Co-reporter:Gengjia Chen, Meng Xu, Shuang Zhao, Jing Sun, Qianqian Yu, and Jie Liu
ACS Applied Materials & Interfaces October 4, 2017 Volume 9(Issue 39) pp:33645-33645
Publication Date(Web):September 12, 2017
DOI:10.1021/acsami.7b10553
Even though numerous therapeutic methods exist for cancer treatment, many fail to achieve ideal outcomes or have severe side effects. Here, we describe a theranostic nanocarrier system with improved tumor vasculature detection and tumor margin quantification that increases the accuracy and guidance efficiency of phototherapy. Novel pompon-like RuNPs with superb photothermal properties and high encapsulation efficiency were first synthesized via the polyol reducing method. Based on these RuNPs, we then developed a multifunctional theranostic system, pRu-pNIPAM@RBT, composed of poly(N-isopropylacrylamide) as the thermal-response switch and of [Ru(bpy)2(tip)]2+ as the photosensitizer of PDT and the contrast agent of biomedical imaging. We demonstrate that the pRu-pNIPAM@RBT can generate intracellular hyperthermia and reactive oxygen species (ROS) for simultaneous photothermal therapy (PTT) and photodynamic therapy (PDT) by laser activation. In contrast to other studies, our work highlights the integration of quantitative analysis of infrared thermal imaging and PA imaging data, which can distinguish between tumor and healthy tissues and guide the destructive but precise phototherapy and decrease nonspecific tissue injury. Considering the excellent in vivo antitumor phototherapeutic effects, this strategy may help preclinical researchers gain insight into theoretical as well as practical aspects of precision cancer therapy.Keywords: drug controlled release; photoacoustic imaging; phototherapy; ruthenium nanoparticles; theranostic nanomedicine;
Co-reporter:Na Huang, Xu Chen, Xufeng Zhu, Mengmeng Xu, Jie Liu
Biomaterials 2017 Volume 141(Volume 141) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.biomaterials.2017.07.005
Bacterial infection has been a threat to human health, and so early diagnosis and treatment of bacterial infection is an urgent problem that needs to be solved. In this work, a multifunctional theranostic selenium nanoplatform (Se@PEP-Ru NPs) with early imaging diagnosis and efficient treatment of bacterial infections was designed and constructed. First, the antibacterial peptide UBI29-41 (PEP) was linked to functionalized Selenium nanoparticles (NPs), which enhanced the stability of the antimicrobial peptide and also caused the nanocomposites to specifically target bacterial infection. Ruthenium complexes with good antibacterial activity and fluorescence properties were then coated on to their outer layers. It was worth mentioning that, when the resulting nanoprobe was injected into mice by intravenous injection it was found to be sensitive to sites of bacterial infection for selective fluorescence imaging and targeted therapy. Thus, it can be used to distinguish between bacterial infection, inflammation, and tumor-induced tissue infection with high specificity. In the further antibacterial activity experiments, Ruthenium complexes showed synergistic antimicrobial activity with Se NPs, which indicated that the antibacterial activity of Se@PEP-Ru NPs was the strongest that could promote wound healing. Thus, Se@PEP-Ru NPs appears to be a promising antimicrobial with good biocompatibility, excellent selectivity, and potent antimicrobial activity.
Co-reporter:Qianqian Yu;Jing Sun;Xufeng Zhu;Lin Qiu;Mengmeng Xu;Sirun Liu;Jianming Ouyang
Journal of Materials Chemistry B 2017 vol. 5(Issue 30) pp:6081-6096
Publication Date(Web):2017/08/02
DOI:10.1039/C7TB01035D
Photodynamic therapy (PDT), by producing reactive oxygen species (ROS), inhibits cancer cells and is an emerging and pioneering cancer therapeutic modality which can eliminate some of the drawbacks of other traditional anticancer therapies. To combine near-infrared (NIR) mediated PDT, chemotherapy and gene therapy in a synergistic manner, a novel NIR light activated photosensitizer for PDT was designed based on TiO2-coated Fe3O4 nanoparticle core/shell nanocarriers (Fe3O4@TiO2@mTiO2). The chemotherapeutic drug doxorubicin hydrochloride (DOX) was conjugated to the surface of the TiO2 mesopores through pH-reversible hydrazone bond linking and β-catenin siRNA was loaded in the mesopores. Fe3O4@TiO2@mTiO2–DOX/siRNA delivery systems have features and functions of magnetic targeting, fluorescence imaging, MRI diagnosis, and combination therapy through the simultaneous maneuvering of a magnet and NIR-mediated PDT. In vitro, Fe3O4@TiO2@mTiO2–DOX/siRNA effectively silences β-catenin gene, induces tumor cell apoptosis and consequently significantly enhances the cancer suppression effect of the synergistic therapeutic agent. Meanwhile, under NIR irradiation, excess ROS produced can further trigger tumor cell apoptosis. In vivo investigation confirmed that Fe3O4@TiO2@mTiO2–DOX/siRNA exhibited high tumor targeted specificity through MRI and fluorescence imaging, and optimal anti-tumor efficacy. The results verified its significant therapeutic effects on tumors by combination therapy consisting of magnetic targeting and NIR-mediated PDT.
Co-reporter:Licong Yang;Jing Sun;Wenjie Xie;Yanan Liu
Journal of Materials Chemistry B 2017 vol. 5(Issue 30) pp:5954-5967
Publication Date(Web):2017/08/02
DOI:10.1039/C6TB02952C
Inhibition of amyloid β (Aβ) aggregation holds considerable promise as a therapeutic strategy for Alzheimer's disease (AD). However, successful inhibition is hard to achieve due to the blood–brain barrier (BBB) and the non-selective distribution of drugs. Herein, two targeting peptides (LPFFD and TGN) were conjugated to selenium nanoparticles (SeNPs). We found that the concentration ratio of LPFFD to TGN taken as 1 : 1 could form the most effective dual-functional SeNPs (L1T1–SeNPs) for inhibiting Aβ aggregation and crossing the BBB. L1T1–SeNPs can cross the BBB and have a strong affinity toward Aβ species, and thus, they can efficiently suppress extracellular Aβ fibrillation by disrupting hydrophobic and electrostatic interactions that are important for Aβ40 nucleation. Also, L1T1–SeNPs can suppress the Aβ40 fiber mediated generation of reactive oxygen species (ROS) and their corresponding neurotoxicity in PC12 cells. In addition, L1T1–SeNPs exert synergistic effects on the inhibition of Aβ aggregation and cross the BBB efficiently. Collectively, these results demonstrate that dual-functional SeNPs might be a valuable targeting system for inhibiting Aβ aggregation.
Co-reporter:Shuang Zhao;Mengmeng Xu;Chengwen Cao;Qianqian Yu;Yanhui Zhou
Journal of Materials Chemistry B 2017 vol. 5(Issue 33) pp:6908-6919
Publication Date(Web):2017/08/23
DOI:10.1039/C7TB00613F
Co-delivery of gene and drug therapies for cancer treatment remains a major goal of nanocarrier research. In this study, mesoporous silica nanoparticles (MSNs) were used to co-deliver siRNA and doxorubicin (Dox) for redox-controlled release. The present nanocarrier (MSNs-SS-siRNA@Dox) has mesoporous silica cores that can be loaded with Dox, while siRNA connects to the core surface by disulfide linkage and plays a gatekeeper role. Disulfide linkages were also utilized to target intracellular GSH, and their cleavage led to the release of Dox and siRNA. Release of siRNA and Dox was correlated with GSH concentrations, and rapid release at 10 mM GSH reflected reductive cleavage of intermediate disulfide linkages. Subsequent experiments using an in vitro Dox delivery and release assay indicated that MSNs-SS-siRNA@Dox significantly enhanced the accumulation of Dox in cells compared with that after treatment with free Dox. Moreover, MSNs-SS-siRNA@Dox has sufficient efficiency to knock down target protein expression. More importantly, MSNs-SS-siRNA@Dox displayed great potential for tumor targeting and achieved satisfactory therapeutic effects on tumor growth inhibition in vivo. In summary, the present nanoparticles may provide an effective strategy for the design and development of controlled release and co-delivery of siRNA and drugs for cancer therapy.
Co-reporter:Qingchang Chen, Meng Xu, Wenjing Zheng, Taoyuan Xu, Hong Deng, and Jie Liu
ACS Applied Materials & Interfaces 2017 Volume 9(Issue 8) pp:
Publication Date(Web):February 13, 2017
DOI:10.1021/acsami.6b12792
We report here a novel and personalized strategy of selenium/ruthenium nanoparticles modified metal organic frameworks MIL-101(Fe) for delivering pooled small interfering RNAs (siRNAs) to enhance therapy efficacy by silencing multidrug resistance (MDR) genes and interfere with microtubule (MT) dynamics in MCF-7/T (Taxol-resistance) cell. The existence of coordinatively unsaturated metal sites in MIL-101(Fe) can strongly interact with the electron-rich functional groups of cysteine, which can be regarded as the linkage between selenium/ruthenium nanoparticles and MIL-101(Fe). Se@MIL-101 and Ru@MIL-101 loaded with MDR gene-silencing siRNAs via surface coordination can significantly enhance protection of siRNAs against nuclease degradation, increase siRNA cellular uptake, and promote siRNA escape from endosomes/lysosome to silence MDR genes in MCF-7/T cell, resulting in enhanced cytotoxicity through the induction of apoptosis with the signaling pathways of phosphorylation of p53, MAPK, and PI3K/Akt and the dynamic instability of MTs and disrupting normal mitotic spindle formation. Furthermore, in vivo investigation of the nanoparticles on nude mice bearing MCF-7/T cancer xenografts confirmed that Se@MIL-101-(P+V)siRNA nanoparticles can significantly enhance cancer therapeutic efficacy and decrease systemic toxicity in vivo.Keywords: MOF surface modifications; multidrug resistance cancer therapy; selenium/ruthenium nanoparticle; siRNA delivery systems; tubulin polymerization;
Co-reporter:Shuang Zhao, Qianqian Yu, Jiali Pan, Yanhui Zhou, ... Jie Liu
Acta Biomaterialia 2017 Volume 54(Volume 54) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.actbio.2017.02.042
To reduce the side effects and enhance the anti-tumor activities of anticancer drugs in the clinic, the use of nano mesoporous materials, with mesoporous silica (MSN) being the best-studied, has become an effective method of drug delivery. In this study, we successfully synthesized mesoporous selenium (MSe) nanoparticles and first introduced them to the field of drug delivery. Loading MSe with doxorubicin (DOX) is mainly driven by the physical adsorption mechanism of the mesopores, and our results demonstrated that MSe could synergistically enhance the antitumor activity of DOX. Coating the surface of MSe@DOX with Human serum albumin (HSA) generated a unique redox-responsive nanoparticle (HSA-MSe@DOX) that demonstrated glutathione-dependent drug release, increased tumor-targeting effects and enhanced cellular uptake throug nanoparticle interact with SPARC in MCF-7 cells. In vitro, HSA-MSe@DOX prominently induced cancer cell toxicity by synergistically enhancing the effects of MSe and DOX. Moreover, HSA-MSe@DOX possessed tumor-targeting abilities in tumor-bearing nude mice and not only decreased the side effects associated with DOX, but also enhanced its antitumor activity. Therefore, HSA-MSe@DOX is a promising new drug that warrants further evaluation in the treatments of tumors.Statement of significanceTo reduce the side effects and enhance the anti-tumor activities of anticancer drugs, we successfully synthesized mesoporous selenium (MSe) nanoparticles and first introduced them to the field of drug delivery. Loading MSe with doxorubicin (DOX) is mainly driven by the physical adsorption mechanism of the mesopores. Coating the surface of MSe@DOX with Human serum albumin (HSA) generated a unique redox-responsive nanoparticle (HSA-MSe@DOX) that demonstrated glutathione-dependent drug release, increased tumor-targeting effects and enhanced cellular uptake throug nanoparticle interact with SPARC in MCF-7 cells. In vitro and in vivo, HSA-MSe@DOX possessed tumor-targeting abilities and not only decreased the side effects associated with DOX, but also enhanced its antitumor activity. Therefore, HSA-MSe@DOX is a promising new drug that warrants further evaluation in the treatments of tumors.Download high-res image (80KB)Download full-size image
Co-reporter:Ying Liu, Na Huang, Yunfei Yu, Chuping Zheng, Ning Deng and Jie Liu
Journal of Materials Chemistry A 2016 vol. 4(Issue 28) pp:4941-4941
Publication Date(Web):04 Jul 2016
DOI:10.1039/C6TB90091G
Correction for ‘Bioactive SiO2@Ru nanoparticles for osteogenic differentiation of mesenchymal stem cells via activation of Akt signaling pathways’ by Ying Liu et al., J. Mater. Chem. B, 2016, DOI: 10.1039/c5tb01898f.
