Co-reporter:Yichao Wu;Weichen Dai;Xin Chen;Aixin Geng;Yadong Chen;Yong Zhu
RSC Advances (2011-Present) 2017 vol. 7(Issue 82) pp:52180-52186
Publication Date(Web):2017/11/07
DOI:10.1039/C7RA08835C
Histone deacetylase (HDAC) inhibitors are known to induce multiple epigenetic modifications affecting signaling networks and act synergistically with phosphatidylinositol 3-kinase (PI3K) inhibitors for the treatment of cancer. Herein we present a novel design approach for cancer drug development by incorporating HDAC inhibitory functionality into a PI3K inhibitor pharmacophore to construct dual-acting inhibitors. The designed compounds were synthesized and showed inhibitory activities against PI3K and HDAC. The representative dual PI3K/HDAC inhibitors, compounds 12a–j, showed potent antiproliferative activities against K562 and Hut78 in cellular assays. This work may lay the foundation for developing novel dual PI3K/HDAC inhibitors as potential anticancer therapeutics.
Co-reporter:Yanmin Zhang, Danfeng Zhang, Haozhong Tian, Yu Jiao, Zhihao Shi, Ting Ran, Haichun Liu, Shuai Lu, Anyang Xu, Xin Qiao, Jing Pan, Lingfeng Yin, Weineng Zhou, Tao Lu, and Yadong Chen
Molecular Pharmaceutics 2016 Volume 13(Issue 9) pp:3106-3118
Publication Date(Web):August 2, 2016
DOI:10.1021/acs.molpharmaceut.6b00302
Covalent drugs have attracted increasing attention in recent years due to good inhibitory activity and selectivity. Targeting noncatalytic cysteines with irreversible inhibitors is a powerful approach for enhancing pharmacological potency and selectivity because cysteines can form covalent bonds with inhibitors through their nucleophilic thiol groups. However, most human kinases have multiple noncatalytic cysteines within the active site; to accurately predict which cysteine is most likely to form covalent bonds is of great importance but remains a challenge when designing irreversible inhibitors. In this work, FTMap was first applied to check its ability in predicting covalent binding site defined as the region where covalent bonds are formed between cysteines and irreversible inhibitors. Results show that it has excellent performance in detecting the hot spots within the binding pocket, and its hydrogen bond interaction frequency analysis could give us some interesting instructions for identification of covalent binding cysteines. Furthermore, we proposed a simple but useful covalent fragment probing approach and showed that it successfully predicted the covalent binding site of seven targets. By adopting a distance-based method, we observed that the closer the nucleophiles of covalent warheads are to the thiol group of a cysteine, the higher the possibility that a cysteine is prone to form a covalent bond. We believe that the combination of FTMap and our distance-based covalent fragment probing method can become a useful tool in detecting the covalent binding site of these targets.Keywords: covalent binding sites; cysteines; fragment probing; irreversible inhibitors;
Co-reporter:Zhimin Zhang, Shaohua Hou, Hongli Chen, Ting Ran, Fei Jiang, Yuanyuan Bian, Dewei Zhang, Yanle Zhi, Lu Wang, Li Zhang, Hongmei Li, Yanmin Zhang, Weifang Tang, Tao Lu, Yadong Chen
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 12) pp:2931-2935
Publication Date(Web):15 June 2016
DOI:10.1016/j.bmcl.2016.04.034
The bromodomain protein module and histone deacetylase (HDAC), which recognize and remove acetylated lysine, respectively, have emerged as important epigenetic therapeutic targets in cancer treatments. Herein we presented a novel design approach for cancer drug development by combination of bromodomain and HDAC inhibitory activity in one molecule. The designed compounds were synthesized which showed inhibitory activity against bromodomain 4 and HDAC1. The representative dual bromodomain/HDAC inhibitors, compound 11 and 12, showed potent antiproliferative activities against human leukaemia cell line K562 and MV4-11 in cellular assays. This work may lay the foundation for developing dual bromodomain/HDAC inhibitors as potential anticancer therapeutics.
Co-reporter:Xuejin Zhang; Zhen Gong; Jian Li
Journal of Chemical Information and Modeling 2015 Volume 55(Issue 10) pp:2138-2153
Publication Date(Web):September 22, 2015
DOI:10.1021/acs.jcim.5b00177
Intermolecular S···O interactions are very common and are important in biological systems, but until recently, the presence of these contacts in protein–ligand systems largely depended on serendipitous discovery instead of rational design. Here we provide insight into the phenomenon of intermolecular S···O contacts by focusing on three sulfur-containing aromatic rings. Quantum mechanics is employed to characterize the strength and directionality of the S···O interactions and to determine their energy dependence on their geometric parameters. Protein Data Bank mining is performed to systematically determine the occurrence and geometry of intermolecular S···O interactions, and several representative examples are discussed. Three typical cases are investigated using a combined quantum mechanics/molecular mechanics approach to demonstrate the potential of these interactions in improving binding affinities and physiochemical properties. Overall, our work elucidates the structures and energy features of intermolecular S···O interactions and addresses their use in molecular design.
Co-reporter:Weimin Yang, Yadong Chen, Xiang Zhou, Yazhou Gu, Wenqi Qian, Fan Zhang, Wei Han, Tao Lu, Weifang Tang
European Journal of Medicinal Chemistry 2015 Volume 89() pp:581-596
Publication Date(Web):7 January 2015
DOI:10.1016/j.ejmech.2014.10.039
•Compound 6a was designed by combining the scaffolds of UI-125 and Sorafenib.•SAR was explored from the 2-amino-3-purinylpyridine derivatives.•All compounds have B-RafV600E inhibitory activities at nanomolar ranges.•Some compounds showed potent antiproliferation against melanoma A375 cell line.•Compound 20g displayed promising antitumor efficacy in vivo.By combining the scaffolds of UI-125 and Sorafenib, a series of bis-aryl ureas and amides based on 2-amino-3-purinylpyridine moiety were designed and synthesized as novel DFG-out B-RafV600E inhibitors. Among them, 20c–e, 20g and 21h displayed potent antiproliferative activities against melanoma A375 (B-RafV600E) cell lines with IC50 values of 3.190, 2.276, 1.856, 1.632 μM and 1.839 μM, respectively, comparable with the positive control Vemurafenib (IC50 = 3.32 μM). Selected compounds were tested for the ERK inhibition in human melanoma A375 (B-RafV600E) and SK-MEL-2 (B-RafWT) cell lines by Western blot. The results revealed that our compounds inhibited the proliferation of melanoma A375 cells (B-RafV600E) through ERK pathway, without paradoxical activation of ERK in melanoma SK-MEL-2 cells (B-RafWT). Eventually, 20g and 21h were selected to confirm their inhibitory effects on tumor growth in A375 xenograft models in mice. Compound 20g exhibited equivalent antitumor efficacy in vivo (T/C = 44.37%), compared to Sorafenib (T/C = 37.35%), by 23-day repetitive administration of a single dose of 50 mg/kg without significant body weight loss.A series of 2-amino-3-purinylpyridine derivatives were synthesized and screened against B-RafV600E and A375 cell line, leading to the discovery of compound 20g, which exhibited potent A375 antiproliferative effects in vivo.
Co-reporter:Ting Ran, Zhimin Zhang, Kejun Liu, Yi Lu, Huifang Li, Jinxing Xu, Xiao Xiong, Yanmin Zhang, Anyang Xu, Shuai Lu, Haichun Liu, Tao Lu and Yadong Chen
Molecular BioSystems 2015 vol. 11(Issue 5) pp:1295-1304
Publication Date(Web):03 Mar 2015
DOI:10.1039/C4MB00723A
The bromodomain is a key protein–protein interaction module that specifically reads the acetylation marks of histones in epigenetic regulation. Currently, lots of inhibitors targeting the bromodomain have been reported as therapeutic agents. To better understand the interaction mechanism of bromodomain inhibitors, 20 diverse bromodomain inhibitors were studied using a combination of computational methods, including molecular docking, interaction fingerprinting, molecular dynamics simulation and binding free energy calculation. As a result, interactions important for the activity were critically analyzed, and the energy contribution in terms of individual residues was explored. These integrated results provided insights into two hot spots in the active site of the bromodomain, where the hydrophobic hot spot formed by Trp81, Val87, Leu92 and Ile146 played a central role in the interaction, and the hydrogen-bond hot spot mediated by Asn140 exhibited a moderate contribution to the binding affinity of the bromodomain inhibitors. This interaction mechanism study may facilitate the rational design of novel small-molecule bromodomain inhibitors.
Co-reporter:Yanmin Zhang;Yu Jiao;Xiao Xiong;Haichun Liu;Ting Ran;Jinxing Xu
Molecular Diversity 2015 Volume 19( Issue 4) pp:895-913
Publication Date(Web):2015 November
DOI:10.1007/s11030-015-9592-4
The discovery of novel scaffolds against a specific target has long been one of the most significant but challengeable goals in discovering lead compounds. A scaffold that binds in important regions of the active pocket is more favorable as a starting point because scaffolds generally possess greater optimization possibilities. However, due to the lack of sufficient chemical space diversity of the databases and the ineffectiveness of the screening methods, it still remains a great challenge to discover novel active scaffolds. Since the strengths and weaknesses of both fragment-based drug design and traditional virtual screening (VS), we proposed a fragment VS concept based on Bayesian categorization for the discovery of novel scaffolds. This work investigated the proposal through an application on VEGFR-2 target. Firstly, scaffold and structural diversity of chemical space for 10 compound databases were explicitly evaluated. Simultaneously, a robust Bayesian classification model was constructed for screening not only compound databases but also their corresponding fragment databases. Although analysis of the scaffold diversity demonstrated a very unevenly distribution of scaffolds over molecules, results showed that our Bayesian model behaved better in screening fragments than molecules. Through a literature retrospective research, several generated fragments with relatively high Bayesian scores indeed exhibit VEGFR-2 biological activity, which strongly proved the effectiveness of fragment VS based on Bayesian categorization models. This investigation of Bayesian-based fragment VS can further emphasize the necessity for enrichment of compound databases employed in lead discovery by amplifying the diversity of databases with novel structures.
Co-reporter:Haoliang Yuan, Jin Zhuang, Shihe Hu, Huifang Li, Jinxing Xu, Yaning Hu, Xiao Xiong, Yadong Chen, and Tao Lu
Journal of Chemical Information and Modeling 2014 Volume 54(Issue 9) pp:2544-2554
