Co-reporter:Andreas F. Tillack, Lewis E. Johnson, Bruce E. Eichinger, and Bruce H. Robinson
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 9) pp:4362-4374
Publication Date(Web):July 19, 2016
DOI:10.1021/acs.jctc.6b00219
We have developed an approach to coarse-grained (CG) modeling of the van der Waals (vdW) type of interactions among molecules by representing groups of atoms within those molecules in terms of ellipsoids (rather than spheres). Our approach systematically translates an arbitrary underlying all-atom (AA) representation of a molecular system to a multisite ellipsoidal potential within the family of Gay–Berne type potentials. As the method enables arbitrary levels of coarse-graining, or even multiple levels of coarse-graining within a single simulation, we describe the method as a Level of Detail (LoD) model. The LoD model, as integrated into our group’s Metropolis Monte Carlo computational package, is also capable of reducing the complexity of the molecular electrostatics by means of a multipole expansion of charges obtained from an AA force field (or directly from electronic structure calculations) of the charges within each ellipsoid. Electronic polarizability may additionally be included. The present CG representation does not include transformation of bonded interactions; ellipsoids are connected at the fully atomistic bond sites by freely rotating links that are constrained to maintain a constant distance. The accuracy of the method is demonstrated for three distinct types of self-assembling or self-organizing molecular systems: (1) the interaction between benzene and perfluorobenzene (dispersion interactions), (2) linear hydrocarbon chains (a system with large conformational flexibility), and (3) the self-organization of ethylene carbonate (a highly polar liquid). Lastly, the method is applied to the interaction of large (∼100 atom) molecules, which are typical of organic nonlinear optical chromophores, to demonstrate the effect of different CG models on molecular assembly.
Co-reporter:Bruce H. Robinson, Lewis E. Johnson, and Bruce E. Eichinger
The Journal of Physical Chemistry B 2015 Volume 119(Issue 7) pp:3205-3212
Publication Date(Web):January 7, 2015
DOI:10.1021/jp507736r
Orientational order parameters are useful metrics for characterizing the probability distribution for vector-valued quantities such as the dipole moment or optical axis of molecules in materials such as liquid crystals and organic glasses. These parameters are the moments of the underlying orientational probability distribution. Many molecular systems can be characterized using a single centrosymmetric (even) moment. For dipolar systems, an applied electric or magnetic field can break the symmetry of the system, leading to nonzero acentric (odd) moments. For complex systems, it is difficult to characterize the nature of the bulk structures and to quantitatively understand the relationship between acentric and centrosymmetric moments. We have found that it is useful to relate the moments of the distribution in terms of an apparent dimensionality of the ordering process. Here we show that the idea of noninteger dimensionality, originally introduced by Stillinger, provides a useful method to characterize the relation between centrosymmetric and acentric orientational order parameters. Applying dimensional constraints is equivalent to removing rotational degrees of freedom or constraining rotation within a restricted volume. Simulations based on simple examples—using restoring potentials on arrays of independent dipoles—and on complex many-body Monte Carlo simulations of dipolar spheroids are described. An analysis of the results illustrates the utility of fractional dimensionality to describe ordering in materials.
Co-reporter:Lewis E. Johnson, Stephanie J. Benight, Robin Barnes, and Bruce H. Robinson
The Journal of Physical Chemistry B 2015 Volume 119(Issue 16) pp:5240-5250
Publication Date(Web):March 30, 2015
DOI:10.1021/acs.jpcb.5b00009
The Stockmayer fluid, composed of dipolar spheres, has a well-known isotropic–ferroelectric phase transition at high dipole densities. However, there has been little investigation of the ferroelectric transition in nearly spherical fluids at dipole densities corresponding to those found in many polar solvents and in guest–host organic electro-optic materials. In this work, we examine the transition to ordered phases of low-aspect-ratio spheroids under both unperturbed and poled conditions, characterizing both the static dielectric response and thermodynamic properties of spheroidal systems. Spontaneous ferroelectric ordering was confined to a small region of aspect ratios about unity, indicating that subtle changes in sterics can have substantial influence on the behavior of coarse-grained liquid models. Our results demonstrate the importance of molecular shape in obtaining even qualitatively correct dielectric responses and provide an explanation for the success of the Onsager model as a phenomenological representation for the dielectric behavior of polar organic liquids.
