Co-reporter:Kun Li, Lei Li, Shuxiang Wang, Xiaojing Li, Tianyi Ma, Dan Liu, Yongkui Jing, Linxiang Zhao
European Journal of Medicinal Chemistry 2017 Volume 126(Volume 126) pp:
Publication Date(Web):27 January 2017
DOI:10.1016/j.ejmech.2016.09.089
•Novel AKBA derivatives were synthesized as anti-prostate cancer agents.•These compounds showed improved anti-proliferative activity over AKBA.•8f caused cell cycle arrest in G2/M and induced apoptosis.•8f decreased the levels of anti-apoptosis and cell cycle regulating protein.•These compounds represented a novel class of Pin1 inhibitors.A series of novel acetyl-11-keto-β-boswellic acid (AKBA) derivatives with a different electron-withdrawing group on ring A and a nitrogen heterocycle at C-24 were designed and synthesized. These semi-synthetic compounds showed improved anti-proliferative activity against prostate cancer cells over AKBA. Compound 8f bearing 2-cyano-3,11-dioxo moiety and piperazine was the most potent to inhibit growth of prostate cancer PC-3 (IC50 = 0.04 μM) and LNCaP (IC50 = 0.27 μM) cell lines. 8f caused cell cycle arrest in G2/M and induced apoptosis. 8f decreased the protein levels of anti-apoptosis protein Mcl-1, c-FLIP and cell cycle regulating protein cyclin D1. 8f inhibited the activity of Pin1, a peptidyl-prolyl cis–trans isomerase to stabilize cyclin D1. 8f represented a compound with improved anti-proliferative effects for prostate cancer therapy working through new mechanisms.Download high-res image (210KB)Download full-size image
Co-reporter:Shenglin Luan, Qi Ge, Yedong Chen, Mingyang Dai, Jinyu Yang, Kun Li, Dan Liu, Linxiang Zhao
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 9(Issue 9) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.bmcl.2017.03.028
Myeloid cell leukemia-1 (Mcl-1) is an important antiapoptotic protein functioning through protein-protein interactions. We discovered LSL-A6 (2-((2-carbamoyl-1-(3-(4-methoxyphenoxy)propyl)-1H-indol-6-yl)oxy)acetic acid) with a novel N-substituted indole scaffold to interfere Mcl-1 binding as a novel Mcl-1 inhibitor. Molecular modeling indicated that this compound binds with Mcl-1 by interaction with P2 and R263 hot-spots. Structure modification focused on several moieties including indole core, hydrophobic tail and acidic chain were conducted and structure-activity relationship was analyzed. The most potent compound 24d which exhibited Ki value of 110 nM for interfering Mcl-1 binding was obtained after hit-to-lead modification.Download high-res image (71KB)Download full-size image
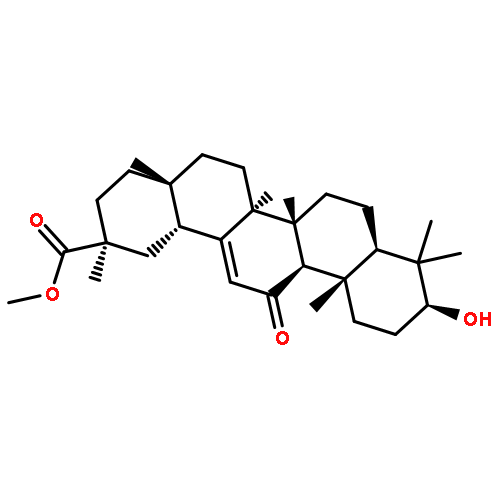
Co-reporter:Kun Li, Tianyi Ma, Jingjing Cai, Min Huang, Hongye Guo, Di Zhou, Shenglin Luan, Jinyu Yang, Dan Liu, Yongkui Jing, Linxiang Zhao
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 20(Issue 20) pp:
Publication Date(Web):15 October 2017
DOI:10.1016/j.bmc.2017.08.002
Twenty-six conjugates of 18β-glycyrrhetinic acid derivatives with 3-(1H-benzo[d]imidazol-2-yl)propanoic acid were designed and synthesized as Pin1 inhibitors. Most of these semi-synthetic compounds showed improved Pin1 inhibitory activity and anti-proliferative effects against prostate cancer cells as compared to 3-(1H-benzo[d]imidazol-2-yl)propanoic acid and GA. Compounds 10a and 12i were the most potent to inhibit growth of prostate cancer PC-3 with GI50 values of 7.80 μM and 3.52 μM, respectively. The enzyme inhibition ratio of nine compounds at 10 μM was over 90%. Structure-activity relationships indicated that both appropriate structure at ring C of GA and suitable length of linker between GA skeleton and benzimidazole moiety had significant impact on improving activity. Western blot assay revealed that 10a decreased the level of cell cycle regulating protein cyclin D1. Thus, these compounds might represent a novel anti-proliferative agent working through Pin1 inhibition.Download high-res image (98KB)Download full-size image
Co-reporter:Shenglin Luan, Hang Zhong, Xuan Zhao, Jinyu Yang, Yongkui Jing, Dan Liu, Linxiang Zhao
European Journal of Medicinal Chemistry 2017 Volume 141(Volume 141) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.ejmech.2017.10.023
•Twenty DHA-derived compounds were synthesized and assessed for their anticancer activity.•Compound 6f exhibited elevated antiproliferative activity against various cancer cell lines.•All compounds exhibited unexpected sensitivity in Adriamycin-resistant MCF-7 cells.•Compound 5f arrested MCF-7 and MCF-7/Adr cells in G0/G1 phase.•Preliminary pharmacokinetic properties of 5f and 6f were investigated in rats.Twenty hybrid compounds, tethering dihydroartemisinin (DHA) with diaryl-pyrazoline/diaryl-pyrazole through ether linkage, were synthesized based on hybridization strategy and assessed for their anticancer activity. The representative compound 6f exhibited significantly elevated antiproliferative activity compared with DHA against a panel of cancer cell lines. Unexpected sensitivity of 6f in Adriamycin-resistant MCF-7 cells (MCF-7/Adr) inspired subsequent research on anticancer activity of these DHA derivatives against breast cancer cell lines. All the novel compounds exhibited potent activity in three breast cancer cells including MDA-MB-231, MCF-7 and MCF-7/Adr. Most of them exerted almost 10-fold higher potency in MCF-7/Adr than in MCF-7 and MDA-MB-231 cells. Compound 5f, the most potent compound against MCF-7/Adr (GI50 = 18 nM), was used for mechanism research which revealed that compound 5f arrested MCF-7 and MCF-7/Adr cells in G0/G1 phase with decreased levels of cyclin D1 and increased levels of p27. Preliminary pharmacokinetic properties of 5f and 6f were investigated in rats after a single intravenous administration. The high sensitivity of MCF-7/Adr indicates that these compounds have potential to be developed as therapeutic agents to treat drug-resistant cancer.Download high-res image (197KB)Download full-size image
