Co-reporter:Bo-Yen Lin, Connor J. Easley, Chia-Hsun Chen, Po-Chen Tseng, Ming-Zer Lee, Pin-Hao Sher, Juen-Kai Wang, Tien-Lung Chiu, Chi-Feng Lin, Christopher J. Bardeen, and Jiun-Haw Lee
ACS Applied Materials & Interfaces March 29, 2017 Volume 9(Issue 12) pp:10963-10963
Publication Date(Web):March 9, 2017
DOI:10.1021/acsami.6b16397
A new concept for organic light-emitting diodes (OLEDs) is presented, which is called exciplex-sensitized triplet–triplet annihilation (ESTTA). The exciplex formed at the organic heterojunction interface of 4,4′,4″-tris(N-3-methyphenyl-N-phenyl-amino) triphenylamine and 9,10-bis(2′-naphthyl) anthracene (ADN) is used to sensitize the triplet–triplet annihilation (TTA) process on the ADN molecules. This results in a turn-on voltage (2.2 V) of the blue emission from the OLED below the bandgap (2.9 eV). From the transient electroluminescence measurement, blue emission totally came from the TTA process without direct recombination on the ADN molecules. The blue singlet exciton from the TTA process can be quenched by energy transfer to the exciplex, as revealed by transient photoluminescence measurements. This can be prevented by blocking the energy transfer path and improving the radiative recombination rate of blue emission. With the insertion of the “triplet diffusion and singlet blocking (TDSB)” layer and the incorporation of the dopant material, an ESTTA-OLED with external quantum efficiency of 5.1% was achieved, which consists of yellow and blue emission coming from the exciplex and ESTTA process, respectively.Keywords: energy transfer; exciplex; organic light-emitting diode; Triplet−triplet annihilation; upconversion;
Co-reporter:Rabih O. Al-Kaysi;Fei Tong;Maram Al-Haidar;Lingyan Zhu
Chemical Communications 2017 vol. 53(Issue 17) pp:2622-2625
Publication Date(Web):2017/02/23
DOI:10.1039/C6CC08999B
When a suspension of the tert-butyl ester of 4-fluoroanthracene-9-carboxylic acid (4F9AC) was slowly hydrolyzed, highly branched photomechanical microcrystals of 4F9AC were grown. Exposure to UV light caused the branches to undergo a reversible sweeping motion that could be used to move and concentrate silica microspheres on a surface.
Co-reporter:Jonathan J. Burdett, Geoffrey B. Piland, Christopher J. Bardeen
Chemical Physics Letters 2016 Volume 661() pp:287
Publication Date(Web):16 September 2016
DOI:10.1016/j.cplett.2016.07.042
Co-reporter:Geoffrey B. Piland
The Journal of Physical Chemistry C 2016 Volume 120(Issue 11) pp:5883-5889
Publication Date(Web):March 7, 2016
DOI:10.1021/acs.jpcc.5b12021
The combination of CdSe semiconductor nanocrystals with 9-anthracene carboxylic acid ligands can sensitize triplet–triplet annihilation on the emitter molecule diphenylanthracene. This hybrid system has recently been shown to upconvert visible light (532 nm) to ultraviolet light (420 nm) (Huang, Z., et al. Nano Lett. 2015, 15, 5552–5557). In the current paper, time-resolved photoluminescence measurements are used to characterize the kinetics of the energy transfer from the CdSe exciton state to the triplet state of the anthracene ligand. We find that 9-anthracene carboxylic acid binds to CdSe according to Poisson statistics with a maximum number of 2–3 ligands per nanocrystal. The CdSe-to-ligand energy transfer rate is 1.5 × 107 s–1. The overall efficiency of the energy transfer appears to be limited by the presence of fast nonradiative decay channels in the nanocrystals and the low coverage of anthracene ligands, resulting from the specific ligand exchange conditions used in this paper. Possible strategies for improving this component of the hybrid upconversion system are discussed in light of these results.
Co-reporter:Geoffrey B. Piland and Christopher J. Bardeen
The Journal of Physical Chemistry C 2016 Volume 120(Issue 43) pp:25158-25160
Publication Date(Web):October 19, 2016
DOI:10.1021/acs.jpcc.6b07021
Co-reporter:Lingyan Zhu;Rabih O. Al-Kaysi
Angewandte Chemie International Edition 2016 Volume 55( Issue 25) pp:7073-7076
Publication Date(Web):
DOI:10.1002/anie.201511444
Abstract
Photomechanical molecular crystals can undergo a variety of light-induced motions, including expansion, bending, twisting, and jumping. The use of more complex crystal shapes may provide ways to turn these motions into useful work. To generate such shapes, pH-driven reprecipitation has been used to grow branched microcrystals of the anthracene derivative 4-fluoroanthracenecarboxylic acid. When these microcrystals are illuminated with light of λ=405 nm, an intermolecular [4+4] photodimerization reaction drives twisting and bending of the individual branches. These deformations drive a rotation of the overall crystal that can be repeated over multiple exposures to light. The magnitude and direction of this rotation vary because of differences in the crystal shape, but a typical branched crystal undergoes a 50° net rotation after 25 consecutive irradiations for 1 s. The ability of these crystals to undergo ratchet-like rotation is attributed to their chiral shape.
Co-reporter:Lingyan Zhu;Rabih O. Al-Kaysi
Angewandte Chemie 2016 Volume 128( Issue 25) pp:7189-7192
Publication Date(Web):
DOI:10.1002/ange.201511444
Abstract
Photomechanical molecular crystals can undergo a variety of light-induced motions, including expansion, bending, twisting, and jumping. The use of more complex crystal shapes may provide ways to turn these motions into useful work. To generate such shapes, pH-driven reprecipitation has been used to grow branched microcrystals of the anthracene derivative 4-fluoroanthracenecarboxylic acid. When these microcrystals are illuminated with light of λ=405 nm, an intermolecular [4+4] photodimerization reaction drives twisting and bending of the individual branches. These deformations drive a rotation of the overall crystal that can be repeated over multiple exposures to light. The magnitude and direction of this rotation vary because of differences in the crystal shape, but a typical branched crystal undergoes a 50° net rotation after 25 consecutive irradiations for 1 s. The ability of these crystals to undergo ratchet-like rotation is attributed to their chiral shape.
Co-reporter:Zhiyuan Huang, Xin Li, Melika Mahboub, Kerry M. Hanson, Valerie M. Nichols, Hoang Le, Ming L. Tang, and Christopher J. Bardeen
Nano Letters 2015 Volume 15(Issue 8) pp:5552-5557
Publication Date(Web):July 10, 2015
DOI:10.1021/acs.nanolett.5b02130
The ability to upconvert two low energy photons into one high energy photon has potential applications in solar energy, biological imaging, and data storage. In this Letter, CdSe and PbSe semiconductor nanocrystals are combined with molecular emitters (diphenylanthracene and rubrene) to upconvert photons in both the visible and the near-infrared spectral regions. Absorption of low energy photons by the nanocrystals is followed by energy transfer to the molecular triplet states, which then undergo triplet–triplet annihilation to create high energy singlet states that emit upconverted light. By using conjugated organic ligands on the CdSe nanocrystals to form an energy cascade, the upconversion process could be enhanced by up to 3 orders of magnitude. The use of different combinations of nanocrystals and emitters shows that this platform has great flexibility in the choice of both excitation and emission wavelengths.
Co-reporter:Chad D. Cruz; Peter R. Christensen; Eric L. Chronister; David Casanova; Michael O. Wolf
Journal of the American Chemical Society 2015 Volume 137(Issue 39) pp:12552-12564
Publication Date(Web):September 2, 2015
DOI:10.1021/jacs.5b05457
Symmetric dimers have the potential to optimize energy transfer and charge separation in optoelectronic devices. In this paper, a combination of optical spectroscopy (steady-state and time-resolved) and electronic structure theory is used to analyze the photophysics of sulfur-bridged terthiophene dimers. This class of dimers has the unique feature that the interchromophore (intradimer) electronic coupling can be modified by varying the oxidation state of the bridging sulfur from sulfide (S), to sulfoxide (SO), to sulfone (SO2). Photoexcitation leads to the formation of a delocalized charge resonance state (S1) that relaxes quickly (<10 ps) to a charge-transfer state (S1*). The amount of charge-transfer character in S1* can be enhanced by increasing the oxidation state of the bridging sulfur group as well as the solvent polarity. The S1* state has a decreased intersystem crossing rate when compared to monomeric terthiophene, leading to an enhanced photoluminescence quantum yield. Computational results indicate that electrostatic screening by the bridging sulfur electrons is the key parameter that controls the amount of charge-transfer character. Control of the sulfur bridge oxidation state provides the ability to tune interchromophore interactions in covalent assemblies without altering the molecular geometry or solvent polarity. This capability provides a new strategy for the design of functional supermolecules with applications in organic electronics.
