Co-reporter:Daniel G. Silva, J. Robert Gillespie, Ranae M. Ranade, Zackary M. Herbst, Uyen T. T. Nguyen, Frederick S. Buckner, Carlos A. Montanari, and Michael H. Gelb
ACS Medicinal Chemistry Letters July 13, 2017 Volume 8(Issue 7) pp:766-766
Publication Date(Web):June 27, 2017
DOI:10.1021/acsmedchemlett.7b00202
The present work describes the synthesis of 22 new imidazopyridine analogues arising from medicinal chemistry optimization at different sites on the molecule. Seven and 12 compounds exhibited an in vitro EC50 ≤ 1 μM against Trypanosoma cruzi (T. cruzi) and Trypanosoma brucei (T. brucei) parasites, respectively. Based on promising results of in vitro activity (EC50 < 100 nM), cytotoxicity, metabolic stability, protein binding, and pharmacokinetics (PK) properties, compound 20 was selected as a candidate for in vivo efficacy studies. This compound was screened in an acute mouse model against T.cruzi (Tulahuen strain). After established infection, mice were dosed twice a day for 5 days, and then monitored for 6 weeks using an in vivo imaging system (IVIS). Compound 20 demonstrated parasite inhibition comparable to the benznidazole treatment group. Compound 20 represents a potential lead for the development of drugs to treat trypanosomiasis.Keywords: Anti-infectives; imidazopyridine; Trypanosoma brucei; Trypanosoma cruzi; Trypanosomiasis;
Co-reporter:Michael H. Gelb, C. Ronald Scott, Frantisek Turecek, Hsuan-Chieh Liao
Molecular Genetics and Metabolism Reports 2017 Volume 12(Volume 12) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.ymgmr.2017.05.004
Co-reporter:Naveen Kumar Chennamaneni, Arun Babu Kumar, Mariana Barcenas, Zdeněk Spáčil, C. Ronald Scott, František Tureček, and Michael H. Gelb
Analytical Chemistry 2014 Volume 86(Issue 9) pp:4508
Publication Date(Web):April 2, 2014
DOI:10.1021/ac5004135
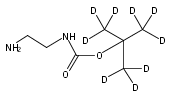
Tandem mass spectrometry for the multiplex and quantitative analysis of enzyme activities in dried blood spots on newborn screening cards has emerged as a powerful technique for early assessment of lysosomal storage diseases. Here we report the design and process-scale synthesis of substrates for the enzymes α-l-iduronidase, iduronate-2-sulfatase, and N-acetylgalactosamine-4-sulfatase that are used for newborn screening of mucopolysaccharidosis types I, II, and VI. The products contain a bisamide unit that is hypothesized to readily protonate in the gas phase, which improves detection sensitivity by tandem mass spectrometry. The products contain a benzoyl group, which provides a useful site for inexpensive deuteration, thus facilitating the preparation of internal standards for the accurate quantification of enzymatic products. Finally, the reagents are designed with ease of synthesis in mind, thus permitting scale-up preparation to support worldwide newborn screening of lysosomal storage diseases. The new reagents provide the most sensitive assay for the three lysosomal enzymes reported to date as shown by their performance in reactions using dried blood spots as the enzyme source. Also, the ratio of assay signal to that measured in the absence of blood (background) is superior to all previously reported mucopolysaccharidosis types I, II, and VI assays.
Co-reporter:Hari Babu Tatipaka ; J. Robert Gillespie ; Arnab K. Chatterjee ; Neil R. Norcross ; Matthew A. Hulverson ; Ranae M. Ranade ; Pendem Nagendar ; Sharon A. Creason ; Joshua McQueen ; Nicole A. Duster ; Advait Nagle ; Frantisek Supek ; Valentina Molteni ; Tanja Wenzler ; Reto Brun ; Richard Glynne ; Frederick S. Buckner ;Michael H. Gelb
Journal of Medicinal Chemistry 2014 Volume 57(Issue 3) pp:828-835
Publication Date(Web):December 19, 2013
DOI:10.1021/jm401178t
A phenotypic screen of a compound library for antiparasitic activity on Trypanosoma brucei, the causative agent of human African trypanosomiasis, led to the identification of substituted 2-(3-aminophenyl)oxazolopyridines as a starting point for hit-to-lead medicinal chemistry. A total of 110 analogues were prepared, which led to the identification of 64, a substituted 2-(3-aminophenyl)imidazopyridine. This compound showed antiparasitic activity in vitro with an EC50 of 2 nM and displayed reasonable druglike properties when tested in a number of in vitro assays. The compound was orally bioavailable and displayed good plasma and brain exposure in mice. Compound 64 cured mice infected with Trypanosoma brucei when dosed orally down to 2.5 mg/kg. Given its potent antiparasitic properties and its ease of synthesis, compound 64 represents a new lead for the development of drugs to treat human African trypanosomiasis.
Co-reporter:Mariana Barcenas, Chang Xue, Tatyana Marushchak-Vlaskin, C. Ronald Scott, Michael H. Gelb, and František Tureček
Analytical Chemistry 2014 Volume 86(Issue 15) pp:7962
Publication Date(Web):July 14, 2014
DOI:10.1021/ac501994b
We report new substrates for quantitative enzyme activity measurements of human palmitoyl protein thioesterase (PPT1) and tripeptidyl peptidase (TPP1) in dried blood spots from newborns using tandem mass spectrometry. Deficiencies in these enzyme activities due to inborn errors of metabolism cause neuronal ceroid lipofuscinoses. The assays use synthetic compounds that were designed to mimic the natural substrates. Incubation produces nanomole quantities of enzymatic products per a blood spot that are quantified by tandem mass spectrometry using synthetic internal standards and selected reaction monitoring. The assays utilize a minimum steps for sample workup and can be run in a duplex format for the detection of neuronal ceroid lipofuscinoses or potentially multiplexed with other mass spectrometry-based assays for newborn screening of lysosomal storage disorders.
Co-reporter:Praveen Kumar Suryadevara, Kishore Kumar Racherla, Srinivas Olepu, Neil R. Norcross, Hari Babu Tatipaka, Jennifer A. Arif, Joseph D. Planer, Galina I. Lepesheva, Christophe L.M.J. Verlinde, Frederick S. Buckner, Michael H. Gelb
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 23) pp:6492-6499
Publication Date(Web):1 December 2013
DOI:10.1016/j.bmcl.2013.08.015
New dialkylimidazole based sterol 14α-demethylase inhibitors were prepared and tested as potential anti-Trypanosoma cruzi agents. Previous studies had identified compound 2 as the most potent and selective inhibitor against parasite cultures. In addition, animal studies had demonstrated that compound 2 is highly efficacious in the acute model of the disease. However, compound 2 has a high molecular weight and high hydrophobicity, issues addressed here. Systematic modifications were carried out at four positions on the scaffold and several inhibitors were identified which are highly potent (EC50 <1 nM) against T. cruzi in culture. The halogenated derivatives 36j, 36k, and 36p, display excellent activity against T. cruzi amastigotes, with reduced molecular weight and lipophilicity, and exhibit suitable physicochemical properties for an oral drug candidate.
Co-reporter:Brian J. Wolfe, Farideh Ghomashchi, Tim Kim, Cynthia A. Abam, Martin Sadilek, Rhona Jack, Jerry N. Thompson, C. Ronald Scott, Michael H. Gelb, and Frantisek Turecek
Bioconjugate Chemistry 2012 Volume 23(Issue 3) pp:557
Publication Date(Web):February 28, 2012
DOI:10.1021/bc200609x
The clinical phenotype of Sanfilippo Syndrome is caused by one of four enzyme deficiencies that are associated with a defect in mucopolysaccharide metabolism. The four subtypes (A, B, C, and D) are each caused by an enzyme deficiency involved in the degradation of heparan sulfate. We have developed a highly efficient synthesis of the substrates and internal standards required for the enzymatic assay of each of the four enzymes. The synthesis of the substrates involves chemical modification of a common intermediate. The substrates and internal standards allow the measurement of the enzymes relevant to heparan N-sulfatase (type A); N-acetyl-α-glucosaminidase (type B); acetyl-CoA:α-glucosamide N-acetyltransferase (type C); and N-acetylglucosamine 6-sulfatase (type D). The internal standards are similar to the substrates and allow for the accurate quantification of the enzyme assays using tandem mass spectrometry. The synthetic substrates incorporate a coumarin moiety and can also be used in fluorometric enzyme assays. We confirm that all four substrates can detect the appropriate Sanfilippo Syndrome in fibroblast lysates, and the measured enzyme activities are distinctly lower by a factor of 10 when compared to fibroblast lysates from unaffected persons.
Co-reporter:Rob C. Oslund and Michael H. Gelb
Biochemistry 2012 Volume 51(Issue 43) pp:
Publication Date(Web):September 28, 2012
DOI:10.1021/bi301140b
We explored the inhibition mode of group IIA secreted phospholipase A2 (GIIA sPLA2) selective inhibitors and tested their ability to inhibit GIIA sPLA2 activity as chemical conjugates with hyaluronic acid (HA). Analogues of a benzo-fused indole sPLA2 inhibitor were developed in which the carboxylate group on the inhibitor scaffold, which has been shown to coordinate to a Ca2+ ligand in the enzyme active site, was replaced with other functionality. Replacing the carboxylate group with amine, amide, or hydroxyl groups had no effect on human GIIA (hGIIA) sPLA2 inhibition potency but dramatically lowered inhibition potency against hGV and hGX sPLA2s. An alkylation protection assay was used to probe active site binding of carboxylate and noncarboxylate inhibitors in the presence and absence of Ca2+ and/or lipid vesicles. We observed that carboxylate-containing inhibitors bind the hGIIA sPLA2 active site with low nanomolar affinity, but only when Ca2+ is present. Noncarboxylate, GIIA sPLA2 selective inhibitors also bind the hGIIA sPLA2 active site in the nanomolar range. However, binding for GIIA sPLA2 selective inhibitors was dependent on the presence of a lipid membrane and not Ca2+. These results indicate that GIIA sPLA2 selective inhibitors exert their inhibitory effects by binding to the hGIIA sPLA2 active site. An HA-linked GIIA inhibitor conjugate was developed using peptide coupling conditions and found to be less potent and selective against hGIIA sPLA2 than the unconjugated inhibitor. Compounds reported in this study are some of the most potent and selective GIIA sPLA2 active site binding inhibitors reported to date.
