Co-reporter:Paul A. Wender, Christian Ebner, Brandon D. Fennell, Fuyuhiko Inagaki, and Birte Schröder
Organic Letters November 3, 2017 Volume 19(Issue 21) pp:5810-5810
Publication Date(Web):October 16, 2017
DOI:10.1021/acs.orglett.7b02765
The previously unexplored metal-catalyzed [5 + 2] cycloadditions of vinylcyclopropanes (VCPs) and electron-rich alkynes (ynol ethers) have been found to provide a highly efficient, direct route to dioxygenated seven-membered rings, a common feature of numerous natural and non-natural targets and building blocks for synthesis. The reactions proceed in high yield at room temperature and tolerate a broad range of functionalities. Substituted VCPs were found to react with high regioselectivity.
Co-reporter:Colin J. McKinlay;Jessica R. Vargas;Timothy R. Blake;Jonathan W. Hardy;Masamitsu Kanada;Christopher H. Contag;Robert M. Waymouth
PNAS 2017 Volume 114 (Issue 4 ) pp:E448-E456
Publication Date(Web):2017-01-24
DOI:10.1073/pnas.1614193114
Functional delivery of mRNA to tissues in the body is key to implementing fundamentally new and potentially transformative
strategies for vaccination, protein replacement therapy, and genome editing, collectively affecting approaches for the prevention,
detection, and treatment of disease. Broadly applicable tools for the efficient delivery of mRNA into cultured cells would
advance many areas of research, and effective and safe in vivo mRNA delivery could fundamentally transform clinical practice.
Here we report the step-economical synthesis and evaluation of a tunable and effective class of synthetic biodegradable materials:
charge-altering releasable transporters (CARTs) for mRNA delivery into cells. CARTs are structurally unique and operate through
an unprecedented mechanism, serving initially as oligo(α-amino ester) cations that complex, protect, and deliver mRNA and
then change physical properties through a degradative, charge-neutralizing intramolecular rearrangement, leading to intracellular
release of functional mRNA and highly efficient protein translation. With demonstrated utility in both cultured cells and
animals, this mRNA delivery technology should be broadly applicable to numerous research and therapeutic applications.
Co-reporter:Nancy L. Benner, Xiaoyu Zang, Daniel C. Buehler, Valerie A. Kickhoefer, Michael E. Rome, Leonard H. Rome, and Paul A. Wender
ACS Nano 2017 Volume 11(Issue 1) pp:
Publication Date(Web):December 28, 2016
DOI:10.1021/acsnano.6b07440
Vault nanoparticles represent promising vehicles for drug and probe delivery. Innately found within human cells, vaults are stable, biocompatible nanocapsules possessing an internal volume that can encapsulate hundreds to thousands of molecules. They can also be targeted. Unlike most nanoparticles, vaults are nonimmunogenic and monodispersed and can be rapidly produced in insect cells. Efforts to create vaults with modified properties have been, to date, almost entirely limited to recombinant bioengineering approaches. Here we report a systematic chemical study of covalent vault modifications, directed at tuning vault properties for research and clinical applications, such as imaging, targeted delivery, and enhanced cellular uptake. As supra-macromolecular structures, vaults contain thousands of derivatizable amino acid side chains. This study is focused on establishing the comparative selectivity and efficiency of chemically modifying vault lysine and cysteine residues, using Michael additions, nucleophilic substitutions, and disulfide exchange reactions. We also report a strategy that converts the more abundant vault lysine residues to readily functionalizable thiol terminated side chains through treatment with 2-iminothiolane (Traut’s reagent). These studies provide a method to doubly modify vaults with cell penetrating peptides and imaging agents, allowing for in vitro studies on their enhanced uptake into cells.Keywords: cell-penetrating transporter; chemical modification; imaging; nanoparticle; protein cage; vaults;
Co-reporter:Colin J. McKinlay; Robert M. Waymouth
Journal of the American Chemical Society 2016 Volume 138(Issue 10) pp:3510-3517
Publication Date(Web):February 22, 2016
DOI:10.1021/jacs.5b13452
The design, synthesis, and biological evaluation of a new family of highly effective cell-penetrating molecular transporters, guanidinium-rich oligophosphoesters, are described. These unique transporters are synthesized in two steps, irrespective of oligomer length, by the organocatalytic ring-opening polymerization (OROP) of 5-membered cyclic phospholane monomers followed by oligomer deprotection. Varying the initiating alcohol results in a wide variety of cargo attachment strategies for releasable or nonreleasable transporter applications. Initiation of oligomerization with a fluorescent probe produces, upon deprotection, a transporter-probe conjugate that is shown to readily enter multiple cell lines in a dose-dependent manner. These new transporters are superior in cell uptake to previously studied guanidinium-rich oligocarbonates and oligoarginines, showing over 2-fold higher uptake than the former and 6-fold higher uptake than the latter. Initiation with a protected thiol gives, upon deprotection, thiol-terminated transporters which can be thiol-click conjugated to a variety of probes, drugs and other cargos as exemplified by the conjugation and delivery of the model probe fluorescein-maleimide and the medicinal agent paclitaxel (PTX) into cells. Of particular significance given that drug resistance is a major cause of chemotherapy failure, the PTX-transporter conjugate, designed to evade Pgp export and release free PTX after cell entry, shows efficacy against PTX-resistant ovarian cancer cells. Collectively this study introduces a new and highly effective class of guanidinium-rich cell-penetrating transporters and methodology for their single-step conjugation to drugs and probes, and demonstrates that the resulting drug/probe-conjugates readily enter cells, outperforming previously reported guanidinium-rich oligocarbonates and peptide transporters.
Co-reporter:Hsiao-Tieh Hsu, Brian M. Trantow, Robert M. Waymouth, and Paul A. Wender
Bioconjugate Chemistry 2016 Volume 27(Issue 2) pp:376
Publication Date(Web):September 14, 2015
DOI:10.1021/acs.bioconjchem.5b00469
The development of abiological catalysts that can function in biological systems is an emerging subject of importance with significant ramifications in synthetic chemistry and the life sciences. Herein we report a biocompatible ruthenium complex [Cp(MQA)Ru(C3H5)]+PF6– 2 (Cp = cyclopentadienyl, MQA = 4-methoxyquinoline-2-carboxylate) and a general analytical method for evaluating its performance in real time based on a luciferase reporter system amenable to high throughput screening in cells and by extension to evaluation in luciferase transgenic animals. Precatalyst 2 activates alloc-protected aminoluciferin 4b, a bioluminescence pro-probe, and releases the active luminophore, aminoluciferin (4a), in the presence of luciferase-transfected cells. The formation and enzymatic turnover of 4a, an overall process selected because it emulates pro-drug activation and drug turnover by an intracellular target, is evaluated in real time by photon counting as 4a is converted by intracellular luciferase to oxyaminoluciferin and light. Interestingly, while the catalytic conversion (activation) of 4b to 4a in water produces multiple products, the presence of biological nucleophiles such as thiols prevents byproduct formation and provides almost exclusively luminophore 4a. Our studies show that precatalyst 2 activates 4b extracellularly, exhibits low toxicity at concentrations relevant to catalysis, and is comparably effective in two different cell lines. This proof of concept study shows that precatalyst 2 is a promising lead for bioorthogonal catalytic activation of pro-probes and, by analogy, similarly activatable pro-drugs. More generally, this study provides an analytical method to measure abiological catalytic activation of pro-probes and, by analogy with our earlier studies on pro-Taxol, similarly activatable pro-drugs in real time using a coupled biological catalyst that mediates a bioluminescent readout, providing tools for the study of imaging signal amplification and of targeted therapy.
Co-reporter:Daryl Staveness; Rana Abdelnabi; Katherine E. Near; Yu Nakagawa; Johan Neyts; Leen Delang; Pieter Leyssen
Journal of Natural Products 2016 Volume 79(Issue 4) pp:680-684
Publication Date(Web):February 22, 2016
DOI:10.1021/acs.jnatprod.5b01017
Chikungunya virus (CHIKV) has been spreading rapidly, with over one million confirmed or suspected cases in the Americas since late 2013. Infection with CHIKV causes devastating arthritic and arthralgic symptoms. Currently, there is no therapy to treat this disease, and the only medications focus on relief of symptoms. Recently, protein kinase C (PKC) modulators have been reported to inhibit CHIKV-induced cell death in cell assays. The salicylate-derived bryostatin analogues described here are structurally simplified PKC modulators that are more synthetically accessible than the natural product bryostatin 1, a PKC modulator and clinical lead for the treatment of cancer, Alzheimer’s disease, and HIV eradication. Evaluation of the anti-CHIKV activity of these salicylate-derived bryostatin analogues in cell culture indicates that they are among the most potent cell-protective agents reported to date. Given that they are more accessible and significantly more active than the parent natural product, they represent new therapeutic leads for controlling CHIKV infection. Significantly, these analogues also provide evidence for the involvement of a PKC-independent pathway. This adds a fundamentally distinct aspect to the importance or involvement of PKC modulation in inhibition of chikungunya virus replication, a topic of recent and growing interest.
Co-reporter:Daryl Staveness; Rana Abdelnabi; Adam J. Schrier; Brian A. Loy; Vishal A. Verma; Brian A. DeChristopher; Katherine E. Near; Johan Neyts; Leen Delang; Pieter Leyssen
Journal of Natural Products 2016 Volume 79(Issue 4) pp:675-679
Publication Date(Web):February 22, 2016
DOI:10.1021/acs.jnatprod.5b01016
Chikungunya virus (CHIKV) is a mosquito-borne alphavirus showing a recent resurgence and rapid spread worldwide. While vaccines are under development, there are currently no therapies to treat this disease, except for over-the-counter (OTC) analgesics, which alleviate the devastating arthritic and arthralgic symptoms. To identify novel inhibitors of the virus, analogues of the natural product bryostatin 1, a clinical lead for the treatment of cancer, Alzheimer’s disease, and HIV eradication, were investigated for in vitro antiviral activity and were found to be among the most potent inhibitors of CHIKV replication reported to date. Bryostatin-based therapeutic efforts and even recent anti-CHIKV strategies have centered on modulation of protein kinase C (PKC). Intriguingly, while the C ring of bryostatin primarily drives interactions with PKC, A- and B-ring functionality in these analogues has a significant effect on the observed cell-protective activity. Significantly, bryostatin 1 itself, a potent pan-PKC modulator, is inactive in these assays. These new findings indicate that the observed anti-CHIKV activity is not solely mediated by PKC modulation, suggesting possible as yet unidentified targets for CHIKV therapeutic intervention. The high potency and low toxicity of these bryologs make them promising new leads for the development of a CHIKV treatment.
Co-reporter:Paul A. Wender, Ryan V. Quiroz, and Matthew C. Stevens
Accounts of Chemical Research 2015 Volume 48(Issue 3) pp:752
Publication Date(Web):March 5, 2015
DOI:10.1021/acs.accounts.5b00004
In 1996, a snapshot of the field of synthesis was provided by many of its thought leaders in a Chemical Reviews thematic issue on “Frontiers in Organic Synthesis”. This Accounts of Chemical Research thematic issue on “Synthesis, Design, and Molecular Function” is intended to provide further perspective now from well into the 21st century. Much has happened in the past few decades. The targets, methods, strategies, reagents, procedures, goals, funding, practices, and practitioners of synthesis have changed, some in dramatic ways as documented in impressive contributions to this issue. However, a constant for most synthesis studies continues to be the goal of achieving function with synthetic economy. Whether in the form of new catalysts, reagents, therapeutic leads, diagnostics, drug delivery systems, imaging agents, sensors, materials, energy generation and storage systems, bioremediation strategies, or molecules that challenge old theories or test new ones, the function of a target has been and continues to be a major and compelling justification for its synthesis. While the targets of synthesis have historically been heavily represented by natural products, increasingly design, often inspired by natural structures, is providing a new source of target structures exhibiting new or natural functions and new or natural synthetic challenges. Complementing isolation and screening approaches to new target identification, design enables one to create targets de novo with an emphasis on sought-after function and synthetic innovation with step-economy. Design provides choice. It allows one to determine how close a synthesis will come to the ideal synthesis and how close a structure will come to the ideal function.In this Account, we address studies in our laboratory on function-oriented synthesis (FOS), a strategy to achieve function by design and with synthetic economy. By starting with function rather than structure, FOS places an initial emphasis on target design, thereby harnessing the power of chemists and computers to create new structures with desired functions that could be prepared in a simple, safe, economical, and green, if not ideal, fashion. Reported herein are examples of FOS associated with (a) molecular recognition, leading to the first designed phorbol-inspired protein kinase C regulatory ligands, the first designed bryostatin analogs, the newest bryologs, and a new family of designed kinase inhibitors, (b) target modification, leading to highly simplified but functionally competent photonucleases—molecules that cleave DNA upon photoactivation, (c) drug delivery, leading to cell penetrating molecular transporters, molecules that ferry other attached or complexed molecules across biological barriers, and (d) new reactivity-regenerating reagents in the form of functional equivalents of butatrienes, reagents that allow for back-to-back three-component cycloaddition reactions, thus achieving structural complexity and value with step-economy. While retrosynthetic analysis seeks to identify the best way to make a target, retrofunction analysis seeks to identify the best targets to make. In essence, form (structure) follows function.
Co-reporter:Brian A. Loy; Adam B. Lesser; Daryl Staveness; Kelvin L. Billingsley; Lynette Cegelski
Journal of the American Chemical Society 2015 Volume 137(Issue 10) pp:3678-3685
Publication Date(Web):February 24, 2015
DOI:10.1021/jacs.5b00886
Protein kinase C (PKC) modulators are currently of great importance in preclinical and clinical studies directed at cancer, immunotherapy, HIV eradication, and Alzheimer’s disease. However, the bound conformation of PKC modulators in a membrane environment is not known. Rotational echo double resonance (REDOR) NMR spectroscopy could uniquely address this challenge. However, REDOR NMR requires strategically labeled, high affinity ligands to determine interlabel distances from which the conformation of the bound ligand in the PKC–ligand complex could be identified. Here we report the first computer-guided design and syntheses of three bryostatin analogues strategically labeled for REDOR NMR analysis. Extensive computer analyses of energetically accessible analogue conformations suggested preferred labeling sites for the identification of the PKC-bound conformers. Significantly, three labeled analogues were synthesized, and, as required for REDOR analysis, all proved highly potent with PKC affinities (∼1 nM) on par with bryostatin. These potent and strategically labeled bryostatin analogues are new structural leads and provide the necessary starting point for projected efforts to determine the PKC-bound conformation of such analogues in a membrane environment, as needed to design new PKC modulators and understand PKC–ligand–membrane structure and dynamics.
Co-reporter:Paul A. Wender; Matthew S. Jeffreys;Andrew G. Raub
Journal of the American Chemical Society 2015 Volume 137(Issue 28) pp:9088-9093
Publication Date(Web):May 11, 2015
DOI:10.1021/jacs.5b04091
