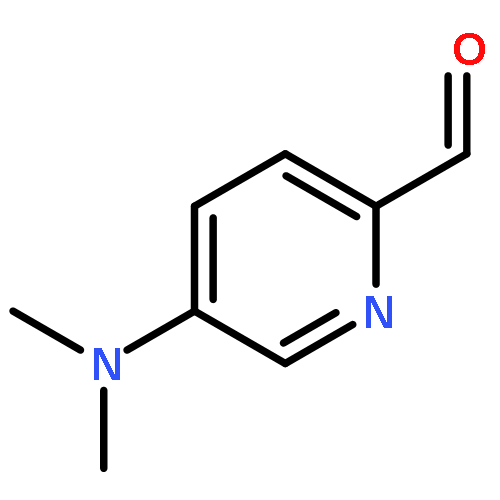
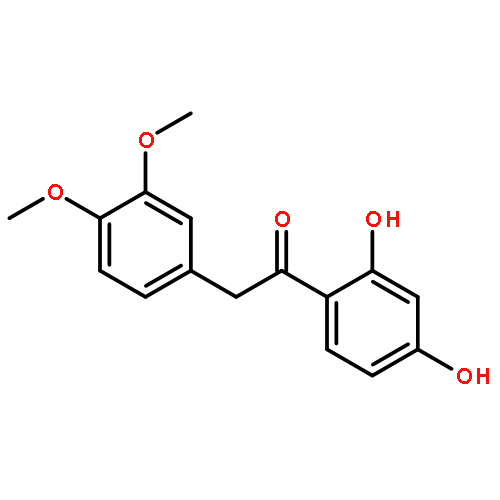
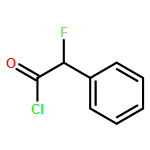
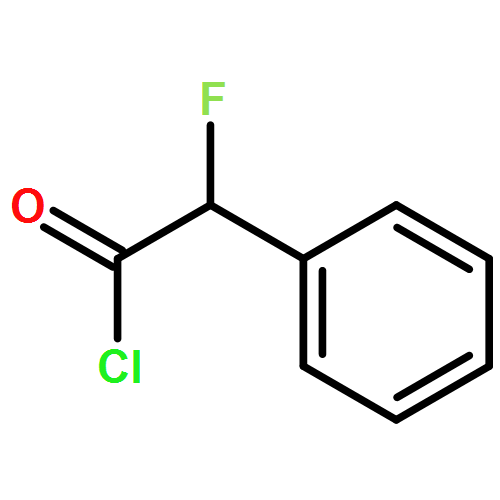
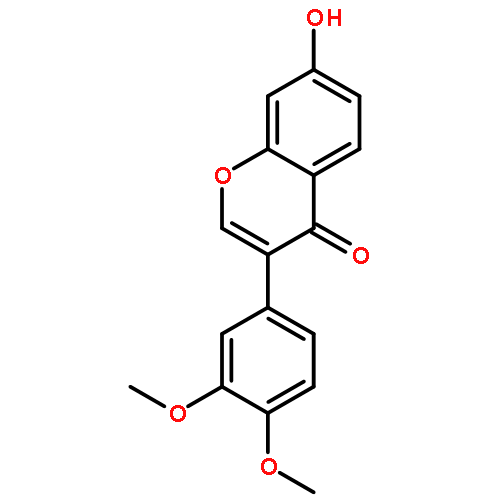
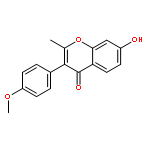
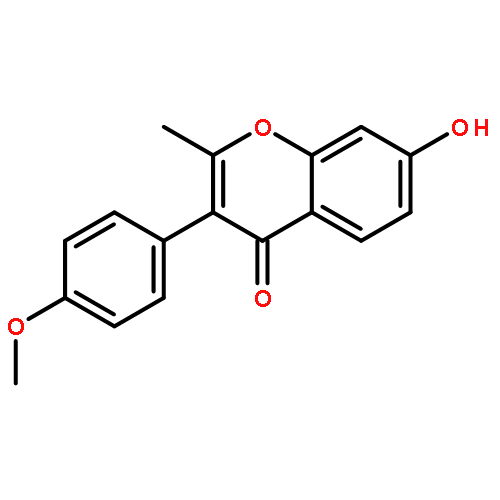
Co-reporter:Mykhaylo S. Frasinyuk;Wen Zhang;Przemyslaw Wyrebek;Tianxin Yu;Xuehe Xu;Vitaliy M. Sviripa;Svitlana P. Bondarenko;Yanqi Xie;Huy X. Ngo;Andrew J. Morris;James L. Mohler;Michael V. Fiandalo;David S. Watt;Chunming Liu
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 36) pp:7623-7629
Publication Date(Web):2017/09/20
DOI:10.1039/C7OB01584D
Cytisine-linked isoflavonoids (CLIFs) inhibited PC-3 prostate and LS174T colon cancer cell proliferation by inhibiting a peroxisomal bifunctional enzyme. A pull-down assay using a biologically active, biotin-modified CLIF identified the target of these agents as the bifunctional peroxisomal enzyme, hydroxysteroid 17β-dehydrogenase-4 (HSD17B4). Additional studies with truncated versions of HSD17B4 established that CLIFs specifically bind the C-terminus of HSD17B4 and selectively inhibited the enoyl CoA hydratase but not the D-3-hydroxyacyl CoA dehydrogenase activity. HSD17B4 was overexpressed in prostate and colon cancer tissues, knocking down HSD17B4 inhibited cancer cell proliferation, suggesting that HSD17B4 is a potential biomarker and drug target and that CLIFs are potential probes or therapeutic agents for these cancers.
Co-reporter:Stefan Kwiatkowski, Vitaliy M. Sviripa, Zhaiyi Zhang, Alison E. Wendlandt, Claudia Höbartner, David S. Watt, Stefan Stamm