Co-reporter:Brian J. Wolfe, Sophie Blanchard, Martin Sadilek, C. Ronald Scott, Frantisek Turecek, and Michael H. Gelb
Analytical Chemistry 2011 Volume 83(Issue 3) pp:1152
Publication Date(Web):December 30, 2010
DOI:10.1021/ac102777s
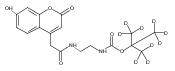
We have developed a tandem mass spectrometry based assay of iduronate-2-sulfatase (IdS) activity for the neonatal detection of mucopolysaccharidosis II (MPS-II, Hunter Syndrome). The assay uses a newly designed synthetic substrate (IdS-S) consisting of α-l-iduronate-2-sulfate, which is glycosidically conjugated to a coumarin and a linker containing a tert-butyloxycarbamido group. A short synthesis of the substrate has been developed that has the potential of being scaled to multigram quantities. Sulfate hydrolysis of IdS-S by IdS found within a 3 mm dried blood spot specifically produces a nonsulfated product (IdS-P) which is detected by electrospray tandem mass spectrometry and quantified using a deuterium-labeled internal standard, both carried out in positive ion mode. Analysis of DBS from 75 random human newborns showed IdS activities in the range of 4.8−16.2 (mean 9.1) μmol/(h L of blood), which were clearly distinguished from the activities measured for 14 MPS-II patients at 0.17−0.52 (mean 0.29) μmol/(h L of blood). The assay shows low blank activity, 0.15 ± 0.03 μmol/(h L of blood). The within-assay coefficient of variation (CV) was 3.1% while the interassay CV was 15%.
Co-reporter:Zdeněk Spáčil, Susan Elliott, Steven L. Reeber, Michael H. Gelb, C. Ronald Scott, and František Tureček
Analytical Chemistry 2011 Volume 83(Issue 12) pp:4822
Publication Date(Web):May 6, 2011
DOI:10.1021/ac200417u
We report a comparative study of triplex tandem mass spectrometry (MS/MS) based assays of lysosomal enzymes in dried blood spots for the early detection of Pompe, Fabry, and Hurler diseases in newborns. Four methods have been evaluated that differed in sample handling and the equipment used. A newly developed method uses assay quenching with acetonitrile to precipitate blood proteins followed by analysis on an LC–electrospray/MS/MS system capable of multiple consecutive sample injections on two parallel chromatographic columns. This method requires 1.5 min per a triplex analysis of enzyme products and internal standards, which matches the throughput of the previously reported flow injection method. LC separation reduces matrix effects and allows for more facile sample workup. The new LC-based method showed figures of merit that were superior to those of the currently used method based on liquid–liquid extraction into ethyl acetate and flow injection into the mass spectrometer. The other methods we investigated for comprehensive comparison involved liquid–liquid extraction into ethyl acetate followed by LC–ESI-MS/MS and acetonitrile quenching followed by direct flow injection. Both methods using acetonitrile quenching were found to be robust and provide good quality data while requiring fewer liquid transfer steps and less disposable material and labor than did the extraction methods. The individual merits of the new methods are discussed to present an evaluated alternative approach to high-throughput analysis in newborn screening laboratories.
Co-reporter:James G. Bollinger, Wallace Thompson, Ying Lai, Rob C. Oslund, Teal S. Hallstrand, Martin Sadilek, Frantisek Turecek and Michael H. Gelb
Analytical Chemistry 2010 Volume 82(Issue 16) pp:6790
Publication Date(Web):July 22, 2010
DOI:10.1021/ac100720p
Combined liquid chromatography−electrospray ionization-tandem mass spectrometry (LC−ESI-MS/MS) is a powerful method for the analysis of oxygenated metabolites of polyunsaturated fatty acids including eicosanoids. Here we describe the synthesis of a new derivatization reagent N-(4-aminomethylphenyl)pyridinium (AMPP) that can be coupled to eicosanoids via an amide linkage in quantitative yield. Conversion of the carboxylic acid of eicosanoids to a cationic AMPP amide improves sensitivity of detection by 10- to 20-fold compared to negative mode electrospray ionization detection of underivatized analytes. This charge reversal derivatization allows detection of cations rather than anions in the electrospray ionization mass spectrometer, which enhances sensitivity. Another factor is that AMPP amides undergo considerable collision-induced dissociation in the analyte portion rather than exclusively in the cationic tag portion, which allows isobaric derivatives to be distinguished by tandem mass spectrometry, and this further enhances sensitivity and specificity. This simple derivatization method allows prostaglandins, thromboxane B2, leukotriene B4, hydroxyeicosatetraenoic acid isomers, and arachidonic acid to be quantified in complex biological samples with limits of quantification in the 200−900 fg range. One can anticipate that the AMPP derivatization method can be extended to other carboxylic acid analytes for enhanced sensitivity detection.
Co-reporter:Trisha A. Duffey, Martin Sadilek, C. Ronald Scott, Frantisek Turecek, and Michael H. Gelb
Analytical Chemistry 2010 Volume 82(Issue 22) pp:9587
Publication Date(Web):October 20, 2010
DOI:10.1021/ac102090v
We report a new assay of N-acetylgalactosamine-4-sulfatase (aryl sulfatase B) activity in dried blood spots (DBS) for the early detection of mucopolysaccharidosis VI (Maroteaux−Lamy syndrome) in newborn screening. The assay uses a synthetic substrate consisting of N-acetylgalactosamine-4-sulfate moiety glycosidically linked to a hydrophobic residue and furnished with a tert-butyloxycarbamido group as a marker for specific mass spectrometric fragmentation. Incubation with aryl sulfatase B present in DBS converts the substrate to a desulfated product which is detected by electrospray tandem mass spectrometry and quantified using a homologous internal standard. Assay and workup procedures were optimized to be compatible with the work flow in newborn screening laboratories. Analysis of DBS from human newborns showed clear distinction of aryl sulfatase B activity from 89 healthy individuals where it ranged between 1.4 and 16.9 μmol/(h L of blood), with an average activity of 7.4 μmol/(h L of blood), and an MPS-VI patient that had an activity of 0.12 μmol/(h L of blood). Results are also reported for the aryl sulfatase B assay in DBS from groups of normal felines and felines affected with MPS-VI.
Co-reporter:James M. Kraus ; Hari Babu Tatipaka ; Sarah A. McGuffin ; Naveen Kumar Chennamaneni ; Mandana Karimi ; Jenifer Arif ; Christophe L. M. J. Verlinde ; Frederick S. Buckner ;Michael H. Gelb
Journal of Medicinal Chemistry 2010 Volume 53(Issue 10) pp:3887-3898
Publication Date(Web):April 29, 2010
DOI:10.1021/jm9013136
We previously reported that the cancer drug clinical candidate tipifarnib kills the causative agent of Chagas disease, Trypanosoma cruzi, by blocking ergosterol biosynthesis at the level of inhibition of lanosterol 14α-demethylase. Tipifarnib is an inhibitor of human protein farnesyltransferase. We synthesized tipifarnib analogues that no longer bind to protein farnesyltransferase and display increased potency for killing parasites. This was achieved in a structure-guided fashion by changing the substituents attached to the phenyl group at the 4-position of the quinoline ring of tipifarnib and by replacing the amino group by OMe. Several compounds that kill Trypanosoma cruzi at subnanomolar concentrations and are devoid of protein farnesyltransferase inhibition were discovered. The compounds are shown to be advantageous over other lanosterol 14α-demethylase inhibitors in that they show only modest potency for inhibition of human cytochrome P450 (3A4). Since tipifarnib displays high oral bioavailability and acceptable pharmacokinetic properties, the newly discovered tipifarnib analogues are ideal leads for the development of drugs to treat Chagas disease.
Co-reporter:Trisha A. Duffey, Tanvir Khaliq, C. Ronald Scott, Frantisek Turecek, Michael H. Gelb
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 20) pp:5994-5996
Publication Date(Web):15 October 2010
DOI:10.1016/j.bmcl.2010.08.080
In continued efforts to develop enzymatic assays for lysosomal storage diseases appropriate for newborn screening laboratories we have synthesized novel and specific enzyme substrates for Maroteaux–Lamy (MPS VI) and Morquio A (MPS IVA) diseases. The sulfated monosaccharide derivatives were found to be converted to product by the respective enzyme in blood from healthy patients but not by blood from patients with the relevant lysosomal storage disease. The latter result shows that the designed substrates are highly selective for the respective enzymes.
Co-reporter:James M. Kraus ; Christophe L. M. J. Verlinde ; Mandana Karimi ; Galina I. Lepesheva ; Michael H. Gelb ;Frederick S. Buckner
Journal of Medicinal Chemistry 2009 Volume 52(Issue 6) pp:1639-1647
Publication Date(Web):February 24, 2009
DOI:10.1021/jm801313t
