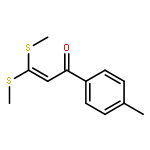
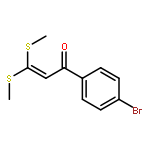
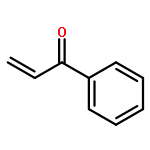
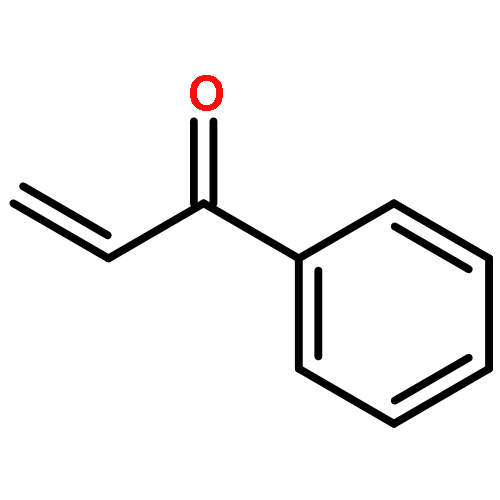
Co-reporter:Qingfu Wang, Huining Chai, and Zhengkun Yu
Organometallics September 25, 2017 Volume 36(Issue 18) pp:3638-3638
Publication Date(Web):September 8, 2017
DOI:10.1021/acs.organomet.7b00587
Dimeric pincer-type ruthenium(II)-NNN complexes bearing an unsymmetrical pyrazolyl-pyridylamino-pyridine ligand were prepared and characterized by NMR, elemental analysis, and X-ray single crystal structural determination. These complexes exhibited very high catalytic activity for both transfer hydrogenation of ketones and acceptorless dehydrogenation of secondary alcohols, achieving TOF values up to 1.9 × 106 h–1 in the transfer hydrogenation of ketones. The high catalytic activity of the Ru(II) complex catalysts is attributed to the presence of the unprotected NH functionality in the ligand and hemilabile unsymmetrical coordination environment around the central metal atoms in the complex.
Co-reporter:Ping Wu, Kaikai Wu, Liandi Wang, and Zhengkun Yu
Organic Letters October 6, 2017 Volume 19(Issue 19) pp:
Publication Date(Web):September 22, 2017
DOI:10.1021/acs.orglett.7b02751
Iron-promoted difunctionalization of α,α-diaryl and α-aryl-α-alkyl allylic alcohols has been efficiently achieved by means of N-(phenylseleno)phthalimide (N-PSP) under mild conditions. An in situ generated phenylselenium cation (PhSe+) was added to the olefinic C═C bond to initiate the regioselective phenylselenylation with concomitant 1,2-aryl migration, following a migration preference contrary to the well-known radical pathway. Hydrazonation of the resultant alkene difunctionalization products, that is, α-aryl-β-phenylselenyl ketones, and subsequent copper-catalyzed dehydroselenylation efficiently afforded functionalized 2-pyrazoline derivatives.
Co-reporter:Quannan Wang, Xiaoge Yang, Ping Wu, and Zhengkun Yu
Organic Letters November 17, 2017 Volume 19(Issue 22) pp:6248-6248
Publication Date(Web):November 2, 2017
DOI:10.1021/acs.orglett.7b03223
Visible-light-induced direct C–H arylation of S,S-functionalized internal alkenes, that is, α-oxo ketene dithioacetals and analogues, has been efficiently realized with aryldiazonium salts (ArN2BF4) as coupling partners and Ru(bpy)3Cl2·6H2O as photosensitizer at ambient temperature. The strategy to activate the internal olefinic C–H bond by both the alkylthio and electron-withdrawing functional groups was investigated. The synthetic protocol was successfully applied to the synthesis of all-carbon tetrasubstituted alkenes including tamoxifen.
Co-reporter:Fei Huang, Zhuqing Liu, Quannan Wang, Jiang Lou, and Zhengkun Yu
Organic Letters July 7, 2017 Volume 19(Issue 13) pp:
Publication Date(Web):June 14, 2017
DOI:10.1021/acs.orglett.7b01668
Efficient copper-catalyzed formal carbene migratory insertion into the olefinic C═C bonds of internal olefins, that is, α-oxo ketene N,S-acetals, has been achieved by means of N-tosylhydrazones of ketones as the carbene precursors. Iminofuran derivatives were obtained and further transformed to the corresponding 2(3H)-furanones and 4-oxobutanoates (γ-ketoesters), respectively, under mild conditions. In a similar fashion, α-thioxo ketene N,S-acetals reacted with N-tosylhydrazones of ketones to afford iminothiophenes. It is suggested that formal carbene migratory insertion into the olefinic C═C bond is involved in the overall catalytic cycle, demonstrating a new type of carbene insertion reaction for five-membered heterocycle construction.
Co-reporter:Tingting Liu, Huining Chai, Liandi Wang, and Zhengkun Yu
Organometallics August 14, 2017 Volume 36(Issue 15) pp:2914-2914
Publication Date(Web):July 7, 2017
DOI:10.1021/acs.organomet.7b00356
Dinuclear ruthenium(II)-NNN complexes were efficiently assembled by means of coordinatively unsaturated 16-electron mononuclear ruthenium(II)-pyrazolyl-imidazolyl-pyridine complex and 4,4′-linked bipyridine ligands. The diruthenium(II)-NNN complex assembled through 4,4′-(CH2)3-bipyridine exhibited exceptionally high catalytic activity for the transfer hydrogenation (TH) of ketones in refluxing 2-propanol and reached TOF values up to 1.4 × 107 h–1, demonstrating a remarkable cooperative effect from the ruthenium(II)-NNN functionalities.
Co-reporter:Quannan Wang;Jiang Lou;Ping Wu;Kaikai Wu
Advanced Synthesis & Catalysis 2017 Volume 359(Issue 17) pp:2981-2998
Publication Date(Web):2017/09/04
DOI:10.1002/adsc.201700654
AbstractMediated by a catalytic amount of FeCl3, the C–H alkylation of S,S-functionalized internal olefins, i.e., α-oxo ketene dithioacetals and their analogues, was efficiently achieved using simple ethers and toluene derivatives as the coupling partners, di-tert-butyl peroxide (DTBP) as the oxidant, and DABCO⋅6 H2O as the additive. The alkylthio functionality is essential for the internal olefinic C–H bond to undergo such an alkylation with the O-adjacent C(sp3)–H bonds of the ethers and the benzylic C–H bonds of the toluene derivatives, respectively. Tetrasubstituted olefins were thus synthesized and further transformed to highly substituted pyrazoles and isoxazoles. The strategy to activate an internal olefinic C–H bond by polarizing its parent olefinic C=C bond with both the dialkylthio group and an electron-withdrawing functionality was investigated. The mechanistic studies suggest a radical pathway for the C(sp2)–H/C(sp3)–H cross-coupling reactions. The present protocol provides a convenient route to tetrasubstituted olefins.
Co-reporter:Quanbin Jiang, Tenglong Guo, and Zhengkun Yu
The Journal of Organic Chemistry 2017 Volume 82(Issue 4) pp:
Publication Date(Web):January 24, 2017
DOI:10.1021/acs.joc.6b02772
Copper-catalyzed borylation of β-trifluoromethyl-α,β-unsaturated ketones was efficiently achieved by means of bis(pinacolato)diboron (B2pin2), affording the enantioenriched products in good yields with high enantioselectivities. CuI and (R,S)-Josiphos consist of the most efficient catalyst system under mild conditions. In the absence of the chiral ligand, the reactions could be performed more efficiently to form β-ketone derivatives which were directly borylated and indirectly trifluoromethylated at the β-carbon atom of the α,β-unsaturated ketone substrates. The present protocol provides a promising method to access a stereogenic carbon center bearing both CF3 and organoboron functional groups.
Co-reporter:Zhuqing Liu;Fei Huang;Jiang Lou;Quannan Wang
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 26) pp:5535-5540
Publication Date(Web):2017/07/05
DOI:10.1039/C7OB01234A
Copper-promoted direct C–H alkoxylation of S,S-functionalized internal olefins, that is, α-oxo ketene dithioacetals, was efficiently achieved with alcohols as the alkoxylating agents, (diacetoxyiodo)benzene (PhI(OAc)2) as the oxidant, and benzoquinone (BQ) as the co-oxidant. The alkoxylated olefins were thus constructed and applied for the synthesis of alkoxylated N-heterocycles. The polarization of the olefinic carbon–carbon double bond by the electron-donating dialkylthio and electron-withdrawing α-oxo functionalities plays a crucial role in making such C–H alkoxylation reactions to occur under mild conditions. Mechanistic studies implicate a single-electron-transfer (SET) reaction pathway involved in the overall catalytic cycle.