Co-reporter:Jiachen Wen, Yu Bao, Qun Niu, Jinyu Yang, Yinbo Fan, Jinhua Li, Yongkui Jing, Linxiang Zhao, Dan Liu
European Journal of Medicinal Chemistry 2016 Volume 109() pp:350-359
Publication Date(Web):15 February 2016
DOI:10.1016/j.ejmech.2016.01.013
•A collection of novel thiol-based HDAC inhibitors was developed.•Compound 15k and its analogs showed potent inhibition activity against HDAC1–3 and HDAC6.•A sulfur-masked prodrug strategy was investigated for HDAC inhibitor.•Disulfide 18 was identified as potent anti-tumor agent in vitro and in vivo.In this study, a collection of N-(6-mercaptohexyl)-3-substituted-1H-pyrazole-5-carboxamide HDAC inhibitors was developed. Among them, 15k was identified as the most potent inhibitor against total HDACs with IC50 of 0.008 μM. Further isoenzyme assays revealed that 15k and its analogs have a preference for HDAC1–3 (class I) and HDAC6 (class IIb) isoforms. The enzyme-based potencies of 15k were 2- to 11-fold higher than those of Vorinostat. The disulfide prodrug 18 was found to be potent cytotoxic agent against a panel of seven tumor cells, causing hyper-acetylation of histone and non-histone proteins in cellular level. In addition, 18 demonstrated a notable in vivo anti-tumor activity in HCT-116 xenografted model. This study provides further possibility of developing novel thiol-based HDAC inhibitors for the treatment of cancer.
Co-reporter:Hang Zhong, Xuan Zhao, Zhizhong Zuo, Jingwei Sun, Yao Yao, Tao Wang, Dan Liu, Linxiang Zhao
European Journal of Medicinal Chemistry 2016 Volume 108() pp:720-729
Publication Date(Web):27 January 2016
DOI:10.1016/j.ejmech.2015.10.040
•A series of novel DHA derivatives characterized by 10-β-aryl substitution was synthesized.•Novel compounds were more potent than positive control adriamycin against MDR cancer cells.•P-gp-overexpressed MCF-7/Adr cells revealed collateral sensitivity towards novel compounds.•MDCK-MDR1 assay indicated 3d and 5c could not be pumped by P-gp.•Compound 5c aroused cell cycle arrest at G1 phase in MCF-7 and MCF-7/Adr cells.A series of 10-β-phenyl ethers of dihydroartemisinin (DHA) with piperazine substitutions were synthesized with the goal of overcoming multidrug resistance in cancer therapy. These novel compounds exerted significant antiproliferative activities in breast cancer MCF-7 and MCF-7/Adr cell lines at the submicromolar level and were shown to be approximately 100- to 300-times more potent than the lead compound DHA. Remarkably, the P-gp-overexpressed MCF-7/Adr cell line showed collateral sensitivity towards these derivatives. Furthermore, compounds 3d and 5c, with the highest selectivity for MCF-7/Adr towards MCF-7 cells, were free from P-gp efflux in a MDCK-MDR1 assay. Flow cytometry and western blot assays suggested that the antiproliferative effects of 5c were associated with cell cycle arrest at G1 phase through the downregulation of Cyclin D1 and Cyclin B1.A series of novel 10-phenyl ethers with an N-aryl piperazine of dihydroartemisinin were synthesized and evaluated for their antiproliferative activities against human breast cancer MCF-7 and multidrug-resistant MCF-7/Adr cell lines. MCF-7/Adr cells revealed collateral sensitivity towards all novel artemisinin-phenyl ethers.
Co-reporter:Yin-bo Fan, Min Huang, Yu Cao, Ping Gong, Wen-bing Liu, Shu-yu Jin, Jia-chen Wen, Yong-kui Jing, Dan Liu and Lin-xiang Zhao
RSC Advances 2016 vol. 6(Issue 29) pp:24091-24096
Publication Date(Web):22 Feb 2016
DOI:10.1039/C6RA01159D
Usnic acid (UA) is a secondary metabolite of lichens with a unique dibenzofuran scaffold. The growth inhibitory and apoptotic effects of UA as well as the potential mechanisms of action were determined in human acute myeloid leukemia HL-60 cells and chronic myeloid leukemia K562 cells. UA inhibits growth and induces apoptosis in both cell lines with HL-60 cells more responsive. The apoptotic effects are associated with a decrease in the levels of the anti-apoptotic protein Mcl-1. UA inhibits mitogen-activated protein kinase-interacting kinase 1/eukaryotic translation-initiation factor 4E (Mnk1/eIF4E) and the proviral integration site of moloney murine leukemia virus-1/eIF4E-binding protein 1 (Pim-1/4E-BP1) signallings in both cell lines. A kinase inhibition assay reveals that UA is a potent Pim-1 inhibitor with limited activity to inhibit Mnk1. Considering the important role of Pim-1 in myeloid leukemia, UA represents a lead compound for developing effective therapeutics for myeloid leukemia treatment.
Co-reporter:Kun Li, Ying Li, Di Zhou, Yinbo Fan, Hongye Guo, Tianyi Ma, Jiachen Wen, Dan Liu, Linxiang Zhao
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 8) pp:1889-1897
Publication Date(Web):15 April 2016
DOI:10.1016/j.bmc.2016.03.016
In this work, a series of quinoline derivatives were designed and synthesized as antitumor agents. Most quinolines showed potent anti-proliferative activity against human prostatic cancer PC-3 cell line. Among which, 9d, 9f and 9g were the most effective compounds with GI50 values of 2.60, 2.81 and 1.29 μM, respectively. Structure–activity relationship analysis indicated that the secondary amine linked quinoline and pyridine ring played an important role in the anti-proliferative effects. Mechanistic studies revealed that 9g was a potential Pim-1 kinase inhibitor with abilities of cell cycle arrest and apoptosis induction. Considering of the increased activity of Pim-1 in prostate cancer, such compounds have potential to be developed as anti-prostate cancer agents.
