Co-reporter:Fang Wang, Sabine Becker, Mikael A. Minier, Andrei Loas, Megan N. Jackson, and Stephen J. Lippard
Inorganic Chemistry September 18, 2017 Volume 56(Issue 18) pp:11050-11050
Publication Date(Web):September 5, 2017
DOI:10.1021/acs.inorgchem.7b01418
We introduce a novel platform to mimic the coordination environment of carboxylate-bridged diiron proteins by tethering a small, dangling internal carboxylate, (CH2)nCOOH, to phenol-imine macrocyclic ligands (H3PIMICn). In the presence of an external bulky carboxylic acid (RCO2H), the ligands react with [Fe2(Mes)4] (Mes = 2,4,6-trimethylphenyl) to afford dinuclear [Fe2(PIMICn)(RCO2)(MeCN)] (n = 4–6) complexes. X-ray diffraction studies revealed structural similarities between these complexes and the reduced diiron active sites of proteins such as Class I ribonucleotide reductase (RNR) R2 and soluble methane monooxygenase hydroxylase. The number of CH2 units of the internal carboxylate arm controls the diiron core geometry, affecting in turn the anodic peak potential of the complexes. As functional synthetic models, these complexes facilitate the oxidation of C–H bonds in the presence of peroxides and oxo transfer from O2 to an internal phosphine moiety.
Co-reporter:Christine E. Tinberg;Woon Ju Song;Viviana Izzo
Biochemistry March 22, 2011 Volume 50(Issue 11) pp:1788-1798
Publication Date(Web):Publication Date (Web): March 2, 2011
DOI:10.1021/bi200028z
Phenol hydroxylase (PH) and toluene/o-xylene monooxygenase (ToMO) from Pseudomonas sp. OX1 require three or four protein components to activate dioxygen for the oxidation of aromatic substrates at a carboxylate-bridged diiron center. In this study, we investigated the influence of the hydroxylases, regulatory proteins, and electron-transfer components of these systems on substrate (phenol; NADH) consumption and product (catechol; H2O2) generation. Single-turnover experiments revealed that only complete systems containing all three or four protein components are capable of oxidizing phenol, a major substrate for both enzymes. Under ideal conditions, the hydroxylated product yield was ∼50% of the diiron centers for both systems, suggesting that these enzymes operate by half-sites reactivity mechanisms. Single-turnover studies indicated that the PH and ToMO electron-transfer components exert regulatory effects on substrate oxidation processes taking place at the hydroxylase actives sites, most likely through allostery. Steady state NADH consumption assays showed that the regulatory proteins facilitate the electron-transfer step in the hydrocarbon oxidation cycle in the absence of phenol. Under these conditions, electron consumption is coupled to H2O2 formation in a hydroxylase-dependent manner. Mechanistic implications of these results are discussed.
Co-reporter:Chanan D. Sessler, Martin Rahm, Sabine Becker, Jacob M. Goldberg, Fang Wang, and Stephen J. Lippard
Journal of the American Chemical Society July 12, 2017 Volume 139(Issue 27) pp:9325-9325
Publication Date(Web):June 2, 2017
DOI:10.1021/jacs.7b04457
The CF2H group, a potential surrogate for the OH group, can act as an unusual hydrogen bond donor, as confirmed by crystallographic, spectroscopic, and computational methods. Here, we demonstrate the bioisosterism of the OH and CF2H groups and the important roles of CF2–H···O hydrogen bonds in influencing intermolecular interactions and conformational preferences. Experimental evidence, corroborated by theory, reveals the distinctive nature of CF2H hydrogen bonding interactions relative to their normal OH hydrogen bonding counterparts.
Co-reporter:Imogen A. Riddell;Samuel G. Awuah
PNAS 2017 Volume 114 (Issue 5 ) pp:950-955
Publication Date(Web):2017-01-31
DOI:10.1073/pnas.1615327114
Cisplatin is the most commonly used anticancer drug for the treatment of testicular germ cell tumors (TGCTs). The hypersensitivity
of TGCTs to cisplatin is a subject of widespread interest. Here, we show that high-mobility group box protein 4 (HMGB4), a
protein preferentially expressed in testes, uniquely blocks excision repair of cisplatin-DNA adducts, 1,2-intrastrand cross-links,
to potentiate the sensitivity of TGCTs to cisplatin therapy. We used CRISPR/Cas9-mediated gene editing to knockout the HMGB4
gene in a testicular human embryonic carcinoma and examined cellular responses. We find that loss of HMGB4 elicits resistance
to cisplatin as evidenced by cell proliferation and apoptosis assays. We demonstrate that HMGB4 specifically inhibits repair
of the major cisplatin-DNA adducts in TGCT cells by using the human TGCT excision repair system. Our findings also reveal
characteristic HMGB4-dependent differences in cell cycle progression following cisplatin treatment. Collectively, these data
provide convincing evidence that HMGB4 plays a major role in sensitizing TGCTs to cisplatin, consistent with shielding of
platinum-DNA adducts from excision repair.
Co-reporter:A. Loas;S. J. Lippard
Journal of Materials Chemistry B 2017 vol. 5(Issue 45) pp:8929-8933
Publication Date(Web):2017/11/22
DOI:10.1039/C7TB02666H
We report the first Cu(II)-based fluorescent sensors for ratiometric detection of nitric oxide. The two probes operate by energy transfer between hydroxycoumarin and fluorescein chromophores, contain polyproline or piperazine as rigid spacers, and elicit a rapid and selective ratiometric response upon direct reaction with nitric oxide.
Co-reporter:Timothy C. Johnstone, Kogularamanan Suntharalingam, and Stephen J. Lippard
Chemical Reviews 2016 Volume 116(Issue 5) pp:3436
Publication Date(Web):February 11, 2016
DOI:10.1021/acs.chemrev.5b00597
The platinum drugs, cisplatin, carboplatin, and oxaliplatin, prevail in the treatment of cancer, but new platinum agents have been very slow to enter the clinic. Recently, however, there has been a surge of activity, based on a great deal of mechanistic information, aimed at developing nonclassical platinum complexes that operate via mechanisms of action distinct from those of the approved drugs. The use of nanodelivery devices has also grown, and many different strategies have been explored to incorporate platinum warheads into nanomedicine constructs. In this Review, we discuss these efforts to create the next generation of platinum anticancer drugs. The introduction provides the reader with a brief overview of the use, development, and mechanism of action of the approved platinum drugs to provide the context in which more recent research has flourished. We then describe approaches that explore nonclassical platinum(II) complexes with trans geometry or with a monofunctional coordination mode, polynuclear platinum(II) compounds, platinum(IV) prodrugs, dual-threat agents, and photoactivatable platinum(IV) complexes. Nanoparticles designed to deliver platinum(IV) complexes will also be discussed, including carbon nanotubes, carbon nanoparticles, gold nanoparticles, quantum dots, upconversion nanoparticles, and polymeric micelles. Additional nanoformulations, including supramolecular self-assembled structures, proteins, peptides, metal–organic frameworks, and coordination polymers, will then be described. Finally, the significant clinical progress made by nanoparticle formulations of platinum(II) agents will be reviewed. We anticipate that such a synthesis of disparate research efforts will not only help to generate new drug development ideas and strategies, but also will reflect our optimism that the next generation of approved platinum cancer drugs is about to arrive.
Co-reporter:Sunghee Kim; Mikael A. Minier; Andrei Loas; Sabine Becker; Fang Wang
Journal of the American Chemical Society 2016 Volume 138(Issue 6) pp:1804-1807
Publication Date(Web):February 2, 2016
DOI:10.1021/jacs.5b12825
To elucidate the factors that impart selectivity for nitroxyl (HNO) over nitric oxide (NO), thiols, and H2S in metal-based fluorescent probes, we investigated five Cu(II)-cyclam (14-N4) derivatives. Upon exposure to NO gas at pH 7, no changes occur in the UV–vis spectra of any of the complexes. Addition of Angeli’s salt to generate HNO promotes reduction of Cu(II) only in the case of [CuII(14-N4-Ts)(OTf)2], which has the most positive reduction potential of the series. To gain insight into the observed reactivity, we prepared the Cu(II) complex of the mixed thia/aza 14-N2S2 ligand. [CuII(14-N2S2)(OTf)2] reacts reversibly with HNO at pH 7, although nonselectively over thiols and H2S. The recurrent sensing of HNO uncovered with the study of Cu(II) azamacrocyclic complexes is a remarkable feature that opens the door for the design of a new generation of metal-based probes.
Co-reporter:Imogen A. Riddell, Keli Agama, Ga Young Park, Yves Pommier, and Stephen J. Lippard
ACS Chemical Biology 2016 Volume 11(Issue 11) pp:2996
Publication Date(Web):September 20, 2016
DOI:10.1021/acschembio.6b00565
Drugs capable of trapping topoisomerase II (Top2), an essential enzyme that cleaves DNA to remove naturally occurring knots and tangles, can serve as potent anticancer agents. The monofunctional platinum agent phenanthriplatin, cis-[Pt(NH3)2(phenanthridine)Cl](NO3), is shown here to trap Top2 in addition to its known modes of inhibition of DNA and RNA polymerases. Its potency therefore combines diverse modes of action by which phenanthriplatin kills cancer cells. The observation that phenanthriplatin can act as a Top2 poison highlights opportunities to design nonclassical platinum anticancer agents with this novel mechanism of action. Such complexes have the potential to overcome current limitations with chemotherapy, such as resistance, and to provide treatment options for cancers that do not respond well to classical agents. Covalent DNA-platinum lesions implicated in Top2 poisoning are distinctive from those generated by known therapeutic topoisomerase poisons, which typically exert their action by reversible binding at the interface of Top2-DNA cleavage complexes.
Co-reporter:Yao-Rong Zheng, Kogularamanan Suntharalingam, Peter M. Bruno, Wei Lin, Weixue Wang, Michael T. Hemann, Stephen J. Lippard
Inorganica Chimica Acta 2016 Volume 452() pp:125-129
Publication Date(Web):1 October 2016
DOI:10.1016/j.ica.2016.03.021
•A hexanuclear platinum cage has anti-proliferative effects against human cancer cells.•Unlike classical Pt anticancer agents, this cage interacts with DNA non-covalently.•The cage intercalates into DNA and promotes DNA condensation.•In cancer cells, the cage induces DNA damage and triggers apoptosis and senescence.The cellular response evoked by a hexanuclear platinum complex, Pt6L4 (1), is reported. Compound 1, a 3-nm octahedral cage formed by self-assembly of six Pt(II) centers and four 2,4,6-tris(4-pyridyl)-1,3,5-triazine ligands (L), exhibits promising in vitro potency against a panel of human cancer cell lines. Unlike classical platinum-based anticancer agents, 1 interacts with DNA in a non-covalent, intercalative manner and promotes DNA condensation. In cancer cells, 1 induces DNA damage, upregulates p53, its phosphorylated form phospho-p53 and its downstream effector, p21, as well as both apoptosis and senescence.This study provides an in-depth mechanistic investigation of the origin of antiproliferative activity for a self-assembled platinum cage. Unlike classical platinum-based anticancer agents, this construct interacts with DNA in a non-covalent, intercalative manner and promotes DNA condensation.
Co-reporter:Anna E. Czapar, Yao-Rong Zheng, Imogen A. Riddell, Sourabh Shukla, Samuel G. Awuah, Stephen J. Lippard, and Nicole F. Steinmetz
ACS Nano 2016 Volume 10(Issue 4) pp:4119
Publication Date(Web):March 16, 2016
DOI:10.1021/acsnano.5b07360
Phenanthriplatin, cis-[Pt(NH3)2Cl(phenanthridine)](NO3), is a cationic monofunctional DNA-binding platinum(II) anticancer drug candidate with unusual potency and cellular response profiles. Its in vivo efficacy has not yet been demonstrated, highlighting the need for a delivery system. Here we report tobacco mosaic virus (TMV) as a delivery system for phenanthriplatin. TMV forms hollow nanotubes with a polyanionic interior surface; capitalizing on this native structure, we developed a one-step phenanthriplatin loading protocol. Phenanthriplatin release from the carrier is induced in acidic environments. This delivery system, designated PhenPt-TMV, exhibits matched efficacy in a cancer cell panel compared to free phenanthriplatin. In vivo tumor delivery and efficacy were confirmed by using a mouse model of triple negative breast cancer. Tumors treated with PhenPt-TMV were 4× smaller than tumors treated with free phenanthriplatin or cisplatin, owing to increased accumulation of phenanthriplatin within the tumor tissue. The biology-derived TMV delivery system may facilitate translation of phenanthriplatin into the clinic.Keywords: cancer therapy; drug delivery; metals in medicine; nanotechnology; tobacco mosaic virus
Co-reporter:Melissa L. Zastrow, Robert J. Radford, Wen Chyan, Charles T. Anderson, Daniel Y. Zhang, Andrei Loas, Thanos Tzounopoulos, and Stephen J. Lippard
ACS Sensors 2016 Volume 1(Issue 1) pp:32
Publication Date(Web):September 23, 2015
DOI:10.1021/acssensors.5b00022
Chelatable, or mobile, forms of zinc play critical signaling roles in numerous biological processes. Elucidating the action of mobile Zn(II) in complex biological environments requires sensitive tools for visualizing, tracking, and manipulating Zn(II) ions. A large toolbox of synthetic photoinduced electron transfer (PET)-based fluorescent Zn(II) sensors are available, but the applicability of many of these probes is limited by poor zinc sensitivity and low dynamic ranges owing to proton interference. We present here a general approach for acetylating PET-based probes containing a variety of fluorophores and zinc-binding units. The new sensors provide substantially improved zinc sensitivity and allow for incubation of live cells and tissue slices with nM probe concentrations, a significant improvement compared to the μM concentrations that are typically required for a measurable fluorescence signal. Acetylation effectively reduces or completely quenches background fluorescence in the metal-free sensor. Binding of Zn(II) selectively and quickly mediates hydrolytic cleavage of the acetyl groups, providing a large fluorescence response. An acetylated blue coumarin-based sensor was used to carry out detailed analyses of metal binding and metal-promoted acetyl hydrolysis. Acetylated benzoresorufin-based red-emitting probes with different zinc-binding sites are effective for sensing Zn(II) ions in live cells when applied at low concentrations (∼50–100 nM). We used green diacetylated Zinpyr1 (DA-ZP1) to image endogenous mobile Zn(II) in the molecular layer of mouse dorsal cochlear nucleus (DCN), confirming that acetylation is a suitable approach for preparing sensors that are highly specific and sensitive to mobile zinc in biological systems.Keywords: auditory synapses; fluorescence; metalloneurochemistry; zinc sensors; ZnT3
Co-reporter:Malay Patra, Samuel G. Awuah, and Stephen J. Lippard
Journal of the American Chemical Society 2016 Volume 138(Issue 38) pp:
Publication Date(Web):August 27, 2016
DOI:10.1021/jacs.6b06937
Glycoconjugation is a promising strategy for specific targeting of cancer. In this study, we investigated the effect of d-glucose substitution position on the biological activity of glucose–platinum conjugates (Glc-Pts). We synthesized and characterized all possible positional isomers (C1α, C1β, C2, C3, C4, and C6) of a Glc-Pt. The synthetic routes presented here could, in principle, be extended to prepare glucose conjugates with different active ingredients, other than platinum. The biological activities of the compounds were evaluated both in vitro and in vivo. We discovered that varying the position of substitution of d-glucose alters not only the cellular uptake and cytotoxicity profile but also the GLUT1 specificity of resulting glycoconjugates, where GLUT1 is glucose transporter 1. The C1α- and C2-substituted Glc-Pts (1α and 2) accumulate in cancer cells most efficiently compared to the others, whereas the C3-Glc-Pt (3) is taken up least efficiently. Compounds 1α and 2 are more potent compared to 3 in DU145 cells. The α- and β-anomers of the C1-Glc-Pt also differ significantly in their cellular uptake and activity profiles. No significant differences in uptake of the Glc-Pts were observed in non-cancerous RWPE2 cells. The GLUT1 specificity of the Glc-Pts was evaluated by determining the cellular uptake in the absence and in the presence of the GLUT1 inhibitor cytochalasin B, and by comparing their anticancer activity in DU145 cells and a GLUT1 knockdown cell line. The results reveal that C2-substituted Glc-Pt 2 has the highest GLUT1-specific internalization, which also reflects the best cancer-targeting ability. In a syngeneic breast cancer mouse model overexpressing GLUT1, compound 2 showed antitumor efficacy and selective uptake in tumors with no observable toxicity. This study thus reveals the synthesis of all positional isomers of d-glucose substitution for platinum warheads with detailed glycotargeting characterization in cancer.
Co-reporter:Dr. Malay Patra;Dr. Timothy C. Johnstone;Dr. Kogularamanan Suntharalingam; Stephen J. Lippard
Angewandte Chemie 2016 Volume 128( Issue 7) pp:2596-2600
Publication Date(Web):
DOI:10.1002/ange.201510551
Abstract
Three rationally designed glucose–platinum conjugates (Glc–Pts) were synthesized and their biological activities evaluated. The Glc–Pts, 1–3, exhibit high levels of cytotoxicity toward a panel of cancer cells. The subcellular target and cellular uptake mechanism of the Glc–Pts were elucidated. For uptake into cells, Glc–Pt 1 exploits both glucose and organic cation transporters, both widely overexpressed in cancer. Compound 1 preferentially accumulates in and annihilates cancer, compared to normal epithelial, cells in vitro.
Co-reporter:Dr. Imogen A. Riddell;Dr. Timothy C. Johnstone;Dr. Ga Young Park ; Stephen J. Lippard
Chemistry - A European Journal 2016 Volume 22( Issue 22) pp:7574-7581
Publication Date(Web):
DOI:10.1002/chem.201600236
Abstract
The monofunctional platinum anticancer agent phenanthriplatin generates covalent adducts with the purine bases guanine and adenine. Preferential nucleotide binding was investigated by using a polymerase stop assay and linear DNA amplification with a 163-base pair DNA double helix. Similarly to cisplatin, phenanthriplatin forms the majority of adducts at guanosine residues, but significant differences in both the number and position of platination sites emerge when comparing results for the two complexes. Notably, the monofunctional complex generates a greater number of polymerase-halting lesions at adenosine residues than does cisplatin. Studies with 9-methyladenine reveal that, under abiological conditions, phenanthriplatin binds to the N1 or N7 position of 9-methyladenine in approximately equimolar amounts. By contrast, comparable reactions with 9-methylguanine afforded only the N7-bound species. Both of the 9-methyladenine linkage isomers (N1 and N7) exist as two diastereomeric species, arising from hindered rotation of the aromatic ligands about their respective platinum–nitrogen bonds. Eyring analysis of rate constants extracted from variable-temperature NMR spectroscopic data revealed that the activation energies for ligand rotation in the N1-bound platinum complex and the N7-linkage isomers are comparable. Finally, a kinetic analysis indicated that phenanthriplatin reacts more rapidly, by a factor of eight, with 9-methylguanine than with 9-methyladenine, suggesting that the distribution of lesions formed on double-stranded DNA is kinetically controlled. In addition, implications for the potent anticancer activity of phenanthriplatin are discussed herein.
Co-reporter:Dr. Malay Patra;Dr. Timothy C. Johnstone;Dr. Kogularamanan Suntharalingam; Stephen J. Lippard
Angewandte Chemie International Edition 2016 Volume 55( Issue 7) pp:2550-2554
Publication Date(Web):
DOI:10.1002/anie.201510551
Abstract
Three rationally designed glucose–platinum conjugates (Glc–Pts) were synthesized and their biological activities evaluated. The Glc–Pts, 1–3, exhibit high levels of cytotoxicity toward a panel of cancer cells. The subcellular target and cellular uptake mechanism of the Glc–Pts were elucidated. For uptake into cells, Glc–Pt 1 exploits both glucose and organic cation transporters, both widely overexpressed in cancer. Compound 1 preferentially accumulates in and annihilates cancer, compared to normal epithelial, cells in vitro.
Co-reporter:Pablo Rivera-Fuentes and Stephen J. Lippard
Accounts of Chemical Research 2015 Volume 48(Issue 11) pp:2927
Publication Date(Web):November 9, 2015
DOI:10.1021/acs.accounts.5b00388
Nitroxyl (HNO) is a biological signaling agent that displays distinctive reactivity compared to nitric oxide (NO). As a consequence, these two reactive nitrogen species trigger different physiological responses. Selective detection of HNO over NO has been a challenge for chemists, and several fluorogenic molecular probes have been recently developed with that goal in mind. Common constructs take advantage of the HNO-induced reduction of Cu(II) to Cu(I). The sensing mechanism of such probes relies on the ability of the unpaired electron in a d orbital of the Cu(II) center to quench the fluorescence of a photoemissive ligand by either an electron or energy transfer mechanism. Experimental and theoretical mechanistic studies suggest that proton-coupled electron transfer mediates this process, and careful tuning of the copper coordination environment has led to sensors with optimized selectivity and kinetics.The current optical probes cover the visible and near-infrared regions of the spectrum. This palette of sensors comprises structurally and functionally diverse fluorophores such as coumarin (blue/green emission), boron dipyrromethane (BODIPY, green emission), benzoresorufin (red emission), and dihydroxanthenes (near-infrared emission). Many of these sensors have been successfully applied to detect HNO production in live cells. For example, copper-based optical probes have been used to detect HNO production in live mammalian cells that have been treated with H2S and various nitrosating agents. These studies have established a link between HSNO, the smallest S-nitrosothiol, and HNO. In addition, a near-infrared HNO sensor has been used to perform multicolor/multianalyte microscopy, revealing that exogenously applied HNO elevates the concentration of intracellular mobile zinc. This mobilization of zinc ions is presumably a consequence of nitrosation of cysteine residues in zinc-chelating proteins such as metallothionein.Future challenges for the optical imaging of HNO include devising probes that can detect HNO reversibly, especially because ratiometric imaging can only report equilibrium concentrations when the sensing event is reversible. Another important aspect that needs to be addressed is the creation of probes that can sense HNO in specific subcellular locations. These tools would be useful to identify the organelles in which HNO is produced in mammalian cells and probe the intracellular signaling networks in which this reactive nitrogen species is involved. In addition, near-infrared emitting probes might be applied to detect HNO in thicker specimens, such as acute tissue slices and even live animals, enabling the investigation of the roles of HNO in physiological or pathological conditions in multicellular systems.
Co-reporter:Weixue Wang, Alexandria D. Liang, and Stephen J. Lippard
Accounts of Chemical Research 2015 Volume 48(Issue 9) pp:2632
Publication Date(Web):August 21, 2015
DOI:10.1021/acs.accounts.5b00312
A fundamental goal in catalysis is the coupling of multiple reactions to yield a desired product. Enzymes have evolved elegant approaches to address this grand challenge. A salient example is the biological conversion of methane to methanol catalyzed by soluble methane monooxygenase (sMMO), a member of the bacterial multicomponent monooxygenase (BMM) superfamily.sMMO is a dynamic protein complex of three components: a hydroxylase, a reductase, and a regulatory protein. The active site, a carboxylate-rich non-heme diiron center, is buried inside the 251 kDa hydroxylase component. The enzyme processes four substrates: O2, protons, electrons, and methane. To couple O2 activation to methane oxidation, timely control of substrate access to the active site is critical. Recent studies of sMMO, as well as its homologues in the BMM superfamily, have begun to unravel the mechanism. The emerging and unifying picture reveals that each substrate gains access to the active site along a specific pathway through the hydroxylase. Electrons and protons are delivered via a three-amino-acid pore located adjacent to the diiron center; O2 migrates via a series of hydrophobic cavities; and hydrocarbon substrates reach the active site through a channel or linked set of cavities. The gating of these pathways mediates entry of each substrate to the diiron active site in a timed sequence and is coordinated by dynamic interactions with the other component proteins. The result is coupling of dioxygen consumption with hydrocarbon oxidation, avoiding unproductive oxidation of the reductant rather than the desired hydrocarbon.To initiate catalysis, the reductase delivers two electrons to the diiron(III) center by binding over the pore of the hydroxylase. The regulatory component then displaces the reductase, docking onto the same surface of the hydroxylase. Formation of the hydroxylase-regulatory component complex (i) induces conformational changes of pore residues that may bring protons to the active site; (ii) connects hydrophobic cavities in the hydroxylase leading from the exterior to the diiron active site, providing a pathway for O2 and methane, in the case of sMMO, to the reduced diiron center for O2 activation and substrate hydroxylation; (iii) closes the pore, as well as a channel in the case of four-component BMM enzymes, restricting proton access to the diiron center during formation of “Fe2O2” intermediates required for hydrocarbon oxidation; and (iv) inhibits undesired electron transfer to the Fe2O2 intermediates by blocking reductase binding during O2 activation. This mechanism is quite different from that adopted by cytochromes P450, a large class of heme-containing monooxygenases that catalyze reactions very similar to those catalyzed by the BMM enzymes. Understanding the timed enzyme control of substrate access has implications for designing artificial catalysts. To achieve multiple turnovers and tight coupling, synthetic models must also control substrate access, a major challenge considering that nature requires large, multimeric, dynamic protein complexes to accomplish this feat.