Co-reporter:Quanbin Jiang, Tenglong Guo, Kaikai Wu and Zhengkun Yu
Chemical Communications 2016 vol. 52(Issue 14) pp:2913-2915
Publication Date(Web):13 Jan 2016
DOI:10.1039/C5CC10361D
Rhodium(III)-catalyzed conjugate addition of aromatic and olefinic C–H bonds to CF3-substituted unsaturated ketones was efficiently achieved. Both arene and olefin substrates bearing a chelate assisted-directing group were coupled with a variety of β-trifluoromethyl-α,β-unsaturated ketones with excellent atom-economy, high yields, and broad substrate scopes.
Co-reporter:Kaikai Wu, Wei He, Chenglin Sun, Zhengkun Yu
Tetrahedron Letters 2016 Volume 57(Issue 36) pp:4017-4020
Publication Date(Web):7 September 2016
DOI:10.1016/j.tetlet.2016.07.037
•Pt–Sn/γ-Al2O3 catalyzed β-alkylation of secondary alcohols with primary alcohols.•The reaction underwent through a hydrogen borrowing strategy.•Atom-economical and environmentally benign C–C bond formation is presented.Heterogeneous bimetallic Pt–Sn/γ-Al2O3 (0.5 wt% Pt, molar ratio Pt:Sn = 1:3) was successfully utilized as the catalyst for direct β-alkylation of secondary alcohols with primary alcohols under solvent-free conditions. β-Alkylated secondary alcohols were obtained in moderate to high yields with water formed as the by-product through a hydrogen borrowing pathway. The present protocol provides a concise atom-economical and environmentally benign method for C–C bond formation.
Co-reporter:Kaikai Wu, Wei He, Chenglin Sun, Zhengkun Yu
Tetrahedron 2016 Volume 72(Issue 51) pp:8516-8521
Publication Date(Web):22 December 2016
DOI:10.1016/j.tet.2016.11.029
•Pt-Sn/γ-Al2O3 catalyzed N-alkylation of amines with alcohols to yield higher-order amines through a borrowing hydrogen strategy under solvent-free conditions.•The Pt-Sn/g-Al2O3 catalyst has exhibited very high catalytic activity towards a wide range of amines and alcohols, and can be conveniently recycled without Pt metal leaching.•The present protocol was applied for the synthesis of N-phenylbenzylamine on a kilogram scale of the substrates.Synthesis of secondary and tertiary amines has been efficiently realized from the N-alkylation of amines with alcohols by means of heterogeneous bimetallic Pt-Sn/γ-Al2O3 catalyst (0.5 wt % Pt, molar ratio Pt:Sn = 1:3) through a borrowing hydrogen strategy. The Pt-Sn/γ-Al2O3 catalyst has exhibited very high catalytic activity towards a wide range of amines and alcohols, and can be conveniently recycled without Pt metal leaching. The present protocol was applied for the synthesis of N-phenylbenzylamine in 96% isolated yield from aniline and benzyl alcohol on a 2.1 kg scale of the substrates, demonstrating its potential applicability for higher-order amine synthesis.
Co-reporter:Qingfu Wang, Kaikai Wu, and Zhengkun Yu
Organometallics 2016 Volume 35(Issue 9) pp:1251-1256
Publication Date(Web):April 21, 2016
DOI:10.1021/acs.organomet.6b00130
A Ru(III)-NNN complex bearing a pyridyl-supported pyrazolyl-imidazolyl ligand was synthesized and utilized as the catalyst for the direct β-alkylation of secondary alcohols with primary alcohols. β-Alkylated secondary alcohols were obtained in moderate to high yields with water formed as the byproduct through a hydrogen borrowing pathway. The present protocol provides a concise atom-economical and environmentally benign method for C–C bond formation.
Co-reporter:Fei Huang;Zhuqing Liu;Dr. Zhengkun Yu
Angewandte Chemie International Edition 2016 Volume 55( Issue 3) pp:862-875
Publication Date(Web):
DOI:10.1002/anie.201507521
Abstract
Transition-metal-catalyzed C-alkylation of ketones and secondary alcohols, with alcohols, avoids use of organometallic or environmentally unfriendly alkylating agents by means of borrowing hydrogen (BH) or hydrogen autotransfer (HA) activation of the alcohol substrates. Water is formed as the only by-product, thus making the BH process atom-economical and environmentally benign. Diverse homogeneous and heterogeneous transition-metal catalysts, ketones, and alcohols can be used for this transformation, thus rendering the BH process promising for replacing those procedures that use traditional alkylating agents. This Minireview summarizes the advances during the last five years in transition-metal-catalyzed BH α-alkylation of ketones, and β-alkylation of secondary alcohols with alcohols. A discussion on the application of the BH strategy for C−C bond formation is included.
Co-reporter:Fei Huang;Zhuqing Liu;Dr. Zhengkun Yu
Angewandte Chemie 2016 Volume 128( Issue 3) pp:872-885
Publication Date(Web):
DOI:10.1002/ange.201507521
Abstract
Die übergangsmetallkatalysierte C-Alkylierung von Ketonen und sekundären Alkoholen mit Alkoholen umgeht den Einsatz von metallorganischen oder umweltschädlichen Alkylierungsreagentien über die Borrowing-Hydrogen(BH)- oder Hydrogen-Autotransfer(HA)-Aktivierung der Alkoholsubstrate. Da Wasser als einziges Begleitprodukt entsteht, ist das BH-Verfahren atomökonomisch und umweltfreundlich. Viele homogene und heterogene Übergangsmetallkatalysatoren, Ketone und Alkohole sind in dieser Reaktion einsetzbar, sodass das BH-Verfahren aussichtsreich scheint, die derzeitigen Verfahren zu ersetzen, die herkömmliche Alkylierungsreagentien verwenden. Dieser Kurzaufsatz fasst die Fortschritte in der übergangsmetallkatalysierten α-Alkylierung von Ketonen und β-Alkylierung von sekundären Alkoholen mit Alkoholen mithilfe der BH-Technik während der letzten fünf Jahre zusammen. Auch die Eignung der BH-Strategie zur C-C-Bindungsbildung wird diskutiert.
Co-reporter:Qin Yang, Qingfu Wang and Zhengkun Yu
Chemical Society Reviews 2015 vol. 44(Issue 8) pp:2305-2329
Publication Date(Web):09 Feb 2015
DOI:10.1039/C4CS00496E
Transition metal-catalyzed substitution of alcohols by N-nucleophiles (or N-alkylation of amines and related compounds with alcohols) avoids the use of alkylating agents by means of borrowing hydrogen (BH) activation of the alcohol substrates. Water is produced as the only by-product, which makes the “BH” processes atom-economic and environmentally benign. Diverse types of homogeneous organometallic and heterogeneous transition metal catalysts, and substrates such as N-nucleophiles including amines, amides, sulfonamides and ammonia, and various alcohols, can be used for this purpose, demonstrating the promising potential of “BH” processes to replace the procedures using traditional alkylating agents in pharmaceutical and chemical industries. Borrowing hydrogen activation of alcohols for C–N bond formation has recently been paid more and more attention, and a lot of new and novel procedures and examples have been documented. This critical review summarizes the recent advances in “BH” substitution of alcohols by N-nucleophiles since 2009. “Semi-BH” N-alkylation processes with or without an external hydrogen acceptor are also briefly presented. Suitable discussion of the “BH” strategy provides new principles for establishing green processes to replace the relevant traditional synthetic methods for C–N bond formation.
Co-reporter:Ping Wu, Liandi Wang, Kaikai Wu, and Zhengkun Yu
Organic Letters 2015 Volume 17(Issue 4) pp:868-871
Publication Date(Web):January 30, 2015
DOI:10.1021/ol503731s
Brønsted acid p-TsOH·H2O-catalyzed hydrovinylation of styrenes with internal olefins α-oxo ketene dithioacetals was efficiently achieved in the presence of N-phenylselenophthalimide (N-PSP), regioselectively affording Markovnikov phenylselenative hydrovinylation products through PhSe transfer of the phenylseleno ketene dithioacetal intermediates. Radical reduction of the resultant PhSe-alkyl-substituted ketene dithioacetals with AIBN/n-Bu3SnH gave the corresponding anti-Markovnikov hydrovinylation products formally from the linear codimerization of styrenes and the internal olefins.
