Co-reporter:Andreas Phanopoulos;Mark Warren;Andrew J. P. White;Andrew Horton
Dalton Transactions 2017 vol. 46(Issue 7) pp:2077-2080
Publication Date(Web):2017/02/14
DOI:10.1039/C6DT04703C
Addition of 2 equiv. of CuI to a [Cu]4 multimetallic complex results in cluster formation leading to the isolation of a rare bicapped tetrahedral [Cu6I2] cluster that is stabilised by two conformationally constrained polynucleating ligands.
Co-reporter:Wenyi Chen;Dr. Thomas N. Hooper;Jamues Ng;Dr. Andrew J. P. White;Dr. Mark R. Crimmin
Angewandte Chemie International Edition 2017 Volume 56(Issue 41) pp:12687-12691
Publication Date(Web):2017/10/02
DOI:10.1002/anie.201706378
AbstractThrough serendipitous discovery, a palladium bis(phosphine) complex was identified as a catalyst for the selective transformation of sp2C−F and sp2C−H bonds of fluoroarenes and heteroarenes to sp2C−Al bonds (19 examples, 1 mol % Pd loading). The carbon–fluorine bond functionalization reaction is highly selective for the formation of organoaluminium products in preference to hydrodefluorination products (selectivity=4.4:1 to 27:1). Evidence is presented for a tandem catalytic process in which hydrodefluorination is followed by sp2C−H alumination.
Co-reporter:Alexra Hicken;Dr. Andrew J. P. White;Dr. Mark R. Crimmin
Angewandte Chemie International Edition 2017 Volume 56(Issue 47) pp:15127-15130
Publication Date(Web):2017/11/20
DOI:10.1002/anie.201709072
AbstractA series of heterobimetallic complexes containing three-center, two-electron Au−H−Cu bonds have been prepared from addition of a parent gold hydride to a bent d10 copper(I) fragment. These highly unusual heterobimetallic complexes represent a missing link in the widely investigated series of neutral and cationic coinage metal hydride complexes containing Cu−H−Cu and M−H−M+ moieties (M=Cu, Ag). The well-defined heterobimetallic hydride complexes act as precatalysts for the conversion of CO2 into HCO2Bpin with HBpin as the reductant. The selectivity of the heterobimetallic complexes for the catalytic production of a formate equivalent surpasses that of the parent monomeric Group 11 complexes.
Co-reporter:Alexra Hicken;Dr. Andrew J. P. White;Dr. Mark R. Crimmin
Angewandte Chemie 2017 Volume 129(Issue 47) pp:15323-15326
Publication Date(Web):2017/11/20
DOI:10.1002/ange.201709072
AbstractA series of heterobimetallic complexes containing three-center, two-electron Au−H−Cu bonds have been prepared from addition of a parent gold hydride to a bent d10 copper(I) fragment. These highly unusual heterobimetallic complexes represent a missing link in the widely investigated series of neutral and cationic coinage metal hydride complexes containing Cu−H−Cu and M−H−M+ moieties (M=Cu, Ag). The well-defined heterobimetallic hydride complexes act as precatalysts for the conversion of CO2 into HCO2Bpin with HBpin as the reductant. The selectivity of the heterobimetallic complexes for the catalytic production of a formate equivalent surpasses that of the parent monomeric Group 11 complexes.
Co-reporter:Sebastian D. Pike;Adrian B. Chaplin
Chemical Communications 2017 vol. 53(Issue 26) pp:3615-3633
Publication Date(Web):2017/03/28
DOI:10.1039/C6CC09575E
Fluorobenzenes, in particular fluorobenzene (FB) and 1,2-difluorobenzene (1,2-DiFB), are increasingly becoming recognised as versatile solvents for conducting organometallic chemistry and transition-metal-based catalysis. The presence of fluorine substituents reduces the ability to donate π-electron density from the arene and consequently fluorobenzenes generally bind weakly to metal centres, allowing them to be used as essentially non-coordinating solvents or as readily displaced ligands. In this context, examples of well-defined complexes of fluorobenzenes are discussed, including trends in binding strength with increasing fluorination and different substitution patterns. Compared to more highly fluorinated benzenes, FB and 1,2-DiFB typically demonstrate greater chemical inertness, however, C–H and C–F bond activation reactions can be induced using appropriately reactive transition metal complexes. Such reactions are surveyed, including catalytic examples, not only to provide perspective for the use of FB and 1,2-DiFB as innocent solvent media, but also to highlight opportunities for their exploitation in contemporary organic synthesis.
Co-reporter:Michael J. Butler
Chemical Communications 2017 vol. 53(Issue 8) pp:1348-1365
Publication Date(Web):2017/01/24
DOI:10.1039/C6CC05702K
The preparation and applications of heterobimetallic complexes continue to occupy researchers in the fields of organometallic, main group, and coordination chemistry. This interest stems from the promise these complexes hold as precursors to materials, reagents in synthesis and as new catalysis. Here we survey and organise the state-of-the-art understanding of the TM–H–M linkage (M = Mg, Zn, Al, Ga). We discuss the structure and bonding in these complexes, their known reactivity, and their largely unrealised potential in catalysis.