Publication Date(Web):September 2, 2014
DOI:10.1021/ci500268s
c-Met has been considered as an attractive target for developing antitumor agents. The highly selective c-Met inhibitors provide invaluable opportunities for the combination with other therapies safely to achieve the optimal efficacy. In this work, a series of triazolopyrazine c-Met inhibitors with exquisitely selectivity were investigated using a combination of molecular docking, three-dimensional quantitative structure–activity relationship (3D-QSAR), and molecular dynamics simulation. Comparative molecular field analysis (CoMFA) and comparative molecular similarity index analysis (CoMSIA) models were developed to reveal the structural determinants for c-Met inhibition. Both models were validated to have high reliability and predictability, and contour map analysis suggested feature requirements for different substituents on the scaffold. It is worth noting that an important hydrogen bond rich region was identified in the unique narrow channel, which is distinct from other kinases. Molecular dynamics simulations and binding free energy calculations provided further support that suitable groups in this hydrogen bond rich region made great contributions to the binding of ligands. Moreover, hydrogen bonds with residues of the narrow channel were also indicated to be essential to improve the activity and selectivity. This study will facilitate the discovery and optimization of novel c-Met inhibitors with higher activity and selectivity.
Co-reporter:Zeng Wu, Ming Yan, Shi-He Hu, Zhi-Cheng Yu, Yong Zhu, Ya-Dong Cheng, Hai-Chun Liu, Yan-Min Zhang, Si-Hui Yao, Wei-Fang Tang, Tao Lu
Chinese Chemical Letters 2014 Volume 25(Issue 2) pp:351-354
Publication Date(Web):February 2014
DOI:10.1016/j.cclet.2013.11.006
A series of novel indole derivatives were designed and synthesized and their inhibitory activity against B-Raf and HepG2 cell were also described. Among them, compounds 7a and 7b exhibited excellent potency, which showed the potential for further research as lead compounds.A series of novel indole derivatives were designed and synthesized and their inhibitory activity against B-Raf and HepG2 cells were also described. Among them, compounds 7a and 7b exhibited excellent potency, which showed the potential for further research as lead compounds.
Co-reporter:Yanmin Zhang, Shangyan Yang, Yu Jiao, Haichun Liu, Haoliang Yuan, Shuai Lu, Ting Ran, Sihui Yao, Zhipeng Ke, Jinxing Xu, Xiao Xiong, Yadong Chen, and Tao Lu
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 12) pp:3163-3177
Publication Date(Web):November 22, 2013
DOI:10.1021/ci400429g
In recent years, various virtual screening (VS) tools have been developed, and many successful screening campaigns have been showcased. However, whether by conventional molecular docking or pharmacophore screening, the selection of virtual hits is based on the ranking of compounds by scoring functions or fit values, which remains the bottleneck of VS due to insufficient accuracy. As the limitations of individual methods persist, a comprehensive comparison and integration of different methods may provide insights into selecting suitable methods for VS. Here, we evaluated the performance of molecular docking, fingerprint-based 2D similarity and multicomplex pharmacophore in an individual and a combined manner, through a retrospective VS study on VEGFR-2 inhibitors. An integrated two-layer workflow was developed and validated through VS of VEGFR-2 inhibitors against the DUD-E database, which demonstrated improved VS performance through a ligand-based method ECFP_4, followed by molecular docking, and then a strict multicomplex pharmacophore. Through a retrospective comparison with six published papers, this integrated approach outperformed 43 out of 45 methods, indicating a great effectiveness. This kind of integrated VS approach can be extended to other targets for the screening and discovery of inhibitors.
Co-reporter:Haoliang Yuan;Wenting Tai;Shihe Hu
Journal of Computer-Aided Molecular Design 2013 Volume 27( Issue 10) pp:897-915
Publication Date(Web):2013 October
DOI:10.1007/s10822-013-9687-x
Fragment-based drug design has emerged as an important methodology for lead discovery and drug design. Different with other studies focused on fragment library design and active fragment identification, a fragment-based strategy was developed in combination with three-dimensional quantitative structure–activity relationship (3D-QSAR) for structural optimization in this study. Based on a validated scaffold or fragment hit, a series of structural optimization was conducted to convert it to lead compounds, including 3D-QSAR modelling, active site analysis, fragment-based structural optimization and evaluation of new molecules. 3D-QSAR models and active site analysis provided sufficient information for confirming the SAR and pharmacophoric features for fragments. This strategy was evaluated through the structural optimization on a c-Met inhibitor scaffold 5H-benzo[4,5]cyclohepta[1,2-b]pyridin-5-one, which resulted in an c-Met inhibitor with high inhibitory activity. Our study suggested the effectiveness of this fragment-based strategy and the druggability of our newly explored active region. The reliability of this strategy indicated it could also be applied to facilitate lead optimization of other targets.
Co-reporter:Shuai Lu;Shan-Liang Sun;Hai-Chun Liu;Ya-Dong Chen;Hao-Liang Yuan;Yi-Ping Gao;Pei Yang
Chemical Biology & Drug Design 2012 Volume 80( Issue 2) pp:328-339
Publication Date(Web):
DOI:10.1111/j.1747-0285.2012.01412.x
Polo-like kinase 1 is an important and attractive oncological target that plays a key role in mitosis and cytokinesis. A combined pharmacophore- and docking-based virtual screening was performed to identify novel polo-like kinase 1 inhibitors. A total of 34 hit compounds were selected and tested in vitro, and some compounds showed inhibition of polo-like kinase 1 and human tumor cell growth. The most potent compound (66) inhibited polo-like kinase 1 with an IC50 value of 6.99 μm. The docked binding models of two hit compounds were discussed in detail. These compounds contained novel chemical scaffolds and may be used as foundations for the development of novel classes of polo-like kinase 1 inhibitors.
Co-reporter:Botao Xin, Weifang Tang, Yue Wang, Guowu Lin, Haichun Liu, Yu Jiao, Yong Zhu, Haoliang Yuan, Yadong Chen, Tao Lu
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 14) pp:4783-4786
Publication Date(Web):15 July 2012
DOI:10.1016/j.bmcl.2012.05.053
β-Carboline family of compounds is a large group of alkaloids widely distributed in nature and exhibits broad-spectrum anti-tumor activities. We designed and synthesized two series of novel 1-carboxamide- and 6-sulfonamide-substituted β-carboline derivatives 7a–p and 12a–b, and their wild type B-Raf kinase inhibitory activities were described. Most compounds showed moderate to excellent inhibitory activities. Among them, 1-carboxamide-6-(N-(3-(dimethylamino)propyl)-sulfamoyl)-β-carboline, 7e exhibited potent activity (IC50 = 1.62 μM), showing the potential for further investigation as a lead compound.Two series of novel 1-carboxamide and 6-sulfonamide substituted β-carboline derivatives 7a–o, 12a–b were designed and synthesized and their wild type B-Raf kinase inhibitory activities were described. Compound 1-carboxamide-6-(N-(3-(dimethylamino)propyl)sulfamoyl)-β-carboline, 7e exhibited potent activity (IC50 = 1.62 μM), showing the potential for further investigation as a lead compound.
Co-reporter:Yanmin Zhang;Haichun Liu;Yu Jiao;Haoliang Yuan;Fengxiao Wang
Molecular Diversity 2012 Volume 16( Issue 4) pp:787-802
Publication Date(Web):2012 November
DOI:10.1007/s11030-012-9405-y
Vascular endothelial growth factor (VEGF) and its receptor tyrosine kinase VEGFR-2 or kinase insert domain receptor (KDR) have been identified as promising targets for novel anticancer agents. To achieve new potent inhibitors of KDR, we conducted molecular fragment replacement (MFR) studies for the understanding of 3D-QSAR modeling and the docking investigation of arylphthalazines and 2-((1H-Azol-1-yl)methyl)-N-arylbenzamides-based KDR inhibitors. Two favorable 3D-QSAR models (CoMFA with q2, 0.671; r2, 0.969; CoMSIA with q2, 0.608; r2, 0.936) have been developed to predict the biological activity of new compounds. The new molecular database generated by MFR was virtually screened using Glide (docking) and further evaluated with CoMFA prediction, protein–ligand interaction fingerprint (PLIF) and ADMET analysis. 44 N-(pyridin-4-ylmethyl)aniline derivatives as novel potential KDR inhibitors were finally obtained. In this paper, the work flow developed could be applied to de novo drug design and virtual screening potential KDR inhibitors, and use hit compounds to further optimize and design new potential KDR inhibitors.
Co-reporter:Yong Zhu, Hui-Fang Li, Shuai Lu, Yi-Xuan Zheng, Zeng Wu, Wei-Fang Tang, Xiang Zhou, Tao Lu
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 5) pp:1777-1791
Publication Date(Web):May 2010
DOI:10.1016/j.ejmech.2010.01.010
A three dimensional (3D) chemical feature based pharmacophore model was developed for selective histone deacetylase 1 (HDAC1) inhibitors, which provides an efficient way to discuss the isoform selectivity of HDAC inhibitors. In contrast to the classical pan-HDAC pharmacophore, two hydrophobic features (HY and HYAr2) were found in the chemical feature based pharmacophore model, which might be responsible for the selectivity of HDAC1 inhibitions. Molecular docking also highlighted the two hydrophobic features, which are located in the internal cavity adjacent to the active site. The results contribute to our understanding of the molecular mechanism underlying the selectivity of HDAC1 inhibitors and suggest a possible target region to design novel selective HDAC1 inhibitors.A selective pharmacophore model was developed based on a series of selective HDAC1 inhibitors. Two hydrophobic features (HY and HYAr2) were responsible for the selectivity of HDAC1 inhibitions.
Co-reporter:Tian Zhu, Yu Jiao, Ya-Dong Chen, Xuan Wang, Hui-Fang Li, Lu-Yong Zhang, Tao Lu
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 7) pp:2346-2350
Publication Date(Web):1 April 2008
DOI:10.1016/j.bmcl.2008.02.068
A three-dimensional pharmacophore model was developed based on 25 currently available Raf-1 kinase inhibitors. The best pharmacophore hypothesis (Hypo1), consisting of four chemical features (one hydrogen-bond acceptor, one hydrogen-bond donor, and two hydrophobic groups), has a correlation coefficient of 0.972. The results of our study provide a valuable tool in designing new leads with desired biological activity by virtual screening.The best pharmacophore hypothesis (Hypo1), consisting of four chemical features, and the high active compound (compound 6) can be well mapped onto the Hypo1 model.

