Co-reporter:Bei-Hua Bao, An Kang, Yang Zhao, Qi Shen, Jun-Song Li, Liu-Qing Di, Jian-Xin Li
Journal of Chromatography B 2017 Volume 1052(Volume 1052) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.jchromb.2017.03.008
•SND-117 might serve as a candidate for rheumatoid arthritis management.•Optimized HPLC–MS/MS method for quantification of SND-117 in rat plasma.•The approach was successfully applied to the pharmacokinetic study of SND-117.Rheumatoid arthritis (RA), a chronic systemic inflammatory disorder affects many adults. Sinomenine, a natural product, has been clinically available for the treatment of RA in China. SND-117, a sinomenine derivative with much more potent activity, might serve as a candidate for anti-arthritis. The aim of the present study was to develop a sensitive and rapid high performance liquid chromatography tandem mass spectrometry (HPLC–MS/MS) method for quantification of SND-117 in rat plasma and to understand its absolute bioavailability. The HPLC–MS/MS method was developed and fully validated for determination of SND-117 in rat plasma, and the pharmacokinetic differences were investigated after different administration routes. The pharmacokinetics parameters were calculated by non-compartment model with DAS 3.0 software. After the oral or intravenous administration of different doses of SND-117, the time to peak is 1.5 h, half-life time is 8–10 h. The absolute oral bioavailability of SND-117 in rats was 9.60%. The results showed that SND-117 in rats was quickly absorbed, slowly eliminate, and the kinetics were linear. This method was suitable for pharmacokinetic studies of SNA-117 in rats.
Co-reporter:Jing Wu, Bei-Hua Bao, Qi Shen, Yu-Chao Zhang, Qing Jiang and Jian-Xin Li
MedChemComm 2016 vol. 7(Issue 2) pp:371-377
Publication Date(Web):23 Dec 2015
DOI:10.1039/C5MD00482A
Osteoporosis is a major public health problem in our aging society. In the present study, a series of novel oleanolic acid (OA) derivatives were synthesized via modifications in the A-ring and C-28 position of OA, and their anti-bone resorption activities were evaluated. Screening results revealed that most of the derivatives inhibited RANKL-induced osteoclast formation from RAW264.7 cells. Among the derivatives, 6g exhibited a potent inhibitory activity with an IC50 of 24 nM. Furthermore, 6g prevented ovariectomy-induced bone loss in rats. The preliminary mechanism investigation demonstrated that 6g suppressed the protein and mRNA expressions of c-Fos and NFATc1, and downregulated the mRNA of other osteoclastogenic markers. The results suggested that the OA derivatives might serve as potential leads for the development of novel agents for the treatment of osteoporosis.
Co-reporter:Dan Zhao;Qi Shen
Advanced Synthesis & Catalysis 2015 Volume 357( Issue 2-3) pp:339-344
Publication Date(Web):
DOI:10.1002/adsc.201400827
Co-reporter:Dan Zhao, Teng Wang, Qi Shen and Jian-Xin Li
Chemical Communications 2014 vol. 50(Issue 33) pp:4302-4304
Publication Date(Web):03 Mar 2014
DOI:10.1039/C4CC01444H
An n-Bu4NI catalyzed domino reaction that involves selective dual amination of sp3 C–H bonds has been developed. The protocol affords a facile and efficient approach to the synthesis of imidazo[1,5-c]quinazolines under mild conditions.
Co-reporter:Dan Zhao, Teng Wang and Jian-Xin Li
Chemical Communications 2014 vol. 50(Issue 49) pp:6471-6474
Publication Date(Web):02 May 2014
DOI:10.1039/C4CC02648A
A novel metal-free synthesis of quinazolinones via dual amination of sp3 C–H bonds was developed. The sp3 carbon in methylarenes or adjacent to a heteroatom in DMSO, DMF or DMA was used as the one carbon synthon.
Co-reporter:Hai-Jian Fu ; Yu-Ren Zhou ; Bei-Hua Bao ; Meng-Xuan Jia ; Yang Zhao ; Lei Zhang ; Jian-Xin Li ; Hai-Lang He ;Xian-Mei Zhou
Journal of Medicinal Chemistry 2014 Volume 57(Issue 11) pp:4692-4709
Publication Date(Web):May 20, 2014
DOI:10.1021/jm5002293
Tryptophan hydroxylase 1 (Tph-1), the principal enzyme for peripheral serotonin biosynthesis, provides a novel target to design anabolic agents for osteoporosis. Here, we present a design, synthesis of a novel series of ursolic acid derivatives under the guidance of docking technique, and bioevaluation of the derivatives using RBL2H3 cells and ovariectomized (OVX) rats. Of the compounds, 9a showed a potent inhibitory activity on serotonin biosynthesis. Further investigations revealed that 9a, as an efficient Tph-1 binder identified by SPR (estimated KD: 6.82 μM), suppressed the protein and mRNA expressions of Tph-1 and lowered serotonin contents in serum and gut without influence on brain serotonin. Moreover, oral administration of 9a elevated serum level of N-terminal propeptide of procollagen type 1 (P1NP), a bone formation marker, and improved bone microarchitecture without estrogenic side effects in ovariectomized rats. Collectively, 9a may serve as a new candidate for bone anabolic drug discovery.