Co-reporter:Lewis E. Johnson, Larry R. Dalton, and Bruce H. Robinson
Accounts of Chemical Research 2014 Volume 47(Issue 11) pp:3258
Publication Date(Web):June 26, 2014
DOI:10.1021/ar5000727
ConspectusOrganic glasses containing chromophores with large first hyperpolarizabilities (β) are promising for compact, high-bandwidth, and energy-efficient electro-optic devices. Systematic optimization of device performance requires development of materials with high acentric order and enhanced hyperpolarizability at operating wavelengths. One essential component of the design process is the accurate calculation of optical transition frequencies and hyperpolarizability. These properties can be computed with a wide range of electronic structure methods implemented within commercial and open-source software packages. A wide variety of methods, especially hybrid density-functional theory (DFT) variants have been used for this purpose. However, in order to provide predictions useful to chromophore designers, a method must be able to consistently predict the relative ordering of standard and novel materials. Moreover, it is important to distinguish between the resonant and nonresonant contribution to the hyperpolarizabiliy and be able to estimate the trade-off between improved β and unwanted absorbance (optical loss) at the target device’s operating wavelength.Therefore, we have surveyed a large variety of common methods for computing the properties of modern high-performance chromophores and compared these results with prior experimental hyper-Rayleigh scattering (HRS) and absorbance data. We focused on hybrid DFT methods, supplemented by more computationally intensive Møller–Plesset (MP2) calculations, to determine the relative accuracy of these methods. Our work compares computed hyperpolarizabilities in chloroform relative to standard chromophore EZ-FTC against HRS data versus the same reference.We categorized DFT methods used by the amount of Hartree–Fock (HF) exchange energy incorporated into each functional. Our results suggest that the relationship between percentage of long-range HF exchange and both βHRS and λmax is nearly linear, decreasing as the fraction of long-range HF exchange increases. Mild hybrid DFT methods are satisfactory for prediction of λmax. However, mild hybrid methods provided qualitatively incorrect predictions of the relative hyperpolarizabilities of three high-performance chromophores. DFT methods with approximately 50% HF exchange, and especially the Truhlar M062X functional, provide superior predictions of relative βHRS values but poorer predictions of λmax. The observed trends for these functionals, as well as range-separated hybrids, are similar to MP2, though predicting smaller absolute magnitudes for βHRS.Frequency dependence for βHRS can be calculated using time-dependent DFT and HF methods. However, calculation quality is sensitive not only to a method’s ability to predict static hyperpolarizability but also to its prediction of optical resonances. Due to the apparent trade-off in accuracy of prediction of these two properties and the need to use static finite-field methods for MP2 and higher-level hyperpolarizability calculations in most codes, we suggest that composite methods could greatly improve the accuracy of calculations of β and λmax.
Co-reporter:Wenkel Liang, Xiaosong Li, Larry R. Dalton, Bruce H. Robinson, and Bruce E. Eichinger
The Journal of Physical Chemistry B 2011 Volume 115(Issue 43) pp:12566-12570
Publication Date(Web):September 19, 2011
DOI:10.1021/jp2069896
The dipole moments of highly polar molecules measured in solution are usually smaller than the molecular dipole moments that are calculated with reaction field methods, whereas vacuum values are routinely calculated in good agreement with available vapor phase data. Whether from Onsager’s theory (or variations thereof) or from quantum mechanical methods, the calculated molecular dipoles in solution are found to be larger than those measured. The reason, of course, is that experiments measure the net dipole moment of solute together with the polarized (perturbed) solvent “cloud” surrounding it. Here we show that the reaction field charges that are generated in the quantum mechanical self-consistent reaction field (SCRF) method give a good estimate of the net dipole moment of the solute molecule together with the moment arising from the reaction field charges. This net dipole is a better description of experimental data than the vacuum dipole moment and certainly better than the bare dipole moment of the polarized solute molecule.
