Co-reporter:Ying Gao, Tan Su, Liang Zhao, Yun Geng, Yong Wu, Min Zhang, Zhong-Min Su
Organic Electronics 2017 Volume 50(Volume 50) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.orgel.2017.07.024
•1 only exhibits a prompt emission whereas 2 has a prominent TADF characteristic.•HOMO and LUMO distributions of 1 and 2 are similar and kr of 1 is even greater than that of 2.•2 has the smaller ΔEST and larger SOC strength than 1.•2 has a smaller energy gap between S1/T1 MECP and S1 than 1.Density functional theory calculation was performed to analyze the critical factors determining whether molecule has a thermally activated delayed fluorescence (TADF) characteristic by comparing two similar molecules (1 and 2), which have a little difference in structure but opposite TADF performance. Some parameters relevant to TADF property were evaluated in this work. The results indicate that although the HOMO and LUMO distributions of 1 and 2 are similar and the radiative decay rate (kr) of 1 is even greater than that of 2, 2 has the smaller singlet-triplet energy splitting (ΔEST) and larger spin-orbital coupling (SOC) strength than 1. Besides, 2 has a smaller energy gap between S1/T1 minimum energy crossing point (MECP) and S1 than 1, indicating that it is easy for 2 to realize reverse intersystem crossing (RISC) from T1 to S1. Therefore, a comprehensive comparison between 1 and 2 suggests that the parameters including ΔEST, SOC and MECP between S1 and T1 are fairly important factors determining the TADF characteristic of 2.The results indicate that the HOMO and LUMO distributions of 1 and 2 are similar and the radiative decay rate (kr) of 1 is even greater than that of 2. However, 2 has smaller singlet-triplet energy splitting (ΔEST) and larger spin-orbital coupling (SOC) strength than 1. Besides, 2 has a smaller energy gap between S1/T1 minimum energy crossing point (MECP) and S1 than 1, indicating that it is easy for 2 to realize reverse intersystem crossing (RISC) from T1 to S1. Therefore, the parameters including ΔEST, SOC and MECP between S1 and T1 are fairly important factors determining the TADF characteristic of 2.Download high-res image (238KB)Download full-size image
Co-reporter:Ying Gao, Yun Geng, Yong Wu, Min Zhang, Zhong-Min Su
Dyes and Pigments 2017 Volume 145(Volume 145) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.dyepig.2017.04.001
•The effected of modifying connected bridge on TADF performances was investigated.•The kr values of compound 2–5 are higher than that of compound 1.•The values of compound 2–5 are noticeably increased compared with compound 1.•The compound 2 has a comparable ΔEST with compound 1 and larger kr and .One thermally activated delayed fluorescence (TADF) molecule DCBPy (compound 1) has been reported. Based on it, four compounds 2–5 have been designed by modifying the connected bridge between electron-donor (D) and electron-acceptor (A) units. The calculated normal mode reorganization energy (λ) for the non-radiative decay process shows that the λ of compounds 2–5 in the high-frequency region is noticeably reduced compared with compound 1, suggesting that the high-frequency CO stretching vibration is hindered through modifying the connected bridge between D and A units. Moreover, the radiative decay rate constant (kr) values of compound 2–5 are nearly one order of magnitude higher than that of pristine compound 1. Besides, for TADF material, the reverse intersystem crossing (RISC) is dependent on a small singlet-triplet energy gap (ΔEST) and large spin-orbital coupling matrix elements . For our designed compounds, modifying the connected bridges noticeably increase their values, although the ΔEST values of compound 2–5 are greater than that of compound 1. Especially, compound 2 has a comparable ΔEST with compound 1 and larger kr and than compound 1, which exhibits the best TADF efficiency among these compounds. As a consequence, for this kind of DCBPy emitter, modifying the connected bridge between D and A units is a valid approach to improve their TADF performances.Previously, extensive efforts have been devoted to designing highly performance TADF material via varying the electron-donator (D) and electron-acceptor (A) units and tried the best to find a matching combination of D and A units with high external quantum efficiency. In present work, we have investigated the effect of modifying the connected bridge between D and A units on their electronic properties. Based on the reported thermally