Co-reporter:Ying Liu, Na Huang, Yunfei Yu, Chuping Zheng, Ning Deng and Jie Liu
Journal of Materials Chemistry A 2016 vol. 4(Issue 25) pp:4389-4401
Publication Date(Web):21 Dec 2015
DOI:10.1039/C5TB01898F
The surface chemistry of materials has an interactive influence on cell behavior. It is now well established that surface chemistry can affect cell adhesion, proliferation, and differentiation. Although amino (NH2)-terminated surfaces generated by the modification of nanoparticles with silane can promote osteogenic differentiation of mesenchymal stem cells (MSCs), how silica surfaces with ruthenium nanoparticles (SiO2@Ru) act on MSCs remains largely unknown. A concentration of 5 μg mL−1 aminopropyltriethoxysilane (APTS)-modified SiO2 nanoparticles (SiO2–NH2) or SiO2@Ru was nontoxic to MSCs, based on MTT and apoptosis assays. In addition, SiO2–NH2 and SiO2@Ru did not affect the surface phenotype or morphology of MSCs. SiO2@Ru can be used to trigger the differentiation of MSCs into osteocytes, minimising the need for exogenous biological supplementation. TEM images revealed that SiO2@Ru might interact with proteins located in the cytoplasm, which would have a further impact on subsequent cellular signaling pathways. Activation of Akt signaling pathways was observed in MSCs cultured with SiO2@Ru and these enhancement effects could be blocked by the Akt inhibitor LY294002. SiO2@Ru exhibited in vitro osteocompatibility that surpassed that of SiO2–NH2, as well as supporting the proliferation and differentiation of MSCs. This demonstrates the potential of SiO2@Ru for use in bone regeneration.
Co-reporter:Yanhui Zhou, Qianqian Yu, Xiuying Qin, Dhairya Bhavsar, Licong Yang, Qingchang Chen, Wenjing Zheng, Lanmei Chen, and Jie Liu
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 24) pp:15000-15012
Publication Date(Web):May 27, 2015
DOI:10.1021/acsami.5b02261
Functionalization can promote the uptake of nanoparticles into cancer cells via receptor-mediated endocytosis, enabling them to exert their therapeutic effects. In this paper, epigallocatechin gallate (EGCG), which has a high binding affinity to 67 kDa laminin receptor (67LR) overexpressed in HCC cells, was employed in the present study to functionalized ruthenium nanoparticles (RuNPs) loaded with luminescent ruthenium complexes to achieve antiliver cancer efficacy. [Ru(bpy)2(4-B)] (ClO4)2·2H2O (RuBB)-loaded EGCG-RuNPs (bpy = 2,2'-bipyridine) showed small particle size with narrow distribution, better stability, and high selectivity between liver cancer and normal cells. The internalization of RuBB-loaded EGCG-RuNPs was inhibited by 67LR-blocking antibody or laminin, suggesting that 67LR-mediated endocytosis played an important role in the uptake into HCC cells. Moreover, transmission electron microscopy and confocal microscopic images showed that RuBB-loaded EGCG-RuNPs accumulated in the cytoplasm of SMMC-7721 cells. Furthermore, our results indicated that the EGCG-functionalized nanoparticles displayed enhanced anticancer effects in a target-specific manner. Concentrations of RuBB-loaded EGCG-RuNPs, nontoxic in normal L-02 cells, showed direct reactive oxygen species-dependent cytotoxic, pro-apoptotic, and anti-invasive effects in SMMC-7721 cells. Furthermore, in vivo animal study demonstrated that RuBB-loaded EGCG-RuNPs possessed high antitumor efficacy on tumor-bearing nude mice. It is encouraging to conclude that the multifunctional RuNPs may form the basis of new strategies on the treatment of liver cancer and other malignancies.
Co-reporter:Tiantian Yin, Wenjie Xie, Jing Sun, Licong Yang, and Jie Liu
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 30) pp:19291
Publication Date(Web):July 14, 2016
DOI:10.1021/acsami.6b05089
The structural changes of amyloid-beta (Aβ) from nontoxic monomers into neurotoxic aggregates are implicated with pathogenesis of Alzheimer’s disease (AD). Over the past decades, weak disaggregation ability and low permeability to the blood–brain barrier (BBB) may be the main obstacles for major Aβ aggregation blockers. Here, we synthesized penetratin (Pen) peptide loaded poly(ethylene glycol) (PEG)-stabilized gold nanostars (AuNS) modified with ruthenium complex (Ru@Pen@PEG-AuNS), and Ru(II) complex as luminescent probes for tracking drug delivery. We revealed that Ru@Pen@PEG-AuNS could obviously inhibit the formation of Aβ fibrils as well as dissociate preformed fibrous Aβ under the irradiation of near-infrared (NIR) due to the NIR absorption characteristic of AuNS. More importantly, this novel design could be applied in medicine as an appropriate nanovehicle, being highly biocompatible and hemocompatible. In addition, Ru@Pen@PEG-AuNS had excellent neuroprotective effect on the Aβ-induced cellular toxicity by applying NIR irradiation. Meanwhile, Pen peptide could effectively improve the delivery of nanoparticles to the brain in vitro and in vivo, which overcame the major limitation of Aβ aggregation blockers. These consequences illustrated that the enhanced BBB permeability and efficient photothermolysis of Ru@Pen@PEG-AuNS are promising agents in AD therapy.Keywords: Alzheimer’s disease; amyloid-beta; blood−brain barrier; gold nanostars; penetratin peptide
Co-reporter:Licong Yang, Tiantian Yin, Yanan Liu, Jing Sun, Yanhui Zhou, Jie Liu
Acta Biomaterialia 2016 Volume 46() pp:177-190
Publication Date(Web):December 2016
DOI:10.1016/j.actbio.2016.09.010
Abstract
Metal ions promote Alzheimer’s disease (AD) pathogenesis by accelerating amyloid-β (Aβ) aggregation and inducing formation of neurotoxic reactive oxygen species (ROS) such as hydrogen peroxide (H2O2). Although metal chelators can block these effects, their therapeutic potential is marred by their inability to cross the blood-brain barrier (BBB) and by their non-specific interactions with metal ions necessary for normal cellular processes, which could result in adverse side effects. To overcome these limitations, we created a novel gold nanoparticle-capped mesoporous silica (MSN-AuNPs) based H2O2-responsive controlled release system for targeted delivery of the metal chelator CQ. In this system, CQ is released only upon exposure to conditions in which H2O2 levels are high, such as those in Aβ plaques. The conjugation of AuNPs on the surface of MSN did not affect their ability to cross the BBB. The AuNPs also help in decrease the Aβ self-assembly, due to this, MSN-CQ-AuNPs were more efficient than MSN-CQ in inhibiting Cu2+-induced Aβ40 aggregation. Furthermore, MSN-CQ-AuNPs reduced the cell membrane disruption, microtubular defects and ROS-mediated apoptosis induced by Aβ40-Cu2+ complexes. The high BBB permeability, efficient anti-Aβ aggregation, and good biocompatibility of MSN-CQ-AuNPs, together with the specific conditions necessary for its release of CQ, demonstrate its potential for future biomedical applications.
Statement of Significance
Due to the low ability to cross the blood-brain barrier (BBB) and non-specific interactions with metal ions necessary for normal cellular processes of metal chelator or Aβ inhibitors, we created a novel gold nanoparticle-capped mesoporous silica (MSN-AuNPs)-based H2O2-responsive controlled release system for targeted delivery of the metal chelator CQ and AuNPs (Aβ inhibitor). In this system, CQ and AuNPs are released only upon exposure to conditions in which H2O2 levels are high, such as those in Aβ plaques. The AuNPs on the surface of MSN also help in decrease the Aβ self-assembly, due to this, MSN-CQ-AuNPs were more efficient than MSN-CQ in inhibiting Cu2+-induced Aβ40 aggregation. Furthermore, MSN-CQ-AuNPs reduced the cell membrane disruption, microtubular defects and ROS-mediated apoptosis induced by Aβ40-Cu2+ complexes. Our data suggest that this controlled release system may have widespread application in the field of medicine for Alzheimer’s disease.
Co-reporter:Wenjing Zheng, Tiantian Yin, Qingchang Chen, Xiuying Qin, Xiaoquan Huang, Shuang Zhao, Taoyuan Xu, Lanmei Chen, Jie Liu
Acta Biomaterialia 2016 Volume 31() pp:197-210
Publication Date(Web):February 2016
DOI:10.1016/j.actbio.2015.11.041
Abstract
Drug resistance mediated by P-glycoprotein (P-gp) and class III β-tubulin (β-tubulin III) is a major barrier in microtubule-targeting cancer chemotherapy. In this study, layered double hydroxide nanoparticles (LDHs) were employed to simultaneously deliver selenium (Se) and pooled small interfering RNAs (siRNAs) to achieve therapeutic efficacy. LDH-supported Se nanoparticles (Se@LDH) were compacted with siRNAs (anti-P-gp and anti-β-tubulin III) via electrostatic interactions, which could protect siRNA from degradation. Se@LDH showed excellent abilities to deliver siRNA into cells, including enhancing siRNA internalization, and promoting siRNA escape from endosomes. siRNA transfection experiments further confirmed a higher gene silencing efficiency of Se@LDH than LDH. Interestingly, we found Se@LDH may be a microtubule (MT) stabilizing agent which could inhibit cell proliferation by blocking cell cycle at G2/M phase, disrupting normal mitotic spindle formation and inducing cell apoptosis. When complexed with different specific siRNAs, Se@LDH/siRNA nanoparticles, especially the Se@LDH-pooled siRNAs, exhibit an efficient gene-silencing effect that significantly downregulate the expression of P-gp and β-tubulin III. Moreover, Se@LDH-pooled siRNAs could induce cell apoptosis, change cell morphology and increase cellular ROS levels through change the expression of Bcl-2/Bax, activation of caspase-3, PI3K/AKT/mTOR and MAPK/ERK pathways. These results suggested that co-delivery of Se and pooled siRNAs may be a promising strategy for overcoming the drug resistance mediated by P-gp and β-tubulin III in drug-resistant breast cancers.
Co-reporter:Xin-Yuan Sun, Jian-Ming Ouyang, Poonam Bhadja, Qin Gui, Hua Peng, and Jie Liu
Journal of Agricultural and Food Chemistry 2016 Volume 64(Issue 42) pp:7911-7920
Publication Date(Web):October 5, 2016
DOI:10.1021/acs.jafc.6b03323
This study aimed to investigate the protective effects of degraded soybean polysaccharides (DSP) on oxidatively damaged African green monkey kidney epithelial (Vero) cells. Low DSP concentration (10 μg/mL) elicited an evident protective effect on H2O2-induced cell injury (0.3 mmol/L). The cell viabilities of the H2O2-treated group and the DSP-protected group were 57.3 and 93.1%, respectively. The cell viability decreased to 88.3% when the dosage was increased to 100 μg/mL. DSP protected Vero cells from H2O2-mediated oxidative damage by enhancing cellular superoxide dismutase activity and total antioxidant capacity and by decreasing malonaldehyde content and lactate dehydrogenase release. The H2O2-treated cells stimulated the aggregation of calcium oxalate monohydrate crystals. DSP could also reduce the crystal size, decrease the attached crystal content, and prevent the cell aggregation by alleviating oxidative injury and lipid peroxidation, enhancing antioxidant capacity, and decreasing hyaluronan expression on cellular surfaces. The internalization ability of the injured cells was improved after these cells were exposed to DSP solution. The regulation ability of DSP-repaired cells on calcium oxalate dihydrate formation, crystal attachment, aggregation, and internalization was lower than that of normal cells but was higher than that of the injured cells. DSP may be a potential green drug to prevent calcium oxalate (CaOx) stone formation because DSP could protect cells from oxidative damage and inhibit CaOx crystal formation.Keywords: CaOx crystallization; cell protection; crystal attachment and internalization; oxidative damage; soybean polysaccharide;
Co-reporter:Xiaonian Zhang, Zhujuan Huang, Shaofeng Wu, Ruishan Lin, Jie Liu, Ning Su
Inorganic Chemistry Communications 2016 Volume 72() pp:1-6
Publication Date(Web):October 2016
DOI:10.1016/j.inoche.2016.07.018
•Δ/Λ-OMe exhibited broad inhibition on human cancer cells, Λ-OMe show better anti-tumor potency than that of Δ-OMe in MGC-803.•Λ-OMe can inhibit telomerase activity by altering the expression levels of the telomere-associated proteins, TRF1 and TRF2.•Λ-OMe can arrest the cell cycle at the G0/G1 phase by increasing the expression of proliferation-related gene P21.•Λ-OMe altered the Bax/Bcl-2 ratio and triggered apoptosis, which occurred in a mitochondria-dependent manner.There has been a vast increase in telomerase inhibition research over the past several years, which was demonstrated as an attractive anti-tumor strategy. Our previous study found that the chiral ruthenium complex, [Ru(phen)2p-MOPIP]2 + (Phen = 1,10-phenanthroline, p-MOPIP = 2-(4-methoxyphenyl)-imidazo[4,5f] Markman (2003), Janaratne et al. (2007) phenanthroline) (dl-OMe) and its enantiomer Δ/Λ-[Ru(phen)2p-MOPIP]2 + (Δ/Λ-OMe) could bind to and stabilize G-quadruplex DNA structure in telomeres, and inhibit telomerase activity. In this study, cytotoxic activity of these Ru complexes was studied by MTT assay. The anti-tumor mechanisms of Λ-OMe were investigated using TRAP assay, Western blot analysis, flow cytometry, Hochest staining, and RT-PCR. Results showed that among several Ru complexes, Λ-OMe demonstrated a better anti-tumor activity against gastric cancer cell line (MGC-803), and had less effect on normal gastric epithelial cell. Λ-OMe effectively inhibited the cell growth by inhibiting cellular telomerase activity, triggering cell cycle arrest, and inducing apoptosis of MGC-803 cells. The inhibitory effect on telomerase activity was associated with the altered expression of telomere-related proteins TRF1 and TRF2. Cell-cycle arrest was associated with increased levels of P21 mRNA. Apoptosis of MGC-803 cell was triggered by modulating the expression of apoptosis-related genes Bax, Bcl-2, and caspase-3. Overall, the results suggest that Λ-OMe may be a new promising agent for human gastric cancer therapy.The chiral ruthenium complex Λ-[Ru(phen)2p-MOPIP]2 +(Λ-OMe) can significantly inhibit the proliferation of human gastric cancer MGC-803 cells by inhibiting telomerase activity, arresting cell cycle at the G0/G1 phase, altering the bax/bcl-2 ratio and triggering apoptosis.