![7-(3,5-Dimethylisoxazol-4-yl)-8-methoxy-1-((R)-1-(pyridin-2-yl)ethyl)-1H-imidazo[4,5-c]quinolin-2(3H)-one](http://img.cochemist.com/ccimg/1300100/1300031-49-5.png)
![7-(3,5-Dimethylisoxazol-4-yl)-8-methoxy-1-((R)-1-(pyridin-2-yl)ethyl)-1H-imidazo[4,5-c]quinolin-2(3H)-one](http://img.cochemist.com/ccimg/1300100/1300031-49-5_b.png)
![Methyl N-[(2s)-1-[[4-[3-[5-chloro-2-fluoro-3-(methanesulfonamido)phenyl]-1-propan-2-ylpyrazol-4-yl]pyrimidin-2-yl]amino]propan-2-yl]carbamate](http://img.cochemist.com/ccimg/1269500/1269440-17-6.png)
![Methyl N-[(2s)-1-[[4-[3-[5-chloro-2-fluoro-3-(methanesulfonamido)phenyl]-1-propan-2-ylpyrazol-4-yl]pyrimidin-2-yl]amino]propan-2-yl]carbamate](http://img.cochemist.com/ccimg/1269500/1269440-17-6_b.png)
![(S)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-70-4.png)
![(S)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-70-4_b.png)