activated delayed fluorescence (TADF) molecule DCBPy (compound 1), four compounds 2–5 have been designed by modifying the connected bridge between D and A units. For predicting the accurate singlet-triplet energy gap (ΔEST), the tuning range-separated functional has been utilized to calculate ΔEST. The calculated normal mode reorganization energy (λ) for the non-radiative decay process displays that the λ of compounds 2–5 in the high-frequency region is noticeably reduced compared with compound 1, suggesting that the high-frequency CO stretching vibration is hindered through modifying the connected bridge between D and A units. Moreover, the radiative decay rate constant (kr) values of compound 2–5 are one order of magnitude higher than that of pristine compound 1. Besides, for our designed molecules, modifying the connected bridges noticeably increase their spin-orbital coupling matrix element () values, although the ΔEST values of compound 2–5 are greater than that of compound 1. As a consequence, for this kind of DCBPy compounds, modifying the connected bridge between D and A units maybe a valid approach to improve their TADF performances.Download high-res image (258KB)Download full-size image
Co-reporter:Ying-Chen Duan;Yong Wu;Xin-Yao Ren;Liang Zhao;Min Zhang;Guang-Yan Sun;Zhong-Min Su
Dalton Transactions 2017 vol. 46(Issue 34) pp:11491-11502
Publication Date(Web):2017/08/29
DOI:10.1039/C7DT02684F
Two reported Ir(III) complexes 1a and 1b containing oxazoline and imidazoline in ancillary ligand, respectively, were investigated by DFT/TD-DFT method. In order to obtain full-color display materials, we designed a group of Ir(III) complexes 2a–3d based on 1a, which exhibits higher quantum efficiency in phosphorescence, by introducing electron-donating/electron-withdrawing moieties to different positions of the ancillary ligand to adjust emission color. In addition to calculating the radiation rate and analyzing its determining factors, we also estimated nonradiative ability by evaluating the spin–orbit coupling matrix element between the ground state (S0) and the lowest triplet state (T1) as well as the reorganization energy from T1 to S0 to estimate quantum efficiency more accurately. In particular, an in-depth analysis on the contribution of each vibration mode to reorganization energy helped us to identify the effect of substituents on the nonradiative process. Besides, charge injection/transfer properties and energy relation of the states related to exciton quenching via the triplet metal-centered state were also examined, which provide an estimation on the OLED performance of our designed complexes. Overall, we expect 2b and 3c to be more efficient blue-emitting emitters than 1a and 3a and 3b to be efficient green and red emitters, respectively.
Co-reporter:Qing-Qing Pan;Shuang-Bao Li;Ying-Chen Duan;Yong Wu;Ji Zhang;Liang Zhao;Zhong-Min Su
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 46) pp:31227-31235
Publication Date(Web):2017/11/29
DOI:10.1039/C7CP05938H
The interface characteristic is a crucial factor determining the power conversion efficiency of organic solar cells (OSCs). In this work, our aim is to conduct a comparative study on the interface characteristics between the very famous non-fullerene acceptor, ITIC, and a fullerene acceptor, PC71BM by combining molecular dynamics simulations with density functional theory. Based on some typical interface models of the acceptor ITIC or PC71BM and the donor PBDB-T selected from MD simulation, besides the evaluation of charge separation/recombination rates, the relative positions of Frenkel exciton (FE) states and the charge transfer states along with their oscillator strengths are also employed to estimate the charge separation abilities. The results show that, when compared with those for the PBDB-T/PC71BM interface, the CT states are more easily formed for the PBDB-T/ITIC interface by either the electron transfer from the FE state or direct excitation, indicating the better charge separation ability of the former. Moreover, the estimation of the charge separation efficiency manifests that although these two types of interfaces have similar charge recombination rates, the PBDB-T/ITIC interface possesses the larger charge separation rates than those of the PBDB-T/PC71BM interface. Therefore, the better match between PBDB-T and ITIC together with a larger charge separation efficiency at the interface are considered to be the reasons for the prominent performance of ITIC in OSCs.