Co-reporter:Yanhui Zhou, Meng Xu, Yanan Liu, Yan Bai, Yuqian Deng, Jie Liu, Lanmei Chen
Colloids and Surfaces B: Biointerfaces 2016 Volume 144() pp:118-124
Publication Date(Web):1 August 2016
DOI:10.1016/j.colsurfb.2016.04.004
•The green synthesis GA-Se/RuNPs (60 nm) has been achieved using GA as both a reducing and a capping agent.•GA-Se/RuNPs effectively inhibited the growth of HeLa cells.•GA-Se/RuNPs also effectively inhibited migration and invasion in HeLa cells.Methods for the synthesis of nanoparticles (NPs) for biomedical applications ideally involve the use of nontoxic reducing and capping agents, and more importantly, enable control over the shape and size of the particles. As such, we used gallic acid (GA) as both a reducing and a capping agent in a simple and “green” synthesis of stable Se/Rualloy NPs (GA-Se/RuNPs). The diameter and morphology of the Se/Ru alloy NPs were regulated by GA concentration, and the presence of Ru was found to be a key factor in regulating and controlling the size of GA-Se/RuNPs. Moreover, GA-Se/RuNPs suppressed HeLa cell proliferation through the induction of apoptosis at concentrations that were nontoxic in normal cells. Furthermore, GA-Se/RuNPs effectively inhibited migration and invasion in HeLa cells via the inhibition of MMP-2 and MMP-9 proteins. Our findings confirm that bimetallic (Se/Ru) NPs prepared via GA-mediated synthesis exhibit enhanced anticancer effects.Bimetallic (Se/Ru) NPs prepared via GA-mediated synthesis effectively inhibited migration and invasion in HeLa cells.
Co-reporter:Dongdong Sun, Nuan Li, Weiwei Zhang, Zhiwei Zhao, Zhipeng Mou, Donghui Huang, Jie Liu, Weiyun Wang
Colloids and Surfaces B: Biointerfaces 2016 Volume 148() pp:116-129
Publication Date(Web):1 December 2016
DOI:10.1016/j.colsurfb.2016.08.052
•The PLGA@QT NPs effectively inhibited Zn2+-induced Aβ fibrils formation and toxicity.•That injection of PLGA@QT NPs into AD mice ameliorated cognition and memory impairments.•The H&E study unambiguously show negligible toxicity to the mice.Dysfunctional interaction of amyloid-β (Aβ) with excess metal ions is proved to be related to the etiology of Alzheimer’s disease (AD). Hence, disruption of these metal-peptide interactions using nanoparticles (NPs) holds considerable promise as a therapeutic strategy to combat this incurable disease. Given that quercetin is a natural product, the biocompatibility and small size essential for permeating the blood-brain barrier make it a potential therapeutic drug candidate for treating AD. Nanocarriers formulated with the US Food and Drug Administration-approved biocompatible and biodegradable polymer PLGA are being widely explored for the controlled delivery of therapeutic drugs, proteins, peptides, oligonucleotides, and genes. With this background, the present study was undertaken to investigate the effects of PLGA-functionalized quercetin (PLGA@QT) NPs on inhibited and disassembled Aβ42 fibrils and the PLGA@QT NPs have low cytotoxicity when tested on SH-SY5Y cells in vitro. As expected, the cytotoxicity studies of the PLGA@QT NPs led to a concentration-related behaviour on the SH-SY5Y human neuroblastoma cells. And, it has demonstrated that PLGA@QT NPs can inhibit the neurotoxicity of Zn2+-Aβ42 system and enhance the viability of neuron cells. The results from behavioral tests indicate that injection of PLGA@QT NPs into APP/PS1 mice ameliorate cognition and memory impairments. Most encouragingly, the in vivo systemic toxicity of PLGA@QT NPs examined by histological analysis in major organs did not show any signs of adverse effect to mice. Thus, the prepared quercetin based nanoscale drug delivery carrier efficiently enhanced the therapeutic index and reduced the side effects. Our findings are highly encouraging, providing substantial evidence of the safety of PLGA@QT NPs for biomedical application. We expect these findings will be relevant for other NPs for treatment of AD and have broad implications in NP-based studies and applications.In this study, we designed PLGA-functionalized quercetin (PLGA@QT) NPs as Aβ42 aggregation inhibitor. Using a combination of in vitro and in vivo experiments, we have shown that PLGA@QT NPs are able to reduce toxic Zn2+-induced Aβ42 aggregates, improved inhibition efficacy, and reduced cytotoxicity. Most importantly, the amelioration of Aβ-induced spatial learning and memory impairment by PLGA@QT NPs could be linked, at least in part, suggesting that the PLGA@QT NPs may be a potential candidate for AD treatment.
Co-reporter:Xianbo Zhou, Jing Sun, Tiantian Yin, Fangling Le, Licong Yang, Yanan Liu and Jie Liu
Journal of Materials Chemistry A 2015 vol. 3(Issue 39) pp:7764-7774
Publication Date(Web):03 Sep 2015
DOI:10.1039/C5TB00731C
Chiral molecules, which selectively target and inhibit amyloid β-peptide (Aβ) aggregation, have potential use as therapeutic agents for the treatment of Alzheimer's disease (AD). Here we use cysteine enantiomer-modified SeNPs (abbreviated as D/LSeNPs) to demonstrate that surface chirality strongly influences the formation of Aβ aggregates in the presence of metal ions, such as Zn2+ or Cu2+. The two chiral molecule modified nanoparticles could inhibit the formation of Aβ fibrils by binding Aβ, thus blocking the formation of Aβ fibrils and blocking the metal binding sites. Of the two enantiomers, D/SeNPs appear to more effectively inhibit Aβ fibril formation due to a greater affinity for Aβ than that of L/SeNPs. Additionally, D/LSeNPs appeared to reduce Aβ and metal ion-induced neurotoxicity. Treatment with D/LSeNPs can also decrease the levels of intracellular reactive oxygen species, and stabilize the mitochondrial membrane potential. Of the two enantiomers, D/SeNPs were more effective in protecting the cells than L/SeNPs, and this could be due to D/SeNPs being selectively absorbed by PC12 cells, maintaining cellular redox potentials, and protecting cells against oxidative stress to a greater extent than L/SeNPs. From these results, it appears that chiral molecules can bring novel insight into better drug treatment designs for Alzheimer's disease.
Co-reporter:Xianbo Zhou, Chengwen Cao, Qingchang Chen, Qianqian Yu, Yanan Liu, Tiantian Yin and Jie Liu
Journal of Materials Chemistry A 2015 vol. 3(Issue 35) pp:7055-7067
Publication Date(Web):12 Aug 2015
DOI:10.1039/C5TB00487J
Human islet amyloid polypeptide (hIAPP) was found as amyloid aggregate deposits in the pancreatic islets of patients with type-2 diabetes and studies showed that insulin and its derivatives were the potent inhibitors of hIAPP aggregation. However, several emerging therapies with this goal showed limited success due to the instability and inefficiency of insulin derivatives. Nanosized graphene oxide (nGO) possesses high stability and affinity toward aromatic rings. In this study, an insulin-derived peptide, EALYLV, was stabilized by loading on nGO@PEG to inhibit aggregation and hIAPP-induced cytotoxicity. The results showed that nGO@PEG@EALYLV (abbreviated as nGO@PEG@E) can effectively inhibit the aggregation of hIAPP via electrostatic adsorption and specific binding to the active sites of hIAPP. We further evaluated the protective effect of nGO@PEG@E on INS-1 cells in the presence of hIAPP. Treatment with nGO@PEG@E could significantly elevate the viability of INS-1 cells, decrease the level of intracellular reactive oxygen species, and stabilize mitochondrial membrane potential. All the results indicated that nGO@PEG@E could inhibit the aggregation of hIAPP, which reduces its cytotoxicity.
Co-reporter:Xu Chen, Xiaoquan Huang, Chuping Zheng, Yanan Liu, Taoyuan Xu and Jie Liu
Journal of Materials Chemistry A 2015 vol. 3(Issue 35) pp:7020-7029
Publication Date(Web):20 May 2015
DOI:10.1039/C5TB00280J
Graphene oxide (GO) has attracted great interest in many different areas, as a delivery vehicle for antibacterial agents, and has shown high potential. Although silver nanoparticles (AgNPs) have a strong antibacterial effect, the biological application of AgNPs is often hindered by their aggregation and low stability. In this study, we developed an approach of polyoxyethylene bis(amine) (PEG) directed AgNPs grown on GO, then we combined the two materials to prepare a series of functionalized GO bearing different sized AgNPs, and studied the size effects of AgNPs on growth inhibition of Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus). We evaluated the antibacterial effect of GO@PEG@AgNPs on E. coli and S. aureus by various methods such as minimum inhibitory concentration (MIC) experiment, cell wall/membrane integrity assay and scanning electron microscopy (SEM) characterisation of bacterial morphology. The GO@PEG@AgNPs composites exhibited markedly higher antibacterial efficacy than AgNPs alone. The smallest GO@PEG@AgNPs (10 nm) particularly demonstrated higher antibacterial activity than other sizes (30, 50, and 80 nm). We believe that these findings contribute to great potential application as a regulated graphene-based antibacterial solution.
Co-reporter:Wenjing Zheng, Chengwen Cao, Yanan Liu, Qianqian Yu, Chuping Zheng, Dongdong Sun, Xiaofan Ren, Jie Liu
Acta Biomaterialia 2015 Volume 11() pp:368-380
Publication Date(Web):1 January 2015
DOI:10.1016/j.actbio.2014.08.035
Abstract
Multidrug resistance (MDR) is a major barrier against effective cancer treatment. Dual-delivering a therapeutic small interfering RNA (siRNA) and chemotherapeutic agents has been developed to reverse drug resistance in tumor cells. In this study, amine-terminated generation 5 polyamidoamine (PAMAM) dendrimers (G5.NH2)-modified selenium nanoparticles (G5@Se NP) were synthesized for the systemic dual-delivery of mdr1 siRNA and cisplatin (cis-diamminedichloroplatinum-(II), DDP), which was demonstrated to enhance siRNA loading, releasing efficiency and gene-silencing efficacy. When the mdr1 siRNA was conjugated with G5@Se NP via electrostatic interaction, a significant down-regulation of P-glycoprotein and multidrug resistance-associated protein expression was observed; G5@Se-DDP-siRNA arrested A549/DDP cells at G1 phase and led to enhanced cytotoxicity in A549/DDP cells through induction of apoptosis involving the AKT and ERK signaling pathways. Interestingly, G5@Se-DDP NP were much less reactive than DDP in the reactions with both MT and GSH, indicating that loading of DDP in a nano-delivery system could effectively prevent cell detoxification. Furthermore, animal studies demonstrated that the new delivery system of G5@Se-DDP-siRNA significantly enhanced the anti-tumor effect on tumor-bearing nude mice, with no appreciable abnormality in the major organs. These results suggest that G5@Se NP could be a potential platform to combine chemotherapy and gene therapy technology in the treatment of human disease.