Co-reporter:Alexandria Deliz Liang
Journal of the American Chemical Society 2015 Volume 137(Issue 33) pp:10520-10523
Publication Date(Web):August 12, 2015
DOI:10.1021/jacs.5b07055
Toluene/o-xylene monooxygenase (ToMO) is a non-heme diiron protein that activates O2 for subsequent arene oxidation. ToMO utilizes four protein components, a catalytic hydroxylase, a regulatory protein, a Rieske protein, and a reductase. O2 activation and substrate hydroxylation in the presence of all four protein components is examined. These studies demonstrate the importance of native reductants by revealing reactivity unobserved when dithionite and mediators are used as the reductant. This reactivity is compared with that of other O2-activating diiron enzymes.
Co-reporter:Samuel G. Awuah; Yao-Rong Zheng; Peter M. Bruno; Michael T. Hemann
Journal of the American Chemical Society 2015 Volume 137(Issue 47) pp:14854-14857
Publication Date(Web):November 12, 2015
DOI:10.1021/jacs.5b10182
Expression of indoleamine-2,3-dioxygenase (IDO), an immunosuppressive enzyme in human tumors, leads to immune evasion and tumor tolerance. IDO is therefore a tumor immunotherapeutic target, and several IDO inhibitors are currently undergoing clinical trials. IDO inhibitors can enhance the efficacy of common cancer chemotherapeutics. Here we investigate Pt(IV)-(D)-1-methyltryptophan conjugates 1 and 2 for combined immunomodulation and DNA cross-link-triggered apoptosis for cancer “immuno-chemotherapy”. Compound 2 effectively kills hormone-dependent, cisplatin-resistant human ovarian cancer cells, inhibiting IDO by transcriptional deregulation of the autocrine-signaling loop IDO-AHR-IL6, which blocks kynurenine production and promotes T-cell proliferation. Additionally, 1 and 2 display low toxicity in mice and are stable in blood. To our knowledge, this construct is the first Pt drug candidate with immune checkpoint blockade properties.
Co-reporter:Kogularamanan Suntharalingam; Samuel G. Awuah; Peter M. Bruno; Timothy C. Johnstone; Fang Wang; Wei Lin; Yao-Rong Zheng; Julia E. Page; Michael T. Hemann
Journal of the American Chemical Society 2015 Volume 137(Issue 8) pp:2967-2974
Publication Date(Web):February 20, 2015
DOI:10.1021/ja511978y
Rhenium(V) oxo complexes of general formula [ReO(OMe)(N^N)Cl2], where N^N = 4,7-diphenyl-1,10-phenanthroline, 1, or 3,4,7,8-tetramethyl-1,10-phenanthroline, 2, effectively kill cancer cells by triggering necroptosis, a non-apoptotic form of cell death. Both complexes evoke necrosome (RIP1-RIP3)-dependent intracellular reactive oxygen species (ROS) production and propidium iodide uptake. The complexes also induce mitochondrial membrane potential depletion, a possible downstream effect of ROS production. Apparently, 1 and 2 are the first rhenium complexes to evoke cellular events consistent with programmed necrosis in cancer cells. Furthermore, 1 and 2 display low acute toxicity in C57BL/6 mice and reasonable stability in fresh human blood.
Co-reporter:Pablo Rivera-Fuentes, Alexandra T. Wrobel, Melissa L. Zastrow, Mustafa Khan, John Georgiou, Thomas T. Luyben, John C. Roder, Kenichi Okamoto and Stephen J. Lippard
Chemical Science 2015 vol. 6(Issue 3) pp:1944-1948
Publication Date(Web):23 Jan 2015
DOI:10.1039/C4SC03388D
Imaging mobile zinc in acidic environments remains challenging because most small-molecule optical probes display pH-dependent fluorescence. Here we report a reaction-based sensor that detects mobile zinc unambiguously at low pH. The sensor responds reversibly and with a large dynamic range to exogenously applied Zn2+ in lysosomes of HeLa cells, endogenous Zn2+ in insulin granules of MIN6 cells, and zinc-rich mossy fiber boutons in hippocampal tissue from mice. This long-wavelength probe is compatible with the green-fluorescent protein, enabling multicolor imaging, and facilitates visualization of mossy fiber boutons at depths of >100 μm, as demonstrated by studies in live tissue employing two-photon microscopy.
Co-reporter:Andrei Loas, Robert J. Radford, Alexandria Deliz Liang and Stephen J. Lippard
Chemical Science 2015 vol. 6(Issue 7) pp:4131-4140
Publication Date(Web):19 May 2015
DOI:10.1039/C5SC00880H
We describe a modular, synthetically facile solid-phase approach aimed at separating the fluorescent reporter and binding unit of small-molecule metal-based sensors. The first representatives contain a lysine backbone functionalized with a tetramethylrhodamine fluorophore, and they operate by modulating the oxidation state of a copper ion ligated to an [N4] (cyclam) or an [N2O] (quinoline-phenolate) moiety. We demonstrate the selectivity of their Cu(II) complexes for sensing nitroxyl (HNO) and thiols (RSH), respectively, and investigate the mechanism responsible for the observed reactivity in each case. The two lysine conjugates are cell permeable in the active, Cu(II)-bound forms and retain their analyte selectivity intracellularly, even in the presence of interfering species such as nitric oxide, nitrosothiols, and hydrogen sulfide. Moreover, we apply the new probes to discriminate between distinct levels of intracellular HNO and RSH generated upon stimulation of live HeLa cells with ascorbate and hydrogen sulfide, respectively. The successful implementation of the lysine-based sensors to gain insight into biosynthetic pathways validates the method as a versatile tool for producing libraries of analogues with minimal synthetic effort.
Co-reporter:Yao-Rong Zheng, Kogularamanan Suntharalingam, Timothy C. Johnstone and Stephen J. Lippard
Chemical Science 2015 vol. 6(Issue 2) pp:1189-1193
Publication Date(Web):24 Nov 2014
DOI:10.1039/C4SC01892C
This report presents a novel strategy that facilitates delivery of multiple, specific payloads of Pt(IV) prodrugs using a well-defined supramolecular system. This delivery system comprises a hexanuclear Pt(II) cage that can host four Pt(IV) prodrug guest molecules. Relying on host–guest interactions between adamantyl units tethered to the Pt(IV) molecules and the cage, four prodrugs could be encapsulated within one cage. This host–guest complex, exhibiting a diameter of about 3 nm, has been characterized by detailed NMR spectroscopic measurements. Owing to the high positive charge, this nanostructure exhibits high cellular uptake. Upon entering cells and reacting with biological reductants such as ascorbic acid, the host–guest complex releases cisplatin, which leads to cell cycle arrest and apoptosis. The fully assembled complex displays cytotoxicity comparable to that of cisplatin against a panel of human cancer cell lines, whereas the cage or the Pt(IV) guest alone exhibit lower cytotoxicity. These findings indicate the potential of utilising well-defined supramolecular constructs for the delivery of prodrug molecules.
Co-reporter:Daniel Y. Zhang, Maria Azrad, Wendy Demark-Wahnefried, Christopher J. Frederickson, Stephen J. Lippard, and Robert J. Radford
ACS Chemical Biology 2015 Volume 10(Issue 2) pp:385
Publication Date(Web):November 10, 2014
DOI:10.1021/cb500617c
Small-molecule fluorescent sensors are versatile agents for detecting mobile zinc in biology. Capitalizing on the abundance of validated mobile zinc probes, we devised a strategy for repurposing existing intensity-based sensors for quantitative applications. Using solid-phase peptide synthesis, we conjugated a zinc-sensitive Zinpyr-1 derivative and a zinc-insensitive 7-hydroxycoumarin derivative onto opposite ends of a rigid P9K peptide scaffold to create HcZ9, a ratiometric fluorescent probe for mobile zinc. A plate reader-based assay using HcZ9 was developed, the accuracy of which is comparable to that of atomic absorption spectroscopy. We investigated zinc accumulation in prostatic cells and zinc levels in human seminal fluid. When normal and tumorigenic cells are bathed in zinc-enriched media, cellular mobile zinc is buffered and changes slightly, but total zinc levels increase significantly. Quantification of mobile and total zinc levels in human seminal plasma revealed that the two are positively correlated with a Pearson’s coefficient of 0.73.
Co-reporter:Kathrin H. Hopmann; Jeanet Conradie; Espen Tangen; Zachary J. Tonzetich; Stephen J. Lippard;Abhik Ghosh
Inorganic Chemistry 2015 Volume 54(Issue 15) pp:7362-7367
Publication Date(Web):July 23, 2015
DOI:10.1021/acs.inorgchem.5b00901
A density functional theory (DFT) study of {CoNO}8 cobalt nitrosyl complexes containing the [n,n]tropocoronand ligand (TC-n,n) has revealed a sharp reduction of singlet–triplet gaps as the structures change from near-square-pyramidal (for n = 3) to trigonal-bipyramidal with an equatorial NO (for n = 5, 6). An experimental reinvestigation of [Co(TC-3,3)(NO)] has confirmed that it is not paramagnetic, as originally reported, but diamagnetic, like all other {CoNO}8 complexes. Furthermore, DFT calculations indicate a substantial singlet–triplet gap of about half an eV or higher for this complex. At the other end of the series, low-energy, thermally accessible triplet states are predicted for [Co(TC-6,6)(NO)]. Enhanced triplet-state reactivity may well provide a partial explanation for the failure to isolate this compound as a stable species.
Co-reporter:Julia Kozhukh, Mikael A. Minier, and Stephen J. Lippard
Inorganic Chemistry 2015 Volume 54(Issue 2) pp:418-424
Publication Date(Web):December 22, 2014
DOI:10.1021/ic5009279
The preparation and characterization of two mononuclear cobalt(III) tropocoronand complexes, [Co(TC-5,5)](BF4) and [Co(TC-6,6)](BPh4), are reported. The cobalt(III) centers exist in rare pseudotetrahedral conformations, with twist angles of 65° and 74° for the [Co(TC-5,5]+ and [Co(TC-6,6)]+ species, respectively. Structural and electrochemical characteristics are compared with those of newly synthesized [Ga(TC-5,5)](GaCl4) and [Ga(TC-6,6)](GaCl4) analogues. The spin state of the pseudotetrahedral [Co(TC-6,6)](BPh4) compound was determined to be S = 2, a change in spin state from the value of S = 1 that occurs in the square-planar and distorted square-planar complexes, [Co(TC-3,3)](X) (X = BPh4, BAr′4) and [Co(TC-4,4)](BPh4), respectively.
Co-reporter:Timothy C. Johnstone, Sarah M. Alexander, Justin J. Wilson and Stephen J. Lippard
Dalton Transactions 2015 vol. 44(Issue 1) pp:119-129
Publication Date(Web):20 Oct 2014
DOI:10.1039/C4DT02627F
A series of Pt(IV) prodrugs has been obtained by oxidative halogenation of either cisplatin or carboplatin. Iodobenzene dichloride is a general reagent that cleanly provides prodrugs bearing axial chlorides without the need to prepare intervening Pt(IV) intermediates or handle chlorine gas. Elemental bromine and iodine afford Pt(IV) compounds as well, although in the case of the iodine-mediated oxidation of carboplatin, an amido-bridged Pt(IV) side product also formed. A detailed analysis of the changes in spectroscopic and structural parameters induced by varying the axial halide is presented. A number of recurring motifs are observed in the solid state structures of these compounds.
Co-reporter:Mikael A. Minier and Stephen J. Lippard
Dalton Transactions 2015 vol. 44(Issue 41) pp:18111-18121
Publication Date(Web):29 Sep 2015
DOI:10.1039/C5DT02138C
A series of asymmetrically carboxylate-bridged diiron(II) complexes featuring fluorine atoms as NMR spectroscopic probes, [Fe2(PIM)(Ar4F-PhCO2)2] (10), [Fe2(F2PIM)(ArTolCO2)2] (11), and [Fe2(F2PIM)(Ar4F-PhCO2)2] (12), were prepared and characterized by X-ray crystallography, Mössbauer spectroscopy, and VT 19F NMR spectroscopy. These complexes are part of a rare family of syn N-donor diiron(II) compounds, [Fe2(X2PIM)(RCO2)2], that are structurally very similar to the active site of the hydroxylase enzyme component of reduced methane monooxygenase (MMOHred). Solution characterization of these complexes demonstrates that they undergo intramolecular carboxylate rearrangements, or carboxylate shifts, a dynamic feature relevant to the reactivity of the diiron centers in bacterial multicomponent monooxygenases.
Co-reporter:Timothy C. Johnstone, Stephen J. Lippard
Inorganica Chimica Acta 2015 Volume 424() pp:254-259
Publication Date(Web):1 January 2015
DOI:10.1016/j.ica.2014.08.047
•A new synthetic approach to preparing and using iodo-bridged platinum(II) dimers.•New method is orders of magnitude faster and avoids common side products.•Interpretation of the dimer cleavage mechanism that reconciles previous debate.•Spectroscopic and crystallographic characterization of [Pt(NH2C6H11)I(μ-I)]2.Mixed amine/ammine motifs are important features in newer generation platinum anticancer agents, including the Pt(IV) prodrug satraplatin. One synthetic route that can be used to access platinum molecules with such structures exploits the trans effect during NH3-mediated cleavage of iodo-bridged platinum(II) dimers of the form [Pt(Am)I(μ-I)]2, where Am is an amine. A clear picture of the nature of these dimers that is consistent with the reactivity they exhibit has remained elusive. Moreover, technical aspects of this chemistry have impeded its more widespread use. We present here an improved strategy that permits isolation and use of [Pt(Am)I(μ-I)]2, where Am is cyclohexylamine, within minutes as opposed to weeks, as previously reported. A detailed spectroscopic, crystallographic, and chromatographic investigation of this intermediate in the synthesis of satraplatin is also presented with a discussion of the ability of both cis and trans isomers of the dimer to produce exclusively cis-[Pt(NH2C6H11)(NH3)I2] upon treatment with NH3.The chemistry of the iodo-bridged intermediate in the synthesis of satraplatin has been investigated.
Co-reporter:Charles T. Anderson;Robert J. Radford;Melissa L. Zastrow;Thanos Tzounopoulos;Daniel Y. Zhang;Ulf-Peter Apfel
PNAS 2015 Volume 112 (Issue 20 ) pp:E2705-E2714
Publication Date(Web):2015-05-19
DOI:10.1073/pnas.1503348112
Many excitatory synapses contain high levels of mobile zinc within glutamatergic vesicles. Although synaptic zinc and glutamate
are coreleased, it is controversial whether zinc diffuses away from the release site or whether it remains bound to presynaptic
membranes or proteins after its release. To study zinc transmission and quantify zinc levels, we required a high-affinity
rapid zinc chelator as well as an extracellular ratiometric fluorescent zinc sensor. We demonstrate that tricine, considered
a preferred chelator for studying the role of synaptic zinc, is unable to efficiently prevent zinc from binding low-nanomolar
zinc-binding sites, such as the high-affinity zinc-binding site found in NMDA receptors (NMDARs). Here, we used ZX1, which
has a 1 nM zinc dissociation constant and second-order rate constant for binding zinc that is 200-fold higher than those for
tricine and CaEDTA. We find that synaptic zinc is phasically released during action potentials. In response to short trains
of presynaptic stimulation, synaptic zinc diffuses beyond the synaptic cleft where it inhibits extrasynaptic NMDARs. During
higher rates of presynaptic stimulation, released glutamate activates additional extrasynaptic NMDARs that are not reached
by synaptically released zinc, but which are inhibited by ambient, tonic levels of nonsynaptic zinc. By performing a ratiometric
evaluation of extracellular zinc levels in the dorsal cochlear nucleus, we determined the tonic zinc levels to be low nanomolar.
These results demonstrate a physiological role for endogenous synaptic as well as tonic zinc in inhibiting extrasynaptic NMDARs
and thereby fine tuning neuronal excitability and signaling.
Co-reporter:Justin J. Wilson and Stephen J. Lippard
Chemical Reviews 2014 Volume 114(Issue 8) pp:4470
Publication Date(Web):November 27, 2013
DOI:10.1021/cr4004314
Co-reporter:Weixue Wang
Journal of the American Chemical Society 2014 Volume 136(Issue 6) pp:2244-2247
Publication Date(Web):January 29, 2014
DOI:10.1021/ja412351b
The regulatory component (MMOB) of soluble methane monooxygenase (sMMO) has a unique N-terminal tail not found in regulatory proteins of other bacterial multicomponent monooxygenases. This N-terminal tail is indispensable for proper function, yet its solution structure and role in catalysis remain elusive. Here, by using double electron–electron resonance (DEER) spectroscopy, we show that the oxidation state of the hydroxylase component, MMOH, modulates the conformation of the N-terminal tail in the MMOH–2MMOB complex, which in turn facilitates catalysis. The results reveal that the N-terminal tail switches from a relaxed, flexible conformational state to an ordered state upon MMOH reduction from the diiron(III) to the diiron(II) state. This observation suggests that some of the crystallographically observed allosteric effects that result in the connection of substrate ingress cavities in the MMOH–2MMOB complex may not occur in solution in the diiron(III) state. Thus, O2 may not have easy access to the active site until after reduction of the diiron center. The observed conformational change is also consistent with a higher binding affinity of MMOB to MMOH in the diiron(II) state, which may allow MMOB to displace more readily the reductase component (MMOR) from MMOH following reduction.
Co-reporter:Yunbo Jiang ; Takahiro Hayashi ; Hirotoshi Matsumura ; Loi H. Do ; Amit Majumdar ; Stephen J. Lippard ;Pierre Moënne-Loccoz
Journal of the American Chemical Society 2014 Volume 136(Issue 36) pp:12524-12527
Publication Date(Web):August 12, 2014
DOI:10.1021/ja504343t
Two non-heme iron–nitrosyl species, [Fe2(N-Et-HPTB)(O2CPh)(NO)2](BF4)2 (1a) and [Fe2(N-Et-HPTB)(DMF)2(NO)(OH)](BF4)3 (2a), are characterized by FTIR and resonance Raman spectroscopy. Binding of NO is reversible in both complexes, which are prone to NO photolysis under visible light illumination. Photoproduction of N2O occurs in high yield for 1a but not 2a. Low-temperature FTIR photolysis experiments with 1a in acetonitrile do not reveal any intermediate species, but in THF at room temperature, a new {FeNO}7 species quickly forms under illumination and exhibits a ν(NO) vibration indicative of nitroxyl-like character. This metastable species reacts further under illumination to produce N2O. A reaction mechanism is proposed, and implications for NO reduction in flavodiiron proteins are discussed.
Co-reporter:Yao-Rong Zheng, Kogularamanan Suntharalingam, Timothy C. Johnstone, Hyunsuk Yoo, Wei Lin, Jamar G. Brooks, and Stephen J. Lippard
Journal of the American Chemical Society 2014 Volume 136(Issue 24) pp:8790-8798
Publication Date(Web):June 6, 2014
DOI:10.1021/ja5038269
Albumin is the most abundant protein in human serum and drugs that are administered intravenously inevitably interact with it. We present here a series of platinum(IV) prodrugs designed specifically to enhance interaction with human serum albumin (HSA) for drug delivery. This goal is achieved by asymmetrically functionalizing the axial ligands of the prodrug so as to mimic the overall features of a fatty acid. Systematic variation of the length of the aliphatic tail tunes the cellular uptake and, consequently, the cytotoxicity of cis,cis,trans-[Pt(NH3)2Cl2(O2CCH2CH2COOH)(OCONHR)], 4, where R is a linear alkyl group. Investigation of an analogue bearing a fluorophore conjugated to the succinate ligand confirmed that these compounds are reduced by biological reductants with loss of the axial ligands. Intracellular release of cisplatin from 4 was further confirmed by observing the characteristic effects of cisplatin on the cell cycle and morphology following treatment with the prodrug. The most potent member of series 4, for which R is a hexadecyl chain, interacts with HSA in a 1:1 stoichiometry to form the platinum-protein complex 7. The interaction is non-covalent and extraction with octanol completely removes the prodrug from an aqueous solution of HSA. Construct 7 is robust and can be isolated following fast protein liquid chromatography. The nature of the tight interaction was investigated computationally, and these studies suggest that the prodrug is buried below the surface of the protein. Consequently, complexation to HSA is able to reduce the rate of reduction of the prodrug by ascorbate. The lead compound from series 4 also exhibited significant stability in whole human blood, attributed to its interaction with HSA. This favorable redox profile, in conjunction with the established nonimmunogenicity, biocompatibility, and enhanced tumor accumulation of HSA, produces a system that holds significant therapeutic potential.
Co-reporter:Weixue Wang ; Roxana E. Iacob ; Rebecca P. Luoh ; John R. Engen
Journal of the American Chemical Society 2014 Volume 136(Issue 27) pp:9754-9762
Publication Date(Web):June 17, 2014
DOI:10.1021/ja504688z
The hydroxylation or epoxidation of hydrocarbons by bacterial multicomponent monooxygenases (BMMs) requires the interplay of three or four protein components. How component protein interactions control catalysis, however, is not well understood. In particular, the binding sites of the reductase components on the surface of their cognate hydroxylases and the role(s) that the regulatory proteins play during intermolecular electron transfer leading to the hydroxylase reduction have been enigmatic. Here we determine the reductase binding site on the hydroxylase of a BMM enzyme, soluble methane monooxygenase (sMMO) from Methylococcus capsulatus (Bath). We present evidence that the ferredoxin domain of the reductase binds to the canyon region of the hydroxylase, previously determined to be the regulatory protein binding site as well. The latter thus inhibits reductase binding to the hydroxylase and, consequently, intermolecular electron transfer from the reductase to the hydroxylase diiron active site. The binding competition between the regulatory protein and the reductase may serve as a control mechanism for regulating electron transfer, and other BMM enzymes are likely to adopt the same mechanism.
Co-reporter:Timothy C. Johnstone
Journal of the American Chemical Society 2014 Volume 136(Issue 5) pp:2126-2134
Publication Date(Web):January 13, 2014
DOI:10.1021/ja4125115
The monofunctional platinum complex cis-[Pt(NH3)2Cl(Am)]+, also known as phenanthriplatin, where Am is the N-heterocyclic base phenanthridine, has promising anticancer activity. Unlike bifunctional compounds such as cisplatin, phenanthriplatin can form only one covalent bond to DNA. Another distinguishing feature is that phenanthriplatin is chiral. Rotation about the Pt–N bond of the phenanthridine ligand racemizes the complex, and the question arises as to whether this process is sufficiently slow under physiological conditions to impact its DNA-binding properties. Here we present the results of NMR spectroscopic, X-ray crystallographic, molecular dynamics, and density functional theoretical investigations of diastereomeric phenanthriplatin analogs in order to probe the internal dynamics of phenanthriplatin. These results reveal that phenanthriplatin rapidly racemizes under physiological conditions. The information also facilitated the interpretation of the NMR spectra of small molecule models of phenanthriplatin-platinated DNA. These studies indicate, inter alia, that one diastereomeric form of the complexes cis-[Pt(NH3)2(Am)(R-Gua)]2+, where R-Gua is 9-methyl- or 9-ethylguanine, is preferred over the other, the origin of which stems from an intramolecular interaction between the carbonyl oxygen of the platinated guanine base and a cis-coordinated ammine. The relevance of this finding to the DNA-damaging properties of phenanthriplatin and its biological activity is discussed.