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 3) pp:965-968
Publication Date(Web):1 February 2016
DOI:10.1016/j.bmcl.2015.12.054
Phosphorylation and dephosphorylation of splicing factors play a key role in pre-mRNA splicing events, and cantharidin and norcantharidin analogs inhibit protein phosphatase-1 (PP1) and change alternative pre-mRNA splicing. Targeted inhibitors capable of selectively inhibiting PP-1 could promote exon 7 inclusion in the survival-of-motorneuron-2 gene (SMN2) and shift the proportion of SMN2 protein from a dysfunctional to a functional form. As a prelude to the development of norcantharidin-tethered oligonucleotide inhibitors, the synthesis a norcantharidin-tethered guanosine was developed in which a suitable tether prevented the undesired cyclization of norcantharidin monoamides to imides and possessed a secondary amine terminus suited to the synthesis of oligonucleotides analogs. Application of this methodology led to the synthesis of a diastereomeric mixture of norcantharidin-tethered guanosines, namely bisammonium (1R,2S,3R,4S)- and (1S,2R,3S,4R)-3-((4-(2-(((((2R,3R,4R,5R)-5-(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-2-(hydroxymethyl)-4-methoxytetrahydrofuran-3-yl)oxy)oxidophosphoryl)oxy)ethyl)-phenethyl)(methyl)carbamoyl)-7-oxabicyclo[2.2.1]heptane-2-carboxylate, which showed activity in an assay for SMN2 pre-mRNA splicing.A tethered isocantharidin-guanosine adduct that affects PP1-inhibition promotes alternative splicing in the survival-of-motoneuron-2 (SMN2) gene.
