Co-reporter:Takahiro Horibe, Shuhei Ohmura, and Kazuaki Ishihara
Organic Letters October 20, 2017 Volume 19(Issue 20) pp:5525-5525
Publication Date(Web):September 28, 2017
DOI:10.1021/acs.orglett.7b02613
Chlorocyclization of tryptamine derivatives has been developed with the use of a diphenyl diselenide–iodine cooperative catalyst. Various tryptamine derivatives can be smoothly converted to the corresponding C3a-chlorohexahydropyrrolo[2,3-b]indoles. Additionally, we demonstrate the formal total syntheses of (−)-psychotriasine and (−)-acetylardeemin by introducing nucleophiles to the C3a position of the products.
Co-reporter:Muhammet Uyanik, Takeshi Yasui, and Kazuaki Ishihara
The Journal of Organic Chemistry November 17, 2017 Volume 82(Issue 22) pp:11946-11946
Publication Date(Web):September 19, 2017
DOI:10.1021/acs.joc.7b01941
Highly enantioselective oxidative dearomatization of 2-naphthol derivatives was achieved for the first time by using conformationally flexible organoiodine catalysts derived from 2-aminoalcohol as a chiral source. Moreover, with the use of these catalysts, excellent enantioselectivities were also achieved for 1-naphthol derivatives, which had previously been obtained with only lower enantioselectivities. Furthermore, the product obtained from the present reaction could be transformed to a highly functionalized spirolactone in high yield and with excellent stereoselectivity.
Co-reporter:Manabu Hatano, Keisuke Nishikawa, and Kazuaki Ishihara
Journal of the American Chemical Society June 28, 2017 Volume 139(Issue 25) pp:8424-8424
Publication Date(Web):May 30, 2017
DOI:10.1021/jacs.7b04795
A chiral magnesium potassium binaphthyldisulfonate cluster, as a chiral Brønsted acid catalyst, was shown to catalyze an enantioselective cycloaddition of styrenes with aldimines for the first time. The strong Brønsted acidity of the catalyst precursors, which might dissolve drying agents and take up the leached Mg2+ and K+, serendipitously led to good enantioselectivity. Mechanistic aspects were supported by X-ray and ESI-MS analysis of the catalyst and a kinetics study of the reaction. Useful transformations to optically active 1,3-amino alcohols on a gram scale were also demonstrated.
Co-reporter:Yanhui Lu;Ke Wang; Dr. Kazuaki Ishihara
Asian Journal of Organic Chemistry 2017 Volume 6(Issue 9) pp:1191-1194
Publication Date(Web):2017/09/01
DOI:10.1002/ajoc.201700194
AbstractTwo novel boronic acid–base complexes were developed as reusable homogeneous catalysts for amide condensation. First, quaternary ammonium resin-bound boronates were shown to be effective catalysts that might release free boronic acids during reactions and could be regenerated afterwards. Next, a 1:1 mixture of 3,5-dinitro-4-tolueneboronic acid and 4-(N,N-dimethylamino)pyridine N-oxide (DMAPO) or 4-(N,N,-dimethylamino)pyridine (DMAP) cooperatively promoted amide formation and precipitated as an acid–base solid complex after cooling. These inexpensive and reusable complexes may be useful in large-scale process chemistry.
Co-reporter:Yanhui Lu;Ke Wang; Dr. Kazuaki Ishihara
Asian Journal of Organic Chemistry 2017 Volume 6(Issue 9) pp:1111-1111
Publication Date(Web):2017/09/01
DOI:10.1002/ajoc.201700311
Boronic acid catalyst shuttle: In general, recovering homogeneous catalysts at the end of a chemical reaction is not easy. Easily recoverable homogeneous catalysts were developed for the dehydrative condensation reaction between carboxylic acids and amines to give amides: resin-bound boronates, and complexes between a boronic acid and DMAP or DMAP N-oxide. Soluble active boronic acids are released from these solids, and work as homogeneous catalysts during the reaction, and are recovered by decantation after the reaction completion. Their behavior resembles a space shuttle. More information can be found in the Communication by Kazuaki Ishihara et al. on page 1191 in Issue 9, 2017 (DOI: 10.1002/ajoc.201700194).
Co-reporter:Kazuaki Ishihara and Yanhui Lu
Chemical Science 2016 vol. 7(Issue 2) pp:1276-1280
Publication Date(Web):06 Nov 2015
DOI:10.1039/C5SC03761A
Arylboronic acid and 4-(N,N-dimethylamino)pyridine N-oxide (DMAPO) cooperatively catalyse the dehydrative condensation reaction between carboxylic acids and amines to give the corresponding amides under azeotropic reflux conditions. This cooperative use is much more effective than their individual use as catalysts, and chemoselectively promotes the amide condensation of (poly)conjugated carboxylic acids. The present method is practical and scalable, and has been applied to the synthesis of sitagliptin and a drug candidate.
Co-reporter:Manabu Hatano, Mai Mizuno, and Kazuaki Ishihara
Organic Letters 2016 Volume 18(Issue 18) pp:4462-4465
Publication Date(Web):September 7, 2016
DOI:10.1021/acs.orglett.6b01774
Regioselective synthetic methods were developed for 1,4- and 1,6-conjugate additions of Grignard reagent-derived organozinc(II)ates to malonate-derived polyconjugated esters. By taking advantage of the tight ion-pair control of organozinc(II)ates, it was possible to switch between 1,4- and 1,6-conjugate additions by introducing a terminal ethoxy moiety in the conjugation.
Co-reporter:Yasuhiro Sawamura, Yoshihiro Ogura, Hidefumi Nakatsuji, Akira Sakakura and Kazuaki Ishihara
Chemical Communications 2016 vol. 52(Issue 36) pp:6068-6071
Publication Date(Web):18 Mar 2016
DOI:10.1039/C6CC00229C
Chiral phosphite–urea bifunctional catalysts have been developed for the enantioselective bromocyclization of 2-geranylphenols with N-bromophthalimide (NBP) for the first time. The chiral triaryl phosphite moiety activates NBP to generate a bromophosphonium ion. On the other hand, the urea moiety interacts with a hydroxyl group of the substrate through hydrogen bonding interactions. Enantioselectivity is effectively induced through two-point attractive interactions between the catalyst and the substrate.
Co-reporter:Dr. Manabu Hatano;Katsuya Yamakawa;Tomoaki Kawai;Dr. Takahiro Horibe;Dr. Kazuaki Ishihara
Angewandte Chemie International Edition 2016 Volume 55( Issue 12) pp:4021-4025
Publication Date(Web):
DOI:10.1002/anie.201510682
Abstract
A highly enantioselective cyanosilylation of ketones was developed by using a chiral lithium(I) phosphoryl phenoxide aqua complex as an acid/base cooperative catalyst. The pentacoordinate silicate generated in situ from Me3SiCN/LiCN acts as an extremely reactive cyano reagent. Described is a 30 gram scale reaction and the synthesis of the key precursor to (+)-13-hydroxyisocyclocelabenzine.
Co-reporter:Dr. Manabu Hatano;Katsuya Yamakawa;Tomoaki Kawai;Dr. Takahiro Horibe;Dr. Kazuaki Ishihara
Angewandte Chemie International Edition 2016 Volume 55( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/anie.201601600
Co-reporter:Dr. Manabu Hatano;Katsuya Yamakawa;Tomoaki Kawai;Dr. Takahiro Horibe;Dr. Kazuaki Ishihara
Angewandte Chemie 2016 Volume 128( Issue 12) pp:4089-4093
Publication Date(Web):
DOI:10.1002/ange.201510682
Abstract
A highly enantioselective cyanosilylation of ketones was developed by using a chiral lithium(I) phosphoryl phenoxide aqua complex as an acid/base cooperative catalyst. The pentacoordinate silicate generated in situ from Me3SiCN/LiCN acts as an extremely reactive cyano reagent. Described is a 30 gram scale reaction and the synthesis of the key precursor to (+)-13-hydroxyisocyclocelabenzine.
Co-reporter:Dr. Manabu Hatano;Katsuya Yamakawa;Tomoaki Kawai;Dr. Takahiro Horibe;Dr. Kazuaki Ishihara
Angewandte Chemie 2016 Volume 128( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/ange.201601600
Co-reporter:Manabu Hatano; Yuta Goto; Atsuto Izumiseki; Matsujiro Akakura
Journal of the American Chemical Society 2015 Volume 137(Issue 42) pp:13472-13475
Publication Date(Web):October 12, 2015
DOI:10.1021/jacs.5b08693
BBr3–chiral phosphoric acid complexes are highly effective and practical Lewis acid-assisted Brønsted acid (LBA) catalysts for promoting the enantioselective Diels–Alder (DA) reaction of α-substituted acroleins and α-CF3 acrylate. In particular, the DA reaction of α-substituted acroleins with 1,2-dihydropyridines gave the corresponding optically active isoquinuclidines with high enantioselectivities. Moreover, transformations to the key intermediates of indole alkaloids, catharanthine and allocatharanthine, are demonstrated.
Co-reporter:Manabu Hatano, Kenji Yamashita, and Kazuaki Ishihara
Organic Letters 2015 Volume 17(Issue 10) pp:2412-2415
Publication Date(Web):April 28, 2015
DOI:10.1021/acs.orglett.5b00927
Highly practical synthetic methods were developed for the C- and N-selective Grignard addition reactions of N-4-MeOC6H4-protected α-aldimino esters in the presence or absence of zinc(II) chloride. Diastereoselective C-alkyl addition, tandem C-alkyl addition–N-alkylation, and some transformations to synthetically useful optically active azacycles were demonstrated.
Co-reporter:Kazuaki Ishihara and Yoshihiro Ogura
Organic Letters 2015 Volume 17(Issue 24) pp:6070-6073
Publication Date(Web):December 4, 2015
DOI:10.1021/acs.orglett.5b03093
The highly enantioselective cyano-alkoxycarbonylation of α-oxoesters with alkyl cyanoformates is promoted by a new chiral Brønsted acid–Lewis base cooperative organocatalyst. The present catalysis can be performed at room temperature under nitrogen or air.
Co-reporter:Dr. Manabu Hatano;Kenji Yamashita;Mai Mizuno;Orie Ito;Dr. Kazuaki Ishihara
Angewandte Chemie 2015 Volume 127( Issue 9) pp:2745-2749
Publication Date(Web):
DOI:10.1002/ange.201408916
Abstract
Since umpolung α-imino esters contain three electrophilic centers, regioselective alkyl addition with traditional organometallic reagents has been a serious problem in the practical synthesis of versatile chiral α-amino acid derivatives. An unusual C-alkyl addition to α-imino esters using a Grignard reagent (RMgX)-derived zinc(II)ate was developed. Zinc(II)ate complexes consist of a Lewis acidic [MgX]+ moiety, a nucleophilic [R3Zn]− moiety, and 2 [MgX2]. Therefore, the ionically separated [R3Zn]− selectively attacks the imino carbon atom ,which is most strongly activated by chelation of [MgX]+. In particular, chiral β,γ-alkynyl-α-imino esters can strongly promote highly regio- and diastereoselective C-alkylation because of structural considerations, and the corresponding optically active α-quaternary amino acid derivatives are obtained within 5 minutes in high to excellent yields.
Co-reporter:Dr. Manabu Hatano;Kenji Yamashita;Mai Mizuno;Orie Ito;Dr. Kazuaki Ishihara
Angewandte Chemie International Edition 2015 Volume 54( Issue 9) pp:2707-2711
Publication Date(Web):
DOI:10.1002/anie.201408916
Abstract
Since umpolung α-imino esters contain three electrophilic centers, regioselective alkyl addition with traditional organometallic reagents has been a serious problem in the practical synthesis of versatile chiral α-amino acid derivatives. An unusual C-alkyl addition to α-imino esters using a Grignard reagent (RMgX)-derived zinc(II)ate was developed. Zinc(II)ate complexes consist of a Lewis acidic [MgX]+ moiety, a nucleophilic [R3Zn]− moiety, and 2 [MgX2]. Therefore, the ionically separated [R3Zn]− selectively attacks the imino carbon atom ,which is most strongly activated by chelation of [MgX]+. In particular, chiral β,γ-alkynyl-α-imino esters can strongly promote highly regio- and diastereoselective C-alkylation because of structural considerations, and the corresponding optically active α-quaternary amino acid derivatives are obtained within 5 minutes in high to excellent yields.
Co-reporter:Masahiro Hori ; Akira Sakakura
Journal of the American Chemical Society 2014 Volume 136(Issue 38) pp:13198-13201
Publication Date(Web):September 8, 2014
DOI:10.1021/ja508441t
We developed 1,3-dipolar cycloadditions of azomethine imines with propioloylpyrazoles catalyzed by a chiral copper(II) complex of 3-(2-naphthyl)-l-alanine amide. The asymmetric environment created by intramolecular π–cation interaction and the N-alkyl group of the chiral ligand gives the corresponding adducts in high yields with excellent enantioselectivity. This is the first successful method for the catalytic enantioselective 1,3-dipolar cycloaddition of azomethine imines with internal alkyne derivatives to give fully substituted pyrazolines.
Co-reporter:Kazuaki Ishihara, Hiroki Yamada and Matsujiro Akakura
Chemical Communications 2014 vol. 50(Issue 48) pp:6357-6360
Publication Date(Web):11 Apr 2014
DOI:10.1039/C4CC01445F
The first enantioselective Diels–Alder reaction of 1,2-dihydropyridines with α-acyloxyacroleins catalyzed by a chiral primary ammonium salt has been developed and it offers more efficient routes to key synthetic intermediates of alkaloids, for which the direct preparations were unavailable before. The asymmetric induction can be understood through the optimized geometry of an iminium salt aqua complex derived from the catalyst and the dienophile.
Co-reporter:Dr. Manabu Hatano;Dr. Kazuaki Ishihara
Asian Journal of Organic Chemistry 2014 Volume 3( Issue 4) pp:352-365
Publication Date(Web):
DOI:10.1002/ajoc.201300256
Abstract
Chiral Brønsted acid catalysts, which are derived from commercially available (R)- or (S)- 1,1′-bi-2-naphthol (BINOL), have found widespread application as chiral organocatalysts and chiral ligands for metal species. The Brønsted acidity of these catalysts is considered to be associated with their catalytic activity. Therefore, due to the rapid development of asymmetric organocatalysis, chiral 1,1′-binaphthyl-2,2′-disulfonic acid (BINSA) and the corresponding chiral binaphthyl disulfonimides are highly attractive as stronger chiral Brønsted acid catalysts than carboxylic acids, phosphoric acids, and phosphoramides. This Focus Review summarizes the latest achievements in chiral BINSA chemistry, particularly in the field of asymmetric organocatalysis.
Co-reporter:Yasuhiro Sawamura;Hidefumi Nakatsuji;Matsujiro Akakura;Akira Sakakura
Chirality 2014 Volume 26( Issue 7) pp:356-360
Publication Date(Web):
DOI:10.1002/chir.22297
ABSTRACT
Nucleophilic phosphite–urea cooperative catalysts are highly efficient for the bromonium-induced cyclization of 2-geranylphenols. Phosphite–N,N’-dimethylurea catalysts also show moderate activity, probably due to the steric effect of their bent conformation. Chirality 26:355–359, 2014. © 2014 Wiley Periodicals, Inc.
Co-reporter:Yuki Matsumura;Takahiro Suzuki;Dr. Akira Sakakura;Dr. Kazuaki Ishihara
Angewandte Chemie International Edition 2014 Volume 53( Issue 24) pp:6131-6134
Publication Date(Web):
DOI:10.1002/anie.201402934
Abstract
The first diastereo- and enantioselective inverse electron demand hetero-Diels–Alder reaction of β,γ-unsaturated α-ketoesters with allylsilanes is described. Chiral copper(II) catalysts successfully activate the β,γ-unsaturated α-ketoesters and promote the reaction with allylsilanes with excellent enantioselectivities. This process represents a new entry to chiral oxanes.
Co-reporter:Dr. Hidefumi Nakatsuji;Yasuhiro Sawamura;Dr. Akira Sakakura;Dr. Kazuaki Ishihara
Angewandte Chemie International Edition 2014 Volume 53( Issue 27) pp:6974-6977
Publication Date(Web):
DOI:10.1002/anie.201400946
Abstract
Chiral triaryl phosphates promote the enantioselective iodolactonization of 4-substituted 4-pentenoic acids to give the corresponding iodolactones in high yields with high enantioselectivity. N-Chlorophthalimide (NCP) is employed as a Lewis acidic activator and oxidant of I2 for the present iodolactonization. In combination with 1.5 equivalents of NCP, only 0.5 equivalents of I2 are sufficient to generate the iodinating reagent.
Co-reporter:Muhammet Uyanik;Hiroki Hayashi
Science 2014 Volume 345(Issue 6194) pp:
Publication Date(Web):
DOI:10.1126/science.1254976
Iodine blooms as an oxidation catalyst