Co-reporter:Tiantian Yin, Licong Yang, Yanan Liu, Xianbo Zhou, Jing Sun, Jie Liu
Acta Biomaterialia 2015 Volume 25() pp:172-183
Publication Date(Web):1 October 2015
DOI:10.1016/j.actbio.2015.06.035
Abstract
The blood–brain barrier (BBB) is a formidable gatekeeper toward exogenous substances, playing an important role in brain homeostasis and maintaining a healthy microenvironment for complex neuronal activities. However, it also greatly hinders drug permeability into the brain and limits the management of brain diseases. The development of new drugs that show improved transport across the BBB represents a promising strategy for Alzheimer’s disease (AD) intervention. Whereas, previous study of receptor-mediated endogenous BBB transport systems has focused on a strategy of using transferrin to facilitate brain drug delivery system, a system that still suffers from limitations including synthesis procedure, stability and immunological response. In the present study, we synthetised sialic acid (SA)-modified selenium (Se) nanoparticles conjugated with an alternative peptide-B6 peptide (B6-SA-SeNPs, a synthetic selenoprotein analogue), which shows high permeability across the BBB and has the potential to serve as a novel nanomedicine for disease modification in AD. Laser-scanning confocal microscopy, flow cytometry analysis and inductively coupled plasma-atomic emission spectroscopy ICP-AES revealed high cellular uptake of B6-SA-SeNPs by cerebral endothelial cells (bEnd.3). The transport efficiency of B6-SA-SeNPs was evaluated in a Transwell experiment based on in vitro BBB model. It provided direct evidence for B6-SA-SeNPs crossing the BBB and being absorbed by PC12 cells. Moreover, inhibitory effects of B6-SA-SeNPs on amyloid-β peptide (Aβ) fibrillation could be demonstrated in PC12 cells and bEnd3 cells. B6-SA-SeNPs could not only effectively inhibit Aβ aggregation but could disaggregate preformed Aβ fibrils into non-toxic amorphous oligomers. These results suggested that B6-SA-SeNPs may provide a promising platform, particularly for the application of nanoparticles in the treatment of brain diseases.
Statement of Significance
Alzheimer’s disease (AD) is the world’s most common form of dementia characterized by intracellular neurofibrillary tangles in the brain. Over the past decades, the blood–brain barrier (BBB) limits access of therapeutic or diagnostic agents into the brain, which greatly hinders the development of new drugs for treating AD. In this work, we evaluated the efficiency of B6-SA-SeNPs across BBB and investigated the interactions between B6-SA-SeNPs and amyloid-β peptide (Aβ). We confirm that B6-SA-SeNPs could provide a promising platform because of its high brain delivery efficiency, anti-amyloid properties and anti-oxidant properties, which may serve as a novel nanomedicine for the application in the treatment of brain diseases.
Co-reporter:Qingchang Chen, Qianqian Yu, Yanan Liu, Dhairya Bhavsar, Licong Yang, Xiaofan Ren, Dongdong Sun, Wenjing Zheng, Jie Liu, Lan-mei Chen
Nanomedicine: Nanotechnology, Biology and Medicine 2015 Volume 11(Issue 7) pp:1773-1784
Publication Date(Web):October 2015
DOI:10.1016/j.nano.2015.04.011
Herein, chiral selenium nanoparticles (L-SeNPs/D-SeNPs) modified with a dinuclear Ruthenium (II) complex were used to effectively deliver siRNA targeting the MDR1 gene. In this co-delivery system, the luminescent dinuclear Ruthenium (II) complex was developed to act as a gene carrier and anti-tumor drug, while offering luminescent imaging to follow the intracellular trafficking. Interestingly, Ru@L-SeNPs exhibited a stronger protein and pDNA affinity than Ru@D-SeNPs, indicating that chirality may have an effect on pDNA/siRNA binding and biocompatibility. Cisplatin-resistant A549R cells treated with Ru@L-SeNPs-siRNA demonstrated significant downregulation of P-glycoprotein (P-gp) expression, resulting in unprecedented enhanced cytotoxicity through the induction of apoptosis with the involvement of phosphorylation of p53, MAPK and PI3K/Akt signaling pathways. In vivo investigation confirmed that Ru@L-SeNPs-siRNA nanoparticles exhibited high tumor-targeted fluorescence, enhanced anti-tumor efficacy, and decreased systemic toxicity. These results suggest that Ru@L-SeNPs are promising vectors for the delivery of siRNA and for real-time tracking of treatment.From the Clinical EditorIn this study, the authors designed bi-functional selenium nanoparticles with specific chirality to deliver siRNA, for targeting tumor MDR1 gene. The underlying ruthenium (II) complex could also offer fluorescence for real-time imaging. This new system has been shown to have enhanced efficacy against drug resistant tumor cells in both in-vitro and in-vivo experiments.Ru@L-SeNPs-siRNA showed more active cellular uptake and anti-tumour effect in vitro/in vivo.
Co-reporter:Chuping Zheng;Jinsheng Wang;Yanan Liu;Qianqian Yu;Ying Liu;Ning Deng
Advanced Functional Materials 2014 Volume 24( Issue 43) pp:6872-6883
Publication Date(Web):
DOI:10.1002/adfm.201401263
Stem cells have generated a great deal of excitement in tissue engineering and regenerative medicine, and it is important to understand the interaction mechanisms between nanomaterials and mesenchymal stem cells (MSCs) for biomedical applications. In this study, ruthenium (II) functional selenium nanoparticles (Ru@Se) are used for stem cell research. Specifically, Ru@Se are compared with citric acid selenium nanoparticles (Cit@Se)to identify their effects on MSCs differentiation and associated molecular mechanism. These data suggest that the effective adsorbing abilities of Ru@Se and Cit@Se allow them to act as preconcentration materials for osteogenic chemical inducers, which accelerates MSCs differentiation into osteoblasts. Further results suggest that selenium nanoparticles enhance the differentiation of MSCs toward osteogenic lineage over adipocytes by promoting osteogenic transcription and attenuating adipogenic transcription. Ru@Se and Cit@Se exert these effects by activating Smad-dependent BMP signaling pathway, which regulates the expression of relevant genes to induce osteogenic differentiation.
Co-reporter:Licong Yang, Qingchang Chen, Ying Liu, Jingnan Zhang, Dongdong Sun, Yanhui Zhou and Jie Liu
Journal of Materials Chemistry A 2014 vol. 2(Issue 14) pp:1977-1987
Publication Date(Web):26 Feb 2014
DOI:10.1039/C3TB21586E
Amyloid β (Aβ) aggregates are considered as possible targets for therapy of Alzheimer's disease (AD). Metal ions play an important role in amyloid aggregation and neurotoxicity in the AD pathogenesis. Disruption of the interactions between these metal ions and peptides holds considerable promise as a therapeutic strategy for AD treatment. In this study, L-Cys-modified Se/Ru nanoparticles (NPs) have been designed as Aβ-binding units to inhibit metal-induced Aβ aggregation. L-Cys was used as both the reducing agent and surface modifier in the formation of SeNPs, RuNPs and Se/RuNPs. We found that RuNPs and Se/RuNPs have a strong affinity toward Aβ species and efficiently suppress extracellular Aβ40 self-assembly and Zn2+-induced fibrillization. Also, Se/RuNPs can suppress the Zn2+–Aβ40 mediated generation of reactive oxygen species (ROS) and their corresponding neurotoxicity in PC12 cells. Intriguingly, SeNPs do not have the same ability as Se/RuNPs. In addition, Se/RuNPs also decrease intracellular Aβ40 fibrillization, but this process does not involve the lysosomal pathway. These results suggest that ruthenium significantly enhances the activity of Se/RuNPs binding to Aβ40. This interaction would block the Zn2+ binding to Aβ40 peptides and lower the concentration of the free monomer, thus decreasing fibrillization. Owing to this, Se/RuNPs may represent a new strategy in AD treatment.
Co-reporter:Jingnan Zhang, Xianbo Zhou, Qianqian Yu, Licong Yang, Dongdong Sun, Yanhui Zhou, and Jie Liu
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 11) pp:8475
Publication Date(Web):April 23, 2014
DOI:10.1021/am501341u
Alzheimer’s disease (AD), the most common neurodegenerative disease, is caused by an accumulation of amyloid-β (Aβ) plaque deposits in the brains. Evidence is increasingly showing that epigallocatechin-3-gallate (EGCG) can partly protect cells from Aβ-mediated neurotoxicity by inhibiting Aβ aggregation. In order to better understand the process of Aβ aggregation and amyloid fibril disaggregation and reduce the cytotoxicity of EGCG at high doses, we attached EGCG onto the surface of selenium nanoparticles (EGCG@Se). Given the low delivery efficiency of EGCG@Se to the targeted cells and the involvement of selenoprotein in antioxidation and neuroprotection, which are the key factors for preventing the onset and progression of AD, we synthesized EGCG-stabilized selenium nanoparticles coated with Tet-1 peptide (Tet-1-EGCG@Se, a synthetic selenoprotein analogue), considering the affinity of Tet-1 peptide to neurons. We revealed that Tet-1-EGCG@Se can effectively inhibit Aβ fibrillation and disaggregate preformed Aβ fibrils into nontoxic aggregates. In addition, we found that both EGCG@Se and Tet-1-EGCG@Se can label Aβ fibrils with a high affinity, and Tet-1 peptides can significantly enhance the cellular uptake of Tet-1-EGCG@Se in PC12 cells rather than in NIH/3T3 cells.Keywords: amyloid; epigallocatechin-3-gallate; fibril; oligomer; selenium nanoparticles;
Co-reporter:Licong Yang, Jingnan Zhang, Chuan Wang, Xiuying Qin, Qianqian Yu, Yanhui Zhou and Jie Liu
Metallomics 2014 vol. 6(Issue 3) pp:518-531
Publication Date(Web):16 Dec 2013
DOI:10.1039/C3MT00237C
Angiogenesis is crucial for tumor growth. Thus, inhibiting angiogenesis represents a promising avenue for preventing tumor growth. This study investigated the anti-angiogenesis and anti-tumor effects of 8-hydroxyquinoline ruthenium(II) complexes [Ru(bpy)2(8-HQ)]+ (BQ) and [Ru(phen)2(8-HQ)]+ (PQ). The results showed that both compounds, especially PQ, suppressed the proliferation, migration, invasion, tube formation and microvessel growth of endothelial cells in vitro. PQ also inhibited tumor growth of human hepatocellular liver carcinoma cells (HepG2) in a mouse xenograft tumor model in vivo. To understand the mechanisms of how ruthenium(II) complexes disrupt bFGF-induced angiogenesis and tumor growth, we have shown that (1) both compounds can interfere with the binding of bFGF to its cell surface receptors, thereby suppressing activation of bFGF-mediated signaling cascades; (2) PQ can induce tumor cell apoptosis. These effects might inhibit angiogenesis and tumor cell proliferation in tumor tissue. Taken together, our findings reveal that 8-hydroxyquinoline ruthenium(II) complexes are specific inhibitors of bFGF-mediated angiogenesis, and may be a viable drug candidate in anti-angiogenesis and anti-tumor therapies.
Co-reporter:Qianqian Yu, Yanan Liu, Lei Xu, Chuping Zheng, Fangling Le, Xiuying Qin, Yanyu Liu, Jie Liu
European Journal of Medicinal Chemistry 2014 Volume 82() pp:82-95
Publication Date(Web):23 July 2014
DOI:10.1016/j.ejmech.2014.05.040
•We present two novel Ru(II) complexes with hydrophobic ancillary ligands RPD and RBD.•RPD enter the HeLa cells through non-endocytotic, but energy-dependent mechanism.•RPD induces HeLa cell apoptosis through mitochondria-mediated pathway.•RPD lock a telomeric DNA into a G-quadruplex conformation.Studies have shown that ruthenium complexes have relatively strong anticancer activity, cell uptake of drugs have a crucial impact on the pharmacological activity, using autofluorescence of ruthenium complexes could effectively track cancer cells and drug distribution, transport accurately in real time. In this work, we present the synthesis and detailed characterization of two novel Ru(II) complexes with hydrophobic ancillary ligands, namely [Ru(bpy)2(5-idip)]2+ (RBD) and [Ru(phen)2(5-idip)]2+ (RPD) (5-idip = 2-indole-[4,5-f][1,10]phenanthroline). We have shown that RPD can enter the HeLa cells efficiently through non-endocytotic, but energy-dependent mechanism and first accumulated in lysosomes, and then escape from the lysosomes and localize within the nuclei, efficiently lead to the inhibition of DNA transcription and translation and induced cell apoptosis. Further studies on the mechanism of apoptosis in HeLa cells demonstrate that RPD is able to induce mitochondria-mediated apoptosis in HeLa cells through activation of initiator caspase-9 and down-stream effector caspase-3 and -7 and cleavage of PARP. We have also demonstrated that RPD bind to telomeric G-quadruplex DNA effectively and selectively, together with increased p21 and p16 expression. Our findings suggest that RPD induces HeLa cell apoptosis through mitochondria-mediated pathway and inhibition of telomerase activity. RPD may be a candidate for further evaluation as a chemotherapeutic agent for human cancers.Two Ruthenium(II) polypyridyl complexes exhibit anticancer activity by mitochondria mediated apoptosis and telomerase inhibition.