![2-[(4S)-6-(4-CHLOROPHENYL)-8-METHOXY-1-METHYL-4H-[1,2,4]TRIAZOLO[4,3-A][1,4]BENZODIAZEPIN-4-YL]-N-ETHYLACETAMIDE](http://img.cochemist.com/ccimg/1261000/1260907-17-2.png)
![2-[(4S)-6-(4-CHLOROPHENYL)-8-METHOXY-1-METHYL-4H-[1,2,4]TRIAZOLO[4,3-A][1,4]BENZODIAZEPIN-4-YL]-N-ETHYLACETAMIDE](http://img.cochemist.com/ccimg/1261000/1260907-17-2_b.png)
![1-[3-(6,7-dimethoxyquinazolin-4-yl)oxyphenyl]-3-[5-(1,1,1-trifluoro-2-methylpropan-2-yl)-1,2-oxazol-3-yl]urea](http://img.cochemist.com/ccimg/1189000/1188910-76-0.png)
![1-[3-(6,7-dimethoxyquinazolin-4-yl)oxyphenyl]-3-[5-(1,1,1-trifluoro-2-methylpropan-2-yl)-1,2-oxazol-3-yl]urea](http://img.cochemist.com/ccimg/1189000/1188910-76-0_b.png)


![Benzenesulfonamide, N-[3-(4-chlorophenyl)-1-phenyl-2-propen-1-ylidene]-4-methyl-](/data/chemimg/144700/1116669-73-8.png)
![Benzenesulfonamide, N-[3-(4-chlorophenyl)-1-phenyl-2-propen-1-ylidene]-4-methyl-](/data/chemimg/144700/1116669-73-8_b.png)
![2-[(1R)-1-[(6-AMINO-5-CHLOROPYRIMIDINE-4-CARBONYL)AMINO]ETHYL]-N-[5-CHLORO-4-(TRIFLUOROMETHYL)PYRIDIN-2-YL]-1,3-THIAZOLE-5-CARBOXAMIDE](http://img.cochemist.com/ccimg/1096800/1096708-71-2.png)
![2-[(1R)-1-[(6-AMINO-5-CHLOROPYRIMIDINE-4-CARBONYL)AMINO]ETHYL]-N-[5-CHLORO-4-(TRIFLUOROMETHYL)PYRIDIN-2-YL]-1,3-THIAZOLE-5-CARBOXAMIDE](http://img.cochemist.com/ccimg/1096800/1096708-71-2_b.png)




![6-(4-Methyl-1-piperazinyl)-N-(5-methyl-1H-pyrazol-3-yl)-2-[(1E)-2-phenylethenyl]-4-pyrimidinamine](http://img.cochemist.com/ccimg/934400/934353-76-1.png)
![6-(4-Methyl-1-piperazinyl)-N-(5-methyl-1H-pyrazol-3-yl)-2-[(1E)-2-phenylethenyl]-4-pyrimidinamine](http://img.cochemist.com/ccimg/934400/934353-76-1_b.png)




![1-Cyclopropyl-3-(3-(5-(morpholinomethyl)-1H-benzo[d]imidazol-2-yl)-1H-pyrazol-4-yl)urea](http://img.cochemist.com/ccimg/896500/896466-04-9.png)
![1-Cyclopropyl-3-(3-(5-(morpholinomethyl)-1H-benzo[d]imidazol-2-yl)-1H-pyrazol-4-yl)urea](http://img.cochemist.com/ccimg/896500/896466-04-9_b.png)
![1,2,3-Cyclohexanetriol,4-[3-[(4-ethylphenyl)methyl]phenyl]-6-(hydroxymethyl)-,(1R,2R,3S,4S,6R)-](http://img.cochemist.com/ccimg/875200/875111-69-6.png)
![1,2,3-Cyclohexanetriol,4-[3-[(4-ethylphenyl)methyl]phenyl]-6-(hydroxymethyl)-,(1R,2R,3S,4S,6R)-](http://img.cochemist.com/ccimg/875200/875111-69-6_b.png)
![1,2,3-Cyclohexanetriol,4-(hydroxymethyl)-6-[3-[(4-methoxyphenyl)methyl]phenyl]-,(1S,2R,3R,4R,6S)-](http://img.cochemist.com/ccimg/875200/875110-92-2.png)
![1,2,3-Cyclohexanetriol,4-(hydroxymethyl)-6-[3-[(4-methoxyphenyl)methyl]phenyl]-,(1S,2R,3R,4R,6S)-](http://img.cochemist.com/ccimg/875200/875110-92-2_b.png)
![2-Propynoic acid, [4-(trifluoromethyl)phenyl]methyl ester](http://img.cochemist.com/ccimg/873800/873785-23-0.png)
![2-Propynoic acid, [4-(trifluoromethyl)phenyl]methyl ester](http://img.cochemist.com/ccimg/873800/873785-23-0_b.png)



![Piperidine, 1-methyl-4-[[4-nitro-2-(trifluoromethyl)phenoxy]methyl]-](http://img.cochemist.com/ccimg/853300/853297-69-5.png)
![Piperidine, 1-methyl-4-[[4-nitro-2-(trifluoromethyl)phenoxy]methyl]-](http://img.cochemist.com/ccimg/853300/853297-69-5_b.png)
![Benzenamine, 4-[(1-methyl-4-piperidinyl)methoxy]-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/853300/853297-68-4.png)
![Benzenamine, 4-[(1-methyl-4-piperidinyl)methoxy]-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/853300/853297-68-4_b.png)






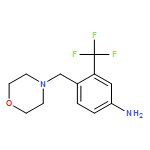
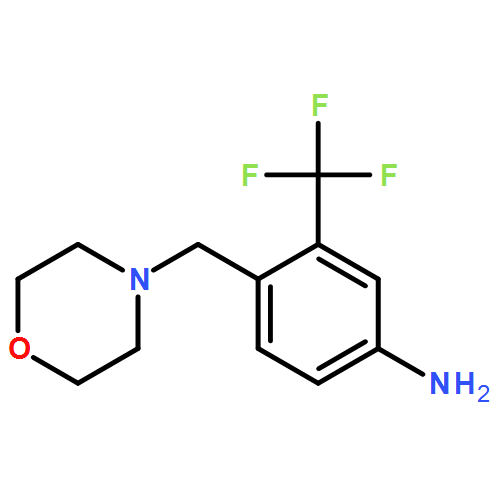
![Morpholine, 4-[[4-nitro-2-(trifluoromethyl)phenyl]methyl]-](/data/chemimg/1933600/694499-28-0.png)
![Morpholine, 4-[[4-nitro-2-(trifluoromethyl)phenyl]methyl]-](/data/chemimg/1933600/694499-28-0_b.png)