Most catalysts for organic oxidation chemistry—whether biochemical or artificial—contain a transition metal like iron or palladium. Uyanik et al. now show that iodine can take the place of a metal in catalyzing efficient oxidative ring closures to make chromans—hexagonal rings incorporating oxygen that are perhaps best known as a constituent of the vitamin E structure (see the Perspective by Nachtsheim). The iodine is added as a salt with a chiral cation, which directs the reaction to form just one of two possible mirror-image variants of the product. Key to the success of the system was the addition of a base, which maintained the viability of an unstable, partially oxidized iodine intermediate critical to the reaction cycle. The results bode well for more general use of iodine salts as asymmetric oxidation-reduction catalysts.
Science, this issue p. 291; see also p. 270
Co-reporter:Muhammet Uyanik and Kazuaki Ishihara
ACS Catalysis 2013 Volume 3(Issue 4) pp:513
Publication Date(Web):February 22, 2013
DOI:10.1021/cs300821u
The Baeyer–Villiger (BV) oxidation of carbonyl compounds to the corresponding esters or lactones is one of the most important transformations. We recently introduced a highly efficient and selective LiB(C6F5)4- or Ca[B(C6F5)4]2-catalyzed BV oxidation of ketones with aqueous hydrogen peroxide to give the corresponding lactones in high yield. In this perspective article, we focus on our discovery and the development of BV oxidation reactions and cascade oxidative transformations through representative metal catalysts and organocatalysts.Keywords: Baeyer−Villiger oxidation; hydrogen peroxide; organocatalysis; tetraaryl borate salts; transition-metal catalysis
Co-reporter:Yasuhiro Sawamura, Hidefumi Nakatsuji, Akira Sakakura and Kazuaki Ishihara
Chemical Science 2013 vol. 4(Issue 11) pp:4181-4186
Publication Date(Web):02 Aug 2013
DOI:10.1039/C3SC51432C
Nucleophilic phosphite–urea cooperative high-turnover catalysts have been designed for the highly selective bromocyclization of homogeranylarenes. The introduction of a urea moiety and bulky aryl groups in the catalyst inhibits decomposition of the catalyst and the generation of byproducts. Only 0.5 mol% of the catalyst successfully promotes the bromocyclization of 4-homogeranyltoluene to give the desired product in 96% yield.
Co-reporter:Manabu Hatano and Kazuaki Ishihara
Chemical Communications 2013 vol. 49(Issue 20) pp:1983-1997
Publication Date(Web):17 Jan 2013
DOI:10.1039/C2CC38204K
A facile, atom-economical, and chemoselective esterification is crucial in modern organic synthesis, particularly in the areas of pharmaceutical, polymer, and material science. However, a truly practical catalytic transesterification of carboxylic esters with various alcohols has not yet been well established, since, with many conventional catalysts, the substrates are limited to 1°- and cyclic 2°-alcohols. In sharp contrast, if we take advantage of the high catalytic activities of La(Oi-Pr)3, La(OTf)3, and La(NO3)3 as ligand-free catalysts, ligand-assisted or additive-enhanced lanthanum(III) catalysts can be highly effective acid–base combined catalysts in transesterification. A highly active dinuclear La(III) catalyst, which is prepared in situ from lanthanum(III) isopropoxide and 2-(2-methoxyethoxy)ethanol, is effective for the practical transesterification of methyl carboxylates, ethyl acetate, weakly reactive dimethyl carbonate, and much less-reactive methyl carbamates with 1°-, 2°-, and 3°-alcohols. As the second generation, nearly neutral “lanthanum(III) nitrate alkoxide”, namely La(OR)m(NO3)3−m, has been developed. This catalyst is prepared in situ from inexpensive, stable, low-toxic lanthanum(III) nitrate hydrate and methyltrioctylphosphonium methyl carbonate, and is highly useful in the non-epimerized transesterification of α-substituted chiral carboxylic esters, even under azeotropic reflux conditions. In these practical La(III)-catalyzed transesterifications, colorless esters can be obtained in small- to large-scale synthesis without the need for inconvenient work-up or careful purification procedures.
Co-reporter:Risa Yamashita, Akira Sakakura, and Kazuaki Ishihara
Organic Letters 2013 Volume 15(Issue 14) pp:3654-3657
Publication Date(Web):June 26, 2013
DOI:10.1021/ol401537f
Primary alkylboronic acids such as methylboronic acid and butylboronic acid are highly active catalysts for the dehydrative amide condensation of α-hydroxycarboxylic acids. The catalytic activities of these primary alkylboronic acids are much higher than those of the previously reported arylboronic acids. The present method was easily applied to a large-scale synthesis, and 14 g of an amide was obtained in a single reaction.
Co-reporter:Masayuki Sakuma, Akira Sakakura, and Kazuaki Ishihara
Organic Letters 2013 Volume 15(Issue 11) pp:2838-2841
Publication Date(Web):May 15, 2013
DOI:10.1021/ol401313d
Chiral phosphonium salts induce the kinetic resolution of racemic α-substituted unsaturated carboxylic acids through asymmetric protolactonization. Both the lactones and the recovered carboxylic acids are obtained with high enantioselectivities and high S (= kfast/kslow) values. Asymmetric protolactonization also leads to the desymmetrization of achiral carboxylic acids. Notably, chiral phosphonous acid diester not only induced the enantioselectivity but also promoted protolactonization.
Co-reporter:Dr. Manabu Hatano;Dr. Takahiro Horibe;Kenji Yamashita;Dr. Kazuaki Ishihara
Asian Journal of Organic Chemistry 2013 Volume 2( Issue 11) pp:952-956
Publication Date(Web):
DOI:10.1002/ajoc.201300190
Co-reporter:Dr. Manabu Hatano;Takahiro Horibe ;Dr. Kazuaki Ishihara
Angewandte Chemie 2013 Volume 125( Issue 17) pp:4647-4651
Publication Date(Web):
DOI:10.1002/ange.201300938
Co-reporter:Dr. Manabu Hatano;Takahiro Horibe ;Dr. Kazuaki Ishihara
Angewandte Chemie 2013 Volume 125( Issue 17) pp:
Publication Date(Web):
DOI:10.1002/ange.201302124
Co-reporter:Dr. Muhammet Uyanik;Dr. Takeshi Yasui;Dr. Kazuaki Ishihara
Angewandte Chemie 2013 Volume 125( Issue 35) pp:9385-9388
Publication Date(Web):
DOI:10.1002/ange.201303559
Co-reporter:Dr. Manabu Hatano;Takahiro Horibe ;Dr. Kazuaki Ishihara
Angewandte Chemie International Edition 2013 Volume 52( Issue 17) pp:4549-4553
Publication Date(Web):
DOI:10.1002/anie.201300938
Co-reporter:Dr. Manabu Hatano;Takahiro Horibe ;Dr. Kazuaki Ishihara
Angewandte Chemie International Edition 2013 Volume 52( Issue 17) pp:
Publication Date(Web):
DOI:10.1002/anie.201302124
Co-reporter:Yoshihiro Ogura;Dr. Matsujiro Akakura;Dr. Akira Sakakura;Dr. Kazuaki Ishihara
Angewandte Chemie International Edition 2013 Volume 52( Issue 32) pp:8299-8303
Publication Date(Web):
DOI:10.1002/anie.201303572
Co-reporter:Dr. Muhammet Uyanik;Dr. Takeshi Yasui;Dr. Kazuaki Ishihara
Angewandte Chemie International Edition 2013 Volume 52( Issue 35) pp:9215-9218
Publication Date(Web):
DOI:10.1002/anie.201303559
Co-reporter:Manabu Hatano, Takuya Ozaki, Keisuke Nishikawa, and Kazuaki Ishihara
The Journal of Organic Chemistry 2013 Volume 78(Issue 20) pp:10405-10413
Publication Date(Web):September 26, 2013
DOI:10.1021/jo401848z
We developed a practical synthesis of optically pure 3,3′-diaryl-1,1′-binaphthyl-2,2′-disulfonic acids (i.e., (R)- or (S)-3,3′-Ar2-BINSAs) from the parent chiral sulfonimides via stepwise N–S bond cleavage of the sulfonimides and the resultant sulfonamides. This unusual synthesis, which provides arylsulfonic acids from arylsulfonamides, is valuable since common methods particularly give amines with the decomposition of sulfone groups during deprotection.
Co-reporter:Manabu Hatano, Sho Kamiya and Kazuaki Ishihara
Chemical Communications 2012 vol. 48(Issue 76) pp:9465-9467
Publication Date(Web):08 Aug 2012
DOI:10.1039/C2CC34987F
In situ generated lanthanum(III) nitrate alkoxide is a highly active and nearly neutral transesterification catalyst, which can promote non-epimerized transesterification of α-substituted chiral carboxylic esters under reflux conditions.
Co-reporter:Manabu Hatano, Takuya Ozaki, Yoshihiro Sugiura and Kazuaki Ishihara
Chemical Communications 2012 vol. 48(Issue 41) pp:4986-4988
Publication Date(Web):16 Apr 2012
DOI:10.1039/C2CC31530K
A highly effective catalytic enantioselective direct aminal synthesis was developed. Chiral ammonium 1,1′-binaphthyl-2,2′-disulfonates, which were prepared in situ from (R)-BINSA and achiral amines, promoted the enantioselective addition of primary amides to aromatic aldimines.
Co-reporter:Manabu Hatano and Kazuaki Ishihara
Chemical Communications 2012 vol. 48(Issue 36) pp:4273-4283
Publication Date(Web):22 Mar 2012
DOI:10.1039/C2CC00046F
The potential of supramolecular catalysts to realize anomalous regio- and/or stereoselectivity in organic synthesis is highly attractive. To date, there have been a few examples of non-polymeric and non-covalent chiral supramolecular catalysts that induce practical enantioselectivity. In this regard, a metal–organic framework (MOF) may be one of the most important techniques for constructing conformationally rigid supramolecular catalysts. However, it is not easy to use the MOF technique to fine-tune a much more precise cage in catalysts for anomalous purposes. To establish high catalytic activity with anomalous regio- and/or stereoselectivity, in principle, an artificial cage should be conformationally flexible, like an active pocket in an enzyme with an induced-fit function. In this feature article, we focus on the anomalous endo/exo-selective Diels–Alder reaction, and overview the development of the successive catalysts including our recent highly active, conformationally flexible, and chiral supramolecular catalysts. The evolution from ‘ready-made’ single-molecule catalysts to ‘tailor-made’ supramolecular catalysts could offer not only high enantioselectivity but also high anomalous endo/exo-selectivities due to substrate-specific characteristics, as with enzymes.
Co-reporter:Akira Sakakura, Hiroki Yamada, and Kazuaki Ishihara
Organic Letters 2012 Volume 14(Issue 12) pp:2972-2975
Publication Date(Web):May 25, 2012
DOI:10.1021/ol300921f
A catalytic and enantioselective Diels–Alder reaction of α-(carbamoylthio)acroleins induced by an organoammonium salt of chiral triamine is described. α-(Carbamoylthio)acroleins are designed and synthesized as new sulfur-containing dienophiles for the first time. The Diels–Alder reaction affords chiral tertiary thiol precursors with up to 91% ee.
Co-reporter:Dr. Akira Sakakura;Hiroki Yamada;Dr. Kazuaki Ishihara
Asian Journal of Organic Chemistry 2012 Volume 1( Issue 2) pp:133-137
Publication Date(Web):
DOI:10.1002/ajoc.201200054
Co-reporter:Dr. Akira Sakakura;Hiroki Yamada;Dr. Kazuaki Ishihara
Asian Journal of Organic Chemistry 2012 Volume 1( Issue 2) pp:
Publication Date(Web):
DOI:10.1002/ajoc.201290006
Co-reporter:Dr. Muhammet Uyanik;Daisuke Nakashima;Dr. Kazuaki Ishihara
Angewandte Chemie 2012 Volume 124( Issue 36) pp:9227-9230
Publication Date(Web):
DOI:10.1002/ange.201204286
Co-reporter:Dr. Muhammet Uyanik;Dr. Kazuaki Ishihara
ChemCatChem 2012 Volume 4( Issue 2) pp:177-185
Publication Date(Web):
DOI:10.1002/cctc.201100352
Abstract
This Concept highlights the discovery and development of oxidative coupling reactions catalyzed by the hypoiodite (IO−) or iodite (OIO−) ion, which are generated in situ from iodide (I−) ion with hydrogen peroxide or tert-butyl hydroperoxide as an environmentally benign oxidant. The most important features of these catalytic systems are 1) metal-free oxidation, 2) milder reaction conditions, 3) high chemoselectivity (they tolerate a wide range of various functional groups), 4) operational simplicity, and 5) water or tert-butyl alcohol as the only byproduct derived from the co-oxidant used.
Co-reporter:Dr. Muhammet Uyanik;Daisuke Nakashima;Dr. Kazuaki Ishihara
Angewandte Chemie International Edition 2012 Volume 51( Issue 36) pp:9093-9096
Publication Date(Web):
DOI:10.1002/anie.201204286
Co-reporter:Akira Sakakura and Kazuaki Ishihara
Chemical Society Reviews 2011 vol. 40(Issue 1) pp:163-172
Publication Date(Web):06 Sep 2010
DOI:10.1039/B924478F
The rational design of small but highly functional artificial catalysts is very important for practical organic synthesis. Asymmetric Lewis acid catalyses with non-covalent secondary interactions have been developed for enantioselective reactions. This tutorial review describes the concept, design and examples of asymmetric Cu(II) catalyses for cycloaddition reactions based on intramolecular π–cation or n–cation interactions between the copper(II) cation and auxiliary Lewis basic sites of the chiral ligands.
Co-reporter:Akira Sakakura, Masayuki Sakuma, and Kazuaki Ishihara
Organic Letters 2011 Volume 13(Issue 12) pp:3130-3133
Publication Date(Web):May 19, 2011
DOI:10.1021/ol201032t
Chiral Lewis base-assisted Brønsted acids (Chiral LBBAs) have been designed as new organocatalysts for biomimetic enantioselective cyclization. A salt of a chiral phosphonous acid diester with FSO3H catalyzes the enantioselective cyclization of 2-geranylphenols to give the desired trans-fused cyclized products with high diastereo- and enantioselectivities (up to 98:2 dr and 93% ee).
Co-reporter:Manabu Hatano, Sho Kamiya, Katsuhiko Moriyama, and Kazuaki Ishihara
Organic Letters 2011 Volume 13(Issue 3) pp:430-433
Publication Date(Web):December 22, 2010
DOI:10.1021/ol102754y
A practical transesterification of less reactive dimethyl carbonate and much less reactive methyl carbamates with primary (1°), secondary (2°), and tertiary (3°) alcohols was established with the use of a lanthanum(III) complex, which was prepared in situ from lanthanum(III) isopropoxide (3 mol %) and 2-(2-methoxyethoxy)ethanol (6 mol %). In particular, corresponding carbonates and carbamates obtained were of synthetic utility from the viewpoint of the selective protection and/or deprotection of 1°-, 2°-, and 3°-alcohols.
Co-reporter:Manabu Hatano, Yoshiro Furuya, Takumi Shimmura, Katsuhiko Moriyama, Sho Kamiya, Toshikatsu Maki, and Kazuaki Ishihara
Organic Letters 2011 Volume 13(Issue 3) pp:426-429
Publication Date(Web):December 22, 2010
DOI:10.1021/ol102753n
The transesterification of an equimolar mixture of carboxylic esters and primary (1°), secondary (2°), and tertiary (3°) alcohols in hydrocarbon solvents was promoted with high efficiency by a lanthanum(III) complex, which was prepared in situ from lanthanum(III) isopropoxide (1 mol %) and 2-(2-methoxyethoxy)ethanol (2 mol %). The present La(III) catalyst was highly effective for the chemoselective transesterification in the presence of competitive 1°- and 2°-amines. Remarkably, esters were obtained in good to excellent yields as colorless materials without an inconvenient workup procedure.
Co-reporter:Akira Sakakura;Shuhei Umemura
Advanced Synthesis & Catalysis 2011 Volume 353( Issue 11-12) pp:
Publication Date(Web):
DOI:10.1002/adsc.201100252
Abstract
The desymmetrization of meso-glycerol derivatives bearing a 3-pyrroline-1-carbonyl (Pyroc) directing group is demonstrated through an enantioselective acylation reaction promoted by L-histidine-derived bifunctional catalysts. The desired monoacylated products are obtained in good yields (up to 74%) with high enantioselectivities (up to 99% ee).
Co-reporter:Manabu Hatano, Riku Gouzu, Tomokazu Mizuno, Hitoshi Abe, Toshihide Yamada and Kazuaki Ishihara
Catalysis Science & Technology 2011 vol. 1(Issue 7) pp:1149-1158
Publication Date(Web):01 Jul 2011
DOI:10.1039/C1CY00108F
A highly practical, catalytic enantioselective alkyl and aryl addition to aldehydes and ketones with organozinc reagents, which were prepared in situ from commercially available Grignard reagents or arylboronic acids, was developed. A chiral phosphoramide ligand was essential for promoting the addition reactions in high yields with high enantioselectivities.
Co-reporter:Manabu Hatano, Tomokazu Mizuno, Kazuaki Ishihara