Co-reporter:Zhi-Wen Zhao, Qing-Qing Pan, Shuang-Bao Li, Yu-Ai Duan, Yun Geng, Min Zhang, Zhong-Min Su
Journal of Molecular Graphics and Modelling 2017 Volume 77(Volume 77) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.jmgm.2017.07.027
•Explaining the effect of fluorine and cyano substitution in polymer donor materials from a theoretical perspective.•The substituent of cyano groups and fluorine in polymer 3 and 4 performs better balance between Voc and Jsc in OSCs.•The designed molecules 3 and 4 with higher ratios of rates kinter-CT/kinter-CR) are promising donor materials.A series of polymer donor materials 1-5 based on diketopyrrolopyrrole and thiophene unit which have been widely used in organic solar cells (OSCs) were investigated based on quantum chemical calculations. The effect of fluorine and cyano substitutions in polymer donor materials was focused on. Based on the investigation on electronic structures and optical properties of the reported molecules 1 and 2 and the analysis on some parameters relevant to charge dissociation ability at donor/acceptor interface constituted by 1 and 2 with PC61BM such as intermolecular charge transfer and recombination, driving force and Coulombic bound energy, we explained why fluorine substitution can improve OPV efficiency through strengthening eletron-withdrawing ability from a theoretical perspective. Then we designed cyano-substituted polymers 3-5 with the aim of obtaining better photovoltaic donor materials. The results reveal that our attempt to design donor materials which can balance large open-circuit voltage (Voc) and high short-circuit current (Jsc) in OSCs has worked out. It is worth noting that the substitutions of fluorine and cyano groups synergistically reduce energy gap and HOMO energy level of polymers 3 and 4. Moreover, 3/PC61BM and 4/PC61BM heterojunctions show over 107 and 104 times higher than 1/PC61BM on the ratios of intermolecular charge transfer and recombination rates (kinter-CT/kinter-CR). Thus, our work here may provide an efficient strategy to design promising donor materials in OPVs and we hope it could be useful in the future experimental synthesis.Download high-res image (140KB)Download full-size image
Co-reporter:Yong Wu, Guo-Gang Shan, Hai-Bin Li, Shui-Xing Wu, Xin-Yao Ren, Yun Geng and Zhong-Min Su
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 6) pp:4771-4771
Publication Date(Web):09 Jan 2015
DOI:10.1039/C4CP90185A
Correction for ‘Theoretical study and design of multifunctional phosphorescent platinum(II) complexes containing triarylboron moieties for efficient OLED emitters’ by Yong Wu et al., Phys. Chem. Chem. Phys., 2015, DOI: 10.1039/c4cp04919e.
Co-reporter:Yong Wu, Guo-Gang Shan, Hai-Bin Li, Shui-Xing Wu, Xin-Yao Ren, Yun Geng and Zhong-Min Su
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 4) pp:2438-2446
Publication Date(Web):01 Dec 2014
DOI:10.1039/C4CP04919E
The geometries, electronic structures, photophysical properties and spin–orbit coupling (SOC) effects in the radiative process for the recently synthesized complexes (Bppy)Pt(acac) (1) and (BNppy)Pt(acac) (2) as well as the designed complexes 3–6 were investigated by DFT and TD-DFT calculations, to reveal the influences of the functional ligands on charge injection ability and phosphorescence efficiency of emitters. It is found that compared with electron acceptor complex 1, complexes 2–6 have lower ionization potentials and comparable high electronic affinities, which are suited for bipolar luminescent materials. The results also demonstrated that Bppy complexes 1, 5 and 6 have more 3MLCT compositions in T1 emitting states compared with BNppy complexes 2–4, which results in strong SOC and fast kr. Thus, the phosphorescence efficiency of 1 is higher than that of 2. In addition, 5 and 6 have the balanced charge transport and better hole injection ability when the hole-transporting ligand is incorporated to 1. Therefore, 5 and 6 can server as promising candidates for efficient multifunctional phosphorescent OLED emitters owing to their ambipolar characters, balanced charge carrier injection/transport features and high phosphorescence quantum efficiency.
