Co-reporter:Daniel E. Mitchell, Guy Clarkson, David J. Fox, Rebecca A. Vipond, Peter Scott, and Matthew I. Gibson
Journal of the American Chemical Society July 26, 2017 Volume 139(Issue 29) pp:9835-9835
Publication Date(Web):July 17, 2017
DOI:10.1021/jacs.7b05822
Antifreeze proteins are produced by extremophile species to control ice formation and growth, and they have potential applications in many fields. There are few examples of synthetic materials which can reproduce their potent ice recrystallization inhibition property. We report that self-assembled enantiomerically pure, amphipathic metallohelicies inhibited ice growth at just 20 μM. Structure–property relationships and calculations support the hypothesis that amphipathicity is the key motif for activity. This opens up a new field of metallo-organic antifreeze protein mimetics and provides insight into the origins of ice-growth inhibition.
Co-reporter:Rebecca A. Kaner, Simon J. Allison, Alan D. Faulkner, Roger M. Phillips, David I. Roper, Samantha L. Shepherd, Daniel H. Simpson, Nicholas R. Waterfield and Peter Scott
Chemical Science 2016 vol. 7(Issue 2) pp:951-958
Publication Date(Web):26 Oct 2015
DOI:10.1039/C5SC03677A
A range of new helicate-like architectures have been prepared via highly diastereoselective self-assembly using readily accessible starting materials. Six pairs of enantiomers [Fe2L3]Cl4·nH2O (L = various bidentate ditopic ligands NN–NN) show very good water solubility and stability. Their activity against a range of cancer cell lines in vitro is structure-dependent and gives IC50 values as low as 40 nM. In an isogenic pair of HCT116 colorectal cancer cells, preferential activity was observed against cell lines that lack functional p53. Selectivity is also excellent, and against healthy human retinal pigment epithelial (ARPE19) and lung fibroblast (WI38) cells IC50 values are nearly three orders of magnitude higher. Cisplatin is unselective in the same tests. The compounds also appear to have low general toxicity in a number of models: there is little if any antimicrobial activity against methicillin-resistant Staphylococcus aureus and Escherichia coli; Acanthamoeba polyphaga is unaffected at 25 μg mL−1 (12.5 μM); Manduca sexta larvae showed clear evidence of systemic distribution of the drug, and rather than any observation of adverse effects they exhibited a significant mean weight gain vs. controls. Investigation of the mode of action revealed no significant interaction of the molecules with DNA, and stimulation of substantial cell death by apoptosis.
Co-reporter:Lihong Li, Guy J. Clarkson, Martin R. Lees, Suzanne E. Howson, Sze-yin Tan, Scott S. Turner, and Peter Scott
Organometallics 2015 Volume 34(Issue 11) pp:2543-2549
Publication Date(Web):May 15, 2015
DOI:10.1021/om501218a
Attempts to synthesize complexes of Fe and Mn(II) with 2-amidopyrrolyl ligands (N–O) were unsuccessful, and only small amounts of the trivalent tris complexes M(N–O)3 were detected, although unusually in the case of Fe(III) a fac structure is observed. In contrast the 2-benzoylpyrrolyl systems give M(II) complexes, and in all instances thus far where Na+ is present, a scorpionate fac-[MII(N–O)3]− unit self-assembles into sandwich anions [MII(N–O)3Na(O–N)3MII]− in which the central metal is efficiently encapsulated by interdigitation of the aryl units. Extended structures are readily made through the use of a 2-(4-pyridinoyl)pyrrolylamide ligand. When Li+ is used, the scorpionate ligand is not assembled, and instead [M(N–O)2] units give rhombic 2D grids. The Fe system displays spin-crossover at 120 K.
Co-reporter:Viktor Brabec, Suzanne E. Howson, Rebecca A. Kaner, Rianne M. Lord, Jaroslav Malina, Roger M. Phillips, Qasem M. A. Abdallah, Patrick C. McGowan, Alison Rodger and Peter Scott
Chemical Science 2013 vol. 4(Issue 12) pp:4407-4416
Publication Date(Web):10 Sep 2013
DOI:10.1039/C3SC51731D
Enantiomers of a relatively rigid DNA-binding metallo-helix are shown to have comparable activity to that of cisplatin against the cell lines MCF7 (human breast adenocarcinoma) and A2780 (human ovarian carcinoma) but are ca five times more active against the cisplatin-resistant A2780cis. The cell-line HCT116 p53+/+ (human colon carcinoma) is highly sensitive giving IC50 values in the nM range, far lower than the cisplatin control. The hypothesis that the biological target of such metallohelices is DNA is probed by various techniques. Tertiary structure changes in ct-DNA (formation of loops and intramolecular coiling) on exposure to the compounds are demonstrated by atomic force microscopy and supported by circular/linear dichroism in solution. Selectivity for 5′-CACATA and 5′-CACTAT segments is shown by DNase I footprinting. Various three- and four-way oligonucleotide junctions are stabilised, and remarkably only the Λ metallo-helix enantiomer stabilizes T-shaped 3WJs during gel electrophoresis; this is despite the lack of a known helix binding site. In studies with oligonucleotide duplexes with bulges it is also shown for the first time that the metallo-helix binding strength and the number of binding sites are dependent on the size of the bulge. In contrast to all the above, flexible metallo-helices show little propensity for structured or selective DNA binding, and while for A2780 the cancer cell line cytotoxicity is retained the A2780cis strain shows significant resistance. For all compounds in the study, H2AX FACS assays on HCT116 p53+/+ showed that no significant DNA damage occurs. In contrast, cell cycle analysis shows that the DNA binders arrest cells in the G2/mitosis phase, and while all compounds cause apoptosis, the DNA binders have the greater effect. Taken together these screening and mechanistic results are consistent with the more rigid helices acting via a DNA binding mechanism while the flexible assemblies do not.
Co-reporter:Suzanne E. Howson, Guy J. Clarkson, Alan D. Faulkner, Rebecca A. Kaner, Michael J. Whitmore and Peter Scott
Dalton Transactions 2013 vol. 42(Issue 42) pp:14967-14981
Publication Date(Web):01 Aug 2013
DOI:10.1039/C3DT51725J
Single diastereomer, diamagnetic, octahedral Fe(II) tris chelate complexes are synthesised that contain three pendant pyridine proligands pre-organised for coordination to a second metal. They bind Cu(I) and Ag(I) with coordination geometry depending on the identity of the metal and the detail of the ligand structure, but for example homohelical (ΔFe,ΔCu) configured systems with unusual trigonal planar Cu cations are formed exclusively in solution as shown by VT-NMR and supported by DFT calculations. Similar heterobimetallic tris(triazole) complexes are synthesised via clean CuAAC reactions at a tris(alkynyl) complex, although here the configurations of the two metals differ (ΔFe,ΛCu), leading to the first optically pure heterohelicates. A second series of Fe complexes perform less well in either strategy as a result of lack of preorganisation.
Co-reporter:Suzanne E. Howson, Nikola P. Chmel, Guy J. Clarkson, Robert J. Deeth, Daniel H. Simpson and Peter Scott
Dalton Transactions 2012 vol. 41(Issue 15) pp:4477-4483
Publication Date(Web):13 Jan 2012
DOI:10.1039/C2DT12378A
Optically pure phenylethaniminopyridine (SC-L) tris-chelates of Fe(II) and other first row transition metal systems have previously been shown to give exclusively the fac structures in the solid state. Here it is shown by powder X-ray diffraction that the complex [CuL3][ClO4]2 crystallises exclusively as the mer isomer, although – for a given absolute configuration of the ligand – of the same helicity (Δ/Λ) as that displayed by the other metal complexes. The similar ligand RC-LFF, which contains a peripheral 19F spin label, gave [CuLFF3][ClO4]2 which also adopts exclusively the mer structure in the crystal, but is shown by NMR spectroscopy to have a fac:mer ratio of 1:6 in solution at low temperature. Molecular mechanics calculations for a number of isomers and conformers are consistent with the presence of such a mixture of isomers in solution for both complexes. The origin of the difference in behaviour between Fe(II) and Cu(II) is the presence of a Jahn–Teller distortion (and the generally longer M–N bonds) in the Cu(II) complexes. This disturbs intra-ligand π-stacking, leading to the poor fac/mer stereoselectivity while leaving enantioselectivity Δ/Λ apparently unaffected.
Co-reporter:Lihong Li, Guy J. Clarkson, David J. Evans, Martin R. Lees, Scott S. Turner and Peter Scott
Chemical Communications 2011 vol. 47(Issue 47) pp:12646-12648
Publication Date(Web):31 Oct 2011
DOI:10.1039/C1CC15574A
Two Fe(II) coordination polymers formed from isomeric ligands give a diamond-like 3D network exhibiting a gradual SCO and a 2D hard magnet with a large coercive field.
Co-reporter:Nikola Paul Chmel, Guy J. Clarkson, Alessandro Troisi, Scott S. Turner, and Peter Scott
Inorganic Chemistry 2011 Volume 50(Issue 9) pp:4039-4046
Publication Date(Web):April 4, 2011
DOI:10.1021/ic102522k
A new family of optically pure tetrathiafulvalenium and tetraselenafulvalenium salts, D3[MIII(S,S-EDDS)]2·nH2O (where D = TTF, TSF; M = Co, Fe, Cr; EDDS = ethylenediaminedisuccinato), were synthesized electrochemically. These phases are semiconductors with conductivities between 6.9 × 10−6 and 1.3 × 10−5 S·cm−1 (Eaca. 0.3 eV) for TTF and 2.8 × 10−4 to 2.8 × 10−5 S·cm−1 (Eaca. 0.1 eV) for TSF compounds. While some crystals suffer from twinning, other well resolved structures consist of TTF/TSF stacks which, under the influence of the chiral anion, exhibit a periodic undulation described by an elliptical helix. The crystallographic data, along with computational work, indicate charge localization in the semiconducting motifs.
Co-reporter:Lihong Li, Jan M. Becker, Laura E. N. Allan, Guy J. Clarkson, Scott S. Turner, and Peter Scott
Inorganic Chemistry 2011 Volume 50(Issue 13) pp:5925-5935
Publication Date(Web):June 2, 2011
DOI:10.1021/ic102291r
The complexes FeL2 [L = bidentate Schiff base ligands obtained from (R)-(+)-α-phenylethanamine and 4-substituted salicylaldehydes, substituent R = H, tBu, NO2, OMe, CN, OH] react with ditopic proligands 1,4-pyrazine (pz) or 4,4′-bipyridine (bpy), to give a family of optically pure Fe(II) polymeric chain complexes of formula {FeL2(μ-pz)}∞ and {FeL2(μ-bpy)}∞. Crystallographic studies show that a range of structures are formed including unidirectional and bidirectional linear polymers and canted zigzag chains. Interchain interactions via π-contacts and hydrogen bonding are also observed. SQuID magnetometry studies on all of the complexes reveal antiferromagnetic interactions, the magnitudes of which are rationalized on the basis of substituent electronic properties and bridging ligand identity. For complexes with bridging pz, the antiferromangnetic interaction is enhanced by electron-releasing substituents on the Fe units, and this is accompanied by a contraction in the intrachain distance. For complexes bridged with the longer bpy the intrachain antiferromagnetic couplings are much weaker as a result of the longer intrachain distance. The magnetic data for this series of chain complexes follow a Bonner–Fisher 1D chain model, alongside a zero field splitting (ZFS) model for Fe(II) (S = 2) as appropriate. The intrachain antiferromagnetic coupling J values, g-factors, and the axial ZFS parameter D were obtained.
Co-reporter:Suzanne E. Howson and Peter Scott
Dalton Transactions 2011 vol. 40(Issue 40) pp:10268-10277
Publication Date(Web):15 Sep 2011
DOI:10.1039/C1DT11423A
Methods by which helical bimetallic assemblies may be accessed as single isomers are described and reviewed. While there are a number of notable successes, the conclusion is made that no existing approach satisfies the criteria which will allow these fascinating compounds to emerge from the synthetic research laboratory environment to practical application in e.g. healthcare. For this to be achieved the compounds must be optically pure and non-racemising, soluble and chemically stable in water, readily available on a large scale, chemically diverse and synthetically robust.
Co-reporter:Nikola Paul Chmel, Laura E. N. Allan, Jan M. Becker, Guy J. Clarkson, Scott S. Turner and Peter Scott
Dalton Transactions 2011 vol. 40(Issue 8) pp:1722-1731
Publication Date(Web):21 Jan 2011
DOI:10.1039/C0DT01184C
Optically pure anionic complexes of pyridinecarboxamide ligands, N2,N6-bis((R)-α-methylbenzyl)pyridine-2, 6-dicarboxamide H2(R,R-L1) and N2,N6-bis((S)-1-methoxypropan-2-yl)pyridine-2, 6-dicarboxamide H2(S,S-L2) have been synthesised and fully characterised. The complexes: (18-crown-6)K[CoIII(R,R-L1)2], (18-crown-6)K[FeIII(R,R-L1)2] and K[CoIII(S,S-L2)2]·3H2O show interesting extended structures from 0D discrete units through 1D zigzag chains to 2D honeycomb layers. The complex anions were used in the synthesis of radical cation salts with tetrathiafulvalene (TTF). The salts (TTF)[CoIII(R,R-L1)2] and (TTF)[CoIII(S,S-L2)2]·EtOAc were characterised by single crystal X-ray diffraction and conductivity measurements. Both compounds comprise mono-oxidised TTF molecules and exhibit similar layered structures with no direct TTF stacking but in which phenyl substituents from the complex anion or co-crystallised ethyl acetate alternate with TTF+ units. Solution spectroscopic and cyclic voltammetric evidence points to the formation of soluble assemblies between TTF+ and the counterion which correspond to the stoichiometry observed by crystallography and other methods in the solid state.
Co-reporter:Suzanne E. Howson and Peter Scott
Dalton Transactions 2011 vol. 40(Issue 16) pp:4332-4333
Publication Date(Web):15 Mar 2011
DOI:10.1039/C0DT01529F
Intramolecular π-stacking plays a role in diastereomerically pure Fe(II) complexes of certain pyridine/imines, but contrary to a recent report, no such interaction is present in the Cu(II) analogues and the stereoselectivity is as yet unknown.
Co-reporter:Suzanne E. Howson, Laura E. N. Allan, Nikola P. Chmel, Guy J. Clarkson, Robert J. Deeth, Alan D. Faulkner, Daniel H. Simpson and Peter Scott
Dalton Transactions 2011 vol. 40(Issue 40) pp:10416-10433
Publication Date(Web):11 Jul 2011
DOI:10.1039/C1DT10588D
One-pot reactions of 2-pyridinecarboxaldehyde, chiral phenylethanamines and Fe(II) give single diastereomer facdiimine complexes at thermodynamic equilibrium so that no chiral separations are required (d.r. > 200:1). The origins of this stereoselectivity are partly steric and partly a result of the presence of three sets of inter-ligand parallel-offset π-stacking interactions. Mn(II), Co(II), Co(III), Ni(II) and Zn(II) give similar fac structures, alongside the imidazole analogues for Fe(II). While most of the complexes are paramagnetic, the series of molecular structures allows us to assess the influence of the π-stacking present, and there is a strong correlation between this and the M–N bond length. Fe(II) is close to optimal. For the larger Zn(II) ion, very weak π-stacking leads to poorer measured stereoselectivity (NMR) but this is improved with increased solvent polarity. The mechanism of stereoselection is further investigated via DFT calculations, chiroptical spectroscopy and the use of synthetic probes.
Co-reporter:Giles W. Theaker, Colin Morton, and Peter Scott
Macromolecules 2011 Volume 44(Issue 6) pp:1393-1404
Publication Date(Web):February 14, 2011
DOI:10.1021/ma102835p
Zirconium salicylaldiminates in the presence of AliBu3/[Ph3C][B(C6F5)4] are inactive for the homopolymerization of p-tert-butylstyrene (TBS), but the addition of hexene leads to a highly active system. Analysis of the polymers by NMR and MALDI indicates surprisingly that most of the product chains contain no hexene, while some contain a single hexene unit. Addition of dihydrogen also leads to the production of poly(TBS) without substantial reduction in molecular weight and with main-chain end-group unsaturation. These and other observations lead us to two related mechanisms for the hexene- and dihydrogen-promoted reactions. Synthetic and catalytic studies indicate that the active species is not a typical bis(salicylaldiminato) compound but an amido/alkoxido system. An authentic compound of this class is highly active for the TBS polymerization even in the absence of added aluminum alkyl.