Co-reporter:Andreas Phanopoulos;Alice H. M. Leung;Shuhui Yow;David Palomas;Andrew J. P. White;Klaus Hellgardt;Andrew Horton
Dalton Transactions 2017 vol. 46(Issue 7) pp:2081-2090
Publication Date(Web):2017/02/14
DOI:10.1039/C6DT04246E
The reaction of a series of dinucleating bis(β-diketiminate) pro-ligands with mesitylcopper in the presence and absence of mono and diphosphines has allowed the isolation of a new series of dicopper(I) complexes. Inclusion of trans-1,2-cyclohexyl (1), 2,6-pyridyl (2), and 2,2′-oxydiaryl (3) spacers between the β-diketiminate units has been studied. The isolation of three new copper(I) phosphine complexes [1·Cu2(PPh3)2], [2·Cu2(PPh3)2] and [3·Cu2(PPh3)2] is reported. While these compounds display large Cu⋯Cu separations of 5.4–7.9 Å in the solid state, solution data are consistent with a large degree of conformational freedom. Modification of the monophosphine to a diphosphine, DPPE, allowed the isolation of the novel 11-membered bimetallic macrocycle [2·Cu2(DPPE)] containing both a binucleating nitrogen based ligand and a chelating diphosphine. While acetonitrile adducts of this series could also be generated in situ, under forcing conditions reaction of the 2,6-pyridyl bridged ligand with mesityl copper led to the formation [2·Cu2]2. This latter complex is a dimer of dicopper(I) units in which the bis(β-diketiminate) ligand now binds four copper(I) centers through not only the expected κ2-N,N′-chelation but also κ1- and η2-binding of the central pyridine through orthogonal Cu–N and Cu–arene interactions. Reversible coordination of alkenes, pyridine and quinoline to the copper cluster was identified allowing the isolation and structural characterisation of a further series of dinuclear complexes [2·Cu2(pyridine)2], [2·Cu2(cyclopentene)2] and [2·Cu2(norbornene)2]. Solution studies allow quantification of the reversible binding event through a van't Hoff analysis. Both solution and the solid state data suggest a weak anagostic interaction exists in the latter two alkene complexes of copper(I). The new complexes have been characterized by X-ray diffraction, multinuclear NMR spectroscopy and CHN analysis.
Co-reporter:Clare Bakewell, Andrew J. P. White, and Mark R. Crimmin
Journal of the American Chemical Society 2016 Volume 138(Issue 39) pp:12763-12766
Publication Date(Web):September 16, 2016
DOI:10.1021/jacs.6b08104
Addition of the carbon–fluorine bond of a series of perfluorinated and polyfluorinated arenes across the Mg–Mg bond of a simple coordination complex proceeds rapidly in solution. The reaction results in the formation of a new carbon–magnesium bond and a new fluorine–magnesium bond and is analogous to Grignard formation in homogeneous solution.
Co-reporter:Michael J. Butler;Dr. Andrew J. P. White ;Dr. Mark R. Crimmin
Angewandte Chemie International Edition 2016 Volume 55( Issue 24) pp:6951-6953
Publication Date(Web):
DOI:10.1002/anie.201601758
Abstract
Reaction of a zinc/zirconium heterobimetallic complex with 1,5-cyclooctadiene (1,5-COD) results in slow isomerization to 1,3-cyclooctadiene (1,3-COD), along with the formation of a new complex that includes a cyclooctyne ligand bridging two metal centers. While analogous magnesium/zirconium and aluminum/zirconium heterobimetallic complexes are competent for the catalytic isomerization of 1,5-COD to 1,3-COD, only in the case of the zinc species is the cyclooctyne adduct observed.
Co-reporter:Michael J. Butler;Dr. Andrew J. P. White ;Dr. Mark R. Crimmin
Angewandte Chemie 2016 Volume 128( Issue 24) pp:7065-7067
Publication Date(Web):
DOI:10.1002/ange.201601758
Abstract
Reaction of a zinc/zirconium heterobimetallic complex with 1,5-cyclooctadiene (1,5-COD) results in slow isomerization to 1,3-cyclooctadiene (1,3-COD), along with the formation of a new complex that includes a cyclooctyne ligand bridging two metal centers. While analogous magnesium/zirconium and aluminum/zirconium heterobimetallic complexes are competent for the catalytic isomerization of 1,5-COD to 1,3-COD, only in the case of the zinc species is the cyclooctyne adduct observed.