Co-reporter:Heyuan Liu, Valerie M. Nichols, Li Shen, Setarah Jahansouz, Yuhan Chen, Kerry M. Hanson, Christopher J. Bardeen and Xiyou Li
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 9) pp:6523-6531
Publication Date(Web):28 Jan 2015
DOI:10.1039/C4CP05444J
A covalently linked tetracene dimer has been prepared and its molecular structure is characterized by 1H NMR and MALDI-TOF mass spectroscopy, and elemental analysis. The minimized molecular structure reveals that the tetracene subunits in a dimer adopt a “face-to-face” stacked configuration. Its absorption spectrum differs significantly from that of the monomeric counterpart in solution, suggesting the presence of strong interactions between the two tetracene subunits. In solution, the fluorescence spectrum is dominated by a band at around 535 nm, due to an oxidative impurity. In the longer wavelength range, a short-lived lower energy emission can be identified as the intrinsic emission of the dimer. In a polystyrene matrix or at low temperatures, the lifetime of the lower energy emission lengthens and it becomes more prominent. We suggest that the interactions between the two tetracene subunits produce a short-lived, lower energy “excimer-like” state. The fluorescence decays show no observable dependence on an applied magnetic field, and no obvious evidence of significant singlet fission is found in this dimer. This research suggests that even though there are strong electronic interactions between the tetracene subunits in the dimer, singlet fission cannot be achieved efficiently, probably because the formation of “excimer-like” states competes effectively with singlet fission.
Co-reporter:Kerry M. Hanson, Swathi Narayanan, Valerie M. Nichols and Christopher J. Bardeen
Photochemical & Photobiological Sciences 2015 vol. 14(Issue 9) pp:1607-1616
Publication Date(Web):26 May 2015
DOI:10.1039/C5PP00074B
The photodegradation of the ultraviolet (UV) filter octyl methoxycinnamate (OMC) is investigated in both dilute solution and in aggregated form. In dilute solution, the ratio of trans and cis isomers achieved at the photostationary state is solvent-dependent because of variations in the isomerization quantum yield. The two isomeric forms at the photostationary state are highly resistant to further photodegradation and no other UVA-absorbing species are formed. Aggregation of OMC, either in a neat film or in aqueous colloidal suspensions, leads to irreversible photodegradation of the molecule and the formation of multiple photoproducts. In addition to previously identified photoproducts like the UVB-absorbing cis and trans isomers and photodimers, we find photoproduct species whose absorption extends into the UVA. Characterization of the photophysical properties of these species indicates that they have long-lived excited-states (τf > 1 ns, 400 nm), unlike the isomeric forms of OMC (τf < 30 ps, 266 nm), and that excitation at 405 nm can sensitize the formation of singlet oxygen. These results show that the environment of OMC affects the photochemistry of the molecule and that the environmental conditions must be taken into account when considering the molecule's stability. In particular, aggregation of OMC molecules results in complex photochemistry that can produce species whose absorption extends into UVA and are capable of generating reactive oxygen species.
Co-reporter:Geoffrey B. Piland
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 10) pp:1841-1846
Publication Date(Web):April 27, 2015
DOI:10.1021/acs.jpclett.5b00569
The dependence of exciton dynamics on the crystalline morphology of tetracene is investigated using time-resolved photoluminescence. Single crystals exhibit relatively slow singlet decays with times that range from 130 to 300 ps depending on the sample. This decay has an activation energy of ∼450 cm–1 over the temperature range of 200–400 K. Single-crystal samples also exhibit more pronounced quantum beats due to the triplet pair spin coherences. Polycrystalline thin films grown by thermal evaporation have singlet decay times on the order of 70–90 ps with a much weaker temperature dependence. Many thin-film samples also exhibit a red-shifted excimer-like emission. When a polycrystalline thin film is thermally annealed to produce larger crystal domains, single-crystal behavior is recovered. We hypothesize that the different dynamics arise from the ability of singlet excitons in the thin films to sample regions with defects or packing motifs that accelerate singlet fission.
Co-reporter:Valerie M. Nichols
The Journal of Physical Chemistry C 2015 Volume 119(Issue 23) pp:12856-12864
Publication Date(Web):May 20, 2015
DOI:10.1021/acs.jpcc.5b03353
The excited-state dynamics of diindenoperylene (DIP) are investigated in dilute solution and in a solid film at room temperature using picosecond photoluminescence and femtosecond transient absorption measurements. In solution, DIP undergoes a rapid (0.89 ns) internal conversion back to its ground state, with no detectable formation of triplet or other long-lived states. In the solid state, multiple emissive species are formed. The time-resolved photoluminescence signal is dominated by an intrinsic exciton that decays on a time scale of 166 ps. Emission from lower energy excimer-like species then persists for >10 ns. Transient absorption experiments indicate that the majority of the excited-state population relaxes to the ground state on the 166 ps time scale, but a smaller fraction (<10%) survives in longer-lived trap or defect states. The rapid internal conversion leads to transient heating that results in a derivative line shape in the transient absorption signal at longer delays. DIP does not appear to support long-lived singlet exciton states or singlet fission. The implications of these results for the function of DIP in organic solar cells are discussed.
Co-reporter:Taehyung Kim ; Lingchao Zhu ; Leonard J. Mueller
Journal of the American Chemical Society 2014 Volume 136(Issue 18) pp:6617-6625
Publication Date(Web):April 11, 2014
DOI:10.1021/ja412216z
The solid-state photodimerization of 9-methylanthracene is used as a model system to investigate how crystal morphology and reaction dynamics affect photomechanical deformations of single microcrystals. By varying the crystallization conditions, two different crystal shapes, microneedles and microribbons, are grown on a clean water surface. The microribbons twist under irradiation, while the microneedles bend. In both shapes, the maximum deformation occurs at roughly the midpoint of the reaction, while further dimerization causes the crystals return to their original shapes. Powder X-ray diffraction patterns establish that the needles and ribbons have the same crystal orientation and that the photoreaction proceeds in a crystal-to-crystal manner. NMR spin–lattice relaxation measurements are consistent with the rapid formation of large (>100 nm) dimer crystal domains. Simultaneous measurement of the needle bending and monomer fluorescence signal allows us to correlate the bending with the reaction progress. The behavior is qualitatively reproduced by a model in which the motion is driven by strain between spatially distinct reactant and product domains, also called heterometry. We consider several different mechanisms that could give rise to these spatially distinct domains. The ability to control the photoinduced crystal deformation by manipulating crystal shape and solid-state reaction kinetics suggests that photoreactive molecular crystals may be useful for generating well-defined motions on small length scales.
Co-reporter:Lingyan Zhu, Fei Tong, Christopher Salinas, Muhanna K. Al-Muhanna, Fook S. Tham, David Kisailus, Rabih O. Al-Kaysi, and Christopher J. Bardeen
Chemistry of Materials 2014 Volume 26(Issue 20) pp:6007
Publication Date(Web):September 15, 2014
DOI:10.1021/cm502866e
Four fluorinated derivatives of 9-anthracene carboxylic acid (9AC), a molecule that shows a reversible photomechanical response in its crystal form, have been synthesized and characterized. The spectroscopic properties and crystal structures of 4-fluoro-9-anthracene carboxylic acid (4F-9AC), 2-fluoro-9-anthracene carboxylic acid (2F-9AC), 10-fluoro-9-anthracene carboxylic acid (10F-9AC), and 2,6-difluoro-9-anthracene carboxylic acid (2,6DF-9AC) are all very similar to those of 9AC. However, their photomechanical properties vary widely. 405 nm light was used to induce [4 + 4] photodimerization and a mechanical response in crystalline microneedles and ribbons. Both the photodimer dissociation rate and the mechanical recovery varied by more than an order of magnitude, with 4F-9AC exhibiting the most rapid recovery time, on the order of 30 s. Nanoindentation measurements show that this crystal has a slightly reduced elastic modulus and a significantly reduced hardness, making it less brittle than the 9AC crystal. Large 4F-9AC crystals remain intact after irradiation, without fragmenting, while microneedles can undergo more than 100 mechanical bending cycles. Given the similarity of the crystal packing in all five molecules, the improved photomechanical properties must arise from subtle changes in intermolecular interactions or possibly differences in disorder. These results demonstrate that it is possible to significantly improve the properties of photomechanical materials through small modifications of the molecular structure.