Co-reporter:Dan Zhao, Yu-Ren Zhou, Qi Shen and Jian-Xin Li
RSC Advances 2014 vol. 4(Issue 13) pp:6486-6489
Publication Date(Web):03 Jan 2014
DOI:10.1039/C3RA46363J
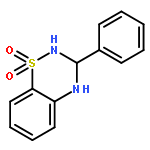
An iron-catalyzed one-pot one-step oxidative system has been successfully developed in the conversion of primary alcohols into nitrogen-containing heterocycles, such as quinazolinone, quinazoline and 3,4-dihydro-2H-1,2,4-benzothiadiazine 1,1-dioxide derivatives.
Co-reporter:Jing Wu, Qi Shen, Yue Wang, Dan Zhao, Chen Peng, and Jian-Xin Li
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 8) pp:911
Publication Date(Web):June 11, 2014
DOI:10.1021/ml500181e
Labeling of a small bioactive molecule with fluorescent probe has been becoming an essential tool in cell biology to reveal the subcellular distribution and the location of a molecular target. QOA-8a is a novel molecule with potent antiosteoporotic effect in vivo. To investigate the molecular mechanism of QOA-8a, novel fluorescence-tagged chemical probes as bioactive as their parent molecule were designed and synthesized. The fluorescent compound 12 showed a more potent inhibitory activity on RANKL-induced osteoclastogenesis at 2 μM compared with that of QOA-8a. Microscopy experiments revealed that almost all of probe 12 accumulated in the fusing region, with little in the osteoclast precursors or the mature osteoclasts during osteoclast formation. The result suggests the location of the binding target of QOA-8a, which might greatly narrow down the search field of the target protein(s).Keywords: Fluorescent derivative probes; osteoclasts; subcellular localization
Co-reporter:Dan Zhao, Qi Shen, Yu-Ren Zhou and Jian-Xin Li
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 35) pp:5908-5912
Publication Date(Web):15 Jul 2013
DOI:10.1039/C3OB41083H
A facile alkenylation of quinazolines with unactivated terminal alkynes has been achieved in the presence of KOtBu without the aid of any transition metal catalysts. The reaction is carried out under very mild conditions and shows a high stereoselectivity. A possible radical-based mechanism is also explored.
Co-reporter:Jie Jin, Peng Teng, Hai-Liang Liu, Jing Wu, Yu-Mei Liu, Qiang Xu, Jian-Xin Li
European Journal of Medicinal Chemistry 2013 Volume 62() pp:280-288
Publication Date(Web):April 2013
DOI:10.1016/j.ejmech.2012.12.051
Sinomenine (1) is currently used for the treatment of rheumatoid arthritis (RA) in China and there is still room for the improvement of its efficacy. In present study, capillary based microfluidic system was effectively applied for the syntheses of two novel series of sinomenine derivatives. The Heck reactions in microreactor gave much higher conversions compared to the batch ones. The two-step synthesis of the isoxazoline in microreactor greatly shortened the reaction time without any isolation of intermediates. The inhibitory activity of synthesized compounds on the TNF-α-induced nuclear factor kappa B (NF-κB) activation was evaluated in vitro. Among the compounds, 3c and 3g showed the potent inhibitory activity. Furthermore, 3g exhibited the antiinflammatory effect in vivo.Graphical abstractNovel sinomenine derivatives were synthesized with the continuous flow capillary microreactor and their antiinflammatory activities were evaluated in vitro and in vivo.Highlights► Continuous flow capillary microreactor was effectively used. ► Two novel series of sinomenine derivative were synthesized with the microreactor. ► The antiinflammatory activities of synthesized compounds were evaluated in vitro and in vivo. ► 3g inhibited NF-κB activation and mouse paw edema.