Co-reporter:Olga Ekkert, Andrew J. P. White, Harold Toms and Mark R. Crimmin
Chemical Science 2015 vol. 6(Issue 10) pp:5617-5622
Publication Date(Web):03 Jul 2015
DOI:10.1039/C5SC01309G
We report the addition of M–H bonds (M = Al, Zn, Mg) to a Rh(III) intermediate generated from the reductive elimination of triethylsilane from [Cp*Rh(H)2(SiEt3)2]. A series of new heterobimetallic complexes possessing Rh–M bonds have been isolated and characterised by a number of spectroscopic (1H, 29Si, 13C, 103Rh NMR, infrared, and X-ray diffraction) and computational techniques (NBO and QTAIM analysis). Experimental and computational data are consistent with cleavage of the M–H bond upon addition to rhodium with formation of new Rh–M and Rh–H bonds. Upon photolysis the Al analogue of this series undergoes a further elimination reaction producing triethylsilane and a highly unusual Rh2Al2H4 containing cluster proposed to contain an Al(I) bridging ligand.
Co-reporter:Mark R. Crimmin, Michael J. Butler and Andrew J. P. White
Chemical Communications 2015 vol. 51(Issue 88) pp:15994-15996
Publication Date(Web):14 Sep 2015
DOI:10.1039/C5CC07140B
Addition of fluoroarenes, fluoroalkanes or benzofuran to [{(2,6-iPr2C6H3NCMe)2CH}Al] results in facile oxidative addition of either a C–F or C–O bond to the Al(I) centre.
Co-reporter:David Palomas, Christos Kalamaras, Peter Haycock, Andrew J. P. White, Klaus Hellgardt, Andrew Horton and Mark R. Crimmin
Catalysis Science & Technology 2015 vol. 5(Issue 8) pp:4108-4115
Publication Date(Web):18 Jun 2015
DOI:10.1039/C5CY00462D
A series of multimetallic copper(II) complexes have been re-investigated for methane oxidation with H2O2. The preparation and properties of trinuclear copper(II) complexes of the form [Cu3(triazole)n(OH2)12−n] (n = 8, 10) are reported. While these complexes are trimeric in the solid-state, 1H NMR studies suggest that facile ligand dissociation occurs in solution. The oxidation of cyclohexane with H2O2 catalyzed by [Cu3(triazole)n(OH2)12−n] (n = 8, 10) is compared against a literature known oxo-centered tetrameric cluster (Angew. Chem., Int. Ed., 2005, 44, 4345) and these catalysts display moderate activities. The series have also been investigated in methane oxidation at 30 bar and 40 °C. Analytical techniques including a solvent suppression 1H NMR method have been applied to quantify the liquid- and gas-phase products. The multi-metallic copper(II) complexes and copper(II) nitrate control samples produce only methanol and CO2. While TONs for methanol production range from 1.4–4.6 in all cases approximately 50 times the amount of CO2 is produced relative to methanol. We conclude that selectivity is a determining factor in methane oxidation under these conditions and should be considered in future studies.
Co-reporter:Adi E. Nako, Andrew J. P. White and Mark R. Crimmin
Dalton Transactions 2015 vol. 44(Issue 28) pp:12530-12534
Publication Date(Web):17 Jun 2015
DOI:10.1039/C5DT02144H
A series of bis(σ-B–H) complexes of copper(I) have been prepared by displacement of arene solvent from a β-diketiminate copper(I) complex by four-coordinate boranes, H3B–L (L = NMe3, lutidine). In the presence of the same copper arene complex, the secondary amine–borane H3B–NMe2H undergoes dehydrogenation. We provide evidence for formation of a heterogengous catalyst from decomposition of the solution species.
Co-reporter:Adi E. Nako, Wenyi Chen, Andrew J. P. White, and Mark R. Crimmin
Organometallics 2015 Volume 34(Issue 17) pp:4369-4375
Publication Date(Web):August 20, 2015
DOI:10.1021/acs.organomet.5b00607
The scope of the catalytic dehydrocoupling of primary and secondary amines with phenylsilanes has been investigated using [Y{N(SiMe3)2}3] and a four-coordinate analogue bearing a cyclometalated phosphonium methylide ligand. Inclusion of the phosphorus-based ligand on yttrium results in increased substrate scope in comparison to the tris(amide) analogue. While reversible C–H bond activation of the cyclometalated ligand was observed in stoichiometric experiments, D-labeling experiments and DFT calculations suggest that reversible ligand activation is not involved in silazane formation under catalytic conditions. We suggest that the extended reaction scope with the four-coordinate yttrium phosphonium methylide complex relative to the three-coordinate yttrium (tris)amide complex is a result of differences in the ease of amine inhibition of catalysis.
Co-reporter:Adi E. Nako, Sarah J. Gates, Nicole Schädel, Andrew J. P. White and Mark R. Crimmin
Chemical Communications 2014 vol. 50(Issue 67) pp:9536-9538
Publication Date(Web):07 Jul 2014
DOI:10.1039/C4CC04484C
We report [Y{N(SiMe3)2}3] as a precatalyst for the dehydrocoupling of sterically demanding amines with β-diketiminate stabilised aluminium dihydrides. While simple fluorinated anilines readily undergo Al–H/N–H dehydrocoupling under thermal conditions, catalytic methods are required to achieve reasonable rates of reaction for ortho-substituted anilines or hindered aliphatic amines.