Co-reporter:Wei Du, Hai-Bin Li, Dong-Mei Gu, Yong Wu, Guang-Yan Sun, Yun Geng and Zhong-Min Su
RSC Advances 2015 vol. 5(Issue 121) pp:100169-100175
Publication Date(Web):10 Nov 2015
DOI:10.1039/C5RA17237C
In this work, we designed a series of ruthenium sensitizers DX2–DX5 derived from a phosphine-coordinated ruthenium sensitizer DX1 with a surprisingly high short-circuit photocurrent density (Jsc) of 26.8 mA cm−2 for dye sensitized solar cells (DSSCs), with the aim of enhancing the light harvesting ability in the near-infrared (NIR) region and further increasing the Jsc. Density functional theory (DFT) and relativistic time-dependent DFT calculations have been performed to evaluate the optical and photovoltaic properties of these Ru dyes, taking the effect of spin–orbit coupling (SOC) into consideration. The intrinsic causes for varied Jsc and open-circuit photovoltage (Voc) have been systematically discussed through investigating the light harvesting efficiency, electron injection driving force, dye regeneration driving force, electronic coupling and conduction band energy shift. The calculated results reveal that the designed DX5 has increased light harvesting efficiency in the NIR region and a higher conduction band energy shift compared with other sensitizers. That is, DX5 may have improved Jsc and Voc, which makes DX5 serve as a promising sensitizer for future DSSC applications.
Co-reporter:Jian-Zhao Zhang, Ji Zhang, Hai-Bin Li, Yong Wu, Hong-Liang Xu, Min Zhang, Yun Geng, Zhong-Min Su
Journal of Power Sources 2014 Volume 267() pp:300-308
Publication Date(Web):1 December 2014
DOI:10.1016/j.jpowsour.2014.05.085
•The different performance of reported dyes was rationalized by DFT calculations.•Present a comprehensive strategy to design high-efficiency dye.•Designed dyes 6 and 7 are promising candidates to further improve efficiency.Factors associated with short circuit current density (Jsc) and open circuit photovoltage (Voc) of dye sensitized solar cells (DSSCs) have been analyzed through DFT and TDDFT calculations to explore the origin of the significant performance differences with only tiny structure difference (1.24% for 1 and 8.21% for 2) (Advanced Functional Materials 2012, 22, 1291–1302). Our results reveal that the insertion of phenyl ring in 2 enlarges the distance between the dye cation hole and the semiconductor surface and makes the benzothiadiazole (BTDA) unit, which has strong interaction with the electrolyte, far away from the semiconductor, resulting in a decreased charge recombination rate compared with that of 1. However, the insertion of phenyl ring also results in a distortion of the molecular structure, leading to a decreased light harvesting ability. Hence, two dyes (6 and 7) derived from 2 with better conjugation degree, farther position of BTDA unit and longer molecular length have been designed to keep the advantages and overcome the disadvantages of 2 simultaneously. The results demonstrate that we get the desired properties of dyes through reasonable molecular design, and these two dyes could be promising candidates in DSSC field and further improve the performance of the cell.
Co-reporter:Hai-Bin Li, Ji Zhang, Yong Wu, Jun-Ling Jin, Yu-Ai Duan, Zhong-Min Su, Yun Geng
Dyes and Pigments 2014 Volume 108() pp:106-114
Publication Date(Web):September 2014
DOI:10.1016/j.dyepig.2014.04.029
•The π-linker effect on p-type DSSC performance was evaluated theoretically in detail.•The calculated charge transfer index is correlated well with the electron lifetime of dyes.•The high IPCE of O7 is due to the efficient regeneration and improved LHE.•The designed dyes all show the red-shifted absorption and larger LHE than O7.•Dye 4 is a promising candidate for the p-type sensitizer.The geometries, electronic structures, absorption spectra and the character of excited state for the reported p-type sensitizers O2, O6 and O7 and designed 1–5 with different π-linkers were investigated by DFT and TDDFT calculations, to reveal the influences of π-linkers on the performance of DSSCs. It is found that the higher IPCE of O7 as compared to O6 stems from the improved light harvesting efficiency (LHE) and the efficient regeneration. And, for O2 and O6 with comparable IPCE, the slightly increased Jsc for O2 can be mainly ascribed to the better spectrum matching with solar spectrum. The results also revealed that the calculated charge transfer index (DCT, the metric of photoinduced electron–hole separation) for reported sensitizers is well correlated with the measured electron lifetime pertaining to Voc. Importantly, dye 4 may be a promising candidate for the p-type sensitizer due to the red-shifted absorption, larger LHE and longer DCT in comparison with prototype O7.The performance of p-type sensitizers used in DSSCs was evaluated and predicted by DFT and TDDFT calculations.
