Co-reporter:Lei Gong, Marianne Wenzel, and Eric Meggers
Accounts of Chemical Research 2013 Volume 46(Issue 11) pp:2635
Publication Date(Web):June 3, 2013
DOI:10.1021/ar400083u
An octahedral metal complex with 6 different monodentate ligands can form 15 diastereomers as pairs of enantiomers. As a result, the elaborate stereochemistry of octahedral coordination geometries provides tremendous opportunities in the fields of catalysis, the materials sciences, and the life sciences. The demand for enantiomerically pure coordination complexes for tasks related to the selective molecular recognition of biomacromolecules led us to develop synthetic methods to control the absolute stereochemistry at octahedral metal centers. A few years ago our laboratory therefore embarked on a project exploring new and general synthetic strategies for the asymmetric synthesis of inert octahedral transition metal complexes. We initially used the example of thermally inert ruthenium polypyridyl complexes and developed a family of chiral bidentate ligands, including salicyloxazolines, (mercaptophenyl)oxazolines, sulfinylphenols, N-acetylsulfinamides, a phosphinohydroxybinaphthyl, and even the amino acid proline to serve as chiral auxiliaries for asymmetric coordination chemistry. All these chiral auxiliaries strongly coordinate to ruthenium(II) in a bidentate, deprotonated fashion, allowing them to control the absolute metal-centered configuration in the course of subsequent ligand exchange reactions. Finally, we can remove them from the metal without any loss of chiral information and without leaving a chemical trace. A key feature of these chiral auxiliary ligands is their switchable binding strength. A chelate effect ensures that the chiral ligands coordinate very tightly to the metal center, placing their carbon-based, sulfur-based, or axial chirality in a well-defined position close to the metal center to efficiently establish the absolute metal-centered configuration. At the same time a coordinating phenolate, carboximidate, carboxylate, or thiophenolate moiety makes the coordination reversible by weakening the binding strength through protonation or methylation. Following this strategy, we synthesized a large number of homoleptic, bis-heteroleptic, and tris-heteroleptic ruthenium polypyridyl complexes in an asymmetric fashion with enantiomeric ratios that routinely reached or exceeded 96:4. Our approach should serve as a blueprint for the asymmetric synthesis of different classes of ruthenium complexes and chiral coordination complexes of other metals
Co-reporter:Liang-An Chen, Xiaobing Ding, Lei Gong and Eric Meggers
Dalton Transactions 2013 vol. 42(Issue 16) pp:5623-5626
Publication Date(Web):08 Mar 2013
DOI:10.1039/C3DT00015J
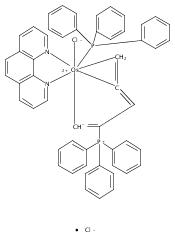
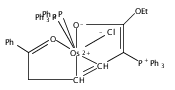
(R)-4-(Alkylthiomethyl)-5,5-dimethyl-2-(2′-hydroxyphenyl)-2-oxazolines are demonstrated to be highly suitable chiral auxiliaries for the two-step conversion of the half-sandwich complex [Ru(η6-C6H6)(bpy)Cl]Cl, bpy = 2,2′-bipyridine, into Δ-configured ruthenium polypyridyl complexes. The tailored thioether substituent at the oxazoline ring is essential for this conversion and not only promotes the removal of the benzene moiety but also controls the absolute metal-centered configuration. Applied to osmium, this strategy resulted in the first highly asymmetric synthesis of Δ-[Os(bpy)3](PF6)2.
Co-reporter:Liang-An Chen, Xiaobing Ding, Lei Gong and Eric Meggers
Dalton Transactions 2013 - vol. 42(Issue 16) pp:NaN5626-5626
Publication Date(Web):2013/03/08
DOI:10.1039/C3DT00015J
(R)-4-(Alkylthiomethyl)-5,5-dimethyl-2-(2′-hydroxyphenyl)-2-oxazolines are demonstrated to be highly suitable chiral auxiliaries for the two-step conversion of the half-sandwich complex [Ru(η6-C6H6)(bpy)Cl]Cl, bpy = 2,2′-bipyridine, into Δ-configured ruthenium polypyridyl complexes. The tailored thioether substituent at the oxazoline ring is essential for this conversion and not only promotes the removal of the benzene moiety but also controls the absolute metal-centered configuration. Applied to osmium, this strategy resulted in the first highly asymmetric synthesis of Δ-[Os(bpy)3](PF6)2.