Co-reporter:Laura E. N. Allan ; Guy J. Clarkson ; David J. Fox ; Andrew L. Gott
Journal of the American Chemical Society 2010 Volume 132(Issue 43) pp:15308-15320
Publication Date(Web):October 12, 2010
DOI:10.1021/ja106588m
The mechanism of hydroamination/cyclization of primary aminoalkenes by catalysts based on Cp*LZr(NMe2)2 (L = κ2-salicyloxazoline) is investigated in a range of kinetic, stoichiometric, and structural studies. The rate law is found to be d[substrate]/dt = k[catalyst]1[substrate]0 for all catalysts and aminoalkenes studied. The overall rate is similar for formation of five- and six-membered rings, and a substantial KIE (kH/kD) is observed, indicating the involvement of N−H bond-breaking in a rate-determining step (RDS) which is not ring-closure. Remarkably, the reaction proceeds at the same rate in THF as it does in toluene, but added non-cyclizable amine slows the reaction, indicating that while the metal is not acting as a Lewis acid in the RDS, the activated substrate is involved. Also in contrast to other catalysts, increasing steric bulk improves the rate, and the origins of this are investigated by X-ray crystallography. Thermodynamic parameters extracted from eight independent kinetic studies indicate moderate ordering (ΔS⧧ = −13 to −23 cal/K·mol) and substantial overall bond disruption (ΔH⧧ = 17 to 21 kcal/mol) in the rate-determining transition state. Secondary amines are unreactive, as is a catalyst with a single aminolyzable site, thus excluding an amido mechanism. A catalytic cycle involving rate-determining formation of a reactive imido species is proposed. Stoichiometric steps in the process are shown to be feasible and have appropriate rates by synthetic and in situ NMR spectroscopic studies. The fate of the catalyst in the absence of excess amine (at the end of the catalytic reaction) is conversion to a metallacyclic species arising from CH activation of a peripheral substituent.
Co-reporter:Jan M. Becker, James Barker, Guy J. Clarkson, Remy van Gorkum, Gursharon K. Johal, Richard I. Walton and Peter Scott
Dalton Transactions 2010 vol. 39(Issue 9) pp:2309-2326
Publication Date(Web):14 Jan 2010
DOI:10.1039/B905706D
A number of bis(salicylaldiminato) (Schiff base) complexes of Fe(II) i.e. L2Fe with a variety of ligand substituents and functionality are synthesised, usually by salt metathesis but on one occasion via the (sal)2Fe complex, and characterised by standard methods. Two molecular structure determinations indicate the expected tetrahedral geometry. Treatment with dry oxygen yielded the Fe(III) complexes [(FeL2)2-μ-O], generally in high yield and in the absence of hydroxylated impurities; one bulky complex L2Fe gave no oxidation product for steric reasons, and one hydroxyl-substituted compound gave an insoluble product. Five molecular structures of these [(FeL2)2-μ-O] compounds were determined. All were homochiral in the sense that the absolute configuration at Fe was the same for both atoms. Three of the structures – those based on optically pure salicylaldimine ligands – were diastereomeric. For two, the diastereomers observed are those predicted on the basis of steric considerations, while a third highly distorted and congested structure represents the unexpected diastereomer. Powder X-ray diffraction confirms high phase purity of the bulk samples (i.e. they are diastereomerically pure to the level detectable). These investigations, coupled with molecular structure and NMR studies on model gallium complexes, indicate that the diastereoselection arises from cooperative interactions across the oxo bridge.
Co-reporter:Cynthia Paul Sebli, Suzanne E. Howson, Guy J. Clarkson and Peter Scott
Dalton Transactions 2010 vol. 39(Issue 18) pp:4447-4454
Publication Date(Web):01 Apr 2010
DOI:10.1039/C000815J
The 2-pyridinehydrazones (from condensation of pyridine-2-carbaldehyde and hydrazines) have previously been noted to have poor ligating ability as a result of a sterically demanding planar conformation. Destabilisation of this conformation is achieved through simple use of the ketohydrazones, and as a result the diamagnetic chiral tris-bidentate diimine complexes fac-[FeL3]2+ are readily isolated. In the solid state, inter-ligand π–π stacking and complex/counter-ion H-bonding are apparent, and these features persist in solution according to dynamic NMR spectra, which also indicate extremely high stereoselectivity for the fac isomers (>200:1). The compounds crystallise as conglomerates, and time-resolved CD spectra of non-racemic samples indicate a high degree of persistence of chirality (racemisation t1/2ca 77 min). Variations of solvent and counter-ion indicate that H-bonding is unimportant in determining the structure of the cation. The fac-selectivity arises in the induction of a chiral conformation in the coordinated ligand, and the fact that such non-planar ligands can only be accommodated about the Fe(II) centre if they all have the same absolute configuration. Adding a hydrazine N-methyl group increases the steric demand further, while retaining the novel non-planar conformation, and as a result paramagnetic chiral bis-bidentate complexes such as [FeL72(CH3CN)2]2+ are readily available.
Co-reporter:Nikola Paul Chmel, Suzanne E. Howson, Laura E. N. Allan, James Barker, Guy J. Clarkson, Scott S. Turner and Peter Scott
Dalton Transactions 2010 vol. 39(Issue 11) pp:2919-2927
Publication Date(Web):09 Feb 2010
DOI:10.1039/B924787D
The first organic-soluble, optically and diastereomerically pure EDDS metal complexes have been synthesised. A number of synthetic approaches were attempted, but finally the tetraphenylphosphonium series emerged as providing readily accessible compounds of trivalent Cr, Fe and Co in reasonable yields via the silver salts without the need to perform ion-exchange chromatography. The species PPh4[MIII(S,S-EDDS)] are very soluble in methanol, acetonitrile and even THF but isolation was facilitated by addition of stoichiometric water giving the highly crystalline but still conveniently soluble title compounds. The structures of the three isomorphous crystals comprise H2O-bridged extended hydrogen bonded structures with large channels occupied by the counterion molecules. The magnetic properties and circular dichroism spectra are reported along with comparative data for water-soluble NH4[FeIII(S,S-EDDS)]. Phase purity (and hence diastereomeric purity) in the paramagnetic systems is assessed through powder XRD. The practical utility of this type of compound was confirmed by optical resolution of (±)-[RuII(bpy)3]Cl2.
Co-reporter:Suzanne E. Howson, Laura E. N. Allan, Nikola P. Chmel, Guy J. Clarkson, Remy van Gorkum and Peter Scott
Chemical Communications 2009 (Issue 13) pp:1727-1729
Publication Date(Web):04 Feb 2009
DOI:10.1039/B821573A
Optically pure, single diastereomer fac-tris(diimine) complexes of Fe(II) are available from a remarkably facile one-pot procedure using a range of readily available (R)-2-phenylglycinol derivatives.
Co-reporter:Giles W. Theaker;Colin Morton
Journal of Polymer Science Part A: Polymer Chemistry 2009 Volume 47( Issue 12) pp:3111-3117
Publication Date(Web):
DOI:10.1002/pola.23417
Abstract
Homogeneous metallocene and CGC catalyzed copolymerizations of ethene (E) and various substituted styrenes are examined. It is found that those with p-tert-butylstyrene (TBS) occur with dramatically higher degrees of incorporation and overall productivity than those observed under the same conditions with styrene. It is argued that that the σ-donating effect of the tert-butyl substituent is responsible for this performance, effecting a destabilization of an otherwise dormant benzylic species following 2,1-insertion of the styrene. It was found possible to produce what is essentially TBS-alt-E (44% TBS with no sequential TBS units) using a standard constrained geometry catalyst at impressively high productivity [1.5 × 106 g/(molTi.barE.h)] and unusually low concentration of TBS (0.7 M). © 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 3111–3117, 2009
Co-reporter:Andrew L. Gott, Adam J. Clarke, Guy J. Clarkson and Peter Scott
Chemical Communications 2008 (Issue 12) pp:1422-1424
Publication Date(Web):15 Feb 2008
DOI:10.1039/B718373A
Chiral-at-metal half-sandwich diamide complexes catalyse enantioselective cyclohydroamination of aminoalkenes at unexpectedly high rates given their high coordination number and steric bulk; substantial evidence is presented which argues against the established σ-bond insertion process and is strongly indicative of an imido [2+2] cycloaddition mechanism.
Co-reporter:Giles W. Theaker, Colin Morton and Peter Scott
Dalton Transactions 2008 (Issue 48) pp:6883-6885
Publication Date(Web):10 Nov 2008
DOI:10.1039/B818829G
A group of readily available zirconium catalysts incapable of ethene-co-styrene polymerization are remarkably active and selective for the production of the new polymer ethene-co-tert-butylstyrenevia a single site mechanism.
Co-reporter:Andrew L. Gott, Guy J. Clarkson, Robert J. Deeth, Max. L. Hammond, Colin Morton and Peter Scott
Dalton Transactions 2008 (Issue 22) pp:2983-2990
Publication Date(Web):01 May 2008
DOI:10.1039/B803831G
DFT calculations indicate that contrary to a prior report, N-heterocycle-augmented constrained geometry ligands e.g.Cp′-SiMe2-NR (Cp′ = Me4C5, R = 2-pyridine) should be capable of binding both atoms of the diazaallyl fragment at zirconium. This was confirmed by a molecular structure of a previously reported complex. Similar R = 2-oxazoline complexes were also shown to be feasible, although an additional N,O binding mode was accessible. The proligands HCp′-SiMe2-NHR (R = chiral non-racemic 2-oxazoline) were readily synthesised in high yield via base mediated reaction of 2-aminooxazolines and Cp′-SiMe2Cl. Subsequent reaction with Zr(NMe2)4 gave, rather than the desired complexes, configurationally stable chiral-at-zirconum guanidinate/alkoxide chelate products; the aminooxazolinate units had undergone ring-opening and migratory insertion of –NMe2. Trends in the level diastereoselection follow the steric demand of the oxazoline substituent, with the larger groups (tBu, iPr) giving single diastereomers. The modest performance of these guanidinate compounds in enantioslective catalytic cyclohydroamination of aminoalkenes follows the expected trends for metal accessibility in a σ-amido insertative mechanism.
Co-reporter:Andrew L. Gott, Adam J. Clarke, Guy J. Clarkson, Ian J. Munslow, Andrew R. Wade and Peter Scott
Organometallics 2008 Volume 27(Issue 12) pp:2706-2714
Publication Date(Web):May 22, 2008
DOI:10.1021/om8002027
A series of half-sandwich complexes [Cp*MLX2] (L = four optically pure κ2-salicyloxazolines, M = Ti, Zr; X = Cl, Me) have been prepared via salt elimination and protonolysis routes. X-ray crystallographic analyses demonstrate that the complexes contain stereogenic metal centers as a result of diastereoselective coordination of the N−O ligand. This chirality persists in solution; NMR spectroscopic investigations revealed that for most members of both the Ti and Zr series a single diastereomeric species is present at all accessible temperatures. Systems with incomplete diastereoselection are also accessible by notionally moving the C-stereogenic center on the oxazoline ring to a position where its chirality is less well expressed in the structure of the complex. In one such case (for M = Zr) a lower limit barrier to epimerization of 80 kJ mol−1 is estimated at high temperature, while for an analogous Ti complex no exchange between epimers could be observed on the NMR chemical shift time scale. In contrast, unsubstituted cyclopentadienyl series CpTiLX2 gives diastereomeric mixtures in most cases and undergoes thermally accessible exchange between epimers.
Co-reporter:Robyn K. J. Bott, Max Hammond, Peter N. Horton, Simon J. Lancaster, Manfred Bochmann and Peter Scott
Dalton Transactions 2005 (Issue 22) pp:3611-3613
Publication Date(Web):14 Oct 2005
DOI:10.1039/B509807F
Octahedral titanium and zirconium complexes based on salicyloxazoline ligands with sterically demanding ortho-substituents provide a new family of extremely active ethene polymerization catalysts [up to 108 g PE (mol bar h)−1] which are in some cases “single site”.
Co-reporter:Paul D. Knight, Guy Clarkson, Max L. Hammond, Brian S. Kimberley, Peter Scott
Journal of Organometallic Chemistry 2005 Volume 690(Issue 23) pp:5125-5144
Publication Date(Web):15 November 2005
DOI:10.1016/j.jorganchem.2005.03.043
Four salicylaldimine derivatives H2L4–7 of 2,2′-diamino-6,6′-dimethylbiphenyl, where the CN bond is sterically protected by substituents on the phenol ring, form alkyls of zirconium, cis-α-[ZrL4–7(CH2Ph)2]. Rather than decomposing via the established pathway of 1,2-migratory insertion of an alkyl group to imine, they undergo a radical mechanism. This is evidenced by the large number of products observed, kinetic and thermodynamic data (Rice-Herfeld, 3/2 order, positive ΔS‡), response to steric factors, and the fact that switching to a less stable radical leaving group inhibits the reaction. In contrast, the 1,2-migratory insertion is a clean, first-order intramolecular process with negative ΔS‡. The steric modification of the ligands nevertheless transforms an inactive precatalyst into a stable system for the polymerisation of ethene. Closely related unbridged salicylaldimine catalysts are known to be highly active catalysts, but in most cases they appear to suffer from high temperature instability. The first examples of zirconium alkyls of this class are isolated, and it is found that they are inherently much more resistant to decomposition by either pathway (migratory insertion or radical). Structural studies are used to interpret this variance in behaviour; the biaryl-bridged complexes are pre-organised for both reactions, while the unbridged systems would have to undergo significant ordering prior to activation. Correspondingly, the unbridged systems are not noticeably affected by the same steric modification of the ligand, and it is concluded that the more likely mechanism of catalyst death in the latter is ligand loss (i.e. transfer to aluminium from co-catalyst).Steric blocking of migratory insertion in the title complexes opens up a new radical pathway. Detailed kinetic studies lead to the synthesis of a stable alkyl and a long-lived catalyst for ethene polymerisation. Studies on unbridged iminophenolate catalysts indicate that 1,2-migratory insertion is probably not responsible for their high temperature instability.
Co-reporter:Edward J. Crust, Ian J. Munslow, Peter Scott
Journal of Organometallic Chemistry 2005 Volume 690(Issue 14) pp:3373-3382
Publication Date(Web):15 July 2005
DOI:10.1016/j.jorganchem.2005.04.019
Amination of 1-bromo-2-methylpyridine with trans-1,2-diaminocyclohexane gives the corresponding bis(aminopyridine) H2L1. Conversion of the same diamine to the N,N′-bis(amino-4,4-dimethylthiazoline) H2L2 is also completed in three steps. The analogous aminooxazoline is however inaccessible, although the aminocyclohexane analogue is prepared readily. The proligand H2L1 forms bis(aminopyridinato) alkyl complexes of the type [ZrL1R2] (R = CH2Ph, CH2But). The molecular structure of the neopentyl complex shows that the chiral backbone leads to a puckering of the N4Zr coordination sphere, which contrasts with the related cyclohexyl-bridged Schiff-base complexes which are essentially planar. [ZrL2(CH2But)2] – the first aminothiazolinato complex – is formed similarly. A comparison of the structures of [ZrL1(CH2But)2] and [ZrL2(CH2But)2] indicates that the latter has a fully delocalised N–C–N system, rather similar to a bis(amidinate). Reaction of H2L2 with [Ti(NMe2)4] gives [TiL2(NMe2)2] which appears to be C2-symmetric like the above complexes according to NMR spectra, but has one uncoordinated thiazoline unit in the solid state. This is a result of increased ring strain at the smaller titanium metal centre.Bis(aminopyridinato) and bis(aminothiazolinato) ligands based on 1,2-diaminocyclohexane give complexes of zirconium in which the N4M unit is distorted from planarity by the presence of the chiral backbone. For the smaller metal Ti, X-ray crystallography shows that the constrained architecture leads to coordination of three N atoms only.