Co-reporter:Ji Zhang, Jian-Zhao Zhang, Hai-Bin Li, Yong Wu, Yun Geng and Zhong-Min Su
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 45) pp:24994-25003
Publication Date(Web):30 Sep 2014
DOI:10.1039/C4CP03355H
Ten porphyrin sensitizers with different electron-withdrawing groups derived from the best sensitizer SM315 were investigated by means of the density functional theory (DFT) and time-dependent DFT calculations. To this end, major factors affecting the performance of the cell, including light harvesting, electron injection, dye regeneration, and conduction band energy shift are taken into consideration. Especially, the calculated distance (r) from the electron recapture center to the semiconductor surface is used to probe the charge recombination process. In addition, considering the complexity of the porphyrin sensitizers' absorption, the maximum short circuit current density (Jmaxsc) is determined for investigating the light harvesting ability quantitatively. We find that when compared to SM315 with 2,1,3-benzothiadiazole, 1 with naphtho[1,2-c:5,6-c]bis[1,2,5]thiadiazole shows better performance due to both larger Jmaxsc and r, and 7 with diketopyrrolopyrrole could also be a promising candidate due to the much larger Jmaxsc and comparable r.
Co-reporter:Xin-Yao Ren, Yong Wu, Guo-Gang Shan, Li Wang, Yun Geng and Zhong-Min Su
RSC Advances 2014 vol. 4(Issue 107) pp:62197-62208
Publication Date(Web):24 Oct 2014
DOI:10.1039/C4RA07041K
A density functional theory/time-depended density functional theory was used to investigate a series of heteroleptic Ir(III) complexes (1–4) employing azadipyrromethene and closely related dipyrromethene derivatives as N^N ancillary ligands, in an effort to explore the underlying reasons of non-radiative behaviour of 1 and further adjust the photophysical properties by the modification of N^N ancillary ligands. The results reveal that the non-emissive phenomenon of 1 can be attributed to the weak 3ILCT character of the emissive excited state and large structure distortion, as well as the small Topt1–Sopt0 energy gap. Upon tailoring the N^N ancillary ligands, the geometry distortion of 2–4 becomes obviously smaller in comparison with 1, accordingly, the spectrum properties are also markedly affected. For instance, the enlargement of frontier molecular orbital energy gaps from 1 to 4 results in the blue-shift of their absorption and emission spectra, which is considered to be dominated by the ancillary ligand, while there is a little contribution from the Ir(III) center. Importantly, further analysis on the quantum yield (ΦPL) of these complexes also indicates a vital role of N^N ancillary ligands. It is intriguing to note that the designed complex 4 without pendant phenyl rings substituent in the ancillary ligand, possesses an efficient indirect spin-orbital coupling route, larger transition electric dipole moment (μS3), higher Topt1–Sopt0 energy gap and smaller S3–T1 splitting energy (ΔE(S3–T1)), which ensure its higher ΦPL compared to other complexes.