![1H-Pyrido[3,4-b]indole, 2,3,4,9-tetrahydro-1,4-diphenyl-, (1R,4R)-rel-](http://img.cochemist.com/ccimg/874000/873977-16-3.png)
![1H-Pyrido[3,4-b]indole, 2,3,4,9-tetrahydro-1,4-diphenyl-, (1R,4R)-rel-](http://img.cochemist.com/ccimg/874000/873977-16-3_b.png)
![1-BUTANOL, 3-[(PHENYLMETHYL)THIO]-, (3R)-](http://img.cochemist.com/ccimg/871300/871210-84-3.png)
![1-BUTANOL, 3-[(PHENYLMETHYL)THIO]-, (3R)-](http://img.cochemist.com/ccimg/871300/871210-84-3_b.png)
![1H-Indole, 3-[(1S)-2-nitro-1-phenylethyl]-](http://img.cochemist.com/ccimg/863400/863301-32-0.png)
![1H-Indole, 3-[(1S)-2-nitro-1-phenylethyl]-](http://img.cochemist.com/ccimg/863400/863301-32-0_b.png)


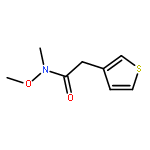
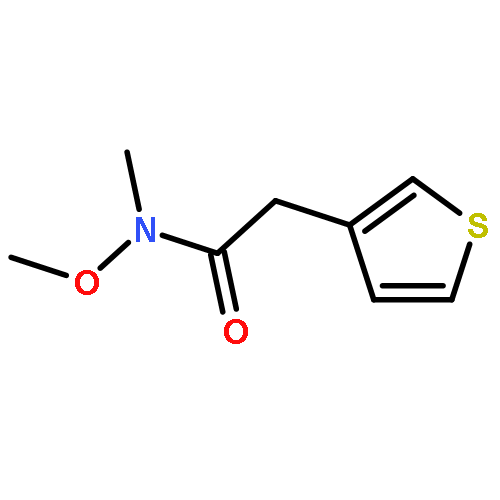


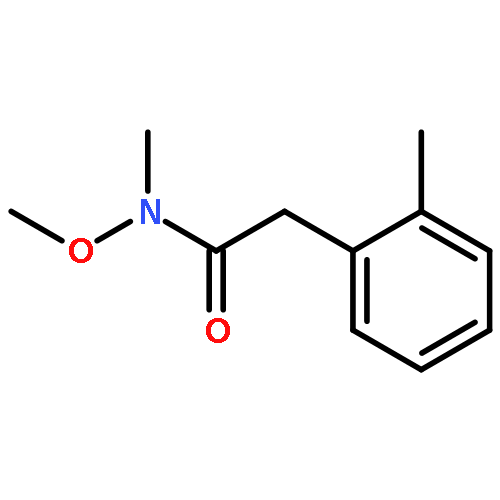
![1H-PYRAZOLE, 1-[(2E)-1-OXO-2-BUTENYL]-](http://img.cochemist.com/ccimg/624800/624738-96-1.png)
![1H-PYRAZOLE, 1-[(2E)-1-OXO-2-BUTENYL]-](http://img.cochemist.com/ccimg/624800/624738-96-1_b.png)
![Thiophene, 2-[(1S)-1-methyl-2-nitroethyl]-](http://img.cochemist.com/ccimg/615600/615553-05-4.png)
![Thiophene, 2-[(1S)-1-methyl-2-nitroethyl]-](http://img.cochemist.com/ccimg/615600/615553-05-4_b.png)
![BENZENE, 1-METHOXY-4-[(1R)-1-METHYL-2-NITROETHYL]-](http://img.cochemist.com/ccimg/615600/615552-98-2.png)
![BENZENE, 1-METHOXY-4-[(1R)-1-METHYL-2-NITROETHYL]-](http://img.cochemist.com/ccimg/615600/615552-98-2_b.png)
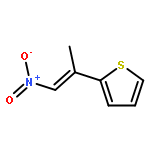
![Benzene, 1-methoxy-4-[(1E)-1-methyl-2-nitroethenyl]-](http://img.cochemist.com/ccimg/615600/615552-91-5.png)
![Benzene, 1-methoxy-4-[(1E)-1-methyl-2-nitroethenyl]-](http://img.cochemist.com/ccimg/615600/615552-91-5_b.png)
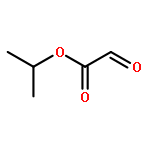
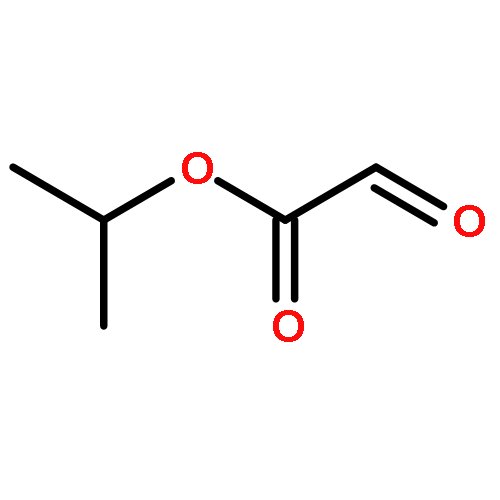
![Acetic acid, [(4-methoxyphenyl)imino]-, 1-methylethyl ester, (2E)-](http://img.cochemist.com/ccimg/350100/350032-06-3.png)
![Acetic acid, [(4-methoxyphenyl)imino]-, 1-methylethyl ester, (2E)-](http://img.cochemist.com/ccimg/350100/350032-06-3_b.png)