Co-reporter:Mykhaylo S. Frasinyuk, Galyna P. Mrug, Svitlana P. Bondarenko, Vitaliy M. Sviripa, Wen Zhang, Xianfeng Cai, Michael V. Fiandalo, James L. Mohler, Chunming Liu and David S. Watt
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 46) pp:11292-11301
Publication Date(Web):23 Sep 2015
DOI:10.1039/C5OB01828E
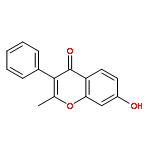
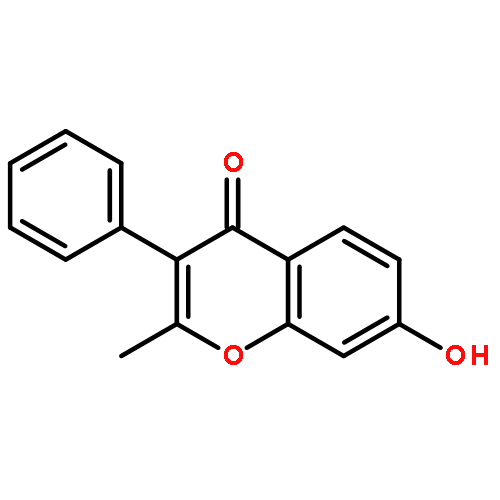
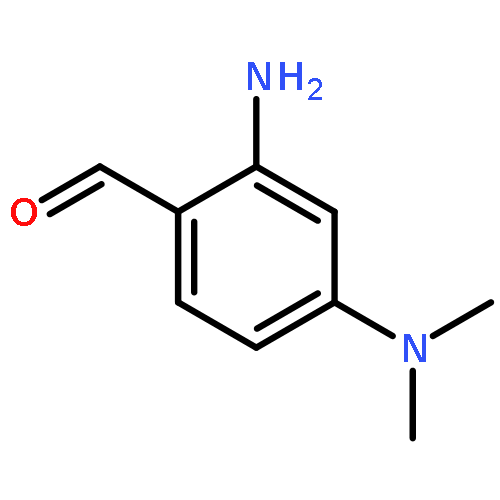
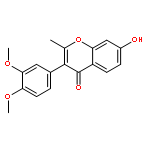
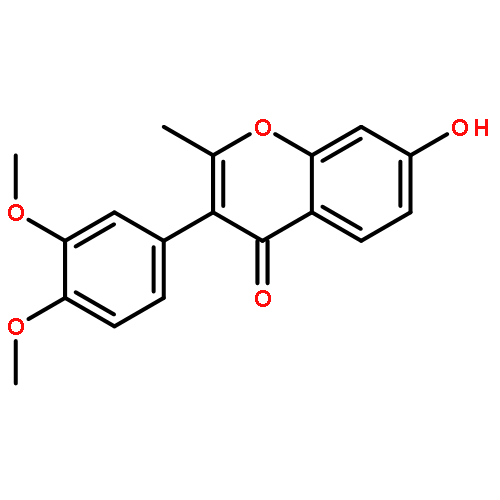
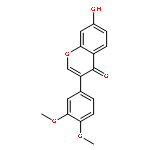
The regiospecific Mannich aminomethylation of 7-hydroxyisoflavonoids using bis(N,N-dimethylamino)methane afforded C-8 substituted N,N-dimethylaminomethyl adducts, and the regioselective aminomethylation of 5-hydroxy-7-methoxyisoflavonoids afforded predominantly the C-6 substituted N,N-dimethylaminomethyl adducts. Acetylation of these C-6 or C-8 Mannich bases with potassium acetate in acetic anhydride provided access to the corresponding acetoxymethyl derivatives that were subsequently converted to hydroxymethyl- and methoxymethyl-substituted 5-hydroxy- or 7-hydroxyisoflavonoids related to naturally occurring flavonoids. The C-8 acetoxymethyl, hydroxymethyl or methoxymethyl-substituted isoflavonoids possessed promising inhibitory potency in the low micromolar range in a prostate cancer PC-3 cell proliferation assay.