Co-reporter:Alexandra T. Wrobel ; Timothy C. Johnstone ; Alexandria Deliz Liang ; Stephen J. Lippard ;Pablo Rivera-Fuentes
Journal of the American Chemical Society 2014 Volume 136(Issue 12) pp:4697-4705
Publication Date(Web):February 24, 2014
DOI:10.1021/ja500315x
The first near-infrared fluorescent turn-on sensor for the detection of nitroxyl (HNO), the one-electron reduced form of nitric oxide (NO), is reported. The new copper-based probe, CuDHX1, contains a dihydroxanthene (DHX) fluorophore and a cyclam derivative as a Cu(II) binding site. Upon reaction with HNO, CuDHX1 displays a five-fold fluorescence turn-on in cuvettes and is selective for HNO over thiols and reactive nitrogen and oxygen species. CuDHX1 can detect exogenously applied HNO in live mammalian cells and in conjunction with the zinc-specific, green-fluorescent sensor ZP1 can perform multicolor/multianalyte microscopic imaging. These studies reveal that HNO treatment elicits an increase in the concentration of intracellular mobile zinc.
Co-reporter:Kogularamanan Suntharalingam, Wei Lin, Timothy C. Johnstone, Peter M. Bruno, Yao-Rong Zheng, Michael T. Hemann, and Stephen J. Lippard
Journal of the American Chemical Society 2014 Volume 136(Issue 41) pp:14413-14416
Publication Date(Web):September 23, 2014
DOI:10.1021/ja508808v
The effect of a newly developed osmium(VI) nitrido complex, 1, on breast cancer stem cells (CSCs) is reported. The complex displays selective toxicity for HMLER breast cancer cells enriched with CD44-positive, CSC-like cells over the same cells having reduced CSC character. Remarkably, 1 also reduces the proportion of CSCs within a heterogeneous breast cancer cell population and irreversibly inhibits the formation of free-floating mammospheres to an extent similar to that of salinomycin, a natural product that targets CSCs. Detailed mechanistic studies reveal that in breast cancer cells 1 induces DNA damage and endoplasmic reticulum stress, the latter being responsible for the CSC selectivity. The anti-CSC properties of 1 provide a strong impetus for the development of new metal-based compounds to target CSCs and to treat chemotherapy-resistant and relapsed tumors.
Co-reporter:Robert J. Radford, Wen Chyan and Stephen J. Lippard
Chemical Science 2014 vol. 5(Issue 11) pp:4512-4516
Publication Date(Web):2014/08/12
DOI:10.1039/C4SC01280A
Fluorescein-based sensors are the most widely applied class of zinc probes but display adventitious localization in live cells. We present here a peptide-based localization strategy that affords precision in targeting of fluorescein-based zinc sensors. By appending the zinc-selective, reaction-based probe Zinpyr-1 diacetate (DA-ZP1) to the N-terminus of two different targeting peptides we achieve programmable localization and avoid unwanted sequestration within acidic vesicles. Furthermore, this approach can be generalized to other fluorescein-based sensors. When appended to a mitochondrial targeting peptide, the esterase-activated profluorophore 2′,7′-dichlorofluorescein diacetate can be used effectively at concentrations four-times lower than previously reported for analogous, non-acetylated derivatives. These results demonstrate on-resin or in-solution esterification of fluorescein to be an effective strategy to facilitate peptide-based targeting in live cells.
Co-reporter:Kogularamanan Suntharalingam, Ying Song and Stephen J. Lippard
Chemical Communications 2014 vol. 50(Issue 19) pp:2465-2468
Publication Date(Web):11 Dec 2013
DOI:10.1039/C3CC48740G
We report two platinum(IV) complexes conjugated with a vitamin E analog, α-tocopherol succinate (α-TOS). One of the conjugates displays the activity of both cisplatin and α-TOS in cancer cells, causing damage to DNA and mitochondria simultaneously. Accordingly, it serves as a promising dual-targeting anticancer agent.
Co-reporter:Amit Majumdar ; Ulf-Peter Apfel ; Yunbo Jiang ; Pierre Moënne-Loccoz
Inorganic Chemistry 2014 Volume 53(Issue 1) pp:167-181
Publication Date(Web):December 20, 2013
DOI:10.1021/ic4019585
A new, DMF-coordinated, preorganized diiron compound [Fe2(N-Et-HPTB)(DMF)4](BF4)3 (1) was synthesized, avoiding the formation of [Fe(N-Et-HPTB)](BF4)2 (10) and [Fe2(N-Et-HPTB)(μ-MeCONH)](BF4)2 (11), where N-Et-HPTB is the anion of N,N,N′,N′-tetrakis[2-(1-ethylbenzimidazolyl)]-2-hydroxy-1,3-diaminopropane. Compound 1 is a versatile reactant from which nine new compounds have been generated. Transformations include solvent exchange to yield [Fe2(N-Et-HPTB)(MeCN)4](BF4)3 (2), substitution to afford [Fe2(N-Et-HPTB)(μ-RCOO)](BF4)2 (3, R = Ph; 4, RCOO = 4-methyl-2,6-diphenyl benzoate]), one-electron oxidation by (Cp2Fe)(BF4) to yield a Robin–Day class II mixed-valent diiron(II,III) compound, [Fe2(N-Et-HPTB)(μ-PhCOO)(DMF)2](BF4)3 (5), two-electron oxidation with tris(4-bromophenyl)aminium hexachloroantimonate to generate [Fe2(N-Et-HPTB)Cl3(DMF)](BF4)2 (6), reaction with (2,2,6,6-tetramethylpiperidin-1-yl)oxyl to form [Fe5(N-Et-HPTB)2(μ-OH)4(μ-O)(DMF)2](BF4)4 (7), and reaction with dioxygen to yield an unstable peroxo compound that decomposes at room temperature to generate [Fe4(N-Et-HPTB)2(μ-O)3(H2O)2](BF4)·8DMF (8) and [Fe4(N-Et-HPTB)2(μ-O)4](BF4)2 (9). Compound 5 loses its bridging benzoate ligand upon further oxidation to form [Fe2(N-Et-HPTB)(OH)2(DMF)2](BF4)3 (12). Reaction of the diiron(II,III) compound 5 with dioxygen was studied in detail by spectroscopic methods. All compounds (1–12) were characterized by single-crystal X-ray structure determinations. Selected compounds and reaction intermediates were further examined by a combination of elemental analysis, electronic absorption spectroscopy, Mössbauer spectroscopy, EPR spectroscopy, resonance Raman spectroscopy, and cyclic voltammetry.
Co-reporter:Andrei Loas, Robert J. Radford, and Stephen J. Lippard
Inorganic Chemistry 2014 Volume 53(Issue 13) pp:6491-6493
Publication Date(Web):June 10, 2014
DOI:10.1021/ic500732z
We report the synthesis and photophysical properties of ZBR4 and ZR1, two resorufin-based ditopic probes for mobile zinc. Upon binding Zn2+, the sensors display 14- and 41-fold enhancements of their red fluorescence emission, respectively. In contrast to ZR1 and other members of the ZBR family, which accumulate in the endoplasmic reticulum, ZBR4 spontaneously localizes to the mitochondria of HeLa cells. The modular approach in designing the constructs facilitates a homologation strategy aimed at tuning the zinc-binding and intracellular targeting properties of future probes.
Co-reporter:Eric Victor and Stephen J. Lippard
Inorganic Chemistry 2014 Volume 53(Issue 10) pp:5311-5320
Publication Date(Web):April 28, 2014
DOI:10.1021/ic500586g
Previous studies provide evidence that [4Fe–4S] clusters serve as targets of reactive nitrogen oxide species in biology. The products of this reaction range from dinitrosyliron complexes, [Fe(NO)2L2]−, to Roussin’s black anion, [Fe4S3(NO)7]−. To date, the pathways by which these reactions occur have not been fully elucidated. In this study, we prepared the site-differentiated complexes [Fe4S4(LS3)L′]2– (LS3 = 1,3,5-tris(4,6-dimethyl-3-mercaptophenylthio)-2,4,6-tris(p-tolylthio)benzene; L′ = Cl, SEt, SPh, N3, 2-SPyr, Tp, S2CNEt2) to serve as synthetic models for biological [4Fe–4S] clusters and studied their reactivity toward NO(g) and Ph3CSNO. The products were characterized by X-ray crystallography, mass spectrometry, and IR, electron paramagnetic resonance (EPR), and 1H NMR spectroscopy. In all cases reported here, the reactions proceed via formation of the S = 1/2 species [Fe4S4(NO)4]−, which ultimately converts to EPR-silent [Fe4S3(NO)7]−.
Co-reporter:Eric Victor, Sunghee Kim, and Stephen J. Lippard
Inorganic Chemistry 2014 Volume 53(Issue 24) pp:12809-12821
Publication Date(Web):November 24, 2014
DOI:10.1021/ic501765g
To explore the release of nitrogen oxide gases from reaction solutions, we developed a series of colorimetric sensors based on the cis-nitrogen-donating ligand bis(1-methylimidazol-2-yl)phenylmethoxymethane (BIPhMe). The complexes M(BIPhMe)X2, where M is Fe2+ or Co2+ and X is Cl–, Br–, or I–, were prepared and structurally and spectroscopically characterized. The reactivity of these complexes toward NO(g) and NO2(g) in solution was explored. These complexes were then incorporated into test strips and syringes to provide devices that can qualitatively, and in the case of the syringes quantitatively, detect NO(g) and NO2(g) in a reaction headspace without additional equipment.
Co-reporter:Kogularamanan Suntharalingam, Justin J. Wilson, Wei Lin and Stephen J. Lippard
Metallomics 2014 vol. 6(Issue 3) pp:437-443
Publication Date(Web):22 Jan 2014
DOI:10.1039/C3MT00364G
The therapeutic index and cellular mechanism of action of [Pt(BDIQQ)]Cl, a monocationic, square-planar platinum(II) complex, are reported. [Pt(BDIQQ)]Cl was used to treat several cell lines, including wild type and cisplatin-resistant ovarian carcinoma cells (A2780 and A2780CP70) and non-proliferating lung carcinoma cells (A549). [Pt(BDIQQ)]Cl selectively kills cancer cells over healthy cells and exhibits no cross-resistance with cisplatin. The mechanism of cell killing was established through detailed cell-based assays. [Pt(BDIQQ)]Cl exhibits dual-threat capabilities, targeting nuclear DNA and mitochondria simultaneously. [Pt(BDIQQ)]Cl induces DNA damage, leading to p53 enrichment, mitochondrial membrane potential depolarisation, and caspase-mediated apoptosis. [Pt(BDIQQ)]Cl also accumulates in the mitochondria, resulting in direct mitochondrial damage. Flow cytometric studies demonstrated that [Pt(BDIQQ)]Cl has no significant effect on cell cycle progression. Remarkably, p53-status is a not a determinant of [Pt(BDIQQ)]Cl activity. In p53-null cells, [Pt(BDIQQ)]Cl induces cell death through mitochondrial dysfunction. Cancers with p53-null status could therefore be targeted using [Pt(BDIQQ)]Cl.
Co-reporter:Alexandria Deliz Liang, Alexandra T. Wrobel, and Stephen J. Lippard
Biochemistry 2014 Volume 53(Issue 22) pp:
Publication Date(Web):May 29, 2014
DOI:10.1021/bi500387y
Toluene/o-xylene monooxygenase (ToMO) is a bacterial multicomponent monooxygenase capable of oxidizing aromatic substrates. The carboxylate-rich diiron active site is located in the hydroxylase component of ToMO (ToMOH), buried 12 Å from the surface of the protein. A small, hydrophilic pore is the shortest pathway between the diiron active site and the protein exterior. In this study of ToMOH from Pseudomonas sp. OX1, the functions of two residues lining this pore, N202 and Q228, were investigated using site-directed mutagenesis. Steady-state characterization of WT and the three mutant enzymes demonstrates that residues N202 and Q228 are critical for turnover. Kinetic isotope effects and pH profiles reveal that these residues govern the kinetics of water egress and prevent quenching of activated oxygen intermediates formed at the diiron active site. We propose that this activity arises from movement of these residues, opening and closing the pore during catalysis, as seen in previous X-ray crystallographic studies. In addition, N202 and Q228 are important for the interactions of the reductase and regulatory components to ToMOH, suggesting that they bind competitively to the hydroxylase. The role of the pore in the hydroxylase components of other bacterial multicomponent monooxygenases within the superfamily is discussed in light of these conclusions.
Co-reporter:Alexandria Deliz Liang and Stephen J. Lippard
Biochemistry 2014 Volume 53(Issue 47) pp:
Publication Date(Web):November 17, 2014
DOI:10.1021/bi500892n
The multicomponent protein toluene/o-xylene monooxygenase (ToMO) activates molecular oxygen to oxidize aromatic hydrocarbons. Prior to dioxygen activation, two electrons are injected into each of two diiron(III) units of the hydroxylase, a process that involves three redox active proteins: the ToMO hydroxylase (ToMOH), Rieske protein (ToMOC), and an NADH oxidoreductase (ToMOF). In addition to these three proteins, a small regulatory protein is essential for catalysis (ToMOD). Through steady state and pre-steady state kinetics studies, we show that ToMOD attenuates electron transfer from ToMOC to ToMOH in a concentration-dependent manner. At substoichiometric concentrations, ToMOD increases the rate of turnover, which we interpret to be a consequence of opening a pathway for oxygen transport to the catalytic diiron center in ToMOH. Excess ToMOD inhibits steady state catalysis in a manner that depends on ToMOC concentration. Through rapid kinetic assays, we demonstrate that ToMOD attenuates formation of the ToMOC–ToMOH complex. These data, coupled with protein docking studies, support a competitive model in which ToMOD and ToMOC compete for the same binding site on the hydroxylase. These results are discussed in the context of other studies of additional proteins in the superfamily of bacterial multicomponent monooxygenases.
Co-reporter:Eric Victor;Mikael A. Minier
European Journal of Inorganic Chemistry 2014 Volume 2014( Issue 33) pp:5640-5645
Publication Date(Web):
DOI:10.1002/ejic.201402543
Abstract
Nitric oxide is released during the immune response by the host during bacterial infection. To counteract this response, bacteria have evolved nitric oxide reductases to convert NO to N2O. Some of these nitric oxide reductases contain a flavodiiron active site that has bridging carboxylates and hydroxides. Only a handful of synthetic complexes currently exist as models for the protein reactivity. Here, we report the reaction of [Fe2(μ-OH)(μ-Ph4DBA)(TMEDA)2(OTf)] (4) with NO(g) and Ph3CSNO to prepare the dinitrosyl-triiron complex [Fe3(μ-OH)2(μ-Ph4DBA)2(TMEDA)2(NO)2](OTf) (5). The reaction was monitored by UV/Vis and ReactIR spectroscopy, and compound 5 was characterized by X-ray crystallography, 57Fe Mössbauer spectroscopy, Evan's method, and FTIR spectroscopy. The IR spectrum of compound 5 compares favorably to experimental spectroscopic data obtained for the proposed mononitrosylated intermediate of the protein.
Co-reporter:Dr. Pablo Rivera-Fuentes ; Stephen J. Lippard
ChemMedChem 2014 Volume 9( Issue 6) pp:1238-1243
Publication Date(Web):
DOI:10.1002/cmdc.201400014
Abstract
A reversible, reaction-based sensor for biological mobile zinc was designed, prepared, and characterized. The sensing mechanism of this probe is based on the zinc-induced, ring-opening reaction of spirobenzopyran to give a cyanine fluorophore that emits in the deep-red region of the electromagnetic spectrum. This probe is not activated by protons and operates efficiently in aqueous solution at pH 7 and high ionic strength. The mechanism of this reaction was studied by using a combination of kinetics experiments and DFT calculations. The biocompatibility of the probe was demonstrated in live HeLa cells.
Co-reporter:Mikael A. Minier and Stephen J. Lippard
Organometallics 2014 Volume 33(Issue 6) pp:1462-1466
Publication Date(Web):March 5, 2014
DOI:10.1021/om5000503
Ethylzinc 2,6-bis(p-tolyl)benzoate converts between two forms in solution. Through NMR spectroscopic techniques and X-ray crystallography, the species in equilibrium were identified as [Zn2(ArTolCO2)2(Et)2(THF)2] (1), [Zn2(ArTolCO2)3(Et)(THF)] (2), and diethylzinc (ArTol = 2,6-bis(p-tolyl)phenyl). The equilibrium provides a model for understanding the speciation between doubly and triply m-terphenylcarboxylate bridged diiron(II) and mononuclear iron(II) complexes. Evidence is presented for the occurrence of coordinatively unsaturated trigonal zinc species in solution. Both 1 and 2 decompose in air to form the T-symmetric oxozinc carboxylate [Zn4O(ArTolCO2)6] (3).
Co-reporter:Ga Young Park;Young-Sam Lee;Timothy C. Johnstone;Mark T. Gregory;Wei Yang
PNAS 2014 Volume 111 (Issue 25 ) pp:9133-9138
Publication Date(Web):2014-06-24
DOI:10.1073/pnas.1405739111
Platinum drugs are a mainstay of anticancer chemotherapy. Nevertheless, tumors often display inherent or acquired resistance
to platinum-based treatments, prompting the search for new compounds that do not exhibit cross-resistance with current therapies.
Phenanthriplatin, cis-diamminephenanthridinechloroplatinum(II), is a potent monofunctional platinum complex that displays a spectrum of activity
distinct from those of the clinically approved platinum drugs. Inhibition of RNA polymerases by phenanthriplatin lesions has
been implicated in its mechanism of action. The present study evaluates the ability of phenanthriplatin lesions to inhibit
DNA replication, a function disrupted by traditional platinum drugs. Phenanthriplatin lesions effectively inhibit DNA polymerases
ν, ζ, and κ and the Klenow fragment. In contrast to results obtained with DNA damaged by cisplatin, all of these polymerases
were capable of inserting a base opposite a phenanthriplatin lesion, but only Pol η, an enzyme efficient in translesion synthesis,
was able to fully bypass the adduct, albeit with low efficiency. X-ray structural characterization of Pol η complexed with
site-specifically platinated DNA at both the insertion and +1 extension steps reveals that phenanthriplatin on DNA interacts
with and inhibits Pol η in a manner distinct from that of cisplatin-DNA adducts. Unlike cisplatin and oxaliplatin, the efficacies
of which are influenced by Pol η expression, phenanthriplatin is highly toxic to both Pol η+ and Pol η− cells. Given that
increased expression of Pol η is a known mechanism by which cells resist cisplatin treatment, phenanthriplatin may be valuable
in the treatment of cancers that are, or can easily become, resistant to cisplatin.
Co-reporter:Kenichi Okamoto;Zhen Huang;Christian R. Goldsmith;John Georgiou;Mustafa Khan;Thomas T. Luyben;John C. Roder
PNAS 2014 Volume 111 (Issue 18 ) pp:6786-6791
Publication Date(Web):2014-05-06
DOI:10.1073/pnas.1405154111
Mossy fiber termini in the hippocampus accumulate Zn2+, which is released with glutamate from synaptic vesicles upon neural excitation. Understanding the spatiotemporal regulation
of mobile Zn2+ at the synaptic level is challenging owing to the difficulty of visualizing Zn2+ at individual synapses. Here we describe the use of zinc-responsive fluorescent probes together with two-photon microscopy
to image Zn2+ dynamics mediated by NMDA receptor-dependent long-term potentiation induction at single mossy fiber termini of dentate gyrus
neurons in adult mouse hippocampal slices. The membrane-impermeant fluorescent Zn2+ probe, 6-CO2H-ZAP4, was loaded into presynaptic vesicles in hippocampal mossy fiber termini upon KCl-induced depolarization, which triggers
subsequent endocytosis and vesicular restoration. Local tetanic stimulation decreased the Zn2+ signal observed at individual presynaptic sites, indicating release of the Zn2+ from vesicles in synaptic potentiation. This synapse-level two-photon Zn2+ imaging method enables monitoring of presynaptic Zn2+ dynamics for improving the understanding of physiological roles of mobile Zn2+ in regular and aberrant neurologic function.
Co-reporter:Timothy C. Johnstone ; Sarah M. Alexander ; Wei Lin
Journal of the American Chemical Society 2013 Volume 136(Issue 1) pp:116-118
Publication Date(Web):December 23, 2013
DOI:10.1021/ja411742c
The effect of the novel and potent monofunctional platinum(II) agent phenanthriplatin on Escherichia coli and bacteriophage λ lysogens is reported. E. coli filamentation was observed by light microscopy when cells were grown in the presence of phenanthriplatin, cis-[Pt(NH3)2(Am)Cl]+ where Am is phenanthridine. Treatment of lysogenic bacteria with this compound resulted in lysis and the production of viral particles, as indicated by plaque formation in a bacterial lawn. The results obtained with phenanthriplatin are contextualized by comparison with those obtained using cisplatin as well as other, less active, monofunctional compounds such as [Pt(NH3)3Cl]+ and cis-[Pt(NH3)2(py)Cl]+, where py is pyridine. The ability of phenanthriplatin to induce bacterial filamentation and initiate lysis in lysogenic bacteria corroborates the hypothesis that the biological activity of this complex is mediated by its interaction with DNA.
Co-reporter:Matthew W. Kellinger ; Ga Young Park ; Jenny Chong ; Stephen J. Lippard ;Dong Wang
Journal of the American Chemical Society 2013 Volume 135(Issue 35) pp:13054-13061
Publication Date(Web):August 8, 2013
DOI:10.1021/ja405475y
Transcription inhibition by platinum anticancer drugs is an important component of their mechanism of action. Phenanthriplatin, a cisplatin derivative containing phenanthridine in place of one of the chloride ligands, forms highly potent monofunctional adducts on DNA having a structure and spectrum of anticancer activity distinct from those of the parent drug. Understanding the functional consequences of DNA damage by phenanthriplatin for the normal functions of RNA polymerase II (Pol II), the major cellular transcription machinery component, is an important step toward elucidating its mechanism of action. In this study, we present the first systematic mechanistic investigation that addresses how a site-specific phenanthriplatin-DNA d(G) monofunctional adduct affects the Pol II elongation and transcriptional fidelity checkpoint steps. Pol II processing of the phenanthriplatin lesion differs significantly from that of the canonical cisplatin-DNA 1,2-d(GpG) intrastrand cross-link. A majority of Pol II elongation complexes stall after successful addition of CTP opposite the phenanthriplatin-dG adduct in an error-free manner, with specificity for CTP incorporation being essentially the same as for undamaged dG on the template. A small portion of Pol II undergoes slow, error-prone bypass of the phenanthriplatin-dG lesion, which resembles DNA polymerases that similarly switch from high-fidelity replicative DNA processing (error-free) to low-fidelity translesion DNA synthesis (error-prone) at DNA damage sites. These results provide the first insights into how the Pol II transcription machinery processes the most abundant DNA lesion of the monofunctional phenanthriplatin anticancer drug candidate and enrich our general understanding of Pol II transcription fidelity maintenance, lesion bypass, and transcription-derived mutagenesis. Because of the current interest in monofunctional, DNA-damaging metallodrugs, these results are of likely relevance to a broad spectrum of next-generation anticancer agents being developed by the medicinal inorganic chemistry community.