![Benzene, 1-[(1Z)-2-bromo-2-nitroethenyl]-4-methyl-](http://img.cochemist.com/ccimg/147100/147089-79-0.png)
![Benzene, 1-[(1Z)-2-bromo-2-nitroethenyl]-4-methyl-](http://img.cochemist.com/ccimg/147100/147089-79-0_b.png)

![Cyclohexane, [(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/132900/132839-80-6.png)
![Cyclohexane, [(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/132900/132839-80-6_b.png)


![Acetic acid, [(4-methoxyphenyl)imino]-, ethyl ester, (2E)-](http://img.cochemist.com/ccimg/120500/120419-96-7.png)
![Acetic acid, [(4-methoxyphenyl)imino]-, ethyl ester, (2E)-](http://img.cochemist.com/ccimg/120500/120419-96-7_b.png)












![Benzenamine, N-[[4-(trifluoromethyl)phenyl]methylene]-](http://img.cochemist.com/ccimg/79200/79128-83-9.png)
![Benzenamine, N-[[4-(trifluoromethyl)phenyl]methylene]-](http://img.cochemist.com/ccimg/79200/79128-83-9_b.png)


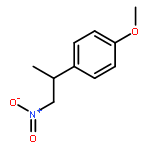
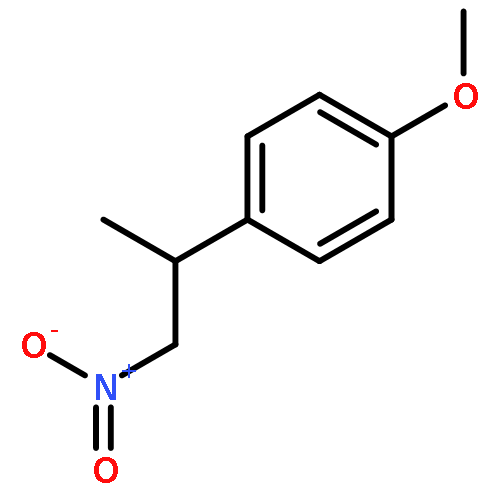