Co-reporter:Dan Zhao, Min-Xue Zhu, Yue Wang, Qi Shen and Jian-Xin Li
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 37) pp:6246-6249
Publication Date(Web):02 Aug 2013
DOI:10.1039/C3OB41488D
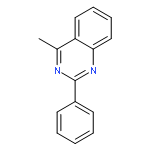
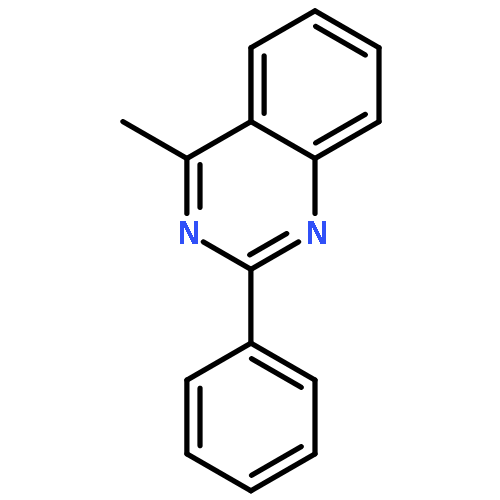
An efficient and selective Pd(0)-catalyzed sp3 C–H bond arylation–oxidation of 4-methylquinazolines is reported. The method enables the introduction of arylketone at the benzylic position of 4-methylquinazolines without the use of an additional directing group, and atmospheric oxygen is used as the sole oxidant.
Co-reporter:Dan Zhao, Yue Wang, Min-Xue Zhu, Qi Shen, Lei Zhang, Yun Du and Jian-Xin Li
RSC Advances 2013 vol. 3(Issue 26) pp:10272-10276
Publication Date(Web):14 Feb 2013
DOI:10.1039/C3RA40695D
A green, simple, and efficient protocol for the selective methylenation via CuCl2/oxygen-mediated C–H (sp3) oxidation and C–N cleavage using tetramethylethylenediamine (TMEDA) as a carbon source has been developed. The reactions were achieved in green solvent water under atmospheric conditions. The protocol exhibited a broad substrate scope including indoles, anilines, pyrroles and 1,3-dicarbonyls. Furthermore, two key intermediates of the reaction were successfully identified and the mechanism was explored.
Co-reporter:Qi Shen, Lei Zhang, Yu-Ren Zhou, Jian-Xin Li
Tetrahedron Letters 2013 Volume 54(Issue 49) pp:6725-6728
Publication Date(Web):4 December 2013
DOI:10.1016/j.tetlet.2013.09.118
Oxidant-dependent Cu-catalyzed alkynylation and aminomethylation reactions have been achieved under facile and mild conditions. TMEDA coupled with various terminal alkynes via C–H bond cleavage in good yields using atmospheric oxygen as an oxidant. Switching from air to TBHP afforded aminomethylation products of terminal alkynes through C–C bond cleavage of TMEDA. The protocol provided a novel strategy to prepare bi/tridentate N-ligand.Highly efficient alkynylation of TMEDA has been achieved under copper(II) catalysis for the first time. The chelation effect of diamines to copper was observed as the key factor to promoting this transformation. Meanwhile, TMEDA can act as an aminomethyl donor for aminomethylation of alkynes using TBHP as oxidant.
Co-reporter:Lei Zhang, Chen Peng, Dan Zhao, Yue Wang, Hai-Jian Fu, Qi Shen and Jian-Xin Li
Chemical Communications 2012 vol. 48(Issue 47) pp:5928-5930
Publication Date(Web):23 Apr 2012
DOI:10.1039/C2CC32009F
Base-switched methylenation and formylation using tetramethylethylenediamine (TMEDA) as a carbon source have been achieved under mild conditions, catalyzed by CuCl2, with atmospheric oxygen as oxidant. Bisindolylmethanes, diphenylmethanes and 3-formylindoles were synthesized with excellent regioselectivity and good yield.