Co-reporter:Chi-Feng Lin, Valerie M. Nichols, Yung-Chih Cheng, Christopher J. Bardeen, Mau-Kuo Wei, Shun-Wei Liu, Chih-Chien Lee, Wei-Cheng Su, Tien-Lung Chiu, Hsieh-Cheng Han, Li-Chyong Chen, Chin-Ti Chen, Jiun-Haw Lee
Solar Energy Materials and Solar Cells 2014 Volume 122() pp:264-270
Publication Date(Web):March 2014
DOI:10.1016/j.solmat.2013.12.006
•High IQE SubPc/C60 OPV device was fabricated by inserting mCP between ITO and SubPc.•Exciton and electron blocking resulted in the high IQE.•The device showed high efficiency of 5.08%, with VOC of 1.09 V and IQE of 95.6%.A planar heterojunction organic solar cell (OSC) with high internal quantum efficiency (IQE=95.6% at 590 nm) was demonstrated based on boron subphthalocyanine chloride (SubPc) and C60 as the electron donor and acceptor materials. A thin layer of N,N-dicarbazolyl-3,5-benzene (mCP), acting as an electron and exciton blocking layer, was inserted between the ITO anode and the SubPc layer to prevent leakage current and exciton quenching. In the optimized device, a mCP thickness of 3 nm was thick enough to block the excitons (as suggested by a high IQE and longer-lived photoluminescence), resulting in an open circuit voltage of 1.09 V, a short circuit current of 7.87 mA/cm2, a fill factor of 59, and a power conversion efficiency of 5.08%. Increasing the mCP thickness resulted in increased serial resistance and SubPc crystallization that reduced the fill factor, VOC, and JSC. Due to the narrow absorption band of SubPC (71 nm at the full-width-at-half-maximum), this high performance (both in peak IQE and VOC) device has the potential to be used as a subcell in a spectral splitting solar cell module.
Co-reporter:Chi-Feng Lin, Valerie Nichols, Yung-Chih Cheng, Christopher J. Bardeen, Mau-Kuo Wei, Shun-Wei Liu, Chih-Chien Lee, Wei-Cheng Su, Tien-Lung Chiu, Hsieh-Cheng Han, Li-Chyong Chen, Chin-Ti Chen, Jiun-Haw Lee
Solar Energy Materials and Solar Cells 2014 Volume 128() pp:300
Publication Date(Web):September 2014
DOI:10.1016/j.solmat.2014.05.025
Co-reporter:Geoffrey B. Piland, Jonathan J. Burdett, Tzu-Yao Hung, Po-Hsun Chen, Chi-Feng Lin, Tien-Lung Chiu, Jiun-Haw Lee, Christopher J. Bardeen
Chemical Physics Letters 2014 Volume 601() pp:33-38
Publication Date(Web):9 May 2014
DOI:10.1016/j.cplett.2014.03.075
•Fluorescence experiments are conducted on a tetracene/LiF/Si sample.•The fluorescence intensity of tetracene increases as LiF thickness increases.•Prompt fluorescence lifetimes decrease as the tetracene thickness increases.•No evidence is found for triplet energy transfer from tetracene to the Si.Tetracene, a molecule that undergoes singlet fission, is deposited on Si with variable thickness LiF spacer layers. In agreement with earlier work (Hayashi et al., 1983 [10]), the fluorescence intensity of the tetracene greatly increases as the LiF thickness approaches 100 nm. This increase is partly due to a 30% increase in the prompt fluorescence decay time but mostly results from weaker coupling of the luminescence into the Si substrate. A decrease in the prompt fluorescence lifetime is observed as the tetracene thickness is increased on bare Si. We find no evidence for triplet energy transfer to the Si.
Co-reporter:Dr. Taehyung Kim;Dr. Lingyan Zhu; Dr. Rabih O. Al-Kaysi; Dr. Christopher J. Bardeen
ChemPhysChem 2014 Volume 15( Issue 3) pp:400-414
Publication Date(Web):
DOI:10.1002/cphc.201300906
Abstract
Organic molecules can transform photons into Angstrom-scale motions by undergoing photochemical reactions. Ordered media, for example, liquid crystals or molecular crystals, can align these molecular-scale motions to produce motion on much larger (micron to millimeter) length scales. In this Review, we describe the basic principles that underlie organic photomechanical materials, starting with a brief survey of molecular photochromic systems that have been used as elements of photomechanical materials. We then describe various options for incorporating these active elements into a solid-state material, including dispersal in a polymer matrix, covalent attachment to a polymer chain, or self-assembly into molecular crystals. Particular emphasis is placed on ordered media, such as liquid-crystal elastomers and molecular crystals, that have been shown to produce motion on large (micron to millimeter) length scales. We also discuss other mechanisms for generating photomechanical motion that do not involve photochemical reactions, such as photothermal expansion and photoinduced charge transfer. Finally, we identify areas for future research, ranging from the study of basic phenomena in solid-state photochemistry, to molecular and host matrix design, and the optimization of photoexcitation conditions. The ultimate realization of photon-fueled micromachines will likely involve advances spanning the disciplines of chemistry, physics and engineering.
Co-reporter:Fei Tong, Chad D. Cruz, Sebastian R. Jezowski, Xiaoquan Zhou, Lingyan Zhu, Rabih O. Al-Kaysi, Eric L. Chronister, and Christopher J. Bardeen
The Journal of Physical Chemistry A 2014 Volume 118(Issue 28) pp:5349-5354
Publication Date(Web):June 19, 2014
DOI:10.1021/jp504771b
9-tert-Butylanthracene undergoes a photochemical reaction to form its strained Dewar isomer, which thermally back-reacts to reform the original molecule. When 9-tert-butylanthracene is dissolved in a polymer host, we find that both the forward and reverse isomerization rates are pressure-dependent. The forward photoreaction rate, which reflects the sum of contributions from photoperoxidation and Dewar isomerization, decreases by a factor of 1000 at high pressure (1.5 GPa). The back-reaction rate, on the other hand, increases by a factor of ∼3 at high pressure. Despite being highly strained and higher volume, the back-reaction reaction rate of the Dewar isomer is at least 100× less sensitive to pressure than that of the bi(anthracene-9,10-dimethylene) photodimer studied previously by our group. These results suggest that the high pressure sensitivity of the bi(anthracene-9,10-dimethylene) photodimer reaction is not just due to the presence of strained four-membered rings but instead relies on the unique molecular geometry of this molecule.
Co-reporter:Geoffrey B. Piland, Jonathan J. Burdett, Robert J. Dillon, and Christopher J. Bardeen
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 13) pp:2312-2319
Publication Date(Web):June 12, 2014
DOI:10.1021/jz500676c
Singlet fission, in which an initially excited singlet state spontaneously splits into a pair of triplet excitons, is a process that can potentially boost the efficiency of solar energy conversion. The separate electronic bands in organic semiconductors make them especially useful for dividing a high-energy singlet exciton into a pair of lower-energy triplet excitons. Recent experiments illustrate the role of spin coherence in fission, while kinetic models are used to describe how triplet and singlet states interact on longer time scales. Despite insights gained from recent experiments, the detailed structure and dynamics of the electronic states involved in the initial step of singlet fission remain active areas of investigation. On longer time scales, finding ways to efficiently harvest the triplet excitons will be an important challenge for making devices based on this phenomenon. A full understanding of singlet fission requires consideration of a sequence of photophysical events (decoherence, relaxation, and diffusion) occurring on different time scales.
Co-reporter:Jonathan J. Burdett and Christopher J. Bardeen
Accounts of Chemical Research 2013 Volume 46(Issue 6) pp:1312
Publication Date(Web):January 29, 2013
DOI:10.1021/ar300191w
Singlet fission (SF) is a spin-allowed process in which an excited singlet state spontaneously splits into a pair of triplet excitons. This relaxation pathway is of interest as a mechanism for increasing the efficiency of photovoltaic solar cells, since ionization of the triplets could produce two charge carriers per absorbed photon. In this Account, we summarize our recent work on trying to understand how SF occurs using both covalent and noncovalent assemblies of tetracene. We first give a brief overview of the SF process and discuss why tetracene, where the singlet and triplet pair energies are nearly degenerate, is a particularly useful molecule for studying this process. Then we describe our experiments, beginning with the study of phenylene-linked tetracene dimers as covalent analogs for the crystal form, where SF is known to be very efficient. We found that only 2–3% of the initially excited singlets underwent SF in these dimers. These results motivated us to study crystalline tetracene in more detail. Transient absorption and photoluminescence experiments on polycrystalline thin films provided evidence for a delocalized singlet exciton that decays with a complicated temperature-dependence, but we were unable to unambiguously identify the signature of the triplet pair formed by SF. Then, using ultrathin single crystals, we observed quantum beats in the delayed fluorescence arising from recombination of spin-coherent triplet pairs. Analyzing these quantum beats revealed that SF proceeds through a direct one-step process occurring within 200 ps at room temperature. The product of this reaction is a pair of unperturbed triplets that have negligible interaction with each other.Looking at the overall SF process in tetracene, remaining issues that need to be clarified include the role of exciton diffusion, the temperature dependence of the SF rate, and how to use insights gained from the solid-state studies to generate design principles for high-efficiency covalent systems. Our experiments provide a good illustration of why the polyacenes, and tetracene in particular, play an important role as systems for the study of SF.
Co-reporter:Robert J. Dillon ; Geoffrey B. Piland
Journal of the American Chemical Society 2013 Volume 135(Issue 46) pp:17278-17281
Publication Date(Web):October 30, 2013
DOI:10.1021/ja409266s
The dynamics of singlet fission (SF) are studied in monoclinic and orthorhombic crystals of 1,6-diphenyl-1,3,5-hexatriene. Picosecond time-resolved fluorescence measurements and the presence of a strong magnetic field effect indicate that up to 90% of the initially excited singlets undergo SF in both forms. The initial SF and subsequent triplet pair dissociation rates are found to be more rapid in the monoclinic crystal by factors of 1.5 and 3.5, respectively. These results provide clear evidence that molecular organization affects the rates of triplet pair formation and separation, both important parameters for determining the ultimate utility of a SF material.