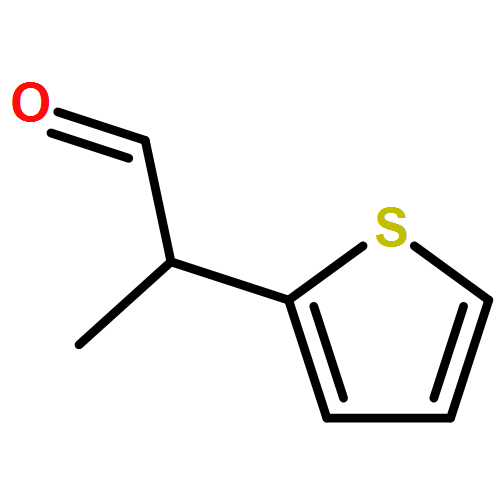


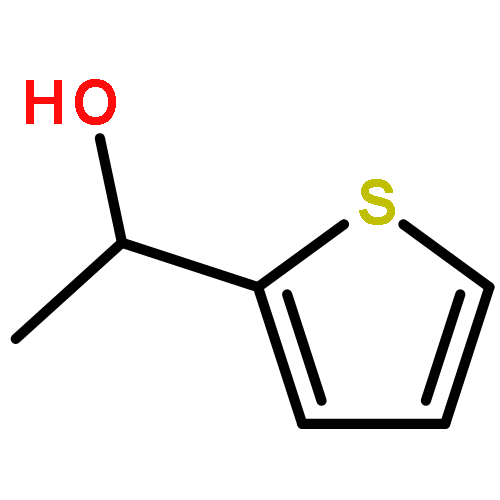
![Butanoic acid, 3-[(phenylmethyl)thio]-, methyl ester](http://img.cochemist.com/ccimg/61500/61452-42-4.png)
![Butanoic acid, 3-[(phenylmethyl)thio]-, methyl ester](http://img.cochemist.com/ccimg/61500/61452-42-4_b.png)

![Benzene, [(nitromethyl)thio]-](http://img.cochemist.com/ccimg/60600/60595-16-6.png)
![Benzene, [(nitromethyl)thio]-](http://img.cochemist.com/ccimg/60600/60595-16-6_b.png)
![Benzene, 1-[(1Z)-2-bromo-2-nitroethenyl]-4-chloro-](http://img.cochemist.com/ccimg/59100/59019-41-9.png)
![Benzene, 1-[(1Z)-2-bromo-2-nitroethenyl]-4-chloro-](http://img.cochemist.com/ccimg/59100/59019-41-9_b.png)
![Benzene, 1-[(1Z)-2-bromo-2-nitroethenyl]-4-methoxy-](http://img.cochemist.com/ccimg/59100/59019-40-8.png)
![Benzene, 1-[(1Z)-2-bromo-2-nitroethenyl]-4-methoxy-](http://img.cochemist.com/ccimg/59100/59019-40-8_b.png)


![2-Phenylbenzo[d]oxazol-5-amine](http://img.cochemist.com/ccimg/41400/41373-37-9.png)
![2-Phenylbenzo[d]oxazol-5-amine](http://img.cochemist.com/ccimg/41400/41373-37-9_b.png)












![1,3-Benzodioxole,5-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/22600/22568-48-5.png)
![1,3-Benzodioxole,5-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/22600/22568-48-5_b.png)


![Benzene, 1-methoxy-2-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/21300/21298-69-1.png)
![Benzene, 1-methoxy-2-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/21300/21298-69-1_b.png)




![Benzene, [(1Z)-2-bromo-2-nitroethenyl]-](http://img.cochemist.com/ccimg/18400/18315-81-6.png)
![Benzene, [(1Z)-2-bromo-2-nitroethenyl]-](http://img.cochemist.com/ccimg/18400/18315-81-6_b.png)



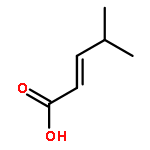
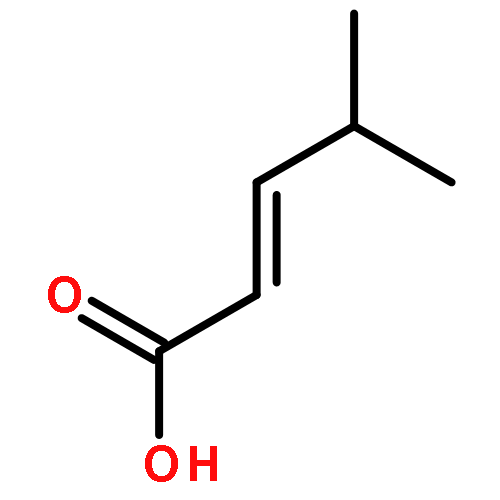
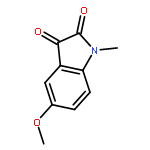
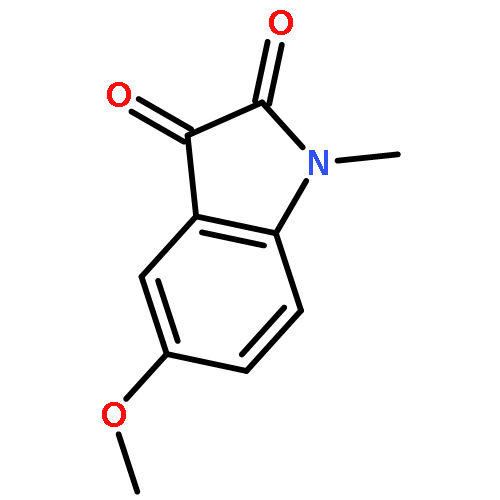