Chagas disease is one of the major neglected diseases of the world. Existing drug therapies are limited, ineffective, and highly toxic. We describe a novel strategy of drug discovery of adapting an existing clinical compound with excellent pharmaceutical properties to target a pathogenic organism. The protein farnesyltransferase (PFT) inhibitor tipifarnib, now in phase III anticancer clinical trials, was previously found to kill Trypanosoma cruzi by blocking sterol 14α-demethylase (14DM). We rationally developed tipifarnib analogues that display reduced affinity for human PFT to reduce toxicity while increasing affinity for parasite 14DM. The lead compound has picomolar activity against cultured T. cruzi and is efficacious in a mouse model of acute Chagas disease.
Co-reporter:Praveen Kumar Suryadevara ; Srinivas Olepu ; Jeffrey W. Lockman ; Junko Ohkanda ; Mandana Karimi ; Christophe L. M. J. Verlinde ; James M. Kraus ; Jan Schoepe ; Wesley C. Van Voorhis ; Andrew D. Hamilton ; Frederick S. Buckner ;Michael H. Gelb
Journal of Medicinal Chemistry 2009 Volume 52(Issue 12) pp:3703-3715
Publication Date(Web):May 22, 2009
DOI:10.1021/jm900030h
We report structure−activity studies of a large number of dialkyl imidazoles as inhibitors of Trypanosoma cruzi lanosterol-14α-demethylase (L14DM). The compounds have a simple structure compared to posaconazole, another L14DM inhibitor that is an anti-Chagas drug candidate. Several compounds display potency for killing T. cruzi amastigotes in vitro with values of EC50 in the 0.4−10 nM range. Two compounds were selected for efficacy studies in a mouse model of acute Chagas disease. At oral doses of 20−50 mg/kg given after establishment of parasite infection, the compounds reduced parasitemia in the blood to undetectable levels, and analysis of remaining parasites by PCR revealed a lack of parasites in the majority of animals. These dialkyl imidazoles are substantially less expensive to produce than posaconazole and are appropriate for further development toward an anti-Chagas disease clinical candidate.
Co-reporter:Naveen Kumar Chennamaneni, Jenifer Arif, Frederick S. Buckner, Michael H. Gelb
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 23) pp:6582-6584
Publication Date(Web):1 December 2009
DOI:10.1016/j.bmcl.2009.10.029
We developed a synthetic route to prepare isoquinoline analogs of the cancer drug clinical candidate tipifarnib. We show that these compounds kill Trypanosoma cruzi amastigotes grown in mammalian host cells at concentrations in the low nanomolar range. These isoquinolines represent new leads for the development of drugs to treat Chagas disease.Isoquinoline-based inhibitors of Trypanosoma cruzi lanosterol 14a-demethylase are prepared and shown to block the growth of the amastigote life cycle state of the parasite in the sub-nanomolar range.
Co-reporter:Sophie Blanchard, Frantisek Turecek, Michael H. Gelb
Carbohydrate Research 2009 Volume 344(Issue 8) pp:1032-1033
Publication Date(Web):26 May 2009
DOI:10.1016/j.carres.2009.03.012
Hunter syndrome (mucopolysaccharidosis-II) is caused by deficiency of the lysosomal enzyme iduronate-2-sulfatase. The assay of this sulfatase requires the use of α-l-iduronate glycosides containing a sulfate at the 2-position. We report a simple, three-step procedure for the introduction of sulfate at the 2-position starting with the methyl ester of α-l-iduronate glycosides. The procedure involves protection of the 2- and 4-hydroxyl groups of the iduronate moiety as the dibutyl stannylene acetal, selective sulfation with sulfur trioxide–trimethylamine, and deprotection of the methyl ester to afford the desired 2-sulfate in 61% overall yield.
Co-reporter:Vivek J. Bulbule ; Kasey Rivas ; Christophe L. M. J. Verlinde ; Wesley C. Van Voorhis ;Michael H. Gelb
Journal of Medicinal Chemistry 2008 Volume 51(Issue 3) pp:384-387
Publication Date(Web):January 17, 2008
DOI:10.1021/jm7013138
We report a series of novel inhibitors of protein farnesyltransferase based on the 2-oxotetrahydroquinoline scaffold. We developed an efficient synthesis of these compounds. These compounds show selective inhibtion of the malaria versus human farnesyltransferase and inhibit the growth of the malaria parasite in the low nanomolar range. Some of the compounds are at least an order of magnitude more stable to metabolic degradation than the corresponding tetrahydroquinolines.
Co-reporter:Rob C. Oslund ; Nathan Cermak ;Michael H. Gelb
Journal of Medicinal Chemistry 2008 Volume 51(Issue 15) pp:4708-4714
Publication Date(Web):July 8, 2008
DOI:10.1021/jm800422v
We report a series of inhibitors of secreted phospholipases A2 (sPLA2s) based on substituted indoles, 6,7-benzoindoles, and indolizines derived from LY315920, a well-known indole-based sPLA2 inhibitor. Using the human group X sPLA2 crystal structure, we prepared a highly potent and selective indole-based inhibitor of this enzyme. Also, we report human and mouse group IIA and IIE specific inhibitors and a substituted 6,7-benzoindole that inhibits nearly all human and mouse sPLA2s in the low nanomolar range.
Co-reporter:Rob C. Oslund, Nathan Cermak, Christophe L.M.J. Verlinde, Michael H. Gelb
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 20) pp:5415-5419
Publication Date(Web):15 October 2008
DOI:10.1016/j.bmcl.2008.09.041
Simplified analogs of YM-26734, a known inhibitor of secreted phospholipase A2 (sPLA2) group IIA, were synthesized and found to also display potent inhibition at low nanomolar concentrations. Analogs were based on the didodecanoylphloroglucinol portion of YM-26734 which contains the predicted active site calcium binding group.Modeling of YM-26734 in the active site of secreted phospholipase A2 group IIA led to the design and synthesis of simplified analogs retaining nanomolar potency.
Co-reporter:Srinivas Olepu, Praveen Kumar Suryadevara, Kasey Rivas, Kohei Yokoyama, Christophe L.M.J. Verlinde, Debopam Chakrabarti, Wesley C. Van Voorhis, Michael H. Gelb
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 2) pp:494-497
Publication Date(Web):15 January 2008
DOI:10.1016/j.bmcl.2007.11.104
A new class of 2-oxo-tetrahydro-1,8-naphthyridine-based protein farnesyltransferase inhibitors were synthesized and found to inhibit protein farnesyltransferase from the malaria parasite with potencies in the low nanomolar range. The compounds were much less potent on mammalian protein prenyltransferases. Two of the compounds block the growth of malaria in culture with potencies in the sub-micromolar range. Some of the compounds were found to be much more metabolically stable than previously described tetrahydroquinoline-based protein farnesyltransferase inhibitors.IC50 on malaria protein farnesyltransferase down to 1 nM, ED50 on malaria parasites down to 175 nM.
Co-reporter:Michael H Gelb
Current Opinion in Chemical Biology 2007 Volume 11(Issue 4) pp:440-445
Publication Date(Web):August 2007
DOI:10.1016/j.cbpa.2007.05.038
The prevalence of resistance to known antimalarial drugs has resulted in the expansion of antimalarial drug discovery efforts. Academic and nonprofit institutions are partnering with the pharmaceutical industry to develop new antimalarial drugs. Several new antimalarial agents are undergoing clinical trials, mainly those resurrected from previous antimalarial drug discovery programs. Novel antimalarials are being advanced through the drug development process, of course, with the anticipated high failure rate typical of drug discovery. Many of these are summarized in this review. Mechanisms for funding antimalarial drug discovery and genomic information to aid drug target selection have never been better. It remains to be seen whether ongoing efforts will be sufficient for reducing malaria burden in the developing world.
Co-reporter:Farideh Ghomashchi;James G. Bollinger;Michael H. Gelb
Journal of Labelled Compounds and Radiopharmaceuticals 2007 Volume 50(Issue 8) pp:729-733
Publication Date(Web):17 JUL 2007
DOI:10.1002/jlcr.1414
With the recent ability to use combined liquid chromatography/electrospray tandem mass spectrometry to analyze for several eicosanoids in biological samples in a single and rapid experiment, heavy isotope-labeled eicosanoids are needed as internal standards in order to quantify eicosanoid analytes. The present study describes a practical preparation of cysteinyl leukotrienes (leukotriene C4, D4 and E4) with three 13C atoms and one 15N atom in the cysteinyl residue. The method involves solid-phase peptide synthesis to make glutathione with heavy isotopes in the cysteinyl residue and reaction of this tripeptide with commercially available leukotriene A4 methyl ester to give labeled leukotriene C4 methyl ester, which is hydrolyzed to labeled leukotriene C4. Labeled leukotriene E4 is prepared in the same way with the use of labeled cysteine. Labeled leukotriene D4 is prepared by treatment of labeled leukotriene C4 with commercially available γ-glutamyl transpeptidase. Copyright © 2007 John Wiley & Sons, Ltd.
Co-reporter:Brian P. Smart, Ying H. Pan, Amanda K. Weeks, James G. Bollinger, Brian J. Bahnson, Michael H. Gelb
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 7) pp:1737-1749
Publication Date(Web):1 April 2004
DOI:10.1016/j.bmc.2004.01.022