Co-reporter:Paul D. Knight, Ian Munslow, Paul N. O'Shaughnessy and Peter Scott
Chemical Communications 2004 (Issue 7) pp:894-895
Publication Date(Web):05 Mar 2004
DOI:10.1039/B401493F
A chiral zirconium alkyl cation catalyses the cyclisation of certain aminoalkenes with enantioselectivity up to 82%, the highest thus far observed for such a process.
Co-reporter:Ian J. Munslow, Andrew R. Wade, Robert J. Deeth and Peter Scott
Chemical Communications 2004 (Issue 22) pp:2596-2597
Publication Date(Web):30 Sep 2004
DOI:10.1039/B409113B
High levels of diastereoselection with respect to chirality-at-metal are achieved at equilibrium for complexes containing a new and available range of diazaallyl ligands.
Co-reporter:Edward J. Crust, Adam J. Clarke, Robert J. Deeth, Colin Morton and Peter Scott
Dalton Transactions 2004 (Issue 23) pp:4050-4058
Publication Date(Web):09 Nov 2004
DOI:10.1039/B413023E
Optically pure 2-alkylaminopyridines (HL) are synthesised readily from bromopyridines and chiral amines [(S)-1,2,3,4-tetrahydro-1-naphthylamine and (S)-(−)-α-methylbenzylamine] using palladium-catalysed amination. Protonolysis reactions of these proligands with ZrX4
(X = NMe2, CH2Ph, CH2But) yield zirconium aminopyridinates, usually of the type [ML2X2], some of which have been characterised by X-ray crystallography. Control of absolute configuration at the metal centre is pursued by investigation of the effects of chiral amine substituent, substitution at the pyridine rings and the identity of co-ligands. Surprisingly the conformationally flexible α-methylbenzyl based aminopyridinato ligands promote much better control of chirality-at-zirconium than do the cyclic tetrahydronaphthyl analogues. One complex of the former class displays complete control of stereochemistry at 193 K; only one diastereomer out of eight possible structures is observed. It is found that there is an excellent correlation between observed selectivities and calculated diastereomer energy differences from DFT. All the complexes studied are in dynamic exchange between diastereomers. The rate of these processes (ΔH‡ca. 40 kJ mol−1) as studied by Selective Polarisation Transfer–Selective Inversion Recovery experiments (SPT-SIR) and lineshape analyses are significantly faster than those for aminopyridines containing bulkier amido substituents (ΔH‡ca. 70 kJ mol−1). This type of dependence on steric effects, and the impact of the trans effect, is consistent with an N-dissociative mechanism, i.e. conversion from six- to five-coordinate structure followed by rapid intramolecular scrambling.
Co-reporter:Edward J. Crust, Ian J. Munslow, Colin Morton and Peter Scott
Dalton Transactions 2004 (Issue 15) pp:2257-2266
Publication Date(Web):29 Jun 2004
DOI:10.1039/B407008A
A range of 2-arylaminopyridines (HL) are synthesised readily from bromopyridines and amines using palladium-catalysed amination. Protonolysis reactions of these proligands with ZrX4
(X = NMe2, CH2Ph, CH2But) yield zirconium complexes of the type [MLnX4−n], several of which have been characterised by X-ray crystallography. Control of metal/ligand stoichiometry and structure is pursued by investigation of the effects on substitution patterns of the pyridine and aryl rings. Some distinct patterns emerged; (i) the 6-methyl position on the pyridine appears to be particularly important with regards to control of stoichiometry, although there are co-ligand effects; (ii) structures of the metal alkyl derivatives [ZrLn(CH2R)4−n] are dominated by aromatic π–π stacking, even when bulky arene substituents are employed at L. This leads to the complexes adopting a C2v-symmetric core; (iii) the amides [ZrL2(NMe2)2] have structures for which aromatic π–π stacking is unfeasible, and correspondingly C2-symmetric or similar structures are adopted. All the structural data presented is consistent with a trans influence order at zirconium Me2N > RCH2 > py.
Co-reporter:Paul N. O'Shaughnessy, Kevin M. Gillespie, Paul D. Knight, Ian J. Munslow and Peter Scott
Dalton Transactions 2004 (Issue 15) pp:2251-2256
Publication Date(Web):28 Jun 2004
DOI:10.1039/B400799A
A group of chiral, dibasic, biaryl-bridged amido proligands containing peripheral methoxyphenyl (anisole) ligation are developed for the synthesis of new amide complexes of yttrium and lanthanum. A potentially tetradentate bis(amidoanisole) system L111 gives, on reaction with [Y{N(SiMe2H)2}3(THF)] a crystallographically-characterised bis complex [YL111(HL111)] presumably as a result of low steric demand, since a more bulky version L222 gives the target [L222Y{N(SiMe2H)2}(THF)]. The molecular structure of the latter reveals a similar cis-α structure to our recently reported Schiff-base analogue. Variable-temperature NMR studies are consistent with low rigidity in the molecular structure. A potentially tridentate, amidoanisolyl/amido proligand L333 gives complexes [L333M{N(SiMe2H)2}(THF)n]
(M = Y, n
= 1; M = La, n
= 2). Chiral non-racemic versions of the above complexes were tested in the hydroamination/cyclisation of 2,2′-dimethylaminopentane to the corresponding pyrrolidine. Activities were relatively low compared to recently reported examples, and ee values were in the range 20–40% despite the well-expressed chirality of the catalysts.
Co-reporter:Paul N. O'Shaughnessy, Paul D. Knight, Colin Morton, Kevin M. Gillespie and Peter Scott
Chemical Communications 2003 (Issue 14) pp:1770-1771
Publication Date(Web):18 Jun 2003
DOI:10.1039/B305105F
Chiral non-racemic complexes [ML{N(SiMe2H)2}(thf)]
(M = Y, La, H2L = salicylaldimine ligands derived from 2,2′-diamino-6,6′-dimethylbiphenyl) are found not to be effective catalysts for the intramolecular hydroamination of aminoalkenes, but new amino/phenoxide ligand designs without reducible functional groups led to long-lived and enantioselective catalysts.
Co-reporter:Paul D Knight, Paul N O'Shaughnessy, Ian J Munslow, Brian S Kimberley, Peter Scott
Journal of Organometallic Chemistry 2003 Volume 683(Issue 1) pp:103-113
Publication Date(Web):7 October 2003
DOI:10.1016/S0022-328X(03)00455-8
Three substituted salicylaldimine derivatives H2L1–3 of 2,2′-diamino-6,6′-dimethylbiphenyl give, under appropriate conditions, isolable alkyls of zirconium [ZrL1–3R2] (R=CH2Ph, CH2But). Two molecular structures confirm their cis-α geometry (C2-symmetric with cis alkyl ligands). They decompose via 1,2-migratory insertion of an alkyl group to imine, followed in some instances by a second similar reaction. The dimeric molecular structure of one such doubly-inserted product is presented. The kinetics of decomposition by this process are studied briefly, and it is noted that the rate increases with increased steric demand of the salicylaldimine unit.Salicylaldimine derivatives H2L of 2,2′-diamino-6,6′-dimethylbiphenyl give isolable alkyls of zirconium [ZrLR2] (R=CH2Ph, CH2But). Molecular structures confirm their cis-α geometry. They decompose via 1,2-migratory insertion of an alkyl group to imine, followed in some instances by a second similar reaction. A brief kinetic study indicates that the rate increases with increased steric demand of R1.
Co-reporter:P.N. O'Shaughnessy, Peter Scott
Tetrahedron: Asymmetry 2003 Volume 14(Issue 14) pp:1979-1983
Publication Date(Web):18 July 2003
DOI:10.1016/S0957-4166(03)00429-4
Arylations of (R)-2,2′-diamino-6,6′-dimethylbiphenyl and the corresponding racemic compound under palladium catalysis gives a range of C2-symmetric secondary amine proligands H2L. The 3,5-di-tert-butylphenyl substituted compound reacts with the silylamides [M{N(SiMe2H)2}3(THF)2] (M=Y, Sm, La) to produce (±)-[ML{N(SiMe2H)2}(THF)2]. However, deprotonation of more sterically hindered proligands H2L proceeded very slowly, but the reaction with the more basic akylamide [Y(NiPr2)3(THF)2] gave [YL1-4(NiPr2)(THF)2] readily. Such catalysts formed in situ from the amine proligand H2L and metal amide, cyclised 2,2′-dimethylaminopent-4-ene to the corresponding pyrrolidine with enantiomeric excesses up to 50%. The alkylamide catalysts gave significantly faster turnover than the silylamides.GraphicN,N′-Bis(2-ethylphenyl)-6,6′-dimethylbiphenyl-2,2′-diamineC30H32N2E.e.=99.5% [by HPLC analysis of a precursor][α]D24.2=−147 (c 0.20, CHCl3)Source of chirality: (R)-(+)-2,2′-diamino-6,6′-dimethylbiphenylAbsolute configuration: RN,N′-Bis(2-cyclohexylphenyl)-6,6′-dimethylbiphenyl-2,2′-diamineC38H44N2E.e.=99.5% [by HPLC analysis of a precursor][α]D24.5=−152 (c 0.20, CHCl3)Source of chirality: (R)-(+)-2,2′-diamino-6,6′-dimethylbiphenylAbsolute configuration: RN,N′-Bis(2-methyl-5-tert-butylphenyl)-6,6′-dimethylbiphenyl-2,2′-diamineC36H44N2E.e.=99.5% [by HPLC analysis of a precursor][α]D24.7=−158 (c 0.20, CHCl3)Source of chirality: (R)-(+)-2,2′-diamino-6,6′-dimethylbiphenylAbsolute configuration: R
Co-reporter:Ian J. Munslow, Adam J. Clarke, Robert J. Deeth, Ian Westmoreland and Peter Scott
Chemical Communications 2002 (Issue 17) pp:1868-1869
Publication Date(Web):01 Aug 2002
DOI:10.1039/B205043A
Only one of eight possible diastereomers of the organometallic chiral-at-metal complex [ZrL2(CH2Ph)2] (L = a bidentate, chiral non-racemic pyrdine alcoholate) is observed by NMR spectroscopy in the slow exchange regime.
Co-reporter:Paul D. Knight, Adam J. Clarke, Brian S. Kimberley, Richard A. Jackson and Peter Scott
Chemical Communications 2002 (Issue 4) pp:352-353
Publication Date(Web):29 Jan 2002
DOI:10.1039/B110423N
Steric blocking of an intramolecular 1,2-migratory insertion reaction of a zirconium salicylaldiminato complex leads to a long-lived catalyst for ethene polymerisation, but promotes a new radical catalyst decomposition mechanism in certain instances; kinetic and thermodynamic parameters for both pathways have been established.
Co-reporter:Paul Roussel, Rita Boaretto, Andrew J. Kingsley, Nathaniel W. Alcock and Peter Scott
Dalton Transactions 2002 (Issue 7) pp:1423-1428
Publication Date(Web):12 Mar 2002
DOI:10.1039/B108584K
Reduction of [U(NN′3)I] [NN′3
= N(CH2CH2NSiMe2But)3] with potassium in pentane gives the purple trivalent monomer [U(NN′3)] in good yield, this compound having previously been synthesised via fractional vacuum sublimation of mixed-valent [{U(NN′3)}2(μ-Cl)]. The magnetic susceptibility of this compound is consistent with the presence of U(III) centres, and this is confirmed by a characteristic near IR spectrum. Its reactions with Lewis bases to give e.g. [U(NN′3)(Py)] and [U(NN′3)(HMPA)] are reported, along with the molecular structure of the latter. The complex [U(NN′3)] is readily oxidised, imido and hydrazido complexes being formed readily by reaction with trimethylsilyl-azide and -diazomethane, respectively. The reaction with methylene trimethylphosproane
however led to the formation of an addition compound [U(NN′3)(CH2PMe3)]. Reaction of this latter complex with air gave a few crystals of the unusual hydroxo complex [U(NN′3)(OH)(CH2PMe3)] which was structurally characterised. Reaction of [U(NN′3)(CH2PMe3)] with trimethylamine N-oxide gave pentavalent [U(NN′3)(O)], or perhaps a dimer thereof. The latter complex reacted with [U(NN′3)] to give the bridging oxo complex [{U(NN′3)}2(μ-O)] which could also be prepared directly by reaction of trimethylamine N-oxide with [U(NN′3)].
Co-reporter:Ian J. Munslow, Kevin M. Gillespie, Robert J. Deeth and Peter Scott
Chemical Communications 2001 (Issue 17) pp:1638-1639
Publication Date(Web):14 Aug 2001
DOI:10.1039/B104964J
Exceptionally high stereoselectivity (ee ⩽ 98%, dr ⩽
99∶1) in the cyclopropanation of alkenes with ethyl diazoacetate
using a non-planar ruthenium(II) Schiff-base precatalyst is a
result of η2C,O binding of the carbenoid ester
intermediate, according to DFT calculations.
Co-reporter:Kevin M. Gillespie, Edward J. Crust, Robert J. Deeth and Peter Scott
Chemical Communications 2001 (Issue 8) pp:785-786
Publication Date(Web):04 Apr 2001
DOI:10.1039/B101415N
The highly enantioselective (⩽98%) aziridination of
cinnamate esters is achieved using the title catalyst system via a
concerted non-polar mechanism involving ancillary binding of carbonyl group
to copper.
Co-reporter:Paul Roussel, William Errington, Nikolas Kaltsoyannis, Peter Scott
Journal of Organometallic Chemistry 2001 Volume 635(1–2) pp:69-74
Publication Date(Web):15 October 2001
DOI:10.1016/S0022-328X(01)00804-X
The synthesis, molecular structure, spectroscopic properties and bonding in a complex of dinitrogen with trivalent uranium are described. The molecule has a side-on U–N2–U core which is supported by U→N2 π donation with no significant N2→U bonding. Steric compression between the spectator triamidoamine ligands prevents the uranium atoms from approaching the N2 ligand at the optimum distance for overlap, and thus the N–N distance is not displaced significantly from that in gaseous dinitrogen.The synthesis, structure and bonding in a complex of dinitrogen with trivalent uranium are described. The side-on U–N2–U core is supported by U→N2 π donation with no significant N2→U bonding. Steric compression between triamidoamine ligands prevents the uranium atoms from approaching N2 for optimum orbital overlap, and thus the N–N distance is not displaced significantly from that in gaseous dinitrogen.
Co-reporter:Colin Morton, Paul O’Shaughnessy and Peter Scott
Chemical Communications 2000 (Issue 21) pp:2099-2100
Publication Date(Web):12 Oct 2000
DOI:10.1039/B006603F
N-Adamantyl-2-aminopyridines (HL) readily form
C2-symmetric aminopyridinato complexes with zirconium
[ZrL2X2] (X = Cl, NMe2, CH2Ph,
CH2But) which are stable with respect to ligand
redistribution and lead to catalysts for ethylene polymerisation with
similar productivity to the related
[Zr(benzamidinate)2X2] system.
Co-reporter:Suzanne E. Howson, Nikola P. Chmel, Guy J. Clarkson, Robert J. Deeth, Daniel H. Simpson and Peter Scott
Dalton Transactions 2012 - vol. 41(Issue 15) pp:NaN4483-4483
Publication Date(Web):2012/01/13
DOI:10.1039/C2DT12378A
Optically pure phenylethaniminopyridine (SC-L) tris-chelates of Fe(II) and other first row transition metal systems have previously been shown to give exclusively the fac structures in the solid state. Here it is shown by powder X-ray diffraction that the complex [CuL3][ClO4]2 crystallises exclusively as the mer isomer, although – for a given absolute configuration of the ligand – of the same helicity (Δ/Λ) as that displayed by the other metal complexes. The similar ligand RC-LFF, which contains a peripheral 19F spin label, gave [CuLFF3][ClO4]2 which also adopts exclusively the mer structure in the crystal, but is shown by NMR spectroscopy to have a fac:mer ratio of 1:6 in solution at low temperature. Molecular mechanics calculations for a number of isomers and conformers are consistent with the presence of such a mixture of isomers in solution for both complexes. The origin of the difference in behaviour between Fe(II) and Cu(II) is the presence of a Jahn–Teller distortion (and the generally longer M–N bonds) in the Cu(II) complexes. This disturbs intra-ligand π-stacking, leading to the poor fac/mer stereoselectivity while leaving enantioselectivity Δ/Λ apparently unaffected.
Co-reporter:Suzanne E. Howson, Laura E. N. Allan, Nikola P. Chmel, Guy J. Clarkson, Robert J. Deeth, Alan D. Faulkner, Daniel H. Simpson and Peter Scott
Dalton Transactions 2011 - vol. 40(Issue 40) pp:NaN10433-10433
Publication Date(Web):2011/07/11
DOI:10.1039/C1DT10588D