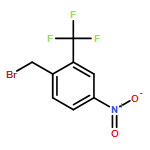
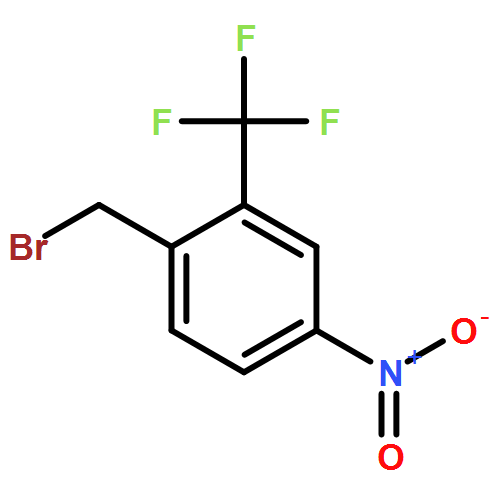
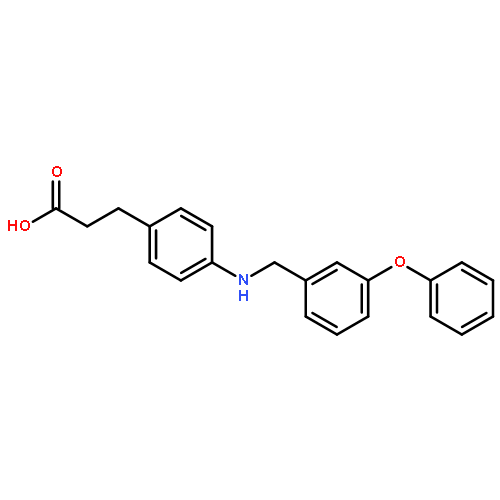
![Benzenepropanoic acid, 4-[[3-(2-methylphenoxy)phenyl]methoxy]-](http://img.cochemist.com/ccimg/692000/691903-08-9.png)
![Benzenepropanoic acid, 4-[[3-(2-methylphenoxy)phenyl]methoxy]-](http://img.cochemist.com/ccimg/692000/691903-08-9_b.png)
![BENZENEPROPANOIC ACID, 4-[[3-(2,6-DIMETHYLPHENOXY)PHENYL]METHOXY]-](http://img.cochemist.com/ccimg/692000/691903-04-5.png)
![BENZENEPROPANOIC ACID, 4-[[3-(2,6-DIMETHYLPHENOXY)PHENYL]METHOXY]-](http://img.cochemist.com/ccimg/692000/691903-04-5_b.png)
![BENZENEPROPANOIC ACID, 4-[[3-(3-METHYLPHENOXY)PHENYL]METHOXY]-](http://img.cochemist.com/ccimg/692000/691902-91-7.png)
![BENZENEPROPANOIC ACID, 4-[[3-(3-METHYLPHENOXY)PHENYL]METHOXY]-](http://img.cochemist.com/ccimg/692000/691902-91-7_b.png)
![BENZENEPROPANOIC ACID, 4-[(2'-METHYL[1,1'-BIPHENYL]-3-YL)METHOXY]-](http://img.cochemist.com/ccimg/692000/691901-94-7.png)
![BENZENEPROPANOIC ACID, 4-[(2'-METHYL[1,1'-BIPHENYL]-3-YL)METHOXY]-](http://img.cochemist.com/ccimg/692000/691901-94-7_b.png)
![Benzenepropanoic acid, 4-[(2'-chloro[1,1'-biphenyl]-3-yl)methoxy]-](http://img.cochemist.com/ccimg/692000/691901-54-9.png)
![Benzenepropanoic acid, 4-[(2'-chloro[1,1'-biphenyl]-3-yl)methoxy]-](http://img.cochemist.com/ccimg/692000/691901-54-9_b.png)
![Benzenepropanoic acid, 4-[(2',6'-dimethyl[1,1'-biphenyl]-3-yl)methoxy]-](/data/chemimg/3373000/691900-43-3.png)
![Benzenepropanoic acid, 4-[(2',6'-dimethyl[1,1'-biphenyl]-3-yl)methoxy]-](/data/chemimg/3373000/691900-43-3_b.png)




![(S)-2-Methoxy-3-(4-(2-(5-methyl-2-phenyloxazol-4-yl)ethoxy)benzo[b]thiophen-7-yl)propanoic acid](http://img.cochemist.com/ccimg/475500/475479-34-6.png)
![(S)-2-Methoxy-3-(4-(2-(5-methyl-2-phenyloxazol-4-yl)ethoxy)benzo[b]thiophen-7-yl)propanoic acid](http://img.cochemist.com/ccimg/475500/475479-34-6_b.png)




![4-amino-5-fluoro-3-[5-(4-methylpiperazin-1-yl)-1h-benzimidazol-2-yl]quinolin-2(1h)-one](http://img.cochemist.com/ccimg/405200/405169-16-6.png)
![4-amino-5-fluoro-3-[5-(4-methylpiperazin-1-yl)-1h-benzimidazol-2-yl]quinolin-2(1h)-one](http://img.cochemist.com/ccimg/405200/405169-16-6_b.png)






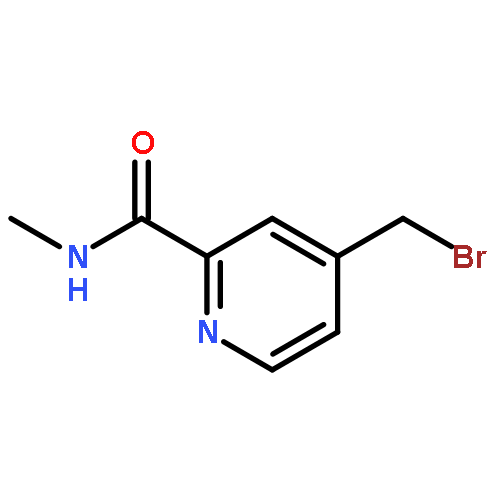
![4-Pyridinecarboxylicacid, 2-[(methylamino)carbonyl]-, ethyl ester](http://img.cochemist.com/ccimg/332100/332013-42-0.png)
![4-Pyridinecarboxylicacid, 2-[(methylamino)carbonyl]-, ethyl ester](http://img.cochemist.com/ccimg/332100/332013-42-0_b.png)



![Piperidine, 1-methyl-4-[4-nitro-2-(trifluoromethyl)phenoxy]-](http://img.cochemist.com/ccimg/325500/325457-78-1.png)
![Piperidine, 1-methyl-4-[4-nitro-2-(trifluoromethyl)phenoxy]-](http://img.cochemist.com/ccimg/325500/325457-78-1_b.png)

![2-phenyl-N-[5-[2-[2-[5-[(2-phenylacetyl)amino]-1,3,4-thiadiazol-2-yl]ethylsulfanyl]ethyl]-1,3,4-thiadiazol-2-yl]acetamide](http://img.cochemist.com/ccimg/314100/314045-39-1.png)
![2-phenyl-N-[5-[2-[2-[5-[(2-phenylacetyl)amino]-1,3,4-thiadiazol-2-yl]ethylsulfanyl]ethyl]-1,3,4-thiadiazol-2-yl]acetamide](http://img.cochemist.com/ccimg/314100/314045-39-1_b.png)
![Piperazine, 1-methyl-4-[4-nitro-2-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/309800/309734-66-5.png)
![Piperazine, 1-methyl-4-[4-nitro-2-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/309800/309734-66-5_b.png)













![Thieno[2,3-d]pyrimidine-2,4-diamine, 5-[2-(2,5-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/152100/152093-77-1.png)
![Thieno[2,3-d]pyrimidine-2,4-diamine, 5-[2-(2,5-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/152100/152093-77-1_b.png)

















![12H-1,12-Ethano-9H-pyrido[1,2,3-lm]pyrrolo[2,3-d]carbazol-9-one,2,3,10,11b,13,13a-hexahydro-14-(2-hydroxyethylidene)-5,6-dimethoxy-,(3aR,11bS,12S,13aS)-](http://img.cochemist.com/ccimg/129800/129724-78-3.png)
![12H-1,12-Ethano-9H-pyrido[1,2,3-lm]pyrrolo[2,3-d]carbazol-9-one,2,3,10,11b,13,13a-hexahydro-14-(2-hydroxyethylidene)-5,6-dimethoxy-,(3aR,11bS,12S,13aS)-](http://img.cochemist.com/ccimg/129800/129724-78-3_b.png)




![Benzamide,N-[(9S,10R,11R,13R)-2,3,10,11,12,13-hexahydro-10-methoxy-9-methyl-1-oxo-9,13-epoxy-1H,9H-diindolo[1,2,3-gh:3',2',1'-lm]pyrrolo[3,4-j][1,7]benzodiazonin-11-yl]-N-methyl-](http://img.cochemist.com/ccimg/120700/120685-11-2.png)
![Benzamide,N-[(9S,10R,11R,13R)-2,3,10,11,12,13-hexahydro-10-methoxy-9-methyl-1-oxo-9,13-epoxy-1H,9H-diindolo[1,2,3-gh:3',2',1'-lm]pyrrolo[3,4-j][1,7]benzodiazonin-11-yl]-N-methyl-](http://img.cochemist.com/ccimg/120700/120685-11-2_b.png)


![Pyrido[1,2-a:3,4-b']diindol-5-ium,12,13-dihydro-13-oxo-, chloride](http://img.cochemist.com/ccimg/114800/114719-57-2.png)
![Pyrido[1,2-a:3,4-b']diindol-5-ium,12,13-dihydro-13-oxo-, chloride](http://img.cochemist.com/ccimg/114800/114719-57-2_b.png)