Co-reporter:Liliia M. Kril, Valery Vilchez, Jieyun Jiang, Lilia Turcios, Changguo Chen, Vitaliy M. Sviripa, Wen Zhang, Chunming Liu, Brett Spear, David S. Watt, Roberto Gedaly
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 18) pp:3897-3899
Publication Date(Web):15 September 2015
DOI:10.1016/j.bmcl.2015.07.040
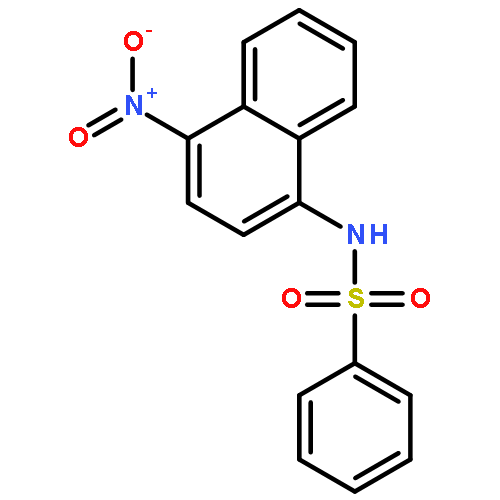
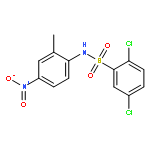
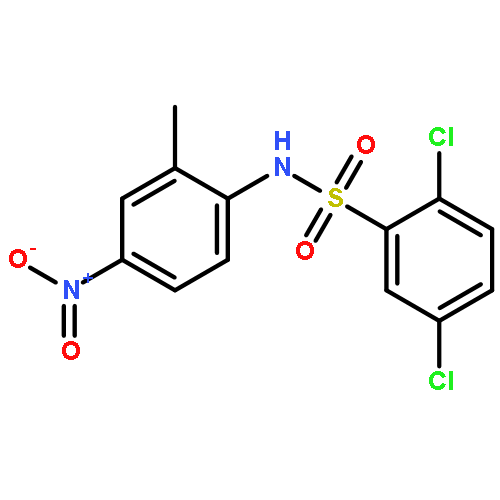
Structure–activity relationships (SAR) in 2,5-dichloro-N-(2-methyl-4-nitrophenyl)benzenesulfonamide (FH535) were examined as part of a program to identify agents that inhibit the Wnt/β-catenin signaling pathway that is frequently upregulated in hepatocellular carcinoma (HCC). FH535 was reported as an inhibitor of both β-catenin in the Wnt signaling pathway and the peroxisome proliferator-activated receptor (PPAR). A β-catenin/T-cell factor (TCF)/Lymphoid-enhancer factor (LEF)-dependent assay (i.e., luciferase-based TOPFlash assay) as well as a [3H]-thymidine incorporation assay were used to explore SAR modifications of FH535. Although replacing the 2,5-dichlorophenylsulfonyl substituent in FH535 with a 2,6-dihalogenation pattern generally produced more biologically active analogs than FH535, other SAR modifications led only to FH535 analogs with comparable or slightly improved activity in these two assays. The absence of a clear SAR pattern in activity suggested a multiplicity of target effectors for N-aryl benzenesulfonamides.
Co-reporter:Vitaliy M. Sviripa ; Wen Zhang ; Andrii G. Balia ; Oleg V. Tsodikov ; Justin R. Nickell ; Florence Gizard ; Tianxin Yu ; Eun Y. Lee ; Linda P. Dwoskin ; Chunming Liu ;David S. Watt
Journal of Medicinal Chemistry 2014 Volume 57(Issue 14) pp:6083-6091
Publication Date(Web):June 20, 2014
DOI:10.1021/jm5004864
Inhibition of the catalytic subunit of the heterodimeric methionine S-adenosyl transferase-2 (MAT2A) with fluorinated N,N-dialkylaminostilbenes (FIDAS agents) offers a potential avenue for the treatment of liver and colorectal cancers where upregulation of this enzyme occurs. A study of structure–activity relationships led to the identification of the most active compounds as those with (1) either a 2,6-difluorostyryl or 2-chloro-6-fluorostyryl subunit, (2) either an N-methylamino or N,N-dimethylamino group attached in a para orientation relative to the 2,6-dihalostyryl subunit, and (3) either an N-methylaniline or a 2-(N,N-dimethylamino)pyridine ring. These modifications led to FIDAS agents that were active in the low nanomolar range, that formed water-soluble hydrochloride salts, and that possessed the desired property of not inhibiting the human hERG potassium ion channel at concentrations at which the FIDAS agents inhibit MAT2A. The active FIDAS agents may inhibit cancer cells through alterations of methylation reactions essential for cancer cell survival and growth.