Co-reporter:Jingnan Zhang, Qianqian Yu, Qian Li, Licong Yang, Lanmei Chen, Yanhui Zhou, Jie Liu
Journal of Inorganic Biochemistry 2014 Volume 134() pp:1-11
Publication Date(Web):May 2014
DOI:10.1016/j.jinorgbio.2013.12.005
•[Ru(ip)3](ClO4)2·2H2O is capable of selectively binding to bcl-2 G-quadruplexes.•[Ru(ip)3](ClO4)2·2H2O can well induce the formations of antiparallel G-quadruplex.•Overlarge ligands of Ru-complexes reduce the induction of bcl-2 G-quadruplexes.•[Ru(ip)3](ClO4)2·2H2O can enter the nuclei of HeLa cells and induce cell apoptosis.Two ruthenium(II) complexes (Ru-complexes) were synthesized and characterized in this study. The selectivity and ability of the complexes to interact with bcl-2 DNA were investigated here. It turned out that [Ru(ip)3](ClO4)2·2H2O (complex 1, ip = 1H-iminazole [4,5-f][1,10] phenanthroline) could induce and stabilize the formations of G-quadruplexes more effectively than [Ru(pip)3](ClO4)2·2H2O (complex 2, pip = 2-phenylimidazo-[4,5-f][1,10]phenanthroline) did. Considering the important role of the Ru-complex ligand in inducing and stabilizing the formations of G-quadruplex in our previous studies, we speculate that the overlarge ligand of complex 2 may block its binding affinity for G-quadruplexes. Complex 1 also induced cell apoptosis in in vitro assays. In general, this study provided potentially important information for further development of the Ru-complexes as good inducers and stabilizers of bcl-2 G-quadruplex DNA for cancer treatment.[Ru(ip)3](ClO4)2·2H2O (complex 1) stabilizes the combination of bcl-2 G-quadruplex to form the antiparallel conformation and causes cell apoptosis mediated by caspase activation
Co-reporter:Ying Liu, Yanan Liu, Licong Yang, Chengwen Cao, Yanhui Zhou and Jie Liu
MedChemComm 2014 vol. 5(Issue 11) pp:1724-1728
Publication Date(Web):15 Sep 2014
DOI:10.1039/C4MD00201F
In this study, two similar ruthenium(II) complexes 1 and 2 were synthesized to explore their selectivity for binding loop isomers of the c-myc G-quadruplex. The results show that both complexes can efficiently bind the c-myc G-quadruplex DNA, although complex 2 exhibited higher binding affinity. Studies of G-quadruplex loop isomers revealed that the c-myc G-quadruplexes on complexes 1 and 2 may contribute to the selectivity of 1:2:1 loop isomers as well as the high affinity of complex 2. This work also found that complex 2 significantly inhibited HeLa cell proliferation and was considerably less toxic to normal cells (Hs-68). Flow cytometric analysis showed that complex 2 induced cell apoptosis by mitochondrial pathways and inhibited the generation of intracellular reactive oxygen species (ROS).
Co-reporter:Dongdong Sun, Yanan Liu, Qianqian Yu, Xiuying Qin, Licong Yang, Yanhui Zhou, Lanmei Chen, Jie Liu
Biomaterials 2014 35(5) pp: 1572-1583
Publication Date(Web):
DOI:10.1016/j.biomaterials.2013.11.007
Co-reporter:Qian Li, Jingnan Zhang, Licong Yang, Qianqian Yu, Qingchang Chen, Xiuying Qin, Fangling Le, Qianling Zhang, Jie Liu
Journal of Inorganic Biochemistry 2014 130() pp: 122-129
Publication Date(Web):
DOI:10.1016/j.jinorgbio.2013.10.006
Co-reporter:Dr. Xiu-Ying Qin;Dr. Ya-Nan Liu;Dr. Qian-Qian Yu;Dr. Li-Cong Yang;Ying Liu;Dr. Yan-Hui Zhou;Dr. Jie Liu
ChemMedChem 2014 Volume 9( Issue 8) pp:1665-1671
Publication Date(Web):
DOI:10.1002/cmdc.201402060
Abstract
A novel copper(II) complex with mixed ligands including β-[(3-formyl-5-methyl-2-hydroxy-benzylidene)amino]propionic acid anion and 1,10′-phenanthroline was synthesized, and its crystal structure was thoroughly characterized. It exerted excellent inducing apoptosis, anti-angiogenesis and antiproliferative properties in vitro. The complex can bind human serum albumin (HSA) at physiological pH conditions. Remarkably, it can induce formation of the mixed parallel/antiparallel G-quadruplex structures in the G-rich sequence of the proximal vascular endothelial growth factor (VEGF) promoter, and stabilize these G-quadruplex structures, which provide an opportunity for anti-angiogenesis chemotherapeutics. Furthermore, the complex showed a strong uptake, and exhibited multiple anticancer functions by inhibiting the expression of p-Akt and p-Erk1/2 proteins and by upregulating the levels of reactive oxygen species (ROS). Because of the reported results, this new copper(II) complex qualifies itself as a potential anticancer drug candidate.
Co-reporter:Lijun Wang;Chuping Zheng;Yanyu Liu;Fangling Le
Biological Trace Element Research 2014 Volume 157( Issue 2) pp:175-182
Publication Date(Web):2014 February
DOI:10.1007/s12011-013-9869-3
A new Ru(II)–Se complex, Ru(bpy)2L2Cl2 (bpy = 2,2′-bipyridine, L = 1,10-phenanthrolineselenazole) (Ru-Se) has been synthesized and characterized. The G-quadruplex DNA-binding properties of the complex and its selenium ligand (Phen-Se) were evaluated by thermal denaturation study, polymerase chain reaction (PCR) stop assay, and telomerase repeat amplification protocol (TRAP). The results showed that the obtained complex could induce and stabilize G-quadruplex structure as well as exhibit potent inhibitory activity against telomerase. In vitro cytotoxicity studies showed that complex Ru-Se inhibited the cancer cell growth through apoptosis. However, the presence of the ligand Phen-Se did not appear to have a significant effect either on G-quadruplex binding or on biological activity. Furthermore, the cell migration assay and the tube formation assay also demonstrated that the complex Ru-Se significantly inhibited human umbilical vascular endothelial cell (HUVEC) proliferation, migration, and tube formation. These findings indicate that the Ru–Se complex may be a potential telomerase inhibitor and a viable drug candidate in antiangiogenesis for anticancer therapies.
Co-reporter:Qianqian Yu, Yanyu Liu, Jingnan Zhang, Fang Yang, Dongdong Sun, Du Liu, Yanhui Zhou and Jie Liu
Metallomics 2013 vol. 5(Issue 3) pp:222-231
Publication Date(Web):11 Jan 2013
DOI:10.1039/C3MT20214C
Two ruthenium(II) polypyridyl complexes [Ru(phen)2(4idip)](ClO4)2 (1) and [Ru(bpy)2(4idip)](ClO4)2 (2) (phen = 1,10-phenanthroline, bpy = 2,2′-bipyridine, 4idip = 4-indoleimidazo[4,5-f][1,10]phenanthroline) designed as telomeric G-quadruplex ligands have been synthesized and characterized. The interaction of human telomeric G-quadruplex DNA (HTG21) with the designed ligands was explored by fluorescence analysis, absorption spectroscopy, continuous variation, circular dichroism spectroscopy, fluorescence resonance energy transfer (FRET) melting assay, polymerase chain reaction (PCR) stop assay, telomerase repeat amplification protocol (TRAP), and visual studies. The results showed that both complexes could induce and stabilize different G-quadruplex structures by using a 1:1 [quadruplex]/[complex] binding mode ratio. Complex 1 exhibited higher interaction ability and better G-quadruplex selectivity than duplex DNA. Furthermore, both ruthenium complexes led to the inhibition of the enzyme telomerase, and complex 1 was the significantly better inhibitor.
Co-reporter:Xiu Ying Qin, Li Cong Yang, Fang Ling Le, Qian Qian Yu, Dong Dong Sun, Ya Nan Liu and Jie Liu
Dalton Transactions 2013 vol. 42(Issue 41) pp:14681-14684
Publication Date(Web):23 Aug 2013
DOI:10.1039/C3DT51548F
The crystals of two binuclear copper-based complexes were obtained. One complex can remarkably induce apoptosis and inhibit angiogenesis to mediate tumour growth at a greater extent. Furthermore, this complex showed a strong energy-dependent and non-endocytotic uptake and exhibited multiple anti-cancer functions by inhibiting the expressions of p-Akt and p-Erk1/2 proteins and by decreasing the levels of reactive oxygen species.
Co-reporter:Yu Xia, Qingchang Chen, Xiuying Qin, Dongdong Sun, Jingnan Zhang and Jie Liu
New Journal of Chemistry 2013 vol. 37(Issue 11) pp:3706-3715
Publication Date(Web):22 Aug 2013
DOI:10.1039/C3NJ00542A
Three ruthenium(II) complexes [Ru(bpy)2(biim)]2+ (1), [Ru(phen)2(biim)]2+ (2) and [Ru(p-mopip)2-(biim)]2+ (3) (where bpy is 2,2′-bipyridine, phen is 1,10-phenanthroline, biim is 2,2′-bisimidazole and p-mopip is 2-(4-methoxyphenyl)-imidazo-[4,5f]phenanthroline), have been synthesized and characterized. The interactions of human telomeric DNA oligomers 5′-G3(T2AG3)3-3′ (HTG21) with ruthenium(II) complexes were investigated via UV-vis, fluorescence resonance energy transfer (FRET) melting assay, polymerase chain reaction (PCR) stop assay, and circular dichroism (CD) measurements. The results indicated that the three ruthenium(II) complexes could stabilize the formation of human telomeric G-quadruplex DNA, and complex 2 was found to be the most efficient. In vitro cytotoxicity assay by MTT also showed that complex 2 was superior to complexes 1 and 3 in inhibiting the growth of cancer cells. Telomeric repeat amplification protocol (TRAP) showed that complexes 2 and 3 led to an inhibition of the telomerase activity, and complex 2 was the significantly better inhibitor. Flow cytometric analysis and evaluation of mitochondrial membrane potential demonstrated that complex 2 inhibited the growth of HeLa cells through induction of apoptotic cell death, as evidenced by the depletion of mitochondrial membrane potential in HeLa cells.
Co-reporter:Fangling Le, Dongdong Sun, Du Liu, Chuping Zheng, Ying Liu, Jie Liu
Inorganic Chemistry Communications 2013 Volume 38() pp:20-27
Publication Date(Web):December 2013
DOI:10.1016/j.inoche.2013.09.060
•Ni(II) complexes have the capability of binding to stabilize G-quadruplex.•Ni(II) complexes can be considered as a new class of highly selective G-quadruplex.•Complex NPH showed a greater ability to induce the stability of G-quadruplex DNA.•Complexes NBH and NPH possessed the greatest inhibitory selectivity against cancer cell lines and display application potential as anticancer agents.In the present investigation, four nickel (II) complexes [Ni(bpy)3]2 + NB, [Ni(phen)3]2 + NP, [Ni(bpy)2(p-ipip)]2 + NBH and [Ni(phen)2(p-ipip)]2 + NPH were synthesized and characterized by electrospray ionization-mass spectrometry, where phen is 1,10-phenanthroline, bpy is 2,2′-bipyridine and p-ipip is 2-(4-indole)-imidazo[4,5f][1,10] phenanthroline. The interactions of human telomeric G-quadruplex DNA with these designed complexes were evaluated by CD spectroscopy, fluorescence resonance energy transfer (FRET) melting assay and FRET competitive binding experiment. The experimental evidence indicated that all complexes could strongly bind to and effectively stabilize the telomeric G-quadruplex DNA. Complex NPH was a better G-quadruplex binder than other complexes. Furthermore, polymerase chain reaction (PCR)-stop assay, gel mobility shift assay and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay demonstrated that complex NPH not only can stabilize dimers forms of the G-quadruplex at low concentrations but also high inhibitory selectivity against cancer cells. The results suggest that complex NPH may be a potential telomerase inhibitor for cancer chemotherapy.The scheme is the illustration of the Ni(II) complex NPH which is able to induce the HTG21 DNA into a mixed-type or hybrid G-quadruple and dependently stabilize the G-quadruplex.
Co-reporter:Dongdong Sun, Yanan Liu, Qianqian Yu, Yanhui Zhou, Rong Zhang, Xiaojia Chen, An Hong, Jie Liu
Biomaterials 2013 34(1) pp: 171-180
Publication Date(Web):
DOI:10.1016/j.biomaterials.2012.09.031
Co-reporter:Chuan Wang;Qianqian Yu;Licong Yang;Yanyu Liu;Dongdong Sun;Yongchao Huang
BioMetals 2013 Volume 26( Issue 3) pp:387-402
Publication Date(Web):2013 June
DOI:10.1007/s10534-013-9622-6
In the present study, the interaction between GC-rich sequence of bcl-2 gene P1 promoter (Pu39) and two ruthenium (II) polypyridyl complexes, [Ru(bpy)2(tip)]2+ (1) and [Ru(phen)2(tip)]2+ (2), was investigated by UV–Visible, fluorescence spectroscopy, circular dichroism, fluorescence resonance energy transfer melting assay and polymerase chain reaction stop assay. Those experimental results indicated that the two complexes can effectively stabilize the G-quadruplex of Pu39. It was found that the complex 2 exhibited greater cytotoxic activity than 1 against human Hela cells and can enter into Hela cells in a short period of time to effectively induce apoptosis of cells. Further experiments found that complexes 1 and 2 had as potent inhibitory effects on ECV-304 cell migration as suramin. Those noteworthy results provide new insights into the development of anticancer agents for targeting G-quadruplex DNA.