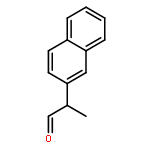
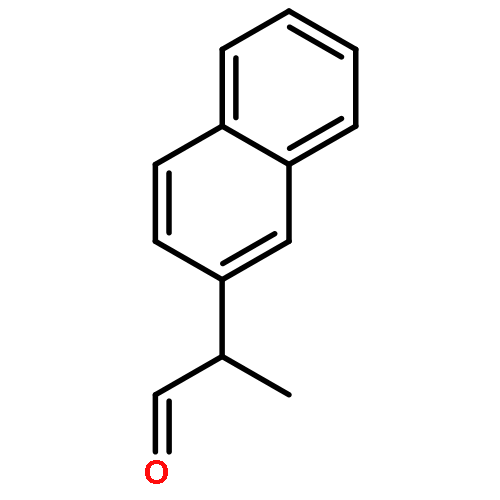



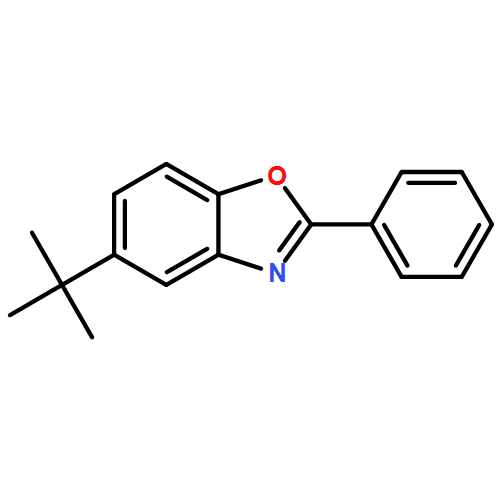
![Benzenamine, N-[(4-bromophenyl)methylene]-](http://img.cochemist.com/ccimg/5900/5877-51-0.png)
![Benzenamine, N-[(4-bromophenyl)methylene]-](http://img.cochemist.com/ccimg/5900/5877-51-0_b.png)




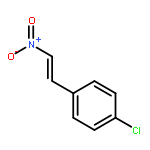












![Benzenamine,N-[(4-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/2400/2362-79-0.png)
![Benzenamine,N-[(4-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/2400/2362-79-0_b.png)
![Benzenamine,N-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/2400/2362-77-8.png)
![Benzenamine,N-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/2400/2362-77-8_b.png)





















































































































































































































































































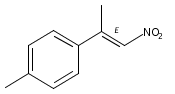
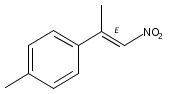

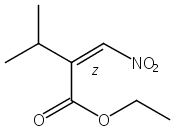





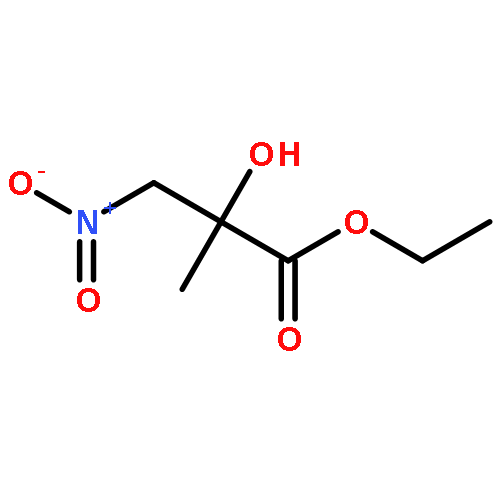
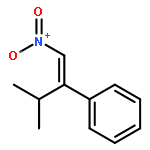