Co-reporter:Sergey V. Matveev, Stefan Kwiatkowski, Vitaliy M. Sviripa, Robert C. Fazio, David S. Watt, Harry LeVine III
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 23) pp:5534-5536
Publication Date(Web):1 December 2014
DOI:10.1016/j.bmcl.2014.09.075
Accumulation of Aβ in the brains of Alzheimer disease (AD) patients reflects an imbalance between Aβ production and clearance from their brains. Alternative cleavage of amyloid precursor protein (APP) by processing proteases generates soluble APP fragments including the neurotoxic amyloid Aβ40 and Aβ42 peptides that assemble into fibrils and form plaques. Plaque-buildup occurs over an extended time-frame, and the early detection and modulation of plaque formation are areas of active research. Radiolabeled probes for the detection of amyloid plaques and fibrils in living subjects are important for noninvasive evaluation of AD diagnosis, progression, and differentiation of AD from other neurodegenerative diseases and age-related cognitive decline. Tritium-labeled (E,E)-1-[3H]-2,5-bis(4′-hydroxy-3′-carbomethoxystyryl)benzene possesses an improved level of chemical stability relative to a previously reported radioiodinated analog for radiometric quantification of Aβ plaque and tau pathology in brain tissue and in vitro studies with synthetic Aβ and tau fibrils.Accumulation of amyloid-β plaque and tau tangle pathologies in Alzheimer’s disease occurs over an extended time-frame, and the early detection and prevention of their formation are active research foci. Tritiated (E,E)-1-[3H]-2,5-bis(4′-hydroxy-3′-carbomethoxystyryl)benzene possesses an improved level of chemical stability relative to a previously reported radioiodinated analog for detecting and quantifying Aβ and tau pathology in brain tissue and in studies of in vitro assembled Aβ and tau.
Co-reporter:Mykhaylo S. Frasinyuk, Stefan Kwiatkowski, Jonathan M. Wagner, Timothy J. Evans, Robert W. Reed, Konstantin V. Korotkov, David S. Watt
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 15) pp:3546-3548
Publication Date(Web):1 August 2014
DOI:10.1016/j.bmcl.2014.05.056
Mycosin protease-1 (MycP1) cleaves ESX secretion-associated protein B (EspB) that is a virulence factor of Mycobacterium tuberculosis, and accommodates an octapeptide, AVKAASLG, as a short peptide substrate. Because peptidoboronic acids are known inhibitors of serine proteases, the synthesis and binding of a boronic acid analog of the pentapeptide cleavage product, AVKAA, was studied using MycP1 variants from Mycobacterium thermoresistible (MycP1mth), Mycobacterium smegmatis (MycP1msm) and M. tuberculosis (MycP1mtu). We synthesized the boropentapeptide, HAlaValLysAlaAlaB(OH)2 (1) and the analogous pinanediol PD-protected HAlaValLysAlaAlaBO2(PD) (2) using an Fmoc/Boc peptide strategy. The pinanediol boropentapeptide 2 displayed IC50 values 121.6 ± 25.3 μM for MycP1mth, 93.2 ± 37.3 μM for MycP1msm and 37.9 ± 5.2 μM for MycP1mtu. Such relatively strong binding creates a chance for crystalizing the complex with 2 and finding the structure of the unknown MycP1 catalytic site that would potentially facilitate the development of new anti-tuberculosis drugs.Boronic acid analogs, HAlaValLysAlaAlaB(OH)2 (1) and the pinanediol PD-protected HAlaValLysAlaAlaBO2(PD) (2), mimic a pentapeptide produced from the biologically active octapeptide substrate, AVKAASLG, for MycP1 protease in Mycobacterium tuberculosis and compound 2 inhibits this protease with an IC50 value of 37.9 μM.