Co-reporter:Dongdong Sun, Rong Zhang, Fang Yuan, Du Liu, Yanhui Zhou and Jie Liu
Dalton Transactions 2012 vol. 41(Issue 6) pp:1734-1741
Publication Date(Web):08 Dec 2011
DOI:10.1039/C1DT11676B
Two arene ruthenium complexes [Ru(η6-C6H6)(p-MOPIP)Cl]+1 and [Ru(η6-C6H6)(p-CFPIP)Cl]+2, where p-MOPIP = 2-(4-methoxyphenyl)-imidazo[4,5f][1,10] phenanthroline and p-CFPIP = 2-(4-trifluoromethylphenyl)-imidazo[4,5f][1,10] phenanthroline, were prepared and the interactions of these compounds with DNA oligomers 5′-G3(T2AG3)3-3′(HTG21) have been studied by UV-vis and circular dichroism (CD) spectroscopy, gel mobility shift assay, fluorescence resonance energy transfer (FRET) melting assay, polymerase chain reaction (PCR) stop assay and telomeric repeat amplification protocol (TRAP) assay. The results show that both complexes can induce the stabilization of quadruplex DNA but complex 1 is a better G-quadruplex binder than complex 2. The two ruthenium complexes tested led to an inhibition of the enzyme telomerase and complex 1 was the significantly better inhibitor. A novel visual method has been developed for making a distinction between G-quadruplex DNA and double DNA by our Ru complexes binding hemin to form the hemin-G-quadruplex DNAzyme. Furthermore, in vitro cytotoxicity studies showed complex 1 exhibited quite potent antitumor activities and the greatest inhibitory selectivity against cancer cell lines.
Co-reporter:Yanyu Liu, Qianqian Yu, Chuan Wang, Dongdong Sun, Yongchao Huang, Yanhui Zhou, Jie Liu
Inorganic Chemistry Communications 2012 Volume 24() pp:104-109
Publication Date(Web):October 2012
DOI:10.1016/j.inoche.2012.08.009
The binding properties of two Ru(II) complexes [Ru(bpy)2(ox)]2 + (RBO) and [Ru(bpy)2(suc)]2 + (RBS) (bpy = 2, 2-bipyridine, ox = oxalate, and suc = succinate) to human serum albumin (HSA) have been studied by fluorescence, UV–vis absorption spectroscopy and circular dichroism (CD) spectroscopy. The fluorescence quenching mechanism was determined to be a static quenching procedure. The Stern–Volmer quenching constant Ksv and corresponding thermodynamic parameters ΔH, ΔS and ΔG were calculated. The number of binding site was about 1. The result of CD showed that the secondary structure of HSA molecules was changed in the presence of the Ru(II) complexes. Furthermore, the anticancer activities of the complexes were evaluated by using the MTT assay, the results indicated that the antiproliferative activity of RBS was higher than that of RBO, and RBS showed a significant antitumor activity through induction of apoptosis in A549 cells.The scheme is the illustration of the Ru(II) complex RBS binding to Human serum albumin (HSA) and its antitumor activity through induction of apoptosis in A549 cells.Highlights► Two Ru(II) complexes have the capability of binding to human serum albumin. ► The mechanism of the bonding might be a static quenching. ► The binding forces are mainly hydrogen bond and Van der Waals force. ► Complex RBS showes apoptosis-inducing activities in A549 cells.
Co-reporter:Dong-dong Sun, Wei-zhang Wang, Jian-wen Mao, Wen-jie Mei, Jie Liu
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 1) pp:102-105
Publication Date(Web):1 January 2012
DOI:10.1016/j.bmcl.2011.11.063
1,10-Phenanthroline has been shown to exhibit anticancer activity. Here, a series of imidazo [4,5f][1,10] phenanthroline derivatives 1−10 were synthesized and their biological activities were further elucidated. We found that 2-(4-Brominephenyl)-imidazo [4,5f][1,10] phenanthroline (compound 3) possessed potent antiproliferation activities again a variety of tumor cell lines using 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Flow cytometric analysis revealed that compound 3 induced both through apoptosis and necrosis in human lung adenocarcinoma cell line, A549. Moreover, compound 3 treatment led to up-regulation of IκBα and down-regulation of p65 and c-myc in A549 cells. Taken together, these results suggested that compound 3 inhibited cell proliferation by suppression of NF-κB activity and down-regulation of c-myc gene expression and may be a candidate for further evaluation as a chemopreventive and chemotherapeutic agent for human cancers, especially for lung cancer.The synthesis and biological evaluation of a series of imidazo [4,5f][1,10] phenanthroline derivatives 1–10 are reported. The active compound 3 displayed potent antitumor activity with IC50 value of approximately 0.85 μM in vitro.
Co-reporter:Dr. Du Liu;Yanan Liu;Chuan Wang; Shuo Shi;Dongdong Sun;Dr. Feng Gao; Qianling Zhang; Jie Liu
ChemPlusChem 2012 Volume 77( Issue 7) pp:
Publication Date(Web):
DOI:10.1002/cplu.201290029
Co-reporter:Dr. Dongdong Sun;Dr. Yanan Liu;Du Liu;Rong Zhang;Xicheng Yang; Jie Liu
Chemistry - A European Journal 2012 Volume 18( Issue 14) pp:4285-4295
Publication Date(Web):
DOI:10.1002/chem.201103156
Abstract
Telomerase inhibition is an attractive strategy for cancer chemotherapy. In the current study, we have synthesized and characterized two chiral ruthenium(II) complexes, namely, Λ-[Ru(phen)2(p-MOPIP)]2+ and Δ-[Ru(phen)2(p-MOPIP)]2+, where phen is 1,10-phenanthroline and p-MOPIP is 2-(4-methoxyphenyl)-imidazo[4,5f][1,10]phenanthroline. The chiral selectivity of the compounds and their ability to discriminate quadruplex DNA were investigated by using UV/Vis, fluorescence spectroscopy, circular dichroism spectroscopy, fluorescence resonance energy transfer melting assay, polymerase chain reaction stop assay and telomerase repeat amplification protocol. The results indicate that the two chiral compounds could induce and stabilize the formation of antiparallel G-quadruplexes of telomeric DNA in the presence or absence of metal cations. We report the remarkable ability of the two complexes Λ-[Ru(phen)2(p-MOPIP)]2+ and Δ-[Ru(phen)2(p-MOPIP)]2+ to stabilize selectively G-quadruplex DNA; the former is a better G-quadruplex binder than the latter. The anticancer activities of these complexes were evaluated by using the MTT assay. Interestingly, the antiproliferative activity of Λ-[Ru(phen)2(p-MOPIP)]2+ was higher than that of Δ-[Ru(phen)2(p-MOPIP)]2+, and Λ-[Ru(phen)2(p-MOPIP)]2+ showed a significant antitumor activity in HepG2 cells. The status of the nuclei in Λ/Δ-[Ru(phen)2(p-MOPIP)]2+-treated HepG2 cells was investigated by using real-time living cell microscopy to determine the effects of Λ/Δ-[Ru(phen)2(p-MOPIP)]2+ on intracellular accumulation. The results show that Λ/Δ-[Ru(phen)2(p-MOPIP)]2+ can be taken up by HepG2 cells and can enter into the cytoplasm as well as accumulate in the nuclei; this suggests that the nuclei were the cellular targets of Λ/Δ-[Ru(phen)2(p-MOPIP)]2+.
Co-reporter:Qian Li, Dongdong Sun, Yanhui Zhou, Du Liu, Qianling Zhang, Jie Liu
Inorganic Chemistry Communications 2012 20() pp: 142-146
Publication Date(Web):
DOI:10.1016/j.inoche.2012.02.037
Co-reporter:Dr. Du Liu;Yanan Liu;Chuan Wang; Shuo Shi;Dongdong Sun;Dr. Feng Gao; Qianling Zhang; Jie Liu
ChemPlusChem 2012 Volume 77( Issue 7) pp:551-562
Publication Date(Web):
DOI:10.1002/cplu.201200039
Abstract
Two ruthenium(II) complexes [Ru(phen)2(tip)](ClO4)2 (1) and [Ru(bpy)2(tip)](ClO4)2 (2; phen=1,10-phenanthroline, bpy=2,2’-bipyridine, tip=2-thiophenimidazo[4,5-f][1,10]phenanthroline) were synthesized and characterized by elemental analysis, 1H NMR spectroscopy, and electrospray ionization-mass spectrometry to explore the role of metal complexes as novel telomeric quadruplex stabilizers. The different quadruplex binding properties of these compounds were evaluated by absorption and emission analyses, circular dichroism spectroscopy, fluorescence resonance energy transfer (FRET) melting assay, NMR spectroscopy, and molecular modeling. The results show that both complexes can well induce and stabilize different G-quadruplex structures using a 1:1 [quadruplex]/[complex] binding mode ratio. Complex 1 exhibits higher interaction ability at 1.43×106 M−1 binding affinity and superior G-quadruplex selectivity over duplex DNA through multiple interaction (mainly intercalating) with the G-quadruplex at the 3’-terminal face. Furthermore, polymerase chain reaction (PCR)-stop assay, electrophoretic mobility shift assay, telomerase repeat amplification protocol, and MTT assay demonstrate that complex 1 not only can stabilize dimer forms of the G-quadruplex at low concentrations but also exhibit better inhibitory activity for telomerase and cancer cells. The results suggest that complex 1 may be a potential telomerase inhibitor for cancer chemotherapy.
Co-reporter:Caiping Tan, Sheng Hu, Jie Liu, Liangnian Ji
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 5) pp:1555-1563
Publication Date(Web):May 2011
DOI:10.1016/j.ejmech.2011.01.074
Two new ruthenium complexes, trans,cis,cis-[RuCl2(DMSO)2(H2biim)] (1) and mer-[RuCl3(DMSO)(H2biim)] (2) (DMSO = dimethyl sulfoxide and H2biim = 2,2′-biimidazole), have been synthesized and fully characterized by single-crystal X-ray analysis. The less stable complex 2 is more cytotoxic against the four human cancer cell lines tested than 1. Further studies show that 1 and 2 exhibit cell growth inhibition by triggering G0/G1 cell cycle arrest and mitochondria-mediated apoptosis. Additionally, complex 2 exerts potent inhibitory effects on the adhesion and migration of human cancer cells comparable to that of NAMI-A ([ImH][trans-[RuCl4(Im)(DMSO-S)], Im = imidazole). Target validation studies show that cyclin-dependent kinases (CDKs), other than DNA, are more likely to be targets of 1 and 2.Highlights► Two new rutheniume--DMSO complexes have been synthesized and characterized. ► Complex 2 is physiologically less stable than complex 1. ► Complex 2 shows higher antiproliferative and anti-metastatic effects than 1 does. ► Cyclin-dependent kinases (CDKs) are possible targets of 1 and 2.
Co-reporter:Yanan Liu;Xiaonian Zhang;Rong Zhang;Tianfeng Chen;Yum-Shing Wong;Wen-Jie Zheng
European Journal of Inorganic Chemistry 2011 Volume 2011( Issue 12) pp:1974-1980
Publication Date(Web):
DOI:10.1002/ejic.201000968
Abstract
Two ruthenium(II)–porphyrin complexes, [(3-Py)Ru(phen)2(tmopp)] [1; phen = phenanthroline, tmopp = 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin] and [(4-Py)Ru(phen)2(tmopp)] (2), have been synthesized and characterized for the first time. It was found that the two ruthenium(II)–porphyrin complexes show significant antitumor activity in HepG2 cells. Flow cytometric analysis showed that complex 1 arrested the cell cycle in the G0/G1 phase and induced apoptosis in HepG2 cells. Fluorescence microscopy and flow cytometric analyses demonstrated that the generation of intracellular reactive oxygen species (ROS) was significantly inhibited in cells treated with either complex. The total antioxidant capacity of the complexes was detected by a 2,2′-azinobis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) assay; this showed that both complexes are good free-radical scavengers. Ruthenium(II)–porphyrin complex 1 also was found to scavenge hydroxy radicals, as measured by the Fenton system. These data demonstrate that ruthenium(II)–porphyrin complexes exhibit antioxidant properties, probably through the involvement of a direct scavenging effect on a hydroxy radical. Taken together, the findings show that ruthenium(II)–porphyrin complexes induce apoptosis in HepG2 cells by inhibiting the generation of ROS and are potential anticancer therapeutic agents.