Co-reporter:Wei Lin ; Daniela Buccella
Journal of the American Chemical Society 2013 Volume 135(Issue 36) pp:13512-13520
Publication Date(Web):July 31, 2013
DOI:10.1021/ja4059487
Zn2+ plays essential roles in biology, and the homeostasis of Zn2+ is tightly regulated in all cells. Subcellular distribution and trafficking of labile Zn2+, and its inter-relation with reactive nitrogen species, are poorly understood due to the scarcity of appropriate imaging tools. We report a new family of red-emitting fluorescent sensors for labile Zn2+, ZBR1–3, based on a benzoresorufin platform functionalized with dipicolylamine or picolylamine-derived metal binding groups. In combination, the pendant amines and fluorophore afford an [N3O] binding motif that resembles that of previously reported fluorescein-based sensors of the Zinpyr family, reproducing well their binding capabilities and yielding comparable Kd values in the sub-nanomolar and picomolar ranges. The ZBR sensors display up to 8.4-fold emission fluorescence enhancement upon Zn2+ binding in the cuvette, with similar responses obtained in live cells using standard wide-field fluorescence microscopy imaging. The new sensors localize spontaneously in the endoplasmic reticulum (ER) of various tested cell lines, allowing for organelle-specific monitoring of zinc levels in live cells. Study of ER zinc levels in neural stem cells treated with a peroxynitrite generator, Sin-1, revealed an immediate decrease in labile Zn2+ thus providing evidence for a direct connection between ER stress and ER Zn2+ homeostasis.
Co-reporter:Kogularamanan Suntharalingam ; Timothy C. Johnstone ; Peter M. Bruno ; Wei Lin ; Michael T. Hemann
Journal of the American Chemical Society 2013 Volume 135(Issue 38) pp:14060-14063
Publication Date(Web):August 20, 2013
DOI:10.1021/ja4075375
The cellular response evoked by antiproliferating osmium(VI) nitrido compounds of general formula OsN(N^N)Cl3 (N^N = 2,2′-bipyridine 1, 1,10-phenanthroline 2, 3,4,7,8-tetramethyl-1,10-phenanthroline 3, or 4,7-diphenyl-1,10-phenanthroline 4) can be tuned by subtle ligand modifications. Complex 2 induces DNA damage, resulting in activation of the p53 pathway, cell cycle arrest at the G2/M phase, and caspase-dependent apoptotic cell death. In contrast, 4 evokes endoplasmic reticulum (ER) stress leading to the upregulation of proteins of the unfolded protein response pathway, increase in ER size, and p53-independent apoptotic cell death. To the best of our knowledge, 4 is the first osmium compound to induce ER stress in cancer cells.
Co-reporter:Robert J. Radford, Wen Chyan and Stephen J. Lippard
Chemical Science 2013 vol. 4(Issue 8) pp:3080-3084
Publication Date(Web):29 May 2013
DOI:10.1039/C3SC50974E
Combining fluorescent zinc sensors with the facile syntheses and biological targeting capabilities of peptides, we created green- and blue-emitting probes that (i) are readily prepared on the solid-phase, (ii) retain the photophysical and zinc-binding properties of the parent sensor, and (iii) can be directed to the extracellular side of plasma membranes in live cells for detection of mobile zinc.
Co-reporter:Simon P. Wisnovsky, Justin J. Wilson, Robert J. Radford, Mark P. Pereira, Maria R. Chan, Rebecca R. Laposa, Stephen J. Lippard, Shana O. Kelley
Chemistry & Biology 2013 Volume 20(Issue 11) pp:1323-1328
Publication Date(Web):21 November 2013
DOI:10.1016/j.chembiol.2013.08.010
•Cisplatin (mtPt) is delivered to mitochondria using a peptide vector•MtPt exhibits cytotoxicity without damaging nuclear DNA•This study demonstrates the specific delivery of a platinum drug to mitochondriaAn analog of the anticancer drug cisplatin (mtPt) was delivered to mitochondria of human cells using a peptide specifically targeting this organelle. mtPt induces apoptosis without damaging nuclear DNA, indicating that mtDNA damage is sufficient to mediate the activity of a platinum-based chemotherapeutic. This study demonstrates the specific delivery of a platinum drug to mitochondria and investigates the effects of directing this agent outside the nucleus.
Co-reporter:Ying Song, Kogularamanan Suntharalingam, Jessica S. Yeung, Maksim Royzen, and Stephen J. Lippard
Bioconjugate Chemistry 2013 Volume 24(Issue 10) pp:1733
Publication Date(Web):August 20, 2013
DOI:10.1021/bc400281a
Pt(IV) anticancer compounds typically operate as prodrugs that are reduced in the hypoxic environment of cancer cells, losing two axial ligands in the process to generate active Pt(II) species. Here we report the synthesis of two fluorescent Pt(IV) prodrugs of cisplatin in order to image and evaluate the Pt(IV) reduction process in simulated and real biological environments. Treatment of the complexes dissolved in PBS buffer with reducing agents typically encountered in cells, glutathione or ascorbate, afforded a 3- to 5-fold fluorescence turn-on owing to reduction and loss of their fluorescein-based axial ligands, which are quenched when bound to platinum. Both Pt(IV) conjugates displayed moderate cytotoxicity against human cancer cell lines, with IC50 values higher than that of cisplatin. Immunoblotting and DNA flow cytometry analyses of one of the complexes, Pt(IV)FL2, revealed that it damages DNA, causes cell cycle arrest in S or G2/M depending on exposure time, and ultimately triggers apoptotic cell death. Fluorescence microscopic studies prove that Pt(IV)FL2 enters cells intact and undergoes reduction intracellularly. The results are best interpreted in terms of a model in which the axial fluorescein ligands are expelled through lysosomes, with the platinum(II) moiety generated in the process binding to genomic DNA, which results in cell death.
Co-reporter:Ulf-Peter Apfel, Daniela Buccella, Justin J. Wilson, and Stephen J. Lippard
Inorganic Chemistry 2013 Volume 52(Issue 6) pp:3285-3294
Publication Date(Web):March 5, 2013
DOI:10.1021/ic302793w
A new family of benzoresorufin-based copper complexes for fluorescence detection of NO and HNO is reported. The copper complexes, CuBRNO1–3, elicit 1.5–4.8-fold emission enhancement in response to NO and HNO. The three sensors differ in the nature of the metal-binding site. The photophysical properties of these sensors are investigated with assistance from density functional theory calculations. The fluorescence turn-on observed upon reaction with HNO is an unexpected result that is discussed in detail. The utility of the new sensors for detecting HNO and NO in HeLa cells and RAW 264.7 macrophages is demonstrated.
Co-reporter:Timothy C. Johnstone and Stephen J. Lippard
Inorganic Chemistry 2013 Volume 52(Issue 17) pp:9915-9920
Publication Date(Web):July 16, 2013
DOI:10.1021/ic4010642
In an effort to expand the therapeutic range of platinum anticancer agents, several new approaches to platinum-based therapy, including nanodelivery, are under active investigation. To better understand the effect of ligand lipophilicity on the encapsulation of Pt(IV) prodrugs within polymer nanoparticles, the series of compounds cis,cis,trans-[Pt(NH3)2Cl2L2] was prepared, where L = acetate, propanoate, butanoate, pentanoate, hexanoate, heptanoate, octanoate, nonanoate, and decanoate. The lipophilicities of these compounds, assessed by reversed-phase HPLC, correlate with the octanol/water partition coefficients of their respective free carboxylic acid ligands, which in turn affect the degree of encapsulation of the Pt(IV) complex within the hydrophobic core of poly(lactic-co-glycolic acid)-block-poly(ethylene glycol) (PLGA-PEG-COOH) nanoparticles. The most lipophilic compound investigated, cis,cis,trans-[Pt(NH3)2Cl2(O2C(CH2)8CH3)2], displayed the best encapsulation. This compound was therefore selected to evaluate the effect of increased platinum concentration on encapsulation. As the platinum concentration was increased, there was an initial increase in encapsulation followed by a decrease due to macroscopic precipitation. Maximal loading occurred when the platinum complex was present at a 40% w/w ratio with respect to polymer during the nanoprecipitation step. Particles formed under these optimal conditions had diameters of approximately 50 nm, as determined by transmission electron microscopy.
Co-reporter:Timothy C. Johnstone ; Justin J. Wilson
Inorganic Chemistry 2013 Volume 52(Issue 21) pp:12234-12249
Publication Date(Web):June 5, 2013
DOI:10.1021/ic400538c
Platinum compounds represent one of the great success stories of metals in medicine. Following the serendipitous discovery of the anticancer activity of cisplatin by Rosenberg, a large number of cisplatin variants have been prepared and tested for their ability to kill cancer cells and inhibit tumor growth. These efforts continue today with increased realization that new strategies are needed to overcome issues of toxicity and resistance inherent to treatment by the approved platinum anticancer agents. One approach has been the use of so-called “non-traditional” platinum(II) and platinum(IV) compounds that violate the structure–activity relationships that governed platinum drug-development research for many years. Another is the use of specialized drug-delivery strategies. Here we describe recent developments from our laboratory involving monofunctional platinum(II) complexes together with a historical account of the manner by which we came to investigate these compounds and their relationship to previously studied molecules. We also discuss work carried out using platinum(IV) prodrugs and the development of nanoconstructs designed to deliver them in vivo.
Co-reporter:Amit Majumdar
Inorganic Chemistry 2013 Volume 52(Issue 23) pp:13292-13294
Publication Date(Web):November 18, 2013
DOI:10.1021/ic4019508
Mononitrosyldiiron complexes having either an [FeII·{FeNO}7] or an [FeIII·{FeNO}7] core formulation have been synthesized by methods that rely on redox-state-induced differentiation of the diiron starting materials in an otherwise symmetrical dinucleating ligand environment. The synthesis, X-ray structures, Mössbauer spectroscopy, cyclic voltammetry, and dioxygen reactivity of [FeIII·{FeNO}7] are described.
Co-reporter:Zhen Huang, Xiao-an Zhang, Miquel Bosch, Sarah J. Smith and Stephen J. Lippard
Metallomics 2013 vol. 5(Issue 6) pp:648-655
Publication Date(Web):28 May 2013
DOI:10.1039/C3MT00103B
We report the characterization of tris(2-pyridylmethyl)amine (TPA) as a membrane-permeable zinc chelator for intercepting biological mobile zinc. Compared to N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN), TPA chelates zinc with faster kinetics in cuvettes, live cells, and brain slices. TPA also is generally less toxic than TPEN in cell culture. Mechanistic analysis indicates that these improvements arise from both the electronic and steric properties of TPA including weaker metal-binding affinity, lower pKa, and smaller size. These results demonstrate that TPA chelation is a valuable addition to the methodologies available for investigating mobile zinc in biology.
Co-reporter:Jennifer M. Hope, Justin J. Wilson and Stephen J. Lippard
Dalton Transactions 2013 vol. 42(Issue 9) pp:3176-3180
Publication Date(Web):06 Nov 2012
DOI:10.1039/C2DT32462H
A platinum(II) complex of a monoanionic, tetradentate β-diketiminate (BDI) ligand with pendant quinoline arms, BDIQQH, is reported. The complex, [Pt(BDIQQ)]Cl, is emissive in DMSO, but non-emissive in aqueous buffer. Upon binding DNA in buffer, however, a 150-fold turn-on in emission intensity occurs. Dynamic light scattering and 1H NMR spectroscopy indicate that [Pt(BDIQQ)]Cl forms non-emissive aggregates in aqueous solution; DNA-binding disperses the aggregates leading to the large emission turn-on response. The cytotoxic activity of the complex, measured in two cancer cell lines, is comparable to or better than that of the established anticancer drug cisplatin.
Co-reporter:Yang Li;Chan Myae Myae Soe;Justin J. Wilson;Suan Lian Tuang;Ulf-Peter Apfel
European Journal of Inorganic Chemistry 2013 Volume 2013( Issue 12) pp:2011-2019
Publication Date(Web):
DOI:10.1002/ejic.201201387
Abstract
A triptycene-based bis(benzimidazole) ester ligand, L3, was designed to enhance the electron-donating ability of the heterocyclic nitrogen atoms relative to those of the first-generation bis(benzoxazole) analogs, L1 and L2. A convergent synthesis of L3 was designed and executed. Three-component titration experiments using UV/Vis spectroscopy revealed that the desired diiron(II) complex could be obtained with a 1:2:1 ratio of L3/Fe(OTf)2(MeCN)2/external carboxylate reactants. X-ray crystallographic studies of two diiron complexes derived in this manner from L3 revealed their formulas to be [Fe2L3(μ-OH)(μ-O2CR)(OTf)2], where R = 2,6-bis(p-tolyl)phenyl (7) or triphenylmethyl (8). The structures are similar to that of a diiron complex derived from L1, [Fe2L1(μ-OH)(μ-O2CArTol)(OTf)2] (9), a notable difference being that, in 7 and 8, the geometry at iron more closely resembles square-pyramidal than trigonal-bipyramidal. Mössbauer spectroscopic analyses of 7 and 8 indicate the presence of high-spin diiron(II) cores. These results demonstrate the importance of substituting benzimidazole for benzoxazole for assembling biomimetic diiron complexes with syn disposition of two N-donor ligands, as found in O2-activating carboxylate-bridged diiron centers in biological systems.
Co-reporter:Maksim Royzen, Justin J. Wilson, Stephen J. Lippard
Journal of Inorganic Biochemistry 2013 Volume 118() pp:162-170
Publication Date(Web):January 2013
DOI:10.1016/j.jinorgbio.2012.08.025
Nitroxyl, or HNO, is involved in a number of important physiological processes, such as vascular relaxation and neuroregulation. Effective imaging tools are required in order to gain a deeper understanding of the in vivo mechanisms of these processes and to identify the endogenous sources of HNO. Here, we further investigate the physical properties of our previously reported fluorescent nitroxyl sensor, [Cu(BOT1)Cl]Cl (J. Am. Chem. Soc.2010, 132, 5536; BOT1 = BODIPY·triazole, a tetradentate ligand). A new high-yielding synthetic procedure for BOT1 is reported. The X-ray crystal structures of two Cu(II) complexes of BOT1 are described. These structural studies show that the BOT1 ligand can form Cu(II) coordination complexes of both square-pyramidal and trigonal–bipyramidal geometries. Cyclic voltammograms of [Cu(BOT1)Cl]Cl were acquired, revealing the presence of a quasi-reversible feature at 130 mV (vs the ferrocene/ferrocenium couple) in MeCN and at − 40 mV (vs Ag/AgCl) in aqueous buffer, which is assigned to the CuII/CuI couple. The reactivity of [Cu(BOT1)Cl]Cl with Angeli's salt, a stable source of HNO, was further investigated. A 1000-fold excess of Angeli's salt elicits an immediate 10-fold emission turn-on response of the sensor, consistent with our previous report. A new observation, reported here, is that the intensity of this turn-on emission diminishes at longer incubation times. Fluorescent imaging of nitroxyl by [Cu(BOT1)Cl]Cl in HeLa cells was carried out. Upon treatment of the cells with Angeli's salt, there was a modest 2-fold intracellular turn-on in emission intensity.[Cu(BOT1)Cl]Cl is a fluorescent probe capable of imaging HNO in solution as well as live cells. The present work describes an optimized synthesis of the probe and further characterization of its photophysical and structural properties.Highlights► Improved synthesis of BOT1. ► X-ray crystal structures of CuII(BOT1) perchlorate and tetrafluoroborate salts. ► Electrochemical properties of CuII(BOT1). ► HNO imaging and localization in live HeLa cells using CuII(BOT1).
Co-reporter:Tsai-Te Lu, Seung Jae Lee, Ulf-Peter Apfel, and Stephen J. Lippard
Biochemistry 2013 Volume 52(Issue 13) pp:
Publication Date(Web):February 27, 2013
DOI:10.1021/bi301674p
The mitochondrial membrane-bound enzyme Clock-1 (CLK-1) extends the average longevity of mice and Caenorhabditis elegans, as demonstrated for Δclk-1 constructs for both organisms. Such an apparent impact on aging and the presence of a carboxylate-bridged diiron center in the enzyme inspired this work. We expressed a soluble human CLK-1 (hCLK-1) fusion protein with an N-terminal immunoglobulin binding domain of protein G (GB1). Inclusion of the solubility tag allowed for thorough characterization of the carboxylate-bridged diiron active site of the resulting GB1-hCLK-1 by spectroscopic and kinetic methods. Both UV–visible and Mössbauer experiments provide unambiguous evidence that GB1-hCLK-1 functions as a 5-demethoxyubiquinone-hydroxylase, utilizing its carboxylate-bridged diiron center. The binding of DMQn (n = 0 or 2) to GB1-hCLK-1 mediates reduction of the diiron center by nicotinamide adenine dinucleotide (NADH) and initiates O2 activation for subsequent DMQ hydroxylation. Deployment of DMQ to mediate reduction of the diiron center in GB1-hCLK-1 improves substrate specificity and diminishes consumption of NADH that is uncoupled from substrate oxidation. Both Vmax and kcat/KM for DMQ hydroxylation increase when DMQ0 is replaced by DMQ2 as the substrate, which demonstrates that an isoprenoid side chain enhances enzymatic hydroxylation and improves catalytic efficiency.
Co-reporter:Timothy C. Johnstone, Nora Kulak, Eric M. Pridgen, Omid C. Farokhzad, Robert Langer, and Stephen J. Lippard
ACS Nano 2013 Volume 7(Issue 7) pp:5675
Publication Date(Web):May 22, 2013
DOI:10.1021/nn401905g
Nanoparticle (NP) therapeutics have the potential to significantly alter the in vivo biological properties of the pharmaceutically active agents that they carry. Here we describe the development of a polymeric NP, termed M-NP, comprising poly(d,l-lactic-co-glycolic acid)-block-poly(ethylene glycol) (PLGA-PEG), stabilized with poly(vinyl alcohol) (PVA), and loaded with a water-soluble platinum(IV) [Pt(IV)] prodrug, mitaplatin. Mitaplatin, c,c,t-[PtCl2(NH3)2(OOCCHCl2)2], is a compound designed to release cisplatin, an anticancer drug in widespread clinical use, and the orphan drug dichloroacetate following chemical reduction. An optimized preparation of M-NP by double emulsion and its physical characterization are reported, and the influence of encapsulation on the properties of the platinum agent is evaluated in vivo. Encapsulation increases the circulation time of Pt in the bloodstream of rats. The biodistribution of Pt in mice is also affected by nanoparticle encapsulation, resulting in reduced accumulation in the kidneys. Finally, the efficacy of both free mitaplatin and M-NP, measured by tumor growth inhibition in a mouse xenograft model of triple-negative breast cancer, reveals that controlled release of mitaplatin over time from the nanoparticle treatment produces long-term efficacy comparable to that of free mitaplatin, which might limit toxic side effects.Keywords: cisplatin; mitaplatin; PLGA-PEG; polymer nanoparticle; Pt(IV) prodrug; triple-negative breast cancer
Co-reporter:Justin J. Wilson, Stephen J. Lippard
Polyhedron 2013 Volume 58() pp:71-78
Publication Date(Web):13 July 2013
DOI:10.1016/j.poly.2012.07.097
Benzyl amine was coupled to the dangling carboxylic acid groups of the platinum(II) complex [Pt(edda)Cl2], where edda = ethylenediamine-N,N′-diacetic acid, to give the diamide-tethered complex [Pt(L)Cl2] (1), where L = ethylenediamine-N,N′-bis(N-benzylacetamide). Complex 1 was oxidized with both PhICl2 and Br2. Oxidation with PhICl2 cleanly afforded the tetrachloride complex, [Pt(L)Cl4] (2), whereas oxidation with Br2 gave rise to several mixed halide complexes of the general formula, [Pt(L)ClxBr4-x], where x = 1, 2, or 3. Complexes 1 and 2 were fully characterized by 1H, 13C, and 195Pt NMR spectroscopy, as well as by ESI-MS. These compounds exist as a mixture of diastereomers that arise from the chirality of the two coordinated nitrogen atoms. Crystal structures of 1, 2, and [Pt(L)ClxBry] (3) are reported. Although refined as the tetrabromide complex [Pt(L)Br4], the crystal structure of 3 is a mixture of species with site-occupancy disorder of chloride and bromide ligands. DFT calculations indicate that the two sets of diastereomers of 1 and 2 are effectively thermoneutral, a conclusion that is also supported by the observation of both members of each pair by NMR spectroscopy. The cytotoxicity of 1 and 2 was measured by the MTT assay in HeLa cells and compared to that of cisplatin. Both exhibit IC50 values close to 50 μM and are therefore substantially less toxic than cisplatin, for which the IC50 is 1 μM.An amide coupling reaction on the dangling carboxylic acids of the compound, [Pt(edda)Cl2], where edda is ethylenediamine-N,N′-diacetic acid, gave the new complex, [Pt(L)Cl2] (1) where L is ethylenediamine-N,N′-bis(N-benzylacetamide). The cytotoxic activity of 1 and its oxidative reactivity with PhICl2 and Br2 were investigated. The use of this general outer-sphere amide bond coupling reaction represents a new route to systematically modified platinum anticancer agents.
Co-reporter:Timothy C. Johnstone, Stephen J. Lippard
Polyhedron 2013 Volume 52() pp:565-575
Publication Date(Web):22 March 2013
DOI:10.1016/j.poly.2012.08.010
X-ray crystallographic analysis of the compound trans-[Pt(NH2C6H11)2I2] revealed the presence of two distinct conformers within one crystal lattice. This compound was studied by variable temperature NMR spectroscopy to investigate the dynamic interconversion between these isomers. The results of this investigation were interpreted using physical (CPK) and computational (molecular mechanics and density functional theory) models. The conversion of the salts [Pt(NH2C6H11)4]X2 into trans-[Pt(NH2C6H11)2X2] (X = Cl, Br, I) was also studied and is discussed here with an emphasis on parallels to the work of Alfred Werner.Spectroscopic and computational investigations of the conformational isomerization involving the title compound are described. Parallels are drawn to the work of Alfred Werner.
Co-reporter:Julia Kozhukh
Journal of the American Chemical Society 2012 Volume 134(Issue 27) pp:11120-11123
Publication Date(Web):June 28, 2012
DOI:10.1021/ja305011g
The reactions of cobalt(II) complexes of tetraazamacrocyclic tropocoronand (TC) ligands with nitric oxide (NO) were investigated. When [Co(TC-5,5)] was allowed to react with NO(g), the {CoNO}8 mononitrosyl [Co(NO)(TC-5,5)] was isolated and structurally characterized. In contrast, a {Co(NO)2}10 species formed when [Co(TC-6,6)] was exposed to NO(g), and the nitrito [Co(NO2)(TC-6,6)] complex was structurally and spectroscopically characterized from the reaction mixture. The {Co(NO)2}10 species was assigned as the bis(cobalt dinitrosyl) complex [Co2(NO)4(TC-6,6)] by spectroscopic comparison with independently synthesized and characterized material. These results provide the first evidence for the influence of tropocoronand ring size on the nitric oxide reactivity of the cobalt(II) complexes.
Co-reporter:Michael D. Pluth and Stephen J. Lippard
Chemical Communications 2012 vol. 48(Issue 98) pp:11981-11983
Publication Date(Web):26 Oct 2012
DOI:10.1039/C2CC37221E
Nitric oxide binds reversibly to the Fe(III) complex of a well-developed tetra-amido macrocyclic ligand. Reaction with NO results in formation of a species consistent with an S = 1 {Fe–NO}6 ground state as characterized by UV-vis, IR, EPR, and Mössbauer spectroscopy. The resultant nitrosyl is labile and dissociates readily upon purging with N2, thus providing a rare example of reversible NO binding to non-heme iron.