Co-reporter:Robert J. Dillon, Ji-Bong Joo, Francisco Zaera, Yadong Yin and Christopher J. Bardeen
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 5) pp:1488-1496
Publication Date(Web):12 Dec 2012
DOI:10.1039/C2CP43666C
The spectroscopic and photocatalytic properties of a series of Au@TiO2 core–shell nanostructures are characterized. The crystallinity of the TiO2 shells was varied by changing the etching and calcination conditions. Measurements of the photoluminescence, transient absorption, and H2 production rate permit us to look for correlations between the spectroscopic and catalytic behaviors. We found that there is a strong effect of crystallinity on the H2 production rate and also the stretched exponential lifetime of the photoluminescence created by short-wavelength (266 and 300 nm) photoexcitation. As the TiO2 crystallinity is increased, the photoluminescence lifetime increases from 22 to 140 ps in a 1 ns detection window, while the H2 production rate increases by a factor of ∼4. There is no discernible effect of crystallinity on the photoluminescence dynamics excited at 350 or 430 nm, or on the electronic dynamics measured by femtosecond transient absorption after excitation at 300 nm. We hypothesize that high-energy photons create reactive and emissive charge-separated states in parallel, and that both species are subject to similar electron–hole recombination processes that depend on sample crystallinity. Based on our observations, it can be concluded that the photoluminescence dynamics may be used to evaluate the potential performance of this class of photocatalysts.
Co-reporter:Jonathan J. Burdett, Geoffrey B. Piland, Christopher J. Bardeen
Chemical Physics Letters 2013 Volume 585() pp:1-10
Publication Date(Web):14 October 2013
DOI:10.1016/j.cplett.2013.08.036
Singlet fission is a photophysical process that has promise for increasing the efficiency of solar cells. The dynamics depend on triplet spin states and can be influenced by external magnetic fields. In 4-electron systems, fission takes an initial singlet state into a superposition of triplet pair states. Direct evidence for this superposition state is provided by quantum beats in the delayed fluorescence of tetracene crystals. The beat frequencies depend on crystal orientation with respect to the magnetic field, consistent with predictions based on solving the full spin Hamiltonian. Magnetic field effects on the kinetics are analyzed in terms of a hybrid quantum-kinetic model. The magnetic field has no effect on the initial fluorescence decay rate but affects the decay after the triplet pair states begin to equilibrate with the singlets. The long-time behavior of the fluorescence decay reflects association and separation of triplet pairs and relaxation into different spin states.
Co-reporter:Dr. Taehyung Kim;Dr. Muhanna K. Al-Muhanna;Dr. Salem D. Al-Suwaidan;Dr. Rabih O. Al-Kaysi;Dr. Christopher J. Bardeen
Angewandte Chemie International Edition 2013 Volume 52( Issue 27) pp:6889-6893
Publication Date(Web):
DOI:10.1002/anie.201302323
Co-reporter:Geoffrey B. Piland, Jonathan J. Burdett, Dharmalingam Kurunthu, and Christopher J. Bardeen
The Journal of Physical Chemistry C 2013 Volume 117(Issue 3) pp:1224-1236
Publication Date(Web):December 22, 2012
DOI:10.1021/jp309286v
Picosecond time-resolved fluorescence experiments are used to study the dynamics of singlet fission in highly disordered films of rubrene. The fluorescence spectral lineshapes are not temperature-dependent, indicating that intermolecular excitonic effects are absent in these films. The temperature-dependent fluorescence decays in the amorphous films are nonexponential, containing both prompt and delayed components. The kinetics are qualitatively consistent with the presence of singlet fission, but to confirm its presence, we examine the effects of magnetic fields on the fluorescence decay. A quantum-kinetic model is developed to describe how magnetic fields perturb the number of triplet pair product states with singlet character and how this in turn affects the singlet state kinetics. Simulations show that the magnetic field effect is very sensitive to mutual chromophore alignment, and the direction of the effect is consistent with a local ordering for rubrene molecules that participate in fission. From our analysis, the dominant fission rate is 0.5 ns–1, about 10 times slower than that observed in polycrystalline tetracene films, but we still estimate that ∼90% of the initially excited singlets undergo fission. Kinetic modeling of our fluorescence decay data and magnetic field dependence reveals that at the low laser intensities used in this experiment geminate triplet pairs do not interact with each other, and that spin–lattice relaxation between triplet sublevels is not complete on the 100 ns time scale. When both exciton fission and fusion are occurring, dynamic measurements in the presence of a magnetic field can elucidate molecular-level details of both processes.
Co-reporter:Valerie M. Nichols, Marco T. Rodriguez, Geoffrey B. Piland, Fook Tham, Vladimir N. Nesterov, W. Justin Youngblood, and Christopher J. Bardeen
The Journal of Physical Chemistry C 2013 Volume 117(Issue 33) pp:16802-16810
Publication Date(Web):July 17, 2013
DOI:10.1021/jp4051116
The photophysical behavior of the polycyclic aromatic hydrocarbon peropyrene is studied both in dilute solution and in the solid state, with the goal of evaluating this molecule as a singlet fission (SF) material. In solution, the fluorescence quantum yield is consistently in the range 0.90–0.95, while the fluorescence lifetime changes from 3.2 to 5.5 ns. Analysis of the solvent dependence of the radiative rate provides evidence that the bright 1Bu singlet state mixes with a second, optically dark state. The presence of a dark state slightly above the 1Bu state in energy is confirmed using two-photon fluorescence excitation spectroscopy. The crystal structure of solid peropyrene consists of a herringbone arrangement of π-stacked molecular pairs, similar to the α-polymorph of perylene. There are two emitting species, centered at approximately 550 and 650 nm, both of which are formed within the 15 ps time resolution of the experiment, and which relax independently via biexponential decays. We find no evidence for rapid SF in the peropyrene crystals, most likely due to the large shift of the singlet state to lower energy where it no longer fulfills the energy condition for SF. These results demonstrate how both energetics and crystal packing influence the ability of a molecule to function as a SF material.
Co-reporter:Sebastian R. Jezowski ; Lingyan Zhu ; Yaobing Wang ; Andrew P. Rice ; Gary W. Scott ; Christopher J. Bardeen ;Eric L. Chronister
Journal of the American Chemical Society 2012 Volume 134(Issue 17) pp:7459-7466
Publication Date(Web):April 9, 2012
DOI:10.1021/ja300424h
The anthracene cyclophane bis-anthracene (BA) can undergo a [4 + 4] photocycloaddition reaction that results in a photodimer with two cyclobutane rings. We find that the subsequent dissociation of the dimer, which involves the rupture of two carbon–carbon bonds, is strongly accelerated by the application of mild pressures. The reaction kinetics of the dimer dissociation in a Zeonex (polycycloolefin) polymer matrix were measured at various pressures and temperatures. Biexponential reaction kinetics were observed for all pressures, consistent with the presence of two different isomers of bis(anthracene). One of the rates showed a strong dependence on pressure, yielding a negative activation volume for the dissociation reaction of ΔV⧧ = −16 Å3. The 93 kJ/mol activation energy for the dissociation reaction at ambient pressure is lowered by more than an order of magnitude from 93 to 7 kJ/mol with the application of modest pressure (0.9 GPa). Both observations are consistent with a transition state that is stabilized at higher pressures, and a mechanism for this is proposed in terms of a two-step process where a flattening of the anthracene rings precedes rupture of the cyclobutane rings. The ability to catalyze covalent bond breakage in isolated small molecules using compressive forces may present opportunities for the development of materials that can be activated by acoustic shock or stress.
Co-reporter:Jonathan J. Burdett
Journal of the American Chemical Society 2012 Volume 134(Issue 20) pp:8597-8607
Publication Date(Web):April 24, 2012
DOI:10.1021/ja301683w
A detailed analysis of the oscillations seen in the delayed fluorescence of crystalline tetracene is presented in order to study the mechanism of singlet fission. Three quantum beat frequencies of 1.06 ± 0.05, 1.82 ± 0.05, and 2.92 ± 0.06 GHz are resolved, which are damped on a time scale of 20 ns. The effects of sample morphology, excitation wavelength, and temperature are examined. A density matrix model for singlet fission is developed that quantitatively describes the frequencies, amplitudes, and damping of the oscillations. The model assumes a direct coupling of the initially excited singlet exciton to the triplet pair manifold. There is no electronic coherence between the singlet and triplet pair states, but the rapid singlet decay time of ∼200 ps in solution-grown single crystals provides the impulsive population transfer necessary to create a coherent superposition of three zero-field triplet pair states |xx⟩, |yy⟩, and |zz⟩ with overall singlet character. This superposition of the three states gives rise to the three quantum beat frequencies seen in the experiment. Damping of the quantum beats results from both population exchange between triplet and singlet manifolds and pure dephasing between the triplet pair states. By lowering the temperature and slowing the SF rate, the visibility of the oscillations decreases. There is no evidence of magnetic dipole–dipole coupling between the product triplets. Our model provides good overall agreement with the data, supporting the conclusion that singlet fission in tetracene proceeds through the “direct” mechanism without strong electronic coupling between the singlet and triplet pair states.