One-pot reactions of 2-pyridinecarboxaldehyde, chiral phenylethanamines and Fe(II) give single diastereomer facdiimine complexes at thermodynamic equilibrium so that no chiral separations are required (d.r. > 200:1). The origins of this stereoselectivity are partly steric and partly a result of the presence of three sets of inter-ligand parallel-offset π-stacking interactions. Mn(II), Co(II), Co(III), Ni(II) and Zn(II) give similar fac structures, alongside the imidazole analogues for Fe(II). While most of the complexes are paramagnetic, the series of molecular structures allows us to assess the influence of the π-stacking present, and there is a strong correlation between this and the M–N bond length. Fe(II) is close to optimal. For the larger Zn(II) ion, very weak π-stacking leads to poorer measured stereoselectivity (NMR) but this is improved with increased solvent polarity. The mechanism of stereoselection is further investigated via DFT calculations, chiroptical spectroscopy and the use of synthetic probes.
Co-reporter:Jan M. Becker, James Barker, Guy J. Clarkson, Remy van Gorkum, Gursharon K. Johal, Richard I. Walton and Peter Scott
Dalton Transactions 2010 - vol. 39(Issue 9) pp:NaN2326-2326
Publication Date(Web):2010/01/14
DOI:10.1039/B905706D
A number of bis(salicylaldiminato) (Schiff base) complexes of Fe(II) i.e. L2Fe with a variety of ligand substituents and functionality are synthesised, usually by salt metathesis but on one occasion via the (sal)2Fe complex, and characterised by standard methods. Two molecular structure determinations indicate the expected tetrahedral geometry. Treatment with dry oxygen yielded the Fe(III) complexes [(FeL2)2-μ-O], generally in high yield and in the absence of hydroxylated impurities; one bulky complex L2Fe gave no oxidation product for steric reasons, and one hydroxyl-substituted compound gave an insoluble product. Five molecular structures of these [(FeL2)2-μ-O] compounds were determined. All were homochiral in the sense that the absolute configuration at Fe was the same for both atoms. Three of the structures – those based on optically pure salicylaldimine ligands – were diastereomeric. For two, the diastereomers observed are those predicted on the basis of steric considerations, while a third highly distorted and congested structure represents the unexpected diastereomer. Powder X-ray diffraction confirms high phase purity of the bulk samples (i.e. they are diastereomerically pure to the level detectable). These investigations, coupled with molecular structure and NMR studies on model gallium complexes, indicate that the diastereoselection arises from cooperative interactions across the oxo bridge.
Co-reporter:Nikola Paul Chmel, Suzanne E. Howson, Laura E. N. Allan, James Barker, Guy J. Clarkson, Scott S. Turner and Peter Scott
Dalton Transactions 2010 - vol. 39(Issue 11) pp:NaN2927-2927
Publication Date(Web):2010/02/09
DOI:10.1039/B924787D
The first organic-soluble, optically and diastereomerically pure EDDS metal complexes have been synthesised. A number of synthetic approaches were attempted, but finally the tetraphenylphosphonium series emerged as providing readily accessible compounds of trivalent Cr, Fe and Co in reasonable yields via the silver salts without the need to perform ion-exchange chromatography. The species PPh4[MIII(S,S-EDDS)] are very soluble in methanol, acetonitrile and even THF but isolation was facilitated by addition of stoichiometric water giving the highly crystalline but still conveniently soluble title compounds. The structures of the three isomorphous crystals comprise H2O-bridged extended hydrogen bonded structures with large channels occupied by the counterion molecules. The magnetic properties and circular dichroism spectra are reported along with comparative data for water-soluble NH4[FeIII(S,S-EDDS)]. Phase purity (and hence diastereomeric purity) in the paramagnetic systems is assessed through powder XRD. The practical utility of this type of compound was confirmed by optical resolution of (±)-[RuII(bpy)3]Cl2.
Co-reporter:Suzanne E. Howson and Peter Scott
Dalton Transactions 2011 - vol. 40(Issue 16) pp:NaN4333-4333
Publication Date(Web):2011/03/15
DOI:10.1039/C0DT01529F
Intramolecular π-stacking plays a role in diastereomerically pure Fe(II) complexes of certain pyridine/imines, but contrary to a recent report, no such interaction is present in the Cu(II) analogues and the stereoselectivity is as yet unknown.
Co-reporter:Viktor Brabec, Suzanne E. Howson, Rebecca A. Kaner, Rianne M. Lord, Jaroslav Malina, Roger M. Phillips, Qasem M. A. Abdallah, Patrick C. McGowan, Alison Rodger and Peter Scott
Chemical Science (2010-Present) 2013 - vol. 4(Issue 12) pp:NaN4416-4416
Publication Date(Web):2013/09/10
DOI:10.1039/C3SC51731D
Enantiomers of a relatively rigid DNA-binding metallo-helix are shown to have comparable activity to that of cisplatin against the cell lines MCF7 (human breast adenocarcinoma) and A2780 (human ovarian carcinoma) but are ca five times more active against the cisplatin-resistant A2780cis. The cell-line HCT116 p53+/+ (human colon carcinoma) is highly sensitive giving IC50 values in the nM range, far lower than the cisplatin control. The hypothesis that the biological target of such metallohelices is DNA is probed by various techniques. Tertiary structure changes in ct-DNA (formation of loops and intramolecular coiling) on exposure to the compounds are demonstrated by atomic force microscopy and supported by circular/linear dichroism in solution. Selectivity for 5′-CACATA and 5′-CACTAT segments is shown by DNase I footprinting. Various three- and four-way oligonucleotide junctions are stabilised, and remarkably only the Λ metallo-helix enantiomer stabilizes T-shaped 3WJs during gel electrophoresis; this is despite the lack of a known helix binding site. In studies with oligonucleotide duplexes with bulges it is also shown for the first time that the metallo-helix binding strength and the number of binding sites are dependent on the size of the bulge. In contrast to all the above, flexible metallo-helices show little propensity for structured or selective DNA binding, and while for A2780 the cancer cell line cytotoxicity is retained the A2780cis strain shows significant resistance. For all compounds in the study, H2AX FACS assays on HCT116 p53+/+ showed that no significant DNA damage occurs. In contrast, cell cycle analysis shows that the DNA binders arrest cells in the G2/mitosis phase, and while all compounds cause apoptosis, the DNA binders have the greater effect. Taken together these screening and mechanistic results are consistent with the more rigid helices acting via a DNA binding mechanism while the flexible assemblies do not.
Co-reporter:Rebecca A. Kaner, Simon J. Allison, Alan D. Faulkner, Roger M. Phillips, David I. Roper, Samantha L. Shepherd, Daniel H. Simpson, Nicholas R. Waterfield and Peter Scott
Chemical Science (2010-Present) 2016 - vol. 7(Issue 2) pp:NaN958-958
Publication Date(Web):2015/10/26
DOI:10.1039/C5SC03677A
A range of new helicate-like architectures have been prepared via highly diastereoselective self-assembly using readily accessible starting materials. Six pairs of enantiomers [Fe2L3]Cl4·nH2O (L = various bidentate ditopic ligands NN–NN) show very good water solubility and stability. Their activity against a range of cancer cell lines in vitro is structure-dependent and gives IC50 values as low as 40 nM. In an isogenic pair of HCT116 colorectal cancer cells, preferential activity was observed against cell lines that lack functional p53. Selectivity is also excellent, and against healthy human retinal pigment epithelial (ARPE19) and lung fibroblast (WI38) cells IC50 values are nearly three orders of magnitude higher. Cisplatin is unselective in the same tests. The compounds also appear to have low general toxicity in a number of models: there is little if any antimicrobial activity against methicillin-resistant Staphylococcus aureus and Escherichia coli; Acanthamoeba polyphaga is unaffected at 25 μg mL−1 (12.5 μM); Manduca sexta larvae showed clear evidence of systemic distribution of the drug, and rather than any observation of adverse effects they exhibited a significant mean weight gain vs. controls. Investigation of the mode of action revealed no significant interaction of the molecules with DNA, and stimulation of substantial cell death by apoptosis.
Co-reporter:Suzanne E. Howson;Laura E. N. Allan;Nikola P. Chmel;Guy J. Clarkson;Remy van Gorkum
Chemical Communications 2009(Issue 13) pp:
Publication Date(Web):2009/03/16
DOI:10.1039/B821573A
Optically pure, single diastereomer fac-tris(diimine) complexes of Fe(II) are available from a remarkably facile one-pot procedure using a range of readily available (R)-2-phenylglycinol derivatives.
Co-reporter:Suzanne E. Howson, Guy J. Clarkson, Alan D. Faulkner, Rebecca A. Kaner, Michael J. Whitmore and Peter Scott
Dalton Transactions 2013 - vol. 42(Issue 42) pp:NaN14981-14981
Publication Date(Web):2013/08/01
DOI:10.1039/C3DT51725J
Single diastereomer, diamagnetic, octahedral Fe(II) tris chelate complexes are synthesised that contain three pendant pyridine proligands pre-organised for coordination to a second metal. They bind Cu(I) and Ag(I) with coordination geometry depending on the identity of the metal and the detail of the ligand structure, but for example homohelical (ΔFe,ΔCu) configured systems with unusual trigonal planar Cu cations are formed exclusively in solution as shown by VT-NMR and supported by DFT calculations. Similar heterobimetallic tris(triazole) complexes are synthesised via clean CuAAC reactions at a tris(alkynyl) complex, although here the configurations of the two metals differ (ΔFe,ΛCu), leading to the first optically pure heterohelicates. A second series of Fe complexes perform less well in either strategy as a result of lack of preorganisation.
Co-reporter:Nikola Paul Chmel, Laura E. N. Allan, Jan M. Becker, Guy J. Clarkson, Scott S. Turner and Peter Scott
Dalton Transactions 2011 - vol. 40(Issue 8) pp:NaN1731-1731
Publication Date(Web):2011/01/21
DOI:10.1039/C0DT01184C
Optically pure anionic complexes of pyridinecarboxamide ligands, N2,N6-bis((R)-α-methylbenzyl)pyridine-2, 6-dicarboxamide H2(R,R-L1) and N2,N6-bis((S)-1-methoxypropan-2-yl)pyridine-2, 6-dicarboxamide H2(S,S-L2) have been synthesised and fully characterised. The complexes: (18-crown-6)K[CoIII(R,R-L1)2], (18-crown-6)K[FeIII(R,R-L1)2] and K[CoIII(S,S-L2)2]·3H2O show interesting extended structures from 0D discrete units through 1D zigzag chains to 2D honeycomb layers. The complex anions were used in the synthesis of radical cation salts with tetrathiafulvalene (TTF). The salts (TTF)[CoIII(R,R-L1)2] and (TTF)[CoIII(S,S-L2)2]·EtOAc were characterised by single crystal X-ray diffraction and conductivity measurements. Both compounds comprise mono-oxidised TTF molecules and exhibit similar layered structures with no direct TTF stacking but in which phenyl substituents from the complex anion or co-crystallised ethyl acetate alternate with TTF+ units. Solution spectroscopic and cyclic voltammetric evidence points to the formation of soluble assemblies between TTF+ and the counterion which correspond to the stoichiometry observed by crystallography and other methods in the solid state.
Co-reporter:Lihong Li, Guy J. Clarkson, David J. Evans, Martin R. Lees, Scott S. Turner and Peter Scott
Chemical Communications 2011 - vol. 47(Issue 47) pp:NaN12648-12648
Publication Date(Web):2011/10/31
DOI:10.1039/C1CC15574A
Two Fe(II) coordination polymers formed from isomeric ligands give a diamond-like 3D network exhibiting a gradual SCO and a 2D hard magnet with a large coercive field.
Co-reporter:Andrew L. Gott, Adam J. Clarke, Guy J. Clarkson and Peter Scott
Chemical Communications 2008(Issue 12) pp:
Publication Date(Web):
DOI:10.1039/B718373A
Co-reporter:Giles W. Theaker, Colin Morton and Peter Scott
Dalton Transactions 2008(Issue 48) pp:NaN6885-6885
Publication Date(Web):2008/11/10
DOI:10.1039/B818829G
A group of readily available zirconium catalysts incapable of ethene-co-styrene polymerization are remarkably active and selective for the production of the new polymer ethene-co-tert-butylstyrenevia a single site mechanism.
Co-reporter:Andrew L. Gott, Guy J. Clarkson, Robert J. Deeth, Max. L. Hammond, Colin Morton and Peter Scott
Dalton Transactions 2008(Issue 22) pp:NaN2990-2990
Publication Date(Web):2008/05/01
DOI:10.1039/B803831G
DFT calculations indicate that contrary to a prior report, N-heterocycle-augmented constrained geometry ligands e.g.Cp′-SiMe2-NR (Cp′ = Me4C5, R = 2-pyridine) should be capable of binding both atoms of the diazaallyl fragment at zirconium. This was confirmed by a molecular structure of a previously reported complex. Similar R = 2-oxazoline complexes were also shown to be feasible, although an additional N,O binding mode was accessible. The proligands HCp′-SiMe2-NHR (R = chiral non-racemic 2-oxazoline) were readily synthesised in high yield via base mediated reaction of 2-aminooxazolines and Cp′-SiMe2Cl. Subsequent reaction with Zr(NMe2)4 gave, rather than the desired complexes, configurationally stable chiral-at-zirconum guanidinate/alkoxide chelate products; the aminooxazolinate units had undergone ring-opening and migratory insertion of –NMe2. Trends in the level diastereoselection follow the steric demand of the oxazoline substituent, with the larger groups (tBu, iPr) giving single diastereomers. The modest performance of these guanidinate compounds in enantioslective catalytic cyclohydroamination of aminoalkenes follows the expected trends for metal accessibility in a σ-amido insertative mechanism.
Co-reporter:Suzanne E. Howson and Peter Scott
Dalton Transactions 2011 - vol. 40(Issue 40) pp:NaN10277-10277
Publication Date(Web):2011/09/15
DOI:10.1039/C1DT11423A
Methods by which helical bimetallic assemblies may be accessed as single isomers are described and reviewed. While there are a number of notable successes, the conclusion is made that no existing approach satisfies the criteria which will allow these fascinating compounds to emerge from the synthetic research laboratory environment to practical application in e.g. healthcare. For this to be achieved the compounds must be optically pure and non-racemising, soluble and chemically stable in water, readily available on a large scale, chemically diverse and synthetically robust.
Co-reporter:Cynthia Paul Sebli, Suzanne E. Howson, Guy J. Clarkson and Peter Scott
Dalton Transactions 2010 - vol. 39(Issue 18) pp:NaN4454-4454
Publication Date(Web):2010/04/01
DOI:10.1039/C000815J
The 2-pyridinehydrazones (from condensation of pyridine-2-carbaldehyde and hydrazines) have previously been noted to have poor ligating ability as a result of a sterically demanding planar conformation. Destabilisation of this conformation is achieved through simple use of the ketohydrazones, and as a result the diamagnetic chiral tris-bidentate diimine complexes fac-[FeL3]2+ are readily isolated. In the solid state, inter-ligand π–π stacking and complex/counter-ion H-bonding are apparent, and these features persist in solution according to dynamic NMR spectra, which also indicate extremely high stereoselectivity for the fac isomers (>200:1). The compounds crystallise as conglomerates, and time-resolved CD spectra of non-racemic samples indicate a high degree of persistence of chirality (racemisation t1/2ca 77 min). Variations of solvent and counter-ion indicate that H-bonding is unimportant in determining the structure of the cation. The fac-selectivity arises in the induction of a chiral conformation in the coordinated ligand, and the fact that such non-planar ligands can only be accommodated about the Fe(II) centre if they all have the same absolute configuration. Adding a hydrazine N-methyl group increases the steric demand further, while retaining the novel non-planar conformation, and as a result paramagnetic chiral bis-bidentate complexes such as [FeL72(CH3CN)2]2+ are readily available.