New reactions and reagents that allow for multiple bond-forming events per synthetic operation are required to achieve structural complexity and thus value with step-, time-, cost-, and waste-economy. Here we report a new class of reagents that function like tetramethyleneethane (TME), allowing for back-to-back [4 + 2] cycloadditions, thereby amplifying the complexity-increasing benefits of Diels–Alder and metal-catalyzed cycloadditions. The parent recursive reagent, 2,3-dimethylene-4-trimethylsilylbutan-1-ol (DMTB), is readily available from the metathesis of ethylene and THP-protected 4-trimethylsilylbutyn-1-ol. DMTB and related reagents engage diverse dienophiles in an initial Diels–Alder or metal-catalyzed [4 + 2] cycloaddition, triggering a subsequent vinylogous Peterson elimination that recursively generates a new diene for a second cycloaddition. Overall, this multicomponent catalytic cascade produces in one operation carbo- and heterobicyclic building blocks for the synthesis of a variety of natural products, therapeutic leads, imaging agents, and materials. Its application to the three step synthesis of a new solvatochromic fluorophore, N-ethyl(6-N,N-dimethylaminoanthracene-2,3-dicarboximide) (6-DMA), and the photophysical characterization of this fluorophore are described.
Co-reporter:Thomas J. L. Mustard, Paul A. Wender, and Paul Ha-Yeon Cheong
ACS Catalysis 2015 Volume 5(Issue 3) pp:1758
Publication Date(Web):January 27, 2015
DOI:10.1021/cs501828e
The origins of differential catalytic reactivities of four Rh(I) catalysts and their derivatives in the (5 + 2) cycloaddition reaction were elucidated using density functional theory. Computed free energy spans are in excellent agreement with known experimental rates. For every catalyst, the substrate geometries in the transition state remained constant (<0.1 Å RMSD for atoms involved in bond-making and -breaking processes). Catalytic efficiency is shown to be a function of how well the catalyst accommodates the substrate transition state geometry and electronics. This shows that the induced fit model for explaining biological catalysis may be relevant to transition metal catalysis. This could serve as a general model for understanding the origins of efficiencies of catalytic reactions.Keywords: catalysis; computations; DFT; ligands; NHC; organometallic; phosphine; rhodium; theory; transition metals
Co-reporter:Paul A. Wender; Melanie A. Huttner; Daryl Staveness; Jessica R. Vargas;Adele F. Xu
Molecular Pharmaceutics 2015 Volume 12(Issue 3) pp:742-750
Publication Date(Web):January 14, 2015
DOI:10.1021/mp500581r
A highly versatile and step-economical route to a new class of guanidinium-rich molecular transporters and evaluation of their ability to complex, deliver, and release siRNA are described. These new drug/probe delivery systems are prepared in only two steps, irrespective of length or composition, using an organocatalytic ring-opening co-oligomerization of glycerol-derived cyclic carbonate monomers incorporating either protected guanidine or lipid side chains. The resultant amphipathic co-oligomers are highly effective vehicles for siRNA delivery, providing an excellent level of target protein suppression (>85%). These new oligocarbonates are nontoxic at levels required for cell penetration and can be tuned for particle size. Relative to the previously reported methyl(trimethylene)carbonate (MTC) scaffold, the ether linkage at C2 in the new transporters markedly enhances the stability of the siRNA/co-oligomer complexes. Both hybrid co-oligomers, containing a mixture of glycerol- and MTC-derived monomers, and co-oligomers containing only glycerol monomers are found to provide tunable control over siRNA complex stability. On the basis of a glycerol and CO2 backbone, these new co-oligomers represent a rapidly tunable and biocompatible siRNA delivery system that is highly effective in suppressing target protein synthesis.
Co-reporter:Xin Hong ; Matthew C. Stevens ; Peng Liu ; Paul A. Wender ;K. N. Houk
Journal of the American Chemical Society 2014 Volume 136(Issue 49) pp:17273-17283
Publication Date(Web):November 7, 2014
DOI:10.1021/ja5098308
Allenes are important 2π building blocks in organic synthesis and engage as 2-carbon components in many metal-catalyzed reactions. Wender and co-workers discovered that methyl substituents on the terminal allene double bond counterintuitively change the reactivities of allenes in [Rh(CO)2Cl]2-catalyzed intermolecular (5 + 2) cycloadditions with vinylcyclopropanes (VCPs). More sterically encumbered allenes afford higher cycloadduct yields, and such effects are also observed in other Rh(I)-catalyzed intermolecular cycloadditions. Through density functional theory calculations (B3LYP and M06) and experiment, we explored this enigmatic reactivity and selectivity of allenes in [Rh(CO)2Cl]2-catalyzed intermolecular (5 + 2) cycloadditions with VCPs. The apparent low reactivity of terminally unsubstituted allenes is associated with a competing allene dimerization that irreversibly sequesters rhodium. With terminally substituted allenes, steric repulsion between the terminal substituents significantly increases the barrier of allene dimerization while the barrier of the (5 + 2) cycloaddition is not affected, and thus the cycloaddition prevails. Computation has also revealed the origin of chemoselectivity in (5 + 2) cycloadditions with allene-ynes. Although simple allene and acetylene have similar reaction barriers, intermolecular (5 + 2) cycloadditions of allene-ynes occur exclusively at the terminal allene double bond. The terminal double bond is more reactive due to the enhanced d−π* backdonation. At the same time, insertion of the internal double bond of an allene-yne has a higher barrier as it would break π conjugation. Substituted alkynes are more difficult to insert compared with acetylene, because of the steric repulsion from the additional substituents. This leads to the greater reactivity of the allene double bond relative to the alkynyl group in allene-ynes.
Co-reporter:Paul A. Wender
Natural Product Reports 2014 vol. 31(Issue 4) pp:433-440
Publication Date(Web):03 Mar 2014
DOI:10.1039/C4NP00013G
This Highlight describes factors that contribute to an ideal synthesis, including economies (step, time, atom, solvent, energy) and orientations (target, diversity, safety, function), and the role of synthesis-informed design directed at function in advancing synthesis and its impact on science.
Co-reporter:Paul A. Wender and Daryl Staveness
Organic Letters 2014 Volume 16(Issue 19) pp:5140-5143
Publication Date(Web):September 19, 2014
DOI:10.1021/ol502492b
Bryostatin 1, in clinical trials or preclinical development for cancer, Alzheimer’s disease, and a first-of-its-kind strategy for HIV/AIDS eradication, is neither readily available nor optimally suited for clinical use. In preceding work, we disclosed a new class of simplified bryostatin analogs designed for ease of access and tunable activity. Here we describe a final step diversification strategy that provides, in only 25 synthetic steps, simplified and tunable analogs with bryostatin-like PKC modulatory activities.
Co-reporter:Paul A. Wender, Yu Nakagawa, Katherine E. Near, and Daryl Staveness
Organic Letters 2014 Volume 16(Issue 19) pp:5136-5139
Publication Date(Web):September 19, 2014
DOI:10.1021/ol502491f
Bryostatin 1 is in clinical trials for the treatment of cancer and Alzheimer’s disease and is a candidate for a first-in-class approach to HIV/AIDS eradication. It is neither readily available nor optimally suited for clinical use. Using a function oriented synthesis strategy, a new class of bryostatin-inspired analogs was designed with a simplified salicylate-derived subunit, enabling step-economical synthesis (23 total steps) of agents exhibiting bryostatin-like affinity to protein kinase C (PKC).
Co-reporter:Paul A. Wender, Fuyuhiko Inagaki, Magnus Pfaffenbach, and Matthew C. Stevens
Organic Letters 2014 Volume 16(Issue 11) pp:2923-2925
Publication Date(Web):May 12, 2014
DOI:10.1021/ol501114q
Conventional allenes have not been effective π-reactive 2-carbon components in many intermolecular cycloadditions including metal-catalyzed [5 + 2] cycloadditions. We report herein that rhodium-catalyzed [5 + 2] cycloadditions of propargyltrimethylsilanes and vinylcyclopropanes provide, after in situ protodesilylation, a highly efficient route to formal allene cycloadducts. Propargyltrimethylsilanes function as safe, easily handled synthetic equivalents of gaseous allenes and hard-to-access monosubstituted allenes. In this one-flask procedure, they provide cycloadducts of what is formally addition to the more sterically encumbered allene double bond.
Co-reporter:Jessica R. Vargas, Erika Geihe Stanzl, Nelson N. H. Teng, and Paul A. Wender
Molecular Pharmaceutics 2014 Volume 11(Issue 8) pp:2553-2565
Publication Date(Web):May 5, 2014
DOI:10.1021/mp500161z
Co-reporter:Erika Geihe Stanzl, Brian M. Trantow, Jessica R. Vargas, and Paul A. Wender
Accounts of Chemical Research 2013 Volume 46(Issue 12) pp:2944
Publication Date(Web):May 22, 2013
DOI:10.1021/ar4000554
All living systems require biochemical barriers. As a consequence, all drugs, imaging agents, and probes have targets that are either on, in, or inside of these barriers. Fifteen years ago, we initiated research directed at more fully understanding these barriers and at developing tools and strategies for breaching them that could be of use in basic research, imaging, diagnostics, and medicine. At the outset of this research and now to a lesser extent, the “rules” for drug design biased the selection of drug candidates mainly to those with an intermediate and narrow log P. At the same time, it was becoming increasingly apparent that Nature had long ago developed clever strategies to circumvent these “rules.” In 1988, for example, independent reports documented the otherwise uncommon passage of a protein (HIV-Tat) across a membrane. A subsequent study implicated a highly basic domain in this protein (Tat49-57) in its cellular entry. This conspicuously contradictory behavior of a polar, highly charged peptide passing through a nonpolar membrane set the stage for learning how Nature had gotten around the current “rules” of transport.As elaborated in our studies and discussed in this Account, the key strategy used in Nature rests in part on the ability of a molecule to change its properties as a function of microenvironment; such molecules need to be polarity chameleons, polar in a polar milieu and relatively nonpolar in a nonpolar environment. Because this research originated in part with the protein Tat and its basic peptide domain, Tat49-57, the field focused heavily on peptides, even limiting its nomenclature to names such as “cell-penetrating peptides,” “cell-permeating peptides,” “protein transduction domains,” and “membrane translocating peptides.” Starting in 1997, through a systematic reverse engineering approach, we established that the ability of Tat49-57 to enter cells is not a function of its peptide backbone, but rather a function of the number and spatial array of its guanidinium groups. These function-oriented studies enabled us and others to design more effective peptidic agents and to think beyond the confines of peptidic systems to new and even more effective nonpeptidic agents. Because the function of passage across a cell membrane is not limited to or even best achieved with the peptide backbone, we referred to these agents by their shared function, “cell-penetrating molecular transporters.” The scope of this molecular approach to breaching biochemical barriers has expanded remarkably in the past 15 years: enabling or enhancing the delivery of a wide range of cargos into cells and across other biochemical barriers, creating new tools for research, imaging, and diagnostics, and introducing new therapies into clinical trials.
Co-reporter:Yasuyuki Ogawa, Phillip P. Painter, Dean J. Tantillo, and Paul A. Wender
The Journal of Organic Chemistry 2013 Volume 78(Issue 1) pp:104-115
Publication Date(Web):November 2, 2012
DOI:10.1021/jo301953h
The Prins cyclization of syn-β-hydroxy allylsilanes and aldehydes gives cis-2,6-disubstituted 4-alkylidenetetrahydropyrans as sole products in excellent yields regardless of the aldehyde (R″) or syn-β-hydroxy allylsilane substituent (R′) used. By reversing the R″ and R′ groups, complementary exocyclic stereocontrol can be achieved. When the anti-β-hydroxy allylsilanes are used, the Prins cyclization gives predominantly cis-2,6-disubstituted 4-alkylidenetetrahydropyrans, now with the opposite olefin geometry in excellent yield. The proposed reaction mechanism and the observed stereoselectivity for these processes are supported by DFT calculations.
Co-reporter:Elizabeth J. Beans;Dennis Fournogerakis;Carolyn Gauntlett;Lars V. Heumann;Rainer Kramer;Matthew D. Marsden;Danielle Murray;Tae-Wook Chun;Jerome A. Zack
PNAS 2013 Volume 110 (Issue 29 ) pp:11698-11703
Publication Date(Web):2013-07-16
DOI:10.1073/pnas.1302634110
Highly active antiretroviral therapy (HAART) decreases plasma viremia below the limits of detection in the majority of HIV-infected
individuals, thus serving to slow disease progression. However, HAART targets only actively replicating virus and is unable
to eliminate latently infected, resting CD4+ T cells. Such infected cells are potentially capable of reinitiating virus replication upon cessation of HAART, thus leading
to viral rebound. Agents that would eliminate these reservoirs, when used in combination with HAART, could thus provide a
strategy for the eradication of HIV. Prostratin is a preclinical candidate that induces HIV expression from latently infected
CD4+ T cells, potentially leading to their elimination through a virus-induced cytopathic effect or host anti-HIV immunity. Here,
we report the synthesis of a series of designed prostratin analogs and report in vitro and ex vivo studies of their activity
relevant to induction of HIV expression. Members of this series are up to 100-fold more potent than the preclinical lead (prostratin)
in binding to cell-free PKC, and in inducing HIV expression in a latently infected cell line and prostratin-like modulation
of cell surface receptor expression in primary cells from HIV-negative donors. Significantly, selected members were also tested
for HIV induction in resting CD4+ T cells isolated from infected individuals receiving HAART and were found to exhibit potent induction activity. These more
potent agents and by extension related tunable analogs now accessible through the studies described herein should facilitate
research and preclinical advancement of this strategy for HIV/AIDS eradication.
Co-reporter:Xiufang Xu ; Peng Liu ; Adam Lesser ; Lauren E. Sirois ; Paul A. Wender ;K. N. Houk
Journal of the American Chemical Society 2012 Volume 134(Issue 26) pp:11012-11025
Publication Date(Web):June 5, 2012
DOI:10.1021/ja3041724
The first theoretical study on the effects of ligands on the mechanism, reactivities, and regioselectivities of Rh(I)-catalyzed (5 + 2) cycloadditions of vinylcyclopropanes (VCPs) and alkynes has been performed using density functional theory (DFT) calculations. Highly efficient and selective intermolecular (5 + 2) cycloadditions of VCPs and alkynes have been achieved recently using two novel rhodium catalysts, [Rh(dnCOT)]+SbF6– and [Rh(COD)]+SbF6–, which provide superior reactivities and regioselectivities relative to that of the previously reported [Rh(CO)2Cl]2 catalyst. Computationally, the high reactivities of the dnCOT and COD ligands are attributed to the steric repulsions that destabilize the Rh-product complex, the catalyst resting state in the catalytic cycle. The regioselectivities of reactions with various alkynes and different Rh catalysts are investigated, and a predictive model is provided that describes substrate–substrate and ligand–substrate steric repulsions, electronic effects, and noncovalent π/π and C–H/π interactions. In the reactions with dnCOT or COD ligands, the first new C–C bond is formed proximal to the bulky substituent on the alkyne to avoid ligand–substrate steric repulsions. This regioselectivity is reversed either by employing the smaller [Rh(CO)2Cl]2 catalyst to diminish the ligand–substrate repulsions or by using aryl alkynes, for which the ligand–substrate interactions become stabilizing due to π/π and C–H/π dispersion interactions. Electron-withdrawing groups on the alkyne prefer to be proximal to the first new C–C bond to maximize metal–substrate back-bonding interactions. These steric, electronic, and dispersion effects can all be utilized in designing new ligands to provide regiochemical control over product formation with high selectivities. The computational studies reveal the potential of employing the dnCOT family of ligands to achieve unique regiochemical control due to the steric influences and dispersion interactions associated with the rigid aryl substituents on the ligand.