Co-reporter:Justin J. Wilson
Journal of Medicinal Chemistry 2012 Volume 55(Issue 11) pp:5326-5336
Publication Date(Web):May 18, 2012
DOI:10.1021/jm3002857
Five cationic platinum(II) complexes of general formula, [Pt(NH3)2(β-diketonate)]X are reported, where X is a noncoordinating anion and β-diketonate = acetylacetonate (acac), 1,1,1,-trifluoroacetylacetonate (tfac), benzoylacetonate (bzac), 4,4,4-trifluorobenzoylacetonate (tfbz), or dibenzoylmethide (dbm), corresponding, respectively, to complexes 1–5. The log P values and the stabilities of 1–5 in aqueous solution were evaluated. The phenyl ring substituents of 3–5 increase the lipophilicity of the resulting complexes, whereas the trifluoromethyl groups of 2 and 4 decrease the stability of the complexes in aqueous solution. The uptake of 1–5 in HeLa cells increases as the lipophilicity of the investigated complex increases. Cancer cell cytotoxicity studies indicate that 1 and 3 are the least active complexes whereas 2, 4, and 5 are comparable in activity to cisplatin.
Co-reporter:Julia Kozhukh and Stephen J. Lippard
Inorganic Chemistry 2012 Volume 51(Issue 13) pp:7346-7353
Publication Date(Web):June 15, 2012
DOI:10.1021/ic3007684
Zinc thiolate complexes containing N2S tridentate ligands were prepared to investigate their reactivity toward reactive nitrogen species, chemistry proposed to occur at the zinc tetracysteine thiolate site of nitric oxide synthase (NOS). The complexes are unreactive toward nitric oxide (NO) in the absence of dioxygen, strongly indicating that NO cannot be the species directly responsible for S-nitrosothiol formation and loss of Zn2+ at the NOS dimer interface in vivo. S-Nitrosothiol formation does occur upon exposure of zinc thiolate solutions to NO in the presence of air, however, or to NO2 or NOBF4, indicating that these reactive nitrogen/oxygen species are capable of liberating zinc from the enzyme, possibly through generation of the S-nitrosothiol. Interaction between simple Zn2+ salts and preformed S-nitrosothiols leads to decomposition of the −SNO moiety, resulting in release of gaseous NO and N2O. The potential biological relevance of this chemistry is discussed.
Co-reporter:Loi H. Do ; Genqiang Xue ; Lawrence Que ; Jr.
Inorganic Chemistry 2012 Volume 51(Issue 4) pp:2393-2402
Publication Date(Web):January 20, 2012
DOI:10.1021/ic202379b
The composition of a (μ-oxo)diiron(III) complex coordinated by tris[(3,5-dimethyl-4-methoxy)pyridyl-2-methyl]amine (R3TPA) ligands was investigated. Characterization using a variety of spectroscopic methods and X-ray crystallography indicated that the reaction of iron(III) perchlorate, sodium hydroxide, and R3TPA affords [Fe2(μ-O)(μ-OH)(R3TPA)2](ClO4)3 (2) rather than the previously reported species [Fe2(μ-O)(OH)(H2O)(R3TPA)2](ClO4)3 (1). Facile conversion of the (μ-oxo)(μ-hydroxo)diiron(III) core of 2 to the (μ-oxo)(hydroxo)(aqua)diiron(III) core of 1 occurs in the presence of water and at low temperature. When 2 is exposed to wet acetonitrile at room temperature, the CH3CN adduct is hydrolyzed to CH3COO–, which forms the compound [Fe2(μ-O)(μ-CH3COO)(R3TPA)2](ClO4)3 (10). The identity of 10 was confirmed by comparison of its spectroscopic properties with those of an independently prepared sample. To evaluate whether or not 1 and 2 are capable of generating the diiron(IV) species [Fe2(μ-O)(OH)(O)(R3TPA)2]3+ (4), which has previously been generated as a synthetic model for high-valent diiron protein oxygenated intermediates, studies were performed to investigate their reactivity with hydrogen peroxide. Because 2 reacts rapidly with hydrogen peroxide in CH3CN but not in CH3CN/H2O, conditions that favor conversion to 1, complex 1 is not a likely precursor to 4. Compound 4 also forms in the reaction of 2 with H2O2 in solvents lacking a nitrile, suggesting that hydrolysis of CH3CN is not involved in the H2O2 activation reaction. These findings shed light on the formation of several diiron complexes of electron-rich R3TPA ligands and elaborate on conditions required to generate synthetic models of diiron(IV) protein intermediates with this ligand framework.
Co-reporter:Julia Kozhukh
Inorganic Chemistry 2012 Volume 51(Issue 17) pp:9416-9422
Publication Date(Web):August 16, 2012
DOI:10.1021/ic3012266
The size-dependent reactivity of cobalt tropocoronands [TC-n,n]2– is manifest in the NO chemistry of the cobalt(III) nitrite complexes [Co(η2-NO2)(TC-n,n)] (n = 4–6), the synthesis and characterization of which are reported for the first time. Complete conversion of [Co(η2-NO2)(TC-4,4)] to the cobalt mononitrosyl [Co(NO)(TC-4,4)] occurs upon exposure to NO(g). In contrast, addition of NO(g) to [Co(η2-NO2)(TC-5,5)] generates both cobalt mono- and dinitrosyl adducts, and addition of nitric oxide to [Co(η2-NO2)(TC-6,6)] converts this complex to the dicobalt tetranitrosyl species [Co2(NO)4(TC-6,6)]. In the latter complex, two tetrahedral cobalt dinitrosyl units are bound to the aminotroponeiminate poles of the [TC-6,6]2– ligand. These results significantly broaden the chemistry of cobalt tropocoronands with nitric oxide and the nitrite anion.
Co-reporter:Justin J. Wilson and Stephen J. Lippard
Inorganic Chemistry 2012 Volume 51(Issue 18) pp:9852-9864
Publication Date(Web):September 4, 2012
DOI:10.1021/ic301289j
Oxidation of the acetate-bridged half-lantern platinum(II) complex cis-[PtII(NH3)2(μ-OAc)2PtII(NH3)2](NO3)2, [1](NO3)2, with iodobenzene dichloride or bromine generates the halide-capped platinum(III) species cis-[XPtIII(NH3)2(μ-OAc)2PtIII(NH3)2X](NO3)2, where X is Cl in [2](NO3)2 or Br in [3](NO3)2, respectively. These three complexes, characterized structurally by X-ray crystallography, feature short (≈2.6 Å) Pt–Pt separations, consistent with formation of a formal metal–metal bond upon oxidation. Elongated axial Pt–X distances occur, reflecting the strong trans influence of the metal–metal bond. The three structures are compared to those of other known dinuclear platinum complexes. A combination of 1H, 13C, 14N, and 195Pt NMR spectroscopy was used to characterize [1]2+–[3]2+ in solution. All resonances shift downfield upon oxidation of [1]2+ to [2]2+ and [3]2+. For the platinum(III) complexes, the 14N and 195Pt resonances exhibit decreased line widths by comparison to those of [1]2+. Density functional theory calculations suggest that the decrease in the 14N line width arises from a diminished electric field gradient at the 14N nuclei in the higher valent compounds. The oxidation of [1](NO3)2 with the alternative oxidizing agent bis(trifluoroacetoxy)iodobenzene affords the novel tetranuclear complex cis-[(O2CCF3)PtIII(NH3)2(μ-OAc)2PtIII(NH3)(μ-NH2)]2(NO3)4, [4](NO3)4, also characterized structurally by X-ray crystallography. In solution, this complex exists as a mixture of species, the identities of which are proposed.
Co-reporter:Yang Li, Justin J. Wilson, Loi H. Do, Ulf-Peter Apfel and Stephen J. Lippard
Dalton Transactions 2012 vol. 41(Issue 31) pp:9272-9275
Publication Date(Web):29 Jun 2012
DOI:10.1039/C2DT31260C
A triptycene-based bis(benzoxazole) diacid ligand H22L2Ph4Ph4 bearing sterically encumbering groups was synthesized. Treatment of H22L2Ph4Ph4 with Fe(OTf)3 afforded a C2-symmetric trinuclear iron(III) complex, [NaFe3(L2Ph4Ph4Ph4)2(μ3-O)(μ-O2CCPh3)2(H2O)3](OTf)2 (8). The triiron core of this complex adopts the well known “basic iron acetate” structure where the heteroleptic carboxylates, comprising two Ph3CCO2− and two (L2Ph4Ph4Ph4)2− ligands, donate the six carboxylate bridges. The (L2Ph4Ph4Ph4)2− ligand undergoes only minor conformational changes upon formation of the complex.
Co-reporter:Semi Park and Stephen J. Lippard
Biochemistry 2012 Volume 51(Issue 34) pp:
Publication Date(Web):August 17, 2012
DOI:10.1021/bi300649v
Proteins in the HMG family are important transcription factors. They recognize cisplatin-damaged DNA lesions with a structure-specific preference and account for more than 70% of all proteins that interact with the cisplatin 1,2-intrastrand d(GpG) cross-link. HMGB4, a new member of the mammalian HMGB protein family expressed preferentially in the testis, was generated recombinantly, and its interactions with cisplatin-modified DNA were investigated in vitro. The binding affinities of the two individual DNA-binding domains of HMGB4 to DNA carrying a cisplatin 1,2-intrastrand d(GpG) cross-link are weaker than those of the DNA-binding domains of HMGB1. Full-length HMGB4, however, has a 28-fold stronger binding affinity (Kd = 4.35 nM) for the platinated adduct compared to that of HMGB1 (Kd = 120 nM), presumably because the former lacks a C-terminal acidic tail. The residue Phe37 plays a critical role in stabilizing the binding complex of HMGB4 with the cisplatin-modified DNA, as it does for HMGB1. Hydroxyl radical footprinting analysis of the HMGB4/platinated DNA complex reveals a footprinting pattern very different from that of HMGB1, however, revealing very little binding asymmetry with respect to the platinated lesion. An in vitro repair assay revealed that HMGB4, at 1 μM, interferes with repair of cisplatin 1,2-intrastrand cross-link damage by >90% compared to control, whereas HMGB1 at the same concentration inhibits repair by 45%. This repair inhibition capability is highly dependent on both the binding affinity and the size of the proteins. The putative role of HMGB4 in the mechanism of action of cisplatin, and especially its potential relevance to the hypersensitivity of testicular germ cell tumors to cisplatin, are discussed.
Co-reporter:Justin J. Wilson, Stephen J. Lippard
Inorganica Chimica Acta 2012 Volume 389() pp:77-84
Publication Date(Web):1 July 2012
DOI:10.1016/j.ica.2011.12.034
The dangling carboxylic acid moiety of the known platinum(II) complex, [Pt(edma)Cl2] (edma = ethylenediaminemonoacetic acid), was functionalized via amide coupling chemistry with benzyl amine and dansyl ethylenediamine to afford the derivatives [Pt(edBz)Cl2] (1) and [Pt(edDs)Cl2] (2). Subsequent oxidation of these platinum(II) complexes with iodobenzene dichloride in DMF yielded the respective platinum(IV) analogs, [Pt(edBz)Cl4] (3) and [Pt(edDs)Cl4] (4). All four platinum complexes were characterized by multinuclear NMR spectroscopy, IR spectroscopy, electrospray ionization mass spectrometry, and elemental analysis. In addition, compounds 1 and 3 were structurally characterized by X-ray crystallography. The photophysical properties of the compounds bearing the fluorescent dansyl moiety, 2 and 4, were evaluated. The emission quantum yields of 2 and 4 in DMF are 27% and 1.6%, respectively. This large difference in emission efficiency indicates that the platinum(IV) center in 4 is more effective at quenching the dansyl-based fluorescence than the platinum(II) center in 2. Time-dependent density functional theory calculations indicate that 4 has several low-lying singlet excited states that energetically lie below the primary radiation-accessible excited state of the dansyl fluorophore. These low-energy excited states may offer non-radiative decay pathways that lower the overall emission quantum yield. Treatment of 4 with biologically relevant reducing agents in pH 7.4 phosphate-buffered saline induces a 6.3-fold increase in emission intensity. These results demonstrate that 4 and future derivatives thereof may be useful for imaging the reduction of platinum(IV) complexes in living systems, chemistry of importance for evolving platinum-based anticancer drug strategies.Graphical abstractA platinum(IV) complex bearing a tethered dansyl fluorophore elicits a turn-on fluorescent response upon conversion to Pt(II) with biological reducing agents.Highlights► Fluorescent complexes of platinum(II) and platinum(IV) were synthesized. ► These complexes were characterized by multinuclear NMR spectroscopy. ► The Pt(IV) complex is much less emissive than the Pt(II) complex. ► Reduction of the Pt(IV) complex elicits a 6.3-fold emission turn-on. ► This work provides a proof of principle for studying Pt(IV) reduction in live cells.
Co-reporter:Nora Graf, Diane R. Bielenberg, Nagesh Kolishetti, Christoph Muus, Jacqueline Banyard, Omid C. Farokhzad, and Stephen J. Lippard
ACS Nano 2012 Volume 6(Issue 5) pp:4530
Publication Date(Web):May 14, 2012
DOI:10.1021/nn301148e
Targeted delivery of therapeutics to tumor neovasculature is potentially a powerful approach for selective cancer treatment. Integrins are heterodimeric transmembrane proteins involved in cell adhesion and cell signaling, and their expression is commonly upregulated in cancers and inflammatory diseases. The αvβ3 integrin is differentially upregulated on angiogenic endothelial cells as well as on many cancer cells. Here we demonstrate the differential targeting of cisplatin prodrug-encapsulated poly(d,l-lactic-co-glycolic acid)-block-polyethylene glycol (PLGA-PEG) nanoparticles (NPs) to the αvβ3 integrin on cancer cells using the cyclic pentapeptide c(RGDfK). Cisplatin is one of the most widely used anticancer drugs, and approaches that can improve its therapeutic index are of broad importance. The RGD-targeted Pt(IV)-encapsulated NPs displayed enhanced cytotoxicity as compared to cisplatin administered in its conventional dosage form in model prostate and breast cancer epithelial cells in vitro. Cytotoxicities were also elevated in comparison to those of previously reported systems, a small molecule Pt(IV)-RGD conjugate and a Pt(IV) nanoscale coordination polymer carrying RGD moieties. This result encouraged us also to evaluate the anticancer effect of the new construct in an animal model. The RGD-targeted PLGA-PEG NPs were more efficacious and better tolerated by comparison to cisplatin in an orthotopic human breast cancer xenograft model in vivo.Keywords: breast cancer xenograft; integrin targeting; polymeric nanoparticle; Pt(IV) complex; RGD peptide
Co-reporter:Nora Graf, Tara E. Mokhtari, Ioannis A. Papayannopoulos, Stephen J. Lippard
Journal of Inorganic Biochemistry 2012 110() pp: 58-63
Publication Date(Web):
DOI:10.1016/j.jinorgbio.2012.02.012
Co-reporter:Ga Young Park;Justin J. Wilson;Ying Song
PNAS 2012 Volume 109 (Issue 30 ) pp:11987-11992
Publication Date(Web):2012-07-24
DOI:10.1073/pnas.1207670109
Monofunctional platinum(II) complexes of general formula cis-[Pt(NH3)2(N-heterocycle)Cl]Cl bind DNA at a single site, inducing little distortion in the double helix. Despite this behavior, these
compounds display significant antitumor properties, with a different spectrum of activity than that of classic bifunctional
cross-linking agents like cisplatin. To discover the most potent monofunctional platinum(II) compounds, the N-heterocycle was systematically varied to generate a small library of new compounds, with guidance from the X-ray structure
of RNA polymerase II (Pol II) stalled at a monofunctional pyriplatin-DNA adduct. In pyriplatin, the N-heterocycle is pyridine. The most effective complex evaluated was phenanthriplatin, cis-[Pt(NH3)2(phenanthridine)Cl]NO3, which exhibits significantly greater activity than the Food and Drug Administration-approved drugs cisplatin and oxaliplatin.
Studies of phenanthriplatin in the National Cancer Institute 60-cell tumor panel screen revealed a spectrum of activity distinct
from that of these clinically validated anticancer agents. The cellular uptake of phenanthriplatin is substantially greater
than that of cisplatin and pyriplatin because of the hydrophobicity of the phenanthridine ligand. Phenanthriplatin binds more
effectively to 5′-deoxyguanosine monophosphate than to N-acetyl methionine, whereas pyriplatin reacts equally well with both reagents. This chemistry supports DNA as a viable cellular
target for phenanthriplatin and suggests that it may avoid cytoplasmic platinum scavengers with sulfur-donor ligands that
convey drug resistance. With the use of globally platinated Gaussia luciferase vectors, we determined that phenanthriplatin inhibits transcription in live mammalian cells as effectively as
cisplatin, despite its inability to form DNA cross-links.
Co-reporter:Christine E. Tinberg and Stephen J. Lippard
Accounts of Chemical Research 2011 Volume 44(Issue 4) pp:280
Publication Date(Web):March 10, 2011
DOI:10.1021/ar1001473
The controlled oxidation of methane to methanol is a chemical transformation of great value, particularly in the pursuit of alternative fuels, but the reaction remains underutilized industrially because of inefficient and costly synthetic procedures. In contrast, methane monooxygenase enzymes (MMOs) from methanotrophic bacteria achieve this chemistry efficiently under ambient conditions. In this Account, we discuss the first observable step in the oxidation of methane at the carboxylate-bridged diiron active site of the soluble MMO (sMMO), namely, the reductive activation of atmospheric O2. The results provide benchmarks against which the dioxygen activation mechanisms of other bacterial multicomponent monooxygenases can be measured.Molecular oxygen reacts rapidly with the reduced diiron(II) cen-ter of the hydroxylase component of sMMO (MMOH). The first spectroscopically characterized intermediate that results from this process is a peroxodiiron(III) species, P*, in which the iron atoms have identical environments. P* converts to a second peroxodiiron(III) unit, Hperoxo, in a process accompanied by the transfer of a proton, probably with the assistance of a residue near the active site. Proton-promoted O−O bond scission and rearrangement of the diiron core then leads to a diiron(IV) unit, termed Q, that is directly responsible for the oxidation of methane to methanol. In one section of this Account, we provide a detailed discussion of these processes, with particular emphasis on possible structures of the intermediates. The geometries of P* and Hperoxo are currently unknown, and recent synthetic modeling chemistry has highlighted the need for further structural characterization of Q, currently assigned as a di(μ-oxo)diiron(IV) “diamond core.”In another section of the Account, we discuss in detail proton transfer during the O2 activation events. The role of protons in promoting O−O bond cleavage, thereby initiating the conversion of Hperoxo to Q, was previously a controversial topic. Recent studies of the mechanism, covering a range of pH values and in D2O instead of H2O, confirmed conclusively that the transfer of protons, possibly at or near the active site, is necessary for both P*-to-Hperoxo and Hperoxo-to-Q conversions. Specific mechanistic insights into these processes are provided.In the final section of the Account, we present our view of experiments that need to be done to further define crucial aspects of sMMO chemistry. Here our goal is to detail the challenges that we and others face in this research, particularly with respect to some long-standing questions about the system, as well as approaches that might be used to solve them.
Co-reporter:Youngmin You ; Sumin Lee ; Taehee Kim ; Kei Ohkubo ; Weon-Sik Chae ; Shunichi Fukuzumi ; Gil-Ja Jhon ; Wonwoo Nam
Journal of the American Chemical Society 2011 Volume 133(Issue 45) pp:18328-18342
Publication Date(Web):October 24, 2011
DOI:10.1021/ja207163r
A new phosphorescent zinc sensor (ZIrF) was constructed, based on an Ir(III) complex bearing two 2-(2,4-difluorophenyl)pyridine (dfppy) cyclometalating ligands and a neutral 1,10-phenanthroline (phen) ligand. A zinc-specific di(2-picolyl)amine (DPA) receptor was introduced at the 4-position of the phen ligand via a methylene linker. The cationic Ir(III) complex exhibited dual phosphorescence bands in CH3CN solutions originating from blue and yellow emission of the dfppy and phen ligands, respectively. Zinc coordination selectively enhanced the latter, affording a phosphorescence ratiometric response. Electrochemical techniques, quantum chemical calculations, and steady-state and femtosecond spectroscopy were employed to establish a photophysical mechanism for this phosphorescence response. The studies revealed that zinc coordination perturbs nonemissive processes of photoinduced electron transfer and intraligand charge-transfer transition occurring between DPA and phen. ZIrF can detect zinc ions in a reversible and selective manner in buffered solution (pH 7.0, 25 mM PIPES) with Kd = 11 nM and pKa = 4.16. Enhanced signal-to-noise ratios were achieved by time-gated acquisition of long-lived phosphorescence signals. The sensor was applied to image biological free zinc ions in live A549 cells by confocal laser scanning microscopy. A fluorescence lifetime imaging microscope detected an increase in photoluminescence lifetime for zinc-treated A549 cells as compared to controls. ZIrF is the first successful phosphorescent sensor that detects zinc ions in biological samples.
Co-reporter:Youngmin You ; Yejee Han ; Yong-Min Lee ; Soo Young Park ; Wonwoo Nam
Journal of the American Chemical Society 2011 Volume 133(Issue 30) pp:11488-11491
Publication Date(Web):July 12, 2011
DOI:10.1021/ja204997c
A phosphorescent sensor based on a multichromophoric iridium(III) complex was synthesized and characterized. The construct exhibits concomitant changes in its phosphorescence intensity ratio and phosphorescence lifetime in response to copper(II) ion. The sensor, which is reversible and selective, is able to quantify copper(II) ions in aqueous media, and it detects intracellular copper ratiometrically.
Co-reporter:Daniela Buccella ; Joshua A. Horowitz
Journal of the American Chemical Society 2011 Volume 133(Issue 11) pp:4101-4114
Publication Date(Web):February 25, 2011
DOI:10.1021/ja110907m
Treatment of aqueous zinc solutions with incremental additions of a ditopic fluorescent sensor of the Zinpyr family, based on pyridine/pyrazine-containing metal recognition units, affords a fluorescence titration curve with a sharp maximum at a sensor:Zn2+ ratio of 0.5 (Zhang, X-a.; Hayes, D.; Smith, S. J.; Friedle, S.; Lippard, S. J. J. Am. Chem. Soc.2008, 130, 15788−15789). This fluorescence response enables the quantification of readily chelatable zinc in biological samples by a simple titration protocol. In the present work a new set of ditopic fluorescence zinc sensors functionalized with pyridine/pyrazine-containing metal chelating units is described, and through detailed studies the principles governing the characteristic OFF−ON−OFF fluorescence behavior and quantification capabilities of the family are delineated. Incorporation of carboxylate/ester groups in the 6 position of the fluorescein allows for control of the spatial distribution of the sensor for selective extra- or intracellular imaging of mobile zinc, without introducing significant changes in zinc-binding properties. A combination of spectrophotometric and potentiometric measurements provided a complete description of the H+- and Zn2+-binding properties of the compounds and their correlation with the observed fluorescence profile. The first zinc-binding event has an apparent affinity, K1′, of 1.9 × 109−3.1 × 109 M−1, whereas for coordination of the second Zn2+ ion, responsible for fluorescence turn-on, the apparent formation constant, K2′, is 5.5 × 107−6.9 × 107 M−1. A detailed chemical and mathematical analysis of the system demonstrated that the difference in emission efficiencies of the dimetalated (LZn2) vs monometalated (LZn) and metal-free (L) forms, a consequence of the combined quenching effects of the two metal-chelating units, significantly influences the shape of the titration curve. The scope of the titration method was investigated mathematically, and a lower boundary for the range of concentrations that can be determined was established as a function of the magnitude of K2′. Our results suggest that the principles governing the response of the ZPP1 series are applicable to other analogues of the Zinpyr family. Moreover, they may guide the design of other ditopic sensors suitable for determining the concentrations of a wide range of mobile metal ions and other chemical signaling agents of relevance in biological systems.