Tetrahedron 2011 67(24) pp: 4417-4424
Publication Date(Web):
DOI:10.1016/j.tet.2011.02.042
Co-reporter:Dr. Muhammet Uyanik;Daisuke Suzuki;Takeshi Yasui;Dr. Kazuaki Ishihara
Angewandte Chemie 2011 Volume 123( Issue 23) pp:5443-5446
Publication Date(Web):
DOI:10.1002/ange.201101522
Co-reporter:Dr. Manabu Hatano;Tomokazu Mizuno;Dr. Atsuto Izumiseki;Ryota Usami;Dr. Takafumi Asai;Dr. Matsujiro Akakura;Dr. Kazuaki Ishihara
Angewandte Chemie 2011 Volume 123( Issue 51) pp:12397-12400
Publication Date(Web):
DOI:10.1002/ange.201106497
Co-reporter:Dr. Muhammet Uyanik;Daisuke Suzuki;Takeshi Yasui;Dr. Kazuaki Ishihara
Angewandte Chemie International Edition 2011 Volume 50( Issue 23) pp:5331-5334
Publication Date(Web):
DOI:10.1002/anie.201101522
Co-reporter:Dr. Manabu Hatano;Tomokazu Mizuno;Dr. Atsuto Izumiseki;Ryota Usami;Dr. Takafumi Asai;Dr. Matsujiro Akakura;Dr. Kazuaki Ishihara
Angewandte Chemie International Edition 2011 Volume 50( Issue 51) pp:12189-12192
Publication Date(Web):
DOI:10.1002/anie.201106497
Co-reporter:Muhammet Uyanik;Hiroaki Okamoto;Takeshi Yasui
Science 2010 Vol 328(5984) pp:1376-1379
Publication Date(Web):11 Jun 2010
DOI:10.1126/science.1188217
Co-reporter:Akira Sakakura ; Masahiro Hori ; Makoto Fushimi
Journal of the American Chemical Society 2010 Volume 132(Issue 44) pp:15550-15552
Publication Date(Web):October 19, 2010
DOI:10.1021/ja1081603
A chiral copper(II) complex of 3-(2-naphthyl)-l-alanine amide successfully catalyzes the enantioselective 1,3-dipolar cycloaddition reaction of nitrones with propioloylpyrazole and acryloylpyrazole derivatives. The asymmetric environment created by intramolecular π−cation interaction gives the corresponding adducts in high yields with excellent enantioselectivity. This is the first successful method for the catalytic enantioselective 1,3-dipolar cycloaddition of nitrones with acetylene derivatives. The 1,3-dipolar cycloadducts can be stereoselectively converted to β-lactams via reductive cleavage of the N−O bond using SmI2.
Co-reporter:Manabu Hatano, Takahiro Horibe and Kazuaki Ishihara
Organic Letters 2010 Volume 12(Issue 15) pp:3502-3505
Publication Date(Web):July 7, 2010
DOI:10.1021/ol101353r
A highly enantioselective direct Mannich-type reaction of aldimines with dialkyl malonates was developed with the use of a Mg(II)-BINOLate salt, which was designed as a cooperative acid—base catalyst that can activate both aldimines and malonates. Optically active β-aminoesters and α-halo-β-aminoesters could be synthesized in high yields and with high enantioselectivities. This inexpensive and practical Mg(II)-BINOLate salt could be used in gram-scale catalysis.
Co-reporter:Manabu Hatano, Orie Ito, Shinji Suzuki and Kazuaki Ishihara
Chemical Communications 2010 vol. 46(Issue 15) pp:2674-2676
Publication Date(Web):09 Feb 2010
DOI:10.1039/B926243A
Highly efficient alkylations and arylations of ketones with Grignard reagents (RMgBr and RMgI) have been developed using catalytic ZnCl2, Me3SiCH2MgCl, and LiCl. Tertiary alcohols were obtained in high yields with high chemoselectivities, while minimizing undesired side products produced by reduction and enolization.
Co-reporter:Manabu Hatano, Tomokazu Mizuno and Kazuaki Ishihara
Chemical Communications 2010 vol. 46(Issue 30) pp:5443-5445
Publication Date(Web):29 Jun 2010
DOI:10.1039/C0CC01301C
A highly practical, catalytic enantioselective 2°-alkyl addition to aldehydes and ketones was developed. Chiral phosphoramide ligand (1) with salt-free and solvent-free di(2°-alkyl)zinc reagents prepared from (2°-alkyl)MgCl was essential.
Co-reporter:Muhammet Uyanik Dr.;Ryota Fukatsu Dr.
Chemistry – An Asian Journal 2010 Volume 5( Issue 3) pp:456-460
Publication Date(Web):
DOI:10.1002/asia.200900609
Co-reporter:Muhammet Uyanik, Takeshi Yasui, Kazuaki Ishihara
Tetrahedron 2010 66(31) pp: 5841-5851
Publication Date(Web):
DOI:10.1016/j.tet.2010.04.060
Co-reporter:Muhammet Uyanik Dr.;Takeshi Yasui Dr.
Angewandte Chemie 2010 Volume 122( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/ange.201000768
Co-reporter:Manabu Hatano, Yoshihiro Sugiura, Kazuaki Ishihara
Tetrahedron: Asymmetry 2010 Volume 21(9–10) pp:1311-1314
Publication Date(Web):17 May 2010
DOI:10.1016/j.tetasy.2010.03.014
A convenient synthesis of chiral 3,3′-disubstituted 1,1′-binaphthyl-2,2′-disulfonic acids (BINSA, 1) was developed. The key was directed ortho-lithiation of BINSA methyl ester 2 with n-BuLi and subsequent reaction with an electrophile. Electrophiles such as Br2, I2, Me3SiOTf, and i-PrOB(Pin) reacted smoothly with 3,3′-dilithiated BINSA methyl ester, and the corresponding 3,3′-dihalo-, 3,3′-bis(trimethylsilyl)-, and 3,3′-diboryl-BINSA derivatives were obtained in yields of 21–78%. This simple synthetic method is highly attractive since the ability to prepare 3,3′-disubstituted BINOLs in advance can be useful.(R)-Dimethyl 1,1′-binaphthalene-2,2′-disulfonateC22H18O6S2[α]D24=+80.8 (c 1.0, CHCl3)Source of chirality: (R)-BINSAAbsolute configuration: (R)(R)-Diethyl 1,1′-binaphthalene-2,2′-disulfonateC24H22O6S2[α]D24=+31.2 (c 1.0, CHCl3)Source of chirality: (R)-BINSAAbsolute configuration: (R)(R)-Dimethyl 3,3′-bis(trimethylsilyl)-1,1′-binaphthalene-2,2′-disulfonateC28H34O6S2Si2[α]D26=+77.7 (c 0.17, CHCl3)Source of chirality: (R)-BINSAAbsolute configuration: (R)Lithium (R)-3,3′-bis(trimethylsilyl)-1,1′-binaphthalene-2,2′-disulfonateC26H28Li2O6S2Si2[α]D28=+95.2 (c 0.29, MeOH)Source of chirality: (R)-BINSAAbsolute configuration: (R)(R)-Dimethyl 3,3′-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,1′-binaphthalene-2,2′-disulfonateC34H40B2O10S2[α]D28=+96.9 (c 1.85, CHCl3).Source of chirality: (R)-BINSAAbsolute configuration: (R)(R)-3,3′-Diborono-1,1′-binaphthyl-2,2′-disulfonic acidC20H16B2O10S2[α]D28=+80.0 (c 1.0, MeOH).Source of chirality: (R)-BINSAAbsolute configuration: (R)
Co-reporter:Muhammet Uyanik Dr.;Takeshi Yasui Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 12) pp:2175-2177
Publication Date(Web):
DOI:10.1002/anie.200907352
Co-reporter:Muhammet Uyanik Dr.;Takeshi Yasui Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/anie.201000768
Co-reporter:Manabu Hatano Dr.;Katsuhiko Moriyama Dr.;Toshikatsu Maki Dr. Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 22) pp:3823-3826
Publication Date(Web):
DOI:10.1002/anie.201000824
Co-reporter:Manabu Hatano Dr.;Katsuhiko Moriyama Dr.;Toshikatsu Maki Dr. Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 22) pp:
Publication Date(Web):
DOI:10.1002/anie.201002202
Co-reporter:Muhammet Uyanik Dr.;Takeshi Yasui Dr.
Angewandte Chemie 2010 Volume 122( Issue 12) pp:2221-2223
Publication Date(Web):
DOI:10.1002/ange.200907352
Co-reporter:Manabu Hatano Dr.;Katsuhiko Moriyama Dr.;Toshikatsu Maki Dr. Dr.
Angewandte Chemie 2010 Volume 122( Issue 22) pp:3911-3914
Publication Date(Web):
DOI:10.1002/ange.201000824
Co-reporter:Manabu Hatano Dr.;Katsuhiko Moriyama Dr.;Toshikatsu Maki Dr. Dr.
Angewandte Chemie 2010 Volume 122( Issue 22) pp:
Publication Date(Web):
DOI:10.1002/ange.201002202
Co-reporter:Manabu Hatano ; Takahiro Horibe
Journal of the American Chemical Society 2009 Volume 132(Issue 1) pp:56-57
Publication Date(Web):December 14, 2009
DOI:10.1021/ja909874b
A highly diastereo- and enantioselective direct Mannich-type reaction of aldimines with 1,3-dicarbonyl compounds using Li(I) BINOLate salts as effective Lewis acid−Brønsted base catalysts has been developed. Li(I) BINOLate salts offered high catalytic activity toward 1,3-dicarbonyl compounds such as diketone, ketoester, ketothioester, ketoamide, and ketolactone. The reactions proceeded at −78 °C within 1−2 h in the presence of 1−10 mol % catalyst, which showed a catalytic activity (turnover frequency = 284 h−1) quite unlike those of other previous catalysts. Anti products were selectively obtained from acyclic ketoesters without epimerization at an α-3°-carbon center, and these are valuable since previous catalysts often gave syn/anti mixtures or the stereochemistry has not yet been determined.
Co-reporter:Akira Sakakura ; Rei Kondo ; Yuki Matsumura ; Matsujiro Akakura
Journal of the American Chemical Society 2009 Volume 131(Issue 49) pp:17762-17764
Publication Date(Web):November 19, 2009
DOI:10.1021/ja906098b
The rational design of bis(oxazoline)-copper(II) catalysts based on postulated intramolecular secondary n-cation interaction for the highly enantioselective Diels−Alder reaction is presented. A theoretical calculation suggested that the n electrons of the 4,4′-sulfonamidomethyl groups successfully interact with the Cu(II) cation and that the counteranions with protons of sulfonamido groups. These secondary interactions might be essential for the high catalytic activity, the broad range of substrates, and the high level of induction of the enantioselectivity.
Co-reporter:Manabu Hatano, Yasushi Hattori, Yoshiro Furuya and Kazuaki Ishihara
Organic Letters 2009 Volume 11(Issue 11) pp:2321-2324
Publication Date(Web):April 30, 2009
DOI:10.1021/ol900680f
A catalytic enantioselective Strecker reaction catalyzed by novel chiral lanthanum(III)−binaphthyl disulfonate complexes was developed. The key to promoting the reactions was a semistoichiometric amount of AcOH or i-PrCO2H, which takes advantage of HCN generation in situ. The corresponding cyanation products were obtained in high yields and with high enantioselectivities.
Co-reporter:Muhammet Uyanik, Ryota Fukatsu and Kazuaki Ishihara
Organic Letters 2009 Volume 11(Issue 15) pp:3470-3473
Publication Date(Web):July 6, 2009
DOI:10.1021/ol9013188
A 2-iodoxybenzenesulfonic acid (IBS)-catalyzed oxidative rearrangement of tertiary allylic alcohols to enones with powdered Oxone in the presence of potassium carbonate and tetrabutylammonium hydrogen sulfate has been developed.
Co-reporter:Muhammet Uyanik and Kazuaki Ishihara
Chemical Communications 2009 (Issue 16) pp:2086-2099
Publication Date(Web):06 Mar 2009
DOI:10.1039/B823399C
Over the past two decades there has been a dramatic increse in the use of hypervalent iodine compounds in synthetic organic chemistry due to their mild and selective oxidizing properties. Hypervalent iodine compounds catalyze various oxidation reactions such as the oxidation of alcohols, α-oxidation of ketones, oxidative spirocyclization of phenols, etc. Very recently, we found that 2-iodoxybenzensulfonic acid (IBS, 7a), which was generated from 2-iodobenzenesulfonic acidin situ, is an extremely active and mild catalyst for the highly chemoselective oxidation of various alcohols with powdered Oxone® to carbonyl compounds such as aldehydes, carboxylic acids, ketones and cycloalkenones under non-aqueous conditions. In this review, we focus on the design of hypervalent iodine catalysts and review the discovery and development of the oxidation of alcohols with the stoichiometric or catalytic use of hypervalent iodine compounds.
Co-reporter:Muhammet Uyanik, Takeshi Yasui, Kazuaki Ishihara
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 14) pp:3848-3851
Publication Date(Web):15 July 2009
DOI:10.1016/j.bmcl.2009.03.148
The hypervalent iodine-catalyzed oxylactonization of ketocarboxylic acids to ketolactones was achieved in the presence of iodobenzene (10 mol %), p-toluenesulfonic acid monohydrate (20 mol %) and meta-chloroperbenzoic acid as a stoichiometric co-oxidant.The hypervalent iodine-catalyzed oxylactonization of ketocarboxylic acids to ketolactones was achieved in the presence of iodobenzene (10 mol %), TsOH (20 mol %) and m-CPBA as stoichiometric co-oxidant.
Co-reporter:Kazuaki Ishihara
Tetrahedron 2009 65(6) pp: 1085-1109
Publication Date(Web):
DOI:10.1016/j.tet.2008.11.004
Co-reporter:Akira Sakakura, Rei Kondo, Shuhei Umemura, Kazuaki Ishihara
Tetrahedron 2009 65(10) pp: 2102-2109
Publication Date(Web):
DOI:10.1016/j.tet.2008.12.074
Co-reporter:Manabu Hatano, Shinji Suzuki, Eri Takagi, Kazuaki Ishihara
Tetrahedron Letters 2009 50(26) pp: 3171-3174
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.01.028
Co-reporter:Akira Sakakura, Shuhei Umemura and Kazuaki Ishihara
Chemical Communications 2008 (Issue 30) pp:3561-3563
Publication Date(Web):06 Jun 2008
DOI:10.1039/B805880F
Efficient total syntheses of fluvibactin and vibriobactin have been achieved via molybdenum(VI) oxide-catalyzed dehydrative cyclization, Sb(OEt)3-catalyzed ester–amide transformation, and WSCI and HOAt-promoted dehydrative amide formation.
Co-reporter:Manabu Hatano;Takumi Ikeno;Tokihiko Matsumura;Shinobu Torii
Advanced Synthesis & Catalysis 2008 Volume 350( Issue 11-12) pp:1776-1780
Publication Date(Web):
DOI:10.1002/adsc.200800314
Abstract
The catalytic enantioselective cyanosilylation of aromatic ketones was developed by using chiral lithium salts of (R)-BINOL- or (S)-BINAM-derived phosphoric acid compounds. In the presence of 10 mol% of chiral conjugate lithium salts, the corresponding tertiary cyanohydrins were obtained in high yields with moderate to high enantioselectivities. This is the first efficient example of asymmetric catalysis using lithium salts of synthetically useful chiral phosphoric acid compounds. A possible catalytic mechanism and transition states are also discussed as a preliminary working hypothesis.
Co-reporter:Manabu Hatano
The Chemical Record 2008 Volume 8( Issue 3) pp:143-155
Publication Date(Web):
DOI:10.1002/tcr.20146
Abstract
A highly enantioselective organozinc (R2Zn) addition to a series of aldehydes and ketones was developed based on conjugate Lewis acid–Lewis base catalysis. Optically active secondary and tertiary alcohols were obtained in high yields with high enantioselectivities without Ti(IV) compounds. Bifunctional chiral 3,3′-diphosphoryl-BINOL ligands were designed and prepared through a phospho-Fries rearrangement as a key step. On the other hand, bifunctional chiral phosphoramide ligands were designed and prepared from L-valine. Mechanistic studies were performed by X-ray analyses of Zn(II) cluster and chiral ligands, a 31P NMR experiment on Zn(II) complexes, and stoichiometric reactions with some chiral or achiral Zn(II) complexes to propose a transition state assembly that includes monomeric active intermediates. © 2008 The Japan Chemical Journal Forum and Wiley Periodicals, Inc. Chem Rec 8: 143–155; 2008: Published online in Wiley InterScience (www.interscience.wiley.com) DOI 10.1002/tcr.20146
Co-reporter:Kazuaki Ishihara, Makoto Fushimi and Matsujiro Akakura
Accounts of Chemical Research 2007 Volume 40(Issue 10) pp:1049
Publication Date(Web):July 28, 2007
DOI:10.1021/ar700083a
We have designed a minimal artificial metalloenzyme that is prepared in situ from Cu(OTf)2 or Cu(NTf2)2 (1.0 equiv) and l-DOPA-derived monopeptide (1.1 equiv) based on the cation–π attractive interaction between copper(II) and the aromatic arm of the ligand, which is postulated on the basis of X-ray diffraction analysis and theoretical calculations. This catalyst (2–10 mol %) is highly effective for not only the enantioselective Diels–Alder reaction with α,β-unsaturated 1-acyl-3,5-dimethylpyrazoles but also the enantioselective Mukaiyama–Michael reaction with these compounds. Products bearing a 3,5-dimethylpyrazolyl auxiliary may be transformed into a range of carboxylic acid derivatives, such as the corresponding carboxylic acids, esters, amides, alcohols, aldehydes, ketones, and β-ketoesters, by known methods. The present results demonstrate that monopeptides are chirally economical and readily tunable ligands compared to bis(oxazoline)s, which have been reported to be notably useful ligands in various enantioselective reactions with bidentate electrophiles.
Co-reporter:Akira Sakakura;Shoko Nakagawa
Nature Protocols 2007 2(7) pp:
Publication Date(Web):2007-07-05
DOI:10.1038/nprot.2007.254