Co-reporter:Olga Ekkert, Sebastian D. A. Strudley, Alisa Rozenfeld, Andrew J. P. White, and Mark R. Crimmin
Organometallics 2014 Volume 33(Issue 24) pp:7027-7030
Publication Date(Web):December 11, 2014
DOI:10.1021/om501113j
[Cp*RhCl(μ-Cl)]2 is reported as a highly efficient and selective precatalyst for the hydrodefluorination of perfluoroarenes using a hydrocarbon-soluble aluminum dihydride as the terminal reductant. Reactions are directed to cleave a C–F bond adjacent to an existing C–H bond with high regioselectivity (98.5–99%). A heterobimetallic complex containing an extremely rare Al–H–Rh functional group has been isolated and shown to be catalytically competent.
Co-reporter:Adi E. Nako, Qian Wen Tan, Andrew J. P. White, and Mark R. Crimmin
Organometallics 2014 Volume 33(Issue 11) pp:2685-2688
Publication Date(Web):May 29, 2014
DOI:10.1021/om500380k
We report the synthesis and isolation of three new σ-complexes of Cu(I) in which E–H (E = Al, Zn) σ-bonds are coordinated to copper. The addition of the main group hydride to a toluene-solvated Cu(I) complex results in reversible ligand exchange, and the Cu(I) σ-complexes have been crystallized. Experimental and computational data provide a wealth of evidence for weak binding of the E–H bond to Cu(I), which can be ascribed to σ-donation from the E–H bond into the 4s orbital of copper and back-donation from copper into the E–H σ* orbital.
Co-reporter:Adi E. Nako, Andrew J. P. White and Mark R. Crimmin
Chemical Science 2013 vol. 4(Issue 2) pp:691-695
Publication Date(Web):06 Nov 2012
DOI:10.1039/C2SC21123H
We report the [Y{N(SiMe3)2}3] catalysed dehydrocoupling of triphenylphosphonium methylide with phenylsilane to form the silylated ylide Ph3PCHSiH2Ph. Attempts to catalyse this reaction with the related group 2 hexamethyldisilazide base [Ca{N(SiMe3)2}2] led to the catalytic formation of the phosphine Ph2PCHSiH2Ph along with Ph2PMe in low selectivity, while group 1 bases [M{N(SiMe3)2}] (M = Li, Na, K) proved ineffective for both transformations. The stoichiometric reactions of Ph3PCH2 with [M{N(SiMe3)2}n] have been investigated and allowed the isolation and characterisation of a cyclometallated phosphonium methylide complex of yttrium.
Co-reporter:Adi E. Nako, Sarah J. Gates, Andrew J. P. White and Mark R. Crimmin
Dalton Transactions 2013 vol. 42(Issue 42) pp:15199-15206
Publication Date(Web):02 Sep 2013
DOI:10.1039/C3DT52148F
The synthesis of a diverse series of hydride complexes of aluminium coordinated by N,N′-chelating ligands is reported. Reaction of [{2,6-iPr2C6H3}NC(Me)CHC(Me)N(H)CH2CH2NMe2] with either LiAlH4 or Me3N·AlH3 allows isolation of the corresponding five-coordinate aluminium dihydride [κ3-{(2,6-iPr2C6H3)NC(Me)CHC(Me)NCH2CH2NMe2}AlH2] (2). The latter complex demonstrates trigonal bipyramidal geometry in the solid-state. Correlation of solid and n-hexane solution infrared spectroscopy data reveals that this coordination is retained in solution. To evaluate the observed coordination geometry, the dissociation of the pendant ligand of 2 was investigated by DFT methods conducted with the M06-2X functional and a hybrid 6,31G+(d,p)/Lanl2DZ basis-set. Reaction of Me3N·AlH3 with both N,N′-bis(di-iso-propylphenyl)ethylenediamine and N,N′-bis(mesityl)ethylenediamine gave [{κ2-(ArNCH2)2}AlH(NMe3)] (Ar = Mes, 3a; Ar = 2,6-di-iso-propylphenyl, 3b) in moderate yields. Removal of NMe3 from 3b by heating under dynamic vacuum allowed the isolation of cis-[AlH{μ-N(Ar)CH2CH2N(Ar)}] (Ar = 2,6-di-iso-propylphenyl, cis-4b22) as a single diastereomer following crystallization. DFT studies in combination with infrared and NMR spectroscopy and single crystal X-ray diffraction data provide a weight of evidence consistent with the robust dimeric structure of cis-4b22 remaining intact in solution. An unusual reaction in which the aluminium dihydride, [κ2-{(2,6-Me2C6H3NHCH2)2CH}AlH2], promotes the P–C bond cleavage of Ph3PCH2 is also reported.