![Phenol, 2-[(4S)-4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]-4,6-bis(1,1-dimethylethyl)-](/data/chemimg/3721600/372137-88-7.png)
![Phenol, 2-[(4S)-4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]-4,6-bis(1,1-dimethylethyl)-](/data/chemimg/3721600/372137-88-7_b.png)


![2-Pyridinamine, N-[(1S)-1,2,3,4-tetrahydro-1-naphthalenyl]-](http://img.cochemist.com/ccimg/830400/830323-16-5.png)
![2-Pyridinamine, N-[(1S)-1,2,3,4-tetrahydro-1-naphthalenyl]-](http://img.cochemist.com/ccimg/830400/830323-16-5_b.png)

![5H-Benzo[cd]pyren-5-one](http://img.cochemist.com/ccimg/4600/4558-16-1.png)
![5H-Benzo[cd]pyren-5-one](http://img.cochemist.com/ccimg/4600/4558-16-1_b.png)


![Phenol, 2-[[[(1R)-1-phenylethyl]imino]methyl]-](http://img.cochemist.com/ccimg/78500/78419-84-8.png)
![Phenol, 2-[[[(1R)-1-phenylethyl]imino]methyl]-](http://img.cochemist.com/ccimg/78500/78419-84-8_b.png)
![3H-Benzo[cd]pyrene, 4,5-dihydro-](http://img.cochemist.com/ccimg/7200/7130-15-6.png)
![3H-Benzo[cd]pyrene, 4,5-dihydro-](http://img.cochemist.com/ccimg/7200/7130-15-6_b.png)