The ester condensation reaction is among the most fundamental organic transformations, and more environmentally benign alternative synthetic approaches to the ones currently used are in strong demand by the chemical industry1. Conventionally, the ester condensation reaction of carboxylic acids with alcohols is catalyzed by Brønsted acids such as HCl, H2SO4, p-toluenesulfonic acid and so on for acid-resistance substrates. For acid-sensitive substrates, weak Brønsted acids such as pyridinium p-toluenesulfonate should be used. However, these have lower catalytic activities and the reactants that can be used are rather limited. With regard to green chemistry, in particular with respect to atom economy and E-factor, several catalytic methods for the ester condensation reaction between equimolar amounts of carboxylic acids and alcohols have been developed2, 3, 4, 5, 6. Conventionally, in fact, esterifications are conducted with an excess of carboxylic acids or alcohols against its reaction counterpart in the presence of an acid catalyst, or with a stoichiometric dehydrating reagent or activated carboxylic acid derivative in the presence of a stoichiometric amount of base. The use of excess amounts of substrates is a wasteful practice in itself. Furthermore, the use of stoichiometric dehydrating reagents or activated carboxylic acid derivatives leads to the formation of significant amounts of undesired by-products. Purifying the crude products from (excess) substrates or from reaction by-products is a rather demanding task—also in financial terms—requiring additional apparati and additional amounts of materials, energy (e.g., for azeotropic reflux) and time. It is therefore evident why the direct catalytic condensation between equimolar amounts of carboxylic acids and alcohols that does not require the presence of dehydrating agents is, at least in principle, such an attractive synthetic goal. Among these 'green' catalytic condensations, metal-free organocatalytic methods are particularly desirable, especially for industrial processes. In 2000, Tanabe and co-workers2 reported that diphenylammonium triflate ([Ph2NH2]+[OTf]−, 1.0–10 mol%) efficiently catalyzed the ester condensation reaction at 80 °C without the need for removal of water. Unfortunately, however, as [Ph2NH2]+[OTf]− is the salt of a superacid (trifluoromethansulfonic acid (TfOH)) and a weak base (Ph2NH), it is a strong Brønsted acid, and as such is difficult to use in the reaction of sterically demanding and acid-sensitive alcohols.In the course of our continuing study on environmentally benign dehydration catalysts, we have developed N,N-dimesitylammonium pentafluorobenzenesulfonate (3) and N-(2,6-diisopropylphenyl)-N-mesitylammonium pentafluorobenzenesulfonate (4) as mild and selective ester condensation catalysts7, 8, 9. A scheme for the synthesis of ammonium catalysts 3 and 4 is shown in Figure 1. N,N-Dimesitylamine (1) is prepared from 2,4,6-trimethylaniline by palladium-catalyzed cross-coupling with 2,4,6-mesitylbromide10, 11. The reaction of 1 with an equimolar amount of pentafluorobenzenesulfonic acid (C6F5SO3H), which is prepared from pentafluorobenzenesulfonyl chloride by hydrolysis, gives ammonium salt 3. This catalyst 3 has been commercially available from Tokyo Chemical Industry Co., Ltd (TCI) since January 2007. Catalyst 4 can be prepared from 2,6-diisopropylaniline by an analogous procedure. It is, in particular, the synthesis of catalyst 4 that is detailed in the PROCEDURE section.C6F5SO3H (pKa(CD3CO2D) = 11.1, H0 = −3.98) is a weaker acid than TfOH (pKa(CD3CO2D) = −0.74, H0 = −14.00), concentrated H2SO4 (pKa(CD3CO2D) = 7.5, H0 = −11.93) and p-toluenesulfonic acid (pKa(CD3CO2D) = 8.5, H0 = −4.5). This means that 3 and 4 are milder acids than the corresponding ammonium triflates, sulfates and tosylates. Nevertheless, 3 and 4 have much higher catalytic activities than Tanabe catalyst ([Ph2NH2]+[OTf]−) (see data in Fig. 2), owing to the hydrophobic environment created around the ammonium protons in the catalyst12. Even though the ester condensation was performed under heating without the removal of water, the reaction proceeded well without any deceleration owing to the generated water.The X-ray single-crystal structures of 3 and [Ph2NH2]+[OTf]− are shown in Figure 3. The crystals obtained were dimeric cyclic ion pairs composed of two diarylammonium cations and two arenesulfonate anions. Interestingly, the dimeric cyclic ion pair of 3 was stabilized by two intermolecular π–π interactions as well as four hydrogen bondings, whereas there was no intermolecular π–π interaction in the ion pair of [Ph2NH2]+[OTf]−. It is conceivable that a 'hydrophobic wall' prevents polar water molecules from gaining access to the active site of the catalysts and thus inhibits the inactivation of the catalyst by water (Fig. 3). Furthermore, the steric bulkiness of the mesityl and pentafluorophenyl groups in the catalyst suppressed the dehydrative elimination of secondary alcohols to produce alkenes.When the ester condensation of 4-phenylbutyric acid with cyclododecanol (5; see Fig. 2) was conducted in the presence of Tanabe catalyst (5 mol%) in heptane under reflux conditions (bath temperature 115 °C), a significant amount of the undesired cyclododecene (7) was produced along with cyclododecyl 4-phenylbutyrate (6) (Fig. 2, graph a). The use of dimesitylammonium triflate ([Mes2NH2]+[OTf]−) showed higher catalytic activity than [Ph2NH2]+[OTf]− and reduced the production of 7 (Fig. 2, graph a). Furthermore, the ester condensation catalyzed by 3 (5 mol%) proceeded more rapidly, and the production of 7 decreased (Fig. 2, graph a). The use of less-polar solvents such as heptane is important. The catalytic activities of 3 and 4 increased in such less-polar solvents, to produce esters in high yields. One of the limitations of the present method is the need for less-polar solvents: it is difficult to perform ester condensation of hydrophilic substrates that cannot dissolve in less-polar solvents.Typically, the ester condensations of 1:1 mixtures of carboxylic acids and alcohols are carried out in the presence of 3 or 4 (1–5 mol%) in heptane by heating at 80 °C without the removal of water. Sterically demanding alcohols are condensed to produce the corresponding esters in high yields. For example, when the ester condensation of 4-phenylbutyric acid with 6-undecanol (1.0 equiv.) was performed in the presence of 4 (5 mol%) in heptane under heating conditions without the removal of water, the corresponding ester was obtained in 88% yield along with 5-undecene (3%) (Fig. 4). In Box 1, a detailed protocol for this reaction is reported. Esterification with 1,2-diols proceeded well to give the corresponding diesters in high yields, while Lewis acidic metal salts were not suitable for use with these diols owing to tight chelation with metal ions13. For example, the condensation between cis-1,2-cyclohexanediol and 4-phenylbutyric acid with 4 gave the corresponding diester in 90% yield, while no esterification product was obtained by heating a mixture of 1,2-butanediol and 1-adamantanecarboxylic acid in toluene in the presence of HfCl4·(THF)2. In addition, we have been able to recover and reuse the bulky diarylammonium pentafluorobenzenesulfonate catalyst immobilized on a polystyrene support without any loss of catalytic activity more than 10 times7, 8. On the contrary, a polystyrene-supported diarylammonium triflate could not be prepared, as the polymer support decomposed with superacidic TfOH.Ester condensation reactions with more reactive primary alcohols proceeded even at ambient temperature (22 °C) without solvents. Several carboxylic acids were esterified with 1.1 equiv. of methanol in good yield in the presence of 3 (1 mol%). For example, when condensation between 4-phenylbutyric acid and methanol (1.1 equiv.) was carried out in the presence of 3 (1 mol%) without the removal of water for 24 h, the corresponding ester was obtained in 95% yield (Fig. 5). This reaction can be carried out by the same protocol as that of the reaction in Figure 4 but without the use of solvent (heptane) and heating. 1-Octanol was also reactive, albeit slightly less reactive than methanol8. Octyl methoxyacetate was obtained in 74% yield under the same conditions described in Figure 5.Synthesis of N-(2,6-diisopropylphenyl)-N-(2,4,6-mesityl)amine (2): Steps 1–7, 1 h; Step 8, 24–48 h; Step 9, 30 min; Step 10, 20 min; Steps 11–15, 2 h; Steps 16–18, 2 h; Steps 19 and 20; 1 hSynthesis of pentafluorobenzenesulfonic acid: Steps 22 and 23, 1 h; Step 24, ~12 h; Step 25, 30 min; Step 26, 30 min; Steps 27 and 28, ~14 hSynthesis of N-(2,6-diisopropylphenyl)-N-(2,4,6-mesityl)ammonium pentafluorobenzenesulfonate (4): Steps 29 and 30, 1.5 h; Step 31, 30 min; Step 32, 30 min; Step 5, ~12 hTroubleshooting advice can be found in Table 1.Typical isolated yield, ~70–95%. 1H NMR (300 MHz, CDCl3) δ 7.10 (s, 3H), 6.76 (s, 2H), 4.68 (br s, 1H), 3.12 (septet, J = 6.9 Hz, 2H), 2.22 (s, 3H), 1.95 (s, 6H), 1.11 (d, J = 6.9 Hz, 12H); 13C NMR (75 MHz, CDCl3) δ 143.3 (s, 1C), 140.4 (s, 1C), 139.1 (s, 1C), 130.0 (s, 2C), 129.0 (s, 1C), 126.3 (s, 2C), 124.2 (s, 2C), 123.2 (s, 2C), 27.9 (s, 2C), 23.4 (s, 4C), 20.4 (s, 1C), 19.3 (s, 2C); IR (KBr, cm−1) 1,484, 1,466, 1,442, 1,340, 1,270. HRMS (FAB) (m/z) [M + H+] calculated for C21H29N 295.2300, found 295.2308.N-(2,6-Diisopropylphenyl)-N-(2,4,6-mesityl)ammonium pentafluorobenzenesulfonate (4)Typical yield, >90%. 1H NMR (300 MHz, CDCl3) δ 7.36 (t, J = 7.8 Hz, 1H), 7.19 (d, J = 8.1 Hz, 2H), 6.73 (s, 2H), 3.11 (septet, J = 6.8 Hz, 2H), 2.20 (s, 3H), 2.15 (s, 6H), 1.07 (d, J = 6.6 Hz, 12H); 13C NMR (125 MHz, CDCl3) δ 143.7 (t, J = 7.8 Hz, 2C), 143.3 (s, 1C), 142.0 (d, J = 255 Hz, 1C), 137.7 (s, 1C), 137.2 (d, J = 252 Hz, 2C), 134.1 (s, 1C), 132.3 (s, 1C), 131.4 (s, 2C), 130.9 (s, 2C), 129.2 (s, 2C), 125.0 (s, 2C), 118.7 (s, 1C), 28.6 (s, 2C), 23.5 (s, 4C), 20.3 (s, 1C), 19.1 (s, 2C); 19F NMR (282 MHz, CDCl3) δ −138.3 (dd, J = 6.2, 21.2 Hz, 2F), −153.0 (t, J = 21.2 Hz, 1F), −162.4 (dt, J = 6.2, 21.2 Hz, 2F). IR (KBr, cm−1) 1,489, 1,247, 1,227, 1,115.
Co-reporter:Akira Sakakura, Mikimoto Katsukawa, Takaomi Hayashi and Kazuaki Ishihara
Green Chemistry 2007 vol. 9(Issue 11) pp:1166-1169
Publication Date(Web):06 Aug 2007
DOI:10.1039/B707974E
A metal-free phosphazenium cation-catalyzed direct dehydrative condensation of phosphoric acid with alcohols has been developed for the environmentally benign synthesis of phosphoric acid monoesters.
Co-reporter:Akira Sakakura;Rei Kondo;Shuhei Umemura
Advanced Synthesis & Catalysis 2007 Volume 349(Issue 10) pp:
Publication Date(Web):17 JUL 2007
DOI:10.1002/adsc.200700068
Bis(2-ethyl-8-quinolinolato)dioxomolybdenum(VI) (9) (1 mol %) shows remarkable catalytic activity for the dehydrative cyclization of cysteine-containing dipeptides 1 to give the corresponding thiazolines 2 with less than 6 % epimerization at the C2-exomethine position. For the dehydrative cyclization of threonine-containing dipeptides 4, 1 mol % of bis(2-phenyl-8-quinolinolato)dioxomolybdenum(VI) (10) gives the corresponding oxazolines 5 with retention of configuration at the 5-position.
Co-reporter:Akira Sakakura;Shuhei Umemura;Rei Kondo
Advanced Synthesis & Catalysis 2007 Volume 349(Issue 4-5) pp:
Publication Date(Web):20 MAR 2007
DOI:10.1002/adsc.200600550
The dehydrative cyclization of N-(o-hydroxybenzoyl)threonine derivative 1a is efficiently promoted by the combined use of molybdenum(VI) oxides and benzoic acids bearing electron-withdrawing substituents. In the presence of ammonium molybdate [(NH4)2MoO4, 10 mol %] and pentafluorobenzoic acid (C6F5CO2H; 10 mol %), dehydrative cyclization of 1a was conducted in toluene under azeotropic reflux conditions to give 2-(o-hydroxyphenyl)oxazoline 2a in 76 % yield. Furthermore, the first total synthesis of the antitumour substance BE-70016 was achieved using the catalytic dehydrative cyclization of 1a as a key reaction.
Co-reporter:Akira Sakakura Dr.;Mikimoto Katsukawa Dr.
Angewandte Chemie 2007 Volume 119(Issue 9) pp:
Publication Date(Web):17 JAN 2007
DOI:10.1002/ange.200604333
Dosierte Veresterung mit Rhenium: Oxorhenium(VII)-Komplexe katalysieren die selektive direkte Kondensation von Phosphorsäure mit einem Alkohol zum Phosphorsäuremonoester im 2- bis 100-mmol-Maßstab (siehe Schema). Diese Methode sollte für die industrielle Synthese von Phosphorsäuremonoestern von Bedeutung sein.
Co-reporter:Shoko Nakagawa;Akira Sakakura Dr.;Hitoshi Watanabe Dr.
Chemistry – An Asian Journal 2007 Volume 2(Issue 4) pp:477-483
Publication Date(Web):15 MAR 2007
DOI:10.1002/asia.200600380
Bulky diarylammonium pentafluorobenzenesulfonates effectively promote dehydration reactions, such as condensation reactions to give esters and the dehydrative cyclization of 1,3,5-triketones. In particular, N-(2,6-diphenylphenyl)-N-mesitylammonium pentafluorobenzenesulfonate shows much higher catalytic activity than C6F5SO3H under reaction conditions without the removal of generated water, even though the former is a weaker acid. Its crystallization gives an aggregated cyclic ion pair, which is composed of two diarylammonium cations, four pentafluorobenzenesulfonate anions, and two oxonium cations. This ion pair is strongly stabilized by four intermolecular and two intramolecular π–π attractive interactions and 10 hydrogen bonds. The extremely high catalytic activity of N-(2,6-diphenylphenyl)-N-mesitylammonium pentafluorobenzenesulfonate in the dehydration reactions may be ascribed to the local hydrophobic environment of the tightly aggregated ammonium salts.
Co-reporter:Akira Sakakura,
Atsushi Ukai
and
Kazuaki Ishihara
Nature 2007 445(7130) pp:900
Publication Date(Web):2007-02-22
DOI:10.1038/nature05553
Polycyclic bio-active natural products that contain halogen atoms have been isolated from a number of different marine organisms1. The biosynthesis of these natural products appears to be initiated by an electrophilic halogenation reaction at a carbon–carbon double bond2, 3, 4 via a mechanism that is similar to a proton-induced olefin polycyclization5, 6, 7, 8. Enzymes such as haloperoxidases generate an electrophilic halonium ion (or its equivalent), which reacts with the terminal carbon–carbon double bond of the polyprenoid, enantioselectively inducing a cyclization reaction that produces a halogenated polycyclic terpenoid. Use of an enantioselective halocyclization reaction is one possible way to chemically synthesize these halogenated cyclic terpenoids; although several brominated cyclic terpenoids have been synthesized via a diastereoselective halocyclization reaction that uses stoichiometric quantities of a brominating reagent9, 10, 11, 12, the enantioselective halocyclization of isoprenoids induced by a chiral promoter has not yet been reported. Here we report the enantioselective halocyclization of simple polyprenoids using a nucleophilic promoter. Achiral nucleophilic phosphorus compounds are able to promote the diastereoselective halocyclization reaction to give a halogenated cyclic product in excellent yields. Moreover, chiral phosphoramidites promote the enantioselective halocyclization of simple polyprenoids with N-iodosuccinimide to give iodinated cyclic products in up to 99% enantiomeric excess and diastereomeric excess. To the best of our knowledge, this is the first successful example of the enantioselective halopolycyclization of polyprenoids.
Co-reporter:Akira Sakakura Dr.;Mikimoto Katsukawa Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 9) pp:
Publication Date(Web):17 JAN 2007
DOI:10.1002/anie.200604333
Come together … over Re: Oxorhenium(VII) complexes catalyze the direct condensation of phosphoric acid with an alcohol to selectively give the corresponding phosphoric acid monoester on a 2–100-mmol scale (see scheme). This method should be useful for the industrially important synthesis of phosphoric acid monoesters.
Co-reporter:Yuka Nakamura;Toshikatsu Maki;Xiaowei Wang;Hisashi Yamamoto