Co-reporter:Jiachen Wen, Qun Niu, Jiang Liu, Yu Bao, Jinyu Yang, Shenglin Luan, Yinbo Fan, Dan Liu, Linxiang Zhao
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 2) pp:375-379
Publication Date(Web):15 January 2016
DOI:10.1016/j.bmcl.2015.12.007
A series of novel thiol-based histone deacetylase (HDAC) inhibitors bearing 3-phenyl-1H-pyrazole-5-carboxamide scaffold as surface recognition motif was designed, synthesized, and evaluated for their HDAC inhibition activity. Among them, 15j (IC50 = 0.08 μM) was identified as a better inhibitor than Vorinostat (IC50 = 0.25 μM) against total HDACs. In addition, Structure–activity relationships (SAR) analyses indicated that (i) compounds with different substituents on pyrazole N-1 position exhibited superior activities than those on pyrazole N-2 position, (ii) variation of functional groups on N-1′-alkyl chain terminus followed the trends of carboxyl group > hydroxyl group ≫ alkyl group, and (iii) methylation on pyrazole C-4 position diminished the HDAC inhibition activity. The SAR will guide us to further refine compounds bearing 3-phenyl-1H-pyrazole-5-carboxamide scaffold to achieve better HDAC inhibitors.
Co-reporter:Jiachen Wen, Yu Bao, Qun Niu, Jiang Liu, Jinyu Yang, Wanqiao Wang, Tao Jiang, Yinbo Fan, Kun Li, Jian Wang, Linxiang Zhao, Dan Liu
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 17) pp:4372-4376
Publication Date(Web):1 September 2016
DOI:10.1016/j.bmcl.2015.12.094
In this study, a concise synthetic method of psammaplin A was achieved from 3-bromo-4-hydroxybenzaldahyde and hydantoin through a four-step synthesis via Knoevenagel condensation, hydrolysis, oximation and amidation in 37% overall yield. A collection of novel psammaplin A analogs focused on the variations of substituents at the benzene ring and modifications at the oxime moiety were synthesized. Among all the synthesized compounds, 5d and 5e showed better HDAC inhibition than psammaplin A and comparable cytotoxicity against four cancer cell lines (PC-3, MCF-7, A549 and HL-60). Molecular docking and dynamics simulation revealed that (i) hydrogen atom of the oxime group interacts with Asp99 of HDAC1 through a water bridged hydrogen bond and (ii) a hydroxyl group is optimal attached on the para-position of benzene, interacting with Glu203 at the entrance to the active site tunnel.
Co-reporter:Yin-Bo Fan, Kun Li, Min Huang, Yu Cao, Ying Li, Shu-Yu Jin, Wen-Bing Liu, Jia-Chen Wen, Dan Liu, Lin-Xiang Zhao
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 4) pp:1224-1228
Publication Date(Web):15 February 2016
DOI:10.1016/j.bmcl.2016.01.032
A novel series of substituted pyrido[3,2-d]-1,2,3-triazines were designed and synthesized as Pim-1 inhibitors through scaffold hopping. Most of the derivatives showed potent in vitro Pim-1 inhibitory activities and anti-proliferative effects toward prostate cancer cells. Among them, 6b, 6h and 6m showed the best Pim-1 inhibitory activity with IC50 values of 0.69, 0.60 and 0.80 μM, respectively. Furthermore, compounds 6b, 6i, 6j and 6m showed strong inhibitory activity to human prostate cancer LNcap and PC-3 cell lines with IC50 values at low micromolar level. Structure–activity relationship analysis revealed that appropriate substitutions at C-6 positions contributed to the kinase inhibition and antiproliferative effects. Moreover, western blot assay suggested that 6j could decrease the levels of p-BAD and p-4E-BP1 in a dose-dependent manner in PC-3 cells. Docking studies showed that 3-N of the scaffold formed a hydrogen bond with Lys67, aromatic 4-aniline formed a key π–π stack with Phe49. Taken together, this study might provide the first sight for developing the pyrido[3,2-d]-1,2,3-triazine scaffold as novel Pim-1 inhibitors.
Co-reporter:Xiangfei Han, Jinling Chen, Mengjuan Jiang, Na Zhang, Kexin Na, Cong Luo, Ruoshi Zhang, Mengchi Sun, Guimei Lin, Rong Zhang, Yan Ma, Dan Liu, and Yongjun Wang
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 49) pp:
Publication Date(Web):November 22, 2016
DOI:10.1021/acsami.6b13057
Recently, nanomedicine without drug carriers has attracted many pharmacists’ attention. A novel paclitaxel–s–s–paclitaxel (PTX–s–s–PTX) conjugate with high drug loading (∼78%, w/w) was synthesized by conjugating paclitaxel to paclitaxel by using disulfide linkage. The conjugate could self-assemble into uniform nanoparticles (NPs) with 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide (DiR) encapsulated within the core of PTX–s–s–PTX NPs for photothermal therapy (PTT). The DiR-loaded self-assembled nanoparticles (DSNs) had a mean diameter of about 150 nm and high stability in biological condition. A disulfide bond is utilized as a redox-responsive linkage to facilitate a rapid release of paclitaxel in tumor cells. DSNs indicated significant cytotoxicity as a result of the synergetic chemo-thermal therapy. DSNs were featured with excellent advantages, including high drug loading, redox-responsive releasing behavior of paclitaxel, capability of loading with photothermal agents, and combinational therapy with PTT. In such a potent nanosystem, prodrug and photothermal strategy are integrated into one system to facilitate the therapy efficiency.Keywords: anticancer; disulfide; high drug loading; photothermal therapy; redox responsive;