![Benzoic acid,4-[[(1,1-dimethylethyl)amino]sulfonyl]-](http://img.cochemist.com/ccimg/100000/99987-05-0.png)
![Benzoic acid,4-[[(1,1-dimethylethyl)amino]sulfonyl]-](http://img.cochemist.com/ccimg/100000/99987-05-0_b.png)




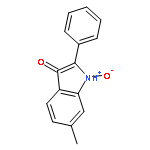
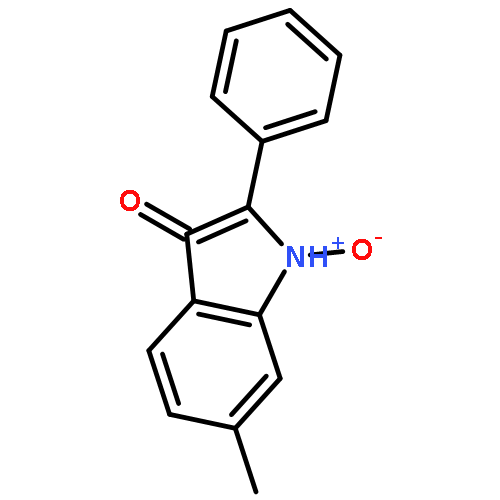
![Ethyl 3-(benzo[d][1,3]dioxol-5-yl)-3-oxopropanoate](http://img.cochemist.com/ccimg/81600/81581-27-3.png)
![Ethyl 3-(benzo[d][1,3]dioxol-5-yl)-3-oxopropanoate](http://img.cochemist.com/ccimg/81600/81581-27-3_b.png)
![N-{2-[(2-METHYL-2-PROPANYL)AMINO]-2-OXOETHYL}-N-PHENYL-1,2,3-THIA<WBR />DIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/79200/79183-44-1.png)
![N-{2-[(2-METHYL-2-PROPANYL)AMINO]-2-OXOETHYL}-N-PHENYL-1,2,3-THIA<WBR />DIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/79200/79183-44-1_b.png)


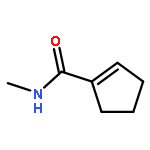
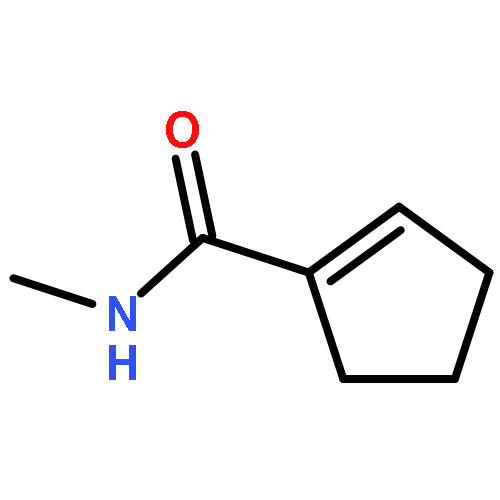




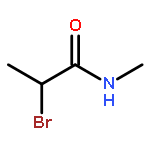
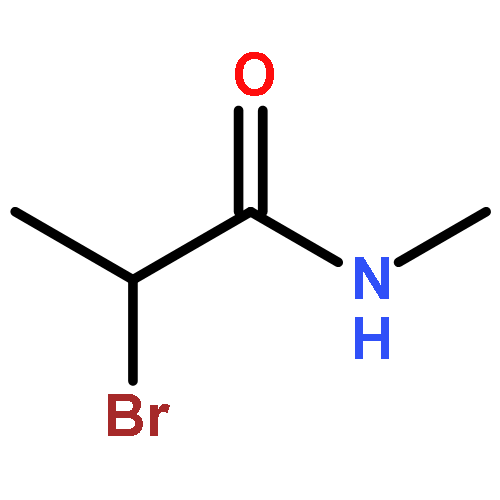
![9H-PYRIDO[3,4-B]INDOLE-3-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/74300/74214-63-4.png)
![9H-PYRIDO[3,4-B]INDOLE-3-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/74300/74214-63-4_b.png)

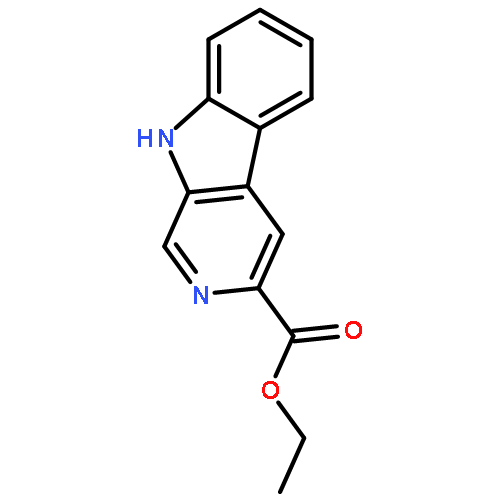




















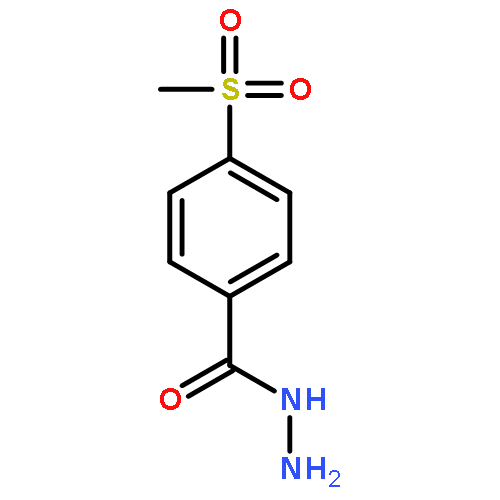
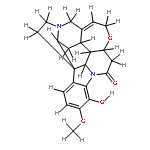
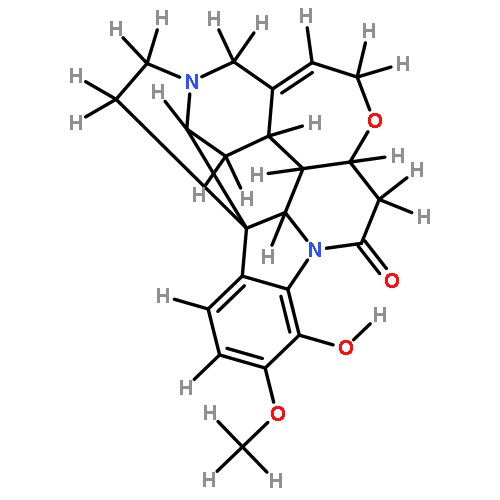
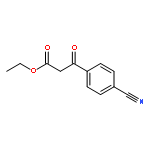



![2,3,4,9-tetrahydro-1H-Pyrido[3,4-b]indole-3-carboxylic acid ethyl ester](http://img.cochemist.com/ccimg/41400/41300-23-6.png)
![2,3,4,9-tetrahydro-1H-Pyrido[3,4-b]indole-3-carboxylic acid ethyl ester](http://img.cochemist.com/ccimg/41400/41300-23-6_b.png)

























![Acetic acid, 2-[1,2-dihydro-2-oxo-1-(phenylmethyl)-3H-indol-3-ylidene]-, ethyl ester, (2E)-](/data/chemimg/1088700/13672-24-7.png)
![Acetic acid, 2-[1,2-dihydro-2-oxo-1-(phenylmethyl)-3H-indol-3-ylidene]-, ethyl ester, (2E)-](/data/chemimg/1088700/13672-24-7_b.png)