Advanced Synthesis & Catalysis 2006 Volume 348(Issue 12-13) pp:
Publication Date(Web):11 AUG 2006
DOI:10.1002/adsc.200606126
N-(Polystyrylbutyl)pyridinium triflylimide is prepared by a coupling reaction of commercially available 4-bromobutyl-polystyrene with pyridine and a subsequent anion exchange reaction with lithium triflylimide. This new polystyrene-bound pyridinium salt is useful as a solid support to immobilize a zirconium(IV)–iron(III) binary metal complex, which is a homogeneous catalyst for the dehydrative ester condensation of an equimolar mixture of carboxylic acids and alcohols. The immobilized zirconium(IV)–iron(III) complex is easily recovered by filtration after complete esterification, and is reusable without any loss of activity.
Co-reporter:Akira Sakakura;Kenji Suzuki
Advanced Synthesis & Catalysis 2006 Volume 348(Issue 16-17) pp:
Publication Date(Web):27 NOV 2006
DOI:10.1002/adsc.200600322
A diammonium salt of chiral 1,1′-binaphthyl-2,2′-diamine (2a) and trifluoromethanesulfonimide (Tf2NH) shows excellent catalytic activity and enantioselectivity for the Diels–Alder reaction of α-acyloxyacroleins. For example, in the presence of 5 mol % of 2a and 9.5 mol % of Tf2NH, the Diels–Alder reaction of α-(cyclohexanecarbonyloxy)acrolein with cyclopentadiene proceeded in EtCN at −75 °C to give the adducts in 88 % yield with 92 % exo and 91 % ee. The electron-donating property of the acyl group of the α-acyloxyacroleins increases the enantioselectivity due to the formation of strong intramolecular hydrogen bonding of the acyl group with a proton of the ammonium group in the transition state. This catalyst can be easily prepared in situ by mixing the commercially available chiral diamine and Tf2NH.
Co-reporter:Yukihiro Hiraiwa;Hisashi Yamamoto
European Journal of Organic Chemistry 2006 Volume 2006(Issue 8) pp:
Publication Date(Web):9 FEB 2006
DOI:10.1002/ejoc.200500845
The silyl Lewis acid induced Mukaiyama aldol reaction proceeds through each catalytic cycle under the influence of their conjugate bases; there is an especially significant difference between the low nucleophilic conjugate bases, –NTf2 and –CTf3, and the relatively high nucleophilic –OTf. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Co-reporter:Manabu Hatano;Takashi Miyamoto
Advanced Synthesis & Catalysis 2005 Volume 347(Issue 11-13) pp:
Publication Date(Web):19 OCT 2005
DOI:10.1002/adsc.200505221
The enantioselective addition of organozinc reagents to aromatic and aliphatic aldehydes 1 gives secondary alcohols 2 with excellent enantioselectivities in high yields through the catalytic use of (R)-3,3′-bis(diphenylphosphinoyl)-BINOL (3) or (R)-3,3′-bis(diphenylthiophosphinoyl)-BINOL (4) without Ti(IV) complexes. The coordination of the O or S atom of a (thio)phosphinoyl group bearing a BINOL backbone to organozinc reagents can efficiently increase the nucleophilicity of the organozinc reagents.
Co-reporter:Muhammet Uyanik, Kazuaki Ishihara, Hisashi Yamamoto
Bioorganic & Medicinal Chemistry 2005 Volume 13(Issue 17) pp:5055-5065
Publication Date(Web):1 September 2005
DOI:10.1016/j.bmc.2005.04.029
Asymmetric total syntheses of acid-sensitive (−)- and (+)-caparrapi oxides (1) and (+)-8-epicaparrapi oxide (2) from farnesol (10) are achieved using Sharpless–Katsuki epoxidation and Lewis acid-assisted chiral Brønsted acid (chiral LBA)-induced polyene cyclization as key steps. The relative configuration of (+)-dysifragin (4) is determined by a single-crystal X-ray diffraction and its total synthesis is accomplished by the diastereoselective epoxidation of (+)-1. Furthermore, (−)-1 can be directly synthesized from (S)-nerolidol (3) and (R)-LBA with 88% ds by reagent control, which overcame substrate control, while (−)-2 is obtained from (R)-3 and (R)-LBA with >99% ds by the double asymmetric induction.
Co-reporter:Aiko Hasegawa Dr.;Hisashi Yamamoto Dr.
Angewandte Chemie 2003 Volume 115(Issue 46) pp:
Publication Date(Web):25 NOV 2003
DOI:10.1002/ange.200352382
Säure und Vitamin E: Trimethylsilyl-Super-Lewis-Säuren wie [C6F5C(Tf)2]− Me3Si+ sind sehr aktive und effektive Katalysatoren für die regioselektive Kondensation von Trimethylhydrochinon mit Isophthyol zur Herstellung von (±)-α-Tocopherol (Vitamin E; siehe Schema, Tf=CF3SO2).
Co-reporter:Aiko Hasegawa Dr.;Hisashi Yamamoto Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 46) pp:
Publication Date(Web):25 NOV 2003
DOI:10.1002/anie.200352382
Acid and vitamin E: Trimethylsilyl super Lewis acids, such as [C6F5C(Tf)2]− Me3Si+ and Tf3CSiMe3, are extremely active and highly effective catalysts for the regioselective condensation of trimethylhydroquinone with isophthyol to afford (±)-α-tocopherol (vitamin E; see scheme, Tf=CF3SO2.
Co-reporter:Yoshiki Koshikari ; Akira Sakakura
Organic Letters () pp:
Publication Date(Web):May 30, 2012
DOI:10.1021/ol301290c
Reverse micelle-type N,N-diarylammonium pyrosulfate (3–5 mol %) efficiently catalyzes the hydrolysis of esters (up to 100 mmol scale) under organic solvent-free conditions. The present method is successfully applied to the hydrolysis of various esters without the decomposition of the base-sensitive moieties and without any loss of optical purity for α-heterosubstituted carboxylic acids.
Co-reporter:Akira Sakakura and Kazuaki Ishihara
Chemical Society Reviews 2011 - vol. 40(Issue 1) pp:NaN172-172
Publication Date(Web):2010/09/06
DOI:10.1039/B924478F
The rational design of small but highly functional artificial catalysts is very important for practical organic synthesis. Asymmetric Lewis acid catalyses with non-covalent secondary interactions have been developed for enantioselective reactions. This tutorial review describes the concept, design and examples of asymmetric Cu(II) catalyses for cycloaddition reactions based on intramolecular π–cation or n–cation interactions between the copper(II) cation and auxiliary Lewis basic sites of the chiral ligands.
Co-reporter:Akira Sakakura, Shuhei Umemura and Kazuaki Ishihara
Chemical Communications 2008(Issue 30) pp:NaN3563-3563
Publication Date(Web):2008/06/06
DOI:10.1039/B805880F
Efficient total syntheses of fluvibactin and vibriobactin have been achieved via molybdenum(VI) oxide-catalyzed dehydrative cyclization, Sb(OEt)3-catalyzed ester–amide transformation, and WSCI and HOAt-promoted dehydrative amide formation.
Co-reporter:Manabu Hatano, Tomokazu Mizuno and Kazuaki Ishihara
Chemical Communications 2010 - vol. 46(Issue 30) pp:NaN5445-5445
Publication Date(Web):2010/06/29
DOI:10.1039/C0CC01301C
A highly practical, catalytic enantioselective 2°-alkyl addition to aldehydes and ketones was developed. Chiral phosphoramide ligand (1) with salt-free and solvent-free di(2°-alkyl)zinc reagents prepared from (2°-alkyl)MgCl was essential.
Co-reporter:Manabu Hatano and Kazuaki Ishihara
Chemical Communications 2012 - vol. 48(Issue 36) pp:NaN4283-4283
Publication Date(Web):2012/03/22
DOI:10.1039/C2CC00046F
The potential of supramolecular catalysts to realize anomalous regio- and/or stereoselectivity in organic synthesis is highly attractive. To date, there have been a few examples of non-polymeric and non-covalent chiral supramolecular catalysts that induce practical enantioselectivity. In this regard, a metal–organic framework (MOF) may be one of the most important techniques for constructing conformationally rigid supramolecular catalysts. However, it is not easy to use the MOF technique to fine-tune a much more precise cage in catalysts for anomalous purposes. To establish high catalytic activity with anomalous regio- and/or stereoselectivity, in principle, an artificial cage should be conformationally flexible, like an active pocket in an enzyme with an induced-fit function. In this feature article, we focus on the anomalous endo/exo-selective Diels–Alder reaction, and overview the development of the successive catalysts including our recent highly active, conformationally flexible, and chiral supramolecular catalysts. The evolution from ‘ready-made’ single-molecule catalysts to ‘tailor-made’ supramolecular catalysts could offer not only high enantioselectivity but also high anomalous endo/exo-selectivities due to substrate-specific characteristics, as with enzymes.
Co-reporter:Manabu Hatano, Orie Ito, Shinji Suzuki and Kazuaki Ishihara
Chemical Communications 2010 - vol. 46(Issue 15) pp:NaN2676-2676
Publication Date(Web):2010/02/09
DOI:10.1039/B926243A
Highly efficient alkylations and arylations of ketones with Grignard reagents (RMgBr and RMgI) have been developed using catalytic ZnCl2, Me3SiCH2MgCl, and LiCl. Tertiary alcohols were obtained in high yields with high chemoselectivities, while minimizing undesired side products produced by reduction and enolization.
Co-reporter:Kazuaki Ishihara, Hiroki Yamada and Matsujiro Akakura
Chemical Communications 2014 - vol. 50(Issue 48) pp:NaN6360-6360
Publication Date(Web):2014/04/11
DOI:10.1039/C4CC01445F
The first enantioselective Diels–Alder reaction of 1,2-dihydropyridines with α-acyloxyacroleins catalyzed by a chiral primary ammonium salt has been developed and it offers more efficient routes to key synthetic intermediates of alkaloids, for which the direct preparations were unavailable before. The asymmetric induction can be understood through the optimized geometry of an iminium salt aqua complex derived from the catalyst and the dienophile.
Co-reporter:Manabu Hatano, Riku Gouzu, Tomokazu Mizuno, Hitoshi Abe, Toshihide Yamada and Kazuaki Ishihara
Catalysis Science & Technology (2011-Present) 2011 - vol. 1(Issue 7) pp:NaN1158-1158
Publication Date(Web):2011/07/01
DOI:10.1039/C1CY00108F
A highly practical, catalytic enantioselective alkyl and aryl addition to aldehydes and ketones with organozinc reagents, which were prepared in situ from commercially available Grignard reagents or arylboronic acids, was developed. A chiral phosphoramide ligand was essential for promoting the addition reactions in high yields with high enantioselectivities.
Co-reporter:Yasuhiro Sawamura, Yoshihiro Ogura, Hidefumi Nakatsuji, Akira Sakakura and Kazuaki Ishihara
Chemical Communications 2016 - vol. 52(Issue 36) pp:NaN6071-6071
Publication Date(Web):2016/03/18
DOI:10.1039/C6CC00229C
Chiral phosphite–urea bifunctional catalysts have been developed for the enantioselective bromocyclization of 2-geranylphenols with N-bromophthalimide (NBP) for the first time. The chiral triaryl phosphite moiety activates NBP to generate a bromophosphonium ion. On the other hand, the urea moiety interacts with a hydroxyl group of the substrate through hydrogen bonding interactions. Enantioselectivity is effectively induced through two-point attractive interactions between the catalyst and the substrate.
Co-reporter:Manabu Hatano, Takuya Ozaki, Yoshihiro Sugiura and Kazuaki Ishihara
Chemical Communications 2012 - vol. 48(Issue 41) pp:NaN4988-4988
Publication Date(Web):2012/04/16
DOI:10.1039/C2CC31530K
A highly effective catalytic enantioselective direct aminal synthesis was developed. Chiral ammonium 1,1′-binaphthyl-2,2′-disulfonates, which were prepared in situ from (R)-BINSA and achiral amines, promoted the enantioselective addition of primary amides to aromatic aldimines.
Co-reporter:Manabu Hatano and Kazuaki Ishihara
Chemical Communications 2013 - vol. 49(Issue 20) pp:NaN1997-1997
Publication Date(Web):2013/01/17
DOI:10.1039/C2CC38204K
A facile, atom-economical, and chemoselective esterification is crucial in modern organic synthesis, particularly in the areas of pharmaceutical, polymer, and material science. However, a truly practical catalytic transesterification of carboxylic esters with various alcohols has not yet been well established, since, with many conventional catalysts, the substrates are limited to 1°- and cyclic 2°-alcohols. In sharp contrast, if we take advantage of the high catalytic activities of La(Oi-Pr)3, La(OTf)3, and La(NO3)3 as ligand-free catalysts, ligand-assisted or additive-enhanced lanthanum(III) catalysts can be highly effective acid–base combined catalysts in transesterification. A highly active dinuclear La(III) catalyst, which is prepared in situ from lanthanum(III) isopropoxide and 2-(2-methoxyethoxy)ethanol, is effective for the practical transesterification of methyl carboxylates, ethyl acetate, weakly reactive dimethyl carbonate, and much less-reactive methyl carbamates with 1°-, 2°-, and 3°-alcohols. As the second generation, nearly neutral “lanthanum(III) nitrate alkoxide”, namely La(OR)m(NO3)3−m, has been developed. This catalyst is prepared in situ from inexpensive, stable, low-toxic lanthanum(III) nitrate hydrate and methyltrioctylphosphonium methyl carbonate, and is highly useful in the non-epimerized transesterification of α-substituted chiral carboxylic esters, even under azeotropic reflux conditions. In these practical La(III)-catalyzed transesterifications, colorless esters can be obtained in small- to large-scale synthesis without the need for inconvenient work-up or careful purification procedures.
Co-reporter:Manabu Hatano, Sho Kamiya and Kazuaki Ishihara
Chemical Communications 2012 - vol. 48(Issue 76) pp:NaN9467-9467
Publication Date(Web):2012/08/08
DOI:10.1039/C2CC34987F
In situ generated lanthanum(III) nitrate alkoxide is a highly active and nearly neutral transesterification catalyst, which can promote non-epimerized transesterification of α-substituted chiral carboxylic esters under reflux conditions.
Co-reporter:Muhammet Uyanik and Kazuaki Ishihara
Chemical Communications 2009(Issue 16) pp:NaN2099-2099
Publication Date(Web):2009/03/06
DOI:10.1039/B823399C
Over the past two decades there has been a dramatic increse in the use of hypervalent iodine compounds in synthetic organic chemistry due to their mild and selective oxidizing properties. Hypervalent iodine compounds catalyze various oxidation reactions such as the oxidation of alcohols, α-oxidation of ketones, oxidative spirocyclization of phenols, etc. Very recently, we found that 2-iodoxybenzensulfonic acid (IBS, 7a), which was generated from 2-iodobenzenesulfonic acidin situ, is an extremely active and mild catalyst for the highly chemoselective oxidation of various alcohols with powdered Oxone® to carbonyl compounds such as aldehydes, carboxylic acids, ketones and cycloalkenones under non-aqueous conditions. In this review, we focus on the design of hypervalent iodine catalysts and review the discovery and development of the oxidation of alcohols with the stoichiometric or catalytic use of hypervalent iodine compounds.
Co-reporter:Kazuaki Ishihara and Yanhui Lu
Chemical Science (2010-Present) 2016 - vol. 7(Issue 2) pp:NaN1280-1280
Publication Date(Web):2015/11/06
DOI:10.1039/C5SC03761A
Arylboronic acid and 4-(N,N-dimethylamino)pyridine N-oxide (DMAPO) cooperatively catalyse the dehydrative condensation reaction between carboxylic acids and amines to give the corresponding amides under azeotropic reflux conditions. This cooperative use is much more effective than their individual use as catalysts, and chemoselectively promotes the amide condensation of (poly)conjugated carboxylic acids. The present method is practical and scalable, and has been applied to the synthesis of sitagliptin and a drug candidate.
Co-reporter:Yasuhiro Sawamura, Hidefumi Nakatsuji, Akira Sakakura and Kazuaki Ishihara
Chemical Science (2010-Present) 2013 - vol. 4(Issue 11) pp:NaN4186-4186
Publication Date(Web):2013/08/02
DOI:10.1039/C3SC51432C
Nucleophilic phosphite–urea cooperative high-turnover catalysts have been designed for the highly selective bromocyclization of homogeranylarenes. The introduction of a urea moiety and bulky aryl groups in the catalyst inhibits decomposition of the catalyst and the generation of byproducts. Only 0.5 mol% of the catalyst successfully promotes the bromocyclization of 4-homogeranyltoluene to give the desired product in 96% yield.