![Benzenesulfonamide, N-[1-(4-chlorophenyl)ethylidene]-4-methyl-](http://img.cochemist.com/ccimg/905300/905272-87-9.png)
![Benzenesulfonamide, N-[1-(4-chlorophenyl)ethylidene]-4-methyl-](http://img.cochemist.com/ccimg/905300/905272-87-9_b.png)
![Benzenesulfonamide, N-[1-(4-fluorophenyl)ethylidene]-4-methyl-](http://img.cochemist.com/ccimg/905300/905272-80-2.png)
![Benzenesulfonamide, N-[1-(4-fluorophenyl)ethylidene]-4-methyl-](http://img.cochemist.com/ccimg/905300/905272-80-2_b.png)
![Benzenesulfonamide, 4-methyl-N-[1-(4-methylphenyl)ethylidene]-](http://img.cochemist.com/ccimg/905300/905272-78-8.png)
![Benzenesulfonamide, 4-methyl-N-[1-(4-methylphenyl)ethylidene]-](http://img.cochemist.com/ccimg/905300/905272-78-8_b.png)



![3-O-[beta-D-glucopyranosyl-(1->2)]-alpha-L-arabinofuranosyl-(1->3)-beta-D-glucuronopyranosyl-21,22-O-diangeloyl-3beta,15alpha,16alpha,21beta,22alpha,24beta,28-heptahydroxyolean-12-ene](http://img.cochemist.com/ccimg/867100/867063-27-2.png)
![3-O-[beta-D-glucopyranosyl-(1->2)]-alpha-L-arabinofuranosyl-(1->3)-beta-D-glucuronopyranosyl-21,22-O-diangeloyl-3beta,15alpha,16alpha,21beta,22alpha,24beta,28-heptahydroxyolean-12-ene](http://img.cochemist.com/ccimg/867100/867063-27-2_b.png)