Co-reporter:Taehyung Kim, Lingchao Zhu, Leonard J. Mueller and Christopher J. Bardeen
CrystEngComm 2012 vol. 14(Issue 22) pp:7792-7799
Publication Date(Web):03 Aug 2012
DOI:10.1039/C2CE25811K
The photochemical dynamics of crystals composed of 4-chlorocinnamic acid (4Cl-CA), whose photochemistry is dominated by an irreversible [2+2] photodimerization reaction, are studied using 13C solid-state NMR, powder X-ray diffraction, and optical and electron microscopy. We find that photoreaction leads to a new crystal phase, but prolonged irradiation leads to an amorphous solid. To investigate effect of crystal morphology on the photoresponsive behavior, molecular crystals with different shapes and sizes are prepared and compared under the same irradiation conditions. Microribbons with submicron thicknesses twist under irradiation, but no response is observed in larger crystals with thicknesses of 5–10 microns. Possible mechanisms to explain these differences are discussed, including differences in defect densities, optical properties, and heat dissipation. We posit that the dominant effect is the dependence of the torsion constant on crystal thickness, which leads to the thinner microribbons being more susceptible to deformation by an internal energy density. This work shows that photo-induced twisting can be observed in photoreactive systems different from the anthracene [4+4] photodimerization studied previously. Our results also suggest that shrinking crystal dimensions to the nanoscale can give rise to new types of photomechanical motion.
Co-reporter:Xiaoquan Zhou, Geoffrey B. Piland, Dharmalingam Kurunthu, Robert J. Dillon, Jonathan J. Burdett, Christopher J. Bardeen
Journal of Luminescence 2012 Volume 132(Issue 11) pp:2997-3003
Publication Date(Web):November 2012
DOI:10.1016/j.jlumin.2012.06.012
Three 2,6 dialkoxyanthracenes with varying ether sidechain lengths (methyl, n-propyl and n-hexyl) are synthesized and characterized. When compared to unsubstituted anthracene, the spectroscopic properties of the alkoxy-anthracenes are significantly different. The oscillator strength appears to be evenly distributed between the La and Lb states, rather than concentrated in the low-energy La state, and the transition dipole moments are rotated by ∼30° in the molecular frame. More importantly, all three derivatives undergo an intramolecular conformational change in the excited state that gives rise to a reshaping of the fluorescence spectrum on the nanosecond timescale. This process has an activation energy of 8±1 kJ/mol. By ∼150 K the fluorescence spectrum reflects only the high energy conformer, whose emission lineshape is similar to that of the unsubstituted anthracene. The temperature-dependent changes in the monomeric fluorescence spectra of these compounds will have to be taken into account in future studies of their solid-state spectroscopy.Highlights► Three 2,6 dialkoxyanthracenes were synthesized and their steady-state spectroscopic properties measured. ► The electronic states and transition dipole strengths of these molecules were calculated using the TDDFT and were compared to those of unsubstituted anthracene. ► Picosecond time-resolved fluorescence measurements in solution showed the existence of two emitting states, and the dependence of their interconversion rate on solvent was determined. ► The activation energy between conformations was obtained from temperature-dependent measurements. ► The identity of the two emissive conformers was discussed, along with their implications for solid-state measurements on these molecules.
Co-reporter:Robert J. Dillon and Christopher J. Bardeen
The Journal of Physical Chemistry A 2012 Volume 116(Issue 21) pp:5145-5150
Publication Date(Web):May 16, 2012
DOI:10.1021/jp302829a
Previous studies of solid-state tetracyanobenzene-based donor–acceptor complexes showed that these materials were highly susceptible to both laser and mechanical damage that complicated the analysis of their electron-transfer kinetics. In this paper, we characterize the optical properties of a pyrene/tetracyanoquinodimethane charge-transfer crystal that is much more robust than the tetracyanobenzene compounds. This donor–acceptor complex has a charge-transfer absorption that extends into the near-infrared, rendering the crystal black. We use time-resolved fluorescence and diffuse reflectance transient absorption to study its dynamics after photoexcitation. We show that the initially excited charge-transfer state undergoes a rapid, monoexponential decay with a lifetime of 290 ps at room temperature. There is no evidence for any long-lived intermediate or dark states; therefore, this decay is attributed to charge recombination back to the ground state. Fluorescence lifetime measurements demonstrate that this process becomes temperature-independent below 60 K, indicative of a thermally activated tunneling mechanism. The subnanosecond charge recombination makes this low-band-gap donor–acceptor material a poor candidate for generating long-lived electron–hole pairs.
Co-reporter:Lingyan Zhu ; Rabih O. Al-Kaysi
Journal of the American Chemical Society 2011 Volume 133(Issue 32) pp:12569-12575
Publication Date(Web):July 12, 2011
DOI:10.1021/ja201925p
9-Anthracenecarboxylic acid, a molecule that undergoes a reversible [4 + 4] photodimerization, is prepared in the form of oriented crystalline microribbons. When exposed to spatially uniform light irradiation, these photoreactive ribbons rapidly twist. After the light is turned off, they relax back to their original shape over the course of minutes. This photoinduced motion can be repeated for multiple cycles. The final twist period and cross-sectional dimensions of individual microribbons are measured using a combination of atomic force and optical microscopies. Analysis of this data suggests that the reversible twisting involves the generation of interfacial strain within the ribbons between unreacted monomer and photoreacted dimer regions, with an interaction energy on the order of 3.4 kJ/mol. The demonstration of reversible twisting without the need for specialized irradiation conditions represents a new type of photoinduced motion in molecular crystals and may provide new modes of operation for photomechanical actuators.
Co-reporter:Lingyan Zhu, Arun Agarwal, Jinfeng Lai, Rabih O. Al-Kaysi, Fook S. Tham, Tarek Ghaddar, Leonard Mueller and Christopher J. Bardeen
Journal of Materials Chemistry A 2011 vol. 21(Issue 17) pp:6258-6268
Publication Date(Web):24 Mar 2011
DOI:10.1039/C1JM10228A
A series of 9-anthroate esters that can form photoresponsive molecular crystal nanorods is prepared and their properties are investigated. All crystal structures that can support a [4 + 4] photodimerization reaction lead to nanorods that undergo photomechanical deformations without fragmentation. In order to determine the molecular-level motions that give rise to the nanorod photomechanical response, the reaction of anthracene-9-carboxylic acid tert-butyl ester is studied in detail using X-ray diffraction and solid-state NMR techniques. The monomer crystal is well-aligned within the nanorod and reacts to form the photodimer crystal according to first-order kinetics. The solid-state reacted dimer crystal is a metastable intermediate that slowly converts into the low energy dimer crystal structure over the course of weeks. Based on single crystal X-ray diffraction studies and solid-state NMR data, this intermediate structure is likely composed of the [4 + 4] photodimer that has not yet undergone the ester group rotations and repacking is necessary to form the lower energy crystal polymorph that is produced directly by crystallization from solution. Our results show that the photomechanical response of these molecular crystal nanostructures is determined by nonequilibrium intermediate states and cannot be predicted based solely on knowledge of the equilibrium reactant and product crystal structures.
Co-reporter:Lingyan Zhu, Rabih O. Al-Kaysi, Robert J. Dillon, Fook S. Tham, and Christopher J. Bardeen
Crystal Growth & Design 2011 Volume 11(Issue 11) pp:4975-4983
Publication Date(Web):September 6, 2011
DOI:10.1021/cg200883b
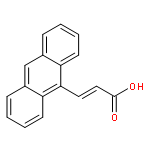
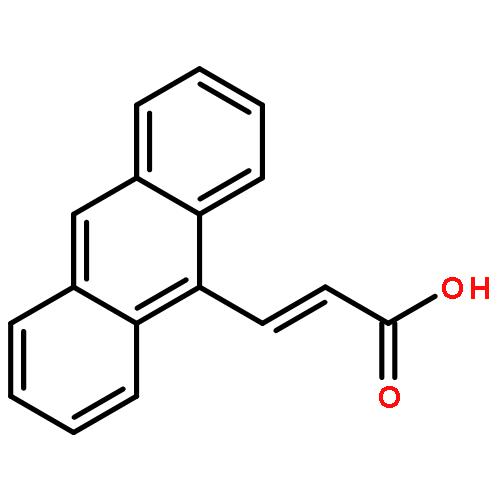
Molecular crystals composed of 9-anthracene carboxylic acid (9AC) can undergo reversible light-induced mechanical motions driven by a [4 + 4] photodimerization reaction. This paper explores the structure, photophysics, and photomechanical response of a family of anthracene carboxylic acid derivatives, with the goal of finding materials that have comparable or improved photomechanical properties. We find that methyl or phenyl substitution at the 10-position leads to a complete loss of photoreactivity due to changes in crystal packing. A series of halogen (F, Cl, Br) 10-substituted 9AC molecules all showed a similar stacked packing motif, but only the fluoro-substituted molecule was photoreactive in the solid. Its photomechanical response was similar to that of 9AC but with a much longer recovery time. Extending the carboxylic acid by adding a vinylene group at the 9-position resulted in crystals that showed good photoreactivity and a lack of fracture but no reversibility. Attempts to self-consistently rationalize observed trends in terms of excited state lifetimes or steric effects were only partly successful. Balancing factors such as electronic relaxation, steric interactions, and crystal packing present a challenge for engineering photoactive solid-state materials based on molecular crystals.