Co-reporter:Lei Zhang, Mei Geng, Peng Teng, Dan Zhao, Xi Lu, Jian-Xin Li
Ultrasonics Sonochemistry 2012 Volume 19(Issue 2) pp:250-256
Publication Date(Web):March 2012
DOI:10.1016/j.ultsonch.2011.07.008
Co-reporter:Cui-Cui He;Yun-Qing Dai;Rong-Rong Hui;Jia Hua;Hong-Juan Chen;Qiao-Yun Luo
Journal of Applied Toxicology 2012 Volume 32( Issue 2) pp:88-97
Publication Date(Web):
DOI:10.1002/jat.1633
ABSTRACT
Cimicifugae Rhizoma, a well-known botanical dietary supplement, has been the subject of intense interest due to its potential application for alleviating menopausal symptom. Although there are clinic data that the Cimicifuga extract should have hepatotoxicity, no evidence on the main chemical components has been reported. Cimicidol-3-O-β-d-xyloside (CX) is one of the main triterpenoids of the rhizome. This work studies the toxicological effects of CX after oral administration (50 mg kg−1 per day) over a 7-day period in female SD rats using metabonomic analyses of 1H NMR spectra of urine, serum and liver tissue extracts. Histopathological studies of liver and analyses of blood biochemical parameter, such as alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, blood urea nitrogen and creatinine revealed that CX had no negative impacts on liver and kidney. However, the metabolic signature of 1H NMR-based urinalysis of daily samples displayed an increment in the levels of taurine, trimethylamine-N-oxide (TMAO), betaine and acetate. Elevated serum levels of creatinine, glucose, alanine, TMAO and betaine and lower levels of lactate were observed. Metabolic profiling on aqueous soluble extracts of liver showed simultaneously increases in succinate, glycogen, choline, glycerophosphorylcholine, TMAO and betaine levels and reduction in valine, glucose and lactate levels. Nevertheless, no changes in any metabonomic level were found in lipid-soluble extracts of liver. These findings indicate that CX has a slight toxicity in liver and kidney via disturbance of the metabolisms of energy and amino acids. The present study provides a reasonable explanation for the clinical hepatotoxicity of Cimicifuga extract. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Zhang Shuang Deng, Dan Zhao, Yi Hu, Jian Xin Li, Kun Zou, Jun Zhi Wang
Chinese Chemical Letters 2012 Volume 23(Issue 3) pp:321-324
Publication Date(Web):March 2012
DOI:10.1016/j.cclet.2012.01.004
Sinomenine is a clinically available drug for the treatment of rheumatoid arthritis (RA). In a continuous research aiming at discovery of sinomenine derivates with better bioactivity, a cross-coupling reaction of sinomenine and 1,2-dihydroxybenzene catalyzed by a fungus Coriolus unicolor afforded an unique CC cross-coupled compound 2, together with (S)-disinomenine and (R)-disinomenine. The structure of 2 was elucidated by MS and NMR spectroscopy. Compound 2 was further assayed for the inhibitory activity on IL-6 overproduction in SW982 cells and exhibited a much more potent activity on IL-6 (96% inhibition) compared with those of (S)-disinomenine and sinomenine (17% and 12% inhibition, respectively).
Co-reporter:Lei Zhang;Zhe Gao;Chen Peng;Zheng-Yang Bin;Dan Zhao;Jing Wu
Molecular Diversity 2012 Volume 16( Issue 3) pp:579-590
Publication Date(Web):2012 August
DOI:10.1007/s11030-012-9390-1
An environmentally friendly and mild Bischler cyclization was developed to access quinazolines with diverse substitution. Based on this method, a library of 53 quinazoline derivatives was prepared and tested in vitro for cytotoxicity and inhibition on T-cell and B-cell proliferation. Compounds 6b, 7b, 17b, 33, and 35 showed higher inhibitory activity on both T-cell and B-cell proliferations, with IC50 values of 6.16, 6.30, 5.43, 2.54, and 9.80 μM on T-cell, respectively. All the tested compounds showed no obvious cytotoxicity at 10 μM concentration. The preliminary structure–activity relationship was concluded revealing that 4-position is the key modification site for potent quinazoline immunosuppressive agent.
Co-reporter:Peng Teng, Hai-Liang Liu, Lei Zhang, Li-Li Feng, Yue Huai, Zhang-Shuang Deng, Yang Sun, Qiang Xu, Jian-Xin Li
European Journal of Medicinal Chemistry 2012 50() pp: 63-74
Publication Date(Web):
DOI:10.1016/j.ejmech.2012.01.036
Co-reporter:Peng Teng, Hai-Liang Liu, Zhang-Shuang Deng, Zhi-Bing Shi, Yun-Mian He, Li-Li Feng, Qiang Xu, Jian-Xin Li
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 10) pp:3096-3104
Publication Date(Web):15 May 2011
DOI:10.1016/j.bmc.2011.04.006
Inhibition of the excessive NO production has been recognized as a potential means for the treatment of rheumatoid arthritis (RA). In order to discover more potent inhibitors and explore the preliminary structure activity relationship, a series of unique stereodimers of sinomenine analogues were designed and synthesized. Their inhibitory activity on NO production and cytotoxicity were evaluated using LPS-activated murine macrophages RAW264.7 assay and MTT method, respectively. Among these compounds, 1a, 2, 2a, 2b, and 4 showed potent inhibitory activity on NO production without obvious cytotoxicity. Furthermore, 2, 2a, and 2b significantly suppressed mRNA expression of iNOS. Interestingly, (S)-dimers displayed a better bioactivity than (R)-dimers. These compounds may sever as lead candidates in the development of novel therapeutic drugs for RA treatment.A series of unique stereodimers of sinomenine analogues were synthesized and evaluated on NO production and cytotoxicity. Dimer 2a and 2b showed the most potent inhibitory activity on NO production without obvious cytotoxicity and also significantly suppressed mRNA expression of iNOS. Interestingly, (S)-dimers displayed a better bioactivity than (R)-dimers.