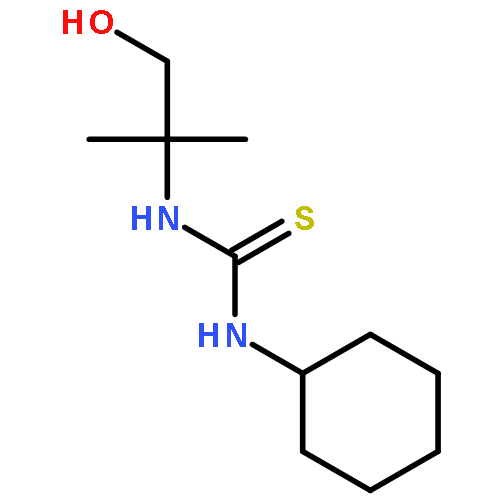
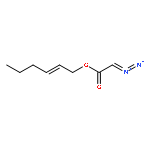
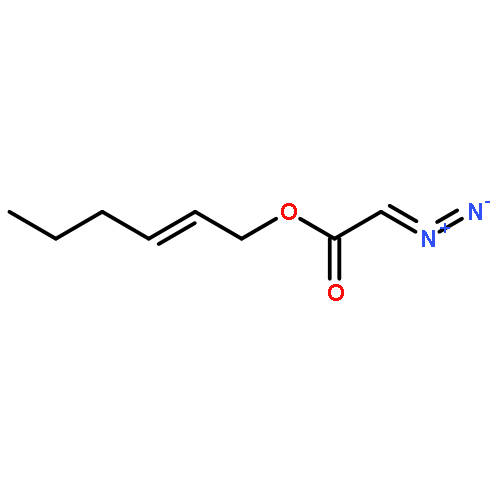