Structure-guided design was employed in a search for potent and selective inhibitors of mammalian secreted phospholipases A2 (sPLA2s). Using the X-ray structures of human groups IIA and X sPLA2s (hGIIA and hGX) as templates, homology structural models were made for the other human and mouse sPLA2s (hGIB, mGIB, mGIIA, mGIIC, hGIID, mGIID, hGIIE, mGIIE, hGIIF, mGIIF, hGV, mGV, and mGX). Me-Indoxam is a previously discovered indole analogue that binds tightly to many sPLA2s, and the X-ray structure of the hGX-Me-Indoxam complex was determined at a resolution of 2.0 Å. Modeling suggests that the residues near the N1-substituent of Me-Indoxam vary significantly among the mammalian sPLA2s, and therefore a library of 83 N1-variants was prepared by parallel synthesis. Several Me-Indoxam analogues bearing a 4-(2-oxy-ethanoic acid) side chain were potent inhibitors (IC50 <0.05 μM) of hGIIA, mGIIA, mGIIC, hGIIE, mGIIE, hGV, and mGV, while they displayed intermediate potency (0.05–5 μM) against hGIB, mGIB, hGX, and mGX, and poorly inhibited (>5 μM) hGIID, mGIID, hGIIF, and mGIIF. Me-Indoxam analogues bearing a 5-(4-oxy-butanoic acid) side chain were generally less potent inhibitors. Although no compounds were found to be highly specific for a single human or mouse sPLA2, combinations of Me-Indoxam analogues were discovered that could be used to distinguish the action of various sPLA2s in cellular events. For example, Me-Indoxam and compound 5 are approximately 5-fold more potent on hGIIA than on hGV, and compound 21 is 10-fold more potent on hGV versus hGIIA.Graphic
Co-reporter:Dora Carrico, Junko Ohkanda, Howard Kendrick, Kohei Yokoyama, Michelle A. Blaskovich, Cynthia J. Bucher, Frederick S. Buckner, Wesley C. Van Voorhis, Debopam Chakrabarti, Simon L. Croft, Michael H. Gelb, Saïd M. Sebti, Andrew D. Hamilton
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 24) pp:6517-6526
Publication Date(Web):15 December 2004
DOI:10.1016/j.bmc.2004.09.020
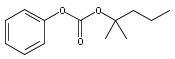
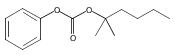
A series of protein farnesyltransferase inhibitor ester prodrugs of FTI-2148 (17) were synthesized in order to evaluate the effects of ester structure modification on antimalarial activity and for further development of a farnesyltransferase inhibitor with in vivo activity. Evaluation against P. falciparum in red blood cells showed that all the investigated esters exhibited significant antimalarial activity, with the benzyl ester 16 showing the best inhibition (ED50 = 150 nM). Additionally, compound 16 displayed in vivo activity and was found to suppress parasitemia by 46.1% at a dose of 50 mg kg−1 day−1 against Plasmodium berghei in mice. The enhanced inhibition potency of the esters is consistent with improved cell membrane permeability compared to that of the free acid. The results of this study suggest that protein farnesyltransferase is a valid antimalarial drug target and that the antimalarial activity of these compounds derives from a balance between the hydrophobic character and the size and conformation of the ester moiety.A series of ester derivatives of 17 with increased lipophilicity were synthesized and tested against P. falciparum in red blood cells, where the benzyl ester derivative 16 exhibited the best inhibition activity (ED50 = 150 nM). Compound 16 showed in vivo antimalarial activity by 46.1% at a daily dose of 50 mg kg−1 using murine malaria models infected with Plasmodium berghei.
Co-reporter:Francesca Clerici, Maria Luisa Gelmi, Kohei Yokoyama, Donato Pocar, Wesley C. Van Voorhis, Frederick S. Buckner, Michael H. Gelb
Bioorganic & Medicinal Chemistry Letters 2002 Volume 12(Issue 16) pp:2217-2220
Publication Date(Web):19 August 2002
DOI:10.1016/S0960-894X(02)00338-4
A series of isothiazole dioxides was synthesized and evaluated as inhibitors of protein farnesyltransferase from the parasite that causes African sleeping sickness (Trypanosoma brucei). The most potent compound in the series inhibited the parasite enzyme with an IC50 of 2 μM and blocked the growth of the bloodstream parasite in vitro with an ED50 of 10 μM. The same compound inhibited rat protein farnesyltransferase and protein geranylgeranyltransferase type I only at much higher concentration.The synthesis and evaluation as Trypanosoma brucei protein farnesyltransferase inhibitors of several isothiazole dioxide derivatives are reported.
Co-reporter:Junko Ohkanda, Jeffrey W. Lockman, Kohei Yokoyama, Michael H. Gelb, Simon L. Croft, Howard Kendrick, Maria Isabel Harrell, Jean E. Feagin, Michelle A. Blaskovich, Said M. Sebti, Andrew D. Hamilton
Bioorganic & Medicinal Chemistry Letters 2001 Volume 11(Issue 6) pp:761-764
Publication Date(Web):26 March 2001
DOI:10.1016/S0960-894X(01)00055-5
Malaria continues to represent a very serious health problem in the tropics. The current methods of clinical treatment are showing deficiencies due to the increased incidence of resistance in the parasite. In the present paper we report the design, synthesis, and evaluation of potential antimalarial agents against a novel target, protein farnesyltransferase. We show that the most potent compounds are active against Plasmodium falciparum in vitro at submicromolar concentrations.Design, synthesis, and evaluation of potential antimalarial agents against a novel target, protein farnesyltransferase are described. The most potent compounds are active against Plasmodium falciparum in vitro at submicromolar concentrations.
Co-reporter:Michael H Gelb, Jeffrey D Scholten, Judith S Sebolt-Leopold
Current Opinion in Chemical Biology 1998 Volume 2(Issue 1) pp:40-48
Publication Date(Web):February 1998
DOI:10.1016/S1367-5931(98)80034-3
A specific set of proteins in eukaryotic cells contain covalently attached carboxy-terminal prenyl groups (15-carbon farnesyl and 20-carbon geranylgeranyl). Many of them are signaling proteins including Ras, heterotrimeric G proteins and Rab proteins. The proteins prenyltransferases which attach prenyl groups to proteins have been well characterized, and an X-ray structure is available for protein farnesyltransferase. Inhibitors of protein farnesyltransferase are showing sufficient promise in preclinical trials as anti-cancer drugs to warrant widespread interest in the pharmaceutical industry.
Co-reporter:Dusko Ilic, James M. Bollinger, Michael Gelb, Theodora M. Mauro
Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids (March 2014) Volume 1841(Issue 3) pp:
Publication Date(Web):March 2014
DOI:10.1016/j.bbalip.2013.11.002
•The epidermal barrier is required for terrestrial life and is established by a lipid barrier.•sPLA2 generates free fatty acids that form stratum corneum lipids and acidity.•sPLA2 controls epidermal barrier function in the first postnatal week.•Several sPLA2 isoforms are expressed in the epidermis.•sPLA II F−/− mice demonstrate a more neutral SC pH and impaired barrier recovery.The mammalian epidermis provides both an interface and a protective barrier between the organism and its environment. Lipid, processed into water-impermeable bilayers between the outermost layers of the epidermal cells, forms the major barrier that prevents water from exiting the organism, and also prevents toxins and infectious agents from entering. The secretory phospholipase 2 (sPLA2) enzymes control important processes in skin and other organs, including inflammation and differentiation. sPLA2 activity contributes to epidermal barrier formation and homeostasis by generating free fatty acids, which are required both for formation of lamellar membranes and also for acidification of the stratum corneum (SC). sPLA2 is especially important in controlling SC acidification and establishment of an optimum epidermal barrier during the first postnatal week. Several sPLA2 isoforms are present in the epidermis. We find that two of these isoforms, sPLA2 IIA and sPLA2 IIF, localize to the upper stratum granulosum and increase in response to experimental barrier perturbation. sPLA2F−/− mice also demonstrate a more neutral SC pH than do their normal littermates, and their initial recovery from barrier perturbation is delayed. These findings confirm that sPLA2 enzymes perform important roles in epidermal development, and suggest that the sPLA2IIF isoform may be central to SC acidification and barrier function. This article is part of a Special Issue entitled The Important Role of Lipids in the Epidermis and their Role in the Formation and Maintenance of the Cutaneous Barrier. Guest Editors: Kenneth R. Feingold and Peter Elias.
Co-reporter:Braden Fitterer, Patricia Hall, Nick Antonishyn, Rajagopal Desikan, ... Denis Lehotay
Molecular Genetics and Metabolism (March 2014) Volume 111(Issue 3) pp:382-389
Publication Date(Web):1 March 2014
DOI:10.1016/j.ymgme.2014.01.002
•We describe the elevated carrier frequency for pathogenic variants in Saskatchewan, Canada.•Several previously unreported HEXB variants are revealed.•Novel hexosaminidase substrates with MS/MS detection are presented.•Variants in the HEXB gene from the 1000 Genomes data set are analyzed.Sandhoff disease is a rare progressive neurodegenerative genetic disorder with a high incidence among certain isolated communities and ethnic groups around the world. Previous reports have shown a high occurrence of Sandhoff disease in northern Saskatchewan. Newborn screening cards from northern Saskatchewan were retrospectively screened in order to investigate the incidence and determine the carrier frequency of Sandhoff disease in these communities. PCR-based screening was conducted for the c.115delG (p.(Val39fs)) variant in the HEXB gene that was previously found in 4 Sandhoff disease patients from this area. The carrier frequency for this allele was estimated to be ~ 1:27. MS/MS-based screening of hexosaminidase activity along with genetic sequencing allowed for the identification of additional variants based on low total hexosaminidase activity and high % hexosaminidase A activity relative to c.115delG carriers. In total 4 pathogenic variants were discovered in the population (c.115delG, c.619A>G, c.1601G>T, and c.1652G>A) of which two are previously unreported (c.1601G>T and c.1652G>A). The combined carrier frequency of these alleles in the study area was estimated at ~ 1:15. Based on the number of cases of Sandhoff disease from this area we estimate the incidence to be ~ 1:390 corresponding to a child being born with the disease every 1–2 years on average. The results from our study were then compared with variants in the HEXB gene from the genomes available from the 1000 Genomes project. A total of 19 HEXB variants were found in the 1092 genomes of which 5 are suspected of having a deleterious effect on hexosaminidase activity. The estimated carrier frequency of Sandhoff disease in Saskatchewan at 1:15 is more than 3 times higher than the carrier frequency in the global sample provided by the 1000 Genomes project at 1:57.