Co-reporter:Tianfeng Chen ; Yanan Liu ; Wen-Jie Zheng ; Jie Liu ;Yum-Shing Wong
Inorganic Chemistry 2010 Volume 49(Issue 14) pp:6366-6368
Publication Date(Web):June 8, 2010
DOI:10.1021/ic100277w
The limitations of cisplatin-based chemotherapy, including high toxicity, undesirable side effects, and drug resistance, have motivated extensive investigations into alternative metal-based cancer therapies. Ruthenium (Ru) possesses several favorable properties suited to rational anticancer drug design and biological applications. In the present study, we synthesized a series of ruthenium polypyridyl complexes containing N,N-chelating ligands, examined their anticancer activities, and elucidated the molecular mechanisms through which they caused the cancer cell death. The results demonstrated that [Ru(phen)2-p-MOPIP](PF6)2·2H2O (RuPOP), a complex with potent antiproliferative activity, is able to induce mitochondria-mediated and caspase-dependent apoptosis in human cancer cells. On the basis of these results, we suggest that RuPOP may be a candidate for further evaluation as a chemopreventive and chemotherapeutic agent for human cancers, especially for melanoma.
Co-reporter:Earl S. Johnston
Science 1920 Vol 52(1352) pp:517-518
Publication Date(Web):26 Nov 1920
DOI:10.1126/science.52.1352.517
Co-reporter:
Science 1918 Vol 47(1218) pp:435-436
Publication Date(Web):03 May 1918
DOI:10.1126/science.47.1218.435
Co-reporter:Weirui Xu, Wenjie Xie, Xiaoquan Huang, Xu Chen, Na Huang, Xin Wang, Jie Liu
Food Chemistry (15 April 2017) Volume 221() pp:
Publication Date(Web):15 April 2017
DOI:10.1016/j.foodchem.2016.10.054
Microorganism breeding is a known cause of food spoilage and disease transmission. Aspergillus niger (A. niger) and Bacillus subtilis (B. subtilis) are examples of microorganisms that cause deterioration of fresh fruits and vegetables during storage, which can be a serious threat to human health. In this work, we synthesized a self-assembled film of graphene oxide (GO) and chitosan (CS) biopolymers with titanium dioxide (TiO2) nanoparticles embedded in its surface. We then characterized its antibacterial and preservative properties. We found that these non-cytotoxic nanometer-scale films, especially when the ratio of graphene oxide, chitosan and titanium dioxide nanoparticles in the nanocomposites is 1:20:4, exhibited high antibacterial activity against the biofilm-forming strains A. niger and B. subtilis. The preservation capacity of the nanocomposites was evaluated by enzymatic experiments. The nanocomposites did not show any cytotoxicity against mammalian somatic cells and plant cells. Altogether, this work demonstrated that the nanocomposites disrupted microbial film formation while avoiding internalization by animal and plant cells. Due to their selectivity and safety, these nanocomposites demonstrate potential as antimicrobial coatings for food preservation.
Co-reporter:Tiantian Yin, Licong Yang, Yanan Liu, Xianbo Zhou, Jing Sun, Jie Liu
Acta Biomaterialia (1 October 2015) Volume 25() pp:172-183
Publication Date(Web):1 October 2015
DOI:10.1016/j.actbio.2015.06.035
The blood–brain barrier (BBB) is a formidable gatekeeper toward exogenous substances, playing an important role in brain homeostasis and maintaining a healthy microenvironment for complex neuronal activities. However, it also greatly hinders drug permeability into the brain and limits the management of brain diseases. The development of new drugs that show improved transport across the BBB represents a promising strategy for Alzheimer’s disease (AD) intervention. Whereas, previous study of receptor-mediated endogenous BBB transport systems has focused on a strategy of using transferrin to facilitate brain drug delivery system, a system that still suffers from limitations including synthesis procedure, stability and immunological response. In the present study, we synthetised sialic acid (SA)-modified selenium (Se) nanoparticles conjugated with an alternative peptide-B6 peptide (B6-SA-SeNPs, a synthetic selenoprotein analogue), which shows high permeability across the BBB and has the potential to serve as a novel nanomedicine for disease modification in AD. Laser-scanning confocal microscopy, flow cytometry analysis and inductively coupled plasma-atomic emission spectroscopy ICP-AES revealed high cellular uptake of B6-SA-SeNPs by cerebral endothelial cells (bEnd.3). The transport efficiency of B6-SA-SeNPs was evaluated in a Transwell experiment based on in vitro BBB model. It provided direct evidence for B6-SA-SeNPs crossing the BBB and being absorbed by PC12 cells. Moreover, inhibitory effects of B6-SA-SeNPs on amyloid-β peptide (Aβ) fibrillation could be demonstrated in PC12 cells and bEnd3 cells. B6-SA-SeNPs could not only effectively inhibit Aβ aggregation but could disaggregate preformed Aβ fibrils into non-toxic amorphous oligomers. These results suggested that B6-SA-SeNPs may provide a promising platform, particularly for the application of nanoparticles in the treatment of brain diseases.Statement of SignificanceAlzheimer’s disease (AD) is the world’s most common form of dementia characterized by intracellular neurofibrillary tangles in the brain. Over the past decades, the blood–brain barrier (BBB) limits access of therapeutic or diagnostic agents into the brain, which greatly hinders the development of new drugs for treating AD. In this work, we evaluated the efficiency of B6-SA-SeNPs across BBB and investigated the interactions between B6-SA-SeNPs and amyloid-β peptide (Aβ). We confirm that B6-SA-SeNPs could provide a promising platform because of its high brain delivery efficiency, anti-amyloid properties and anti-oxidant properties, which may serve as a novel nanomedicine for the application in the treatment of brain diseases.Download high-res image (112KB)Download full-size image
Co-reporter:Ying Liu, Na Huang, Yunfei Yu, Chuping Zheng, Ning Deng and Jie Liu
Journal of Materials Chemistry A 2016 - vol. 4(Issue 25) pp:NaN4401-4401
Publication Date(Web):2015/12/21
DOI:10.1039/C5TB01898F
The surface chemistry of materials has an interactive influence on cell behavior. It is now well established that surface chemistry can affect cell adhesion, proliferation, and differentiation. Although amino (NH2)-terminated surfaces generated by the modification of nanoparticles with silane can promote osteogenic differentiation of mesenchymal stem cells (MSCs), how silica surfaces with ruthenium nanoparticles (SiO2@Ru) act on MSCs remains largely unknown. A concentration of 5 μg mL−1 aminopropyltriethoxysilane (APTS)-modified SiO2 nanoparticles (SiO2–NH2) or SiO2@Ru was nontoxic to MSCs, based on MTT and apoptosis assays. In addition, SiO2–NH2 and SiO2@Ru did not affect the surface phenotype or morphology of MSCs. SiO2@Ru can be used to trigger the differentiation of MSCs into osteocytes, minimising the need for exogenous biological supplementation. TEM images revealed that SiO2@Ru might interact with proteins located in the cytoplasm, which would have a further impact on subsequent cellular signaling pathways. Activation of Akt signaling pathways was observed in MSCs cultured with SiO2@Ru and these enhancement effects could be blocked by the Akt inhibitor LY294002. SiO2@Ru exhibited in vitro osteocompatibility that surpassed that of SiO2–NH2, as well as supporting the proliferation and differentiation of MSCs. This demonstrates the potential of SiO2@Ru for use in bone regeneration.
Co-reporter:Xiaoquan Huang, Gengjia Chen, Jiali Pan, Xu Chen, Na Huang, Xin Wang and Jie Liu
Journal of Materials Chemistry A 2016 - vol. 4(Issue 37) pp:NaN6270-6270
Publication Date(Web):2016/08/18
DOI:10.1039/C6TB01122E
A subcutaneous abscess is a local infection caused by pathogenic bacteria. Due to the emergence of multidrug-resistant (MDR) pathogenic bacteria, there is an urgent need for the development of new approaches to treat subcutaneous abscesses. In recent studies, we determined that ruthenium nanoparticles have high photothermal conversion efficiency and can transfer energy to surrounding oxygen molecules to generate cytotoxic singlet oxygen (ROS). Acetylcholine (Ach) was modified and added to the surface of the ruthenium nanoparticles to form composite nanoparticles (Ach@RuNPs). Acetylcholine played a role in targeting the nanoparticles on bacteria, and promoting their entry into the bacterial cells as determined by bacterial plate assays. Experiments using a mouse subcutaneous abscess model also showed that Ach@RuNPs were targeted on bacteria. Upon irradiation with an 808 nm laser, Ach@RuNPs could function as both a photodynamic therapeutic (PDT) and a photothermal therapeutic (PTT) agent to kill pathogenic bacteria, and repair infected wounds without leaving residual implanted materials. The use of Ach@RuNPs represents an attractive potential treatment approach for subcutaneous abscesses due to their high efficacy and minimal invasiveness.
Co-reporter:Qianqian Yu, Jing Sun, Xufeng Zhu, Lin Qiu, Mengmeng Xu, Sirun Liu, Jianming Ouyang and Jie Liu
Journal of Materials Chemistry A 2017 - vol. 5(Issue 30) pp:NaN6096-6096
Publication Date(Web):2017/06/15
DOI:10.1039/C7TB01035D
Photodynamic therapy (PDT), by producing reactive oxygen species (ROS), inhibits cancer cells and is an emerging and pioneering cancer therapeutic modality which can eliminate some of the drawbacks of other traditional anticancer therapies. To combine near-infrared (NIR) mediated PDT, chemotherapy and gene therapy in a synergistic manner, a novel NIR light activated photosensitizer for PDT was designed based on TiO2-coated Fe3O4 nanoparticle core/shell nanocarriers (Fe3O4@TiO2@mTiO2). The chemotherapeutic drug doxorubicin hydrochloride (DOX) was conjugated to the surface of the TiO2 mesopores through pH-reversible hydrazone bond linking and β-catenin siRNA was loaded in the mesopores. Fe3O4@TiO2@mTiO2–DOX/siRNA delivery systems have features and functions of magnetic targeting, fluorescence imaging, MRI diagnosis, and combination therapy through the simultaneous maneuvering of a magnet and NIR-mediated PDT. In vitro, Fe3O4@TiO2@mTiO2–DOX/siRNA effectively silences β-catenin gene, induces tumor cell apoptosis and consequently significantly enhances the cancer suppression effect of the synergistic therapeutic agent. Meanwhile, under NIR irradiation, excess ROS produced can further trigger tumor cell apoptosis. In vivo investigation confirmed that Fe3O4@TiO2@mTiO2–DOX/siRNA exhibited high tumor targeted specificity through MRI and fluorescence imaging, and optimal anti-tumor efficacy. The results verified its significant therapeutic effects on tumors by combination therapy consisting of magnetic targeting and NIR-mediated PDT.
Co-reporter:Licong Yang, Jing Sun, Wenjie Xie, Yanan Liu and Jie Liu
Journal of Materials Chemistry A 2017 - vol. 5(Issue 30) pp:NaN5967-5967
Publication Date(Web):2017/04/04
DOI:10.1039/C6TB02952C
Inhibition of amyloid β (Aβ) aggregation holds considerable promise as a therapeutic strategy for Alzheimer's disease (AD). However, successful inhibition is hard to achieve due to the blood–brain barrier (BBB) and the non-selective distribution of drugs. Herein, two targeting peptides (LPFFD and TGN) were conjugated to selenium nanoparticles (SeNPs). We found that the concentration ratio of LPFFD to TGN taken as 1:1 could form the most effective dual-functional SeNPs (L1T1–SeNPs) for inhibiting Aβ aggregation and crossing the BBB. L1T1–SeNPs can cross the BBB and have a strong affinity toward Aβ species, and thus, they can efficiently suppress extracellular Aβ fibrillation by disrupting hydrophobic and electrostatic interactions that are important for Aβ40 nucleation. Also, L1T1–SeNPs can suppress the Aβ40 fiber mediated generation of reactive oxygen species (ROS) and their corresponding neurotoxicity in PC12 cells. In addition, L1T1–SeNPs exert synergistic effects on the inhibition of Aβ aggregation and cross the BBB efficiently. Collectively, these results demonstrate that dual-functional SeNPs might be a valuable targeting system for inhibiting Aβ aggregation.
Co-reporter:Licong Yang, Qingchang Chen, Ying Liu, Jingnan Zhang, Dongdong Sun, Yanhui Zhou and Jie Liu
Journal of Materials Chemistry A 2014 - vol. 2(Issue 14) pp:NaN1987-1987
Publication Date(Web):2014/02/26
DOI:10.1039/C3TB21586E
Amyloid β (Aβ) aggregates are considered as possible targets for therapy of Alzheimer's disease (AD). Metal ions play an important role in amyloid aggregation and neurotoxicity in the AD pathogenesis. Disruption of the interactions between these metal ions and peptides holds considerable promise as a therapeutic strategy for AD treatment. In this study, L-Cys-modified Se/Ru nanoparticles (NPs) have been designed as Aβ-binding units to inhibit metal-induced Aβ aggregation. L-Cys was used as both the reducing agent and surface modifier in the formation of SeNPs, RuNPs and Se/RuNPs. We found that RuNPs and Se/RuNPs have a strong affinity toward Aβ species and efficiently suppress extracellular Aβ40 self-assembly and Zn2+-induced fibrillization. Also, Se/RuNPs can suppress the Zn2+–Aβ40 mediated generation of reactive oxygen species (ROS) and their corresponding neurotoxicity in PC12 cells. Intriguingly, SeNPs do not have the same ability as Se/RuNPs. In addition, Se/RuNPs also decrease intracellular Aβ40 fibrillization, but this process does not involve the lysosomal pathway. These results suggest that ruthenium significantly enhances the activity of Se/RuNPs binding to Aβ40. This interaction would block the Zn2+ binding to Aβ40 peptides and lower the concentration of the free monomer, thus decreasing fibrillization. Owing to this, Se/RuNPs may represent a new strategy in AD treatment.