Co-reporter:Benjamin C. Olbricht, Philip A. Sullivan, Peter C. Dennis, Jeffrey T. Hurst, Lewis E. Johnson, Stephanie J. Benight, Joshua A. Davies, Antao Chen, Bruce E. Eichinger, Philip J. Reid, Larry R. Dalton, and Bruce H. Robinson
The Journal of Physical Chemistry B 2011 Volume 115(Issue 2) pp:231-241
Publication Date(Web):December 17, 2010
DOI:10.1021/jp107995t
Organic nonlinear electrooptical (ONLO) chromophores must be acentrically ordered for the ONLO material to have electrooptic (EO) activity. The magnitude of the order is characterized by the acentric order parameter, ⟨cos3 β⟩, where β is the major Euler angle between the main axis of the chromophore and the poling field which imposes the acentric order. The acentric order parameter, which is difficult to measure directly, is related to the centrosymmetric order parameter, defined as ⟨P2⟩ = 1/2(3⟨cos2 β⟩ − 1), through the underlying statistical distribution. We have developed a method to determine centrosymmetric order of the ONLO chromophores when the order is low (i.e., ⟨P2⟩ < 0.1). We have extended the method (begun by Graf et al. J. Appl. Phys. 1994, 75, 3335.) based on the absorption of light to determine the centrosymmetric order parameter induced by a poling field on a thin film sample of ONLO material. We find that the order parameters, analyzed by two different methods, are similar and also consistent with theoretical estimates from modeling of the system using coarse-grained Monte Carlo statistical mechanical methods.
Co-reporter:Denise H. Bale, Bruce E. Eichinger, Wenkel Liang, Xiaosong Li, Larry R. Dalton, Bruce H. Robinson, and Philip J. Reid
The Journal of Physical Chemistry B 2011 Volume 115(Issue 13) pp:3505-3513
Publication Date(Web):March 16, 2011
DOI:10.1021/jp109870y
Experimental and computational studies of the solvent dependence of the first molecular hyperpolarizability (β) for two donor-bridge-acceptor chromophores (CLD-1 and YLD156) are presented. Hyper-Rayleigh scattering (HRS) measurements are performed with 1907 nm excitation in a series of solvents with dielectric constants ranging from ∼2 (toluene) to ∼36 (acetonitrile). For both chromophores an approximately 2-fold increase in β is observed by HRS over this range of dielectric constants. Computational studies employing a polarized continuum model to represent the solvent are capable of reproducing this experimental result. The experimental and computational results are compared to the predictions of the widely employed two-state model (TSM) for β. Surprisingly, for the chromophores studied here the TSM predicts that β should decrease with increasing dielectric constant over the range investigated. The results presented here demonstrate that the TSM provides neither a quantitative nor qualitative description of the solvent dependence of β for CLD-1 and YLD156. The enhancement of β with increased dielectric constant suggests that modification of the dielectric surrounding the chromophore is one path by which the performance of nonlinear optical devices employing these chromophores may be significantly enhanced.