![Octanoic acid, 8-bromo-2-[[(1,1-dimethylethoxy)carbonyl]amino]-, (2S)-](http://img.cochemist.com/ccimg/591800/591773-05-6.png)
![Octanoic acid, 8-bromo-2-[[(1,1-dimethylethoxy)carbonyl]amino]-, (2S)-](http://img.cochemist.com/ccimg/591800/591773-05-6_b.png)
![Carbamic acid, [2-(2-pyridinyldithio)ethyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/536000/535943-48-7.png)
![Carbamic acid, [2-(2-pyridinyldithio)ethyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/536000/535943-48-7_b.png)







![2-Propenamide,N-[4-[[(2,6-dimethoxy-4-pyrimidinyl)amino]sulfonyl]phenyl]-2-methyl-](http://img.cochemist.com/ccimg/288000/287967-58-2.png)
![2-Propenamide,N-[4-[[(2,6-dimethoxy-4-pyrimidinyl)amino]sulfonyl]phenyl]-2-methyl-](http://img.cochemist.com/ccimg/288000/287967-58-2_b.png)


![1,3,5-Triazin-2-amine, 4-phenyl-N-(trimethylsilyl)-6-[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/207200/207116-66-3.png)
![1,3,5-Triazin-2-amine, 4-phenyl-N-(trimethylsilyl)-6-[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/207200/207116-66-3_b.png)






![Carbamic acid, [3-[(chloroacetyl)amino]propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/201300/201282-03-3.png)
![Carbamic acid, [3-[(chloroacetyl)amino]propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/201300/201282-03-3_b.png)








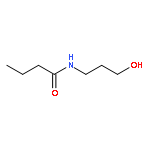
![Ethanethioic acid, S-[3-[(2-aminoethyl)amino]-3-oxopropyl] ester](http://img.cochemist.com/ccimg/160300/160285-97-2.png)
![Ethanethioic acid, S-[3-[(2-aminoethyl)amino]-3-oxopropyl] ester](http://img.cochemist.com/ccimg/160300/160285-97-2_b.png)
![2-Propen-1-one, 3-[4-(2-hydroxyethoxy)phenyl]-1-phenyl-](http://img.cochemist.com/ccimg/151600/151563-81-4.png)
![2-Propen-1-one, 3-[4-(2-hydroxyethoxy)phenyl]-1-phenyl-](http://img.cochemist.com/ccimg/151600/151563-81-4_b.png)




![Carbamic acid, [2-[(chloroacetyl)amino]ethyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/150000/149979-14-6.png)
![Carbamic acid, [2-[(chloroacetyl)amino]ethyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/150000/149979-14-6_b.png)








![Cyclohexanone, 2-[(4-methylphenyl)methylene]-, (2E)-](http://img.cochemist.com/ccimg/131000/130954-47-1.png)
![Cyclohexanone, 2-[(4-methylphenyl)methylene]-, (2E)-](http://img.cochemist.com/ccimg/131000/130954-47-1_b.png)
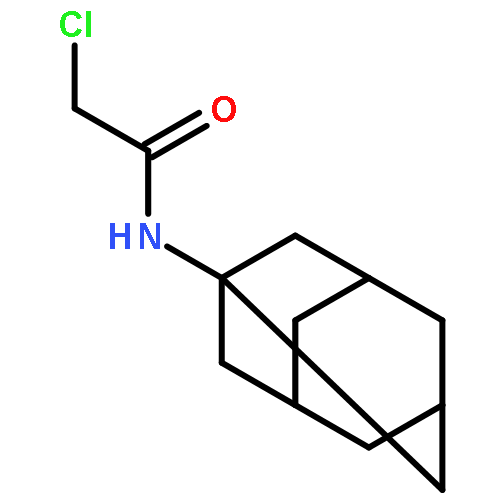
![Butanamide, 2-amino-3-methyl-N-tricyclo[3.3.1.13,7]dec-1-yl-, (S)-](http://img.cochemist.com/ccimg/121100/121009-91-4.png)
![Butanamide, 2-amino-3-methyl-N-tricyclo[3.3.1.13,7]dec-1-yl-, (S)-](http://img.cochemist.com/ccimg/121100/121009-91-4_b.png)
![Imidazo[5,1-d]-1,2,3,5-tetrazine-8-carbonyl chloride, 3,4-dihydro-3-methyl-4-oxo-](http://img.cochemist.com/ccimg/114000/113942-58-8.png)
![Imidazo[5,1-d]-1,2,3,5-tetrazine-8-carbonyl chloride, 3,4-dihydro-3-methyl-4-oxo-](http://img.cochemist.com/ccimg/114000/113942-58-8_b.png)








![1,3,5-TRIAZIN-2-AMINE, 4-METHYL-N-(TRIMETHYLSILYL)-6-[(TRIMETHYLSILYL)OXY]-](http://img.cochemist.com/ccimg/80700/80646-61-3.png)
![1,3,5-TRIAZIN-2-AMINE, 4-METHYL-N-(TRIMETHYLSILYL)-6-[(TRIMETHYLSILYL)OXY]-](http://img.cochemist.com/ccimg/80700/80646-61-3_b.png)
![(5z)-5-[(4-hydroxyphenyl)methylidene]imidazolidine-2,4-dione](http://img.cochemist.com/ccimg/80200/80171-33-1.png)
![(5z)-5-[(4-hydroxyphenyl)methylidene]imidazolidine-2,4-dione](http://img.cochemist.com/ccimg/80200/80171-33-1_b.png)