Co-reporter:Xianbo Zhou, Jing Sun, Tiantian Yin, Fangling Le, Licong Yang, Yanan Liu and Jie Liu
Journal of Materials Chemistry A 2015 - vol. 3(Issue 39) pp:NaN7774-7774
Publication Date(Web):2015/09/03
DOI:10.1039/C5TB00731C
Chiral molecules, which selectively target and inhibit amyloid β-peptide (Aβ) aggregation, have potential use as therapeutic agents for the treatment of Alzheimer's disease (AD). Here we use cysteine enantiomer-modified SeNPs (abbreviated as D/LSeNPs) to demonstrate that surface chirality strongly influences the formation of Aβ aggregates in the presence of metal ions, such as Zn2+ or Cu2+. The two chiral molecule modified nanoparticles could inhibit the formation of Aβ fibrils by binding Aβ, thus blocking the formation of Aβ fibrils and blocking the metal binding sites. Of the two enantiomers, D/SeNPs appear to more effectively inhibit Aβ fibril formation due to a greater affinity for Aβ than that of L/SeNPs. Additionally, D/LSeNPs appeared to reduce Aβ and metal ion-induced neurotoxicity. Treatment with D/LSeNPs can also decrease the levels of intracellular reactive oxygen species, and stabilize the mitochondrial membrane potential. Of the two enantiomers, D/SeNPs were more effective in protecting the cells than L/SeNPs, and this could be due to D/SeNPs being selectively absorbed by PC12 cells, maintaining cellular redox potentials, and protecting cells against oxidative stress to a greater extent than L/SeNPs. From these results, it appears that chiral molecules can bring novel insight into better drug treatment designs for Alzheimer's disease.
Co-reporter:Xianbo Zhou, Chengwen Cao, Qingchang Chen, Qianqian Yu, Yanan Liu, Tiantian Yin and Jie Liu
Journal of Materials Chemistry A 2015 - vol. 3(Issue 35) pp:NaN7067-7067
Publication Date(Web):2015/08/12
DOI:10.1039/C5TB00487J
Human islet amyloid polypeptide (hIAPP) was found as amyloid aggregate deposits in the pancreatic islets of patients with type-2 diabetes and studies showed that insulin and its derivatives were the potent inhibitors of hIAPP aggregation. However, several emerging therapies with this goal showed limited success due to the instability and inefficiency of insulin derivatives. Nanosized graphene oxide (nGO) possesses high stability and affinity toward aromatic rings. In this study, an insulin-derived peptide, EALYLV, was stabilized by loading on nGO@PEG to inhibit aggregation and hIAPP-induced cytotoxicity. The results showed that nGO@PEG@EALYLV (abbreviated as nGO@PEG@E) can effectively inhibit the aggregation of hIAPP via electrostatic adsorption and specific binding to the active sites of hIAPP. We further evaluated the protective effect of nGO@PEG@E on INS-1 cells in the presence of hIAPP. Treatment with nGO@PEG@E could significantly elevate the viability of INS-1 cells, decrease the level of intracellular reactive oxygen species, and stabilize mitochondrial membrane potential. All the results indicated that nGO@PEG@E could inhibit the aggregation of hIAPP, which reduces its cytotoxicity.
Co-reporter:Xu Chen, Xiaoquan Huang, Chuping Zheng, Yanan Liu, Taoyuan Xu and Jie Liu
Journal of Materials Chemistry A 2015 - vol. 3(Issue 35) pp:NaN7029-7029
Publication Date(Web):2015/05/20
DOI:10.1039/C5TB00280J
Graphene oxide (GO) has attracted great interest in many different areas, as a delivery vehicle for antibacterial agents, and has shown high potential. Although silver nanoparticles (AgNPs) have a strong antibacterial effect, the biological application of AgNPs is often hindered by their aggregation and low stability. In this study, we developed an approach of polyoxyethylene bis(amine) (PEG) directed AgNPs grown on GO, then we combined the two materials to prepare a series of functionalized GO bearing different sized AgNPs, and studied the size effects of AgNPs on growth inhibition of Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus). We evaluated the antibacterial effect of GO@PEG@AgNPs on E. coli and S. aureus by various methods such as minimum inhibitory concentration (MIC) experiment, cell wall/membrane integrity assay and scanning electron microscopy (SEM) characterisation of bacterial morphology. The GO@PEG@AgNPs composites exhibited markedly higher antibacterial efficacy than AgNPs alone. The smallest GO@PEG@AgNPs (10 nm) particularly demonstrated higher antibacterial activity than other sizes (30, 50, and 80 nm). We believe that these findings contribute to great potential application as a regulated graphene-based antibacterial solution.
Co-reporter:Ying Liu, Na Huang, Yunfei Yu, Chuping Zheng, Ning Deng and Jie Liu
Journal of Materials Chemistry A 2016 - vol. 4(Issue 28) pp:NaN4941-4941
Publication Date(Web):2016/07/04
DOI:10.1039/C6TB90091G
Correction for ‘Bioactive SiO2@Ru nanoparticles for osteogenic differentiation of mesenchymal stem cells via activation of Akt signaling pathways’ by Ying Liu et al., J. Mater. Chem. B, 2016, DOI: 10.1039/c5tb01898f.
Co-reporter:Dongdong Sun, Rong Zhang, Fang Yuan, Du Liu, Yanhui Zhou and Jie Liu
Dalton Transactions 2012 - vol. 41(Issue 6) pp:NaN1741-1741
Publication Date(Web):2011/12/08
DOI:10.1039/C1DT11676B
Two arene ruthenium complexes [Ru(η6-C6H6)(p-MOPIP)Cl]+1 and [Ru(η6-C6H6)(p-CFPIP)Cl]+2, where p-MOPIP = 2-(4-methoxyphenyl)-imidazo[4,5f][1,10] phenanthroline and p-CFPIP = 2-(4-trifluoromethylphenyl)-imidazo[4,5f][1,10] phenanthroline, were prepared and the interactions of these compounds with DNA oligomers 5′-G3(T2AG3)3-3′(HTG21) have been studied by UV-vis and circular dichroism (CD) spectroscopy, gel mobility shift assay, fluorescence resonance energy transfer (FRET) melting assay, polymerase chain reaction (PCR) stop assay and telomeric repeat amplification protocol (TRAP) assay. The results show that both complexes can induce the stabilization of quadruplex DNA but complex 1 is a better G-quadruplex binder than complex 2. The two ruthenium complexes tested led to an inhibition of the enzyme telomerase and complex 1 was the significantly better inhibitor. A novel visual method has been developed for making a distinction between G-quadruplex DNA and double DNA by our Ru complexes binding hemin to form the hemin-G-quadruplex DNAzyme. Furthermore, in vitro cytotoxicity studies showed complex 1 exhibited quite potent antitumor activities and the greatest inhibitory selectivity against cancer cell lines.
Co-reporter:Xiu Ying Qin, Li Cong Yang, Fang Ling Le, Qian Qian Yu, Dong Dong Sun, Ya Nan Liu and Jie Liu
Dalton Transactions 2013 - vol. 42(Issue 41) pp:NaN14684-14684
Publication Date(Web):2013/08/23
DOI:10.1039/C3DT51548F
The crystals of two binuclear copper-based complexes were obtained. One complex can remarkably induce apoptosis and inhibit angiogenesis to mediate tumour growth at a greater extent. Furthermore, this complex showed a strong energy-dependent and non-endocytotic uptake and exhibited multiple anti-cancer functions by inhibiting the expressions of p-Akt and p-Erk1/2 proteins and by decreasing the levels of reactive oxygen species.













![2-Propenamide, N-hydroxy-3-[3-[(phenylamino)sulfonyl]phenyl]-, (2E)-](http://img.cochemist.com/ccimg/866400/866323-14-0.png)
![2-Propenamide, N-hydroxy-3-[3-[(phenylamino)sulfonyl]phenyl]-, (2E)-](http://img.cochemist.com/ccimg/866400/866323-14-0_b.png)








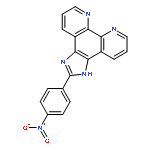
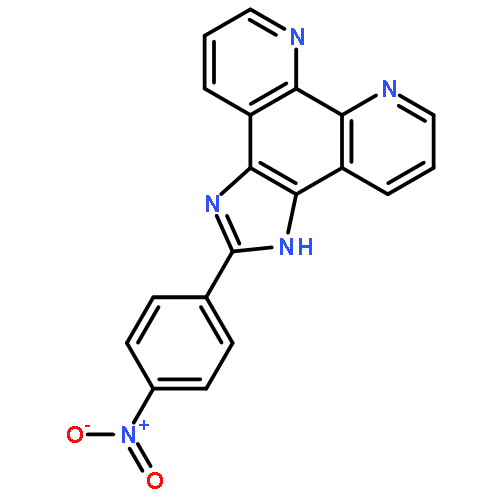









































![1H,10H-Pyrrolo[1,2-c]purine-10,10-diol,2-amino-4-[[(aminocarbonyl)oxy]methyl]-3a,4,5,6,8,9-hexahydro-5-hydroxy-6-imino-,(3aS,4R,10aS)-](http://img.cochemist.com/ccimg/64300/64296-20-4.png)
![1H,10H-Pyrrolo[1,2-c]purine-10,10-diol,2-amino-4-[[(aminocarbonyl)oxy]methyl]-3a,4,5,6,8,9-hexahydro-5-hydroxy-6-imino-,(3aS,4R,10aS)-](http://img.cochemist.com/ccimg/64300/64296-20-4_b.png)
![1H,10H-Pyrrolo[1,2-c]purine-10,10-diol,2,6-diamino-4-[[(aminocarbonyl)oxy]methyl]-3a,4,8,9-tetrahydro-, (3aS,4R,10aS)-](http://img.cochemist.com/ccimg/35600/35523-89-8.png)
![1H,10H-Pyrrolo[1,2-c]purine-10,10-diol,2,6-diamino-4-[[(aminocarbonyl)oxy]methyl]-3a,4,8,9-tetrahydro-, (3aS,4R,10aS)-](http://img.cochemist.com/ccimg/35600/35523-89-8_b.png)













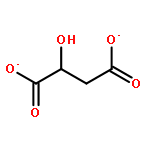
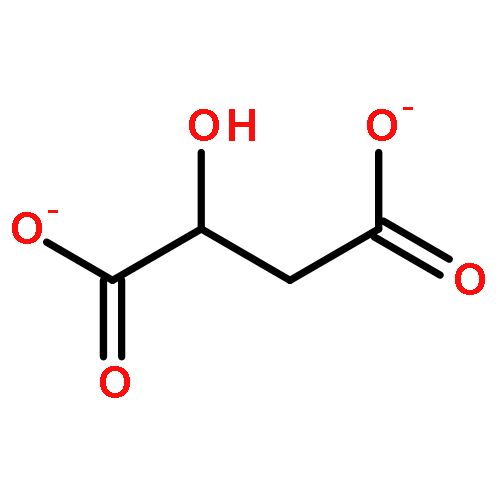












![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)


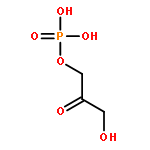
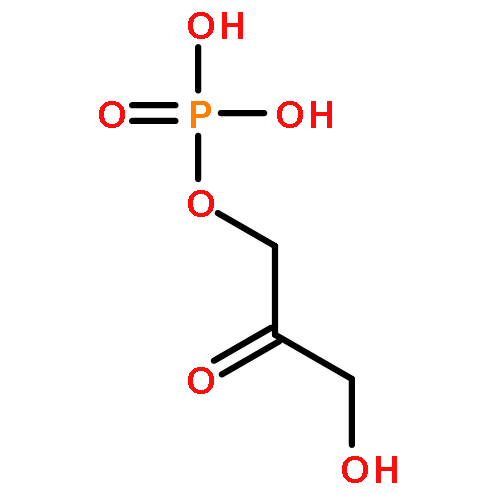






![1H-Imidazo[4,5-f][1,10]phenanthroline, 2-phenyl-](/data/chemimg/133600/171565-44-9.png)
![1H-Imidazo[4,5-f][1,10]phenanthroline, 2-phenyl-](/data/chemimg/133600/171565-44-9_b.png)
![1H-Imidazo[4,5-f][1,10]phenanthroline](/data/chemimg/129500/171565-43-8.png)
![1H-Imidazo[4,5-f][1,10]phenanthroline](/data/chemimg/129500/171565-43-8_b.png)