Co-reporter:Farideh Ghomashchi, Allison Stewart, Ying Hefner, Sasanka Ramanadham, John Turk, Christina C. Leslie, Michael H. Gelb
Biochimica et Biophysica Acta (BBA) - Biomembranes (June 2011) Volume 1808(Issue 6) pp:
Publication Date(Web):June 2011
DOI:10.1016/j.bbamem.2010.11.018
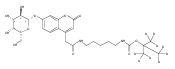
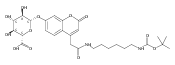
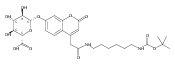
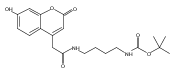
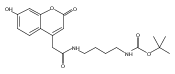
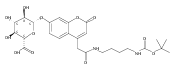
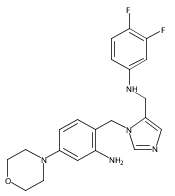
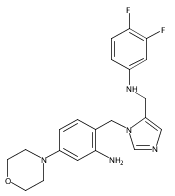
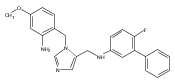
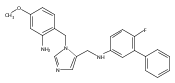
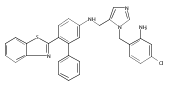
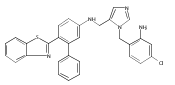
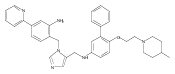
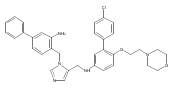
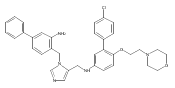
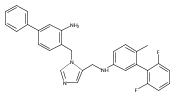
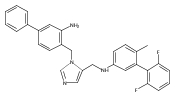
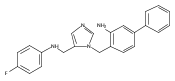
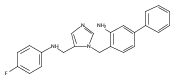
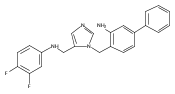
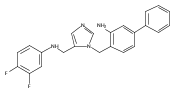
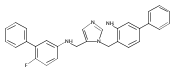
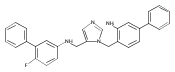
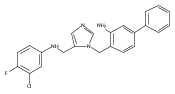
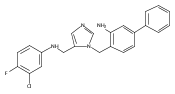
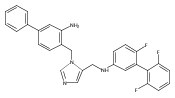
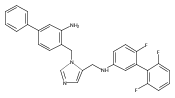
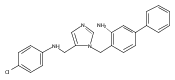
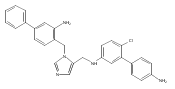
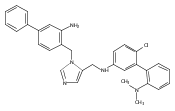
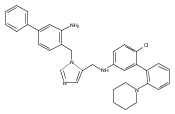
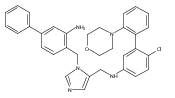
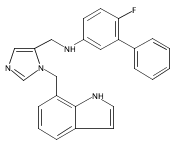
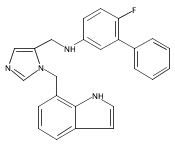
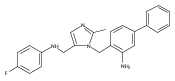
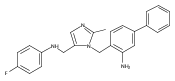
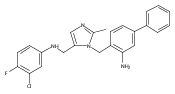
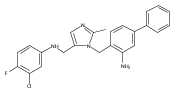
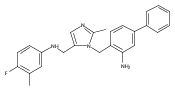
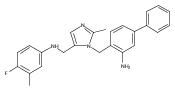
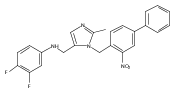
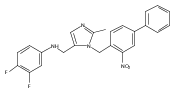
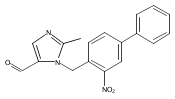
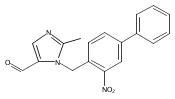
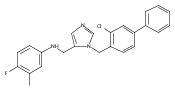
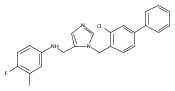
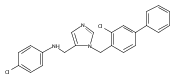
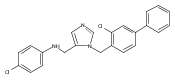
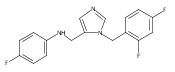
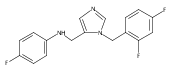
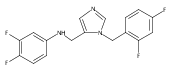
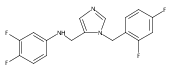
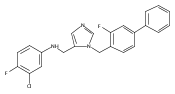
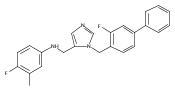
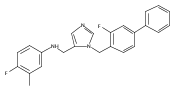
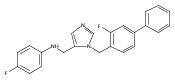
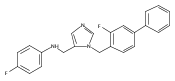
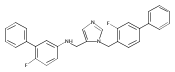
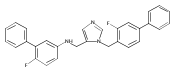
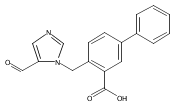
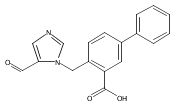
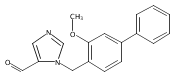
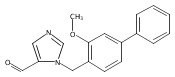
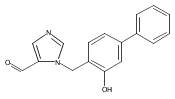
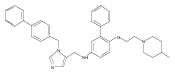
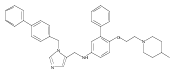
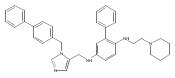
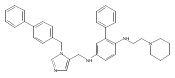
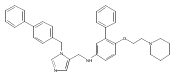
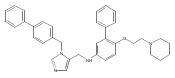
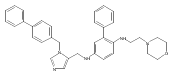
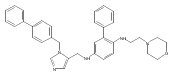
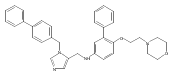
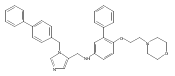
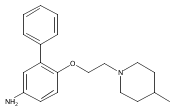
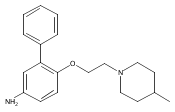
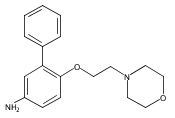
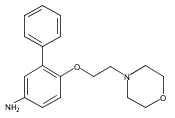
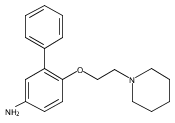
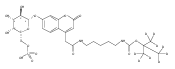
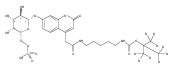
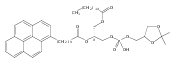
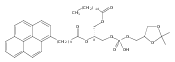
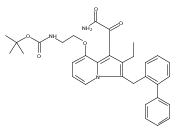
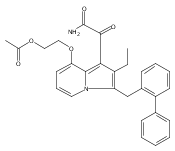
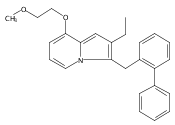
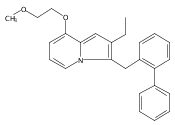
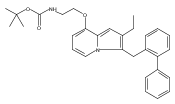
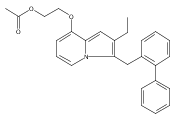
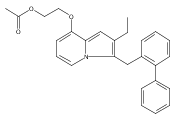
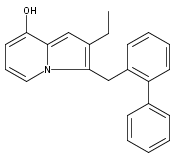
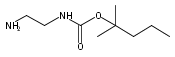
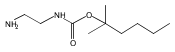
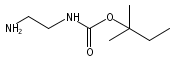
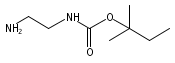
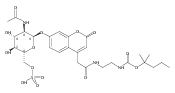
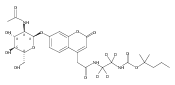
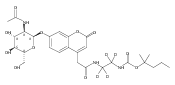
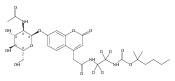
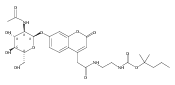
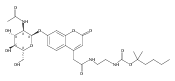
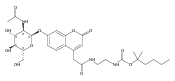
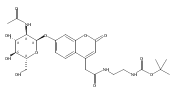
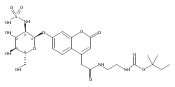
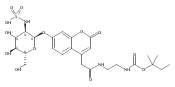
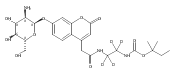
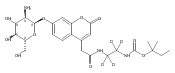
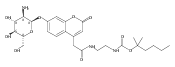
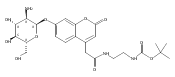
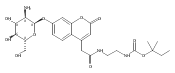
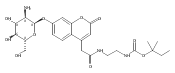
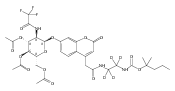
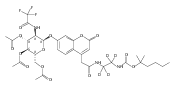
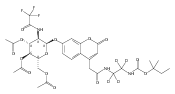
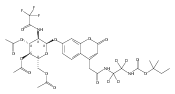
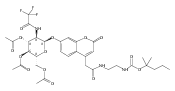
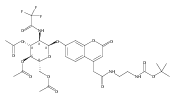
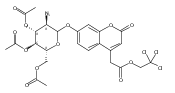
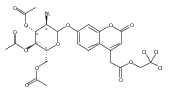
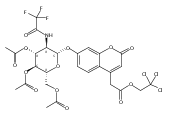
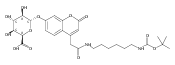
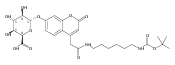
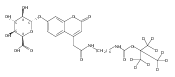
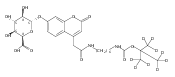
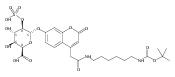
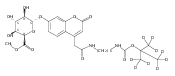
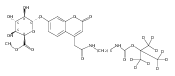
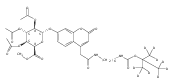
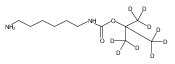
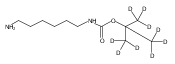
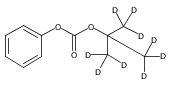
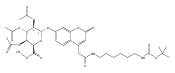
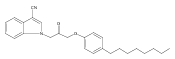
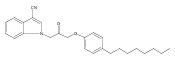
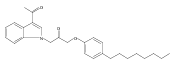
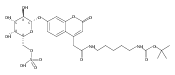
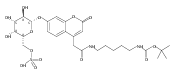
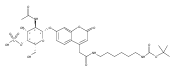
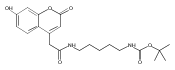
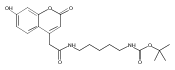
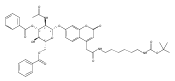
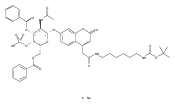
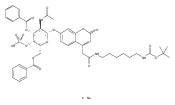
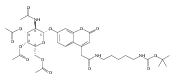
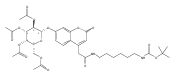
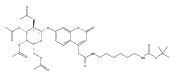
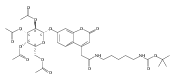
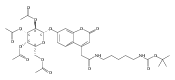
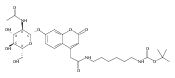
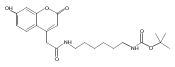
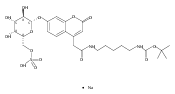
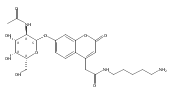
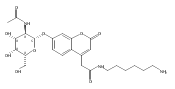
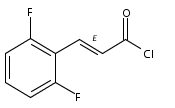
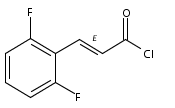
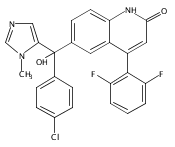
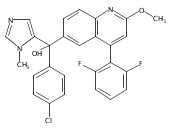
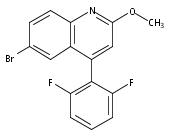
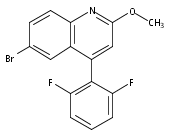
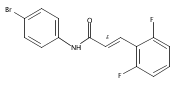
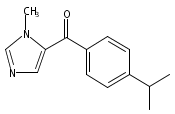
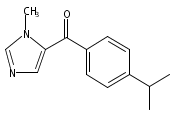
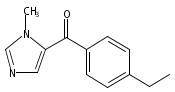
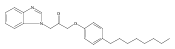
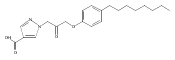
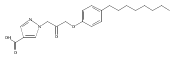
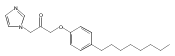
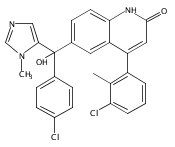
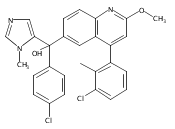
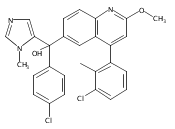
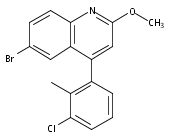
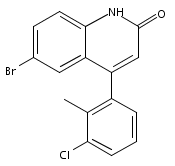
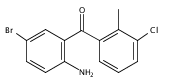
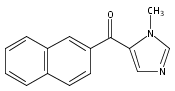
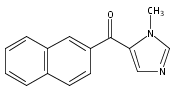
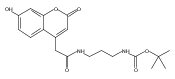
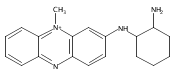
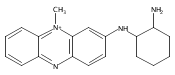
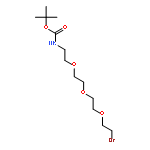
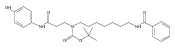
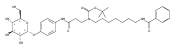
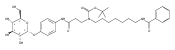
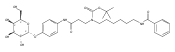
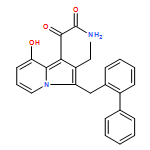
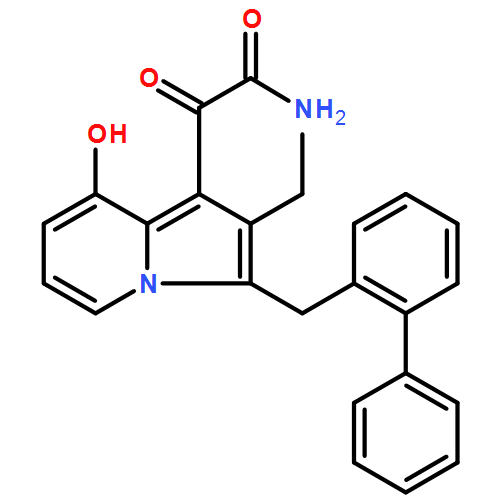
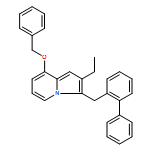
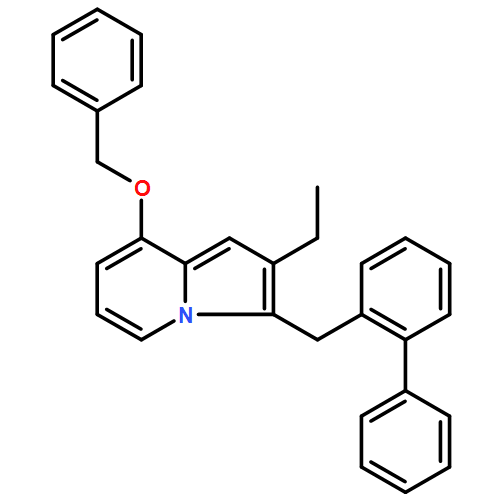
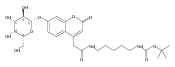
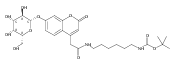
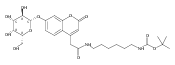
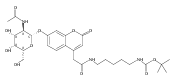
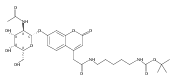
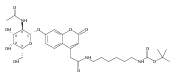
Co-reporter:Andriy Buchynskyy, J. Robert Gillespie, Matthew A. Hulverson, Joshua McQueen, Sharon A. Creason, Ranae M. Ranade, Nicole A. Duster, Michael H. Gelb, Frederick S. Buckner
Bioorganic & Medicinal Chemistry (1 March 2017) Volume 25(Issue 5) pp:1571-1584
Publication Date(Web):1 March 2017
DOI:10.1016/j.bmc.2016.11.019
.jpg)
![2-bromo-3H-Imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/1380300/1380245-88-4.png)
![2-bromo-3H-Imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/1380300/1380245-88-4_b.png)
![Propanal, 3-(1H-imidazo[4,5-b]pyridin-2-ylamino)-](http://img.cochemist.com/ccimg/733100/733053-94-6.png)
![Propanal, 3-(1H-imidazo[4,5-b]pyridin-2-ylamino)-](http://img.cochemist.com/ccimg/733100/733053-94-6_b.png)





