Co-reporter:Robert J. Dillon and Christopher J. Bardeen
The Journal of Physical Chemistry A 2011 Volume 115(Issue 9) pp:1627-1633
Publication Date(Web):February 14, 2011
DOI:10.1021/jp110912d
Charge-transfer molecular crystals are structurally well-defined systems whose electron transfer dynamics can be studied using time-resolved spectroscopy. In this paper, five 1:1 complexes, consisting of 1,2,4,5-tetracyanobenzene as the electron acceptor and durene, 9-methylanthracene, naphthalene, phenanthrene, and pyrene as electron donors, are studied using time-resolved fluorescence and transient absorption in the diffuse reflectance geometry. Two different sample morphologies were studied: single crystals and powders prepared by pulverizing the crystals and diluting them with barium sulfate microparticles. Fluorescence lifetime and transient absorption measurements performed on the crystals and the powders yielded different results. The crystals typically exhibited long-lived monoexponential fluorescence decays, while the powders had shorter multiexponential decays. Exposure of both types of samples to high laser fluence was also shown to induce faster excited state decay dynamics as observed using fluorescence and diffuse reflectance. In addition to the more rapid decays, these molecular crystals exhibited relatively high photobleaching quantum yields on the order of 10−4. Previous work that interpreted picosecond decays in the transient absorption as evidence for rapid recombination and charge dissociation should be re-evaluated based on the susceptibility of this class of compounds to mechanical and photochemical damage.
Co-reporter:Kathryn A. Colby and Christopher J. Bardeen
The Journal of Physical Chemistry A 2011 Volume 115(Issue 26) pp:7574-7581
Publication Date(Web):June 7, 2011
DOI:10.1021/jp202654v
In this paper, we continue our evaluation of Forster-type theories of exciton diffusion in disordered environments. The perylenediimide dye Lumogen Red is used as a donor molecule in two different liquids, CHCl3 and dimethylformamide, and the energy transfer to the acceptor molecule Rhodamine 700 is measured using time-resolved fluorescence decays. The exciton motion is measured over Lumogen Red concentrations ranging from 1 × 10–4 to 5 × 10–2 M, and the results are compared to previous results for exciton diffusion in a solid polymer. Depending on the theoretical approach used to analyze the data, we find that the energy migration in the liquids is a factor of 2–3 faster than in the solid polymer, even after taking molecular translation into account. Measurements for a Lumogen Red concentration of 10 mM in the different host environments yield diffusion constants ranging from 2.2 to 3.1 nm2/ns in the liquids, as compared to 1.1–1.2 nm2/ns in solid poly(methyl methacrylate) (PMMA). The results in the liquids are in good agreement with theoretical predictions and numerical simulations of previous workers, while the results in solid PMMA are 2–3 times slower. This discrepancy is discussed in the context of the rapid energetic averaging present in the liquid environments but absent in the solid matrix, where unfavorable configurations and low energy trapping sites are frozen in by the static disorder.
Co-reporter:Yaobing Wang, Dharmalingam Kurunthu, Gary W. Scott and Christopher J. Bardeen
The Journal of Physical Chemistry C 2010 Volume 114(Issue 9) pp:4153-4159
Publication Date(Web):February 10, 2010
DOI:10.1021/jp9097793
Conjugated polymers blended with graphene represent a possible approach for making organic bulk heterojunction solar cells. In this paper, the time-resolved fluorescence dynamics of poly(3-hexylthiophene-2,5-diyl) (P3HT) and poly[2-methoxy-5-(2′-ethylhexyloxy)-1,4-phenylenevinylene] (MEH-PPV) blended with graphene microsheets derived from chemically reduced graphitic oxide are studied. Both polymers exhibit strong quenching and shortened fluorescence lifetimes when mixed with graphene. The fluorescence quenching function takes the form of e−kQt1/2, where kQ is linearly proportional to the weight fraction of graphene in the blend. We consider two physical models to explain the origin of the fluorescence quenching. The first assumes that energy transfer occurs within a three-dimensional space to molecular scale defects within the graphene according to the standard Forster model with an energy transfer rate proportional to the donor−acceptor separation R−6. The second model assumes a quasi-two-dimensional environment where the energy transfer rate between the donor and graphene sheets is proportional to R−4. Using the second model, an estimate of ∼5 nm is obtained for the critical energy transfer radius for energy transfer between P3HT chains and graphene sheets. This value is in reasonable agreement with theory. Differences between the quenching behavior of graphene in MEH-PPV and P3HT blends are also discussed.
Co-reporter:Kathryn A. Colby, Jonathan J. Burdett, Robert F. Frisbee, Lingyan Zhu, Robert J. Dillon and Christopher J. Bardeen
The Journal of Physical Chemistry A 2010 Volume 114(Issue 10) pp:3471-3482
Publication Date(Web):February 19, 2010
DOI:10.1021/jp910277j
Electronic energy transfer plays an important role in many types of organic electronic devices. Förster-type theories of exciton diffusion provide a way to calculate diffusion constants and lengths, but their applicability to amorphous polymer systems must be evaluated. In this paper, the perylenediimide dye Lumogen Red in a poly(methyl methacrylate) host matrix is used to test theories of exciton motion over Lumogen Red concentrations (CLR’s) ranging from 1 × 10−4 to 5 × 10−2 M. Two experimental quantities are measured. First, time-resolved anisotropy decays in films containing only Lumogen Red provide an estimate of the initial energy transfer rate from the photoexcited molecule. Second, the Lumogen Red lifetime decays in mixed systems where the dyes Malachite Green and Rhodamine 700 act as energy acceptors are measured to estimate the diffusive quenching of the exciton. From the anisotropy measurements, it is found that theory accurately predicts both the CLR−2 concentration dependence of the polarization decay time τpol, as well as its magnitude to within 30%. The theory also predicts that the diffusive quenching rate is proportional to CLRα, where α ranges between 1.00 and 1.33. Experimentally, it is found that α = 1.1 ± 0.2 when Malachite Green is used as an acceptor, and α = 1.2 ± 0.2 when Rhodamine 700 is the acceptor. On the basis of the theory that correctly describes the anisotropy data, the exciton diffusion constant is projected to be 4−9 nm2/ns. By use of several different analysis methods for the quenching data, the experimental diffusion constant is found to be in the range of 0.32−1.20 nm2/ns. Thus the theory successfully describes the early time anisotropy data but fails to quantitatively describe the quenching experiments which are sensitive to motion on longer time scales. The data are consistent with the idea that orientational and energetic disorder leads to a time-dependent exciton migration rate, suggesting that simple diffusion models cannot accurately describe exciton motion within this system.
Co-reporter:Rabih O. Al-Kaysi, Astrid M. Müller, Robert J. Frisbee and Christopher J. Bardeen
Crystal Growth & Design 2009 Volume 9(Issue 4) pp:1780-1785
Publication Date(Web):February 20, 2009
DOI:10.1021/cg800898f
The reaction of single-component molecular crystal nanorods with a second species to form cocrystal nanorods is described. Single-component crystalline nanorods, composed of 9-methylanthracene (9-MA), are grown in a porous anodic aluminum oxide template. These templated rods are then exposed to a suspension of 1,2,4,5-tetracyanobenzene (TCNB) in water, which slowly diffuses into the 9-MA rods over the course of days. The two species form a 1:1 charge-transfer complex within the rods, which are transformed from crystalline 9-MA into cocrystalline 9-MA/TCNB. The cocrystal nanorods are characterized by electron microscopy, X-ray diffraction, and optical spectroscopy, confirming their highly crystalline structure and the formation of the charge-transfer complex. Attempts to grow cocrystal nanorods directly from a mixed solution were unsuccessful, as were attempts to recreate the single crystal-to-crystal reaction in macroscopic crystals. This work demonstrates how organic nanostructures can support structure-preserving chemical transformations that are impossible in larger crystals.