Co-reporter:Guo-Biao Liu, Jian-Liang Xu, Mei Geng, Rui Xu, Rong-Rong Hui, Jian-Wei Zhao, Qiang Xu, Hong-Xi Xu, Jian-Xin Li
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 8) pp:2864-2871
Publication Date(Web):15 April 2010
DOI:10.1016/j.bmc.2010.03.020
A novel series of diphenolic chromone derivatives were synthesized and their inhibitory activity on nitric oxide (NO) production and cytotoxicity were evaluated using LPS-activated murine macrophages RAW264.7 assay and MTT method, respectively. Among these compounds, (5,7-dihydroxy-4-oxo-4H-chromen-3-yl) methyl esters (6b, 6c, 6f, 6g, and 6h) showed quite potent inhibitory activities with IC50 values of 2.20, 3.48, 0.35, 0.80, and 0.61 μM, respectively. The MTT results showed that all of the active compounds exhibited no cytotoxicity at the effective concentrations. The preliminary mechanism of the most potent compounds (6b, 6c, 6f, 6g, and 6h) was further examined based on the RT-PCR results and the compounds 6f, 6g, and 6h inhibited NO production by suppressing the expression of iNOS mRNA in a dose dependent manner. Furthermore, a computational analysis of physicochemical parameters revealed that the most of the compounds possessed drug-like properties.A series of diphenolic chromone derivatives were synthesized and their inhibitory activity on nitric oxide production was evaluated. Three of them exhibited quite potent inhibitory activity without obvious cytotoxicity.
Co-reporter:Jun-Feng Li, Yu Zhao, Min-Min Cai, Xiao-Fei Li, Jian-Xin Li
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 7) pp:2796-2806
Publication Date(Web):July 2009
DOI:10.1016/j.ejmech.2008.12.024
Oleanolic acid with anti-bone resorption effect was an active component discovered in a medicinal plant of Achyranthes bidentata. A series of heterocyclic derivatives of oleanolic acid including indole, pyrazine, quinoxaline, quinoline moieties and their natural amino acid amides were synthesized. Their inhibitory activity on the formation of osteoclast-like multinucleated cells (OCLs) and cytotoxicity of the selected derivatives were evaluated. Among the derivatives, compounds 2a and 8a displayed quite a potent activity even at 200 nM. The structure–activity relationships of the derivatives were also discussed.
Co-reporter:Guo-Biao Liu, Jian-Liang Xu, Cui-Cui He, Gong Chen, Qiang Xu, Hong-Xi Xu, Jian-Xin Li
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 15) pp:5433-5441
Publication Date(Web):1 August 2009
DOI:10.1016/j.bmc.2009.06.043
A series of quinoline derivatives were synthesized and their immunosuppressive activity and cytotoxicity were evaluated with a T-cell functional assay and MTT method, respectively. Most of 5,7-dimethoxyquinolin-4-yl ortho-substituted benzoate derivatives (18, 22, 24, 28, 31 and 37) showed a quite stronger inhibitory activity compared to other analogs. Among the synthesized compounds, 5,7-dimethoxyquinolin-4-yl 2,6-dichlorobenzoate (22) and 5,7-dimethoxyquinolin-4-yl 4-methylbenzenesulfonate (40) exhibited a potent inhibitory activity without significant cytotoxicity at 10 μM concentration. The preliminary mechanism of the active compounds 22 and 40 was further clarified based on the fluorescence activated cell sorter (FACS) assay, and the compounds exerted immunosuppressive activity via inhibiting the T cell activation in a dose dependent manner.
Co-reporter:Jun-Feng Li, Song-Jie Chen, Yu Zhao, Jian-Xin Li
Carbohydrate Research 2009 Volume 344(Issue 5) pp:599-605
Publication Date(Web):31 March 2009
DOI:10.1016/j.carres.2009.01.019
Oleanolic acid, a natural product, possesses an anti-osteoclast formation activity. Targeting at discovery of novel and potent anti-bone resorption agents, 22 glycosides of oleanolic acid derivatives (including d-galactopyranosides, d-glucopyranosides, d-xylopyranoses, d-arabopyranoses and d-glycuronic acids) were synthesized at phase-transfer-catalyzed conditions (K2CO3, Bu4NBr, CH2Cl2–H2O) and their inhibitory activity on the formation of osteoclast-like multinucleated cells (OCLs) induced by 1α, 25-dihydroxy vitamin D3 was evaluated in a co-culture assay system. The structure–activity relationships of these compounds were also discussed.