![2-Pyridinamine, 6-methyl-N-tricyclo[3.3.1.13,7]dec-1-yl-](http://img.cochemist.com/ccimg/314100/314000-43-6.png)
![2-Pyridinamine, 6-methyl-N-tricyclo[3.3.1.13,7]dec-1-yl-](http://img.cochemist.com/ccimg/314100/314000-43-6_b.png)


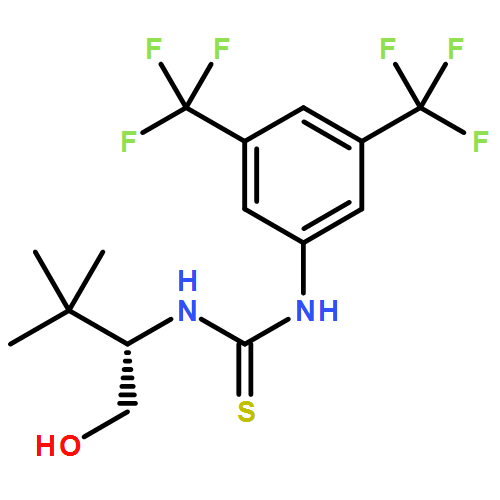
![THIOUREA, N-[(1S)-2-HYDROXY-1-PHENYLETHYL]-N'-PHENYL-](http://img.cochemist.com/ccimg/821800/821775-11-5.png)
![THIOUREA, N-[(1S)-2-HYDROXY-1-PHENYLETHYL]-N'-PHENYL-](http://img.cochemist.com/ccimg/821800/821775-11-5_b.png)
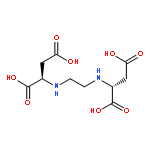

![2-PYRIDINAMINE, N-[3,5-BIS(1,1-DIMETHYLETHYL)PHENYL]-](http://img.cochemist.com/ccimg/758800/758705-80-5.png)
![2-PYRIDINAMINE, N-[3,5-BIS(1,1-DIMETHYLETHYL)PHENYL]-](http://img.cochemist.com/ccimg/758800/758705-80-5_b.png)
![7H-CYCLOPENTA[A]PYRENE, 8,9-DIHYDRO-](http://img.cochemist.com/ccimg/83000/82979-72-4.png)
![7H-CYCLOPENTA[A]PYRENE, 8,9-DIHYDRO-](http://img.cochemist.com/ccimg/83000/82979-72-4_b.png)


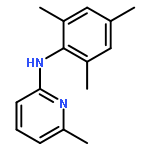
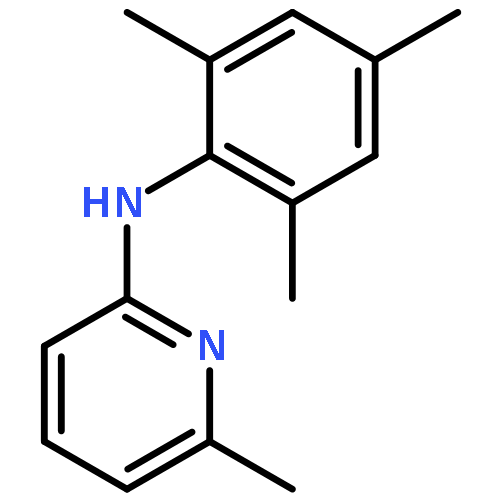
![2-PYRIDINAMINE, 6-METHYL-N-[2-(1-METHYLETHYL)PHENYL]-](http://img.cochemist.com/ccimg/758800/758705-82-7.png)
![2-PYRIDINAMINE, 6-METHYL-N-[2-(1-METHYLETHYL)PHENYL]-](http://img.cochemist.com/ccimg/758800/758705-82-7_b.png)




![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2,3-diphenyl-, cis-](http://img.cochemist.com/ccimg/17900/17879-98-0.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2,3-diphenyl-, cis-](http://img.cochemist.com/ccimg/17900/17879-98-0_b.png)