Co-reporter:Joel M. Hyman;Bahram Parvin;Erika I. Geihe;Brian M. Trantow
PNAS 2012 Volume 109 (Issue 33 ) pp:
Publication Date(Web):2012-08-14
DOI:10.1073/pnas.1202509109
Interest in algae has significantly accelerated with the increasing recognition of their potentially unique role in medical,
materials, energy, bioremediation, and synthetic biological research. However, the introduction of tools to study, control,
or expand the inner-workings of algae has lagged behind. Here we describe a general molecular method based on guanidinium-rich
molecular transporters (GR-MoTrs) for bringing small and large cargos into algal cells. Significantly, this method is shown
to work in wild-type algae that have an intact cell wall. Developed using Chlamydomonas reinhardtii, this method is also successful with less studied algae including Neochloris oleoabundans and Scenedesmus dimorphus thus providing a new and versatile tool for algal research.
Co-reporter: Paul A. Wender;Adam B. Lesser ;Lauren E. Sirois
Angewandte Chemie 2012 Volume 124( Issue 11) pp:2790-2794
Publication Date(Web):
DOI:10.1002/ange.201108270
Co-reporter: Paul A. Wender;Adam B. Lesser ;Lauren E. Sirois
Angewandte Chemie International Edition 2012 Volume 51( Issue 11) pp:2736-2740
Publication Date(Web):
DOI:10.1002/anie.201108270
Co-reporter:Erika I. Geihe;Christina B. Cooley;Matthew K. Kiesewetter;Jeff R. Simon;Justin A. Edward;Roger L. Kaspar;Robyn P. Hickerson;James L. Hedrick;Robert M. Waymouth
PNAS 2012 Volume 109 (Issue 33 ) pp:
Publication Date(Web):2012-08-14
DOI:10.1073/pnas.1211361109
The polyanionic nature of oligonucleotides and their enzymatic degradation present challenges for the use of siRNA in research
and therapy; among the most notable of these is clinically relevant delivery into cells. To address this problem, we designed
and synthesized the first members of a new class of guanidinium-rich amphipathic oligocarbonates that noncovalently complex,
deliver, and release siRNA in cells, resulting in robust knockdown of target protein synthesis in vitro as determined using
a dual-reporter system. The organocatalytic oligomerization used to synthesize these co-oligomers is step-economical and broadly
tunable, affording an exceptionally quick strategy to explore chemical space for optimal siRNA delivery in varied applications.
The speed and versatility of this approach and the biodegradability of the designed agents make this an attractive strategy
for biological tool development, imaging, diagnostics, and therapeutic applications.
Co-reporter:Paul A. Wender ;Adam J. Schrier
Journal of the American Chemical Society 2011 Volume 133(Issue 24) pp:9228-9231
Publication Date(Web):May 27, 2011
DOI:10.1021/ja203034k
The total synthesis of bryostatin 9 was accomplished using a uniquely step-economical and convergent Prins-driven macrocyclization strategy. At 25 linear and 42 total steps, this is currently the most concise and convergent synthesis of a potent bryostatin.
Co-reporter:Paul A. Wender;Brian A. Loy ;Adam J. Schrier
Israel Journal of Chemistry 2011 Volume 51( Issue 3-4) pp:453-472
Publication Date(Web):
DOI:10.1002/ijch.201100020
Abstract
We review in part our computational, design, synthesis, and biological studies on a remarkable class of compounds and their designed analogs that have led to preclinical candidates for the treatment of cancer, a first-in-class approach to Alzheimer’s disease, and a promising strategy to eradicate HIV/AIDS. Because these leads target, in part, protein kinase C (PKC) isozymes, they have therapeutic potential even beyond this striking set of therapeutic indications. This program has given rise to new synthetic methodology and represents an increasingly important direction of synthesis focused on achieving function through synthesis-informed design (function-oriented synthesis).
Co-reporter:Paul A. Wender;Jeremy L. Baryza;Stacey E. Brenner;Brian A. DeChristopher;Brian A. Loy;Adam J. Schrier;Vishal A. Verma
PNAS 2011 108 (17 ) pp:6721-6726
Publication Date(Web):2011-04-26
DOI:10.1073/pnas.1015270108
Modern methods for the identification of therapeutic leads include chemical or virtual screening of compound libraries. Nature’s
library represents a vast and diverse source of leads, often exhibiting exquisite biological activities. However, the advancement
of natural product leads into the clinic is often impeded by their scarcity, complexity, and nonoptimal properties or efficacy
as well as the challenges associated with their synthesis or modification. Function-oriented synthesis represents a strategy
to address these issues through the design of simpler and therefore synthetically more accessible analogs that incorporate
the activity-determining features of the natural product leads. This study illustrates the application of this strategy to
the design and synthesis of functional analogs of the bryostatin marine natural products. It is specifically directed at exploring
the activity-determining role of bryostatin A-ring functionality on PKC affinity and selectivity. The resultant functional
analogs, which were prepared by a flexible, modular synthetic strategy, exhibit excellent affinity to PKC and differential
isoform selectivity. These and related studies provide the basic information needed for the design of simplified and thus
synthetically more accessible functional analogs that target PKC isoforms, major targets of therapeutic interest.
Co-reporter:Paul A. Wender;Jeremy L. Baryza;Stacey E. Brenner;Brian A. DeChristopher;Brian A. Loy;Adam J. Schrier;Vishal A. Verma
PNAS 2011 108 (17 ) pp:6721-6726
Publication Date(Web):2011-04-26
DOI:10.1073/pnas.1015270108
Modern methods for the identification of therapeutic leads include chemical or virtual screening of compound libraries. Nature’s
library represents a vast and diverse source of leads, often exhibiting exquisite biological activities. However, the advancement
of natural product leads into the clinic is often impeded by their scarcity, complexity, and nonoptimal properties or efficacy
as well as the challenges associated with their synthesis or modification. Function-oriented synthesis represents a strategy
to address these issues through the design of simpler and therefore synthetically more accessible analogs that incorporate
the activity-determining features of the natural product leads. This study illustrates the application of this strategy to
the design and synthesis of functional analogs of the bryostatin marine natural products. It is specifically directed at exploring
the activity-determining role of bryostatin A-ring functionality on PKC affinity and selectivity. The resultant functional
analogs, which were prepared by a flexible, modular synthetic strategy, exhibit excellent affinity to PKC and differential
isoform selectivity. These and related studies provide the basic information needed for the design of simplified and thus
synthetically more accessible functional analogs that target PKC isoforms, major targets of therapeutic interest.
Co-reporter:Paul A. Wender;Jeremy L. Baryza;Stacey E. Brenner;Brian A. DeChristopher;Brian A. Loy;Adam J. Schrier;Vishal A. Verma
PNAS 2011 108 (17 ) pp:6721-6726
Publication Date(Web):2011-04-26
DOI:10.1073/pnas.1015270108
Modern methods for the identification of therapeutic leads include chemical or virtual screening of compound libraries. Nature’s
library represents a vast and diverse source of leads, often exhibiting exquisite biological activities. However, the advancement
of natural product leads into the clinic is often impeded by their scarcity, complexity, and nonoptimal properties or efficacy
as well as the challenges associated with their synthesis or modification. Function-oriented synthesis represents a strategy
to address these issues through the design of simpler and therefore synthetically more accessible analogs that incorporate
the activity-determining features of the natural product leads. This study illustrates the application of this strategy to
the design and synthesis of functional analogs of the bryostatin marine natural products. It is specifically directed at exploring
the activity-determining role of bryostatin A-ring functionality on PKC affinity and selectivity. The resultant functional
analogs, which were prepared by a flexible, modular synthetic strategy, exhibit excellent affinity to PKC and differential
isoform selectivity. These and related studies provide the basic information needed for the design of simplified and thus
synthetically more accessible functional analogs that target PKC isoforms, major targets of therapeutic interest.
Co-reporter:Paul Wender
Chemistry – An Asian Journal 2011 Volume 6( Issue 8) pp:
Publication Date(Web):
DOI:10.1002/asia.201100574
No abstract is available for this article.
Co-reporter:Paul A. Wender, Jenny Reuber
Tetrahedron 2011 67(51) pp: 9998-10005
Publication Date(Web):
DOI:10.1016/j.tet.2011.09.058
Co-reporter:Peng Liu ; Lauren E. Sirois ; Paul Ha-Yeon Cheong ; Zhi-Xiang Yu ; Ingo V. Hartung ; Heiko Rieck ◻; Paul A. Wender ;K. N. Houk
Journal of the American Chemical Society 2010 Volume 132(Issue 29) pp:10127-10135
Publication Date(Web):June 30, 2010
DOI:10.1021/ja103253d
The first studies on the regioselectivity of Rh(I)-catalyzed (5 + 2) cycloadditions between vinylcyclopropanes (VCPs) and alkynes have been conducted experimentally and analyzed using density functional theory (DFT). The previously unexplored regiochemical consequences for this catalytic, intermolecular cycloaddition were determined by studying the reactions of several substituted VCPs with a range of unsymmetrical alkynes. Experimental trends were identified, and a predictive model was established. VCPs with terminal substitution on the alkene reacted with high regioselectivity (>20:1), as predicted by a theoretical model in which bulkier alkyne substituents prefer to be distal to the forming C−C bond to avoid steric repulsions. VCPs with substitution at the internal position of the alkene reacted with variable regioselectivity (ranging from >20:1 to a reversed 1:2.3), suggesting a refined model in which electron-withdrawing substituents on the alkyne decrease or reverse sterically controlled selectivity by stabilizing the transition state in which the substituent is proximal to the forming C−C bond.
Co-reporter:Paul A. Wender ; René T. Stemmler ;Lauren E. Sirois
Journal of the American Chemical Society 2010 Volume 132(Issue 8) pp:2532-2533
Publication Date(Web):February 8, 2010
DOI:10.1021/ja910696x
The bicyclo[5.3.0]decane skeleton is one of the most commonly encountered bicyclic subunits in nature and the core scaffold of a wide range of targets of structural, biological, and therapeutic importance. Prompted by the interest in such structures, we report the first studies of metal-catalyzed [5+2] cycloadditions of vinylcyclopropanes (VCPs) and enynones. The resultant efficiently formed dienone cycloadducts serve as substrates for subsequent Nazarov cyclizations and as intermediates for single-operation [5+2]/Nazarov serial reactions and catalytic cascades. In many cases the one-flask process can be carried out in shorter reaction times and with comparable or superior yields to the two-flask procedure. Significantly, a single catalyst can be used to mediate both transformations. These [5+2]/Nazarov reaction sequences and cascades collectively provide strategically novel and facile access to the bicyclo[5.3.0]decane skeleton from simple and readily available components.
Co-reporter:Paul A. Wender, Lauren E. Sirois, René T. Stemmler and Travis J. Williams
Organic Letters 2010 Volume 12(Issue 7) pp:1604-1607
Publication Date(Web):March 2, 2010
DOI:10.1021/ol100337m
A cationic rhodium(I) complex―[(C10H8)Rh(cod)]+ SbF6−―catalyzes the remarkably efficient intermolecular [5 + 2] cycloaddition of vinylcyclopropanes (VCPs) and various alkynes, providing cycloheptene cycloadducts in excellent yields in minutes at room temperature. The efficacy and selectivity of this catalyst are also shown in a novel diversification strategy, affording a cycloadduct library in one step from nine commercially available components.
Co-reporter:Mitchell P. Croatt
European Journal of Organic Chemistry 2010 Volume 2010( Issue 1) pp:19-32
Publication Date(Web):
DOI:10.1002/ejoc.200900929
Abstract
Metal-catalyzed diene-yne, diene-ene, and diene-allene [2+2+1] cycloaddition reactions provide new methods for the facile construction of highly functionalized five-membered rings. These reactions can be conducted with a variety of substrate substitution patterns and functional groups and often in the absence of solvent. The special reactivity of dienes, a key to enabling or enhancing the effectiveness of the [2+2+1] and other reactions, is significantly different from that of alkynes, alkenes, or allenes. For example, the [2+2+1] reaction of a diene-yne is accelerated compared to that of the corresponding ene-yne. An even more dramatic “diene effect” is found with diene-enes and diene-allenes. While bis-enes and ene-allenes are not reported to yield [2+2+1] cycloadducts, the related diene-enes and diene-allenes undergo efficient [2+2+1] cycloadditions, providing new routes to cyclopentanones and alkylidenecyclopentanones. Mechanistic studies suggest that the unique reactivity observed with dienes arises from their participation in the putative rate-determining reductive elimination step by providing an additional energy-lowering coordination site for the transition metal catalyst.
Co-reporter:Paul A. Wender ;Daniel Strand
Journal of the American Chemical Society 2009 Volume 131(Issue 22) pp:7528-7529
Publication Date(Web):May 12, 2009
DOI:10.1021/ja901799s
An efficient cyclocarboamination reaction of nonactivated alkynes with aziridines, catalyzed by Lewis or Bronsted acids, to form 2,3-dihydropyrroles through a formal [3+2] cycloaddition, is described. The reaction provides a wide range of polysubstituted dihydropyrroles in a highly regioselective manner, is scalable, proceeds under mild reaction conditions, and uses low catalyst loadings.
Co-reporter:Christina B. Cooley ; Brian M. Trantow ; Fredrik Nederberg ; Matthew K. Kiesewetter ; James L. Hedrick ; Robert M. Waymouth
Journal of the American Chemical Society 2009 Volume 131(Issue 45) pp:16401-16403
Publication Date(Web):October 27, 2009
DOI:10.1021/ja907363k
A new family of guanidinium-rich molecular transporters featuring a novel oligocarbonate backbone with 1,7-side chain spacing is described. Conjugates can be rapidly assembled irrespective of length in a one-step oligomerization strategy that can proceed with concomitant introduction of probes (or by analogy drugs). The new transporters exhibit excellent cellular entry as determined by flow cytometry and fluorescence microscopy, and the functionality of their drug delivery capabilities was confirmed by the delivery of the bioluminescent small molecule probe luciferin and turnover by its intracellular target enzyme.
Co-reporter:Paul A. Wender and Kate E. Longcore
Organic Letters 2009 Volume 11(Issue 23) pp:5474-5477
Publication Date(Web):November 4, 2009
DOI:10.1021/ol902308v
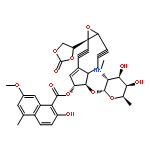
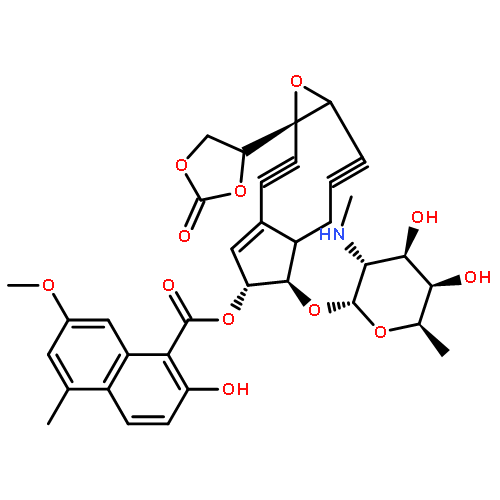
Two new glycosylated macrolactones, apoptolidins E (5) and F (6), were isolated from fermentation of the actinomycete Nocardiopsis sp. and their structures assigned. Lacking the C16 and C20 oxygens of apoptolidin A (1), these macrolides are also the first members of this family to display a 4-O-methyl-l-rhamnose at C9 rather than a 6-deoxy-4-O-methyl-l-glucose.
Co-reporter:Chad A. Lewis, Kate E. Longcore, Scott J. Miller and Paul A. Wender
Journal of Natural Products 2009 Volume 72(Issue 10) pp:1864-1869
Publication Date(Web):September 21, 2009
DOI:10.1021/np9004932
We report the application of peptide-based catalysts to the site-selective modification of apoptolidin A (1), an agent that displays remarkable selectivity for inducing apoptosis in E1A-transformed cell lines. Key to the approach was the development of an assay suitable for the screening of dozens of catalysts in parallel reactions that could be conducted using only microgram quantities of the starting material. Employing this assay, catalysts (e.g., 11 and ent-11) were identified that afforded unique product distributions, distinct from the product mixtures produced when a simple catalyst (N,N-dimethyl-4-aminopyridine (10)) was employed. Preparative reactions were then carried out with the preferred catalysts so that unique, homogeneous apoptolidin analogues could be isolated and characterized. From these studies, three new apoptolidin analogues were obtained (12−14), each differing from the other in either the location of acyl group substituents or the number of acetate groups appended to the natural product scaffold. Biological evaluation of the new apoptolidin analogues was then conducted using growth inhibition assays based on the H292 human lung carcinoma cell line. The new analogues exhibited activities comparable to apoptolidin A.