Co-reporter:Lewis E. Johnson, Robin Barnes, Thomas W. Draxler, Bruce E. Eichinger and Bruce H. Robinson
The Journal of Physical Chemistry B 2010 Volume 114(Issue 25) pp:8431-8440
Publication Date(Web):June 8, 2010
DOI:10.1021/jp1010605
Coarse-grained models of molecular interactions are of interest because they convey the essence of molecular interactions in simple and easy to understand form. However, coarse-grained models fail to adequately predict some material properties, such as the failure of the Stockmayer model to reproduce the dielectric behavior of highly polar liquids. We examine the behavior of the Stockmayer fluid over a range of dipole densities that covers known organic solvents, as well as that of an ellipsoidal Stockmayer-like fluid, using NVT rigid-body Monte Carlo simulations. Both fluids are examined under different electrostatic boundary conditions and ensemble sizes. While the Stockmayer model predicts that liquids of similar dipole density to acetonitrile would be ferroelectric and have a dielectric constant far higher than shown by experiment, the ellipsoidal model provides a better accounting of dielectric behavior. This result bodes well for the use of coarse-grained solvent models for large-scale simulations.
Co-reporter:Stephanie J. Benight, Lewis E. Johnson, Robin Barnes, Benjamin C. Olbricht, Denise H. Bale, Philip J. Reid, Bruce E. Eichinger, Larry R. Dalton, Philip A. Sullivan, and Bruce H. Robinson
The Journal of Physical Chemistry B 2010 Volume 114(Issue 37) pp:11949-11956
Publication Date(Web):August 23, 2010
DOI:10.1021/jp1022423
Identification of electronic intermolecular electrostatic interactions that can significantly enhance poling-induced order is important to the advancement of the field of organic electro-optics. Here, we demonstrate an example of such improvement achieved through exploitation of the interaction of coumarin pendant groups in chromophore-containing macromolecules. Acentric order enhancement is explained in terms of lattice-symmetry effects, where constraint of orientational degrees of freedom alters the relationship between centrosymmetric and acentric order. We demonstrate both experimentally and theoretically that lattice dimensionality can be defined using the relationship between centrosymmetric order and acentric order. Experimentally: Acentric order is determined by attenuated total reflection measurement of electro-optic activity coupled with hyper-Rayleigh scattering measurement of molecular first hyperpolarizability, and centrosymmetric order is determined by the variable angle polarization referenced absorption spectroscopy method. Theoretically: Order is determined from statistical mechanical models that predict the properties of soft condensed matter.
Co-reporter:Philip A. Sullivan, Harrison L. Rommel, Yoshinari Takimoto, Scott R. Hammond, Denise H. Bale, Benjamin C. Olbricht, Yi Liao, John Rehr, Bruce E. Eichinger, Alex K.-Y. Jen, Philip J. Reid, Larry R. Dalton and Bruce H. Robinson
The Journal of Physical Chemistry B 2009 Volume 113(Issue 47) pp:15581-15588
Publication Date(Web):October 16, 2009
DOI:10.1021/jp908057d
For the past three decades, a full understanding of the electro-optic (EO) effect in amorphous organic media has remained elusive. Calculating a bulk material property from fundamental molecular properties, intermolecular electrostatic forces, and field-induced net acentric dipolar order has proven to be very challenging. Moreover, there has been a gap between ab initio quantum-mechanical (QM) predictions of molecular properties and their experimental verification at the level of bulk materials and devices. This report unifies QM-based estimates of molecular properties with the statistical mechanical interpretation of the order in solid phases of electric-field-poled, amorphous, organic dipolar chromophore-containing materials. By combining interdependent statistical and quantum mechanical methods, bulk material EO properties are predicted. Dipolar order in bulk, amorphous phases of EO materials can be understood in terms of simple coarse-grained force field models when the dielectric properties of the media are taken into account. Parameters used in the statistical mechanical modeling are not adjusted from the QM-based values, yet the agreement with the experimentally determined electro-optic coefficient is excellent.