Co-reporter:Kerry M. Hanson
Photochemistry and Photobiology 2009 Volume 85( Issue 1) pp:33-44
Publication Date(Web):
DOI:10.1111/j.1751-1097.2008.00508.x
Abstract
Recent advances in the use of nonlinear optical microscopy (NLOM) in skin microscopy are presented. Nonresonant spectroscopies including second harmonic generation, coherent anti-Stokes Raman and two-photon absorption are described and applications to problems in skin biology are detailed. These nonlinear techniques have several advantages over traditional microscopy methods that rely on one-photon excitation: intrinsic 3D imaging with <1 μm spatial resolution, decreased photodamage to tissue samples and penetration depths up to 1000 μm with the use of near-infrared lasers. Thanks to these advantages, nonlinear optical spectroscopy has become a powerful tool to study the physical and biochemical properties of the skin. Structural information can be obtained using the response of endogenous chemical species in the skin, such as collagen or lipids, indicating that optical biopsy may replace current invasive, time-consuming traditional histology methods. Insertion of specific probe molecules into the skin provides the opportunity to monitor specific biochemical processes such as skin transport, molecular penetration, barrier homeostasis and ultraviolet radiation-induced reactive oxygen species generation. While the field is quite new, it seems likely that the use of NLOM to probe structure and biochemistry of live skin samples will only continue to grow.
Co-reporter:C. J. Bardeen;R. O. Al-Kaysi
Advanced Materials 2007 Volume 19(Issue 9) pp:1276-1280
Publication Date(Web):4 APR 2007
DOI:10.1002/adma.200602741
Crystalline nanorods composed of 9-anthracene carboxylic acid (9-AC) are synthesized using nanoporous Al2O3 templates. When a segment of an isolated rod is exposed to UV-light, localized solid-state photodimerization of the 9-AC molecules induces micrometer-scale bending (see figure). The nanorod reverts back to its original shape after several minutes in the dark at room temperature. The reversible photoinduced shape change could be repeated multiple times.
Co-reporter:Tai-Sang Ahn, Nicholas Wright, Christopher J. Bardeen
Chemical Physics Letters 2007 Volume 446(1–3) pp:43-48
Publication Date(Web):26 September 2007
DOI:10.1016/j.cplett.2007.08.003
Numerical simulations of exciton migration along a one-dimensional lattice via Forster energy transfer are reported. The roles of static Gaussian energetic disorder and transition dipole orientational disorder are investigated. Both disorder mechanisms result in subdiffusive behavior during the initial phase of energy migration, tending asymptotically to normal diffusion. The dependence of the subdiffusion parameters on the inhomogeneous linewidth and intersite spacing is examined. Depending on the exciton lifetime, subdiffusive motion may dominate the exciton displacement behavior, and it becomes problematic to use normal diffusion theory to make even qualitative predictions about the exciton motion.Static energetic and orientational disorder leads to subdiffusive Forster energy migration along a one-dimensional lattice of chromophores.
Co-reporter:Rabih O. Al-Kaysi, Tai Sang Ahn, Astrid M. Müller and Christopher J. Bardeen
Physical Chemistry Chemical Physics 2006 vol. 8(Issue 29) pp:3453-3459
Publication Date(Web):19 Jun 2006
DOI:10.1039/B605925B
The absorption, fluorescence, and photostability of five conjugated chromophores: perylene, 2,5,8,11-tetra-t-butyl perylene (TTBP), perylene orange (PO), perylene red (PR), and a zwitterionic Meisenheimer complex (MHC), are studied as a function of concentration in poly(methyl methacrylate) (PMMA). At 1 mM concentrations, all five molecules exhibit properties consistent with unaggregated chromophores. At higher concentrations, perylene and PO both exhibit excimer formation, while TTBP, PR, and the MHC retain their monomeric fluorescent lineshapes. In these three molecules, however, the fluorescence decay times decrease by 10% (TTBP) to 50% (MHC) at concentrations of 100 mM in PMMA. The fluorescence properties of these highly concentrated samples are sensitive to the sample preparation conditions. In the neat solid where the effective concentration is on the order of 1 M, all three molecules exhibit very fast fluorescence decays, on the order of 150 ps or less, despite the fact that they retain their basic monomeric fluorescence lineshape. In addition to the enhanced nonradiative decay at high concentrations, these three molecules also undergo a concentration-dependent photobleaching. The combined effects of intermolecular nonradiative decay channels and photobleaching appear to be a general obstacle to achieving highly concentrated dye-doped solids.
Co-reporter:Christopher J. Bardeen;Sara K. Davis
Photochemistry and Photobiology 2005 Volume 81(Issue 3) pp:548-555
Publication Date(Web):30 APR 2007
DOI:10.1111/j.1751-1097.2005.tb00224.x
In eukaryotic cell nuclei, double-stranded DNA is found in the form of chromatin, a large fiber made up of DNA complexed to histone proteins. In this article, recent studies using fluorescence techniques to look at the dynamics of chromatin, both in vivo and in vitro, are reviewed. Two-photon counter-propagating fluorescence recovery after patterned photobleaching is used to examine chromatin fluctuations on lengthscales ranging from less than 100 nm to microns. By combining in vivo studies with data on isolated nuclei and by measuring how these fluctuations depend on variables like ionic strength and photochemical cross-linking, it is demonstrated that the relatively large-scale motions of chromatin observed in vivo are consistent with smaller scale modifications of the histone-DNA interaction. This connection may provide a means to use conformational dynamics as an in vivo probe of the biochemical events involved in gene expression.
Co-reporter:Chi-Feng Lin, Valerie M. Nichols, Yung-Chih Cheng, Christopher J. Bardeen, Mau-Kuo Wei, Shun-Wei Liu, Chih-Chien Lee, Wei-Cheng Su, Tien-Lung Chiu, Hsieh-Cheng Han, Li-Chyong Chen, Chin-Ti Chen, Jiun-Haw Lee
Solar Energy Materials and Solar Cells (March 2014) Volume 122() pp:264-270
Publication Date(Web):1 March 2014
DOI:10.1016/j.solmat.2013.12.006
•High IQE SubPc/C60 OPV device was fabricated by inserting mCP between ITO and SubPc.•Exciton and electron blocking resulted in the high IQE.•The device showed high efficiency of 5.08%, with VOC of 1.09 V and IQE of 95.6%.A planar heterojunction organic solar cell (OSC) with high internal quantum efficiency (IQE=95.6% at 590 nm) was demonstrated based on boron subphthalocyanine chloride (SubPc) and C60 as the electron donor and acceptor materials. A thin layer of N,N-dicarbazolyl-3,5-benzene (mCP), acting as an electron and exciton blocking layer, was inserted between the ITO anode and the SubPc layer to prevent leakage current and exciton quenching. In the optimized device, a mCP thickness of 3 nm was thick enough to block the excitons (as suggested by a high IQE and longer-lived photoluminescence), resulting in an open circuit voltage of 1.09 V, a short circuit current of 7.87 mA/cm2, a fill factor of 59, and a power conversion efficiency of 5.08%. Increasing the mCP thickness resulted in increased serial resistance and SubPc crystallization that reduced the fill factor, VOC, and JSC. Due to the narrow absorption band of SubPC (71 nm at the full-width-at-half-maximum), this high performance (both in peak IQE and VOC) device has the potential to be used as a subcell in a spectral splitting solar cell module.
Co-reporter:Geoffrey B. Piland, Christopher J. Bardeen
Chemical Physics Letters (February 2017) Volume 669() pp:
Publication Date(Web):February 2017
DOI:10.1016/j.cplett.2016.12.021
•Time-resolved spectroscopy was used to study rubrene, diphenylhexatriene, and tetracene in the liquid phase.•Singlet fission was found to occur in rubrene with a timescale comparable to the crystalline system.•Diphenylhexatriene was observed to have a rapid singlet loss channel that outcompetes singlet fission.•Tetracene decomposed at the temperature needed to melt the sample which negated the possibly of further study.•Singlet fission and triplet lifetimes were not enhanced in the liquid phase.The effect of high temperature melting on the photophysics of three prototypical singlet fission molecules is investigated. Time-resolved photoluminescence is used to look at the melt phase of the molecules tetracene, diphenylhexatriene and rubrene. Chemical decomposition of tetracene precluded any detailed measurements on this molecule. In the diphenylhexatriene melt, a rapid singlet state nonradiative relaxation process outcompetes singlet fission. In the rubrene melt, singlet fission occurs at a rate similar to that of the crystal, but the decay of the delayed fluorescence is much more rapid. The rapid decay of the delayed fluorescence suggests that either the triplet lifetime is shortened, or the fusion probability decreases, or that both factors are operative at higher temperatures.
Co-reporter:Rabih O. Al-Kaysi, Fei Tong, Maram Al-Haidar, Lingyan Zhu and Christopher J. Bardeen
Chemical Communications 2017 - vol. 53(Issue 17) pp:NaN2625-2625
Publication Date(Web):2017/02/06
DOI:10.1039/C6CC08999B
When a suspension of the tert-butyl ester of 4-fluoroanthracene-9-carboxylic acid (4F9AC) was slowly hydrolyzed, highly branched photomechanical microcrystals of 4F9AC were grown. Exposure to UV light caused the branches to undergo a reversible sweeping motion that could be used to move and concentrate silica microspheres on a surface.