Co-reporter:Paul A. Wender, Mitchell P. Croatt and Björn Kühn
Organometallics 2009 Volume 28(Issue 20) pp:5841-5844
Publication Date(Web):September 30, 2009
DOI:10.1021/om9007373
A novel metal-catalyzed, all-alkene [2+2+2] cycloaddition reaction involving a strained and conformationally restricted bis-ene and a diene is reported.(1) Modification of the catalyst leads to competition with a diene-ene [2+2] reaction, and when an alkyne is used in place of the diene, [2+2+2] and [2+2+2+2] reactions occur involving the bis-ene and 1 or 2 equiv of the alkyne, respectively.
Co-reporter:PaulA. Wender ;JustinP. Christy;AdamB. Lesser ;MarcT. Gieseler
Angewandte Chemie 2009 Volume 121( Issue 41) pp:7823-7826
Publication Date(Web):
DOI:10.1002/ange.200903859
Co-reporter:PaulA. Wender ;JustinP. Christy;AdamB. Lesser ;MarcT. Gieseler
Angewandte Chemie International Edition 2009 Volume 48( Issue 41) pp:7687-7690
Publication Date(Web):
DOI:10.1002/anie.200903859
Co-reporter:Susan L. Mooberry, Michael K. Hilinski, Erin A. Clark and Paul A. Wender
Molecular Pharmaceutics 2008 Volume 5(Issue 5) pp:829-838
Publication Date(Web):July 29, 2008
DOI:10.1021/mp800043n
Laulimalide is a potent microtubule stabilizing agent and a promising anticancer therapeutic lead. The identification of stable, efficacious and accessible analogues is critical to clinically exploiting this novel lead. To determine which structural features of laulimalide are required for beneficial function and thus for accessing superior clinical candidates, a series of side chain analogues were prepared through a last step cross metathesis diversification strategy and their biological activities were evaluated. Five analogues, differing in potency from 233 nM to 7.9 μM, effectively inhibit cancer cell proliferation. Like laulimalide, they retain activity against multidrug resistant cells, stabilize microtubules and cause the formation of aberrant mitotic spindles, mitotic accumulation, Bcl-2 phosphorylation and initiation of apoptosis. Structural modifications in the C23−C27 dihydropyran side chain can be made without changing the overall mechanism of action, but it is clear that this subunit has more than a bystander role.Keywords: antimitotics; Laulimalide; laulimalide analogues; metathesis; microtubule stabilizers; synthetic chemistry;
Co-reporter:Paul A. Wender;Jung-Min Kee;Jeffrey M. Warrington
Science 2008 Volume 320(Issue 5876) pp:649-652
Publication Date(Web):02 May 2008
DOI:10.1126/science.1154690
Abstract
Although antiretroviral therapies have been effective in decreasing active viral loads in AIDS patients, the persistence of latent viral reservoirs prevents eradication of the virus. Prostratin and DPP (12-deoxyphorbol-13-phenylacetate) activate the latent virus and thus represent promising adjuvants for antiviral therapy. Their limited supply and the challenges of accessing related structures have, however, impeded therapeutic development and the search for clinically superior analogs. Here we report a practical synthesis of prostratin and DPP starting from phorbol or crotophorbolone, agents readily available from renewable sources, including a biodiesel candidate. This synthesis reliably supplies gram quantities of the therapeutically promising natural products, hitherto available only in low and variable amounts from natural sources, and opens access to a variety of new analogs.
Co-reporter:Thomas H. Pillow;Christopher H. Contag;Elena A. Dubikovskaya;Steve H. Thorne
PNAS 2008 Volume 105 (Issue 34 ) pp:12128-12133
Publication Date(Web):2008-08-26
DOI:10.1073/pnas.0805374105
Many cancer therapeutic agents elicit resistance that renders them ineffective and often produces cross-resistance to other
drugs. One of the most common mechanisms of resistance involves P-glycoprotein (Pgp)-mediated drug efflux. To address this
problem, new agents have been sought that are less prone to inducing resistance and less likely to serve as substrates for
Pgp efflux. An alternative to this approach is to deliver established agents as molecular transporter conjugates into cells
through a mechanism that circumvents Pgp-mediated efflux and allows for release of free drug only after cell entry. Here we
report that the widely used chemotherapeutic agent Taxol, ineffective against Taxol-resistant human ovarian cancer cell lines,
can be incorporated into a releasable octaarginine conjugate that is effective against the same Taxol-resistant cell lines.
It is significant that the ability of the Taxol conjugates to overcome Taxol resistance is observed both in cell culture and
in animal models of ovarian cancer. The generality and mechanistic basis for this effect were also explored with coelenterazine,
a Pgp substrate. Although coelenterazine itself does not enter cells because of Pgp efflux, its octaarginine conjugate does
so readily. This approach shows generality for overcoming the multidrug resistance elicited by small-molecule cancer chemotherapeutics
and could improve the prognosis for many patients with cancer and fundamentally alter search strategies for novel therapeutic
agents that are effective against resistant disease.
Co-reporter:Elena A. Goun;Lisa R. Jones;Jonathan B. Rothbard;Thomas H. Pillow;Rajesh Shinde;Christopher H. Contag
PNAS 2007 Volume 104 (Issue 25 ) pp:10340-10345
Publication Date(Web):2007-06-19
DOI:10.1073/pnas.0703919104
Many therapeutic leads fail to advance clinically because of bioavailability, selectivity, and formulation problems. Molecular
transporters can be used to address these problems. Molecular transporter conjugates of otherwise poorly soluble or poorly
bioavailable drugs or probes exhibit excellent solubility in water and biological fluids and at the same time an enhanced
ability to enter tissues and cells and with modification to do so selectively. For many conjugates, however, it is necessary
to release the drug/probe cargo from the transporter after uptake to achieve activity. Here, we describe an imaging method
that provides quantification of transporter conjugate uptake and cargo release in real-time in animal models. This method
uses transgenic (luciferase) reporter mice and whole-body imaging, allowing noninvasive quantification of transporter conjugate
uptake and probe (luciferin) release in real time. This process effectively emulates drug-conjugate delivery, drug release,
and drug turnover by an intracellular target, providing a facile method to evaluate comparative uptake of new transporters
and efficacy and selectivity of linker release as required for fundamental studies and therapeutic applications.
Co-reporter:Elena A. Goun;Thomas H. Pillow;Lisa R. Jones;Jonathan B. Rothbard Dr. Dr.
ChemBioChem 2006 Volume 7(Issue 10) pp:
Publication Date(Web):14 SEP 2006
DOI:10.1002/cbic.200600171
Crossing borders. Methods to enhance or control selective passage of therapeutics or probes into or through biological barriers are a key to the future of drug therapy and many fundamental advances in science. One bioinspired strategy for crossing biological barriers is based on the use of molecular transporters, the focus of this minireview and our studies Stanford.
Co-reporter:Paul A. Wender ;Nicole M. Deschamps;Robert Sun
Angewandte Chemie 2006 Volume 118(Issue 24) pp:
Publication Date(Web):9 MAY 2006
DOI:10.1002/ange.200600806
Expandieren: Eine Rhodium(I)-katalysierte C-C-Aktivierung von Allenylcyclobutanen liefert Metallacyclen-Intermediate, die mit CO abgefangen werden. Funktionalisierte mono- wie bicyclische Cycloheptenone werden mit dieser neuen carbonylierenden [6+1]-Cycloaddition höherer Ordnung in guten bis ausgezeichneten Ausbeuten erhalten.
Co-reporter:Paul A. Wender ;Mitchell P. Croatt;Nicole M. Deschamps
Angewandte Chemie 2006 Volume 118(Issue 15) pp:
Publication Date(Web):9 MAR 2006
DOI:10.1002/ange.200600300
Der Kreis wird geschlossen: Die RhI-katalysierten Titelreaktionen verlaufen unter milden Bedingungen und mit guten bis ausgezeichneten Ausbeuten bei einer Katalysatorbeladung von nur 0.1 Mol-%. Die Dienylallene reagieren selektiv über die inneren Doppelbindungen der Allen- und Dieneinheiten, was eine Vielzahl an Substituenten ermöglicht. DCE=1,2-Dichlorethan, TFE=2,2,2-Trifluorethanol.
Co-reporter:Paul A. Wender ;Nicole M. Deschamps;Robert Sun
Angewandte Chemie International Edition 2006 Volume 45(Issue 24) pp:
Publication Date(Web):9 MAY 2006
DOI:10.1002/anie.200600806
Ever expanding: A RhI-catalyzed CC activation reaction of allenylcyclobutanes generates intermediate metallacycles that can be trapped with CO to produce a new [6+1] reaction. Functionalized monocyclic and bicyclic cycloheptenones are obtained in good-to-excellent yields with this [6+1] higher-order carbonylative cycloaddition reaction.
Co-reporter:Paul A. Wender, Mitchell P. Croatt,Nicole M. Deschamps
Angewandte Chemie International Edition 2006 45(15) pp:2459-2462
Publication Date(Web):
DOI:10.1002/anie.200600300
Co-reporter:Jeremy L. Baryza, Stacey E. Brenner, Madeleine L. Craske, Tobias Meyer, Paul A. Wender
Chemistry & Biology 2004 Volume 11(Issue 9) pp:1261-1267
Publication Date(Web):September 2004
DOI:10.1016/j.chembiol.2004.06.014
Structurally simplified analogs of bryostatin 1, a marine natural product in clinical trials for the treatment of cancer, have been shown to be up to 50 times more potent than bryostatin 1 at inducing the translocation of PKCδ-GFP from the cytosol of rat basophilic leukemia (RBL) cells. The end distribution of the protein is similar for all three compounds, despite a significant difference in translocation kinetics. The potency of the compounds for inducing the translocation response appears to be only qualitatively related to their binding affinity for PKC, highlighting the importance of using binding affinity in conjunction with real-time measurements of protein localization for the pharmacological profiling of biologically active agents.
Co-reporter:Susan L. Mooberry;Deborah A. Randall-Hlubek;Sayee G. Hegde;Lei Zhang;Rachel M. Leal;Robert D. Hubbard
PNAS 2004 Volume 101 (Issue 23 ) pp:8803-8808
Publication Date(Web):2004-06-08
DOI:10.1073/pnas.0402759101
Laulimalide is a potent, structurally unique microtubule-stabilizing agent originally isolated from the marine sponge Cacospongia mycofijiensis. Laulimalide exhibits an activity profile different from other microtubule-binding agents, notably including effectiveness
against paclitaxel-resistant cells, but it is intrinsically unstable. Five analogues of laulimalide were designed to exhibit
enhanced chemical stability yet retain its exceptional biological activities. Evaluations of these analogues showed that all
are effective inhibitors of cancer-cell proliferation yet differ substantially in potency with an IC50 range of 0.12–16.5 μM. Although all of the analogues initiated cellular changes similar to laulimalide, including increased
density of interphase microtubules, aberrant mitotic spindles, and ultimately apoptosis, differences among the analogues were
apparent. The two most potent analogues, C16-C17-des-epoxy laulimalide and C20-methoxy laulimalide, appear to have a mechanism of action identical to laulimalide. The C16-C17-des-epoxy, C20-methoxy laulimalide derivative, which incorporates both chemical changes of the most potent analogues, was significantly
less potent and initiated the formation of unique interphase microtubules unlike the parent compound and other analogues.
Two C2-C3-alkynoate derivatives had lower potency, and they initiated abnormal microtubule structures but did not cause micronucleation
or extensive G2/M accumulation. Significantly, paclitaxel- and epothilone-resistant cell lines were less resistant to the laulimalide analogues.
In summary, analogues of laulimalide designed to minimize or eliminate its intrinsic instability have been synthesized, and
some have been found to retain the unique biological activities of laulimalide.
Co-reporter:Paul A. Wender ;Nicole M. Deschamps;Travis J. Williams
Angewandte Chemie 2004 Volume 116(Issue 23) pp:
Publication Date(Web):13 MAY 2004
DOI:10.1002/ange.200454117
Drei-Komponenten-[2+2+1]-Cycloadditionen gelingen mit Dienen statt Alkenen in einer intermolekularen, RhI-katalysierten Variante der Pauson-Khand-Reaktion (siehe Schema). Die höhere Reaktivität der Diene ermöglicht einen effizienten Zugang zu Alkenylcyclopentenonen ausgehend von leicht erhältlichen Ausgangsstoffen. R1, R2=Alkyl, Silyl, Carbonyl; R3=Me, Bn.
Co-reporter:Paul A. Wender ;Nicole M. Deschamps;Travis J. Williams
Angewandte Chemie International Edition 2004 Volume 43(Issue 23) pp:
Publication Date(Web):13 MAY 2004
DOI:10.1002/anie.200454117
Three-component [2+2+1] cycloadditions are facilitated by the use of dienes as activated two-carbon-atom components in a RhI-catalyzed intermolecular variant of the Pauson–Khand reaction (see scheme). The enhanced reactivity of the dienes allows efficient access to a variety of alkenyl cyclopentenones from commercially or readily available starting materials. R1, R2=alkyl, silyl, carbonyl; R3=Me, Bn.
Co-reporter:Paul A. Wender ;Nicole M. Deschamps;Gabriel G. Gamber
Angewandte Chemie 2003 Volume 115(Issue 16) pp:
Publication Date(Web):23 APR 2003
DOI:10.1002/ange.200350949
Die Dien-Einheit spielt die Schlüsselrolle bei der Beschleunigung intramolekularer [2+2+1]-Pauson-Khand-Reaktionen von Dieninen (siehe Schema). In Gegenwart von Kohlenmonoxid, [RhCl(CO)(PPh3)2] und AgSbF6 reagieren Dienine mit guten bis ausgezeichneten Ausbeuten und – im Unterschied zu vergleichbaren Eninen – oft bereits bei Raumtemperatur.
Co-reporter:Paul A. Wender ;Nicole M. Deschamps;Gabriel G. Gamber
Angewandte Chemie International Edition 2003 Volume 42(Issue 16) pp:
Publication Date(Web):23 APR 2003
DOI:10.1002/anie.200350949
The diene plays a key role in the Pauson–Khand [2+2+1] reaction of tethered dienynes (see scheme). These reactions are accelerated by the diene (relative to an alkene) and proceed in good to excellent yields, often at room temperature, in the presence of carbon monoxide, [RhCl(CO)(PPh3)2], and AgSbF6.
Co-reporter:Paul A. Wender, Christina B. Cooley, Erika I. Geihe
Drug Discovery Today: Technologies (Spring 2012) Volume 9(Issue 1) pp:e49-e55
Publication Date(Web):1 March 2012
DOI:10.1016/j.ddtec.2011.07.004
Inspired originally by peptides that traverse biological barriers, research on molecular transporters has since identified the key structural requirements that govern cellular entry, leading to new, significantly more effective and more readily available agents. These new drug delivery systems enable or enhance cellular and tissue uptake, can be targeted and provide numerous additional advantages of significance in imaging, diagnostics and therapy.
Co-reporter:Paul A. Wender, Wesley C. Galliher, Elena A. Goun, Lisa R. Jones, Thomas H. Pillow
Advanced Drug Delivery Reviews (1 March 2008) Volume 60(Issues 4–5) pp:452-472
Publication Date(Web):1 March 2008
DOI:10.1016/j.addr.2007.10.016
The ability of a drug or probe to cross a biological barrier has historically been viewed to be a function of its intrinsic physical properties. This view has largely restricted drug design and selection to agents within a narrow log P range. Molecular transporters offer a strategy to circumvent these restrictions. In the case of guanidinium-rich transporters (GRTs), a typically highly water-soluble conjugate is found to readily pass through the non-polar membrane of a cell and for some across tissue barriers. This activity opens a field of opportunities for the use of GRTs to enable delivery of polar and non-polar drugs or probes as well as to enhance uptake of those of intermediate polarity. The field of transporter enabled or enhanced uptake has grown dramatically in the last decade. Some GRT drug conjugates have been advanced into clinical trials. This review will provide an overview of recent work pertinent to the design and mechanism of uptake of GRTs.
Co-reporter:Paul A. Wender, Alison D. Axtman, Jennifer E. Golden, Jung-Min Kee, Lauren E. Sirois, Ryan V. Quiroz and Matthew C. Stevens
Inorganic Chemistry Frontiers 2014 - vol. 1(Issue 10) pp:NaN1171-1171
Publication Date(Web):2014/10/06
DOI:10.1039/C4QO00228H
The human kinome comprises over 500 protein kinases. When mutated or over-expressed, many play critical roles in abnormal cellular functions associated with cancer, cardiovascular disease and neurological disorders. Here we report a step-economical approach to designed kinase inhibitors inspired by the potent, but non-selective, natural product staurosporine, and synthetically enabled by a novel, complexity-increasing, serialized [5 + 2]/[4 + 2] cycloaddition strategy. This function-oriented synthesis approach rapidly affords tunable scaffolds, and produced a low nanomolar inhibitor of protein kinase C.