Co-reporter:Jian-Xin Li;Jun-Feng Li;Song-Jie Chen ;Zhi-Yong Yu
Chemistry & Biodiversity 2008 Volume 5( Issue 2) pp:
Publication Date(Web):
DOI:10.1002/cbdv.200890026
Abstract
Cimicidol-3-O-β-D-xyloside (1), one of the main components isolated from Cimicifugae rhizoma, is an active component with antiosteoporotic effect. The metabolism of 1 by rat intestinal bacteria was investigated, and two metabolites, 11β-hydroxycimigenol (2) and foetidinol (3), were isolated and characterized by spectroscopic means including two-dimensional NMR. Furthermore, the structures of six intermediates in the bacterial incubation were proposed on the basis of LC/MS analyses, and the possible metabolic pathway of the formation of 2, which passes through a unique sequence including hydrolysis, hydroxylation, and reduction, is discussed in detail.
Co-reporter:Yuan Zhang, Jian-xin Li, Jianwei Zhao, Shao-zhong Wang, Yi Pan, Ken Tanaka, Shigetoshi Kadota
Bioorganic & Medicinal Chemistry Letters 2005 Volume 15(Issue 6) pp:1629-1632
Publication Date(Web):15 March 2005
DOI:10.1016/j.bmcl.2005.01.061
Two series of oleanolic acid derivatives were synthesized and their inhibitory activity on the formation of osteoclast-like multinucleated cells (OCLs) induced by 1α,25-dihydroxy vitamin D3 was evaluated in a co-culture assay system. The structure–activity relationships, together with electronic structure based on the frontier molecular orbitals, for example, HOMO and LUMO, related to different amino acid substituents were studied. Derivatives with proline or phenylalanine showed a tendency to enhance the inhibitory activity.Two series of oleanolic acid derivatives were synthesized and their inhibitory activity on the formation of osteoclast-like multinucleated cells (OCLs) induced by 1α,25-dihydroxy vitamin D3 was evaluated in a co-culture assay system.
Co-reporter:Yu-Ren Zhou, Yang Zhao, Bei-Hua Bao, Jian-Xin Li
International Immunopharmacology (June 2015) Volume 26(Issue 2) pp:423-431
Publication Date(Web):1 June 2015
DOI:10.1016/j.intimp.2015.04.006
•SND-117 reduced arthritis, bone erosion and joint destruction in CIA mice.•SND-117 inhibited the serum levels of IL-1β, IL-6 and TNF-α in vivo.•SND-117 suppressed phosphorylation and nuclear translocation of NF-κB in fibroblast like synovial cells.•SND-117 may possess potential clinic benefits in rheumatoid arthritis management.Rheumatoid arthritis (RA) is a chronic, systemic inflammatory disorder that affects about 1% of the population worldwide. RA is mainly manifested by persistent synovitis and progressive joint destruction. The aim of the present study was to examine the anti-arthritis effects of SND-117, a sinomenine bivalent that is obtained from the structure modification of a clinically available anti-RA drug, sinomenine. The arthritis model (CIA) was established by immunizing DBA/1 mice with type II collagen, and the arthritis scores including inflammation, joint destruction and bone erosion were assessed after booster immunization for 3 weeks. The levels of cytokines such as IL-1β, IL-6 and TNF-α were analyzed by quantitative PCR and ELISA. The TNF-α induced NF-κB activation in fibroblast-like synovial cells (FLSCs) was analyzed by Western blot. SND-117 significantly relieved the inflammatory symptoms of collagen-induced arthritis, reduced bone erosion and joint destruction in CIA mice. The serum levels of IL-1β, IL-6 and TNF-α of CIA mice were markedly decreased by SND-117. SND-117 also strongly inhibited the phosphorylation and nuclear translocation of NF-κB p65 in FLSCs upon TNF-α stimulation. These data demonstrated that SND-117 could effectively block the pathogenesis of collagen-induced arthritis in CIA mice via inhibition of NF-κB signaling, and might provide potential clinic benefits in rheumatoid arthritis management.Download full-size image
Co-reporter:Hai-Jian Fu, Yang Zhao, Yu-Ren Zhou, Bei-Hua Bao, Yun Du, Jian-Xin Li
European Journal of Pharmaceutical Sciences (30 August 2015) Volume 76() pp:33-47
Publication Date(Web):30 August 2015
DOI:10.1016/j.ejps.2015.04.021
Tryptophan hydroxylase 1 (Tph-1) initiates the biosynthesis of peripheral serotonin. As peripheral serotonin suppresses bone formation, inhibitor of Tph-1 provides a useful tool to discover anabolic agents for osteoporosis. In the present study, series of ursolic acid (UA) derivatives were synthesized, and their inhibitory activity on serotonin biosynthesis and cytotoxicity were evaluated. Among the derivatives, 8d with potent inhibitory activity on serotonin was applied for further research. The data revealed that 8d significantly inhibited protein and mRNA expressions of Tph-1, and an SPR study indicated that 8d directly interacted to Tph-1 with a binding affinity of KD = 15.09 μM. Oral administration of 8d significantly prevented bone loss via suppressing serotonin biosynthesis without estrogenic side-effects in ovariectomized (OVX) rats.Graphical abstractSeries of ursolic acid derivatives were synthesized, and their inhibitory activity on serotonin was screened. Compound 8d suppressed Tph-1 and prevented bone loss in an anabolic fashion.Download high-res image (178KB)Download full-size image