Co-reporter:Shuhui Yow ; Adi E. Nako ; Léonard Neveu ; Andrew J. P. White
Organometallics 2013 Volume 32(Issue 19) pp:5260-5262
Publication Date(Web):September 25, 2013
DOI:10.1021/om4008295
The chemoselective C–O bond functionalization of benzofuran with an aluminum dihydride may be catalyzed by zirconocene dichlorides. The reaction proceeds with the formal addition of a C–O bond to, and elimination of dihydrogen from, aluminum. The product of C–O bond alumination reacts with benzaldehyde via insertion of the carbonyl into the newly formed Al–C bond.
Co-reporter:Mark R. Crimmin and Andrew J. P. White
Chemical Communications 2012 vol. 48(Issue 12) pp:1745-1747
Publication Date(Web):03 Jan 2012
DOI:10.1039/C2CC16431K
We report the synthesis of an yttrium phosphonium methylide complex and its reaction with benzophenone to form an excedingly rare hydrocarbon soluble metal-coordinated betaine. While this reaction models the C–C σ-bond formation step of the Wittig reaction under salt-conditions, addition of Ph3PO to the betaine complex results in formation of 1,1-diphenylethene.
Co-reporter:Dr. Shuhui Yow;Sarah J. Gates;Dr. Andrew J. P. White ;Dr. Mark R. Crimmin
Angewandte Chemie International Edition 2012 Volume 51( Issue 50) pp:
Publication Date(Web):
DOI:10.1002/anie.201207036
Co-reporter:Dr. Shuhui Yow;Sarah J. Gates;Dr. Andrew J. P. White ;Dr. Mark R. Crimmin
Angewandte Chemie 2012 Volume 124( Issue 50) pp:
Publication Date(Web):
DOI:10.1002/ange.201207036
Co-reporter:David Palomas, Christos Kalamaras, Peter Haycock, Andrew J. P. White, Klaus Hellgardt, Andrew Horton and Mark R. Crimmin
Catalysis Science & Technology (2011-Present) 2015 - vol. 5(Issue 8) pp:NaN4115-4115
Publication Date(Web):2015/06/18
DOI:10.1039/C5CY00462D
A series of multimetallic copper(II) complexes have been re-investigated for methane oxidation with H2O2. The preparation and properties of trinuclear copper(II) complexes of the form [Cu3(triazole)n(OH2)12−n] (n = 8, 10) are reported. While these complexes are trimeric in the solid-state, 1H NMR studies suggest that facile ligand dissociation occurs in solution. The oxidation of cyclohexane with H2O2 catalyzed by [Cu3(triazole)n(OH2)12−n] (n = 8, 10) is compared against a literature known oxo-centered tetrameric cluster (Angew. Chem., Int. Ed., 2005, 44, 4345) and these catalysts display moderate activities. The series have also been investigated in methane oxidation at 30 bar and 40 °C. Analytical techniques including a solvent suppression 1H NMR method have been applied to quantify the liquid- and gas-phase products. The multi-metallic copper(II) complexes and copper(II) nitrate control samples produce only methanol and CO2. While TONs for methanol production range from 1.4–4.6 in all cases approximately 50 times the amount of CO2 is produced relative to methanol. We conclude that selectivity is a determining factor in methane oxidation under these conditions and should be considered in future studies.
Co-reporter:Olga Ekkert, Andrew J. P. White, Harold Toms and Mark R. Crimmin
Chemical Science (2010-Present) 2015 - vol. 6(Issue 10) pp:NaN5622-5622
Publication Date(Web):2015/07/03
DOI:10.1039/C5SC01309G
We report the addition of M–H bonds (M = Al, Zn, Mg) to a Rh(III) intermediate generated from the reductive elimination of triethylsilane from [Cp*Rh(H)2(SiEt3)2]. A series of new heterobimetallic complexes possessing Rh–M bonds have been isolated and characterised by a number of spectroscopic (1H, 29Si, 13C, 103Rh NMR, infrared, and X-ray diffraction) and computational techniques (NBO and QTAIM analysis). Experimental and computational data are consistent with cleavage of the M–H bond upon addition to rhodium with formation of new Rh–M and Rh–H bonds. Upon photolysis the Al analogue of this series undergoes a further elimination reaction producing triethylsilane and a highly unusual Rh2Al2H4 containing cluster proposed to contain an Al(I) bridging ligand.
Co-reporter:Mark R. Crimmin and Andrew J. P. White
Chemical Communications 2012 - vol. 48(Issue 12) pp:NaN1747-1747
Publication Date(Web):2012/01/03
DOI:10.1039/C2CC16431K
We report the synthesis of an yttrium phosphonium methylide complex and its reaction with benzophenone to form an excedingly rare hydrocarbon soluble metal-coordinated betaine. While this reaction models the C–C σ-bond formation step of the Wittig reaction under salt-conditions, addition of Ph3PO to the betaine complex results in formation of 1,1-diphenylethene.