Co-reporter:Kaikai Wu;Ping Wu;Jiping Chen;Chenglin Sun
Advanced Synthesis & Catalysis 2015 Volume 357( Issue 14-15) pp:3353-3358
Publication Date(Web):
DOI:10.1002/adsc.201500546
Co-reporter:Xiaoge Yang, Kaikai Wu, Zhengkun Yu
Tetrahedron Letters 2015 Volume 56(Issue 30) pp:4490-4493
Publication Date(Web):22 July 2015
DOI:10.1016/j.tetlet.2015.05.102
BF3·OEt2-mediated alkenylation of pyrroles with α-oxo ketene dithioacetals was efficiently realized, affording mono- and disubstituted ketene pyrrolyl acetals. In the cases of using N-unprotected pyrrole, the reactions gave ketene bipyrrolyl acetals as well as N,O-chelated BF2 complexes. Diverse C–S transformations were achieved for the monosubstituted products, yielding N-heterocycles or multisubstituted olefins.
Co-reporter:Tenglong Guo, Fei Huang, Likun Yu, Zhengkun Yu
Tetrahedron Letters 2015 Volume 56(Issue 2) pp:296-302
Publication Date(Web):8 January 2015
DOI:10.1016/j.tetlet.2014.11.114
Indole synthesis is among one of the most important tasks in N-heterocyclic chemistry. Versatile synthetic methods have been developed for the establishment of an indole backbone, but concise and straightforward routes to access indole derivatives have been strongly desired. This digest paper summarizes the major advances in catalytic synthesis of indoles through transition-metal-catalyzed C–H activation during the last five years. Brief discussions are given for possible applications of these synthetic protocols.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Ping Wu, Fei Huang, Jiang Lou, Quannan Wang, Zhuqing Liu, Zhengkun Yu
Tetrahedron Letters 2015 Volume 56(Issue 19) pp:2488-2491
Publication Date(Web):6 May 2015
DOI:10.1016/j.tetlet.2015.03.096
Brønsted acid p-TsOH·H2O-catalyzed phenylselenenylation of α-oxo ketene dithioacetals was efficiently achieved by using N-phenylselenophthalimide (N-PSP) as the phenylseleno reagent. The resultant selenated ketene dithioacetals reacted with styrene in the presence of the same acid to undergo PhSe group transfer, regioselectively affording Markovnikov-type PhSe-alkylated olefins. Their further transformation with phenylboronic acid, guanidine, and hydrazine offers a promising route to access S,Se-incorporated organic compounds.
Co-reporter:Xiaoge Yang;Kaikai Wu;Ping Wu;Dr. Jiping Chen; Chenglin Sun;Dr. Zhengkun Yu
Chemistry - A European Journal 2015 Volume 21( Issue 26) pp:9323-9327
Publication Date(Web):
DOI:10.1002/chem.201501022
Abstract
Brønsted acid-mediated annulation of internal olefins α-oxo ketene dithioacetals to pyrroles was efficiently achieved to afford cyclopenta[b]pyrroles. A pair of Brønsted acids with acid strengths, that is, trifluoroacetic acid, and para-toluenesulfonic acid hydrate, were applied to promote the annulation reactions. The resultant products were readily oxidized to sulfones by meta-chloroperoxybenzoic acid. Subsequent treatment with 1,8-diazabicyclo[5.4.0]undec-7-ene gave desulfurized terminal olefins or [2+2] cycloaddition products from the desulfurized olefin intermediates. The present protocol provides facile access to structurally diverse cyclopenta[b]pyrrole derivatives under mild conditions.
Co-reporter:Xiaoge Yang;Zhuqing Liu; Chenglin Sun;Dr. Jiping Chen;Dr. Zhengkun Yu
Chemistry - A European Journal 2015 Volume 21( Issue 40) pp:14085-14094
Publication Date(Web):
DOI:10.1002/chem.201502280
Abstract
Efficient palladium-catalyzed cross-coupling reactions of the internal olefins α-cyanoketene dithioacetals with a variety of olefins were achieved in dioxane/HOAc/DMSO (9:3:1 v/v/v) under air atmosphere or by means of AgOAc as the terminal oxidant. Electron-deficient terminal olefins reacted to form the linear diene derivatives with air as the oxidant. Styrenes underwent the cross-coupling to give both the linear and branched dienes when using AgOAc as the oxidant. Unactivated cyclic and linear internal olefin substrates both reacted in the presence of a catalytic amount of benzoquinone in air to produce skipped dienes. The typical products were structurally confirmed by X-ray crystallography.
Co-reporter:Quanbin Jiang;Tenglong Guo;Dr. Zhengkun Yu
ChemCatChem 2015 Volume 7( Issue 4) pp:660-665
Publication Date(Web):
DOI:10.1002/cctc.201402823
Abstract
The Cu-catalyzed, one-pot tandem (asymmetric) borylation of β-chloroalkyl aryl ketones via the in situ generated acyclic enones with bis(pinacolato)diboron was achieved efficiently to reach up to 97 % yield and 99 % enantioselectivity for the formal sp3 CCl borylation products. The present methodology provides an efficient alternative route to (chiral) alkylboron compounds.
Co-reporter:Huining Chai, Tingting Liu, Qingfu Wang, and Zhengkun Yu
Organometallics 2015 Volume 34(Issue 21) pp:5278-5284
Publication Date(Web):October 20, 2015
DOI:10.1021/acs.organomet.5b00727
Air- and moisture-stable ruthenium(II) complexes bearing a multisubstituted pyrazolyl-imidazolyl-pyridine ligand were synthesized and structurally characterized by NMR and X-ray single-crystal crystallographic analyses. The substituents on the imidazolyl moiety of the NNN ligand exhibited a remarkable impact on the catalytic activity of the corresponding Ru(II) complexes for transfer hydrogenation of ketones in refluxing 2-propanol, following the order NHTs > Me > H > NO2, to tune the catalytic activity. The highest final TOF value of 345 600 h–1 was reached by means of 0.05 mol % of the Ru(II)-NHTs-substituted NNN complex as the catalyst. The corresponding structurally confirmed RuH complexes are proposed as the catalytically active species.
Co-reporter:Fei Huang, Ping Wu, Liandi Wang, Jiping Chen, Chenglin Sun and Zhengkun Yu
Chemical Communications 2014 vol. 50(Issue 87) pp:13395-13395
Publication Date(Web):26 Sep 2014
DOI:10.1039/C4CC90399D
Correction for ‘Copper-mediated intramolecular oxidative C–H/N–H cross-coupling of α-alkenoyl ketene N,S-acetals to synthesize pyrrolone derivatives’ by Fei Huang et al., Chem. Commun., 2014, DOI: 10.1039/c4cc05837b.
Co-reporter:Fei Huang, Ping Wu, Liandi Wang, Jiping Chen, Chenglin Sun and Zhengkun Yu
Chemical Communications 2014 vol. 50(Issue 83) pp:12479-12481
Publication Date(Web):28 Aug 2014
DOI:10.1039/C4CC05837B
CuCl2 and CuBr2-mediated intramolecular oxidative C–H/N–H cross-coupling/halogenation of β-thioalkyl-substituted α-alkenoyl ketene N,S-acetals occurred efficiently, affording 4-halo-5-thioalkyl-3-pyrrolones. Tunable C–S and C–halo bond transformations of the resultant pyrrolone derivatives led to highly functionalized N-heterocyclic compounds.
Co-reporter:Qin Yang, Ping Wu, Jiping Chen and Zhengkun Yu
Chemical Communications 2014 vol. 50(Issue 48) pp:6337-6339
Publication Date(Web):29 Apr 2014
DOI:10.1039/C4CC02264E
Iron-catalyzed alkylation of internal olefins, that is, α-oxo ketene dithioacetals, was successfully realized by using styrenes as the alkylating reagents. Highly functionalized tetrasubstituted olefins were prepared in moderate to high yields.