Co-reporter:Vitaliy M. Sviripa, Wen Zhang, Liliia M. Kril, Alice X. Liu, Yaxia Yuan, Chang-Guo Zhan, Chunming Liu, David S. Watt
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 15) pp:3638-3640
Publication Date(Web):1 August 2014
DOI:10.1016/j.bmcl.2014.04.113
Co-reporter:Vitaliy Sviripa, Wen Zhang, Michael D. Conroy, Eric S. Schmidt, Alice X. Liu, Johnny Truong, Chunming Liu, David S. Watt
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 6) pp:1600-1603
Publication Date(Web):15 March 2013
DOI:10.1016/j.bmcl.2013.01.096
Adenosine monophosphate-activated kinase (AMPK) plays a central role in regulating energy homeostasis in eukaryotic cells. AMPK also regulates lipid synthesis by inhibiting acetyl-CoA carboxylase (ACC) and regulates mTOR signaling by activating TSC2. Due to its important roles in cell metabolism, AMPK is an attractive target for metabolic diseases, such as type II diabetes and obesity. AMPK activators, such as metformin, that are used for diabetes treatment are also effective anticancer agents. However, the efficacies of many known AMPK activators are relatively low. For example, metformin activates AMPK at millimolar levels. In this study, we identified a novel family of AMPK activators, namely fluorinated N,N′-diarylureas, that activate AMPK at 1–3 μM concentrations. These novel agents strongly inhibit the proliferation of colon cancer cells. We studied the potential mechanisms of these agents, performed a structure–activity relationship (SAR) study and identified several fluorinated N,N′-diarylureas as potent AMPK activators.Halogenated N,N′-diarylureas inhibit mechanistic-target-of-rapamycin (mTOR) signaling by activating AMP-activated protein kinase (AMPK) at 1–3 μM concentrations.
Co-reporter:Sanjib K. Shrestha, Liliia M. Kril, Keith D. Green, Stefan Kwiatkowski, Vitaliy M. Sviripa, Justin R. Nickell, Linda P. Dwoskin, David S. Watt, Sylvie Garneau-Tsodikova
Bioorganic & Medicinal Chemistry (1 January 2017) Volume 25(Issue 1) pp:
Publication Date(Web):1 January 2017
DOI:10.1016/j.bmc.2016.10.009
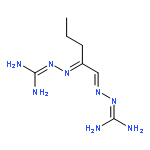
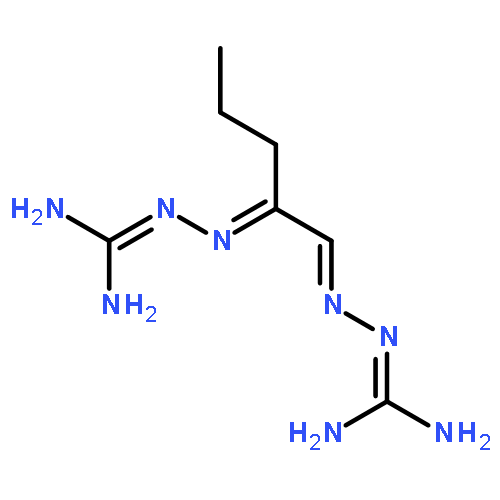
The emergence of multidrug-resistant bacterial and fungal strains poses a threat to human health that requires the design and synthesis of new classes of antimicrobial agents. We evaluated bis(N-amidinohydrazones) and N-(amidino)-N′-aryl-bishydrazones for their antibacterial and antifungal activities against panels of Gram-positive/Gram-negative bacteria as well as fungi. We investigated their potential to develop resistance against both bacteria and fungi by a multi-step resistance-selection method, explored their potential to induce the production of reactive oxygen species, and assessed their toxicity. In summary, we found that these compounds exhibited broad-spectrum antibacterial and antifungal activities against most of the tested strains with minimum inhibitory concentration (MIC) values ranging from <0.5 to >500 μM against bacteria and 1.0 to >31.3 μg/mL against fungi; and in most cases, they exhibited either superior or similar antimicrobial activity compared to those of the standard drugs used in the clinic. We also observed minimal emergence of drug resistance to these newly synthesized compounds by bacteria and fungi even after 15 passages, and we found weak to moderate inhibition of the human Ether-à-go-go-related gene (hERG) channel with acceptable IC50 values ranging from 1.12 to 3.29 μM. Overall, these studies show that bis(N-amidinohydrazones) and N-(amidino)-N′-aryl-bishydrazones are potentially promising scaffolds for the discovery of novel antibacterial and antifungal agents.