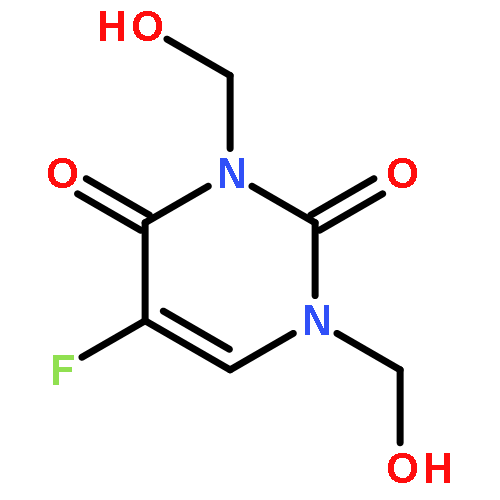

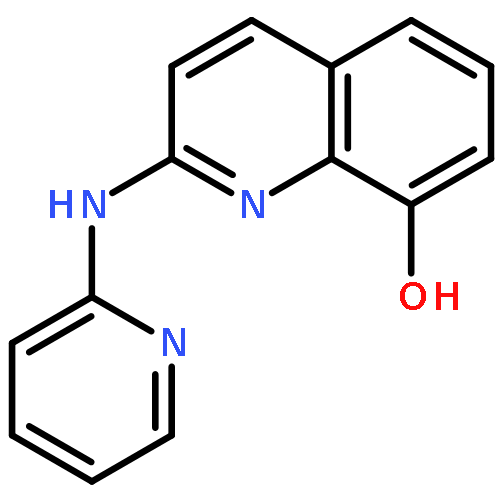


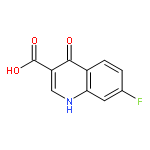
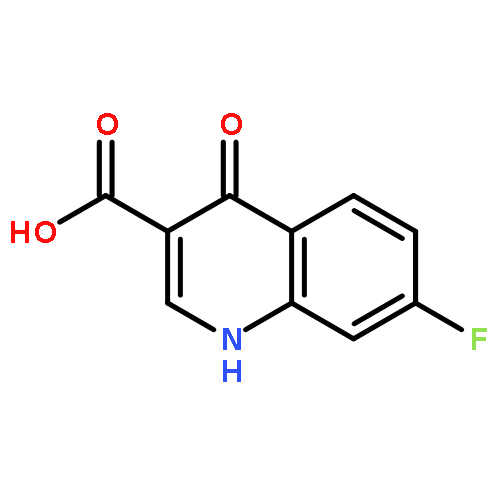








![2-phenoxy-N-(tricyclo[3.3.1.1~3,7~]dec-1-yl)acetamide](http://img.cochemist.com/ccimg/53000/52944-09-9.png)
![2-phenoxy-N-(tricyclo[3.3.1.1~3,7~]dec-1-yl)acetamide](http://img.cochemist.com/ccimg/53000/52944-09-9_b.png)


![2,4-Imidazolidinedione,5-[(4-hydroxy-3-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/52100/52036-16-5.png)
![2,4-Imidazolidinedione,5-[(4-hydroxy-3-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/52100/52036-16-5_b.png)





![3c-((2Xi)-5-hydroxy-2r,3t,8,8-tetramethyl-4-oxo-3,4-dihydro-2H,8H-pyrano[2,3-f]chromen-6-yl)-3t-phenyl-acrylic acid](http://img.cochemist.com/ccimg/36700/36626-19-4.png)
![3c-((2Xi)-5-hydroxy-2r,3t,8,8-tetramethyl-4-oxo-3,4-dihydro-2H,8H-pyrano[2,3-f]chromen-6-yl)-3t-phenyl-acrylic acid](http://img.cochemist.com/ccimg/36700/36626-19-4_b.png)

![Pyrrolo[1,2-a]pyrazine-1,4-dione,hexahydro-3-methyl-, (3S,8aS)-](http://img.cochemist.com/ccimg/36400/36357-32-1.png)
![Pyrrolo[1,2-a]pyrazine-1,4-dione,hexahydro-3-methyl-, (3S,8aS)-](http://img.cochemist.com/ccimg/36400/36357-32-1_b.png)




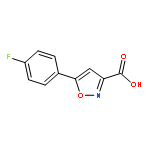

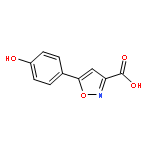



![5-[(3-CHLOROPHENYL)METHYLIDENE]IMIDAZOLIDINE-2,4-DIONE](http://img.cochemist.com/ccimg/28800/28744-91-4.png)
![5-[(3-CHLOROPHENYL)METHYLIDENE]IMIDAZOLIDINE-2,4-DIONE](http://img.cochemist.com/ccimg/28800/28744-91-4_b.png)




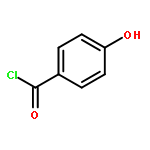
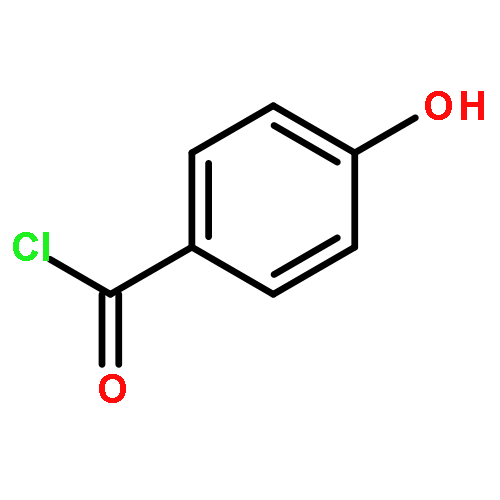
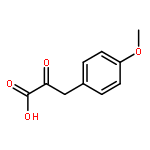
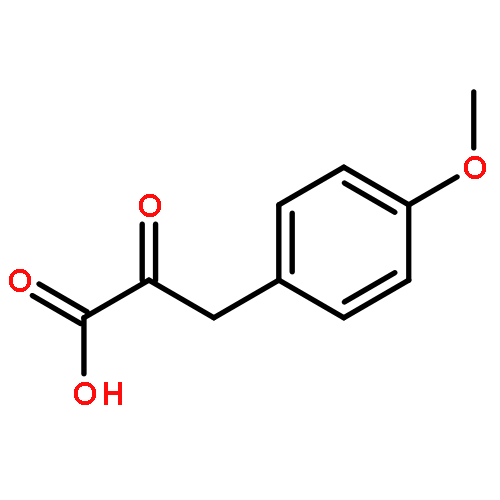