Co-reporter:Adi E. Nako, Sarah J. Gates, Andrew J. P. White and Mark R. Crimmin
Dalton Transactions 2013 - vol. 42(Issue 42) pp:NaN15206-15206
Publication Date(Web):2013/09/02
DOI:10.1039/C3DT52148F
The synthesis of a diverse series of hydride complexes of aluminium coordinated by N,N′-chelating ligands is reported. Reaction of [{2,6-iPr2C6H3}NC(Me)CHC(Me)N(H)CH2CH2NMe2] with either LiAlH4 or Me3N·AlH3 allows isolation of the corresponding five-coordinate aluminium dihydride [κ3-{(2,6-iPr2C6H3)NC(Me)CHC(Me)NCH2CH2NMe2}AlH2] (2). The latter complex demonstrates trigonal bipyramidal geometry in the solid-state. Correlation of solid and n-hexane solution infrared spectroscopy data reveals that this coordination is retained in solution. To evaluate the observed coordination geometry, the dissociation of the pendant ligand of 2 was investigated by DFT methods conducted with the M06-2X functional and a hybrid 6,31G+(d,p)/Lanl2DZ basis-set. Reaction of Me3N·AlH3 with both N,N′-bis(di-iso-propylphenyl)ethylenediamine and N,N′-bis(mesityl)ethylenediamine gave [{κ2-(ArNCH2)2}AlH(NMe3)] (Ar = Mes, 3a; Ar = 2,6-di-iso-propylphenyl, 3b) in moderate yields. Removal of NMe3 from 3b by heating under dynamic vacuum allowed the isolation of cis-[AlH{μ-N(Ar)CH2CH2N(Ar)}] (Ar = 2,6-di-iso-propylphenyl, cis-4b22) as a single diastereomer following crystallization. DFT studies in combination with infrared and NMR spectroscopy and single crystal X-ray diffraction data provide a weight of evidence consistent with the robust dimeric structure of cis-4b22 remaining intact in solution. An unusual reaction in which the aluminium dihydride, [κ2-{(2,6-Me2C6H3NHCH2)2CH}AlH2], promotes the P–C bond cleavage of Ph3PCH2 is also reported.
Co-reporter:Mark R. Crimmin, Michael J. Butler and Andrew J. P. White
Chemical Communications 2015 - vol. 51(Issue 88) pp:NaN15996-15996
Publication Date(Web):2015/09/14
DOI:10.1039/C5CC07140B
Addition of fluoroarenes, fluoroalkanes or benzofuran to [{(2,6-iPr2C6H3NCMe)2CH}Al] results in facile oxidative addition of either a C–F or C–O bond to the Al(I) centre.
Co-reporter:Andreas Phanopoulos, Alice H. M. Leung, Shuhui Yow, David Palomas, Andrew J. P. White, Klaus Hellgardt, Andrew Horton and Mark R. Crimmin
Dalton Transactions 2017 - vol. 46(Issue 7) pp:NaN2090-2090
Publication Date(Web):2017/01/20
DOI:10.1039/C6DT04246E
The reaction of a series of dinucleating bis(β-diketiminate) pro-ligands with mesitylcopper in the presence and absence of mono and diphosphines has allowed the isolation of a new series of dicopper(I) complexes. Inclusion of trans-1,2-cyclohexyl (1), 2,6-pyridyl (2), and 2,2′-oxydiaryl (3) spacers between the β-diketiminate units has been studied. The isolation of three new copper(I) phosphine complexes [1·Cu2(PPh3)2], [2·Cu2(PPh3)2] and [3·Cu2(PPh3)2] is reported. While these compounds display large Cu⋯Cu separations of 5.4–7.9 Å in the solid state, solution data are consistent with a large degree of conformational freedom. Modification of the monophosphine to a diphosphine, DPPE, allowed the isolation of the novel 11-membered bimetallic macrocycle [2·Cu2(DPPE)] containing both a binucleating nitrogen based ligand and a chelating diphosphine. While acetonitrile adducts of this series could also be generated in situ, under forcing conditions reaction of the 2,6-pyridyl bridged ligand with mesityl copper led to the formation [2·Cu2]2. This latter complex is a dimer of dicopper(I) units in which the bis(β-diketiminate) ligand now binds four copper(I) centers through not only the expected κ2-N,N′-chelation but also κ1- and η2-binding of the central pyridine through orthogonal Cu–N and Cu–arene interactions. Reversible coordination of alkenes, pyridine and quinoline to the copper cluster was identified allowing the isolation and structural characterisation of a further series of dinuclear complexes [2·Cu2(pyridine)2], [2·Cu2(cyclopentene)2] and [2·Cu2(norbornene)2]. Solution studies allow quantification of the reversible binding event through a van't Hoff analysis. Both solution and the solid state data suggest a weak anagostic interaction exists in the latter two alkene complexes of copper(I). The new complexes have been characterized by X-ray diffraction, multinuclear NMR spectroscopy and CHN analysis.