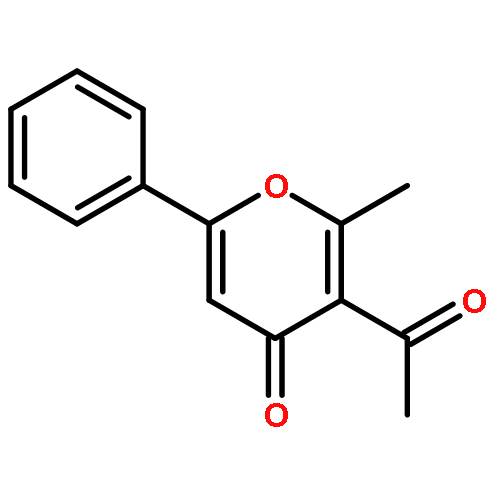




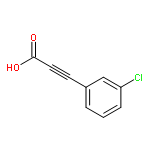
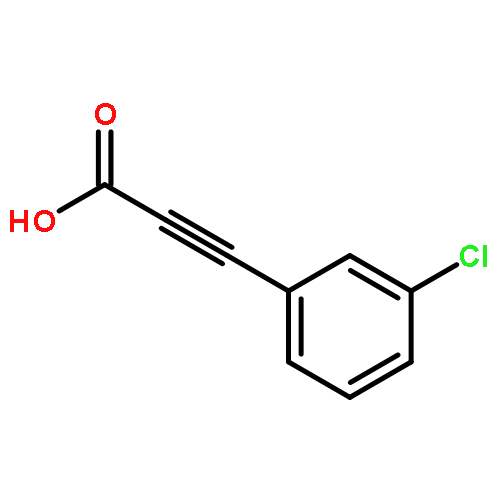
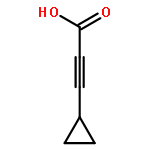
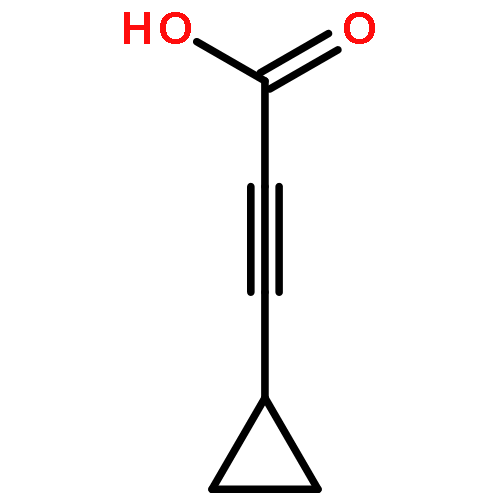






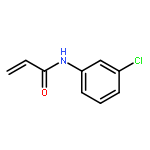


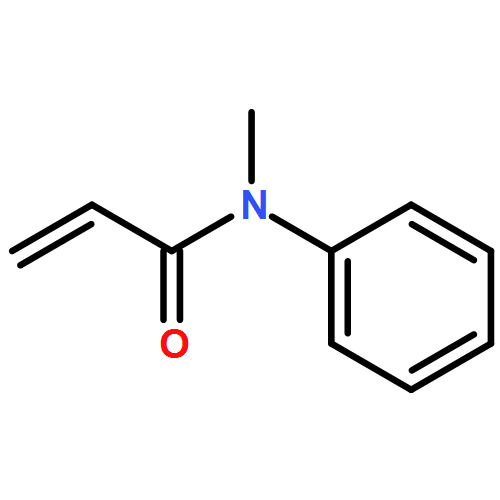
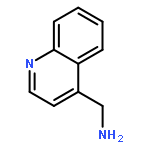


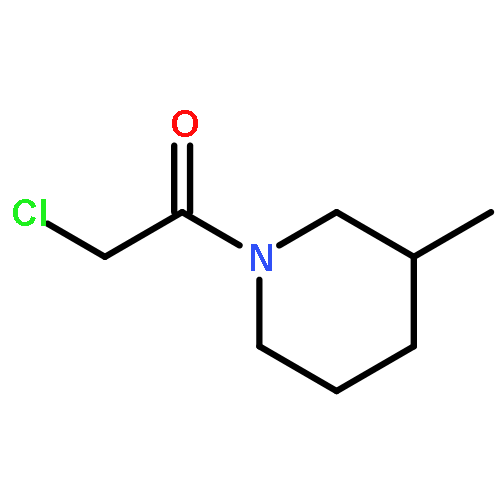
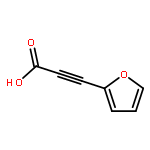
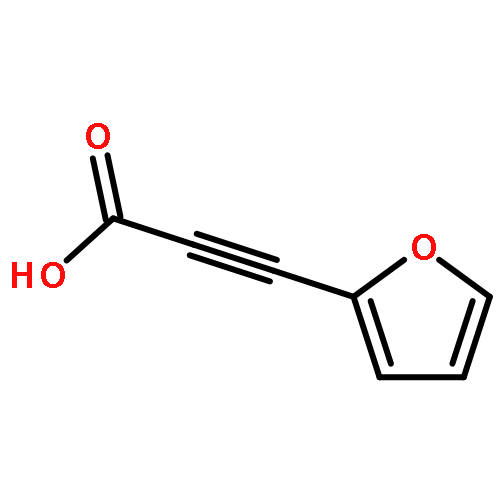


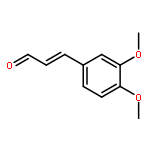



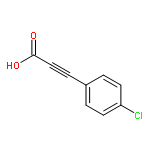
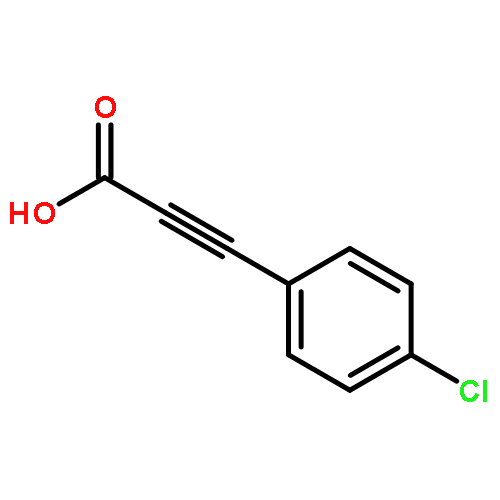
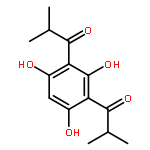
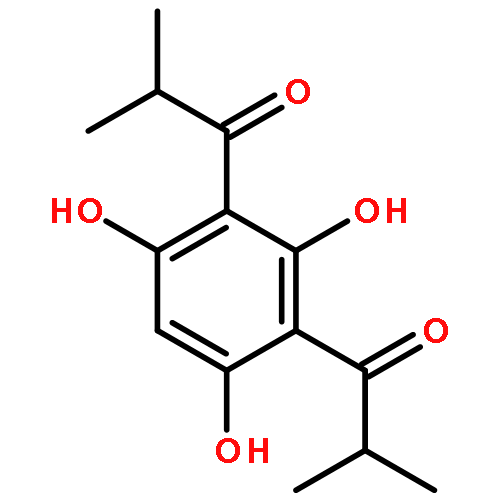



