png)
![8-[5-(5,7-dimethoxy-4-oxo-4H-chromen-2-yl)-2-methoxyphenyl]-5,7-dimethoxy-2-(4-methoxyphenyl)-4H-chromen-4-one](http://img.cochemist.com/ccimg/3800/3778-26-5_b.png)





![1H-Benzimidazole,1-[(4-chlorophenyl)methyl]-2-(1-pyrrolidinylmethyl)-](http://img.cochemist.com/ccimg/500/442-52-4.png)
![1H-Benzimidazole,1-[(4-chlorophenyl)methyl]-2-(1-pyrrolidinylmethyl)-](http://img.cochemist.com/ccimg/500/442-52-4_b.png)

