Co-reporter:Alyssa L. Smith, Pavol Cekan, Greg P. Brewood, Tamara M. Okonogi, Saba Alemayehu, Eric J. Hustedt, Albert S. Benight, Snorri Th. Sigurdsson and Bruce H. Robinson
The Journal of Physical Chemistry B 2009 Volume 113(Issue 9) pp:2664-2675
Publication Date(Web):February 5, 2009
DOI:10.1021/jp808260b
Conformational flexibility in nucleic acids provides a basis for complex structures, binding, and signaling. One-base bulges directly neighboring single-base mismatches in nucleic acids can be present in a minimum of two distinct conformations, complicating the examination of the thermodynamics by calorimetry or UV-monitored melting techniques. To provide additional information about such structures, we demonstrate how electron paramagnetic resonance (EPR) active spin-labeled base analogues, base-specifically incorporated into the DNA, are monitors of the superposition of different bulge-mismatch conformations. EPR spectra provide information about the dynamic environments of the probe. This information is cast in terms of “dynamic signatures” that have an underlying basis in structural variations. By examining the changes in the equilibrium of the different states across a range of temperatures, the enthalpy and entropy of the interconversion among possible conformations can be determined. The DNA constructs with a single bulge neighboring a single-base mismatch (“bulge-mismatches”) may be approximately modeled as an equilibrium between two possible conformations. This structural information provides insight into the local composition of the bulge-mismatch sequences. Experiments on the bulge-mismatches show that basepairing across the helix can be understood in terms of purine and pyrimidine interactions, rather than specific bases. Measurements of the enthalpy and entropy of formation for the bulge-mismatches by differential scanning calorimetry and UV-monitored melting confirm that the formation of bulge-mismatches is in fact more complicated than a simple two-state process, consistent with the base-specific spectral data that bulge-mismatches exist in multiple conformations in the premelting temperature region. We find that the calculations with the nearest-neighbor (NN) model for the two likely conformations do not correlate well with the populations of structures and thermodynamic parameters inferred from the base-specific EPR dynamics probe. We report that the base-specific spin probes are able to identify a bistable, temperature dependent, switching between conformations for a particular complex bulged construct.
Co-reporter:Alyssa L. Smith, Pavol Cekan, David P. Rangel, Snorri Th. Sigurdsson, Colin Mailer and Bruce H. Robinson
The Journal of Physical Chemistry B 2008 Volume 112(Issue 30) pp:9219-9236
Publication Date(Web):July 2, 2008
DOI:10.1021/jp7111704
The weakly bending rod (WBR) model of double-stranded DNA (dsDNA) is adapted to analyze the internal dynamics of dsDNA as observed in electron paramagnetic resonance (EPR) measurements of the spin−lattice relaxation rate, R1e, for spin probes rigidly attached to nucleic acid−bases. The WBR theory developed in this work models dsDNA base-pairs as diffusing rigid cylindrical discs connected by bending and twisting springs whose elastic force constants are κ and α, respectively. Angular correlation functions for both rotational displacement and velocity are developed in detail so as to compute values for R1e due to four relaxation mechanisms: the chemical shift anisotropy (CSA), the electron−nuclear dipolar (END), the spin rotation (SR), and the generalized spin diffusion (GSD) relaxation processes. Measured spin−lattice relaxation rates in dsDNA under 50 bp in length are much faster than those calculated for the same DNAs modeled as rigid rods. The simplest way to account for this difference is by allowing for internal flexibility in models of DNA. Because of this discrepancy, we derive expressions for the spectral densities due to CSA, END, and SR mechanisms directly from a weakly bending rod model for DNA. Special emphasis in this development is given to the SR mechanism because of the lack of such detail in previous treatments. The theory developed in this paper provides a framework for computing relaxation rates from the WBR model to compare with magnetic resonance relaxation data and to ascertain the twisting and bending force constants that characterize DNA.