![Phenol, 4-methoxy-2-[[[(1R)-1-phenylethyl]imino]methyl]-](http://img.cochemist.com/ccimg/393200/393110-01-5.png)
![Phenol, 4-methoxy-2-[[[(1R)-1-phenylethyl]imino]methyl]-](http://img.cochemist.com/ccimg/393200/393110-01-5_b.png)
![Phenol, 4-nitro-2-[[[(1R)-1-phenylethyl]imino]methyl]-](http://img.cochemist.com/ccimg/393200/393110-00-4.png)
![Phenol, 4-nitro-2-[[[(1R)-1-phenylethyl]imino]methyl]-](http://img.cochemist.com/ccimg/393200/393110-00-4_b.png)

![6H-Benzo[cd]pyrene](http://img.cochemist.com/ccimg/200/191-33-3.png)
![6H-Benzo[cd]pyrene](http://img.cochemist.com/ccimg/200/191-33-3_b.png)








![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2,3-diphenyl-, trans-](http://img.cochemist.com/ccimg/17900/17879-99-1.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2,3-diphenyl-, trans-](http://img.cochemist.com/ccimg/17900/17879-99-1_b.png)
![6,6'-Dimethyl-[1,1'-biphenyl]-2,2'-diamine](/data/chemimg/410500/20261-65-8.png)
![6,6'-Dimethyl-[1,1'-biphenyl]-2,2'-diamine](/data/chemimg/410500/20261-65-8_b.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-3-phenyl-, (2R,3S)-rel-](http://img.cochemist.com/ccimg/152000/151909-39-6.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-3-phenyl-, (2R,3S)-rel-](http://img.cochemist.com/ccimg/152000/151909-39-6_b.png)
![Zirconium, dichloro[h10-2,4-cyclopentadien-1-ylidene(1-methylethylidene)-9H-fluoren-9-ylidene]-](http://img.cochemist.com/ccimg/130700/130638-44-7.png)
![Zirconium, dichloro[h10-2,4-cyclopentadien-1-ylidene(1-methylethylidene)-9H-fluoren-9-ylidene]-](http://img.cochemist.com/ccimg/130700/130638-44-7_b.png)

![Phenol, 2-(1,1-dimethylethyl)-6-[(phenylimino)methyl]-](http://img.cochemist.com/ccimg/215100/215033-50-4.png)
![Phenol, 2-(1,1-dimethylethyl)-6-[(phenylimino)methyl]-](http://img.cochemist.com/ccimg/215100/215033-50-4_b.png)
![2-Pyridinamine, 6-methyl-N-[(1S)-1,2,3,4-tetrahydro-1-naphthalenyl]-](http://img.cochemist.com/ccimg/830400/830323-17-6.png)
![2-Pyridinamine, 6-methyl-N-[(1S)-1,2,3,4-tetrahydro-1-naphthalenyl]-](http://img.cochemist.com/ccimg/830400/830323-17-6_b.png)







![7-Azabicyclo[4.1.0]heptane, 7-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/68900/68820-12-2.png)
![7-Azabicyclo[4.1.0]heptane, 7-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/68900/68820-12-2_b.png)
![Phenol,2-[[(4-ethoxyphenyl)imino]methyl]-](http://img.cochemist.com/ccimg/700/637-45-6.png)
![Phenol,2-[[(4-ethoxyphenyl)imino]methyl]-](http://img.cochemist.com/ccimg/700/637-45-6_b.png)
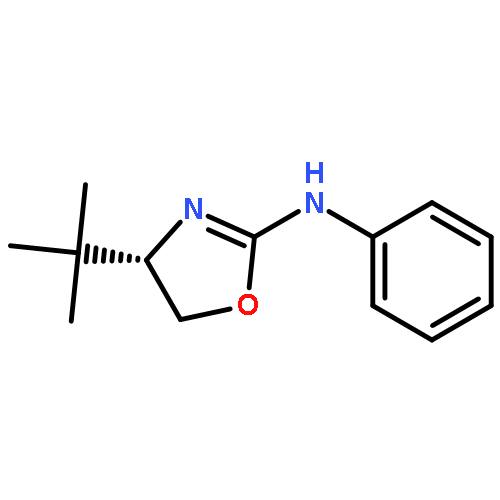
![Thiourea, N-[(1S)-1-(hydroxymethyl)-2,2-dimethylpropyl]-N'-phenyl-](http://img.cochemist.com/ccimg/821800/821775-10-4.png)
![Thiourea, N-[(1S)-1-(hydroxymethyl)-2,2-dimethylpropyl]-N'-phenyl-](http://img.cochemist.com/ccimg/821800/821775-10-4_b.png)


![2-Pyridinamine, 6-methyl-N-[(1S)-1-phenylethyl]-](http://img.cochemist.com/ccimg/830400/830323-19-8.png)
![2-Pyridinamine, 6-methyl-N-[(1S)-1-phenylethyl]-](http://img.cochemist.com/ccimg/830400/830323-19-8_b.png)
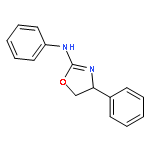
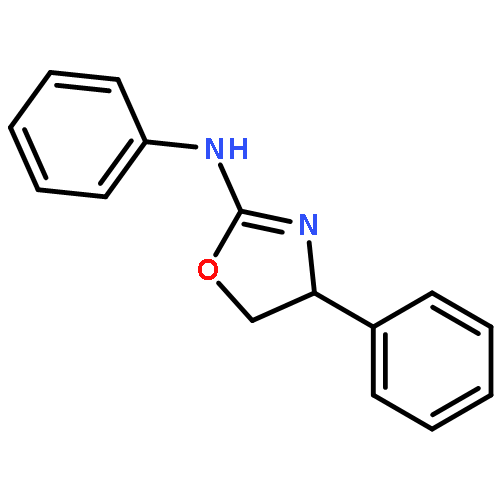
![2-Pyridinamine, N-[(1S)-1-phenylethyl]-](http://img.cochemist.com/ccimg/830400/830323-18-7.png)
![2-Pyridinamine, N-[(1S)-1-phenylethyl]-](http://img.cochemist.com/ccimg/830400/830323-18-7_b.png)


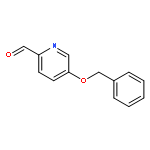
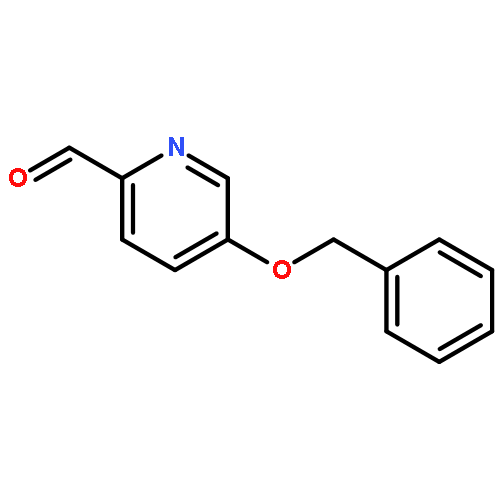
![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[(2,6-dimethoxybenzoyl)amino]-3,3-dimethyl-7-oxo-, (2S,5R,6R)-](http://img.cochemist.com/ccimg/100/61-32-5.png)
![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[(2,6-dimethoxybenzoyl)amino]-3,3-dimethyl-7-oxo-, (2S,5R,6R)-](http://img.cochemist.com/ccimg/100/61-32-5_b.png)



![Aziridine,1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24400/24395-14-0.png)
![Aziridine,1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24400/24395-14-0_b.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-3-phenyl-, trans-](http://img.cochemist.com/ccimg/137600/137595-21-2.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-3-phenyl-, trans-](http://img.cochemist.com/ccimg/137600/137595-21-2_b.png)


![[1,1'-Biphenyl]-2,2'-diamine,6,6'-dimethyl-, (1S)-](/data/chemimg/931200/3685-05-0.png)
![[1,1'-Biphenyl]-2,2'-diamine,6,6'-dimethyl-, (1S)-](/data/chemimg/931200/3685-05-0_b.png)




















































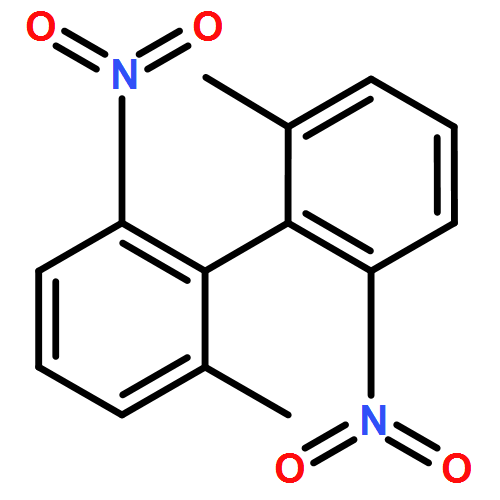
![6H-Benzo[cd]pyren-6-one](http://img.cochemist.com/ccimg/3100/3074-00-8.png)
![6H-Benzo[cd]pyren-6-one](http://img.cochemist.com/ccimg/3100/3074-00-8_b.png)








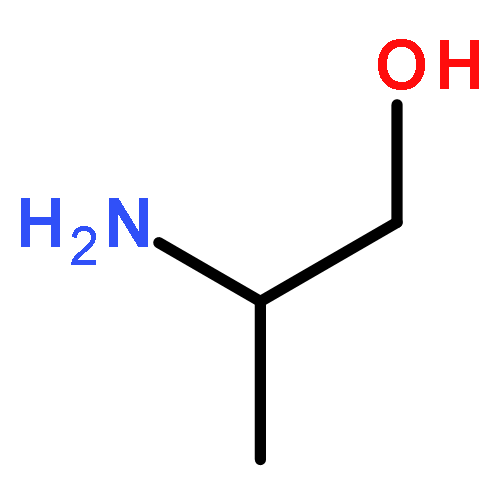
![Benzonitrile,4-[[(2-hydroxyphenyl)methylene]amino]-](http://img.cochemist.com/ccimg/33800/33721-67-4.png)
![Benzonitrile,4-[[(2-hydroxyphenyl)methylene]amino]-](http://img.cochemist.com/ccimg/33800/33721-67-4_b.png)







![Cobaltate(3-),tris[carbonato(2-)-kO,kO']-, sodium (1:3), (OC-6-11)-](http://img.cochemist.com/ccimg/23400/23311-39-9.png)
![Cobaltate(3-),tris[carbonato(2-)-kO,kO']-, sodium (1:3), (OC-6-11)-](http://img.cochemist.com/ccimg/23400/23311-39-9_b.png)