Co-reporter:Mykhaylo S. Frasinyuk, Galyna P. Mrug, Svitlana P. Bondarenko, Vitaliy M. Sviripa, Wen Zhang, Xianfeng Cai, Michael V. Fiandalo, James L. Mohler, Chunming Liu and David S. Watt
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 46) pp:NaN11301-11301
Publication Date(Web):2015/09/23
DOI:10.1039/C5OB01828E
The regiospecific Mannich aminomethylation of 7-hydroxyisoflavonoids using bis(N,N-dimethylamino)methane afforded C-8 substituted N,N-dimethylaminomethyl adducts, and the regioselective aminomethylation of 5-hydroxy-7-methoxyisoflavonoids afforded predominantly the C-6 substituted N,N-dimethylaminomethyl adducts. Acetylation of these C-6 or C-8 Mannich bases with potassium acetate in acetic anhydride provided access to the corresponding acetoxymethyl derivatives that were subsequently converted to hydroxymethyl- and methoxymethyl-substituted 5-hydroxy- or 7-hydroxyisoflavonoids related to naturally occurring flavonoids. The C-8 acetoxymethyl, hydroxymethyl or methoxymethyl-substituted isoflavonoids possessed promising inhibitory potency in the low micromolar range in a prostate cancer PC-3 cell proliferation assay.

![1H-Thieno[3,4-d]imidazole-4-pentanamide, hexahydro-N-[2-[2-[2-[(2-iodoacetyl)amino]ethoxy]ethoxy]ethyl]-2-oxo-, (3aS,4S,6aR)-](/data/chemimg/3748200/292843-75-5.png)