Co-reporter:Qin Yang, Tongyu Xu, and Zhengkun Yu
Organic Letters 2014 Volume 16(Issue 24) pp:6310-6313
Publication Date(Web):December 1, 2014
DOI:10.1021/ol503039j
FeCl3- and FeBr3-mediated tandem carboarylation/cyclization of propargylanilines with diethyl benzaldehyde acetals furnished the tetracyclic core of indeno[2,1-c]quinolines. 5-Tosyl-6,7-dihydro-5H-indeno[2,1-c]quinoline and 7H-indeno[2,1-c]quinoline derivatives were obtained in good to excellent yields, respectively, by tuning the FeX3 loadings and/or reaction temperatures.
Co-reporter:Weiwei Jin;Qin Yang;Ping Wu;Jiping Chen
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 9) pp:2097-2102
Publication Date(Web):
DOI:10.1002/adsc.201400076
Co-reporter:Kaikai Wu;Ping Wu;Lii Wang;Jiping Chen;Chenglin Sun
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 18) pp:3871-3880
Publication Date(Web):
DOI:10.1002/adsc.201400477
Co-reporter:Qingfu Wang, Wangming Du, Tingting Liu, Huining Chai, Zhengkun Yu
Tetrahedron Letters 2014 Volume 55(Issue 9) pp:1585-1588
Publication Date(Web):26 February 2014
DOI:10.1016/j.tetlet.2014.01.072
Highly efficient Oppenauer-type oxidation of secondary alcohols to the corresponding ketones has been realized by means of the ruthenium(II) complex catalysts bearing a 2-(benzoimidazol-2-yl)-6-(3,5-dimethylpyrazol-1-yl)pyridine ligand. The oxidation reaction underwent in the presence of acetone as oxidant under mild conditions, reaching final TOF values up to 3960 h−1. The hemilability of the ligand is attributed to the high catalytic activity of these Ru(II) complexes.
Co-reporter:Zhifeng Mao;Fei Huang;Dr. Haifeng Yu;Dr. Jiping Chen;Dr. Zhengkun Yu;Dr. Zhaoqing Xu
Chemistry - A European Journal 2014 Volume 20( Issue 12) pp:3439-3445
Publication Date(Web):
DOI:10.1002/chem.201305069
Abstract
The functionalization of internal olefins has been a challenging task in organic synthesis. Efficient CuII-catalyzed trifluoromethylation of internal olefins, that is, α-oxoketene dithioacetals, has been achieved by using Cu(OH)2 as a catalyst and TMSCF3 as a trifluoromethylating reagent. The push–pull effect from the polarized olefin substrates facilitates the internal olefinic CH trifluoromethylation. Cyclic and acyclic dithioalkyl α-oxoketene acetals were used as the substrates and various substituents were tolerated. The internal olefinic CH bond cleavage was not involved in the rate-determining step, and a mechanism that involves radicals is proposed based on a TEMPO-quenching experiment of the trifluoromethylation reaction. Further derivatization of the resultant CF3 olefins led to multifunctionalized tetrasubstituted CF3 olefins and trifluoromethylated N-heterocycles.
Co-reporter:Fei Huang, Ping Wu, Liandi Wang, Jiping Chen, Chenglin Sun, and Zhengkun Yu
The Journal of Organic Chemistry 2014 Volume 79(Issue 21) pp:10553-10560
Publication Date(Web):October 16, 2014
DOI:10.1021/jo5014542
CuCl2-mediated intramolecular C–H/C–H cross-dehydrogenative coupling (CDC) of thioalkyl-substituted α-acetyl or α-aroyl ketene N,S-acetals afforded 2-thioalkyl indoles. Tunable C–S bond transformations of the resultant indoles led to highly functionalized N-heterocyclic compounds. A β-thioalkyl is necessary to activate the N,S-acetal substrate and enable the CDC reaction to occur, and the relevant mechanism studies revealed that the CDC reaction follows a radical pathway.
Co-reporter:Wangming Du, Qingfu Wang, Liandi Wang, and Zhengkun Yu
Organometallics 2014 Volume 33(Issue 4) pp:974-982
Publication Date(Web):February 11, 2014
DOI:10.1021/om401144u
Ru(III) and Ru(II) complexes bearing a tridentate 2-(3′,5′-dimethylpyrazol-1′-yl)-6-(3″,5″-bis(trifluoromethyl)pyrazol-1″-yl)pyridine or 2-(benzimidazol-2′-yl)-6-(3″,5″-bis(trifluoromethyl)pyrazol-1″-yl)pyridine ligand were synthesized and applied to the transfer hydrogenation of ketones. The Ru(II) complex was structurally confirmed by the X-ray crystallographic analysis and achieved up to 2150 turnover numbers and final TOFs up to 29700 h–1 in the transfer hydrogenation of ketones. The benzimidazolyl moiety with an unprotected NH functionality in the ligand exhibited an enhancement effect on the catalytic activity of its RuCl3 complex in the ketone reduction reactions, reaching a final TOF value up to 35640 h–1. The controlled experiments have revealed that the compatibility of the trifluoromethylated pyrazolyl and unprotected benzimidazolyl is crucial for the establishment of the highly active catalytic system.
Co-reporter:Liandi Wang, Wei He and Zhengkun Yu
Chemical Society Reviews 2013 vol. 42(Issue 2) pp:599-621
Publication Date(Web):18 Oct 2012
DOI:10.1039/C2CS35323G
C–S bond activation, cleavage and transformations by means of transition metal compounds have recently become more and more important in the petroleum industry and synthetic chemistry. Homogeneous transition metal compounds have been investigated in order to provide the fundamental insight into the C–S bond cleavage in problematic organosulfur compounds such as thiophene, benzo- and dibenzothiophene derivatives. Rendering transition-metal mediated reactions with organosulfur compounds catalytic may provide promising routes to deep hydrodesulfurization of petroleum feedstocks, and offer potentially useful synthetic protocols for cross-couplings and biomimetic organic synthesis. During the last few decades increasing work was documented on C–S bond activation and transformations by means of transition metal compounds. This review summarizes the recent advances in C–S bond cleavage via the insertion of transition metals into the inert C–S bonds of these problematic organosulfur compounds, and transition-metal mediated C–S bond transformations via C–S activation through cross-couplings of thioesters, ketene dithioacetals, sulfonyl chlorides, and other diverse organosulfur compounds.
Co-reporter:Quanbin Jiang;Tenglong Guo;Qingfu Wang;Ping Wu
Advanced Synthesis & Catalysis 2013 Volume 355( Issue 9) pp:1874-1880
Publication Date(Web):
DOI:10.1002/adsc.201200821
Abstract
Highly efficient arylations of β-chloro ketones and their ester and amide derivatives were achieved by means of domino dehydrochlorination/Rh(I)-catalyzed conjugate addition. In situ generated vinyl ketones and their analogues were identified as the reaction intermediates. The present synthetic protocol provides a concise route to (chiral) β-aryl ketones, esters, and amides.
Co-reporter:Qin Yang, Huining Chai, Tingting Liu, Zhengkun Yu
Tetrahedron Letters 2013 Volume 54(Issue 48) pp:6485-6489
Publication Date(Web):27 November 2013
DOI:10.1016/j.tetlet.2013.09.072
Direct synthesis of 1,1-disubstitued 1,3-butadienes has been efficiently realized from the cross-coupling of cyclopropylmethyl N-tosylhydrazones with aromatic bromides by means of PdCl2(MeCN)2 as catalyst. 1,1,4-Trisubstitued 1,3-butadiene derivatives were obtained in up to 70% yields through a one-pot procedure catalyzed by Pd(OAc)2 in the presence of excessive amount of aromatic bromides. The present methodology provides an easy and efficient route to multisubstituted 1,3-butadienes.
Co-reporter:Wangming Du, Ping Wu, Qingfu Wang, and Zhengkun Yu
Organometallics 2013 Volume 32(Issue 10) pp:3083-3090
Publication Date(Web):May 7, 2013
DOI:10.1021/om400298c
Air- and moisture-stable ruthenium(II) complexes bearing a unsymmetrical 2-(benzimidazol-2-yl)-6-(benzotriazol-1-yl)pyridine ligand were synthesized and structurally characterized by NMR analysis and X-ray crystallographic determinations. These complexes have exhibited excellent catalytic activity in the transfer hydrogenation of ketones in refluxing 2-propanol, reaching final TOFs up to 176400 h–1. The corresponding RuH complex was isolated and is proposed as the catalytically active species by controlled experiments. The high catalytic activity of the Ru(II) the complex catalysts is attributed to the hemilabile unsymmetrical coordinating environment around the central metal atom in the complexes and presence of a convertible benzimidazolyl NH functionality in the ligand.