Co-reporter:Arteum D. Bochevarov ; Jianing Li ; Woon Ju Song ; Richard A. Friesner
Journal of the American Chemical Society 2011 Volume 133(Issue 19) pp:7384-7397
Publication Date(Web):April 25, 2011
DOI:10.1021/ja110287y
The methane and toluene monooxygenase hydroxylases (MMOH and TMOH, respectively) have almost identical active sites, yet the physical and chemical properties of their oxygenated intermediates, designated P*, Hperoxo, Q, and Q* in MMOH and ToMOHperoxo in a subclass of TMOH, ToMOH, are substantially different. We review and compare the structural differences in the vicinity of the active sites of these enzymes and discuss which changes could give rise to the different behavior of Hperoxo and Q. In particular, analysis of multiple crystal structures reveals that T213 in MMOH and the analogous T201 in TMOH, located in the immediate vicinity of the active site, have different rotatory configurations. We study the rotational energy profiles of these threonine residues with the use of molecular mechanics (MM) and quantum mechanics/molecular mechanics (QM/MM) computational methods and put forward a hypothesis according to which T213 and T201 play an important role in the formation of different types of peroxodiiron(III) species in MMOH and ToMOH. The hypothesis is indirectly supported by the QM/MM calculations of the peroxodiiron(III) models of ToMOH and the theoretically computed Mössbauer spectra. It also helps explain the formation of two distinct peroxodiiron(III) species in the T201S mutant of ToMOH. Additionally, a role for the ToMOD regulatory protein, which is essential for intermediate formation and protein functioning in the ToMO system, is advanced. We find that the low quadrupole splitting parameter in the Mössbauer spectrum observed for a ToMOHperoxo intermediate can be explained by protonation of the peroxo moiety, possibly stabilized by the T201 residue. Finally, similarities between the oxygen activation mechanisms of the monooxygenases and cytochrome P450 are discussed.
Co-reporter:Loi H. Do
Journal of the American Chemical Society 2011 Volume 133(Issue 27) pp:10568-10581
Publication Date(Web):June 17, 2011
DOI:10.1021/ja2021312
A dinucleating macrocycle, H2PIM, containing phenoxylimine metal-binding units has been prepared. Reaction of H2PIM with [Fe2(Mes)4] (Mes = 2,4,6-trimethylphenyl) and sterically hindered carboxylic acids, Ph3CCO2H or ArTolCO2H (2,6-bis(p-tolyl)benzoic acid), afforded complexes [Fe2(PIM)(Ph3CCO2)2] (1) and [Fe2(PIM)(ArTolCO2)2] (2), respectively. X-ray diffraction studies revealed that these diiron(II) complexes closely mimic the active site structures of the hydroxylase components of bacterial multicomponent monooxygenases (BMMs), particularly the syn disposition of the nitrogen donor atoms and the bridging μ-η1η2 and μ-η1η1 modes of the carboxylate ligands at the diiron(II) centers. Cyclic voltammograms of 1 and 2 displayed quasi-reversible redox couples at +16 and +108 mV vs ferrocene/ferrocenium, respectively. Treatment of 2 with silver perchlorate afforded a silver(I)/iron(III) heterodimetallic complex, [Fe2(μ-OH)2(ClO4)2(PIM)(ArTolCO2)Ag] (3), which was structurally and spectroscopically characterized. Complexes 1 and 2 both react rapidly with dioxygen. Oxygenation of 1 afforded a (μ-hydroxo)diiron(III) complex [Fe2(μ-OH)(PIM)(Ph3CCO2)3] (4), a hexa(μ-hydroxo)tetrairon(III) complex [Fe4(μ-OH)6(PIM)2(Ph3CCO2)2] (5), and an unidentified iron(III) species. Oxygenation of 2 exclusively formed di(carboxylato)diiron(III) compounds, a testimony to the role of the macrocylic ligand in preserving the dinuclear iron center under oxidizing conditions. X-ray crystallographic and 57Fe Mössbauer spectroscopic investigations indicated that 2 reacts with dioxygen to give a mixture of (μ-oxo)diiron(III) [Fe2(μ-O)(PIM)(ArTolCO2)2] (6) and di(μ-hydroxo)diiron(III) [Fe2(μ-OH)2(PIM)(ArTolCO2)2] (7) units in the same crystal lattice. Compounds 6 and 7 spontaneously convert to a tetrairon(III) complex, [Fe4(μ-OH)6(PIM)2(ArTolCO2)2] (8), when treated with excess H2O.
Co-reporter:Loi H. Do, Hongxin Wang, Christine E. Tinberg, Eric Dowty, Yoshitaka Yoda, Stephen P. Cramer and Stephen J. Lippard
Chemical Communications 2011 vol. 47(Issue 39) pp:10945-10947
Publication Date(Web):06 Sep 2011
DOI:10.1039/C1CC13836G
The vibrational spectrum of an η1,η1-1,2-peroxodiiron(III) complex was measured by nuclear resonance vibrational spectroscopy and fit using an empirical force field analysis. Isotopic 18O2 labelling studies revealed a feature involving motion of the {Fe2(O2)}4+ core that was not previously observed by resonance Raman spectroscopy.
Co-reporter:Yang Li, Rui Cao, and Stephen J. Lippard
Organic Letters 2011 Volume 13(Issue 19) pp:5052-5055
Publication Date(Web):August 29, 2011
DOI:10.1021/ol201882v
A novel triptycene-based ligand with a preorganized framework was designed to model carboxylate-bridged diiron active sites in bacterial multicomponent monooxygenase (BMM) hydroxylase enzymes. The synthesis of the bis(benzoxazole)-appended ligand L1 depicted was accomplished in 11 steps. Reaction of L1 with iron(II) triflate and a carboxylate source afforded the desired diiron(II) complex [Fe2L1(μ-OH)(μ-O2CArTol)(OTf)2].
Co-reporter:Andrew G. Tennyson, Stephen J. Lippard
Chemistry & Biology 2011 Volume 18(Issue 10) pp:1211-1220
Publication Date(Web):28 October 2011
DOI:10.1016/j.chembiol.2011.09.009
Nitric oxide (NO) is a gaseous diatomic radical that is involved in a wide range of physiological and pathological functions in biology. Conceptually, the biochemistry of NO can be separated into three stages: generation (stage 1), translocation (stage 2), and action (stage 3). In stage 1 the oxygenase domain of NO synthase converts L-arginine to L-citrulline and NO (g). Owing to its short-lived nature, this molecule is converted into a different nitrogen oxide such as NO2, an organonitrosyl such as a nitrosothiol, or a metal nitrosyl such as a heme-nitrosyl, for transportation in stage 2. Each of these derivatives features unique physical characteristics, chemical reactivity, and biological activity. Upon delivery in stage 3, NO exerts its physiological or pathological function by reaction with biomolecules containing redox-active metals or other residues.
Co-reporter:Zachary J. Tonzetich ; Florent Héroguel ; Loi H. Do
Inorganic Chemistry 2011 Volume 50(Issue 4) pp:1570-1579
Publication Date(Web):January 18, 2011
DOI:10.1021/ic102300d
Several nitrosyl complexes of Fe and Co have been prepared using the sterically hindered Ar-nacnac ligand (Ar-nacnac = anion of [(2,6-diisopropylphenyl)NC(Me)]2CH). The dinitrosyliron complexes [Fe(NO)2(Ar-nacnac)] (1) and (Bu4N)[Fe(NO)2(Ar-nacnac)] (2) react with [FeIII(TPP)Cl] (TPP = tetraphenylporphine dianion) to generate [FeII(NO)(TPP)] and the corresponding mononitrosyliron complexes. The factors governing NO transfer with dinitrosyliron complexes (DNICs) 1 and 2 are evaluated, together with the chemistry of the related mononitrosyliron complex, [Fe(NO)Br(Ar-nacnac)] (4). The synthesis and properties of the related cobalt dinitrosyl [Co(NO)2(Ar-nacnac)] (3) is also discussed for comparison to DNICs 1 and 2. The solid-state structures of several of these compounds as determined by X-ray crystallography are reported.
Co-reporter:Justin J. Wilson and Stephen J. Lippard
Inorganic Chemistry 2011 Volume 50(Issue 7) pp:3103-3115
Publication Date(Web):March 1, 2011
DOI:10.1021/ic2000816
The synthesis, characterization, and cytotoxicity of eight new platinum(IV) complexes having the general formula cis,cis,trans-[Pt(NH3)2Cl2(O2CNHR)2] are reported, where R = tert-butyl (4), cyclopentyl (5), cyclohexyl (6), phenyl (7), p-tolyl (8), p-anisole (9), 4-fluorophenyl (10), or 1-naphthyl (11). These compounds were synthesized by reacting organic isocyanates with the platinum(IV) complex cis,cis,trans-[Pt(NH3)2Cl2(OH)2]. The electrochemistry of the compounds was investigated by cyclic voltammetry. The aryl carbamate complexes 7−11 exhibit reduction peak potentials near −720 mV vs Ag/AgCl, whereas the alkyl carbamate complexes display reduction peak potentials between −820 and −850 mV vs Ag/AgCl. The cyclic voltammograms of cis,cis,trans-[Pt(NH3)2Cl2(O2CCH3)2] (1), cis,cis,trans-[Pt(NH3)2Cl2(O2CCF3)2] (2), and cis-[Pt(NH3)2Cl4] (3) were measured for comparison. Density functional theory studies were undertaken to investigate the electronic structures of 1−11 and to determine their adiabatic electron affinities. A linear correlation (R2 = 0.887) between computed adiabatic electron affinities and measured reduction peak potentials was discovered. The biological activity of 4−11 and, for comparison, cisplatin was evaluated in human lung cancer A549 and normal MRC-5 cells by the MTT assay. The compounds exhibit comparable or slightly better activity than cisplatin against the A549 cells. In MRC-5 cells, all are equally or slightly less cytotoxic than cisplatin, except for 4 and 5, which are more toxic.
Co-reporter:Rui Cao, Brian D. McCarthy, and Stephen J. Lippard
Inorganic Chemistry 2011 Volume 50(Issue 19) pp:9499-9507
Publication Date(Web):August 26, 2011
DOI:10.1021/ic201172r
We describe a multidentate tripodal ligand in which three pendant arms carrying di(2-picolyl)amine units are linked to the ortho positions of a tris(o-xylyl) scaffold, providing N(CH2-o-C6H4CH2N(CH2py)2)3 (L). Reaction of L with CuCl2 in the presence of hexafluorophosphate anion afforded blue cubes of [(CuCl)3L](PF6)3·5H2O (1). Crystallographic studies of 1 revealed that the three symmetry-related arms each coordinate a {CuIICl} unit, and two molecules of 1 are connected to one another through a Cu(μ-Cl)2Cu bridge, extending the molecular structure to form a two-dimensional (2-D) layer. These 2-D layers pack in an ABCABC... fashion with PF6– anions located in between. Reaction of 1 with a stoichiometric amount of perrhenate ion afforded blue plates of [(CuCl)3L](PF6)(ReO4)2·3H2O (2). Compound 2 has the same lattice structure as 1, but the tricopper unit backbone now traps one ReO4– anion through Coulombic interactions. In addition, three molecules of 2 are bridged by a perrhenate ion, forming a Cu3(μ3-ReO4) cluster, to give a different 2-D structure displaying a rare tridentate bridging ReO4– mode. Thus, in addition to classic perrhenate trapping through weak Coulombic interactions, 2 represents an exceptional example in which the ReO4– anion is immobilized in an extended framework through tight covalent interactions. The interlamellar PF6– anions in 1 can be exchanged with other anions including perrhenate, perchlorate, or periodate. The structural similarity between perrhenate and pertechnetate makes these materials of potential interest for pertechnetate trapping.
Co-reporter:Michael D. Pluth, Maria R. Chan, Lindsey E. McQuade, and Stephen J. Lippard
Inorganic Chemistry 2011 Volume 50(Issue 19) pp:9385-9392
Publication Date(Web):September 7, 2011
DOI:10.1021/ic200986v
Fluorescent turn-on probes for nitric oxide based on seminaphthofluorescein scaffolds were prepared and spectroscopically characterized. The Cu(II) complexes of these fluorescent probes react with NO under anaerobic conditions to yield a 20–45-fold increase in integrated emission. The seminaphthofluorescein-based probes emit at longer wavelengths than the parent FL1 and FL2 fluorescein-based generations of NO probes, maintaining emission maxima between 550 and 625 nm. The emission profiles depend on the excitation wavelength; maximum fluorescence turn-on is achieved at excitations between 535 and 575 nm. The probes are highly selective for NO over other biologically relevant reactive nitrogen and oxygen species including NO3–, NO2–, HNO, ONOO–, NO2, OCl–, and H2O2. The seminaphthofluorescein-based probes can be used to visualize endogenously produced NO in live cells, as demonstrated using Raw 264.7 macrophages.
Co-reporter:Woon Ju Song and Stephen J. Lippard
Biochemistry 2011 Volume 50(Issue 23) pp:
Publication Date(Web):May 19, 2011
DOI:10.1021/bi200340f
Site-directed mutagenesis studies of a strictly conserved T201 residue in the active site of toluene/o-xylene monooxygenase hydroxylase (ToMOH) revealed that a single mutation can facilitate kinetic isolation of two distinctive peroxodiiron(III) species, designated T201peroxo and ToMOHperoxo, during dioxygen activation. Previously, we characterized both oxygenated intermediates by UV–vis and Mössbauer spectroscopy, proposed structures from DFT and QM/MM computational studies, and elucidated chemical steps involved in dioxygen activation through the kinetic studies of T201peroxo formation. In this study, we investigate the kinetics of T201peroxo decay to explore the reaction mechanism of the oxygenated intermediates following O2 activation. The decay rates of T201peroxo were monitored in the absence and presence of external (phenol) or internal (tryptophan residue in an I100W variant) substrates under pre-steady-state conditions. Three possible reaction models for the formation and decay of T201peroxo were evaluated, and the results demonstrate that this species is on the pathway of arene oxidation and appears to be in equilibrium with ToMOHperoxo.
Co-reporter:Michael S. McCormick and Stephen J. Lippard
Biochemistry 2011 Volume 50(Issue 51) pp:
Publication Date(Web):December 2, 2011
DOI:10.1021/bi201248b
In all structurally characterized bacterial multicomponent monooxygenase (BMM) hydroxylase proteins, a series of hydrophobic cavities in the α-subunit trace a conserved path from the protein exterior to the carboxylate-bridged diiron active site. This study examines these cavities as a potential route for transport of dioxygen to the active site by crystallographic characterization of a xenon-pressurized sample of the hydroxylase component of phenol hydroxylase from Pseudomonas sp. OX1. Computational analyses of the hydrophobic cavities in the hydroxylase α-subunits of phenol hydroxylase (PHH), soluble methane monooxygenase (MMOH), and toluene/o-xylene monooxygenase (ToMOH) are also presented. The results, together with previous findings from crystallographic studies of xenon-pressurized sMMO hydroxylase, clearly identify the propensity for these cavities to bind hydrophobic gas molecules in the protein interior. This proposed functional role is supported by recent stopped flow kinetic studies of ToMOH variants [Song, W. J., et al. (2011) Proc. Natl. Acad. Sci. U.S.A.108, 14795–14800]. In addition to information about the Xe sites, the structure determination revealed significantly weakened binding of regulatory protein to the hydroxylase in comparison to that in the previously reported structure of PHH, as well as the presence of a newly identified metal-binding site in the α-subunit that adopts a linear coordination environment consistent with Cu(I), and a glycerol molecule bound to Fe1 in a fashion that is unique among hydrocarbon–diiron site adducts reported to date in BMM hydroxylase structures. Finally, a comparative analysis of the α-subunit structures of PHH, MMOH, and ToMOH details proposed routes for the other three BMM substrates, the hydrocarbon, electrons, and protons, comprising cavities, channels, hydrogen-bonding networks, and pores in the structures of their α-subunits.
Co-reporter:Loi H. Do, Stephen J. Lippard
Journal of Inorganic Biochemistry 2011 Volume 105(Issue 12) pp:1774-1785
Publication Date(Web):December 2011
DOI:10.1016/j.jinorgbio.2011.08.025
We present a comprehensive review of research conducted in our laboratory in pursuit of the long-term goal of reproducing the structures and reactivity of carboxylate-bridged diiron centers used in biology to activate dioxygen for the conversion of hydrocarbons to alcohols and related products. This article describes the evolution of strategies devised to achieve these goals and illustrates the challenges in getting there. Particular emphasis is placed on controlling the geometry and coordination environment of the diiron core, preventing formation of polynuclear iron clusters, maintaining the structural integrity of model complexes during reactions with dioxygen, and tuning the ligand framework to stabilize desired oxygenated diiron species. Studies of the various model systems have improved our understanding of the electronic and physical characteristics of carboxylate-bridged diiron units and their reactivity toward molecular oxygen and organic moieties. The principles and lessons that have emerged from these investigations will guide future efforts to develop more sophisticated diiron protein model complexes.Examples of synthetic analogs of carboxylate-bridged diiron cores in non-heme iron proteins that activate dioxygen for transport or hydrocarbon oxidation. A series of specialized ligands have been evolved to stabilize these units. Studies of the physical and chemical properties of the diiron complexes have enriched our understanding of their protein counterparts.
Co-reporter:Semi Park and Stephen J. Lippard
Biochemistry 2011 Volume 50(Issue 13) pp:
Publication Date(Web):February 28, 2011
DOI:10.1021/bi2000214
HMGB1, one of the most abundant nuclear proteins, has a strong binding affinity for cisplatin-modified DNA. It has been proposed that HMGB1 enhances the anticancer efficacy of cisplatin by shielding platinated DNA lesions from repair. Two cysteine residues in HMGB1 domain A form a reversible disulfide bond under mildly oxidizing conditions. The reduced domain A protein binds to a 25-bp DNA probe containing a central 1,2-d(GpG) intrastrand cross-link, the major platinum−DNA adduct, with a 10-fold greater binding affinity than the oxidized domain A. The binding affinities of singly and doubly mutated HMGB1 domain A, respectively deficient in one or both cysteine residues that form the disulfide bond, are unaffected by changes in external redox conditions. The redox-dependent nature of the binding of HMGB1 domain A to cisplatin-modified DNA suggests that formation of the intradomain disulfide bond induces a conformational change that disfavors binding to cisplatin-modified DNA. Hydroxyl radical footprinting analyses of wild-type domain A bound to platinated DNA under different redox conditions revealed identical cleavage patterns, implying that the asymmetric binding mode of the protein across from the platinated lesion is conserved irrespective of the redox state. The results of this study reveal that the cellular redox environment can influence the interaction of HMGB1 with the platinated DNA and suggest that the redox state of the A domain is a potential factor in regulating the role of the protein in modulating the activity of cisplatin as an anticancer drug.
Co-reporter:Dr. Nora Graf;Dr. Wee Han Ang;Dr. Guangyu Zhu;MyatNoeZin Myint ; Stephen J. Lippard
ChemBioChem 2011 Volume 12( Issue 7) pp:1115-1123
Publication Date(Web):
DOI:10.1002/cbic.201000724
Abstract
Resistance of tumor cells to platinum anticancer agents poses a major problem in cancer chemotherapy. One of the mechanisms associated with platinum-based drug resistance is the enhanced capacity of the cell to carry out nucleotide excision repair (NER) on platinum-damaged DNA. Endonucleases XPF and XPG are critical components of NER, responsible for excising the damaged DNA strand to remove the DNA lesion. Here, we investigated possible consequences of down-regulation of XPF and XPG gene expression in osteosarcoma cancer cells (U2OS) and the impact on cellular transcription and DNA repair. We further evaluated the sensitivity of such cells toward the platinum anticancer drugs cisplatin and oxaliplatin.
Co-reporter:Joel Rosenthal, Alexander B. Nepomnyashchii, Julia Kozhukh, Allen J. Bard, and Stephen J. Lippard
The Journal of Physical Chemistry C 2011 Volume 115(Issue 36) pp:17993-18001
Publication Date(Web):August 10, 2011
DOI:10.1021/jp204487r
Two new 2,2′-bipyridine (bpy)-based ligands with ancillary BODIPY chromophores attached at the 4- and 4′-positions were prepared and characterized, which vary in the substitution pattern about the BODIPY periphery by either excluding (BB1) or including (BB2) a β-alkyl substituent. Both absorb strongly throughout the visible region and are strongly emissive. The basic photophysics and electrochemical properties of BB1 and BB2 are comparable to those of the BODIPY monomers on which they are based. The solid-state structures and electronic structure calculations both indicate that there is negligible electronic communication between the BODIPY moieties and the intervening bpy spacers. Electrogenerated chemiluminescence spectra of the two bpy-BODIPY derivatives are similar to their recorded fluorescence profiles and are strongly influenced by substituents on the BODIPY chromophores. These 2,2′-bipyridine derivatives represent a new set of ligands that should find utility in applications, including light-harvesting, photocatalysis, and molecular electronics.
Co-reporter:Woon Ju Song;Grant Gucinski;Matthew H. Sazinsky
PNAS 2011 Volume 108 (Issue 36 ) pp:
Publication Date(Web):2011-09-06
DOI:10.1073/pnas.1106514108
For numerous enzymes reactive toward small gaseous compounds, growing evidence indicates that these substrates diffuse into
active site pockets through defined pathways in the protein matrix. Toluene/o-xylene monooxygenase hydroxylase is a dioxygen-activating enzyme. Structural analysis suggests two possible pathways for
dioxygen access through the α-subunit to the diiron center: a channel or a series of hydrophobic cavities. To distinguish
which is utilized as the O2 migration pathway, the dimensions of the cavities and the channel were independently varied by site-directed mutagenesis
and confirmed by X-ray crystallography. The rate constants for dioxygen access to the diiron center were derived from the
formation rates of a peroxodiiron(III) intermediate, generated upon treatment of the diiron(II) enzyme with O2. This reaction depends on the concentration of dioxygen to the first order. Altering the dimensions of the cavities, but
not the channel, changed the rate of dioxygen reactivity with the enzyme. These results strongly suggest that voids comprising
the cavities in toluene/o-xylene monooxygenase hydroxylase are not artifacts of protein packing/folding, but rather programmed routes for dioxygen
migration through the protein matrix. Because the cavities are not fully connected into the diiron active center in the enzyme
resting state, conformational changes will be required to facilitate dioxygen access to the diiron center. We propose that
such temporary opening and closing of the cavities may occur in all bacterial multicomponent monooxygenases to control O2 consumption for efficient catalysis. Our findings suggest that other gas-utilizing enzymes may employ similar structural
features to effect substrate passage through a protein matrix.
Co-reporter:Nagesh Kolishetti;Omid C. Farokhzad;Shanta Dhar
PNAS 2011 Volume 108 (Issue 5 ) pp:1850-1855
Publication Date(Web):2011-02-01
DOI:10.1073/pnas.1011379108
Targeted delivery and controlled release of inactive platinum (Pt) prodrugs may offer a new approach to improve the efficacy
and tolerability of the Pt family of drugs, which are used to treat 50% of all cancers today. Using prostate cancer (PCa)
as a model disease, we previously described the engineering of aptamer (Apt)-targeted poly(D,L-lactic-co-glycolic acid)-b-poly(ethylene glycol) (PLGA-b-PEG) nanoparticles (NPs) encapsulating a Pt(IV) prodrug c,t,c[Pt(NH3)2-(O2CCH2CH2CH2CH2CH3)2Cl2] (1) (Pt-PLGA-b-PEG-Apt-NP), which target the extracellular domain of the prostate specific membrane antigen (PSMA), for enhanced in vitro
cytotoxicity. Here we demonstrate enhanced in vivo pharmacokinetics (PK), biodistribution, tolerability, and efficacy of Pt-PLGA-b-PEG-Apt-NP (150±15 nm encapsulating ∼5% wt/wt Pt(IV) prodrug) when compared to cisplatin administered in its conventional
form in normal Sprague Dawley rats, Swiss Albino mice, and the PSMA-expressing LNCaP subcutaneous xenograft mouse model of
PCa, respectively. The 10-d maximum tolerated dose following a single i.v. injection of Pt-PLGA-b-PEG-NP in rats and mice was determined at 40 mg/kg and 5 mg/kg, respectively. PK studies with Pt-PLGA-b-PEG-NP revealed prolonged Pt persistence in systemic blood circulation and decreased accumulation of Pt in the kidneys, a
major target site of cisplatin toxicity. Pt-PLGA-b-PEG-Apt-NPs further displayed the significant dose-sparing characteristics of the drug, with equivalent antitumor efficacy
in LNCaP xenografts at 1/3 the dose of cisplatin administered in its conventional form (0.3 mg/kg vs. 1 mg/kg). When considering
the simultaneous improvement in tolerability and efficacy, the Pt-PLGA-b-PEG-Apt NP provides a remarkable improvement in the drug therapeutic index.