Co-reporter:Heyuan Liu, Valerie M. Nichols, Li Shen, Setarah Jahansouz, Yuhan Chen, Kerry M. Hanson, Christopher J. Bardeen and Xiyou Li
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 9) pp:NaN6531-6531
Publication Date(Web):2015/01/28
DOI:10.1039/C4CP05444J
A covalently linked tetracene dimer has been prepared and its molecular structure is characterized by 1H NMR and MALDI-TOF mass spectroscopy, and elemental analysis. The minimized molecular structure reveals that the tetracene subunits in a dimer adopt a “face-to-face” stacked configuration. Its absorption spectrum differs significantly from that of the monomeric counterpart in solution, suggesting the presence of strong interactions between the two tetracene subunits. In solution, the fluorescence spectrum is dominated by a band at around 535 nm, due to an oxidative impurity. In the longer wavelength range, a short-lived lower energy emission can be identified as the intrinsic emission of the dimer. In a polystyrene matrix or at low temperatures, the lifetime of the lower energy emission lengthens and it becomes more prominent. We suggest that the interactions between the two tetracene subunits produce a short-lived, lower energy “excimer-like” state. The fluorescence decays show no observable dependence on an applied magnetic field, and no obvious evidence of significant singlet fission is found in this dimer. This research suggests that even though there are strong electronic interactions between the tetracene subunits in the dimer, singlet fission cannot be achieved efficiently, probably because the formation of “excimer-like” states competes effectively with singlet fission.
Co-reporter:Lingyan Zhu, Fei Tong, Norhan Zaghloul, Omar Baz, Christopher J. Bardeen and Rabih O. Al-Kaysi
Journal of Materials Chemistry A 2016 - vol. 4(Issue 35) pp:NaN8252-8252
Publication Date(Web):2016/08/10
DOI:10.1039/C6TC02517J
Anthracene derivatives with a 1,3-butadiene group attached at the 9-position can undergo E ↔ Z (trans ↔ cis) photoisomerization reactions, providing a route to new photomechanical materials. In this paper, we report the properties of a new anthracene derivative, 2-(3-anthracen-9-yl-allylidene)-malononitrile (9DVAM). The structure, photophysics and photochemistry of 9DVAM are characterized in both dilute solution and in the crystal form. This molecule possesses a strong charge-transfer transition that extends its absorption into the visible region. Photoexcitation of this transition leads to a E → Z isomerization reaction in both dilute solution and in the crystal. Rapid nonradiative relaxation and a high E → Z photoisomerization yield appear to prevent dimerization side reactions in the crystal. Exposure to ultraviolet light can partially reverse this reaction in both liquid solution and in the crystal, allowing the molecule to be cycled back and forth between its cis and trans isomers. This switching between populations can cause reversible shape changes, like bending, in crystalline microribbons and nanowires. The divinylanthracenes provide a new class of photomechanical molecular crystals that complement existing photoreversible materials.
Co-reporter:Fei Tong, Mervin P. Hanson and Christopher J. Bardeen
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 46) pp:NaN31945-31945
Publication Date(Web):2016/10/17
DOI:10.1039/C6CP04459J
In order to develop an improved description of the photomechanical response of 9-methylanthracene (9MA) microcrystals, a detailed study of its solid-state photochemical reaction kinetics is performed. The reaction progress is monitored through the decrease in absorption of an optically microcrystalline thin film. The evolution of the time-dependent photoluminescence during the reaction is also measured. Both the photochemical reaction and nonradiative relaxation rates increase as more photoproduct is formed. In order to analyze the data, an extended version of the Finke–Watzky kinetic model for photochemical reactions is derived, denoted the FW-P model. This extended version enables a systematic analysis of photochemical reaction kinetics in solid-state molecular systems at varying levels of approximation. The FW-P model describes the non-exponential decrease in reactant and also correctly predicts the magnitude of the observed decrease in photoluminescence lifetime over the course of the reaction. The lifetime analysis is complicated by the fact that the microcrystalline 9MA sample contains multiple emitting species, and the extended FW-P model fails to capture the exact dependence of the photoluminescence on product formation. Analysis of the 9MA data indicates that both the photodimerization and the nonradiative relaxation rates can be accelerated by a factor of 10 over the course of the reaction. The results in this paper demonstrate that autocatalytic photodimerization kinetics are present in crystalline 9MA and may influence its photomechanical response.
Co-reporter:Robert J. Dillon, Ji-Bong Joo, Francisco Zaera, Yadong Yin and Christopher J. Bardeen
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 5) pp:NaN1496-1496
Publication Date(Web):2012/12/12
DOI:10.1039/C2CP43666C
The spectroscopic and photocatalytic properties of a series of Au@TiO2 core–shell nanostructures are characterized. The crystallinity of the TiO2 shells was varied by changing the etching and calcination conditions. Measurements of the photoluminescence, transient absorption, and H2 production rate permit us to look for correlations between the spectroscopic and catalytic behaviors. We found that there is a strong effect of crystallinity on the H2 production rate and also the stretched exponential lifetime of the photoluminescence created by short-wavelength (266 and 300 nm) photoexcitation. As the TiO2 crystallinity is increased, the photoluminescence lifetime increases from 22 to 140 ps in a 1 ns detection window, while the H2 production rate increases by a factor of ∼4. There is no discernible effect of crystallinity on the photoluminescence dynamics excited at 350 or 430 nm, or on the electronic dynamics measured by femtosecond transient absorption after excitation at 300 nm. We hypothesize that high-energy photons create reactive and emissive charge-separated states in parallel, and that both species are subject to similar electron–hole recombination processes that depend on sample crystallinity. Based on our observations, it can be concluded that the photoluminescence dynamics may be used to evaluate the potential performance of this class of photocatalysts.
Co-reporter:Lingyan Zhu, Arun Agarwal, Jinfeng Lai, Rabih O. Al-Kaysi, Fook S. Tham, Tarek Ghaddar, Leonard Mueller and Christopher J. Bardeen
Journal of Materials Chemistry A 2011 - vol. 21(Issue 17) pp:NaN6268-6268
Publication Date(Web):2011/03/24
DOI:10.1039/C1JM10228A
A series of 9-anthroate esters that can form photoresponsive molecular crystal nanorods is prepared and their properties are investigated. All crystal structures that can support a [4 + 4] photodimerization reaction lead to nanorods that undergo photomechanical deformations without fragmentation. In order to determine the molecular-level motions that give rise to the nanorod photomechanical response, the reaction of anthracene-9-carboxylic acid tert-butyl ester is studied in detail using X-ray diffraction and solid-state NMR techniques. The monomer crystal is well-aligned within the nanorod and reacts to form the photodimer crystal according to first-order kinetics. The solid-state reacted dimer crystal is a metastable intermediate that slowly converts into the low energy dimer crystal structure over the course of weeks. Based on single crystal X-ray diffraction studies and solid-state NMR data, this intermediate structure is likely composed of the [4 + 4] photodimer that has not yet undergone the ester group rotations and repacking is necessary to form the lower energy crystal polymorph that is produced directly by crystallization from solution. Our results show that the photomechanical response of these molecular crystal nanostructures is determined by nonequilibrium intermediate states and cannot be predicted based solely on knowledge of the equilibrium reactant and product crystal structures.





![[BIS(TRIMETHYLSILYL)]SELENIDE](http://img.cochemist.com/ccimg/4100/4099-46-1.png)
![[BIS(TRIMETHYLSILYL)]SELENIDE](http://img.cochemist.com/ccimg/4100/4099-46-1_b.png)






![Propanedioic acid,2-[(4-hydroxy-3,5-dimethoxyphenyl)methylene]-, 1,3-bis(2-ethylhexyl) ester](http://img.cochemist.com/ccimg/444900/444811-29-4.png)
![Propanedioic acid,2-[(4-hydroxy-3,5-dimethoxyphenyl)methylene]-, 1,3-bis(2-ethylhexyl) ester](http://img.cochemist.com/ccimg/444900/444811-29-4_b.png)


![Poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene]](http://img.cochemist.com/ccimg/138200/138184-36-8.png)
![Poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene]](http://img.cochemist.com/ccimg/138200/138184-36-8_b.png)



















![Dibenzo[cd,lm]perylene](/data/chemimg/1089200/188-96-5.png)
![Dibenzo[cd,lm]perylene](/data/chemimg/1089200/188-96-5_b.png)


![2,3,6,7,12,13,16,17-octahydro-9-(trifluoromethyl)-1H,5H,11H,15H-xantheno[2,3,4-ij:5,6,7-i'j']diquinolizin-18-ium perchlorate](http://img.cochemist.com/ccimg/63600/63561-42-2.png)
![2,3,6,7,12,13,16,17-octahydro-9-(trifluoromethyl)-1H,5H,11H,15H-xantheno[2,3,4-ij:5,6,7-i'j']diquinolizin-18-ium perchlorate](http://img.cochemist.com/ccimg/63600/63561-42-2_b.png)