Co-reporter:Zhifeng Mao, Zhe Wang, Zhaoqing Xu, Fei Huang, Zhengkun Yu, and Rui Wang
Organic Letters 2012 Volume 14(Issue 15) pp:3854-3857
Publication Date(Web):July 13, 2012
DOI:10.1021/ol301517y
Cu(OAc)2-mediated dehydrogenative cross-coupling between two heteroarenes has been realized in the absence of any other additive. A mechanism involving a formal Cu(II) to Cu(0) route by convergent disproportionation of the copper mediator is proposed and has been evidenced by copper mirror formation during the reaction. This synthetic protocol provides a concise and “green” access to unsymmetrical biheteroarenes bearing structural motifs of substantial utility in organic synthesis.
Co-reporter:Weiwei Jin ; Liandi Wang
Organometallics 2012 Volume 31(Issue 15) pp:5664-5667
Publication Date(Web):July 31, 2012
DOI:10.1021/om300602w
Ruthenium(II) complexes bearing a pyrazolyl–pyridyl–pyrazole ligand were synthesized and exhibited exceptionally high catalytic activity in the transfer hydrogenation of ketones in refluxing isopropyl alcohol, reaching final TOFs up to 720 000 h–1. The β-NH functionality of the pyrazole arm in the ligand demonstrated a remarkable acceleration effect on the reaction rate. The unsymmetrical nature (hemilability) and presence of the convertible NH group of the ligand is attributed to the high catalytic activity of the complex catalyst.
Co-reporter:Chao Dai, Zhaoqing Xu, Fei Huang, Zhengkun Yu, and Yan-Feng Gao
The Journal of Organic Chemistry 2012 Volume 77(Issue 9) pp:4414-4419
Publication Date(Web):April 17, 2012
DOI:10.1021/jo202624s
A Lewis acid (AgI, NiII, or FeII) catalyzed, CuII-mediated thiolation reaction between heteroarenes and thiols was achieved with good yield under base-free conditions. DMSO could serve as an effective methylthiolation reagent for the synthesis of heterocyclic methyl thioethers.
Co-reporter:Qin Yang, Liandi Wang, Tenglong Guo, and Zhengkun Yu
The Journal of Organic Chemistry 2012 Volume 77(Issue 18) pp:8355-8361
Publication Date(Web):August 25, 2012
DOI:10.1021/jo301416s
FeCl3·6H2O-catalyzed efficient C3-alkenylation of indoles was realized through the condensation of aldehydes and indole derivatives in the presence of 2 equiv of ethanol at ambient temperature, forming 3-vinylindoles in up to 93% yields. Ethanol promoted formation of the desired products. An obvious solvent effect was observed, and bisindoles were identified as the reaction intermediates.
Co-reporter:Wenjing Ye;Dr. Miao Zhao;Dr. Zhengkun Yu
Chemistry - A European Journal 2012 Volume 18( Issue 35) pp:10843-10846
Publication Date(Web):
DOI:10.1002/chem.201201703
Co-reporter:Wangming Du;Lii Wang;Ping Wu;Dr. Zhengkun Yu
Chemistry - A European Journal 2012 Volume 18( Issue 37) pp:11550-11554
Publication Date(Web):
DOI:10.1002/chem.201201938
Co-reporter:Dr. Zhengkun Yu;Weiwei Jin;Quanbin Jiang
Angewandte Chemie International Edition 2012 Volume 51( Issue 25) pp:6060-6072
Publication Date(Web):
DOI:10.1002/anie.201200963
Abstract
Asymmetric hydrogenation plays an important role in organic synthesis, but that of the challenging substrates such as N-unprotected imines, enamines, and N-heteroaromatic compounds (1H-indoles, 1H-pyrroles, pyridines, quinolines, and quinoxalines) has only received increased attention in the past three years. Considering the interaction modes of a Brønsted acid with a Lewis base, Brønsted acids may be used as the ideal activators of CN bonds. This Minireview summarizes the recent advances in transition-metal-catalyzed, Brønsted acid activated asymmetric hydrogenation of these challenging substrates, thus offering a promising substrate activation strategy for transformations involving CN bonds.
Co-reporter:Dr. Zhengkun Yu;Weiwei Jin;Quanbin Jiang
Angewandte Chemie 2012 Volume 124( Issue 25) pp:6164-6177
Publication Date(Web):
DOI:10.1002/ange.201200963
Abstract
Die asymmetrische Hydrierung spielt in der organischen Synthese eine wichtige Rolle, aber erst in den letzten drei Jahren wurde die Hydrierung von schwierigen Substraten, wie N-ungeschützten Iminen, Enaminen und N-Heteroarenen (1H-Indole, 1H-Pyrrole, Pyridine, Chinoline und Chinoxaline) vermehrt untersucht. Wenn man die Wechselwirkungen einer Brønsted-Säure mit einer Lewis-Base betrachtet, könnten Brønsted-Säuren als ideale Aktivatoren von CN-Bindungen genutzt werden. Dieser Kurzaufsatz fasst aktuelle Entwicklungen bei der übergangsmetallkatalysierten, Brønsted-Säure-aktivierten asymmetrischen Hydrierung dieser anspruchsvollen Substrate zusammen und präsentiert damit eine vielversprechende Substrataktivierungsstrategie für Umwandlungen unter Beteiligung von CN-Bindungen.
Co-reporter:Ning Luo, Zhaoyan Zheng, and Zhengkun Yu
Organic Letters 2011 Volume 13(Issue 13) pp:3384-3387
Publication Date(Web):June 2, 2011
DOI:10.1021/ol201139w
The highly regioselective [3 + 2] cycloaddition of azomethine imines to 1-alkynyl Fischer carbene complexes has been successfully realized under mild conditions. Oxidative demetalation of the newly formed pyrazolo-pyrazolone carbene complexes with pyridine-N-oxide or ceric ammonium nitrate efficiently afforded pyrazolo-pyrazolone derivatives as well as cycloprop-2-enone and trisubstituted 1H-pyrazoles in some cases, providing a novel route to versatile functionalized N,N-bicyclic pyrazolidin-3-ones.
Co-reporter:Weiwei Jin, Haifeng Yu, Zhengkun Yu
Tetrahedron Letters 2011 Volume 52(Issue 44) pp:5884-5887
Publication Date(Web):2 November 2011
DOI:10.1016/j.tetlet.2011.08.168
Multisubstituted pyrazoles were efficiently synthesized by cyclocondensation of β-thioalkyl-α,β-unsaturated ketones with hydrazines under relatively mild conditions. A one-pot synthetic protocol through tandem Liebeskind–Srogl cross-coupling/cyclocondensation using α-oxo ketene dithioacetals as the starting materials was also realized for the same purpose.
Co-reporter:Liandi Wang, Wei He, Kaikai Wu, Songbo He, Chenglin Sun, Zhengkun Yu
Tetrahedron Letters 2011 Volume 52(Issue 52) pp:7103-7107
Publication Date(Web):28 December 2011
DOI:10.1016/j.tetlet.2011.10.100
Direct synthesis of diamines has been efficiently realized from the N-alkylation of amines with diols by means of heterogeneous bimetallic Pt–Sn/γ-Al2O3 catalyst (0.5 wt % Pt, molar ratio Pt:Sn = 1:3) through a ‘Borrowing Hydrogen’ strategy under ligand-free conditions. The present methodology provides an environmentally benign route to diamines.