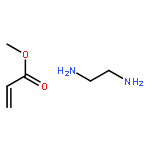
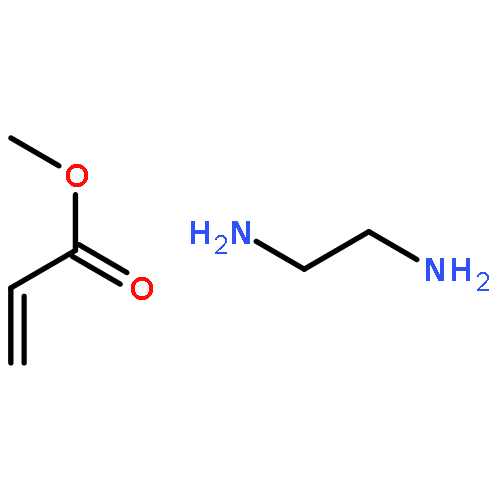
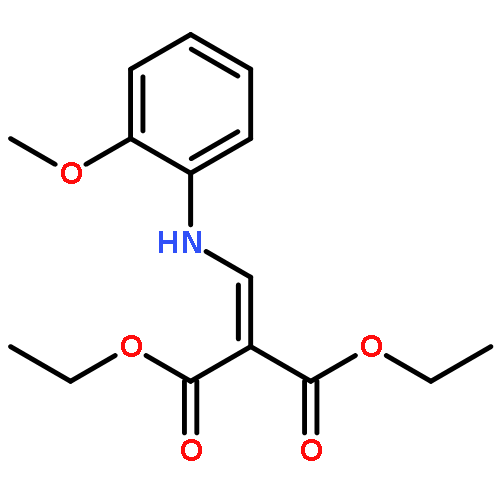
![Propanedioic acid, [[(3-fluorophenyl)amino]methylene]-, diethyl ester](http://img.cochemist.com/ccimg/26900/26832-95-1.png)
![Propanedioic acid, [[(3-fluorophenyl)amino]methylene]-, diethyl ester](http://img.cochemist.com/ccimg/26900/26832-95-1_b.png)




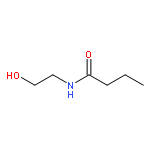
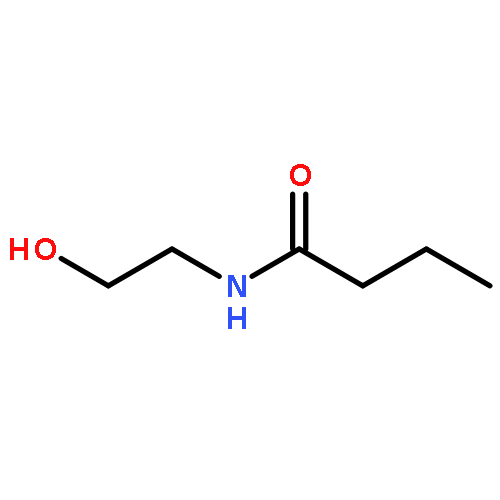
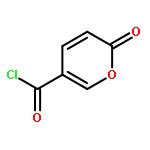

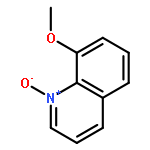
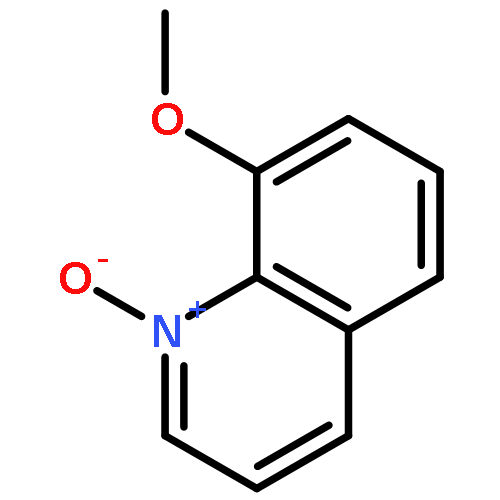




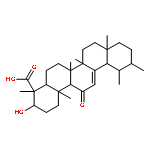





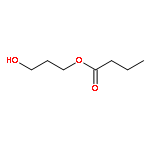
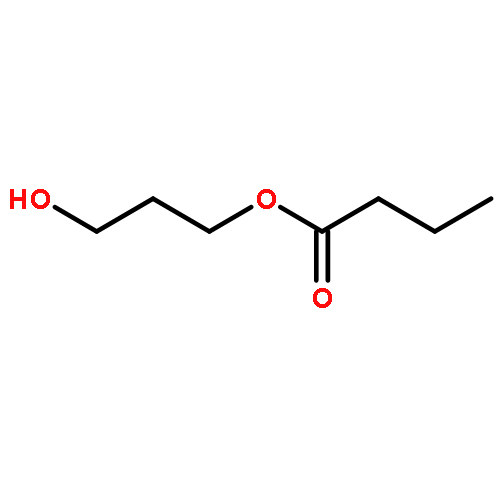