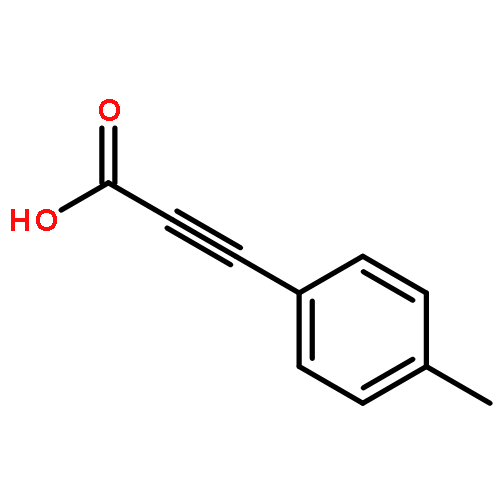




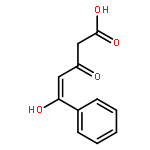
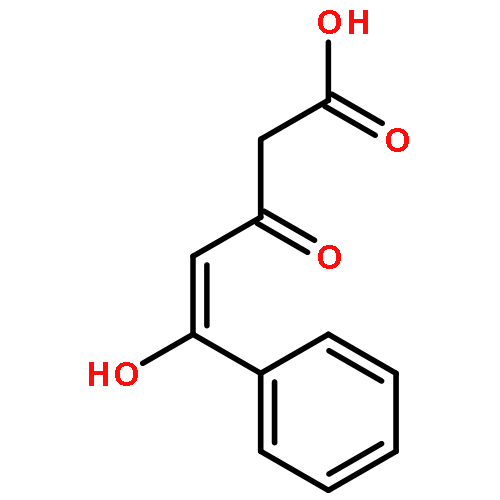
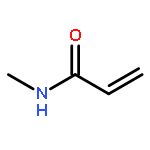
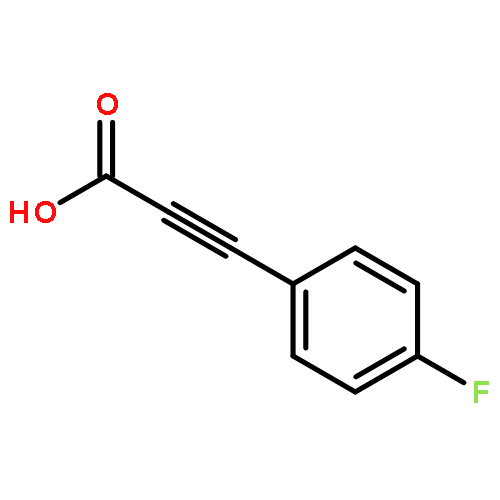
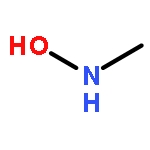
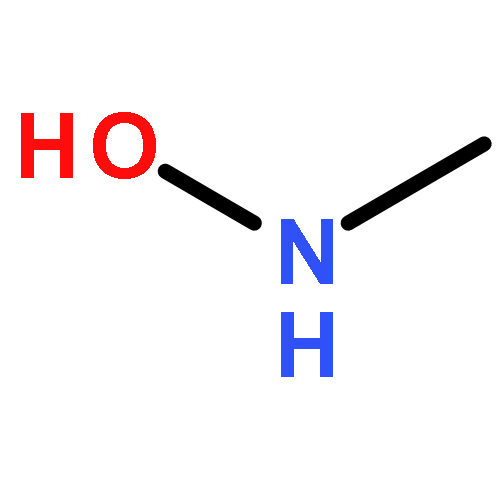










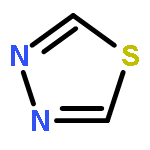
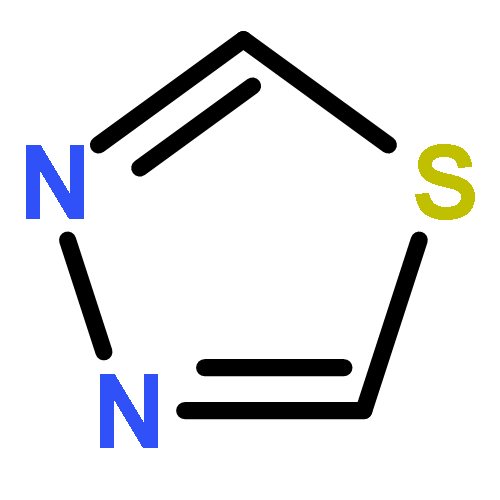
![Pyrazolo[1,5-a]-1,3,5-triazine](http://img.cochemist.com/ccimg/300/274-72-6.png)
![Pyrazolo[1,5-a]-1,3,5-triazine](http://img.cochemist.com/ccimg/300/274-72-6_b.png)
![12H-6a,4-(Ethaniminomethano)indolo[3,2,1-ij]oxepino[2,3,4-de]quinoline-6,12(2H)-dione,4a,5,13,13a,13b,13c-hexahydro-10-hydroxy-16-methyl-, (4aR,6aS,13aS,13bR,13cS)-](http://img.cochemist.com/ccimg/200/125-15-5.png)
![12H-6a,4-(Ethaniminomethano)indolo[3,2,1-ij]oxepino[2,3,4-de]quinoline-6,12(2H)-dione,4a,5,13,13a,13b,13c-hexahydro-10-hydroxy-16-methyl-, (4aR,6aS,13aS,13bR,13cS)-](http://img.cochemist.com/ccimg/200/125-15-5_b.png)


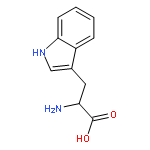
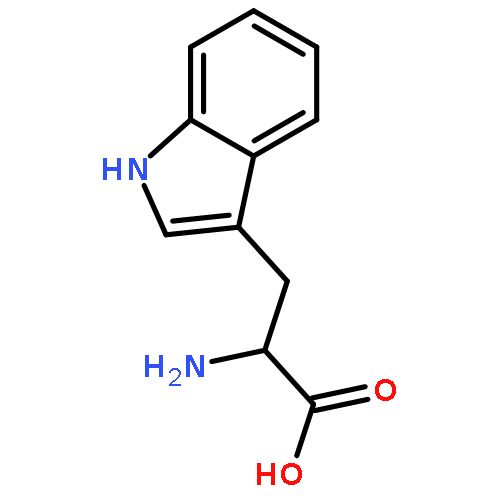
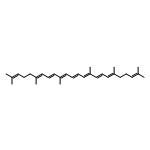













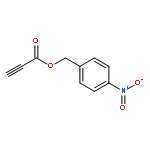
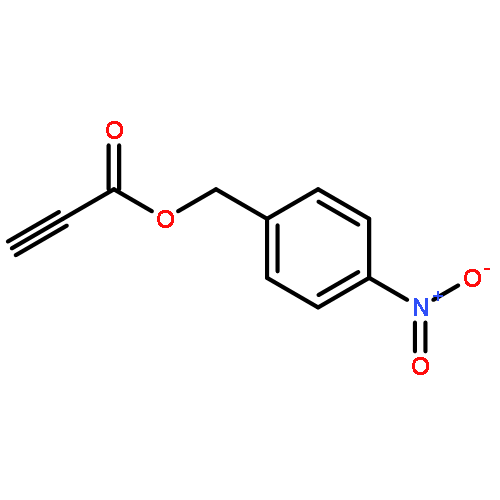










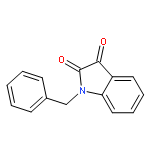
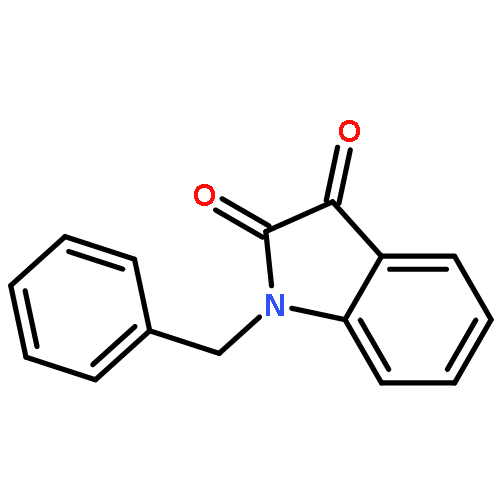










![2-(2,4,6-trimethylphenyl)-2,5,6,7-tetrahydropyrrolo[2,1-c][1,2,4]triazol-4-ium chloride](http://img.cochemist.com/ccimg/862900/862893-81-0.png)
![2-(2,4,6-trimethylphenyl)-2,5,6,7-tetrahydropyrrolo[2,1-c][1,2,4]triazol-4-ium chloride](http://img.cochemist.com/ccimg/862900/862893-81-0_b.png)