![Bicyclo[2.2.1]heptane-2-carboxylic acid, methyl ester, (1R,2R,4S)-rel-](/data/chemimg/1160700/23057-38-7.png)
![Bicyclo[2.2.1]heptane-2-carboxylic acid, methyl ester, (1R,2R,4S)-rel-](/data/chemimg/1160700/23057-38-7_b.png)


![Silane, [[4-(1,1-dimethylethyl)-1-cyclohexen-1-yl]oxy]trimethyl-](http://img.cochemist.com/ccimg/20000/19980-19-9.png)
![Silane, [[4-(1,1-dimethylethyl)-1-cyclohexen-1-yl]oxy]trimethyl-](http://img.cochemist.com/ccimg/20000/19980-19-9_b.png)






![Bicyclo[2.2.1]heptane-2-carboxylic acid, methyl ester, (1R,2S,4S)-rel-](/data/chemimg/1141400/16646-41-6.png)
![Bicyclo[2.2.1]heptane-2-carboxylic acid, methyl ester, (1R,2S,4S)-rel-](/data/chemimg/1141400/16646-41-6_b.png)






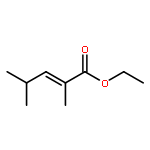
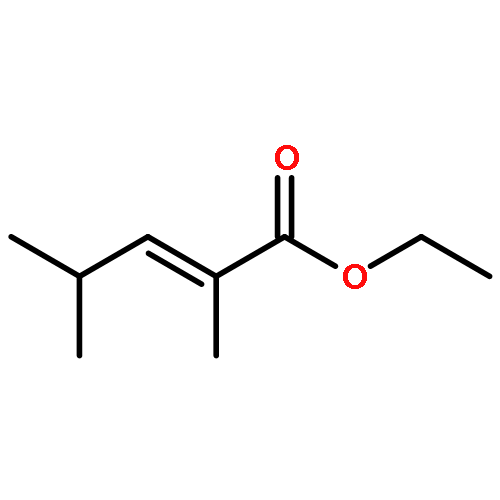