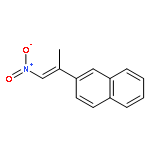
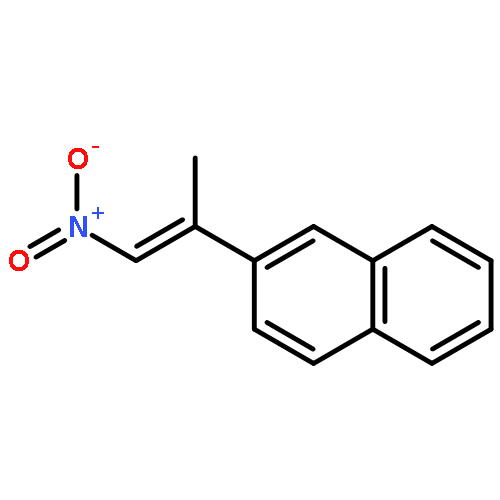






























![Phenol, 2-[(4S)-4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]-](/data/chemimg/120900/131380-91-1.png)
![Phenol, 2-[(4S)-4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]-](/data/chemimg/120900/131380-91-1_b.png)




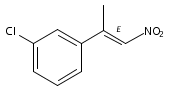
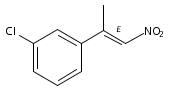
![Benzene, 1-chloro-4-[(1R)-1-methyl-2-nitroethyl]-](http://img.cochemist.com/ccimg/109800/109757-74-6.png)
![Benzene, 1-chloro-4-[(1R)-1-methyl-2-nitroethyl]-](http://img.cochemist.com/ccimg/109800/109757-74-6_b.png)


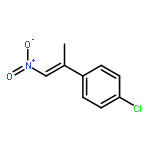
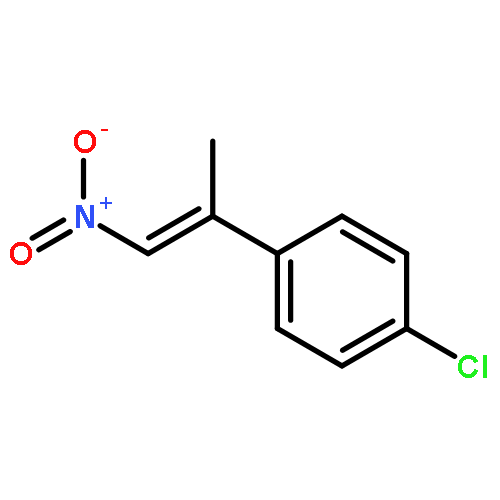













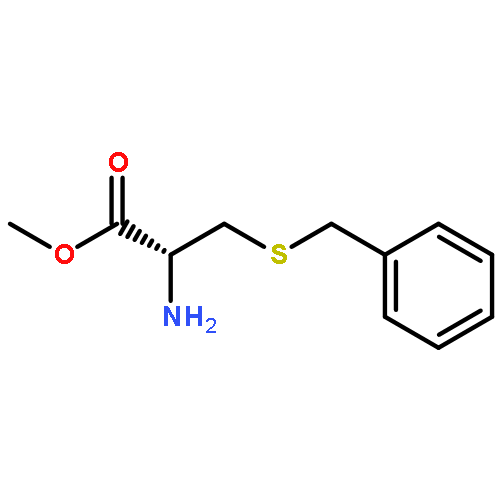




![Benzene, [(1E)-1-methyl-2-nitroethenyl]-](/data/chemimg/1737100/15241-24-4.png)
![Benzene, [(1E)-1-methyl-2-nitroethenyl]-](/data/chemimg/1737100/15241-24-4_b.png)
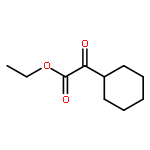

![BENZENE, [2-METHYL-1-(NITROMETHYL)PROPYL]-](http://img.cochemist.com/ccimg/7800/7796-81-8.png)
![BENZENE, [2-METHYL-1-(NITROMETHYL)PROPYL]-](http://img.cochemist.com/ccimg/7800/7796-81-8_b.png)