Co-reporter:Adi E. Nako, Andrew J. P. White and Mark R. Crimmin
Chemical Science (2010-Present) 2013 - vol. 4(Issue 2) pp:NaN695-695
Publication Date(Web):2012/11/06
DOI:10.1039/C2SC21123H
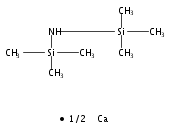
We report the [Y{N(SiMe3)2}3] catalysed dehydrocoupling of triphenylphosphonium methylide with phenylsilane to form the silylated ylide Ph3PCHSiH2Ph. Attempts to catalyse this reaction with the related group 2 hexamethyldisilazide base [Ca{N(SiMe3)2}2] led to the catalytic formation of the phosphine Ph2PCHSiH2Ph along with Ph2PMe in low selectivity, while group 1 bases [M{N(SiMe3)2}] (M = Li, Na, K) proved ineffective for both transformations. The stoichiometric reactions of Ph3PCH2 with [M{N(SiMe3)2}n] have been investigated and allowed the isolation and characterisation of a cyclometallated phosphonium methylide complex of yttrium.
Co-reporter:Adi E. Nako, Andrew J. P. White and Mark R. Crimmin
Dalton Transactions 2015 - vol. 44(Issue 28) pp:NaN12534-12534
Publication Date(Web):2015/06/17
DOI:10.1039/C5DT02144H
A series of bis(σ-B–H) complexes of copper(I) have been prepared by displacement of arene solvent from a β-diketiminate copper(I) complex by four-coordinate boranes, H3B–L (L = NMe3, lutidine). In the presence of the same copper arene complex, the secondary amine–borane H3B–NMe2H undergoes dehydrogenation. We provide evidence for formation of a heterogengous catalyst from decomposition of the solution species.
Co-reporter:Sebastian D. Pike, Mark R. Crimmin and Adrian B. Chaplin
Chemical Communications 2017 - vol. 53(Issue 26) pp:NaN3633-3633
Publication Date(Web):2017/03/17
DOI:10.1039/C6CC09575E
Fluorobenzenes, in particular fluorobenzene (FB) and 1,2-difluorobenzene (1,2-DiFB), are increasingly becoming recognised as versatile solvents for conducting organometallic chemistry and transition-metal-based catalysis. The presence of fluorine substituents reduces the ability to donate π-electron density from the arene and consequently fluorobenzenes generally bind weakly to metal centres, allowing them to be used as essentially non-coordinating solvents or as readily displaced ligands. In this context, examples of well-defined complexes of fluorobenzenes are discussed, including trends in binding strength with increasing fluorination and different substitution patterns. Compared to more highly fluorinated benzenes, FB and 1,2-DiFB typically demonstrate greater chemical inertness, however, C–H and C–F bond activation reactions can be induced using appropriately reactive transition metal complexes. Such reactions are surveyed, including catalytic examples, not only to provide perspective for the use of FB and 1,2-DiFB as innocent solvent media, but also to highlight opportunities for their exploitation in contemporary organic synthesis.
Co-reporter:Michael J. Butler and Mark R. Crimmin
Chemical Communications 2017 - vol. 53(Issue 8) pp:NaN1365-1365
Publication Date(Web):2017/01/03
DOI:10.1039/C6CC05702K
The preparation and applications of heterobimetallic complexes continue to occupy researchers in the fields of organometallic, main group, and coordination chemistry. This interest stems from the promise these complexes hold as precursors to materials, reagents in synthesis and as new catalysis. Here we survey and organise the state-of-the-art understanding of the TM–H–M linkage (M = Mg, Zn, Al, Ga). We discuss the structure and bonding in these complexes, their known reactivity, and their largely unrealised potential in catalysis.
Co-reporter:Adi E. Nako, Sarah J. Gates, Nicole Schädel, Andrew J. P. White and Mark R. Crimmin
Chemical Communications 2014 - vol. 50(Issue 67) pp:NaN9538-9538
Publication Date(Web):2014/07/07
DOI:10.1039/C4CC04484C
We report [Y{N(SiMe3)2}3] as a precatalyst for the dehydrocoupling of sterically demanding amines with β-diketiminate stabilised aluminium dihydrides. While simple fluorinated anilines readily undergo Al–H/N–H dehydrocoupling under thermal conditions, catalytic methods are required to achieve reasonable rates of reaction for ortho-substituted anilines or hindered aliphatic amines.
Co-reporter:Andreas Phanopoulos, Mark Warren, Andrew J. P. White, Andrew Horton and Mark R. Crimmin
Dalton Transactions 2017 - vol. 46(Issue 7) pp:NaN2080-2080
Publication Date(Web):2017/01/20
DOI:10.1039/C6DT04703C
Addition of 2 equiv. of CuI to a [Cu]4 multimetallic complex results in cluster formation leading to the isolation of a rare bicapped tetrahedral [Cu6I2] cluster that is stabilised by two conformationally constrained polynucleating ligands.