Co-reporter:Tamara M. Okonogi;Stephen C. Alley;Eric A. Harwood;Paul B. Hopkins
PNAS 2002 Volume 99 (Issue 7 ) pp:4156-4160
Publication Date(Web):2002-04-02
DOI:10.1073/pnas.072067799
An important component of protein–DNA recognition is the charge neutralization of DNA backbone phosphates and subsequent protein-induced
DNA bending. Replacement of phosphates by neutral methylphosphonates has previously been shown to be a model for protein-induced
bending. In addition to bending, the neutralization process may change the inherent flexibility of the DNA—a feature never
before tested. We have developed a method to measure the differential flexibility of duplex DNA when methylphosphonate substitutions
are made and find that the local flexibility is increased up to 40%. These results imply that backbone-neutralization-dependent
DNA flexibility augments DNA-binding motifs in protein–DNA recognition processes.
.jpg)
![Benzoic acid, 3,5-bis[(2,3,4,5,6-pentafluorophenyl)methoxy]-, [5-[2-[4-[[2-[[3,5-bis(phenylmethoxy)benzoyl]oxy]ethyl]methylamino]phenyl]ethenyl]-2-[2-[4-cyano-5-(dicyanomethylene)-2,5-dihydro-2-phenyl-2-(trifluoromethyl)-3-furanyl]ethenyl]-3-thienyl]methyl ester](/data/chemimg/196900/1374873-48-9.png)
![Benzoic acid, 3,5-bis[(2,3,4,5,6-pentafluorophenyl)methoxy]-, [5-[2-[4-[[2-[[3,5-bis(phenylmethoxy)benzoyl]oxy]ethyl]methylamino]phenyl]ethenyl]-2-[2-[4-cyano-5-(dicyanomethylene)-2,5-dihydro-2-phenyl-2-(trifluoromethyl)-3-furanyl]ethenyl]-3-thienyl]methyl ester](/data/chemimg/196900/1374873-48-9_b.png)
![Benzoic acid, 3,5-bis[(2,3,4,5,6-pentafluorophenyl)methoxy]-, [5-[2-[4-[[2-[[3,5-bis(phenylmethoxy)benzoyl]oxy]ethyl]ethylamino]phenyl]ethenyl]-2-[2-[4-cyano-5-(dicyanomethylene)-2,5-dihydro-2-phenyl-2-(trifluoromethyl)-3-furanyl]ethenyl]-3-thienyl]methyl ester](/data/chemimg/196900/1374873-47-8.png)
![Benzoic acid, 3,5-bis[(2,3,4,5,6-pentafluorophenyl)methoxy]-, [5-[2-[4-[[2-[[3,5-bis(phenylmethoxy)benzoyl]oxy]ethyl]ethylamino]phenyl]ethenyl]-2-[2-[4-cyano-5-(dicyanomethylene)-2,5-dihydro-2-phenyl-2-(trifluoromethyl)-3-furanyl]ethenyl]-3-thienyl]methyl ester](/data/chemimg/196900/1374873-47-8_b.png)