Co-reporter:Xin-Yao Ren, Yong Wu, Li Wang, Liang Zhao, Min Zhang, Yun Geng, Zhong-Min Su
Journal of Molecular Graphics and Modelling 2014 Volume 51() pp:149-157
Publication Date(Web):June 2014
DOI:10.1016/j.jmgm.2014.05.005
•One synthesized and two designed guanidinate-based iridium (III) complexes are investigated.•Phosphorescence quantum efficiency is evaluated with the aid of kr and knr values.•Different cyclometalated ligands can greatly affect the photophysical properties of complexes.•Changing the pyridine ring with diazole groups can significantly enhance the SOC effects.•The designed complex 3 is expected to be a promising candidate for highly efficient guanidinate-based phosphorescence emitter.A density functional theory/time-depended density functional theory was used to investigate the synthesized guanidinate-based iridium(III) complex [(ppy)2Ir{(NiPr)2C(NPh2)}] (1) and two designed derivatives (2 and 3) to determine the influences of different cyclometalated ligands on photophysical properties. Except the conventional discussions on geometric relaxations, absorption and emission properties, many relevant parameters, including spin-orbital coupling (SOC) matrix elements, zero-field-splitting parameters, radiative rate constants (kr) and so on were quantitatively evaluated. The results reveal that the replacement of the pyridine ring in the 2-phenylpyridine ligand with different diazole rings cannot only enlarge the frontier molecular orbital energy gaps, resulting in a blue-shift of the absorption spectra for 2 and 3, but also enhance the absorption intensity of 3 in the lower-energy region. Furthermore, it is intriguing to note that the photoluminescence quantum efficiency (ΦPL) of 3 is significantly higher than that of 1. This can be explained by its large SOC value (n = 3–4) and large transition electric dipole moment (μS3), which could significantly contribute to a larger kr. Besides, compared with 1, the higher emitting energy (ET1) and smaller 2 value for 3 may lead to a smaller non-radiative decay rate. Additionally, the detailed results also indicate that compared to 1 with pyridine ring, 3 with imidazole ring performs a better hole injection ability. Therefore, the designed complex 3 can be expected as a promising candidate for highly efficient guanidinate-based phosphorescence emitter for OLEDs applications.Calculation results indicate that upon changing the main ligand of guanidinate-based complex 1 from 2-phenylpyridine to different phenyl-diazoles, significant SOC effects can be observed, leading to higher kr values compared to 1. The designed complex 3 would be considered as a promising candidate for highly efficient guanidinate-based phosphorescence emitter in OLEDs.
Co-reporter:Yong Wu, Guo-Gang Shan, Hai-Bin Li, Shui-Xing Wu, Xin-Yao Ren, Yun Geng and Zhong-Min Su
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 4) pp:NaN2446-2446
Publication Date(Web):2014/12/01
DOI:10.1039/C4CP04919E
The geometries, electronic structures, photophysical properties and spin–orbit coupling (SOC) effects in the radiative process for the recently synthesized complexes (Bppy)Pt(acac) (1) and (BNppy)Pt(acac) (2) as well as the designed complexes 3–6 were investigated by DFT and TD-DFT calculations, to reveal the influences of the functional ligands on charge injection ability and phosphorescence efficiency of emitters. It is found that compared with electron acceptor complex 1, complexes 2–6 have lower ionization potentials and comparable high electronic affinities, which are suited for bipolar luminescent materials. The results also demonstrated that Bppy complexes 1, 5 and 6 have more 3MLCT compositions in T1 emitting states compared with BNppy complexes 2–4, which results in strong SOC and fast kr. Thus, the phosphorescence efficiency of 1 is higher than that of 2. In addition, 5 and 6 have the balanced charge transport and better hole injection ability when the hole-transporting ligand is incorporated to 1. Therefore, 5 and 6 can server as promising candidates for efficient multifunctional phosphorescent OLED emitters owing to their ambipolar characters, balanced charge carrier injection/transport features and high phosphorescence quantum efficiency.
Co-reporter:Yong Wu, Guo-Gang Shan, Hai-Bin Li, Shui-Xing Wu, Xin-Yao Ren, Yun Geng and Zhong-Min Su
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 6) pp:NaN4771-4771
Publication Date(Web):2015/01/09
DOI:10.1039/C4CP90185A
Correction for ‘Theoretical study and design of multifunctional phosphorescent platinum(II) complexes containing triarylboron moieties for efficient OLED emitters’ by Yong Wu et al., Phys. Chem. Chem. Phys., 2015, DOI: 10.1039/c4cp04919e.