![Phosphine, [(1E)-1,2-diphenylethenyl]diphenyl-](http://img.cochemist.com/ccimg/14500/14447-37-1.png)
![Phosphine, [(1E)-1,2-diphenylethenyl]diphenyl-](http://img.cochemist.com/ccimg/14500/14447-37-1_b.png)

-](http://img.cochemist.com/ccimg/510800/510720-04-4.png)
-](http://img.cochemist.com/ccimg/510800/510720-04-4_b.png)









![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)

















![Zirconium,dichlorobis[(1,2,3,3a,7a-h)-1H-inden-1-yl]-](http://img.cochemist.com/ccimg/12200/12148-49-1.png)
![Zirconium,dichlorobis[(1,2,3,3a,7a-h)-1H-inden-1-yl]-](http://img.cochemist.com/ccimg/12200/12148-49-1_b.png)
![4-Chloro-1-methyl-[1,2,4]triazolo[4,3-a]quinoxaline](http://img.cochemist.com/ccimg/91900/91895-39-5.png)
![4-Chloro-1-methyl-[1,2,4]triazolo[4,3-a]quinoxaline](http://img.cochemist.com/ccimg/91900/91895-39-5_b.png)






![Benzoic acid, 3-(trifluoromethyl)-, [3-(trifluoromethyl)phenyl]methyl ester](http://img.cochemist.com/ccimg/124100/124061-53-6.png)
![Benzoic acid, 3-(trifluoromethyl)-, [3-(trifluoromethyl)phenyl]methyl ester](http://img.cochemist.com/ccimg/124100/124061-53-6_b.png)

























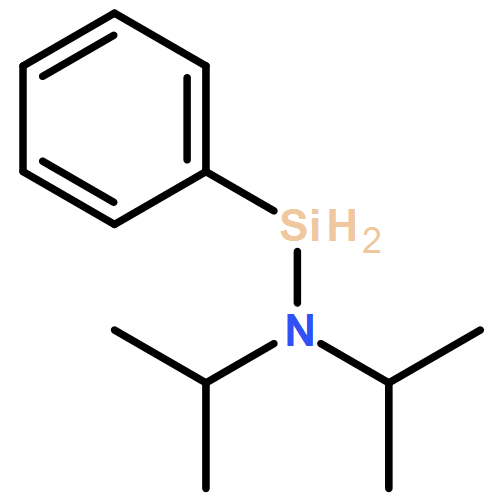


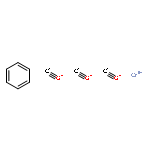
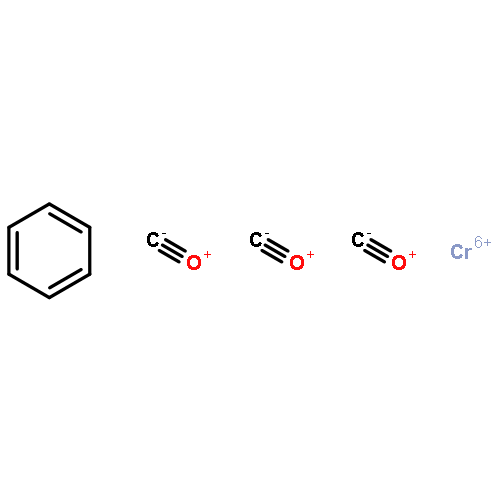
![Tris[N,N-bis(trimethylsilyl)amide]yttrium (III)](http://img.cochemist.com/ccimg/41900/41836-28-6.png)
![Tris[N,N-bis(trimethylsilyl)amide]yttrium (III)](http://img.cochemist.com/ccimg/41900/41836-28-6_b.png)
![3-Penten-2-one, 4-[[2,6-bis(1-methylethyl)phenyl]amino]-](http://img.cochemist.com/ccimg/206000/205993-26-6.png)
![3-Penten-2-one, 4-[[2,6-bis(1-methylethyl)phenyl]amino]-](http://img.cochemist.com/ccimg/206000/205993-26-6_b.png)






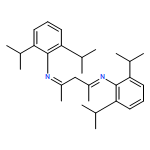
![Benzenamine, N-[3-[[2,6-bis(1-methylethyl)phenyl]amino]-1-methyl-2-buten-1-ylidene]-2,6-bis(1-methylethyl)-](/data/chemimg/120200/181708-81-6.png)
![Benzenamine, N-[3-[[2,6-bis(1-methylethyl)phenyl]amino]-1-methyl-2-buten-1-ylidene]-2,6-bis(1-methylethyl)-](/data/chemimg/120200/181708-81-6_b.png)