Co-reporter:Fanzhi Yang; Zhaoqing Xu;Zhe Wang; Zhengkun Yu; Rui Wang
Chemistry - A European Journal 2011 Volume 17( Issue 23) pp:6321-6325
Publication Date(Web):
DOI:10.1002/chem.201100136
Co-reporter:Wenjing Ye;Miao Zhao;Wangming Du;Quanbin Jiang;Kaikai Wu;Ping Wu;Dr. Zhengkun Yu
Chemistry - A European Journal 2011 Volume 17( Issue 17) pp:4737-4741
Publication Date(Web):
DOI:10.1002/chem.201002039
Co-reporter:Tongyu Xu;Qin Yang;Wenjing Ye;Quanbin Jiang;Dr. Zhaoqing Xu;Dr. Jiping Chen;Dr. Zhengkun Yu
Chemistry - A European Journal 2011 Volume 17( Issue 38) pp:10547-10551
Publication Date(Web):
DOI:10.1002/chem.201101667
Co-reporter:Wei He;Lii Wang; Chenglin Sun;Kaikai Wu;Dr. Songbo He;Dr. Jiping Chen;Ping Wu;Dr. Zhengkun Yu
Chemistry - A European Journal 2011 Volume 17( Issue 47) pp:13308-13317
Publication Date(Web):
DOI:10.1002/chem.201101725
Abstract
Versatile syntheses of secondary and tertiary amines by highly efficient direct N-alkylation of primary and secondary amines with alcohols or by deaminative self-coupling of primary amines have been successfully realized by means of a heterogeneous bimetallic Pt–Sn/γ-Al2O3 catalyst (0.5 wt % Pt, Pt/Sn molar ratio=1:3) through a borrowing-hydrogen strategy. In the presence of oxygen, imines were also efficiently prepared from the tandem reactions of amines with alcohols or between two primary amines. The proposed mechanism reveals that an alcohol or amine substrate is initially dehydrogenated to an aldehyde/ketone or NH-imine with concomitant formation of a [PtSn] hydride. Condensation of the aldehyde/ketone species or deamination of the NH-imine intermediate with another molecule of amine forms an N-substituted imine which is then reduced to a new amine product by the in-situ generated [PtSn] hydride under a nitrogen atmosphere or remains unchanged as the final product under an oxygen atmosphere. The Pt–Sn/γ-Al2O3 catalyst can be easily recycled without Pt metal leaching and has exhibited very high catalytic activity toward a wide range of amine and alcohol substrates, which suggests potential for application in the direct production of secondary and tertiary amines and N-substituted imines.
Co-reporter:Xiaowei Wu, Zhengkun Yu
Tetrahedron Letters 2010 Volume 51(Issue 11) pp:1500-1503
Publication Date(Web):17 March 2010
DOI:10.1016/j.tetlet.2010.01.040
1H-Quinazoline-2,4-diones were efficiently synthesized by selenium-catalyzed carbonylation of o-nitrobenzamides under relatively mild conditions. In situ-generated carbonyl selenide (SeCO) is proposed to initiate the catalytic carbonylation. Thus, a concise transition metal and phosgene-free synthetic route to potentially bioactive-substituted 1H-quinazoline-2,4-dione derivatives has been developed.
Co-reporter:Haifeng Yu Dr.;Weiwei Jin;Chenglin Sun ;Jiping Chen Dr.;Wangmin Du;Songbo He Dr. Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 33) pp:5792-5797
Publication Date(Web):
DOI:10.1002/anie.201002737
Co-reporter:Ning Luo Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 3) pp:787-791
Publication Date(Web):
DOI:10.1002/chem.200902612
Co-reporter:Tongyu Xu;Qin Yang;Dongpo Li;Jinhua Dong Dr. Dr.;Yuxue Li Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 30) pp:9264-9272
Publication Date(Web):
DOI:10.1002/chem.201000686
Abstract
FeCl3⋅6 H2O- and FeBr3-catalyzed Prins cyclization/halogenation of alkynyl aldehyde acetals has been realized with acetyl chloride or bromide as halogen source in dichloromethane to afford 2-(1-halobenzylidene or alkylidene)-substituted five-membered carbo- and heterocycles, and thus provides an alternative route for vinylic CCl and CBr bond formation. Five- to eight-membered cyclic enones were efficiently synthesized by FeCl3⋅6 H2O-catalyzed intramolecular cyclization of alkynyl aldehyde acetals in acetone under mild conditions. An oxocarbonium species generated in situ is proposed to initiate the reaction, and the target products are formed via vinylogous carbenium cation and oxete intermediates according to DFT calculations. Intermolecular reactions of alkynes and aldehyde acetals were also investigated with 20–40 mol % FeCl3⋅6 H2O catalyst, and produced α,β-unsaturated enones and chlorinated indene derivatives. The present protocol has applications in the synthesis of carbo-, oxa- and azacycles.
Co-reporter:Wenjing Ye, Ning Luo and Zhengkun Yu
Organometallics 2010 Volume 29(Issue 4) pp:1049-1052
Publication Date(Web):January 28, 2010
DOI:10.1021/om900997y
Efficient regioselective direct alkenylation of benzo[h]quinoline was realized with cinnamoyl chlorides as the coupling partners via decarbonylation of the chlorides and C−H bond activation by means of [Rh(CO)2Cl]2 as the catalyst in refluxing o-xylene under phosphine-free conditions. For 2-phenylpyridine, [Rh(CO)2Cl]2 or [Rh(COD)Cl]2 efficiently promoted its direct alkenylation with cinnamic anhydrides. Polyarenes were synthesized from [Rh(COD)Cl]2-catalyzed decarbonylative poly(arylation) of isophthaloyl dichloride, terephthaloyl dichloride, or benzene-1,3,5-tricarbonyl chloride with benzo[h]quinoline.
Co-reporter:Haifeng Yu Dr.;Weiwei Jin;Chenglin Sun ;Jiping Chen Dr.;Wangmin Du;Songbo He Dr. Dr.
Angewandte Chemie 2010 Volume 122( Issue 33) pp:5928-5933
Publication Date(Web):
DOI:10.1002/ange.201002737
Co-reporter:Tongyu Xu, Zhengkun Yu and Liandi Wang
Organic Letters 2009 Volume 11(Issue 10) pp:2113-2116
Publication Date(Web):April 10, 2009
DOI:10.1021/ol9005689
FeCl3- and FeBr3-promoted cyclization/halogenation of alkynyl diethyl acetals has been efficiently realized, selectively affording (E)-2-(1-halobenzylidene or alkylidene)-substituted five-membered carbo- and heterocycles which were then efficiently transformed to vinylarenes by Suzuki coupling. The present protocol has provided a new alternative route to vinylic C−Cl and C−Br bond formation.
Co-reporter:Miao Zhao, Zhengkun Yu, Shenggang Yan, Yang Li
Tetrahedron Letters 2009 50(32) pp: 4624-4628
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.05.100
Co-reporter:Fanlong Zeng and Zhengkun Yu
Organometallics 2009 Volume 28(Issue 6) pp:1855-1862
Publication Date(Web):February 23, 2009
DOI:10.1021/om801080p
A family of hemilabile ruthenium(II) NNN complexes bearing a unsymmetrical 2-(benzoimidazol-2-yl)-6-(pyrazol-1-yl)pyridine ligand has been synthesized and exhibited good to excellent catalytic activity in transfer hydrogenation of ketones in refluxing 2-propanol, reaching final TOFs up to 7.2 × 105 h−1 with 0.05 mol % loading. The γ-NH effect of the benzoimidazol-2-yl moiety in the ligand and coordination modes of the metal center in a Ru(II) NNN complex has great influence on the catalytic activity of the complex catalyst in transfer hydrogenation of ketones. It has been demonstrated that one of the structural prerequisites for an active Ru(II) complex catalyst is the coordinatively unsaturated environment around the metal center in the complex or the precatalyst, and the catalytic activity of a complex catalyst can be enhanced by making its metal center cationic. This paper presents a methodology to construct new types of efficient Ru(II) complex catalysts for transfer hydrogenation of ketones.
Co-reporter:Weiqiang Tan, Zhengkun Yu, Bing Liu, Kaikai Wu, Zishuang Liu, Jinzhu Chen
Journal of Organometallic Chemistry 2009 694(2) pp: 199-206
Publication Date(Web):
DOI:10.1016/j.jorganchem.2008.10.021
Co-reporter:Miao Zhao, Zhengkun Yu, Shenggang Yan, Yang Li
Journal of Organometallic Chemistry 2009 694(19) pp: 3068-3075
Publication Date(Web):
DOI:10.1016/j.jorganchem.2009.05.028
Co-reporter:Ning Luo, Zhengkun Yu
Journal of Organometallic Chemistry 2009 694(19) pp: 3058-3067
Publication Date(Web):
DOI:10.1016/j.jorganchem.2009.05.029
Co-reporter:Haifeng Yu Dr. Dr.
Angewandte Chemie 2009 Volume 121( Issue 16) pp:2973-2977
Publication Date(Web):
DOI:10.1002/ange.200900278
Co-reporter:Haifeng Yu Dr. Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 16) pp:2929-2933
Publication Date(Web):
DOI:10.1002/anie.200900278
Co-reporter:Fanlong Zeng
Organometallics 2008 Volume 27(Issue 13) pp:2898-2901
Publication Date(Web):May 23, 2008
DOI:10.1021/om8002043
Rare ruthenium(II) complexes bearing a pyridyl-based pyrazolyl−imidazolyl ligand were synthesized and exhibited exceptionally high catalytic activity in the transfer hydrogenation of ketones in 2-propanol at 82 °C or room temperature, reaching 100% conversion of the substrates and final TOFs up to 7.2 × 105 h−1 with 0.05 mol % catalyst at 82 °C and 55 800 h−1 with 0.1 mol % catalyst at room temperature.