![bicyclo[3.2.0]heptan-6-one](http://img.cochemist.com/ccimg/13800/13756-54-2.png)
![bicyclo[3.2.0]heptan-6-one](http://img.cochemist.com/ccimg/13800/13756-54-2_b.png)




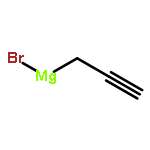
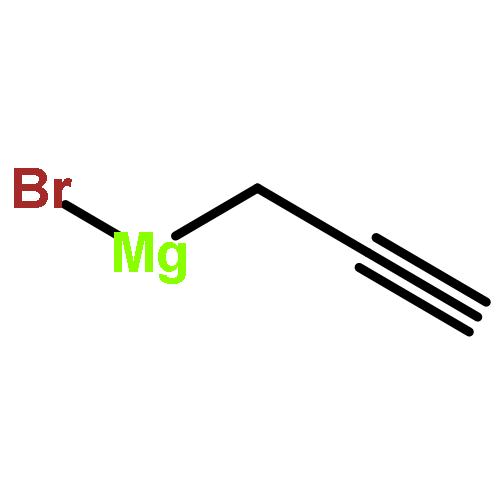


![6H-Benzo[g][1,4]diazonino[7,6,5-cd]indol-6-one,13-ethenyl-1,3,4,5,7,8,10,11,12,13-decahydro-4-(hydroxymethyl)-8,10,13-trimethyl-7,10-bis(1-methylethyl)-,(4S,7S,10R,13R)- (9CI)](http://img.cochemist.com/ccimg/11100/11032-05-6.png)
![6H-Benzo[g][1,4]diazonino[7,6,5-cd]indol-6-one,13-ethenyl-1,3,4,5,7,8,10,11,12,13-decahydro-4-(hydroxymethyl)-8,10,13-trimethyl-7,10-bis(1-methylethyl)-,(4S,7S,10R,13R)- (9CI)](http://img.cochemist.com/ccimg/11100/11032-05-6_b.png)








![7,8-dihydro-6H-Cyclopent[g]isoquinoline](http://img.cochemist.com/ccimg/7200/7193-30-8.png)
![7,8-dihydro-6H-Cyclopent[g]isoquinoline](http://img.cochemist.com/ccimg/7200/7193-30-8_b.png)



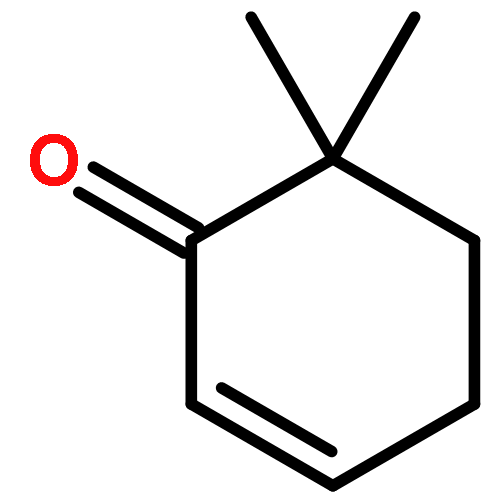


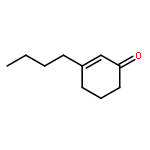
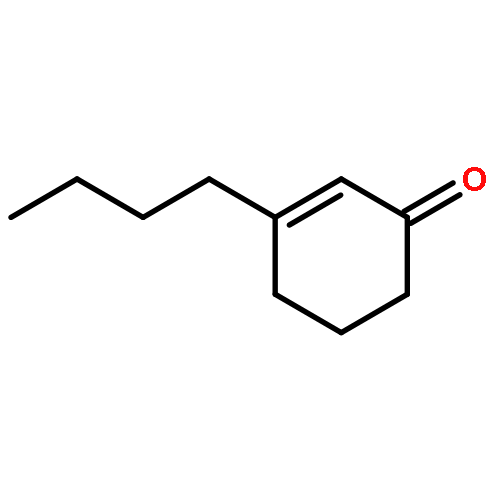


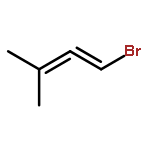







![2H-Benzo[a]quinolizine, 1,3,4,6,7,11b-hexahydro-9,10-dimethoxy-](http://img.cochemist.com/ccimg/4800/4787-30-8.png)
![2H-Benzo[a]quinolizine, 1,3,4,6,7,11b-hexahydro-9,10-dimethoxy-](http://img.cochemist.com/ccimg/4800/4787-30-8_b.png)
![5H-Dibenzo[a,c]cyclononene, 6,7,8,9-tetrahydro-](http://img.cochemist.com/ccimg/4500/4444-45-5.png)
![5H-Dibenzo[a,c]cyclononene, 6,7,8,9-tetrahydro-](http://img.cochemist.com/ccimg/4500/4444-45-5_b.png)




![Methyl bicyclo[2.2.1]hepta-2,5-diene carboxylate](http://img.cochemist.com/ccimg/3700/3604-36-2.png)
![Methyl bicyclo[2.2.1]hepta-2,5-diene carboxylate](http://img.cochemist.com/ccimg/3700/3604-36-2_b.png)
![(7-methoxy-6-methyl-5,8-dioxo-2,3,5,8-tetrahydro-1H-pyrrolo[1,2-a]indol-9-yl)methyl carbamate](http://img.cochemist.com/ccimg/3600/3567-46-2.png)
![(7-methoxy-6-methyl-5,8-dioxo-2,3,5,8-tetrahydro-1H-pyrrolo[1,2-a]indol-9-yl)methyl carbamate](http://img.cochemist.com/ccimg/3600/3567-46-2_b.png)





![Bicyclo[2.2.1]heptane-2-carbonitrile,(1R,2R,4S)-rel-](http://img.cochemist.com/ccimg/3300/3211-90-3.png)
![Bicyclo[2.2.1]heptane-2-carbonitrile,(1R,2R,4S)-rel-](http://img.cochemist.com/ccimg/3300/3211-90-3_b.png)
![ENDO-2-CYANOBICYCLO[2.2.1]HEPTANE](http://img.cochemist.com/ccimg/3300/3211-87-8.png)
![ENDO-2-CYANOBICYCLO[2.2.1]HEPTANE](http://img.cochemist.com/ccimg/3300/3211-87-8_b.png)






![Spiro[5.5]undecan-2-one](http://img.cochemist.com/ccimg/1800/1781-81-3.png)
![Spiro[5.5]undecan-2-one](http://img.cochemist.com/ccimg/1800/1781-81-3_b.png)





![1H-Pyrrolo[1,2-a]indole, 2,3-dihydro-](/data/chemimg/2271300/1421-19-8.png)
![1H-Pyrrolo[1,2-a]indole, 2,3-dihydro-](/data/chemimg/2271300/1421-19-8_b.png)




![Dibenzo[a,c]cyclooctene,5,6,7,8-tetrahydro-](http://img.cochemist.com/ccimg/1100/1082-12-8.png)
![Dibenzo[a,c]cyclooctene,5,6,7,8-tetrahydro-](http://img.cochemist.com/ccimg/1100/1082-12-8_b.png)














![Spiro[2,26-dioxabicyclo[23.3.1]nonacosa-4,18,20-triene-27,2'-[2H]pyran]-3,9,13-trione,22-ethyl-3',4',5',6'-tetrahydro-7,11,14,15-tetrahydroxy-6'-[(2R)-2-hydroxypropyl]-5',6,8,10,12,14,16,28,29-nonamethyl-,(1R,2'R,4E,5'S,6S,6'S,7R,8S,10R,11R,12S,14R,15S,16R,18E,20E,22R,25S,28S,29R)-](http://img.cochemist.com/ccimg/600/579-13-5.png)
![Spiro[2,26-dioxabicyclo[23.3.1]nonacosa-4,18,20-triene-27,2'-[2H]pyran]-3,9,13-trione,22-ethyl-3',4',5',6'-tetrahydro-7,11,14,15-tetrahydroxy-6'-[(2R)-2-hydroxypropyl]-5',6,8,10,12,14,16,28,29-nonamethyl-,(1R,2'R,4E,5'S,6S,6'S,7R,8S,10R,11R,12S,14R,15S,16R,18E,20E,22R,25S,28S,29R)-](http://img.cochemist.com/ccimg/600/579-13-5_b.png)




![Tetracyclo[3.2.0.02,7.04,6]heptane](http://img.cochemist.com/ccimg/300/278-06-8.png)
![Tetracyclo[3.2.0.02,7.04,6]heptane](http://img.cochemist.com/ccimg/300/278-06-8_b.png)
![1H-Naphtho[2,3-d]triazole](http://img.cochemist.com/ccimg/300/269-12-5.png)
![1H-Naphtho[2,3-d]triazole](http://img.cochemist.com/ccimg/300/269-12-5_b.png)
![Bicyclo[2.1.0]pentane](http://img.cochemist.com/ccimg/200/185-94-4.png)
![Bicyclo[2.1.0]pentane](http://img.cochemist.com/ccimg/200/185-94-4_b.png)
![Bicyclo[1.1.0]butane](http://img.cochemist.com/ccimg/200/157-33-5.png)
![Bicyclo[1.1.0]butane](http://img.cochemist.com/ccimg/200/157-33-5_b.png)


![Bicyclo[3.1.1]hept-3-en-2-one,4,6,6-trimethyl-](http://img.cochemist.com/ccimg/100/80-57-9.png)
![Bicyclo[3.1.1]hept-3-en-2-one,4,6,6-trimethyl-](http://img.cochemist.com/ccimg/100/80-57-9_b.png)


![Benzenesulfonamide,N-[(2E)-3-cyclopropyl-2-propenyl]-4-methyl-N-2-propynyl-](http://img.cochemist.com/ccimg/518100/518020-59-2.png)
![Benzenesulfonamide,N-[(2E)-3-cyclopropyl-2-propenyl]-4-methyl-N-2-propynyl-](http://img.cochemist.com/ccimg/518100/518020-59-2_b.png)






![1H-Cyclohepta[c]furan, 3,3a,6,7-tetrahydro-8-phenyl-](http://img.cochemist.com/ccimg/166100/166043-99-8.png)
![1H-Cyclohepta[c]furan, 3,3a,6,7-tetrahydro-8-phenyl-](http://img.cochemist.com/ccimg/166100/166043-99-8_b.png)
![Benzene, [3-[[(2E)-3-cyclopropyl-2-propenyl]oxy]-1-propynyl]-](http://img.cochemist.com/ccimg/166100/166043-88-5.png)
![Benzene, [3-[[(2E)-3-cyclopropyl-2-propenyl]oxy]-1-propynyl]-](http://img.cochemist.com/ccimg/166100/166043-88-5_b.png)
![SILANE, [3-[[(2E)-3-CYCLOPROPYL-2-PROPENYL]OXY]-1-PROPYNYL]TRIMETHYL-](http://img.cochemist.com/ccimg/166100/166043-86-3.png)
![SILANE, [3-[[(2E)-3-CYCLOPROPYL-2-PROPENYL]OXY]-1-PROPYNYL]TRIMETHYL-](http://img.cochemist.com/ccimg/166100/166043-86-3_b.png)