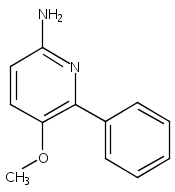
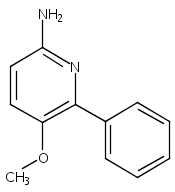
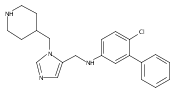
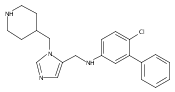






























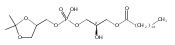
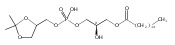
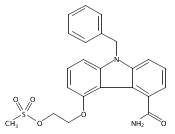
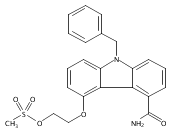






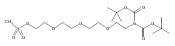
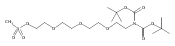
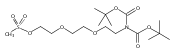

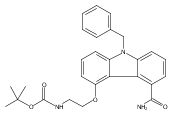
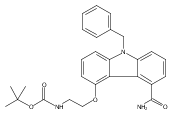
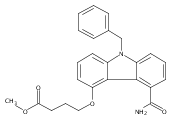
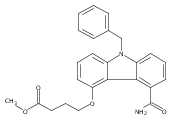
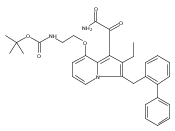






























































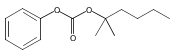
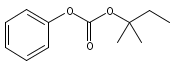
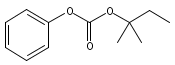
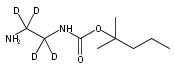




