![1H-Thieno[3,4-d]imidazole-4-pentanamide, hexahydro-N-[2-[2-[2-[(2-iodoacetyl)amino]ethoxy]ethoxy]ethyl]-2-oxo-, (3aS,4S,6aR)-](/data/chemimg/3748200/292843-75-5_b.png)
![5-[2-[3-BROMO-4-[2-(3-CARBOXY-4-HYDROXYPHENYL)ETHENYL]PHENYL]ETHENYL]-2-HYDROXYBENZOIC ACID](http://img.cochemist.com/ccimg/291800/291766-06-8.png)
![5-[2-[3-BROMO-4-[2-(3-CARBOXY-4-HYDROXYPHENYL)ETHENYL]PHENYL]ETHENYL]-2-HYDROXYBENZOIC ACID](http://img.cochemist.com/ccimg/291800/291766-06-8_b.png)
![Benzenamine, 4-[2-(2-chlorophenyl)ethynyl]-N,N-dimethyl-](/data/chemimg/3740800/1445990-50-0.png)
![Benzenamine, 4-[2-(2-chlorophenyl)ethynyl]-N,N-dimethyl-](/data/chemimg/3740800/1445990-50-0_b.png)





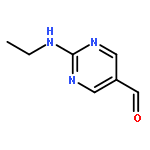
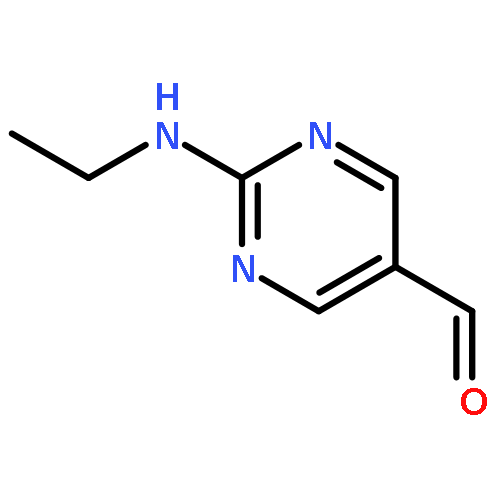


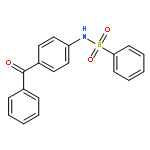
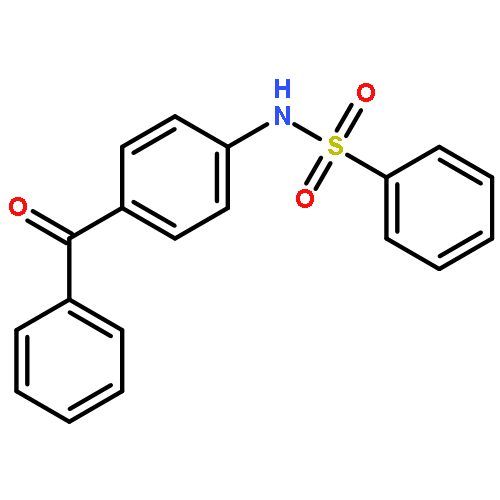
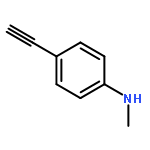
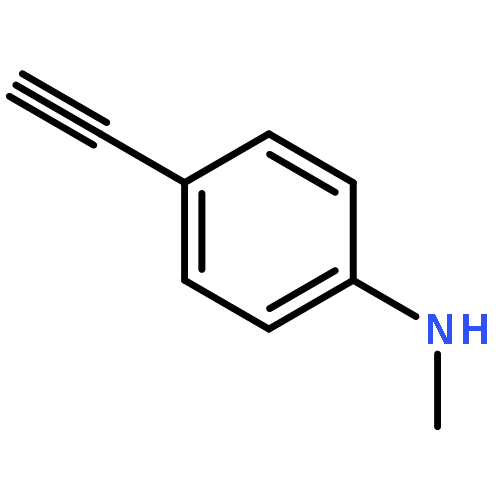
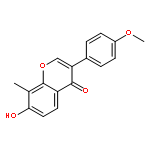
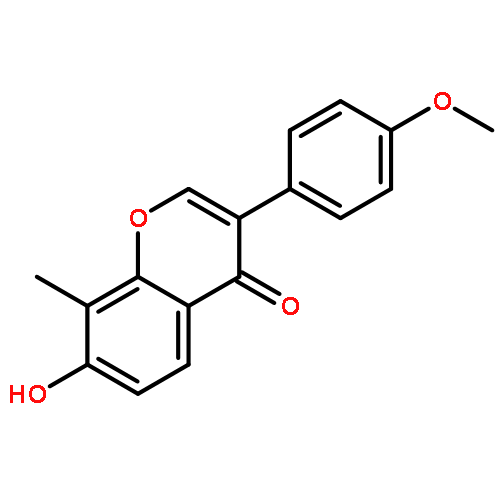




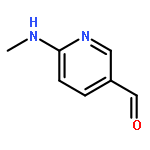
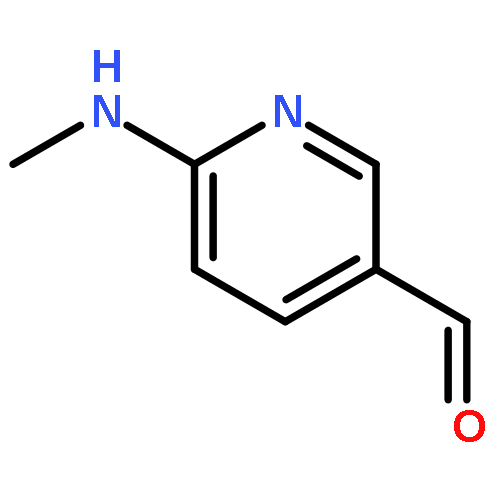
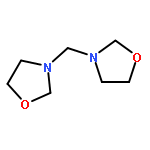

![Phosphonic acid, [(2,6-dichlorophenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/64000/63909-56-8.png)
![Phosphonic acid, [(2,6-dichlorophenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/64000/63909-56-8_b.png)