Co-reporter:J.X. Li, J. Liu, C.C. He, Z.Y. Yu, Y. Du, S. Kadota, H. Seto
Maturitas (20 September 2007) Volume 58(Issue 1) pp:59-69
Publication Date(Web):20 September 2007
DOI:10.1016/j.maturitas.2007.06.001
ObjectiveIncreasing research suggested that Cimicifugae rhizoma might be protective against osteoporosis. In this study, we investigated the effects of three cycloartane-type triterpenoids isolated from Cimicifugae rhizoma, cimicidol-3-O-β-d-xyloside (1), cimicidanol-3-O-β-d-xyloside (2) and acetylacteol-3-O-β-d-xyloside (3) on bone resorption in vitro and bone loss in ovariectomized (OVX) mice.MethodsThe activities of the tested compounds on bone resorption were evaluated using three assays, neonatal mouse parietal bone organ culture, osteoclast-like cells (OCLs) formation and pit formation. The effects on bone mineral density (BMD) and uterine weight were examined using OVX mice. Using LC–MS/MS method, the serum concentrations of the triterpenoids were measured in mice serum collected at 0.5, 1, 3, 6 and 12 h following its oral administration.ResultsAll of the tested compounds exerted the inhibitory effects on bone resorption in bone organ culture, suppressed both of the formation and the resorbing activity of OCLs. Furthermore, a synergistic effect was observed among those compounds. In vivo studies revealed that compounds 1–3 and the mixture of compounds 1–3 prevented the bone loss in OVX mice without affecting uterine weight, and each compound was detected in the mice serum after single oral administration.ConclusionsThe triterpenoids exerted the inhibitory effects on osteoclastic bone resorption through the suppression of both OCLs formation and the resorbing activity of OCLs, and also showed a significant protective effect on BMD in OVX mice. The present results might provide a new pharmacological potential for the treatment of osteoporosis.
Co-reporter:Lei Zhang, Chen Peng, Dan Zhao, Yue Wang, Hai-Jian Fu, Qi Shen and Jian-Xin Li
Chemical Communications 2012 - vol. 48(Issue 47) pp:NaN5930-5930
Publication Date(Web):2012/04/23
DOI:10.1039/C2CC32009F
Base-switched methylenation and formylation using tetramethylethylenediamine (TMEDA) as a carbon source have been achieved under mild conditions, catalyzed by CuCl2, with atmospheric oxygen as oxidant. Bisindolylmethanes, diphenylmethanes and 3-formylindoles were synthesized with excellent regioselectivity and good yield.
Co-reporter:Dan Zhao, Min-Xue Zhu, Yue Wang, Qi Shen and Jian-Xin Li
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 37) pp:NaN6249-6249
Publication Date(Web):2013/08/02
DOI:10.1039/C3OB41488D
An efficient and selective Pd(0)-catalyzed sp3 C–H bond arylation–oxidation of 4-methylquinazolines is reported. The method enables the introduction of arylketone at the benzylic position of 4-methylquinazolines without the use of an additional directing group, and atmospheric oxygen is used as the sole oxidant.
Co-reporter:Dan Zhao, Qi Shen, Yu-Ren Zhou and Jian-Xin Li
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 35) pp:NaN5912-5912
Publication Date(Web):2013/07/15
DOI:10.1039/C3OB41083H
A facile alkenylation of quinazolines with unactivated terminal alkynes has been achieved in the presence of KOtBu without the aid of any transition metal catalysts. The reaction is carried out under very mild conditions and shows a high stereoselectivity. A possible radical-based mechanism is also explored.
Co-reporter:Dan Zhao, Teng Wang, Qi Shen and Jian-Xin Li
Chemical Communications 2014 - vol. 50(Issue 33) pp:NaN4304-4304
Publication Date(Web):2014/03/03
DOI:10.1039/C4CC01444H
An n-Bu4NI catalyzed domino reaction that involves selective dual amination of sp3 C–H bonds has been developed. The protocol affords a facile and efficient approach to the synthesis of imidazo[1,5-c]quinazolines under mild conditions.
Co-reporter:Dan Zhao, Teng Wang and Jian-Xin Li
Chemical Communications 2014 - vol. 50(Issue 49) pp:NaN6474-6474
Publication Date(Web):2014/05/02
DOI:10.1039/C4CC02648A
A novel metal-free synthesis of quinazolinones via dual amination of sp3 C–H bonds was developed. The sp3 carbon in methylarenes or adjacent to a heteroatom in DMSO, DMF or DMA was used as the one carbon synthon.