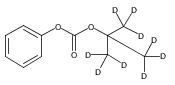
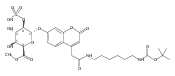
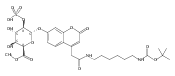

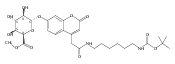















































































































![Morpholine, 4-[(3-chloro-4-fluorobenzo[b]thien-2-yl)carbonyl]-2,6-dimethyl- (9ci)](http://img.cochemist.com/ccimg/588700/588674-15-1.png)
![Morpholine, 4-[(3-chloro-4-fluorobenzo[b]thien-2-yl)carbonyl]-2,6-dimethyl- (9ci)](http://img.cochemist.com/ccimg/588700/588674-15-1_b.png)
![Benzoic acid,4-[3-[5-chloro-2-[2-[[[(2,6-dimethylphenyl)methyl]sulfonyl]amino]ethyl]-1-(diphenylmethyl)-1H-indol-3-yl]propyl]-](http://img.cochemist.com/ccimg/540600/540523-42-0.png)
![Benzoic acid,4-[3-[5-chloro-2-[2-[[[(2,6-dimethylphenyl)methyl]sulfonyl]amino]ethyl]-1-(diphenylmethyl)-1H-indol-3-yl]propyl]-](http://img.cochemist.com/ccimg/540600/540523-42-0_b.png)







![4-{2-[5-chloro-2-(2-{[(3,4-dichlorobenzyl)sulfonyl]amino}ethyl)-1-(diphenylmethyl)-1H-indol-3-yl]ethoxy}benzoic acid](http://img.cochemist.com/ccimg/381700/381683-92-7.png)
![4-{2-[5-chloro-2-(2-{[(3,4-dichlorobenzyl)sulfonyl]amino}ethyl)-1-(diphenylmethyl)-1H-indol-3-yl]ethoxy}benzoic acid](http://img.cochemist.com/ccimg/381700/381683-92-7_b.png)



![N-[1-[2-(2,4-Difluorobenzoyl)benzoyl]-4(R)-(triphenylmethylsulfanyl)pyrrolidin-2(S)-ylmethyl]-4-(2,4-dioxothiazolidin-5-ylidenemethyl)benzamide](http://img.cochemist.com/ccimg/342000/341973-06-6.png)
![N-[1-[2-(2,4-Difluorobenzoyl)benzoyl]-4(R)-(triphenylmethylsulfanyl)pyrrolidin-2(S)-ylmethyl]-4-(2,4-dioxothiazolidin-5-ylidenemethyl)benzamide](http://img.cochemist.com/ccimg/342000/341973-06-6_b.png)










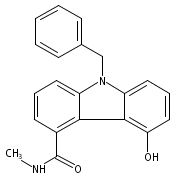
![Acetic acid,[[5-(aminocarbonyl)-9-(phenylmethyl)-9H-carbazol-4-yl]oxy]-, methylester](http://img.cochemist.com/ccimg/246600/246513-46-2.png)
![Acetic acid,[[5-(aminocarbonyl)-9-(phenylmethyl)-9H-carbazol-4-yl]oxy]-, methylester](http://img.cochemist.com/ccimg/246600/246513-46-2_b.png)
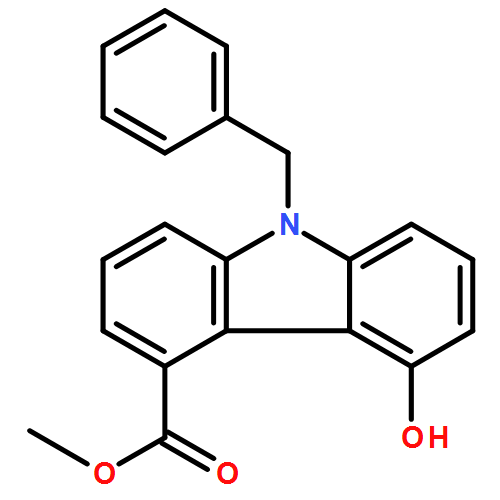
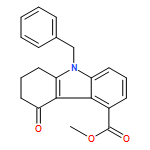
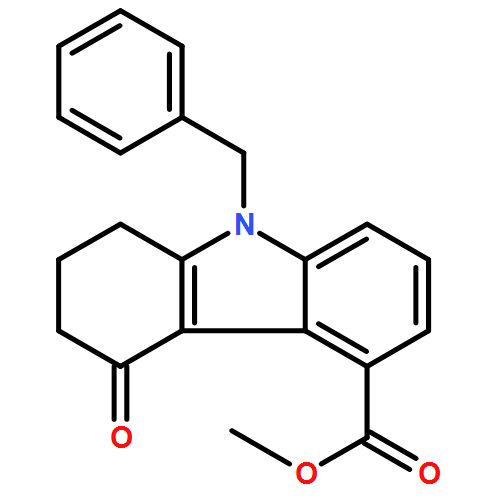
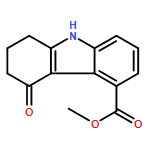

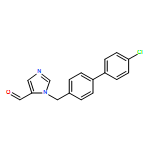
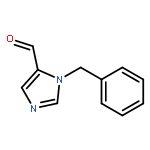
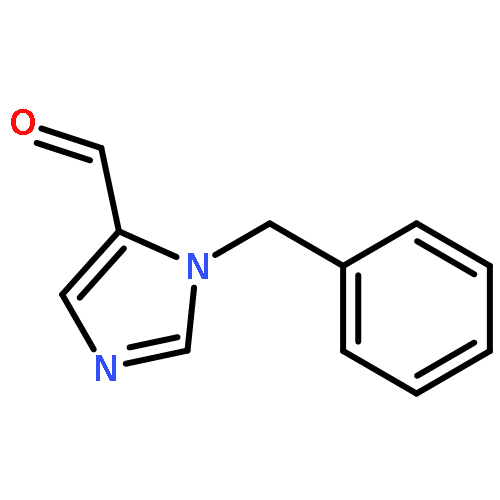
![1H-Imidazole-5-carboxaldehyde, 1-([1,1'-biphenyl]-4-ylmethyl)-](http://img.cochemist.com/ccimg/238800/238765-01-0.png)
![1H-Imidazole-5-carboxaldehyde, 1-([1,1'-biphenyl]-4-ylmethyl)-](http://img.cochemist.com/ccimg/238800/238765-01-0_b.png)







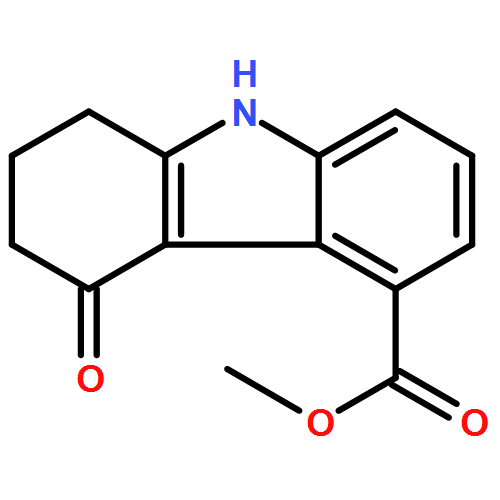
![Acetic acid,[[5-(aminocarbonyl)-9-(phenylmethyl)-9H-carbazol-4-yl]oxy]-](http://img.cochemist.com/ccimg/207400/207340-86-1.png)
![Acetic acid,[[5-(aminocarbonyl)-9-(phenylmethyl)-9H-carbazol-4-yl]oxy]-](http://img.cochemist.com/ccimg/207400/207340-86-1_b.png)











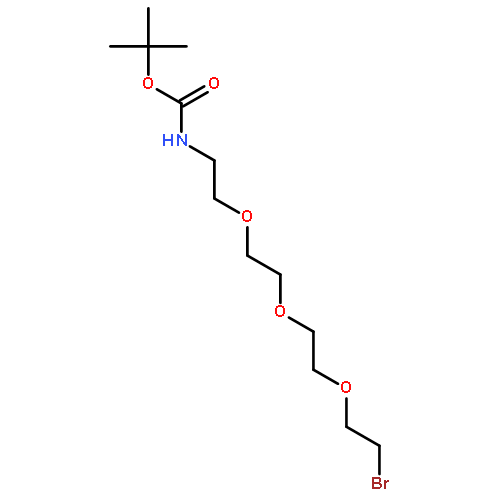
![2-[2-[2-T-BOC-AMINOETHOXY]ETHOXY]ETHYL BROMIDE](http://img.cochemist.com/ccimg/166000/165963-71-3.png)
![2-[2-[2-T-BOC-AMINOETHOXY]ETHOXY]ETHYL BROMIDE](http://img.cochemist.com/ccimg/166000/165963-71-3_b.png)