![1,3-Dioxane-4,6-dione, 2,2-dimethyl-5-[(2E)-3-phenyl-2-propenylidene]-](http://img.cochemist.com/ccimg/548800/548765-13-5.png)
![1,3-Dioxane-4,6-dione, 2,2-dimethyl-5-[(2E)-3-phenyl-2-propenylidene]-](http://img.cochemist.com/ccimg/548800/548765-13-5_b.png)
![[1,1'-Biphenyl]-4-ol, 3',5'-bis(trifluoromethyl)-](http://img.cochemist.com/ccimg/462700/462660-27-1.png)
![[1,1'-Biphenyl]-4-ol, 3',5'-bis(trifluoromethyl)-](http://img.cochemist.com/ccimg/462700/462660-27-1_b.png)
![[1,1':3',1''-Terphenyl]-2'-amine, 3,3'',5,5''-tetramethyl-](/data/chemimg/102900/340187-65-7.png)
![[1,1':3',1''-Terphenyl]-2'-amine, 3,3'',5,5''-tetramethyl-](/data/chemimg/102900/340187-65-7_b.png)




![Phenol, 2-[(dimethylphenylsilyl)methyl]-](http://img.cochemist.com/ccimg/120800/120743-97-7.png)
![Phenol, 2-[(dimethylphenylsilyl)methyl]-](http://img.cochemist.com/ccimg/120800/120743-97-7_b.png)