Co-reporter:Fanlong Zeng and Zhengkun Yu
Organometallics 2008 Volume 27(Issue 22) pp:6025-6028
Publication Date(Web):October 21, 2008
DOI:10.1021/om8006213
Ruthenium(II) complexes bearing a pyridyl-supported pyrazol-3-yl−N-heterocyclic carbene (NNC) ligand were synthesized from the reactions of RuCl3·3H2O with pyridyl-supported pyrazol-3-yl−imidazolium salts in the presence of carbon monoxide. These Ru(II) complexes have exhibited good to excellent catalytic activity in the transfer hydrogenation of ketones in refluxing 2-propanol. Formation of a coordinatively unsaturated Ru(II) center in the precatalyst is attributed to the high catalytic activity of the Ru(II) complexes.
Co-reporter:Zhengkun Yu;Shenggang Yan;Guangtao Zhang;Wei He;Lii Wang;Yu Li;Fanlong Zeng
Advanced Synthesis & Catalysis 2006 Volume 348(Issue 1-2) pp:
Publication Date(Web):19 JAN 2006
DOI:10.1002/adsc.200505224
Using a proazaphosphatrane catalyst, P(RNCH2CH2)3N (R=Me, i-Pr), allylaromatics and allyl phenyl sulfide were selectively isomerized to the corresponding vinyl isomers in yields up to >99% in CH3CN at 40 °C. Efficient transformation of allyl phenyl sulfone at ambient temperature afforded an isomerization/dimerization product in >95% yield. Conjugation of bis-allylmethylene double bond-containing compounds gave the corresponding conjugated isomers for cis,cis-9,12-octadecadienol and its methyl ether in yields up to 97%, and desilylation/conjugation products were obtained from the catalytic reaction of the trimethylsilyl ether of cis,cis-9,12-octadecadienol. The reaction mechanism is discussed based upon the 1H and 31P NMR-monitored reactions in CD3CN or CH3CN under the reaction conditions.
Co-reporter:Xiaodan Zhao;Fanlong Zeng;Jinzhu Chen;Xiaowei Wu;Sizhong Wu;Wen-Jing Xiao;Zhaoyan Zheng
Advanced Synthesis & Catalysis 2005 Volume 347(Issue 6) pp:
Publication Date(Web):9 MAY 2005
DOI:10.1002/adsc.200404380
Reactions of selenium with imines (RR1CNR2) of aldehydes and ketones in the presence of carbon monoxide, water and triethylamine lead to reductive selenation, on aerobic work-up, to afford symmetrical diselenides (RR1CHSe)2 in good to excellent yields. The proposed mechanism suggests that both in situ generated carbonyl selenide (SeCO) and hydrogen selenide (H2Se) are involved in the reaction.
Co-reporter:Jinzhu Chen;Gang Ling;Sizhong Wu;Xiaodan Zhao;Xiaowei Wu;Shiwei Lu
Advanced Synthesis & Catalysis 2004 Volume 346(Issue 11) pp:
Publication Date(Web):29 SEP 2004
DOI:10.1002/adsc.200404077
N-Arylamides were exclusively obtained in moderate to good yields from selenium-catalyzed reactions of nitroaromatics with amides in the presence of CO and mixed organic bases Et3N and DBU.
Co-reporter:Xiaofang Wang;Shiwei Lu
Advanced Synthesis & Catalysis 2004 Volume 346(Issue 8) pp:
Publication Date(Web):5 AUG 2004
DOI:10.1002/adsc.200303244
Selenium-catalyzed carbonylation of nitrobenzene and substituted nitroarenes with CO under atmospheric pressure afforded symmetrical 1,3-diarylureas in yields up to 94%. A mechanism has been proposed to demonstrate the formation of symmetrical ureas.
Co-reporter:Liandi Wang, Wei He and Zhengkun Yu
Chemical Society Reviews 2013 - vol. 42(Issue 2) pp:NaN621-621
Publication Date(Web):2012/10/18
DOI:10.1039/C2CS35323G
C–S bond activation, cleavage and transformations by means of transition metal compounds have recently become more and more important in the petroleum industry and synthetic chemistry. Homogeneous transition metal compounds have been investigated in order to provide the fundamental insight into the C–S bond cleavage in problematic organosulfur compounds such as thiophene, benzo- and dibenzothiophene derivatives. Rendering transition-metal mediated reactions with organosulfur compounds catalytic may provide promising routes to deep hydrodesulfurization of petroleum feedstocks, and offer potentially useful synthetic protocols for cross-couplings and biomimetic organic synthesis. During the last few decades increasing work was documented on C–S bond activation and transformations by means of transition metal compounds. This review summarizes the recent advances in C–S bond cleavage via the insertion of transition metals into the inert C–S bonds of these problematic organosulfur compounds, and transition-metal mediated C–S bond transformations via C–S activation through cross-couplings of thioesters, ketene dithioacetals, sulfonyl chlorides, and other diverse organosulfur compounds.
Co-reporter:Fei Huang, Ping Wu, Liandi Wang, Jiping Chen, Chenglin Sun and Zhengkun Yu
Chemical Communications 2014 - vol. 50(Issue 87) pp:NaN13395-13395
Publication Date(Web):2014/09/26
DOI:10.1039/C4CC90399D
Correction for ‘Copper-mediated intramolecular oxidative C–H/N–H cross-coupling of α-alkenoyl ketene N,S-acetals to synthesize pyrrolone derivatives’ by Fei Huang et al., Chem. Commun., 2014, DOI: 10.1039/c4cc05837b.
Co-reporter:Tenglong Guo, Quanbin Jiang, Fei Huang, Jiping Chen and Zhengkun Yu
Inorganic Chemistry Frontiers 2014 - vol. 1(Issue 6) pp:NaN711-711
Publication Date(Web):2014/06/13
DOI:10.1039/C4QO00122B
Construction of the benzene ring in carbazoles was efficiently realized through a domino dehydrochlorination/alkenylation/cycloaddition–oxidation sequence by means of palladium(II)-catalyzed, copper(II)-mediated reactions of N-protected 2,3-unsubstituted indoles with 3-chloropropiophenones in the presence of a base. 3-Alkenylated indole was confirmed to be formed as the reaction intermediate which then underwent Diels–Alder cycloaddition to the initially in situ generated enone from a 3-chloropropiophenones substrate, and the subsequent dehydrogenative aromatization yielded the carbazole product. The strategy to employ in situ generated enones as the reactive species avoided the use of a large excess of labile substrates and lessened the side reactions.
Co-reporter:Tenglong Guo, Quanbin Jiang and Zhengkun Yu
Inorganic Chemistry Frontiers 2015 - vol. 2(Issue 10) pp:NaN1365-1365
Publication Date(Web):2015/08/12
DOI:10.1039/C5QO00203F
Palladium(II)-catalyzed, copper(II)-mediated indole synthesis was achieved from the reactions of N-substituted simple pyrroles with enones generated in situ from 3-chloropropiophenones. A benzene ring was thus constructed onto a pyrrole backbone, affording substituted indole derivatives. A domino dehydrochlorination/C–H olefination /Diels–Alder cycloaddition/dehydrogenative aromatization sequence was established as the reaction pathway. The present methodology provides a concise route to highly functionalized indole derivatives.
Co-reporter:Huining Chai, Qingfu Wang, Tingting Liu and Zhengkun Yu
Dalton Transactions 2016 - vol. 45(Issue 44) pp:NaN17849-17849
Publication Date(Web):2016/10/10
DOI:10.1039/C6DT03620A
Dinuclear ruthenium(II)–NNN pincer complexes bearing a π linker-supported bis(pyrazolyl-imidazolyl-pyridine) ligand were synthesized and structurally characterized, and they exhibited excellent catalytic activity for the transfer hydrogenation of ketones in refluxing isopropanol, reaching TOF values up to 1.3 × 106 h−1. Compared with the corresponding mononuclear Ru(II)–NNN pincer complexes, the bimetallic complexes could be applied at concentrations as low as 0.03 mol% Ru and demonstrated remarkably enhanced catalytic activity in the transfer hydrogenation reactions of ketones. The high catalytic activity of the diruthenium(II) complexes is attributed to the excellent stability and possible cooperativity of the two coordinated Ru(II) metal centers through the π linker. The present synthetic methodology has established an applicable strategy to construct highly active bimetallic NNN pincer complex catalysts.