![2,4-Imidazolidinedione,5-[(2,4-dichlorophenyl)methylene]-](http://img.cochemist.com/ccimg/6400/6331-80-2.png)
![2,4-Imidazolidinedione,5-[(2,4-dichlorophenyl)methylene]-](http://img.cochemist.com/ccimg/6400/6331-80-2_b.png)





![2,4-Imidazolidinedione,5-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/5400/5349-42-8.png)
![2,4-Imidazolidinedione,5-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/5400/5349-42-8_b.png)










![Pyrrolo[1,2-a]pyrazine-1,4-dione,hexahydro-3-(phenylmethyl)-, (3S,8aS)-](http://img.cochemist.com/ccimg/3800/3705-26-8.png)
![Pyrrolo[1,2-a]pyrazine-1,4-dione,hexahydro-3-(phenylmethyl)-, (3S,8aS)-](http://img.cochemist.com/ccimg/3800/3705-26-8_b.png)





![(3S-trans)-hexahydro-3-isobutylpyrrolo[1,2-a]pyrazine-1,4-dione](http://img.cochemist.com/ccimg/2900/2873-36-1.png)
![(3S-trans)-hexahydro-3-isobutylpyrrolo[1,2-a]pyrazine-1,4-dione](http://img.cochemist.com/ccimg/2900/2873-36-1_b.png)
![Pyrrolo[1,2-a]pyrazine-1,4-dione,hexahydro-3-(1-methylethyl)-, (3S,8aS)-](http://img.cochemist.com/ccimg/2900/2854-40-2.png)
![Pyrrolo[1,2-a]pyrazine-1,4-dione,hexahydro-3-(1-methylethyl)-, (3S,8aS)-](http://img.cochemist.com/ccimg/2900/2854-40-2_b.png)


![Benzo[g]pteridine-2,4(1H,3H)-dione,7,8-dimethyl-](http://img.cochemist.com/ccimg/1100/1086-80-2.png)
![Benzo[g]pteridine-2,4(1H,3H)-dione,7,8-dimethyl-](http://img.cochemist.com/ccimg/1100/1086-80-2_b.png)




![5-METHOXY-2,2-DIMETHYL-6-[(E)-2-METHYLBUT-2-ENOYL]-10-PHENYLPYRANO[2,3-F]CHROMEN-8-ONE](http://img.cochemist.com/ccimg/600/548-27-6.png)
![5-METHOXY-2,2-DIMETHYL-6-[(E)-2-METHYLBUT-2-ENOYL]-10-PHENYLPYRANO[2,3-F]CHROMEN-8-ONE](http://img.cochemist.com/ccimg/600/548-27-6_b.png)





















![N-(Trimethylsilyl)-4-[(trimethylsilyl)oxy]-1,3,5-triazin-2-amine](http://img.cochemist.com/ccimg/52600/52523-35-0.png)
![N-(Trimethylsilyl)-4-[(trimethylsilyl)oxy]-1,3,5-triazin-2-amine](http://img.cochemist.com/ccimg/52600/52523-35-0_b.png)
![10H-Benzo[4,5]cyclohepta[1,2-b]thiophen-10-one, 4,9-dihydro-4-(1-methyl-4-piperidinylidene)-](http://img.cochemist.com/ccimg/34600/34580-13-7.png)
![10H-Benzo[4,5]cyclohepta[1,2-b]thiophen-10-one, 4,9-dihydro-4-(1-methyl-4-piperidinylidene)-](http://img.cochemist.com/ccimg/34600/34580-13-7_b.png)

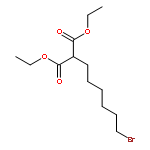
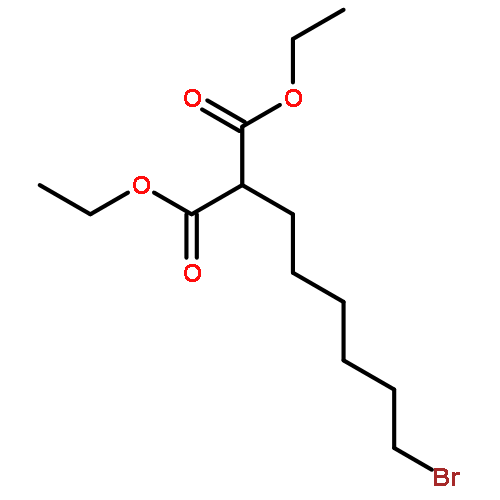


![3,5,9-Trioxa-4-phosphapentacosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxohexadecyl)oxy]-, inner salt, 4-oxide](http://img.cochemist.com/ccimg/2700/2644-64-6.png)
![3,5,9-Trioxa-4-phosphapentacosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxohexadecyl)oxy]-, inner salt, 4-oxide](http://img.cochemist.com/ccimg/2700/2644-64-6_b.png)