![Benzoic acid, 3,5-bis[(2,3,4,5,6-pentafluorophenyl)methoxy]-, 2-[[2-[2-[4-[[2-[[3,5-bis(phenylmethoxy)benzoyl]oxy]ethyl]ethylamino]phenyl]ethenyl]-4,4-dimethyl-6-(2-oxoethylidene)-1-cyclohexen-1-yl]oxy]ethyl ester](/data/chemimg/196900/1374873-46-7.png)
![Benzoic acid, 3,5-bis[(2,3,4,5,6-pentafluorophenyl)methoxy]-, 2-[[2-[2-[4-[[2-[[3,5-bis(phenylmethoxy)benzoyl]oxy]ethyl]ethylamino]phenyl]ethenyl]-4,4-dimethyl-6-(2-oxoethylidene)-1-cyclohexen-1-yl]oxy]ethyl ester](/data/chemimg/196900/1374873-46-7_b.png)
![Benzoic acid, 3,5-bis[(2,3,4,5,6-pentafluorobenzoyl)oxy]-, 2-[[2-[2-[4-[[2-[[3,5-bis(benzoyloxy)benzoyl]oxy]ethyl]ethylamino]phenyl]ethenyl]-6-[3-[4-cyano-5-(dicyanomethylene)-2,5-dihydro-2-phenyl-2-(trifluoromethyl)-3-furanyl]-2-propen-1-ylidene]-4,4-dimethyl-1-cyclohexen-1-yl]oxy]ethyl ester](/data/chemimg/196900/1374873-45-6.png)
![Benzoic acid, 3,5-bis[(2,3,4,5,6-pentafluorobenzoyl)oxy]-, 2-[[2-[2-[4-[[2-[[3,5-bis(benzoyloxy)benzoyl]oxy]ethyl]ethylamino]phenyl]ethenyl]-6-[3-[4-cyano-5-(dicyanomethylene)-2,5-dihydro-2-phenyl-2-(trifluoromethyl)-3-furanyl]-2-propen-1-ylidene]-4,4-dimethyl-1-cyclohexen-1-yl]oxy]ethyl ester](/data/chemimg/196900/1374873-45-6_b.png)
![Benzeneacetonitrile, 4-[1-cyano-4-[4-(dibutylamino)phenyl]-1,3-butadien-1-yl]-α-[3-[4-(dibutylamino)phenyl]-2-propen-1-ylidene]-](/data/chemimg/196900/1373922-08-7.png)
![Benzeneacetonitrile, 4-[1-cyano-4-[4-(dibutylamino)phenyl]-1,3-butadien-1-yl]-α-[3-[4-(dibutylamino)phenyl]-2-propen-1-ylidene]-](/data/chemimg/196900/1373922-08-7_b.png)










![Benzoic acid, 3,5-bis[(2,3,4,5,6-pentafluorophenyl)methoxy]-, 2-[[2-[2-[4-[[2-[[3,5-bis(phenylmethoxy)benzoyl]oxy]ethyl]ethylamino]phenyl]ethenyl]-6-[3-[4-cyano-5-(dicyanomethylene)-2,5-dihydro-2-phenyl-2-(trifluoromethyl)-3-furanyl]-2-propen-1-ylidene]-4,4-dimethyl-1-cyclohexen-1-yl]oxy]ethyl ester](/data/chemimg/196900/1354221-98-9.png)