Co-reporter:Zhuqing Liu, Fei Huang, Jiang Lou, Quannan Wang and Zhengkun Yu
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 26) pp:NaN5540-5540
Publication Date(Web):2017/06/13
DOI:10.1039/C7OB01234A
Copper-promoted direct C–H alkoxylation of S,S-functionalized internal olefins, that is, α-oxo ketene dithioacetals, was efficiently achieved with alcohols as the alkoxylating agents, (diacetoxyiodo)benzene (PhI(OAc)2) as the oxidant, and benzoquinone (BQ) as the co-oxidant. The alkoxylated olefins were thus constructed and applied for the synthesis of alkoxylated N-heterocycles. The polarization of the olefinic carbon–carbon double bond by the electron-donating dialkylthio and electron-withdrawing α-oxo functionalities plays a crucial role in making such C–H alkoxylation reactions to occur under mild conditions. Mechanistic studies implicate a single-electron-transfer (SET) reaction pathway involved in the overall catalytic cycle.
Co-reporter:Tenglong Guo, Quanbin Jiang and Zhengkun Yu
Inorganic Chemistry Frontiers 2016 - vol. 3(Issue 2) pp:NaN281-281
Publication Date(Web):2016/01/12
DOI:10.1039/C6QO90004F
Correction for ‘Palladium-catalyzed oxidative annulation of in situ generated enones to pyrroles: a concise route to functionalized indoles’ by Tenglong Guo et al., Org. Chem. Front., 2015, 2, 1361–1365.
Co-reporter:Quanbin Jiang, Tenglong Guo, Kaikai Wu and Zhengkun Yu
Chemical Communications 2016 - vol. 52(Issue 14) pp:NaN2915-2915
Publication Date(Web):2016/01/13
DOI:10.1039/C5CC10361D
Rhodium(III)-catalyzed conjugate addition of aromatic and olefinic C–H bonds to CF3-substituted unsaturated ketones was efficiently achieved. Both arene and olefin substrates bearing a chelate assisted-directing group were coupled with a variety of β-trifluoromethyl-α,β-unsaturated ketones with excellent atom-economy, high yields, and broad substrate scopes.
Co-reporter:Qin Yang, Ping Wu, Jiping Chen and Zhengkun Yu
Chemical Communications 2014 - vol. 50(Issue 48) pp:NaN6339-6339
Publication Date(Web):2014/04/29
DOI:10.1039/C4CC02264E
Iron-catalyzed alkylation of internal olefins, that is, α-oxo ketene dithioacetals, was successfully realized by using styrenes as the alkylating reagents. Highly functionalized tetrasubstituted olefins were prepared in moderate to high yields.
Co-reporter:Fei Huang, Ping Wu, Liandi Wang, Jiping Chen, Chenglin Sun and Zhengkun Yu
Chemical Communications 2014 - vol. 50(Issue 83) pp:NaN12481-12481
Publication Date(Web):2014/08/28
DOI:10.1039/C4CC05837B
CuCl2 and CuBr2-mediated intramolecular oxidative C–H/N–H cross-coupling/halogenation of β-thioalkyl-substituted α-alkenoyl ketene N,S-acetals occurred efficiently, affording 4-halo-5-thioalkyl-3-pyrrolones. Tunable C–S and C–halo bond transformations of the resultant pyrrolone derivatives led to highly functionalized N-heterocyclic compounds.
Co-reporter:Qin Yang, Qingfu Wang and Zhengkun Yu
Chemical Society Reviews 2015 - vol. 44(Issue 8) pp:NaN2329-2329
Publication Date(Web):2015/02/09
DOI:10.1039/C4CS00496E
Transition metal-catalyzed substitution of alcohols by N-nucleophiles (or N-alkylation of amines and related compounds with alcohols) avoids the use of alkylating agents by means of borrowing hydrogen (BH) activation of the alcohol substrates. Water is produced as the only by-product, which makes the “BH” processes atom-economic and environmentally benign. Diverse types of homogeneous organometallic and heterogeneous transition metal catalysts, and substrates such as N-nucleophiles including amines, amides, sulfonamides and ammonia, and various alcohols, can be used for this purpose, demonstrating the promising potential of “BH” processes to replace the procedures using traditional alkylating agents in pharmaceutical and chemical industries. Borrowing hydrogen activation of alcohols for C–N bond formation has recently been paid more and more attention, and a lot of new and novel procedures and examples have been documented. This critical review summarizes the recent advances in “BH” substitution of alcohols by N-nucleophiles since 2009. “Semi-BH” N-alkylation processes with or without an external hydrogen acceptor are also briefly presented. Suitable discussion of the “BH” strategy provides new principles for establishing green processes to replace the relevant traditional synthetic methods for C–N bond formation.

![Ferrocene,1-[(S)-(dimethylamino)[2-(diphenylphosphino)phenyl]methyl]-2-(diphenylphosphino)-,(2R)- (9CI)](http://img.cochemist.com/ccimg/850500/850444-36-9.png)
![Ferrocene,1-[(S)-(dimethylamino)[2-(diphenylphosphino)phenyl]methyl]-2-(diphenylphosphino)-,(2R)- (9CI)](http://img.cochemist.com/ccimg/850500/850444-36-9_b.png)






















![1H-Pyrrole, 1-methyl-2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/480400/480390-47-4.png)
![1H-Pyrrole, 1-methyl-2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/480400/480390-47-4_b.png)






















































![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2R)-](http://img.cochemist.com/ccimg/155900/155806-35-2.png)
![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2R)-](http://img.cochemist.com/ccimg/155900/155806-35-2_b.png)
![(+)-1,2-BIS[(2S,5S)-2,5-DIMETHYLPHOSPHOLANO]BENZENE](http://img.cochemist.com/ccimg/136800/136735-95-0.png)
![(+)-1,2-BIS[(2S,5S)-2,5-DIMETHYLPHOSPHOLANO]BENZENE](http://img.cochemist.com/ccimg/136800/136735-95-0_b.png)


















![3-Buten-2-one, 4,4-bis[(phenylmethyl)thio]-](http://img.cochemist.com/ccimg/98500/98471-36-4.png)
![3-Buten-2-one, 4,4-bis[(phenylmethyl)thio]-](http://img.cochemist.com/ccimg/98500/98471-36-4_b.png)



![Pyridine, 2-[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/93400/93324-66-4.png)
![Pyridine, 2-[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/93400/93324-66-4_b.png)










![Methanone, [4-(hydroxymethyl)phenyl]phenyl-](/data/chemimg/974300/81449-01-6.png)
![Methanone, [4-(hydroxymethyl)phenyl]phenyl-](/data/chemimg/974300/81449-01-6_b.png)




















































![1H-Pyrrole, 1-[(4-bromophenyl)methyl]-](http://img.cochemist.com/ccimg/23700/23694-48-6.png)
![1H-Pyrrole, 1-[(4-bromophenyl)methyl]-](http://img.cochemist.com/ccimg/23700/23694-48-6_b.png)

















![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)






![Benzene, 1,1'-[(1E)-3-methyl-1-propene-1,3-diyl]bis-](http://img.cochemist.com/ccimg/7400/7302-01-4.png)
![Benzene, 1,1'-[(1E)-3-methyl-1-propene-1,3-diyl]bis-](http://img.cochemist.com/ccimg/7400/7302-01-4_b.png)

































































![1H-Pyrrole, 1-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/23700/23694-47-5.png)
![1H-Pyrrole, 1-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/23700/23694-47-5_b.png)