![2-Cyclopenten-1-ol, 1-(4-hydroxy-2-pentynyl)-2-[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/137600/137527-69-6.png)
![2-Cyclopenten-1-ol, 1-(4-hydroxy-2-pentynyl)-2-[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/137600/137527-69-6_b.png)
![Cyclopropanecarboxaldehyde,2-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-, (1R,2S)-rel-](http://img.cochemist.com/ccimg/124300/124200-38-0.png)
![Cyclopropanecarboxaldehyde,2-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-, (1R,2S)-rel-](http://img.cochemist.com/ccimg/124300/124200-38-0_b.png)
![Benzenepropanoic acid, b-(benzoylamino)-a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-,(2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-6,12b-bis(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahydro-4,11-dihydroxy-4a,8,13,13-tetramethyl-5-oxo-7,11-methano-1H-cyclodeca[3,4]benz[1,2-b]oxet-9-ylester, (aR,bS)-](http://img.cochemist.com/ccimg/114700/114655-02-6.png)
![Benzenepropanoic acid, b-(benzoylamino)-a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-,(2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-6,12b-bis(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahydro-4,11-dihydroxy-4a,8,13,13-tetramethyl-5-oxo-7,11-methano-1H-cyclodeca[3,4]benz[1,2-b]oxet-9-ylester, (aR,bS)-](http://img.cochemist.com/ccimg/114700/114655-02-6_b.png)


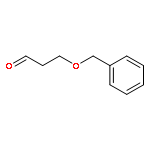
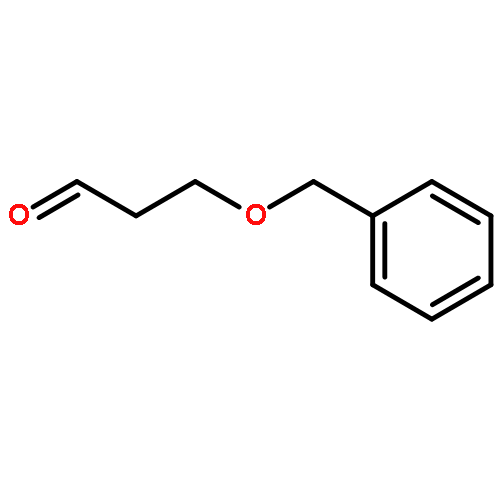
![Pentanal, 3-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-5-(phenylmethoxy)-,(S)-](http://img.cochemist.com/ccimg/106100/106064-52-2.png)
![Pentanal, 3-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-5-(phenylmethoxy)-,(S)-](http://img.cochemist.com/ccimg/106100/106064-52-2_b.png)
![Butanoic acid,1,1'-[(1S,3S,5Z,7R,8E,11S,12S,13E,15S,17R,21R,23R,25S)-1,11,21-trihydroxy-17-[(1R)-1-hydroxyethyl]-5,13-bis(2-methoxy-2-oxoethylidene)-10,10,26,26-tetramethyl-19-oxo-18,27,28,29-tetraoxatetracyclo[21.3.1.13,7.111,15]nonacos-8-ene-12,25-diyl]ester](http://img.cochemist.com/ccimg/102600/102580-64-3.png)
![Butanoic acid,1,1'-[(1S,3S,5Z,7R,8E,11S,12S,13E,15S,17R,21R,23R,25S)-1,11,21-trihydroxy-17-[(1R)-1-hydroxyethyl]-5,13-bis(2-methoxy-2-oxoethylidene)-10,10,26,26-tetramethyl-19-oxo-18,27,28,29-tetraoxatetracyclo[21.3.1.13,7.111,15]nonacos-8-ene-12,25-diyl]ester](http://img.cochemist.com/ccimg/102600/102580-64-3_b.png)
![Butanoic acid,3-methyl-,(1S,3S,5Z,7R,8E,11S,12S,13E,15S,17R,21R,23R,25S)-12-(acetyloxy)-1,11,21-trihydroxy-17-[(1R)-1-hydroxyethyl]-5,13-bis(2-methoxy-2-oxoethylidene)-10,10,26,26-tetramethyl-19-oxo-18,27,28,29-tetraoxatetracyclo[21.3.1.13,7.111,15]nonacos-8-en-25-ylester](http://img.cochemist.com/ccimg/97900/97850-04-9.png)
![Butanoic acid,3-methyl-,(1S,3S,5Z,7R,8E,11S,12S,13E,15S,17R,21R,23R,25S)-12-(acetyloxy)-1,11,21-trihydroxy-17-[(1R)-1-hydroxyethyl]-5,13-bis(2-methoxy-2-oxoethylidene)-10,10,26,26-tetramethyl-19-oxo-18,27,28,29-tetraoxatetracyclo[21.3.1.13,7.111,15]nonacos-8-en-25-ylester](http://img.cochemist.com/ccimg/97900/97850-04-9_b.png)
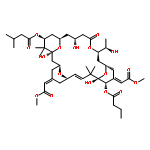
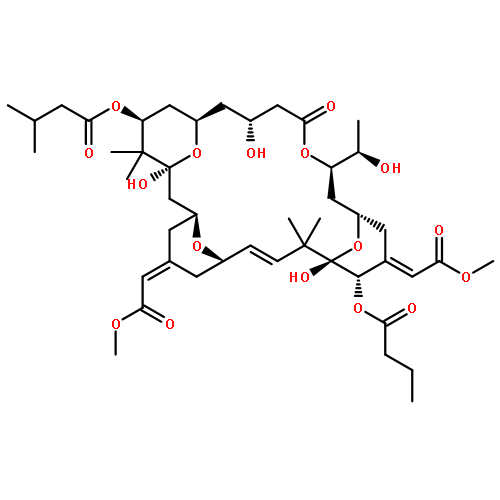

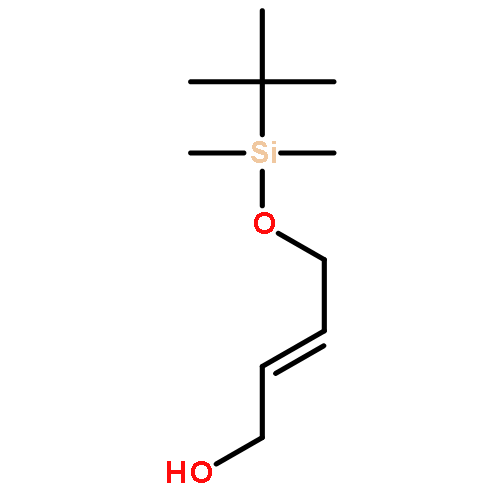


![2,4-Octadienoic acid,(1S,3S,5Z,7R,8E,11S,12S,13E,15S,17R,21R,23R,25S)-1,11,21,25-tetrahydroxy-17-[(1R)-1-hydroxyethyl]-5,13-bis(2-methoxy-2-oxoethylidene)-10,10,26,26-tetramethyl-19-oxo-18,27,28,29-tetraoxatetracyclo[21.3.1.13,7.111,15]nonacos-8-en-12-ylester, (2E,4E)-](http://img.cochemist.com/ccimg/87800/87745-28-6.png)
![2,4-Octadienoic acid,(1S,3S,5Z,7R,8E,11S,12S,13E,15S,17R,21R,23R,25S)-1,11,21,25-tetrahydroxy-17-[(1R)-1-hydroxyethyl]-5,13-bis(2-methoxy-2-oxoethylidene)-10,10,26,26-tetramethyl-19-oxo-18,27,28,29-tetraoxatetracyclo[21.3.1.13,7.111,15]nonacos-8-en-12-ylester, (2E,4E)-](http://img.cochemist.com/ccimg/87800/87745-28-6_b.png)
![4H-1,3a-Ethanocyclopropa[c]pentalen-8-one, 1,1a,5,6-tetrahydro- (9CI)](/data/chemimg/3435300/83043-46-3.png)
![4H-1,3a-Ethanocyclopropa[c]pentalen-8-one, 1,1a,5,6-tetrahydro- (9CI)](/data/chemimg/3435300/83043-46-3_b.png)


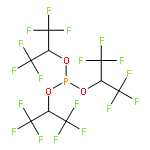
![9,9-DIMETHYLSPIRO[4,5]DECAN-7-ONE](http://img.cochemist.com/ccimg/63900/63858-64-0.png)
![9,9-DIMETHYLSPIRO[4,5]DECAN-7-ONE](http://img.cochemist.com/ccimg/63900/63858-64-0_b.png)
![16aH-1,6:2,6-Diepoxybenz[7,8]oxireno[5,6]azuleno[8,1-bc]oxacyclotridecin-7,16,16a,17-tetrol, 4-[(benzoyloxy)methyl]eicosahydro-17a-(hydroxymethyl)-](/data/chemimg/597800/60796-70-5.png)
![16aH-1,6:2,6-Diepoxybenz[7,8]oxireno[5,6]azuleno[8,1-bc]oxacyclotridecin-7,16,16a,17-tetrol, 4-[(benzoyloxy)methyl]eicosahydro-17a-(hydroxymethyl)-](/data/chemimg/597800/60796-70-5_b.png)


![(1bS,4aR,7aS,7bR,8R,9aS)-4a,7b-dihydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-5-oxo-1,1a,1b,4,4a,5,7a,7b,8,9-decahydro-9aH-cyclopropa[3,4]benzo[1,2-e]azulen-9a-yl phenylacetate](http://img.cochemist.com/ccimg/58900/58821-98-0.png)
![(1bS,4aR,7aS,7bR,8R,9aS)-4a,7b-dihydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-5-oxo-1,1a,1b,4,4a,5,7a,7b,8,9-decahydro-9aH-cyclopropa[3,4]benzo[1,2-e]azulen-9a-yl phenylacetate](http://img.cochemist.com/ccimg/58900/58821-98-0_b.png)

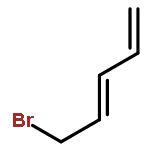

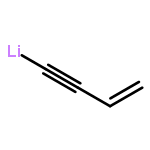
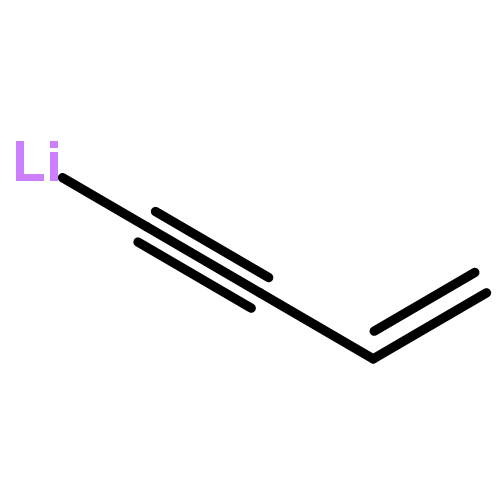





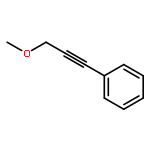
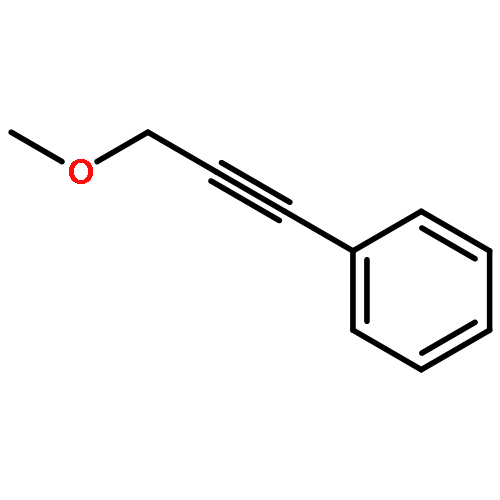
![5H-Cyclopropa[3,4]benz[1,2-e]azulen-5-one,9,9a-bis(acetyloxy)-3-[(acetyloxy)methyl]-1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-4a,7b-dihydroxy-1,1,6,8-tetramethyl-,(1aR,1bS,4aR,7aS,7bS,8R,9R)-](http://img.cochemist.com/ccimg/19900/19891-05-5.png)
![5H-Cyclopropa[3,4]benz[1,2-e]azulen-5-one,9,9a-bis(acetyloxy)-3-[(acetyloxy)methyl]-1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-4a,7b-dihydroxy-1,1,6,8-tetramethyl-,(1aR,1bS,4aR,7aS,7bS,8R,9R)-](http://img.cochemist.com/ccimg/19900/19891-05-5_b.png)