Co-reporter:Rui Cao ; Peter Müller
Journal of the American Chemical Society 2010 Volume 132(Issue 49) pp:17366-17369
Publication Date(Web):November 22, 2010
DOI:10.1021/ja108212v
Multidentate tripodal ligands, N(CH2-m-C6H4-CH2tacn)3 (L1) and N(CH2-o-C6H4-CH2N(CH2py)2)3 (L2), have been devised for assembling high-nuclearity metal clusters. By using the same tripodal platform with different ligand appendages, either triazacyclononanes or dipicolylamines, and functionalizing either the ortho or the meta positions on the tris(xylyl) linker arms, discrete trimetal phosphate units of relevance to phosphate-metabolizing trimetallic centers in biology were prepared. Four such compounds, [(CuIICl)3(HPO4)L1](PF6) (1), [(CuIICl)3(HAsO4)L1](PF6) (2), Na2[MnIII6MnII2(H2O)2(HPO4)6(PO4)4(L1)2] (3), and [CoII3(H2PO4)Cl2(MeCN)L2](PF6)3 (4), all containing three metal centers bound to a central phosphate or arsenate unit bridging oxygen atoms, have been synthesized and structurally characterized. These results demonstrate the propensity of this novel tripodal ligand platform, in the presence of phosphate or arsenate, to assemble {M3(EO4)} units and thus structurally mimic trimetallic active sites of proteins involved in phosphate metabolism. Reactivity studies reveal that the tricopper complex 1 is more efficient than monocopper analogues in catalyzing the hydrolysis of 4-nitrophenyl phosphate.
Co-reporter:Christine E. Tinberg ; Zachary J. Tonzetich ; Hongxin Wang ; Loi H. Do ; Yoshitaka Yoda ; Stephen P. Cramer
Journal of the American Chemical Society 2010 Volume 132(Issue 51) pp:18168-18176
Publication Date(Web):December 6, 2010
DOI:10.1021/ja106290p
Reactions of nitric oxide with cysteine-ligated iron−sulfur cluster proteins typically result in disassembly of the iron−sulfur core and formation of dinitrosyl iron complexes (DNICs). Here we report the first evidence that DNICs also form in the reaction of NO with Rieske-type [2Fe-2S] clusters. Upon treatment of a Rieske protein, component C of toluene/o-xylene monooxygenase from Pseudomonas sp. OX1, with an excess of NO(g) or NO-generators S-nitroso-N-acetyl-d,l-pencillamine and diethylamine NONOate, the absorbance bands of the [2Fe-2S] cluster are extinguished and replaced by a new feature that slowly grows in at 367 nm. Analysis of the reaction products by electron paramagnetic resonance, Mössbauer, and nuclear resonance vibrational spectroscopy reveals that the primary product of the reaction is a thiolate-bridged diiron tetranitrosyl species, [Fe2(μ-SCys)2(NO)4], having a Roussin’s red ester (RRE) formula, and that mononuclear DNICs account for only a minor fraction of nitrosylated iron. Reduction of this RRE reaction product with sodium dithionite produces the one-electron-reduced RRE, having absorptions at 640 and 960 nm. These results demonstrate that NO reacts readily with a Rieske center in a protein and suggest that dinuclear RRE species, not mononuclear DNICs, may be the primary iron dinitrosyl species responsible for the pathological and physiological effects of nitric oxide in such systems in biology.
Co-reporter:Woon Ju Song ; Michael S. McCormick ; Rachel K. Behan ; Matthew H. Sazinsky ; Wei Jiang ; Jeffery Lin ; Carsten Krebs
Journal of the American Chemical Society 2010 Volume 132(Issue 39) pp:13582-13585
Publication Date(Web):September 14, 2010
DOI:10.1021/ja1063795
Toluene/o-xylene monooxygenase hydroxylase (ToMOH), a diiron-containing enzyme, can activate dioxygen to oxidize aromatic substrates. To elucidate the role of a strictly conserved T201 residue during dioxygen activation of the enzyme, T201S, T201G, T201C, and T201V variants of ToMOH were prepared by site-directed mutagenesis. X-ray crystal structures of all the variants were obtained. Steady-state activity, regiospecificity, and single-turnover yields were also determined for the T201 mutants. Dioxygen activation by the reduced T201 variants was explored by stopped-flow UV−vis and Mössbauer spectroscopy. These studies demonstrate that the dioxygen activation mechanism is preserved in all T201 variants; however, both the formation and decay kinetics of a peroxodiiron(III) intermediate, T201peroxo, were greatly altered, revealing that T201 is critically involved in dioxygen activation. A comparison of the kinetics of O2 activation in the T201S, T201C, and T201G variants under various reaction conditions revealed that T201 plays a major role in proton transfer, which is required to generate the peroxodiiron(III) intermediate. A mechanism is postulated for dioxygen activation, and possible structures of oxygenated intermediates are discussed.
Co-reporter:Zachary J. Tonzetich ; Hongxin Wang ; Devrani Mitra ; Christine E. Tinberg ; Loi H. Do ; Francis E. Jenney ; Jr.; Michael W. W. Adams ; Stephen P. Cramer
Journal of the American Chemical Society 2010 Volume 132(Issue 20) pp:6914-6916
Publication Date(Web):April 29, 2010
DOI:10.1021/ja101002f
We have applied 57Fe nuclear resonance vibrational spectroscopy (NRVS) to identify protein-bound dinitrosyl iron complexes. Intense NRVS peaks due to vibrations of the N−Fe−N unit can be observed between 500 and 700 cm−1 and are diagnostic indicators of the type of iron dinitrosyl species present. NRVS spectra for four iron dinitrosyl model compounds are presented and used as benchmarks for the identification of species formed in the reaction of Pyrococcus furiosus ferredoxin D14C with nitric oxide.
Co-reporter:Wee Han Ang ; MyatNoeZin Myint
Journal of the American Chemical Society 2010 Volume 132(Issue 21) pp:7429-7435
Publication Date(Web):May 5, 2010
DOI:10.1021/ja101495v
We have investigated the processing of site-specific Pt−DNA cross-links in live mammalian cells to enhance our understanding of the mechanism of action of platinum-based anticancer drugs. The activity of platinum drugs against cancer is mediated by a combination of processes including cell entry, drug activation, DNA-binding, and transcription inhibition. These drugs bind nuclear DNA to form Pt−DNA cross-links, which arrest key cellular functions, including transcription, and trigger a variety of responses, such as repair. Mechanistic investigations into the processing of specific Pt−DNA cross-links are critical for understanding the effects of platinum−DNA damage, but conventional in vitro techniques do not adequately account for the complex and intricate environment within a live cell. With this limitation in mind, we developed a strategy to study platinum cross-links on plasmid DNAs transfected into live mammalian cells based on luciferase reporter vectors containing defined platinum−DNA lesions that are either globally or site-specifically incorporated. Using cells with either competent or deficient nucleotide excision repair systems, we demonstrate that Pt−DNA cross-links impede transcription by blocking passage of the RNA polymerase complex and that nucleotide excision repair can remove the block and restore transcription. Results are presented for ∼3800-base pair plasmids that are either globally platinated or carry a single 1,2-d(GpG) or 1,3-d(GpTpG) intrastrand cross-link formed by either cis-{Pt(NH3)2}2+ or cis-{Pt(R,R-dach)}2+, where {Pt(NH3)2}2+ is the platinum unit conveyed by cisplatin and carboplatin and R,R-dach is the oxaliplatin ligand, R,R-1,2-diaminocyclohexane.
Co-reporter:Joel Rosenthal
Journal of the American Chemical Society 2010 Volume 132(Issue 16) pp:5536-5537
Publication Date(Web):March 31, 2010
DOI:10.1021/ja909148v
The synthesis, photophysical properties, and a biological application of BODIPY-triazole 1 (BOT1) are described. BOT1 juxtaposes a BODIPY fluorophore with a tripodal ligand platform via a triazole bridge. The triazole linker is afforded by azide−alkyne “click” chemistry and comprises the third arm of the tripodal architecture. BOT1 binds Cu2+ in aqueous solution to form a CuII[BOT1] complex in which emission from the BODIPY moiety is efficiently quenched. This complex exhibits a prompt turn-on response when exposed to nitroxyl, and this emission response is specific to HNO over other reactive nitrogen and oxygen species. Notably, CuII[BOT1] does not fluoresce in the presence of nitric oxide, making this system the first discrete molecular probe capable of detecting HNO over NO under physiologically relevant conditions. Fluorescence microscopy experiments establish that CuII[BOT1] is membrane-permeable and can successfully signal the presence of HNO in live cells.
Co-reporter:Loi H. Do ; Takahiro Hayashi ; Pierre Moënne-Loccoz
Journal of the American Chemical Society 2010 Volume 132(Issue 4) pp:1273-1275
Publication Date(Web):January 7, 2010
DOI:10.1021/ja909718f
Addition of H+ to a synthetic (μ-1,2-peroxo)diiron(III) model complex results in protonation of a carboxylate rather than the peroxo ligand. This conclusion is based on spectroscopic evidence from UV−vis, 57Fe Mössbauer, resonance Raman, infrared, and 1H/19F NMR studies. These results suggest a similar role for protons in the dioxygen activation reactions in soluble methane monooxygenase and related carboxylate-bridged diiron enzymes.
Co-reporter:Michael D. Pluth, Lindsey E. McQuade and Stephen J. Lippard
Organic Letters 2010 Volume 12(Issue 10) pp:2318-2321
Publication Date(Web):April 20, 2010
DOI:10.1021/ol1006289
Two new cell-trappable fluorescent probes for nitric oxide (NO) are reported based on either incorporation of hydrolyzable esters or conjugation to aminodextran polymers. Both probes are highly selective for NO over other reactive oxygen and nitrogen species (RONS). The efficacy of these probes for the fluorescence imaging of nitric oxide produced endogenously in Raw 264.7 cells is demonstrated.
Co-reporter:Youngmin You, Elisa Tomat, Kevin Hwang, Tatjana Atanasijevic, Wonwoo Nam, Alan P. Jasanoff and Stephen J. Lippard
Chemical Communications 2010 vol. 46(Issue 23) pp:4139-4141
Publication Date(Web):10 May 2010
DOI:10.1039/C0CC00179A
A paramagnetic manganese complex of a fluorescein-based probe affords a dual-modality zinc sensor featuring an improved fluorescence dynamic range and an MRI readout.
Co-reporter:Ryan C. Todd, Stephen J. Lippard
Chemistry & Biology 2010 Volume 17(Issue 12) pp:1334-1343
Publication Date(Web):22 December 2010
DOI:10.1016/j.chembiol.2010.10.018
The effects of cisplatin binding to DNA were explored at the nucleosome level to incorporate key features of the eukaryotic nuclear environment. An X-ray crystal structure of a site-specifically platinated nucleosome carrying a 1,3-cis-{Pt(NH3)2}2+-d(GpTpG) intrastrand cross-link reveals the details of how this adduct dictates the rotational positioning of DNA in the nucleosome. Results from in vitro nucleosome mobility assays indicate that a single platinum adduct interferes with ATP-independent sliding of DNA around the octamer core. Data from in vitro transcription experiments suggest that RNA polymerases can successfully navigate along cisplatin-damaged DNA templates that contain nucleosomes, but stall when the transcription elongation complex physically contacts a platinum cross-link located on the template strand. These results provide information about the effects of cisplatin binding to nuclear DNA and enhance our understanding of the mechanism of transcription inhibition by platinum anticancer compounds.Highlights► The first X-ray crystal structure of a cisplatin–DNA adduct at the level of the nucleosome ► The cisplatin 1,3-d(GTG) intrastrand cross-link directs rotational phasing of DNA in nucleosomes ► Pt–DNA adducts retard ATP-independent nucleosome mobility ► Transcription of cisplatin-damaged nucleosomes is blocked at Pt sites on the DNA template
Co-reporter:Taekwan Lee, Xiao-an Zhang, Shanta Dhar, Henryk Faas, Stephen J. Lippard, Alan Jasanoff
Chemistry & Biology 2010 Volume 17(Issue 6) pp:665-673
Publication Date(Web):25 June 2010
DOI:10.1016/j.chembiol.2010.05.009
Magnetic resonance imaging (MRI) with molecular probes offers the potential to monitor physiological parameters with comparatively high spatial and temporal resolution in living subjects. For detection of intracellular analytes, construction of cell-permeable imaging agents remains a challenge. Here we show that a porphyrin-based MRI molecular imaging agent, Mn-(DPA-C2)2-TPPS3, effectively penetrates cells and persistently stains living brain tissue in intracranially injected rats. Chromogenicity of the probe permitted direct visualization of its distribution by histology, in addition to MRI. Distribution was concentrated in cell bodies after hippocampal infusion. Mn-(DPA-C2)2-TPPS3 was designed to sense zinc ions, and contrast enhancement was more pronounced in the hippocampus, a zinc-rich brain region, than in the caudate nucleus, which contains relatively little labile Zn2+. Membrane permeability, optical activity, and high relaxivity of porphyrin-based contrast agents offer exceptional functionality for in vivo imaging.Graphical AbstractFigure optionsDownload full-size imageDownload high-quality image (361 K)Download as PowerPoint slideHighlights► An MRI contrast agent designed for cell permeability and zinc detection labels living brain tissue ►Multimodal imaging and elemental analysis show that injected probe is cytosolically localized ►A brain region with high levels of labile zinc stains more efficiently than a zinc-poor region
Co-reporter:Lindsey E. McQuade
Inorganic Chemistry 2010 Volume 49(Issue 16) pp:7464-7471
Publication Date(Web):July 19, 2010
DOI:10.1021/ic100802q
A series of symmetrical, fluorescein-derived ligands appended with two derivatized 2-methyl-8-aminoquinolines were prepared and spectroscopically characterized. The ligands FL2, FL2E, and FL2A were designed to improve the dynamic range of previously described asymmetric systems, and the copper complex Cu2(FL2E) was constructed as a trappable NO probe that is hydrolyzed intracellularly to form Cu2(FL2A). The ligands themselves are only weakly emissive, and the completely quenched Cu(II) complexes, generated in situ by combining each ligand with 2 equiv of CuCl2, were investigated as fluorescent probes for nitric oxide. Upon introduction of excess NO under anaerobic conditions to buffered solutions of Cu2(FL2), Cu2(FL2E), and Cu2(FL2A), the fluorescence increased by factors of 23 ± 3, 17 ± 2, and 27 ± 3, respectively. The corresponding rate constants for fluorescence turn-on were determined to be 0.4 ± 0.2, 0.35 ± 0.05, and 0.6 ± 0.1 min−1. The probes are highly specific for NO over other biologically relevant reactive oxygen and nitrogen species, as well as Zn(II), the metal ion for which similar probes were designed to detect.
Co-reporter:Espen Tangen ; Jeanet Conradie ; Katherine Franz ; Simone Friedle ; Joshua Telser ; Stephen J. Lippard ;Abhik Ghosh
Inorganic Chemistry 2010 Volume 49(Issue 6) pp:2701-2705
Publication Date(Web):February 18, 2010
DOI:10.1021/ic901860x
Using density functional theory (OLYP/STO-TZP) calculations, we have investigated the electronic structure of [Mn(5,5-tropocoronand)(NO)], a rare paramagnetic {MNO}6 complex. Experimental methods, including magnetic susceptibility measurements and high-field electron paramagnetic resonance spectroscopy, have not provided an unambiguous spin state assignment for this complex. In other respects, however, the compound was fully characterized, including by means of single-crystal X-ray structure determination. The optimized S = 1 OLYP geometry reproduced all key aspects of the trigonal-bipyramidal molecular structure, including a short Mn−N(O) distance (∼1.7 Å) and an essentially linear MnNO angle. In contrast, the S = 0 and S = 2 optimized structures disagreed with the crystal structure in critical respects. Moreover, three different exchange-correlation functionals (OLYP, B3LYP, and B3LYP*) indicated an S = 1 ground state by a clear margin of energy. An examination of the Kohn−Sham MOs of this state indicated a primarily dxz2dyz2dxy1dx2−z21 electronic configuration, where the z axis is identified with the nearly linear MnNO axis. The dy2 orbital is formally unoccupied in this state, interacting, as it does, head-on with two tropocoronand nitrogens lying along the y axis, the pseudo-3-fold axis of the trigonal bipyramid. The doubly occupied dxz and dyz orbitals are in actuality dπ(Fe)−π*(NO)-based π-bonding molecular orbitals, the α and β “components” of which are significantly offset spatially. This offset results in excess minority spin density on the NO unit. Thus, the OLYP/TZP atomic spin populations are Mn, 2.85; N(O), −0.52; and O, −0.35.
Co-reporter:Tatjana Atanasijevic ; Xiao-an Zhang ; Stephen J. Lippard ;Alan Jasanoff
Inorganic Chemistry 2010 Volume 49(Issue 6) pp:2589-2591
Publication Date(Web):February 8, 2010
DOI:10.1021/ic100150e
We introduce a mechanism for ion sensing by MRI in which analytes compete with paramagnetic ions for binding to polydentate chelating agents. Displacement of the paramagnetic ions results in alteration of solvent interaction parameters and consequent changes in relaxivity and MRI contrast. The MRI changes can be tuned by the choice of chelator. As an example, we show that calcium-dependent displacement of Mn2+ ions bound to EGTA and BAPTA results in a T1-weighted MRI signal increase, whereas displacement from calmodulin results in a signal decrease. The changes are ion selective and can be explained using relaxivity theory. The ratio of T2 to T1 relaxivity is also calcium-dependent, indicating the feasibility of “ratiometric” analyte detection, independent of the probe concentration. Measurement of paramagnetic ion displacement effects could be used to determine analyte ion concentrations with spatial resolution in opaque specimens.
Co-reporter:Justin J. Wilson, Juliana Fedoce Lopes and Stephen J. Lippard
Inorganic Chemistry 2010 Volume 49(Issue 11) pp:5303-5315
Publication Date(Web):April 27, 2010
DOI:10.1021/ic100411p
Three new ligands of the general formula [RNHCH(py)2] (py = pyridine; R = tosyl, Ts-dpm; R = dansyl, Ds-dpm; R = 7-nitro-1,2,3-benzoxadiazole, NBD-dpm) have been synthesized and characterized. Reactions of these ligands with cis-[Pt(DMSO)2Cl2] (DMSO = dimethyl sulfoxide) in methanol affords [Pt(Ts-dpm)Cl2] (1), [Pt(Ds-dpm)Cl2] (2), and [Pt(NBD-dpm)Cl2] (3). The crystal structures of these complexes reveal bidentate coordination of the ligands to the Pt center with nonplanar chelate rings. Because of inequivalent substituents on the methine carbon atom of the ligands, distinct exo and endo isomers exist in the three complexes. X-ray analyses indicate that 1 crystallizes in the endo conformation, 2 in the exo conformation, and 3 as a mixture of the two conformers. The 1H NMR and 195Pt NMR spectra of the complexes display two sets of independent signals corresponding to the chemically inequivalent exo and endo conformers. The exo conformer was determined by 2D NMR spectroscopy to be thermodynamically favored for all three complexes. Density functional theory (DFT), time-dependent DFT, and atoms in molecules calculations were carried out for both conformers of 3 to investigate differences in their electronic structures and to explore intramolecular interactions. In the presence of dioxygen, 1 thermally decomposes at 60 °C to form several unidentified products. Compound 2 is thermally stable even in the presence of dioxygen and water but upon light exposure decomposes to form a new platinum(II) species with a 195Pt NMR shift of −2177 ppm. Compound 3 reacts both thermally and photochemically in the presence of dioxygen and trace amounts of water to form both 4-amino-7-nitro-2,1,3-benzoxadiazole and [Pt(dpk)Cl2] (dpk = di-2-pyridyl ketone). Oxidation of 1 and 3 with H2O2 in acetic acid affords a mixture of compounds, two of which contain dpm ligands bound in a tridentate manner to platinum.
Co-reporter:Zachary J. Tonzetich ; Lindsey E. McQuade
Inorganic Chemistry 2010 Volume 49(Issue 14) pp:6338-6348
Publication Date(Web):July 12, 2010
DOI:10.1021/ic9022757
We are pursuing a dual strategy for investigating the chemistry of nitric oxide as a biological signaling agent. In one approach, metal-based fluorescent sensors for the detection of NO in living cells are evaluated, and a sensor based on a copper fluorescein complex has proved to be a valuable lead compound. Sensors of this class permit identification of NO from both inducible and constitutive forms of nitric oxide synthase and facilitate investigation of different NO functions in response to external stimuli. In the other approach, we employ synthetic model complexes of iron−sulfur clusters to probe their reactivity toward nitric oxide as biomimics of the active sites of iron−sulfur proteins. Our studies reveal that NO disassembles the Fe−S clusters to form dinitrosyl iron complexes.
Co-reporter:Lindsey E. McQuade, Michael D. Pluth and Stephen J. Lippard
Inorganic Chemistry 2010 Volume 49(Issue 17) pp:8025-8033
Publication Date(Web):August 12, 2010
DOI:10.1021/ic101054u
The mechanism of the reaction of CuFL1 (FL1 = 2-{2-chloro-6-hydroxy-5-[(2-methylquinolin-8-ylamino)methyl]-3-oxo-3H-xanthen-9-yl}benzoic acid) with nitric oxide (NO) to form the N-nitrosated product FL1-NO in buffered aqueous solutions was investigated. The reaction is first-order in [CuFL1], [NO], and [OH−]. The observed rate saturation at high base concentrations is consistent with a mechanism in which the protonation state of the secondary amine of the ligand is important for reactivity. This information provides a rationale for designing faster-reacting probes by lowering the pKa of the secondary amine. Activation parameters for the reaction of CuFL1 with NO indicate an associative mechanism (ΔS‡ = −120 ± 10 J/mol·K) with a modest thermal barrier (ΔH⧧ = 41 ± 2 kJ/mol; Ea = 43 ± 2 kJ/mol). Variable-pH electron paramagnetic resonance experiments reveal that, as the secondary amine of CuFL1 is deprotonated, electron density shifts to yield a new spin-active species having electron density localized on the deprotonated amine nitrogen atom. This result suggests that FL1-NO formation occurs when NO attacks the deprotonated secondary amine of the coordinated ligand, followed by inner-sphere electron transfer to Cu(II) to form Cu(I) and release of FL1-NO from the metal.
Co-reporter:Lindsey E. McQuade
Inorganic Chemistry 2010 Volume 49(Issue 20) pp:9535-9545
Publication Date(Web):September 17, 2010
DOI:10.1021/ic1012507
The synthesis and spectroscopic characterization of two new, cell-trappable fluorescent probes for Zn(II) are presented. These probes, 2-(4,5-bis(((6-(2-ethoxy-2-oxoethoxy)quinolin-8-yl)amino)methyl)-6-hydroxy-3-oxo-3H-8 xanthen-9-yl)benzoic acid (QZ2E) and 2,2′-((8,8′-(((9-(2-carboxyphenyl)-6-hydroxy-3-oxo-3H-xanthene-4,5-diyl)bis(methylene))bis(azanediyl))bis(quinoline-8,6-diyl))bis(oxy))diacetic acid (QZ2A), are poorly emissive in the off-state but exhibit dramatic increases in fluorescence upon Zn(II) binding (120 ± 10-fold for QZ2E, 30 ± 7-fold for QZ2A). This binding is selective for Zn(II) over other biologically relevant metal cations, toxic heavy metals, and most first-row transition metals and is of appropriate affinity (Kd1(QZ2E) = 150 ± 100 μM, Kd2(QZ2E) = 3.5 ± 0.1 mM, Kd1(QZ2A) = 220 ± 30 μM, Kd2(QZ2A) = 160 ± 80 μM, Kd3(QZ2A) = 9 ± 6 μM) to reversibly bind Zn(II) at physiological levels. In live cells, QZ2E localizes to the Gogli apparatus where it can detect Zn(II). It is cell-membrane-permeable until cleavage of its ester groups by intracellular esterases produces QZ2A, a negatively charged acid form that cannot cross the cell membrane.