![[1,1':3',1''-Terphenyl]-2'-amine](http://img.cochemist.com/ccimg/87700/87666-57-7.png)
![[1,1':3',1''-Terphenyl]-2'-amine](http://img.cochemist.com/ccimg/87700/87666-57-7_b.png)




![[(E)-2-CYCLOPENTYLVINYL]BORONIC ACID](http://img.cochemist.com/ccimg/72900/72836-73-8.png)
![[(E)-2-CYCLOPENTYLVINYL]BORONIC ACID](http://img.cochemist.com/ccimg/72900/72836-73-8_b.png)


![ACETIC ACID, [3,5-BIS(1,1-DIMETHYLETHYL)-2-HYDROXYPHENOXY]-, ETHYL ESTER](http://img.cochemist.com/ccimg/65700/65659-40-7.png)
![ACETIC ACID, [3,5-BIS(1,1-DIMETHYLETHYL)-2-HYDROXYPHENOXY]-, ETHYL ESTER](http://img.cochemist.com/ccimg/65700/65659-40-7_b.png)











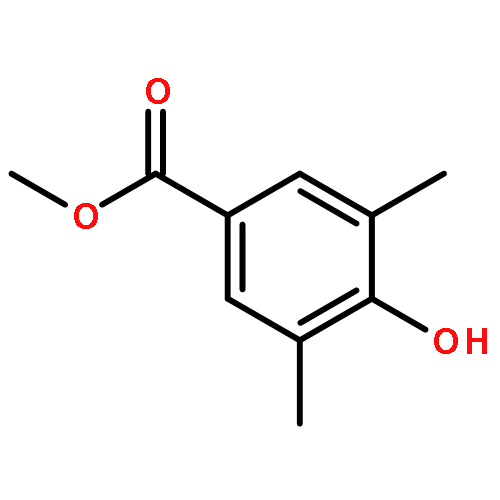
![1H-Naphtho[2,1-b]pyran, 2,3-dihydro-3-phenyl-](http://img.cochemist.com/ccimg/13300/13255-85-1.png)
![1H-Naphtho[2,1-b]pyran, 2,3-dihydro-3-phenyl-](http://img.cochemist.com/ccimg/13300/13255-85-1_b.png)





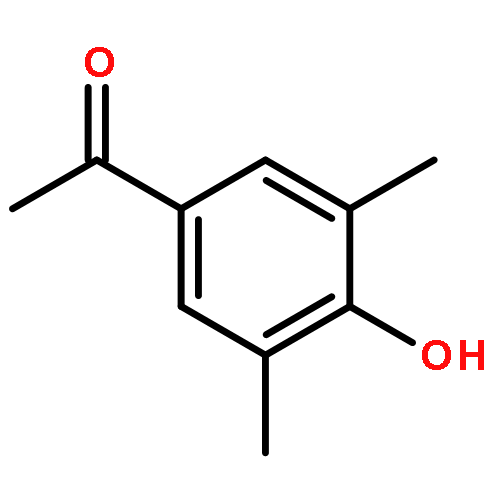


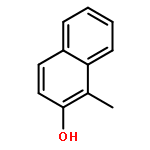
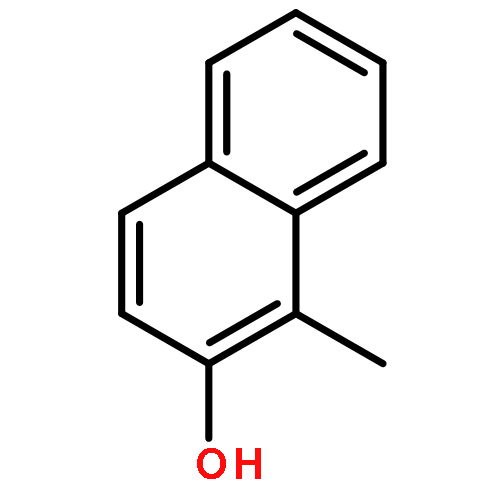



















![3-Cyclobutene-1,2-dione, 3-[[3,5-bis(trifluoromethyl)phenyl]amino]-4-methoxy-](/data/chemimg/144700/1223105-85-8.png)
![3-Cyclobutene-1,2-dione, 3-[[3,5-bis(trifluoromethyl)phenyl]amino]-4-methoxy-](/data/chemimg/144700/1223105-85-8_b.png)





















![2-Cyclohexene-1-carbonitrile, 2-bromo-1-[(trimethylsilyl)oxy]-, (1R)-](http://img.cochemist.com/ccimg/861000/860995-00-2.png)
![2-Cyclohexene-1-carbonitrile, 2-bromo-1-[(trimethylsilyl)oxy]-, (1R)-](http://img.cochemist.com/ccimg/861000/860995-00-2_b.png)
![N-[3,5-Bis(trifluoromethyl)phenyl]-N-[(8a,9S)-6-methoxy-9-cinchonanyl]thiourea](/data/chemimg/103500/852913-16-7.png)
![N-[3,5-Bis(trifluoromethyl)phenyl]-N-[(8a,9S)-6-methoxy-9-cinchonanyl]thiourea](/data/chemimg/103500/852913-16-7_b.png)
![[1,1'-Binaphthalene]-2,2'-diol,3,3'-bis[3,5-bis(trifluoromethyl)phenyl]-, (1R)-](http://img.cochemist.com/ccimg/756500/756491-54-0.png)
![[1,1'-Binaphthalene]-2,2'-diol,3,3'-bis[3,5-bis(trifluoromethyl)phenyl]-, (1R)-](http://img.cochemist.com/ccimg/756500/756491-54-0_b.png)










![Acetic acid, [(4-methoxyphenyl)imino]-, 1-methylethyl ester, (2E)-](http://img.cochemist.com/ccimg/350100/350032-06-3.png)
![Acetic acid, [(4-methoxyphenyl)imino]-, 1-methylethyl ester, (2E)-](http://img.cochemist.com/ccimg/350100/350032-06-3_b.png)






![Bicyclo[2.2.2]oct-5-ene-2-carboxaldehyde, 2-bromo-, (1S,2R,4S)-](http://img.cochemist.com/ccimg/231000/230952-12-2.png)
![Bicyclo[2.2.2]oct-5-ene-2-carboxaldehyde, 2-bromo-, (1S,2R,4S)-](http://img.cochemist.com/ccimg/231000/230952-12-2_b.png)





































![[5-(HYDROXYMETHYL)-4-METHOXY-3-METHYL-2-PYRIDINYL]METHYL ACETATE](http://img.cochemist.com/ccimg/76300/76228-15-4.png)
![[5-(HYDROXYMETHYL)-4-METHOXY-3-METHYL-2-PYRIDINYL]METHYL ACETATE](http://img.cochemist.com/ccimg/76300/76228-15-4_b.png)
![[1,1'-Binaphthalene]-2,2'-diol, 3,3'-diphenyl-, (1R)-](http://img.cochemist.com/ccimg/75700/75684-93-4.png)
![[1,1'-Binaphthalene]-2,2'-diol, 3,3'-diphenyl-, (1R)-](http://img.cochemist.com/ccimg/75700/75684-93-4_b.png)


![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-methyl-, (1S,2R,4S)-](http://img.cochemist.com/ccimg/72300/72203-36-2.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-methyl-, (1S,2R,4S)-](http://img.cochemist.com/ccimg/72300/72203-36-2_b.png)























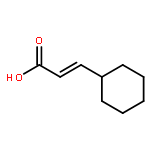











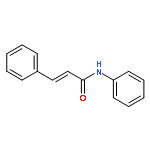
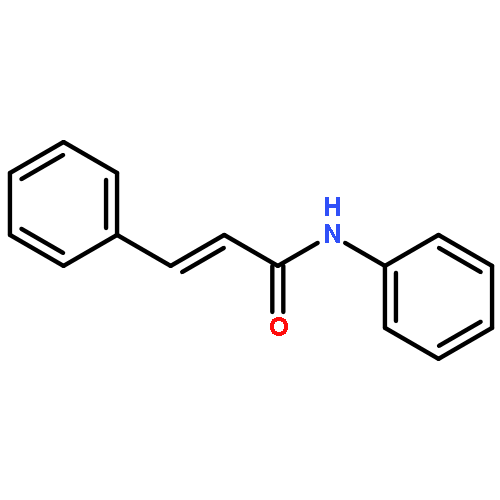


![Benzene, [(2E)-3,7-dimethyl-2,6-octadien-1-yl]-](/data/chemimg/1790100/21488-84-6.png)
![Benzene, [(2E)-3,7-dimethyl-2,6-octadien-1-yl]-](/data/chemimg/1790100/21488-84-6_b.png)
![1-Naphthalenecarboxamide, N-[(1S)-1-phenylethyl]-](http://img.cochemist.com/ccimg/19200/19141-53-8.png)
![1-Naphthalenecarboxamide, N-[(1S)-1-phenylethyl]-](http://img.cochemist.com/ccimg/19200/19141-53-8_b.png)