![(3aR)-3a,10a-Dihydroxy-5-hydroxymethyl-7c-isopropenyl-2,10t-dimethyl-(3ar,6ac,10at,10bt)-3a,4,6a,7,9,10,10a,10b-octahydro-benz[e]azulen-3,8-dion, Crotophorbolon](http://img.cochemist.com/ccimg/18400/18325-99-0.png)
![(3aR)-3a,10a-Dihydroxy-5-hydroxymethyl-7c-isopropenyl-2,10t-dimethyl-(3ar,6ac,10at,10bt)-3a,4,6a,7,9,10,10a,10b-octahydro-benz[e]azulen-3,8-dion, Crotophorbolon](http://img.cochemist.com/ccimg/18400/18325-99-0_b.png)


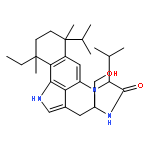












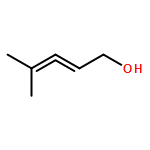

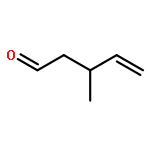
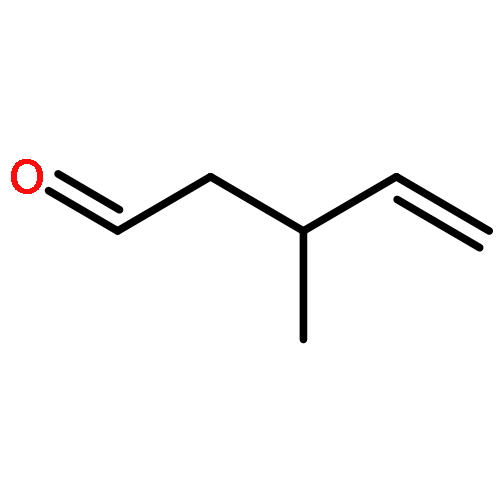
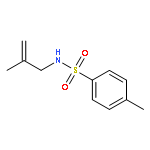
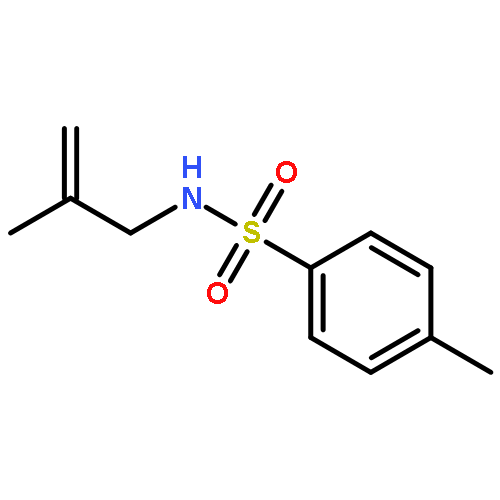


![Thiourea, N'-[3,5-bis(trifluoromethyl)phenyl]-N-cyclohexyl-](/data/chemimg/108000/454203-57-7.png)
![Thiourea, N'-[3,5-bis(trifluoromethyl)phenyl]-N-cyclohexyl-](/data/chemimg/108000/454203-57-7_b.png)
![1H-Cyclohepta[c]furan, 3,3a,6,7-tetrahydro-](http://img.cochemist.com/ccimg/166100/166044-00-4.png)
![1H-Cyclohepta[c]furan, 3,3a,6,7-tetrahydro-](http://img.cochemist.com/ccimg/166100/166044-00-4_b.png)
![Cyclopropane, [(1E)-3-(2-propynyloxy)-1-propenyl]-](http://img.cochemist.com/ccimg/166100/166043-89-6.png)
![Cyclopropane, [(1E)-3-(2-propynyloxy)-1-propenyl]-](http://img.cochemist.com/ccimg/166100/166043-89-6_b.png)
![2-Butynoic acid, 4-[[(2E)-3-cyclopropyl-2-propenyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/166100/166043-87-4.png)
![2-Butynoic acid, 4-[[(2E)-3-cyclopropyl-2-propenyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/166100/166043-87-4_b.png)

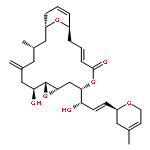
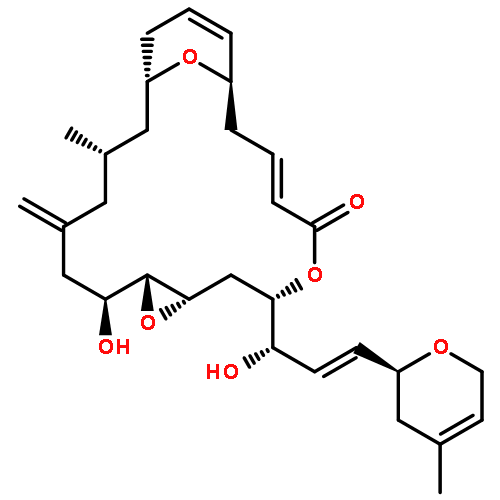
![Butanoic acid,(1S,3S,5Z,7R,8E,11S,12S,13E,15S,17R,21R,23R,25S)-25-(acetyloxy)-1,11,21-trihydroxy-17-[(1R)-1-hydroxyethyl]-5,13-bis(2-methoxy-2-oxoethylidene)-10,10,26,26-tetramethyl-19-oxo-18,27,28,29-tetraoxatetracyclo[21.3.1.13,7.111,15]nonacos-8-en-12-ylester](http://img.cochemist.com/ccimg/102700/102604-78-4.png)
![Butanoic acid,(1S,3S,5Z,7R,8E,11S,12S,13E,15S,17R,21R,23R,25S)-25-(acetyloxy)-1,11,21-trihydroxy-17-[(1R)-1-hydroxyethyl]-5,13-bis(2-methoxy-2-oxoethylidene)-10,10,26,26-tetramethyl-19-oxo-18,27,28,29-tetraoxatetracyclo[21.3.1.13,7.111,15]nonacos-8-en-12-ylester](http://img.cochemist.com/ccimg/102700/102604-78-4_b.png)
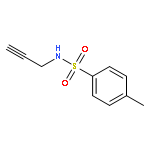
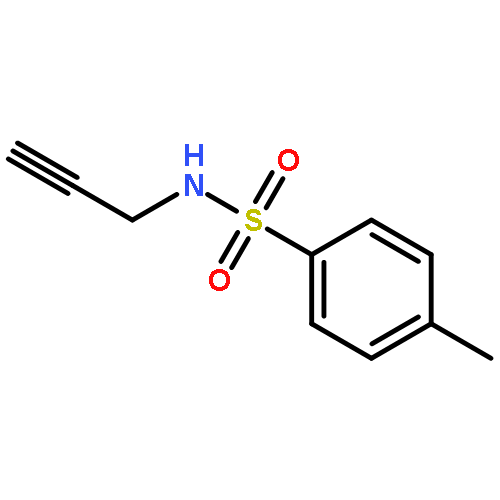
![2,4-Pentadienoic acid,5-phenyl-,(2S,3aR,3bS,3cS,4aR,5S,5aS,8aR,8bR,9R,10R,10aS)-3a,3b,3c,4a,5,5a,8a,9,10,10a-decahydro-5,5a-dihydroxy-4a-(hydroxymethyl)-7,9-dimethyl-10a-(1-methylethenyl)-6-oxo-2-phenyl-6H-2,8b-epoxyoxireno[6,7]azuleno[5,4-e]-1,3-benzodioxol-10-ylester, (2E,4E)-](http://img.cochemist.com/ccimg/34900/34807-41-5.png)
![2,4-Pentadienoic acid,5-phenyl-,(2S,3aR,3bS,3cS,4aR,5S,5aS,8aR,8bR,9R,10R,10aS)-3a,3b,3c,4a,5,5a,8a,9,10,10a-decahydro-5,5a-dihydroxy-4a-(hydroxymethyl)-7,9-dimethyl-10a-(1-methylethenyl)-6-oxo-2-phenyl-6H-2,8b-epoxyoxireno[6,7]azuleno[5,4-e]-1,3-benzodioxol-10-ylester, (2E,4E)-](http://img.cochemist.com/ccimg/34900/34807-41-5_b.png)
![Bicyclo[3.1.1]hept-3-en-2-one,4,6,6-trimethyl-, (1R,5R)-](http://img.cochemist.com/ccimg/18400/18309-32-5.png)
![Bicyclo[3.1.1]hept-3-en-2-one,4,6,6-trimethyl-, (1R,5R)-](http://img.cochemist.com/ccimg/18400/18309-32-5_b.png)


![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0.png)
![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0_b.png)



![Cyclopropane, [(1E)-3-(2-propenyloxy)-1-propenyl]-](http://img.cochemist.com/ccimg/204000/203928-05-6.png)
![Cyclopropane, [(1E)-3-(2-propenyloxy)-1-propenyl]-](http://img.cochemist.com/ccimg/204000/203928-05-6_b.png)

![PHORBOL 12,13-DIBUTYRATE;(1AR,1BS,4AR,7AS,7BS,8R,9R,9AS)-1A,1B,4,4A,5,7A,7B,8,9,9A-DECAHYDRO-4A,7B-DIHYDROXY-3-(HYDROXYMETHYL)-1,1,6,8-TETRAMETHYL-5-OXO-1H-CYCLOPROPA[3,4]BENZ[1,2-E]AZULEN-9,9A-DIYLBUTANOICACIDESTER](http://img.cochemist.com/ccimg/37600/37558-16-0.png)
![PHORBOL 12,13-DIBUTYRATE;(1AR,1BS,4AR,7AS,7BS,8R,9R,9AS)-1A,1B,4,4A,5,7A,7B,8,9,9A-DECAHYDRO-4A,7B-DIHYDROXY-3-(HYDROXYMETHYL)-1,1,6,8-TETRAMETHYL-5-OXO-1H-CYCLOPROPA[3,4]BENZ[1,2-E]AZULEN-9,9A-DIYLBUTANOICACIDESTER](http://img.cochemist.com/ccimg/37600/37558-16-0_b.png)


![[1,1'-Biphenyl]-2-ol,2,2'',2''''-phosphite](http://img.cochemist.com/ccimg/2800/2752-19-4.png)
![[1,1'-Biphenyl]-2-ol,2,2'',2''''-phosphite](http://img.cochemist.com/ccimg/2800/2752-19-4_b.png)


![(1aR,1bS,4aR,7aS,7bR,8R,9aS)-9a-(Acetyloxy)-1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-4a,7b-dihydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-5H-cyclopropa[3,4]benz[1,2-e]azulen-5-one](http://img.cochemist.com/ccimg/60900/60857-08-1.png)
![(1aR,1bS,4aR,7aS,7bR,8R,9aS)-9a-(Acetyloxy)-1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-4a,7b-dihydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-5H-cyclopropa[3,4]benz[1,2-e]azulen-5-one](http://img.cochemist.com/ccimg/60900/60857-08-1_b.png)
![(S)-2-(6-Hydroxybenzo[d]thiazol-2-yl)-4,5-dihydrothiazole-4-carboxylic acid](/data/chemimg/473800/2591-17-5.png)
![(S)-2-(6-Hydroxybenzo[d]thiazol-2-yl)-4,5-dihydrothiazole-4-carboxylic acid](/data/chemimg/473800/2591-17-5_b.png)






![5H-Cyclopropa[3,4]benz[1,2-e]azulen-5-one,1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-4a,7b,9,9a-tetrahydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-,(1aR,1bS,4aR,7aS,7bS,8R,9R,9aS)-](http://img.cochemist.com/ccimg/17700/17673-25-5.png)
![5H-Cyclopropa[3,4]benz[1,2-e]azulen-5-one,1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-4a,7b,9,9a-tetrahydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-,(1aR,1bS,4aR,7aS,7bS,8R,9R,9aS)-](http://img.cochemist.com/ccimg/17700/17673-25-5_b.png)




![BRYOSTATIN 1;(1S,3S,5Z,7R,8E,11S,12S,13E,15S,17R,21R,23R,25S)-25-(ACETYLOXY)-1,11,21-TRIHYDROXY-17-[(1R)-1-HYDROXYETHYL]-5,13-BIS(2-METHOXY-2-OXOETHYLIDENE)-10,10,26,26-TETRAMETHYL-19-OXO-18,27,28,29-TETRAOXATETRACYCLO[21.3.1.13,7.111,15]NONACOS-8-EN-12-Y](http://img.cochemist.com/ccimg/83400/83314-01-6.png)
![BRYOSTATIN 1;(1S,3S,5Z,7R,8E,11S,12S,13E,15S,17R,21R,23R,25S)-25-(ACETYLOXY)-1,11,21-TRIHYDROXY-17-[(1R)-1-HYDROXYETHYL]-5,13-BIS(2-METHOXY-2-OXOETHYLIDENE)-10,10,26,26-TETRAMETHYL-19-OXO-18,27,28,29-TETRAOXATETRACYCLO[21.3.1.13,7.111,15]NONACOS-8-EN-12-Y](http://img.cochemist.com/ccimg/83400/83314-01-6_b.png)