![Benzoic acid, 3,5-bis[(2,3,4,5,6-pentafluorophenyl)methoxy]-, 2-[[2-[2-[4-[[2-[[3,5-bis(phenylmethoxy)benzoyl]oxy]ethyl]ethylamino]phenyl]ethenyl]-6-[3-[4-cyano-5-(dicyanomethylene)-2,5-dihydro-2-phenyl-2-(trifluoromethyl)-3-furanyl]-2-propen-1-ylidene]-4,4-dimethyl-1-cyclohexen-1-yl]oxy]ethyl ester](/data/chemimg/196900/1354221-98-9_b.png)










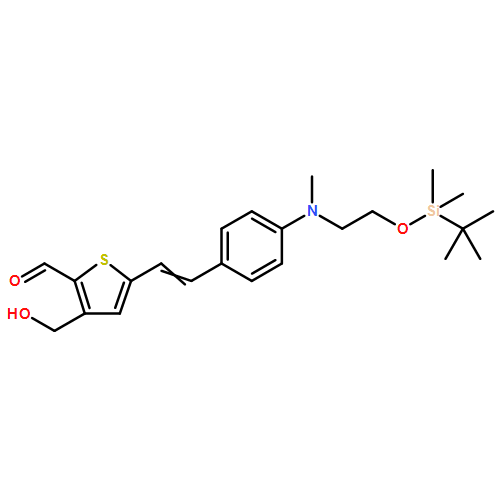
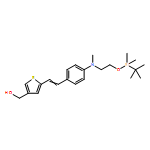
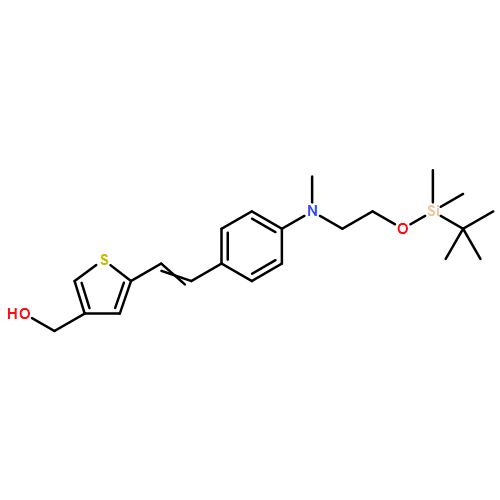
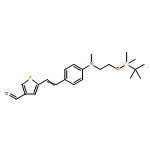
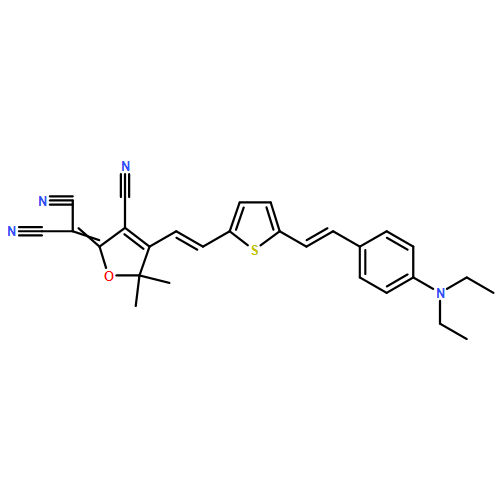


![Propanedinitrile, 2-[3-cyano-4-methyl-5-phenyl-5-(trifluoromethyl)-2(5H)-furanylidene]-](/data/chemimg/119100/436097-14-2.png)
![Propanedinitrile, 2-[3-cyano-4-methyl-5-phenyl-5-(trifluoromethyl)-2(5H)-furanylidene]-](/data/chemimg/119100/436097-14-2_b.png)
![Propanedinitrile,2-[4-[(1E,3E)-3-[3-[(1E)-2-[4-[bis[2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]ethyl]amino]phenyl]ethenyl]-5,5-dimethyl-2-cyclohexen-1-ylidene]-1-propen-1-yl]-3-cyano-5,5-dimethyl-2(5H)-furanylidene]-](http://img.cochemist.com/ccimg/368900/368874-13-9.png)
![Propanedinitrile,2-[4-[(1E,3E)-3-[3-[(1E)-2-[4-[bis[2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]ethyl]amino]phenyl]ethenyl]-5,5-dimethyl-2-cyclohexen-1-ylidene]-1-propen-1-yl]-3-cyano-5,5-dimethyl-2(5H)-furanylidene]-](http://img.cochemist.com/ccimg/368900/368874-13-9_b.png)




![Hexanedioic acid, 1-[[5-[2-[4-[[2-[[1,6-dioxo-6-[4-[[(2-oxo-2H-1-benzopyran-7-yl)oxy]carbonyl]phenoxy]hexyl]oxy]ethyl]methylamino]phenyl]ethenyl]-2-formyl-3-thienyl]methyl] 6-[4-[[(2-oxo-2H-1-benzopyran-7-yl)oxy]carbonyl]phenyl] ester](/data/chemimg/196900/1373922-53-2.png)
![Hexanedioic acid, 1-[[5-[2-[4-[[2-[[1,6-dioxo-6-[4-[[(2-oxo-2H-1-benzopyran-7-yl)oxy]carbonyl]phenoxy]hexyl]oxy]ethyl]methylamino]phenyl]ethenyl]-2-formyl-3-thienyl]methyl] 6-[4-[[(2-oxo-2H-1-benzopyran-7-yl)oxy]carbonyl]phenyl] ester](/data/chemimg/196900/1373922-53-2_b.png)