Co-reporter:Ji Zhang;Jian-Zhao Zhang;Hai-Bin Li;Yong Wu;Zhong-Min Su
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 45) pp:
Publication Date(Web):2014/10/30
DOI:10.1039/C4CP03355H
Ten porphyrin sensitizers with different electron-withdrawing groups derived from the best sensitizer SM315 were investigated by means of the density functional theory (DFT) and time-dependent DFT calculations. To this end, major factors affecting the performance of the cell, including light harvesting, electron injection, dye regeneration, and conduction band energy shift are taken into consideration. Especially, the calculated distance (r) from the electron recapture center to the semiconductor surface is used to probe the charge recombination process. In addition, considering the complexity of the porphyrin sensitizers' absorption, the maximum short circuit current density (Jmaxsc) is determined for investigating the light harvesting ability quantitatively. We find that when compared to SM315 with 2,1,3-benzothiadiazole, 1 with naphtho[1,2-c:5,6-c]bis[1,2,5]thiadiazole shows better performance due to both larger Jmaxsc and r, and 7 with diketopyrrolopyrrole could also be a promising candidate due to the much larger Jmaxsc and comparable r.

![Benzenamine, 4-[2,7-bis(diphenylphosphinyl)-9-phenyl-9H-fluoren-9-yl]-N,N-diphenyl-](/data/chemimg/165100/1198361-98-6.png)
![Benzenamine, 4-[2,7-bis(diphenylphosphinyl)-9-phenyl-9H-fluoren-9-yl]-N,N-diphenyl-](/data/chemimg/165100/1198361-98-6_b.png)











![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,6,12,13-tetrafluoro-](/data/chemimg/128700/946612-30-2.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,6,12,13-tetrafluoro-](/data/chemimg/128700/946612-30-2_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,12-difluoro-](/data/chemimg/128700/942294-90-8.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,12-difluoro-](/data/chemimg/128700/942294-90-8_b.png)



![2,2'-BITHIAZOLE, 5,5'-BIS[4-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/869900/869896-77-5.png)
![2,2'-BITHIAZOLE, 5,5'-BIS[4-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/869900/869896-77-5_b.png)



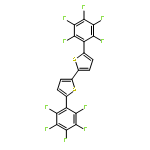
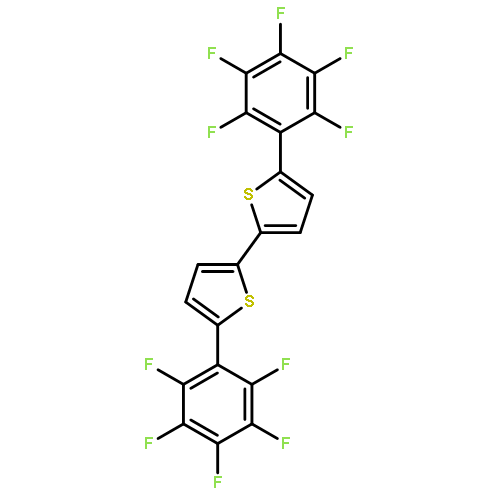
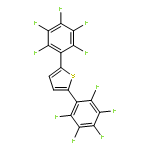
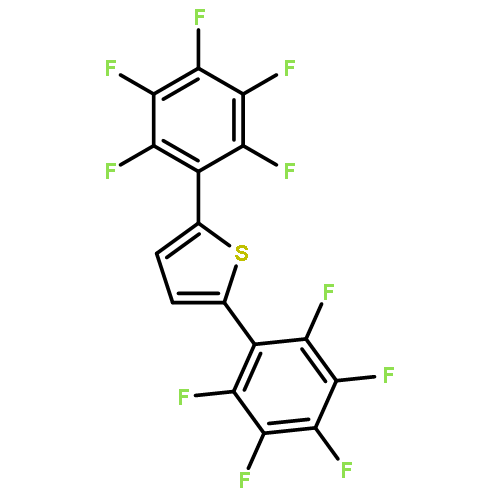
![2,2'-BITHIOPHENE, 5,5'-BIS[4-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/852100/852068-45-2.png)
![2,2'-BITHIOPHENE, 5,5'-BIS[4-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/852100/852068-45-2_b.png)
![1H-PYRROLE, 3-BROMO-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/850000/849939-30-6.png)
![1H-PYRROLE, 3-BROMO-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/850000/849939-30-6_b.png)


![Thieno[3,2-b]thiophene, 2,5-di-9H-fluoren-2-yl-](http://img.cochemist.com/ccimg/807400/807346-21-0.png)
![Thieno[3,2-b]thiophene, 2,5-di-9H-fluoren-2-yl-](http://img.cochemist.com/ccimg/807400/807346-21-0_b.png)


![2,2'-Bithiophene, 5,5'-bis[2,3,5,6-tetrafluoro-4-(2-thienyl)phenyl]-](http://img.cochemist.com/ccimg/587000/586954-33-8.png)