![OLEANA-2,12-DIENO[2,3-D]ISOXAZOL-28-OIC ACID](http://img.cochemist.com/ccimg/218700/218600-48-7.png)
![OLEANA-2,12-DIENO[2,3-D]ISOXAZOL-28-OIC ACID](http://img.cochemist.com/ccimg/218700/218600-48-7_b.png)




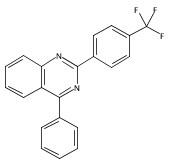
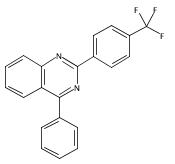













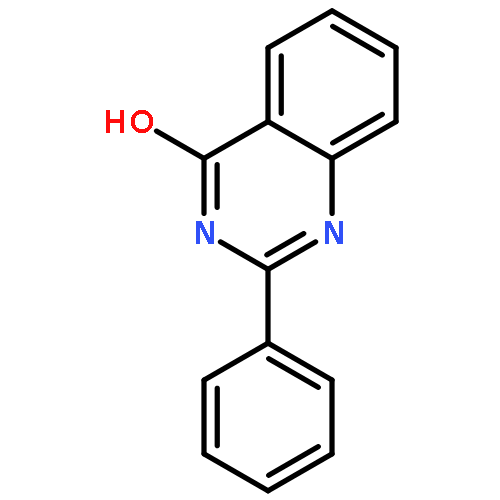
![4(1H)-Quinazolinone, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/83900/83800-83-3.png)
![4(1H)-Quinazolinone, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/83900/83800-83-3_b.png)





![4(3H)-Quinazolinone, 2-[4-(1,1-dimethylethyl)phenyl]-](/data/chemimg/1120600/59455-93-5.png)
![4(3H)-Quinazolinone, 2-[4-(1,1-dimethylethyl)phenyl]-](/data/chemimg/1120600/59455-93-5_b.png)




![[2-(4-methoxy-phenyl)-ethyl]-methyl-amine Hydrochloride](http://img.cochemist.com/ccimg/35900/35803-88-4.png)
![[2-(4-methoxy-phenyl)-ethyl]-methyl-amine Hydrochloride](http://img.cochemist.com/ccimg/35900/35803-88-4_b.png)
![8-METHYLIMIDAZO[1,2-A]PYRIDINE-2-CARBALDEHYDE](http://img.cochemist.com/ccimg/34800/34790-24-4.png)
![8-METHYLIMIDAZO[1,2-A]PYRIDINE-2-CARBALDEHYDE](http://img.cochemist.com/ccimg/34800/34790-24-4_b.png)























![Benzamide,2-[(phenylmethyl)amino]-](http://img.cochemist.com/ccimg/5400/5363-37-1.png)
![Benzamide,2-[(phenylmethyl)amino]-](http://img.cochemist.com/ccimg/5400/5363-37-1_b.png)










![Benzene, [(1,1-dimethylethoxy)methyl]-](http://img.cochemist.com/ccimg/3500/3459-80-1.png)
![Benzene, [(1,1-dimethylethoxy)methyl]-](http://img.cochemist.com/ccimg/3500/3459-80-1_b.png)




















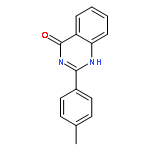
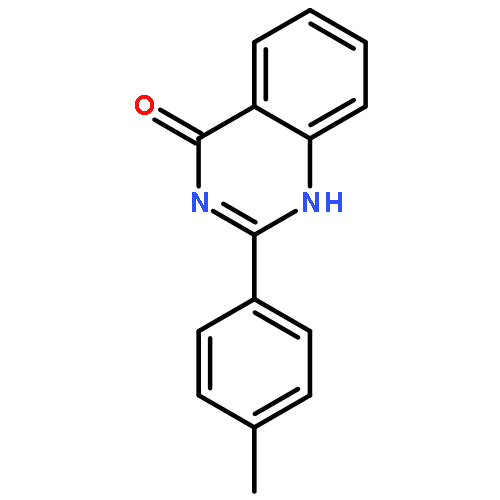




![1-(2,4-DIMETHYLPHENYL)-1H-PYRAZOLO[3,4-D]PYRIMIDIN-4-OL](http://img.cochemist.com/ccimg/7500/7455-77-8.png)
![1-(2,4-DIMETHYLPHENYL)-1H-PYRAZOLO[3,4-D]PYRIMIDIN-4-OL](http://img.cochemist.com/ccimg/7500/7455-77-8_b.png)