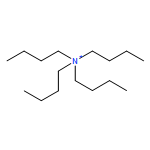








![Phenol, 2-[(2E)-3,7-dimethyl-2,6-octadien-1-yl]-](/data/chemimg/1648300/10232-02-7.png)
![Phenol, 2-[(2E)-3,7-dimethyl-2,6-octadien-1-yl]-](/data/chemimg/1648300/10232-02-7_b.png)


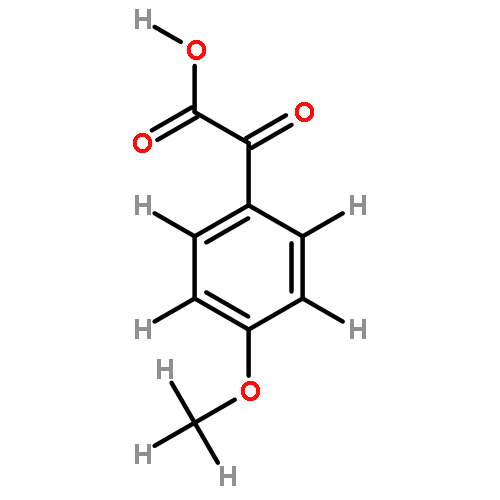
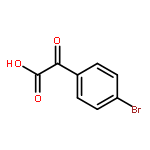
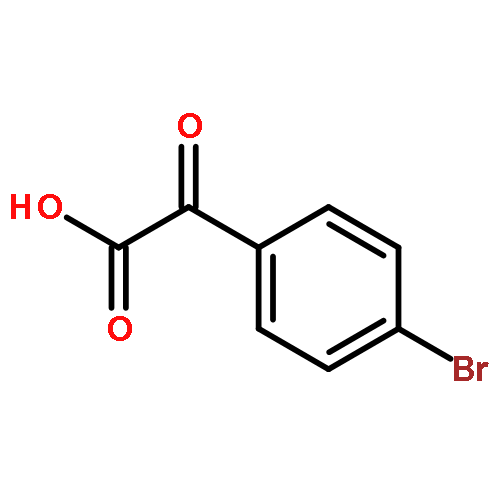



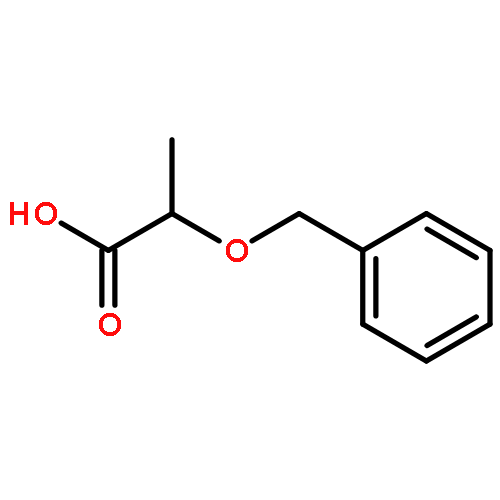
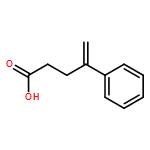
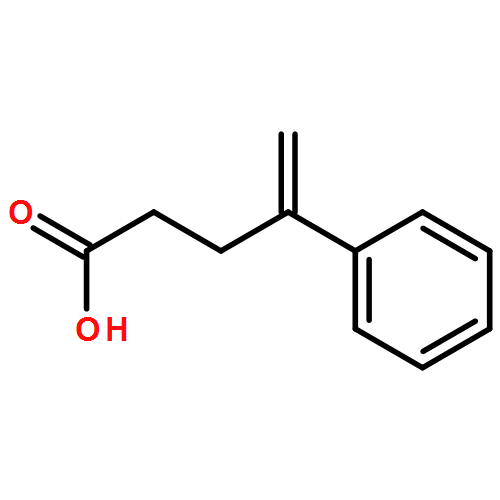


![1-Oxaspiro[4.5]decane-2,6-dione](http://img.cochemist.com/ccimg/4400/4361-37-9.png)
![1-Oxaspiro[4.5]decane-2,6-dione](http://img.cochemist.com/ccimg/4400/4361-37-9_b.png)
![2-OXASPIRO[5.5]UNDECAN-3-ONE](http://img.cochemist.com/ccimg/4400/4361-36-8.png)
![2-OXASPIRO[5.5]UNDECAN-3-ONE](http://img.cochemist.com/ccimg/4400/4361-36-8_b.png)

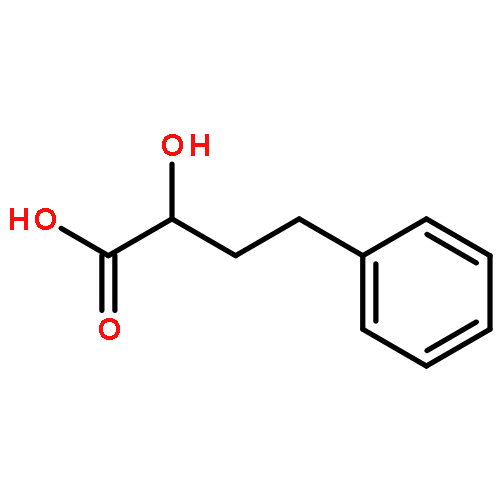


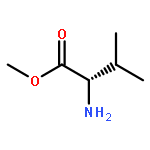
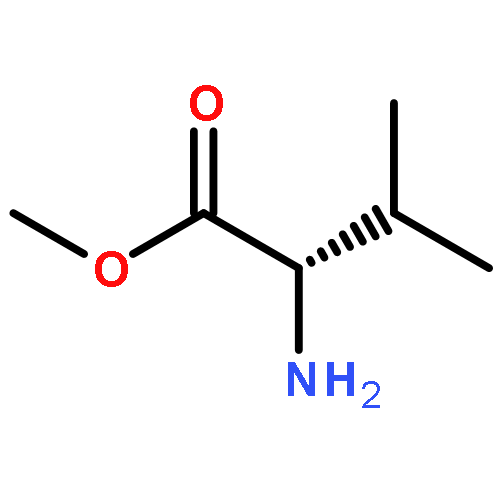











![N-[3-(1-HYDROXYETHYL)PHENYL]-3-METHYLBENZAMIDE](http://img.cochemist.com/ccimg/400/313-50-8.png)
![N-[3-(1-HYDROXYETHYL)PHENYL]-3-METHYLBENZAMIDE](http://img.cochemist.com/ccimg/400/313-50-8_b.png)
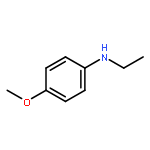
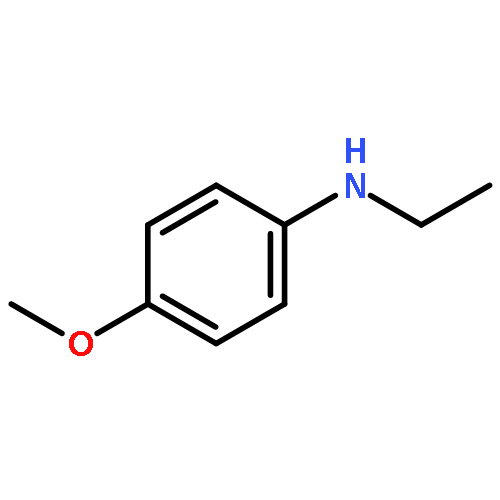


![Phenol, 4-bromo-2-[(2E)-3,7-dimethyl-2,6-octadienyl]-](http://img.cochemist.com/ccimg/746700/746621-28-3.png)
![Phenol, 4-bromo-2-[(2E)-3,7-dimethyl-2,6-octadienyl]-](http://img.cochemist.com/ccimg/746700/746621-28-3_b.png)
![BICYCLO[2.2.2]OCT-5-ENE-2-CARBOXALDEHYDE, 2-METHYL-, (1S,2S,4S)- (9CI)](http://img.cochemist.com/ccimg/426900/426841-45-4.png)
![BICYCLO[2.2.2]OCT-5-ENE-2-CARBOXALDEHYDE, 2-METHYL-, (1S,2S,4S)- (9CI)](http://img.cochemist.com/ccimg/426900/426841-45-4_b.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-ethyl-, (1R,2S,4R)-](http://img.cochemist.com/ccimg/374700/374633-09-7.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-ethyl-, (1R,2S,4R)-](http://img.cochemist.com/ccimg/374700/374633-09-7_b.png)


![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-ethyl-, (1S,2S,4S)- (9CI)](http://img.cochemist.com/ccimg/174800/174757-35-8.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-ethyl-, (1S,2S,4S)- (9CI)](http://img.cochemist.com/ccimg/174800/174757-35-8_b.png)


![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, (1R,2S,4R)-](http://img.cochemist.com/ccimg/96500/96443-41-3.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, (1R,2S,4R)-](http://img.cochemist.com/ccimg/96500/96443-41-3_b.png)





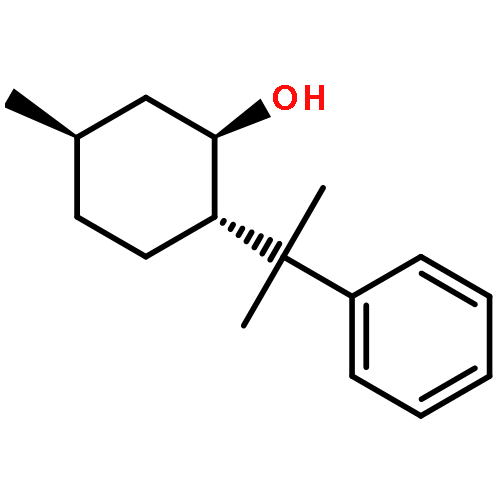
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, (1S,2S,4S)-](http://img.cochemist.com/ccimg/58100/58001-94-8.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, (1S,2S,4S)-](http://img.cochemist.com/ccimg/58100/58001-94-8_b.png)


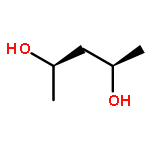
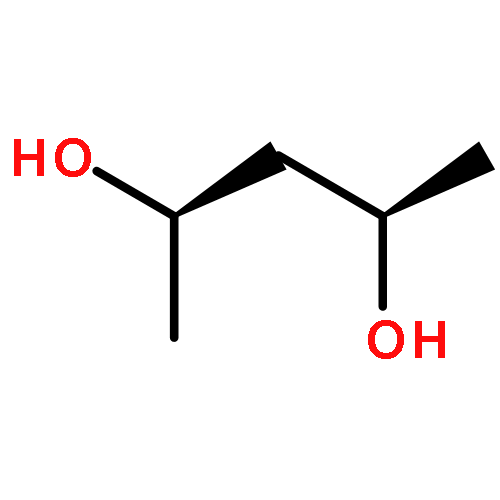




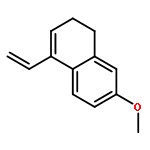
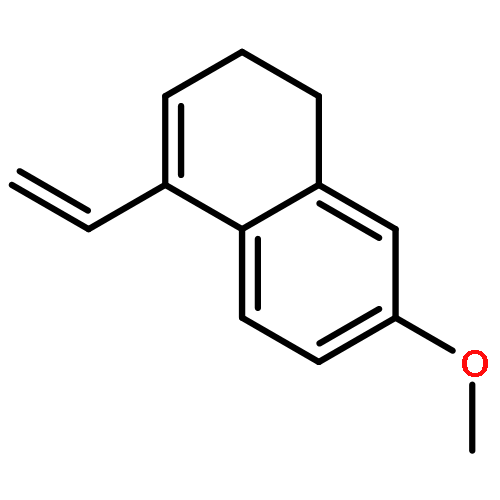

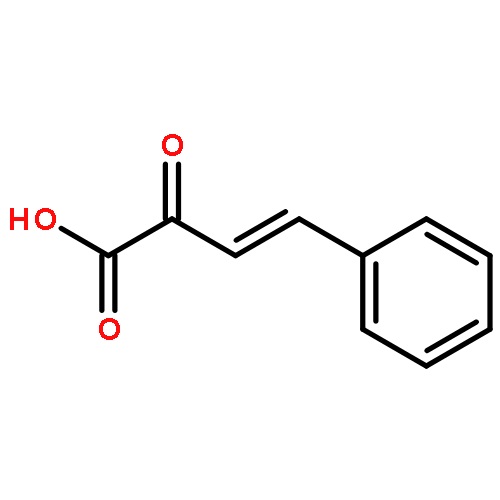
![Urea, N-[3,5-bis(trifluoromethyl)phenyl]-N'-phenyl-](http://img.cochemist.com/ccimg/1700/1644-96-8.png)
![Urea, N-[3,5-bis(trifluoromethyl)phenyl]-N'-phenyl-](http://img.cochemist.com/ccimg/1700/1644-96-8_b.png)


![BICYCLO[2.2.1]HEPT-5-ENE-2-CARBOXALDEHYDE, 2-BROMO-, (1S,2R,4S)-](http://img.cochemist.com/ccimg/151300/151282-94-9.png)
![BICYCLO[2.2.1]HEPT-5-ENE-2-CARBOXALDEHYDE, 2-BROMO-, (1S,2R,4S)-](http://img.cochemist.com/ccimg/151300/151282-94-9_b.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-methyl-, (1S,2S,4S)-](http://img.cochemist.com/ccimg/142600/142506-91-0.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-methyl-, (1S,2S,4S)-](http://img.cochemist.com/ccimg/142600/142506-91-0_b.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-bromo-, (1R,2R,4R)-](http://img.cochemist.com/ccimg/136900/136891-49-1.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-bromo-, (1R,2R,4R)-](http://img.cochemist.com/ccimg/136900/136891-49-1_b.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-methyl-, (1R,2S,4R)-](http://img.cochemist.com/ccimg/72300/72204-08-1.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-methyl-, (1R,2S,4R)-](http://img.cochemist.com/ccimg/72300/72204-08-1_b.png)

{kind=link}