![2,2'-Bithiophene, 5,5'-bis[2,3,5,6-tetrafluoro-4-(2-thienyl)phenyl]-](http://img.cochemist.com/ccimg/587000/586954-33-8_b.png)


![DITHIENO[3,2-B:2',3'-D]THIOPHENE, 2,6-DIMETHYL-](http://img.cochemist.com/ccimg/534000/533930-12-0.png)
![DITHIENO[3,2-B:2',3'-D]THIOPHENE, 2,6-DIMETHYL-](http://img.cochemist.com/ccimg/534000/533930-12-0_b.png)
![Ethenetricarbonitrile, [2,2'-bithiophen]-5-yl-](http://img.cochemist.com/ccimg/440700/440669-03-4.png)
![Ethenetricarbonitrile, [2,2'-bithiophen]-5-yl-](http://img.cochemist.com/ccimg/440700/440669-03-4_b.png)
![Benzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetrone, 2,7-bis(2,2,3,3,4,4,4-heptafluorobutyl)-](/data/chemimg/136100/268724-95-4.png)
![Benzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetrone, 2,7-bis(2,2,3,3,4,4,4-heptafluorobutyl)-](/data/chemimg/136100/268724-95-4_b.png)
![Ethenetricarbonitrile, [2,2':5',2''-terthiophen]-5-yl-](http://img.cochemist.com/ccimg/197300/197295-93-5.png)
![Ethenetricarbonitrile, [2,2':5',2''-terthiophen]-5-yl-](http://img.cochemist.com/ccimg/197300/197295-93-5_b.png)








![1H-Pyrrole, 1-[(4-methylphenyl)sulfonyl]-3-phenyl-](http://img.cochemist.com/ccimg/153700/153633-35-3.png)
![1H-Pyrrole, 1-[(4-methylphenyl)sulfonyl]-3-phenyl-](http://img.cochemist.com/ccimg/153700/153633-35-3_b.png)



![2,9-Di(pyridin-4-yl)anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetraone](http://img.cochemist.com/ccimg/136900/136847-29-5.png)
![2,9-Di(pyridin-4-yl)anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetraone](http://img.cochemist.com/ccimg/136900/136847-29-5_b.png)



![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone,5,6,12,13-tetrachloro-](/data/chemimg/102600/115662-06-1.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone,5,6,12,13-tetrachloro-](/data/chemimg/102600/115662-06-1_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(4-hydroxybutyl)-](http://img.cochemist.com/ccimg/105400/105315-83-1.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(4-hydroxybutyl)-](http://img.cochemist.com/ccimg/105400/105315-83-1_b.png)
![6,1 1-Benzo[b]phenazine- quinone](http://img.cochemist.com/ccimg/97600/97594-66-6.png)
![6,1 1-Benzo[b]phenazine- quinone](http://img.cochemist.com/ccimg/97600/97594-66-6_b.png)

![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-dioctyl-](/data/chemimg/709500/78151-58-3.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-dioctyl-](/data/chemimg/709500/78151-58-3_b.png)








![Indolo[3,2-b]carbazole](/data/chemimg/415800/241-55-4.png)
![Indolo[3,2-b]carbazole](/data/chemimg/415800/241-55-4_b.png)


![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,6,12,13-tetrafluoro-2,9-bis(2,2,3,3,4,4,4-heptafluorobutyl)-](/data/chemimg/128700/946612-26-6.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,6,12,13-tetrafluoro-2,9-bis(2,2,3,3,4,4,4-heptafluorobutyl)-](/data/chemimg/128700/946612-26-6_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,12-difluoro-2,9-bis(2,2,3,3,4,4,4-heptafluorobutyl)-](/data/chemimg/128700/946612-19-7.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,12-difluoro-2,9-bis(2,2,3,3,4,4,4-heptafluorobutyl)-](/data/chemimg/128700/946612-19-7_b.png)