![Carbamic acid, N-[2-(2-bromoethoxy)ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/118800/164332-88-1.png)
![Carbamic acid, N-[2-(2-bromoethoxy)ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/118800/164332-88-1_b.png)
















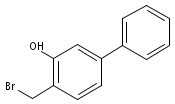
![4-Methyl-[1,1'-biphenyl]-3-ol](http://img.cochemist.com/ccimg/106000/105902-32-7.png)
![4-Methyl-[1,1'-biphenyl]-3-ol](http://img.cochemist.com/ccimg/106000/105902-32-7_b.png)
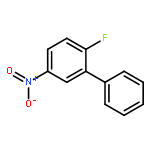
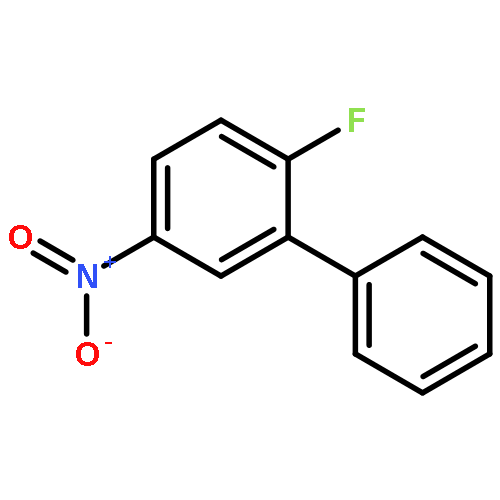
](http://img.cochemist.com/ccimg/102900/102839-00-9.png)
](http://img.cochemist.com/ccimg/102900/102839-00-9_b.png)
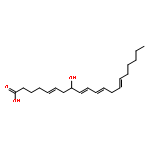
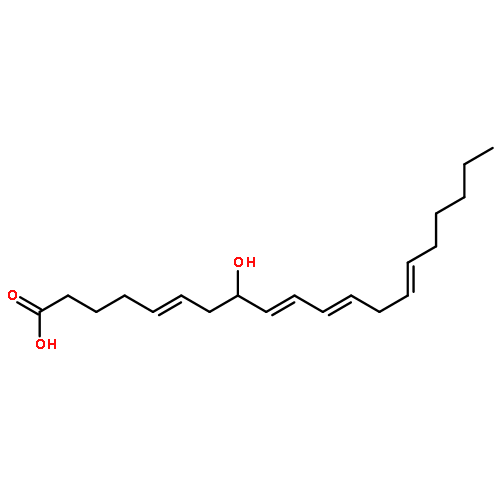


![2-[4-(bromomethyl)phenyl]thiophene](http://img.cochemist.com/ccimg/81500/81443-46-1.png)
![2-[4-(bromomethyl)phenyl]thiophene](http://img.cochemist.com/ccimg/81500/81443-46-1_b.png)















![[(2r)-2-hydroxy-3-phosphonooxypropyl] Octadec-9-enoate](http://img.cochemist.com/ccimg/65600/65528-98-5.png)
![[(2r)-2-hydroxy-3-phosphonooxypropyl] Octadec-9-enoate](http://img.cochemist.com/ccimg/65600/65528-98-5_b.png)






![2-METHYL-2-PROPANYL [1-(4-AMINOPHENYL)-3-PYRROLIDINYL]CARBAMATE](http://img.cochemist.com/ccimg/55500/55400-73-2.png)
![2-METHYL-2-PROPANYL [1-(4-AMINOPHENYL)-3-PYRROLIDINYL]CARBAMATE](http://img.cochemist.com/ccimg/55500/55400-73-2_b.png)










![[(2r)-3-heptadecanoyloxy-2-hydroxypropyl] 2-(trimethylazaniumyl)ethyl Phosphate](http://img.cochemist.com/ccimg/51000/50930-23-9.png)
![[(2r)-3-heptadecanoyloxy-2-hydroxypropyl] 2-(trimethylazaniumyl)ethyl Phosphate](http://img.cochemist.com/ccimg/51000/50930-23-9_b.png)
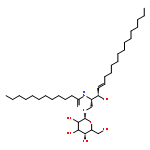
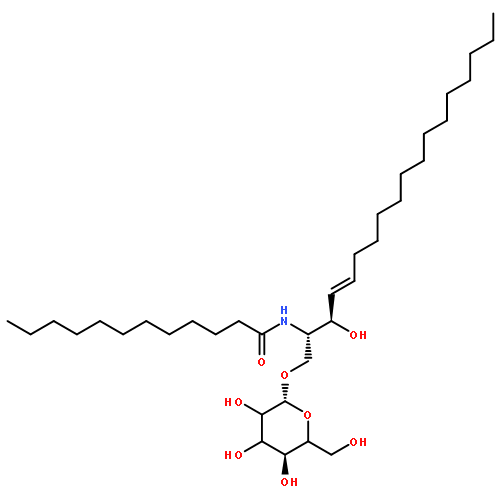
![N-[(E,2S,3R)-3-hydroxy-1-[(2R,5R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoctadec-4-en-2-yl]octanamide](http://img.cochemist.com/ccimg/41700/41613-16-5.png)
![N-[(E,2S,3R)-3-hydroxy-1-[(2R,5R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoctadec-4-en-2-yl]octanamide](http://img.cochemist.com/ccimg/41700/41613-16-5_b.png)
![S-[(7S,8R,9S,10R,13S,14S,17R)-10,13-DIMETHYL-3,5'-DIOXO-1,2,3,4',<WBR />5',6,7,8,9,10,11,12,13,14,15,16-HEXADECAHYDRO-3'H-SPIRO[CYCLOPENT<WBR />A[A]PHENANTHRENE-17,2'-FURAN]-7-YL] ETHANETHIOATE](http://img.cochemist.com/ccimg/39200/39123-23-4.png)
![S-[(7S,8R,9S,10R,13S,14S,17R)-10,13-DIMETHYL-3,5'-DIOXO-1,2,3,4',<WBR />5',6,7,8,9,10,11,12,13,14,15,16-HEXADECAHYDRO-3'H-SPIRO[CYCLOPENT<WBR />A[A]PHENANTHRENE-17,2'-FURAN]-7-YL] ETHANETHIOATE](http://img.cochemist.com/ccimg/39200/39123-23-4_b.png)






![[1,1'-Biphenyl]-2-amine,5-nitro-](http://img.cochemist.com/ccimg/29700/29608-75-1.png)
![[1,1'-Biphenyl]-2-amine,5-nitro-](http://img.cochemist.com/ccimg/29700/29608-75-1_b.png)


![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6.png)
![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6_b.png)
![9-Octadecenoic acid(9Z)-,(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexadecyl)oxy]ethylester](http://img.cochemist.com/ccimg/26700/26662-94-2.png)
![9-Octadecenoic acid(9Z)-,(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexadecyl)oxy]ethylester](http://img.cochemist.com/ccimg/26700/26662-94-2_b.png)


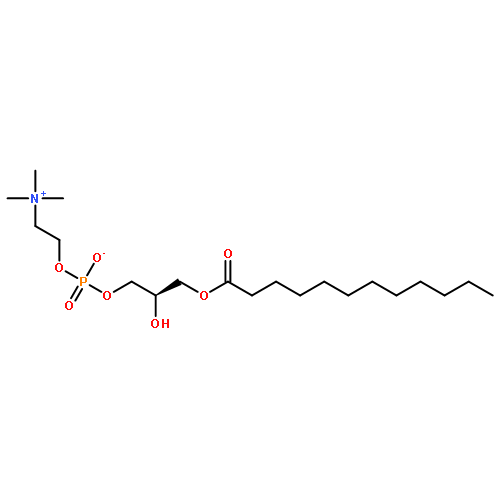


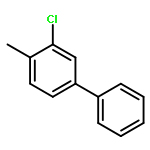
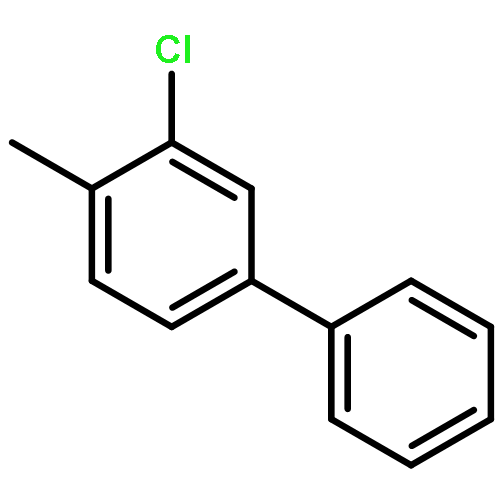




![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)
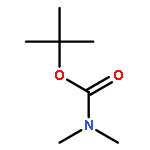



![[1,1'-Biphenyl]-3-carboxylicacid, 4-methyl-](http://img.cochemist.com/ccimg/2900/2840-35-9.png)
![[1,1'-Biphenyl]-3-carboxylicacid, 4-methyl-](http://img.cochemist.com/ccimg/2900/2840-35-9_b.png)






























![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2.png)
![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2_b.png)