Co-reporter:Elisa Tomat
Inorganic Chemistry 2010 Volume 49(Issue 20) pp:9113-9115
Publication Date(Web):September 22, 2010
DOI:10.1021/ic101513a
A xanthene-forming condensation reaction yields rhodol and rhodamine dyes carrying a zinc-binding ligand that includes the aniline-type nitrogen donor of the fluorophores. Upon zinc coordination in neutral aqueous solution, rhodol RF3 behaves as a ratiometric sensor, and rhodamine RA1 acts as a turn-off intensity-based indicator. Both fluorescent compounds bind the divalent zinc cation with micromolar affinity.
Co-reporter:Pingwu Du and Stephen J. Lippard
Inorganic Chemistry 2010 Volume 49(Issue 23) pp:10753-10755
Publication Date(Web):October 28, 2010
DOI:10.1021/ic101569a
We describe ZRL1, a turn-on colorimetric and red fluorescent zinc ion sensor. The Zn2+-promoted ring opening of the rhodamine spirolactam ring in ZRL1 evokes a 220-fold fluorescence turn-on response. In aqueous media, ZRL1 turn-on luminescence is highly selective for Zn2+ ions, with no significant response to other competitive cations, including Na+, K+, Ca2+, Mg2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Cd2+, or Hg2+. In addition to these characteristics, preliminary results indicate that ZRL1 can be delivered to living cells and can be used to monitor changes in intracellular Zn2+ levels.
Co-reporter:Rachel K. Behan and Stephen J. Lippard
Biochemistry 2010 Volume 49(Issue 45) pp:
Publication Date(Web):October 5, 2010
DOI:10.1021/bi101475z
The aging-associated enzyme CLK-1 is proposed to be a member of the carboxylate-bridged diiron family of proteins. To evaluate this hypothesis and characterize the protein, we expressed soluble mouse CLK-1 (MCLK1) in Escherichia coli as a heterologous host. Using Mössbauer and EPR spectroscopy, we established that MCLK1 indeed belongs to this protein family. Biochemical analyses of the in vitro activity of MCLK1 with quinone substrates revealed that NADH can serve directly as a reductant for catalytic activation of dioxygen and substrate oxidation by the enzyme, with no requirement for an additional reductase protein component. The direct reaction of NADH with a diiron-containing oxidase enzyme has not previously been encountered for any member of the protein superfamily.
Co-reporter:Ryan C. Todd, Stephen J. Lippard
Journal of Inorganic Biochemistry 2010 Volume 104(Issue 9) pp:902-908
Publication Date(Web):September 2010
DOI:10.1016/j.jinorgbio.2010.04.005
We report the 1.77-Å resolution X-ray crystal structure of a dodecamer DNA duplex with the sequence 5′-CCTCTGGTCTCC-3′ that has been modified to contain a single engineered 1,2-cis-{Pt(NH3)2}2+-d(GpG) cross-link, the major DNA adduct of cisplatin. These data represent a significant improvement in resolution over the previously published 2.6-Å structure. The ammine ligands in this structure are clearly resolved, leading to improved visualization of the cross-link geometry with respect to both the platinum center and to the nucleobases, which adopt a higher energy conformation. Also better resolved are the deoxyribose sugar puckers, which allow us to re-examine the global structure of platinum-modified DNA. Another new feature of this model is the location of four octahedral [Mg(H2O)6]2+ ions associated with bases in the DNA major groove and the identification of 124 ordered water molecules that participate in hydrogen-bonding interactions with either the nucleic acid or the diammineplatinum(II) moiety.The structure of a cisplatin-modified DNA dodecamer duplex containing a 1,2-cis-{Pt(NH3)2}2+-d(GpG) cross-link has been determined at 1.77-Å resolution. Improved electron density reveals new features not seen in the originally published 2.6 Å structure.
Co-reporter:Christine E. Tinberg and Stephen J. Lippard
Biochemistry 2010 Volume 49(Issue 36) pp:
Publication Date(Web):August 3, 2010
DOI:10.1021/bi1009375
Soluble methane monooxygenase is a bacterial enzyme that converts methane to methanol at a carboxylate-bridged diiron center with exquisite control. Because the oxidizing power required for this transformation is demanding, it is not surprising that the enzyme is also capable of hydroxylating and epoxidizing a broad range of hydrocarbon substrates in addition to methane. In this work we took advantage of this promiscuity of the enzyme to gain insight into the mechanisms of action of Hperoxo and Q, two oxidants that are generated sequentially during the reaction of reduced protein with O2. Using double-mixing stopped-flow spectroscopy, we investigated the reactions of the two intermediate species with a panel of substrates of varying C−H bond strength. Three classes of substrates were identified according to the rate-determining step in the reaction. We show for the first time that an inverse trend exists between the rate constant of reaction with Hperoxo and the C−H bond strength of the hydrocarbon examined for those substrates in which C−H bond activation is rate-determining. Deuterium kinetic isotope effects revealed that reactions performed by Q, but probably not Hperoxo, involve extensive quantum mechanical tunneling. This difference sheds light on the observation that Hperoxo is not a sufficiently potent oxidant to hydroxylate methane, whereas Q can perform this reaction in a facile manner. In addition, the reaction of Hperoxo with acetonitrile appears to proceed by a distinct mechanism in which a cyanomethide anionic intermediate is generated, bolstering the argument that Hperoxo is an electrophilic oxidant that operates via two-electron transfer chemistry.
Co-reporter:Guangyu Zhu, Paul Chang and Stephen J. Lippard
Biochemistry 2010 Volume 49(Issue 29) pp:
Publication Date(Web):June 15, 2010
DOI:10.1021/bi100775t
Poly(ADP-ribose) polymerase-1 (PARP-1) was recently identified as a platinum−DNA damage response protein. To investigate the properties of binding of PARP-1 to different platinum−DNA adducts in greater detail, biotinylated DNA probes containing a site-specific cisplatin 1,2-d(GpG) or 1,3-d(GpTpG) intrastrand cross-link or a cisplatin 5′-GC/5′-GC interstrand cross-link (ICL) were utilized in binding assays with cell-free extracts (CFEs) in vitro. The activated state of PARP-1 was generated by treatment of cells with a DNA-damaging agent or by addition of NAD+ to CFEs. PARP-1 binds with a higher affinity to cisplatin-damaged DNA than to undamaged DNA, and the amount of protein that binds to the most common cisplatin−DNA cross-link, 1,2-d(GpG), is greater than the amount that binds to other types of cisplatin−DNA cross-links. Both DNA damage-activated PARP-1 and unactivated PARP-1 bind to cisplatin-damaged DNA, and both automodified PARP-1 and cleaved PARP-1 bind to cisplatin−DNA lesions. The role of poly(ADP-ribose) (pADPr) in mediating binding of PARP-1 to platinum damage was further investigated. The extent of binding of PARP-1 to the cisplatin 1,2-d(GpG) cross-link decreases upon automodification, and overactivated PARP-1 loses its affinity for the cross-link. Elimination of pADPr facilitates binding of PARP-1 to the cisplatin 1,2-d(GpG) cross-link. PARP-1 also binds to DNA damaged by other platinum compounds, including oxaliplatin and pyriplatin, indicating protein affinity for the damage in an adduct-specific manner rather than recognition of distorted DNA. Our results reveal the unique binding properties for binding of PARP-1 to platinum−DNA damage, providing insights into, and a better understanding of, the cellular response to platinum-based anticancer drugs.
Co-reporter:Jie Ma;Graeme Lowe;Lindsey E. McQuade;Ambarish Ghatpande;Alan Gelperin
PNAS 2010 Volume 107 (Issue 19 ) pp:8525-8530
Publication Date(Web):2010-05-11
DOI:10.1073/pnas.0914794107
We report the visualization of NO production using fluorescence in tissue slices of the mouse main olfactory bulb. This discovery
was possible through the use of a novel, cell-trappable probe for intracellular nitric oxide detection based on a symmetric
scaffold with two NO-reactive sites. Ester moieties installed onto the fluorescent probe are cleaved by intracellular esterases
to yield the corresponding negatively charged, cell-impermeable acids. The trappable probe Cu2(FL2E) and the membrane-impermeable acid derivative Cu2(FL2A) respond rapidly and selectively to NO in buffers that simulate biological conditions, and application of Cu2(FL2E) leads to detection of endogenously produced NO in cell cultures and olfactory bulb brain slices.
Co-reporter:Dong Wang;Guangyu Zhu;Xuhui Huang
PNAS 2010 107 (21 ) pp:9584-9589
Publication Date(Web):2010-05-25
DOI:10.1073/pnas.1002565107
DNA is a major target of anticancer drugs. The resulting adducts interfere with key cellular processes, such as transcription,
to trigger downstream events responsible for drug activity. cis-Diammine(pyridine)chloroplatinum(II), cDPCP or pyriplatin, is a monofunctional platinum(II) analogue of the widely used anticancer
drug cisplatin having significant anticancer properties with a different spectrum of activity. Its novel structure-activity
properties hold promise for overcoming drug resistance and improving the spectrum of treatable cancers over those responsive
to cisplatin. However, the detailed molecular mechanism by which cells process DNA modified by pyriplatin and related monofunctional
complexes is not at all understood. Here we report the structure of a transcribing RNA polymerase II (pol II) complex stalled
at a site-specific monofunctional pyriplatin-DNA adduct in the active site. The results reveal a molecular mechanism of pol
II transcription inhibition and drug action that is dramatically different from transcription inhibition by cisplatin and
UV-induced 1,2-intrastrand cross-links. Our findings provide insight into structure-activity relationships that may apply
to the entire family of monofunctional DNA-damaging agents and pave the way for rational improvement of monofunctional platinum
anticancer drugs.
Co-reporter:Pedro M. Valencia;Nagesh Kolishetti;Robert Langer;Lucy Q. Lin;Shanta Dhar;Rohit Karnik;Omid C. Farokhzad
PNAS 2010 Volume 107 (Issue 42 ) pp:17939-17944
Publication Date(Web):2010-10-19
DOI:10.1073/pnas.1011368107
The genomic revolution has identified therapeutic targets for a plethora of diseases, creating a need to develop robust technologies
for combination drug therapy. In the present work, we describe a self-assembled polymeric nanoparticle (NP) platform to target
and control precisely the codelivery of drugs with varying physicochemical properties to cancer cells. As proof of concept,
we codelivered cisplatin and docetaxel (Dtxl) to prostate cancer cells with synergistic cytotoxicity. A polylactide (PLA)
derivative with pendant hydroxyl groups was prepared and conjugated to a platinum(IV) [Pt(IV)] prodrug, c,t,c-[Pt(NH3)2(O2CCH2CH2COOH)(OH)Cl2] [PLA-Pt(IV)]. A blend of PLA-Pt(IV) functionalized polymer and carboxyl-terminated poly(d,l-lactic-co-glycolic acid)-block-poly(ethylene glycol) copolymer in the presence or absence of Dtxl, was converted, in microfluidic channels, to NPs with
a diameter of ∼100 nm. This process resulted in excellent encapsulation efficiency (EE) and high loading of both hydrophilic
platinum prodrug and hydrophobic Dtxl with reproducible EEs and loadings. The surface of the NPs was derivatized with the
A10 aptamer, which binds to the prostate-specific membrane antigen (PSMA) on prostate cancer cells. These NPs undergo controlled
release of both drugs over a period of 48–72 h. Targeted NPs were internalized by the PSMA-expressing LNCaP cells via endocytosis,
and formation of cisplatin 1,2-d(GpG) intrastrand cross-links on nuclear DNA was verified. In vitro toxicities demonstrated
superiority of the targeted dual-drug combination NPs over NPs with single drug or nontargeted NPs. This work reveals the
potential of a single, programmable nanoparticle to blend and deliver a combination of drugs for cancer treatment.
Co-reporter:Elizabeth M. Nolan and Stephen J. Lippard
Accounts of Chemical Research 2009 Volume 42(Issue 1) pp:193
Publication Date(Web):November 7, 2008
DOI:10.1021/ar8001409
The metalloneurochemistry of Zn(II) is of substantial current interest. Zinc is the second most abundant d-block metal ion in the human brain, and its distribution varies with relatively high concentrations found in the hippocampus. Brain zinc is generally divided into two types, protein-bound and loosely bound, the latter also being termed histochemically observable, chelatable, labile, or mobile zinc. The neurophysiological and neuropathological significance of mobile Zn(II) remains enigmatic. Studies of Zn(II) distribution, translocation, and function in vivo require tools for its detection. Because Zn(II) has a closed-shell d10 configuration and no convenient spectroscopic signature, fluorescence is a well-suited method for monitoring Zn(II) in biological contexts. This Account summarizes work by our laboratory addressing the design, preparation, characterization, and use of small-molecule fluorescent sensors for imaging Zn(II) in living cells and samples of brain tissue. These sensors provide “turn-on” or ratiometric Zn(II) detection in aqueous solution at neutral pH. By making alterations to the Zn(II)-binding unit and fluorophore platform, we have devised sensors with varied photophysical and metal-binding properties. Several of these probes have been applied to image Zn(II) distribution, uptake, and mobilization in a variety of cell types, including neuronal cultures.
Co-reporter:Zachary J. Tonzetich ; Loi H. Do
Journal of the American Chemical Society 2009 Volume 131(Issue 23) pp:7964-7965
Publication Date(Web):May 21, 2009
DOI:10.1021/ja9030159
Reaction of the Rieske cluster model complex (Et4N)2[(N2CHPh)Fe2S2(S2-o-xyl)] (N2CHPh = dianion of 2,2′-(phenylmethylene)bis(3-methylindole); S2-o-xyl = dianion of 1,2-phenylenedimethanethiol) with nitric oxide results in disassembly of the iron−sulfur core and formation of {Fe(NO)2}9 dinitrosyliron complexes (DNICs). Isolation and characterization of these DNICs, including the new compound, (Et4N)[(N2CHPh)Fe(NO)2], demonstrates a homology between the synthetic Riekse cluster and purely thiolate-bound Fe2S2 clusters in reactions involving NO. To model the nitrogen-rich environment of Rieske cluster-derived dinitroysliron species, a new type of neutral {Fe(NO)2}9 DNIC was prepared containing a β-diketiminate ligand. One-electron reduction of this compound affords the isolable {Fe(NO)2}10 DNIC. These compounds represent a rare example of structurally analogous DNIC redox partners.
Co-reporter:Woon Ju Song ; Rachel K. Behan ; Sunil G. Naik ; Boi Hanh Huynh
Journal of the American Chemical Society 2009 Volume 131(Issue 17) pp:6074-6075
Publication Date(Web):April 8, 2009
DOI:10.1021/ja9011782
We report the observation of a novel intermediate in the reaction of a reduced toluene/o-xylene monooxygenase hydroxylase (ToMOHred) T201S variant, in the presence of a regulatory protein (ToMOD), with dioxygen. This species is the first oxygenated intermediate with an optical band in any toluene monooxygenase. The UV−vis and Mössbauer spectroscopic properties of the intermediate allow us to assign it as a peroxodiiron(III) species, T201Speroxo, similar to Hperoxo in methane monooxygenase. Although T201S generates T201Speroxo in addition to optically transparent ToMOHperoxo, previously observed in wild-type ToMOH, this conservative variant is catalytically active in steady-state catalysis and single-turnover experiments and displays the same regiospecificity for toluene and slightly different regiospecificity for o-xylene oxidation.
Co-reporter:Brian A. Wong ; Simone Friedle
Journal of the American Chemical Society 2009 Volume 131(Issue 20) pp:7142-7152
Publication Date(Web):April 30, 2009
DOI:10.1021/ja900980u
The mechanism by which dipicolylamine (DPA) chelate-appended fluorophores respond to zinc was investigated by the synthesis and study of five new analogues of the 2′,7′-dichlorofluorescein-based Zn2+ sensor Zinpyr-1 (ZP1). With the use of absorption and emission spectroscopy in combination with potentiometric titrations, a detailed molecular picture has emerged of the Zn2+ and H+ binding properties of the ZP1 family of sensors. The two separate N3O donor atom sets on ZP1 converge to form binding pockets in which all four heteroatoms participate in coordination to either Zn2+ or protons. The position of the pyridyl group nitrogen atom, 2-pyridyl or 4-pyridyl, has a large impact on the fluorescence response of the dyes to protons despite relatively small changes in pKa values. The fluorescence quenching effects of such multifunctional electron-donating units are often taken as a whole. Despite the structural complexity of ZP1, however, we provide evidence that the pyridyl arms of the DPA appendages participate in the quenching process, in addition to the contribution from the tertiary nitrogen amine atom. Potentiometric titrations reveal ZP1 dissociation constants (Kd) for Zn2+ of 0.04 pM and 1.2 nM for binding to the first and second binding pockets of the ligand, respectively, the second of which correlates with the value observed by fluorescence titration. This result demonstrates that both binding pockets of this symmetric, ditopic sensor need to be occupied in order for full fluorescence turn-on to be achieved. These results have significant implications for the design and implementation of fluorescent sensors for studies of mobile zinc ions in biology.
Co-reporter:Simone Friedle, Jeremy J. Kodanko, Anna J. Morys, Takahiro Hayashi, Pierre Moënne-Loccoz and Stephen J. Lippard
Journal of the American Chemical Society 2009 Volume 131(Issue 40) pp:14508-14520
Publication Date(Web):September 16, 2009
DOI:10.1021/ja906137y
In order to model the syn disposition of histidine residues in carboxylate-bridged non-heme diiron enzymes, we prepared a new dinucleating ligand, H2BPG2DEV, that provides this geometric feature. The ligand incorporates biologically relevant carboxylate functionalities, which have not been explored as extensively as nitrogen-only analogues. Three novel oxo-bridged diiron(III) complexes, [Fe2(μ-O)(H2O)2(BPG2DEV)](ClO4)2 (6), [Fe2(μ-O)(μ-O2CAriPrO)(BPG2DEV)](ClO4) (7), and [Fe2(μ-O)(μ-CO3)(BPG2DEV)] (8), were prepared. Single-crystal X-ray structural characterization confirms that two pyridyl groups are bound syn with respect to the Fe−Fe vector in these compounds. The carbonato-bridged complex 8 forms quantitatively from 6 in a rapid reaction with gaseous CO2 in organic solvents. A common maroon-colored intermediate (λmax = 490 nm; ε = 1500 M−1 cm−1) forms in reactions of 6, 7, or 8 with H2O2 and NEt3 in CH3CN/H2O solutions. Mass spectrometric analyses of this species, formed using 18O-labeled H2O2, indicate the presence of a peroxide ligand bound to the oxo-bridged diiron(III) center. The Mössbauer spectrum at 90 K of the EPR-silent intermediate exhibits a quadrupole doublet with δ = 0.58 mm/s and ΔEQ = 0.58 mm/s. The isomer shift is typical for a peroxodiiron(III) species, but the quadrupole splitting parameter is unusually small compared to those of related complexes. These Mössbauer parameters are comparable to those observed for a peroxo intermediate formed in the reaction of reduced toluene/o-xylene monooxygenase hydroxylase with dioxygen. Resonance Raman studies reveal an unusually low-energy O−O stretching mode in the peroxo intermediate that is consistent with a short diiron distance. Although peroxodiiron(III) intermediates generated from 6, 7, and 8 are poor O-atom-transfer catalysts, they display highly efficient catalase activity, with turnover numbers up to 10 000. In contrast to hydrogen peroxide reactions of diiron(III) complexes that lack a dinucleating ligand, the intermediates generated here could be re-formed in significant quantities after a second addition of H2O2, as observed spectroscopically and by mass spectrometry.
Co-reporter:Wee Han Ang and Stephen J. Lippard
Chemical Communications 2009 (Issue 39) pp:5820-5822
Publication Date(Web):08 Sep 2009
DOI:10.1039/B914843D
Replacement of a single dA nucleotide positioned at a programmed site in a DNA plasmid with its 7-deaza-analog is described together with its complete resistance to restriction enzymatic cleavage.
Co-reporter:Brian A. Wong, Simone Friedle and Stephen J. Lippard
Inorganic Chemistry 2009 Volume 48(Issue 15) pp:7009-7011
Publication Date(Web):July 2, 2009
DOI:10.1021/ic900990w
The spectroscopic and proton- and Zn(II)-binding properties of two new members of the Zinpyr family of fluorescent sensors are reported. In ZP1B and ZP3B, a (2-picolyl)(4-picolyl)amine (2,4-DPA) moiety is installed in place of the di(2-picolyl)amine (2,2-DPA) ligand used in the parent sensors ZP1 and ZP3. This modification has the benefit of both lowering the proton-induced turn-on at physiological pH levels and altering the Zn(II) affinity so as to detect only the most concentrated stores of this ion in biological samples. Comparison of the proton affinities of all four probes, as determined by potentiometric titrations, contributes to our understanding of the solution properties of this family of sensors.
Co-reporter:Loi H. Do and Stephen J. Lippard
Inorganic Chemistry 2009 Volume 48(Issue 22) pp:10708-10719
Publication Date(Web):October 21, 2009
DOI:10.1021/ic901711c
A series of 2-phenoxypyridyl and 2-phenoxyimino ligands, H2LR,R′ [2,2′-(5,5′-(1,2-phenylenebis(ethyne-2,1-diyl))bis(pyridine-5,2-diyl))diphenol, where R = H, Me, or t-Bu, and R′ = H or Ph] and H2BIPSMe,Ph [(3,3′-(1E,1′E)-(3,3′-sulfonylbis(3,1-phenylene)bis(azan-1-yl-1-ylidene))bis(methan-1-yl-1-ylidene)bis(5-methylbiphenyl-2-ol)], were synthesized as platforms for nonheme diiron(II) protein model complexes. UV−vis spectrophotometric studies and preparative-scale reactions of LR,R′ or BIPSMe,Ph, where LR,R′ and BIPSMe,Ph are the deprotonated forms of H2LR,R′ and H2BIPSMe,Ph, respectively, with iron(II) revealed that the presence of sterically protective o-phenol substituents is necessary to obtain discrete dinuclear species. The reaction of LMe,Ph with iron(II) in tetrahydrofuran (THF) afforded the doubly bridged compound [Fe2(LMe,Ph)2(THF)3] (1), which was characterized in the solid state by X-ray crystallography. A large internal cavity in this complex facilitates its rapid reaction with dioxygen, even at −50 °C, to produce the thermodynamically stable [Fe2(μ-O)(LMe,Ph)2] (2) species. Reaction of 18O2 instead of 16O2 with 1 led to a shift in the Fe−O−Fe vibrational frequency from 833 to 798 cm−1, confirming the presence of the (μ-oxo)diiron(III) core and molecular oxygen as the source of the bridging oxo group. The LMe,Ph ligand is robust toward oxidative decomposition and does not display any reversible redox activity.
Co-reporter:Wee Han Ang, William Wesley Brown and Stephen J. Lippard
Bioconjugate Chemistry 2009 Volume 20(Issue 5) pp:1058
Publication Date(Web):April 7, 2009
DOI:10.1021/bc900031a
FDA-approved platinum-based anticancer drugs, cisplatin, carboplatin, and oxaliplatin, are some of the most effective chemotherapies in clinical use. The cytotoxic action of these compounds against cancer requires a combination of processes including cell entry, drug activation, DNA binding, and transcription inhibition resulting in apoptotic cell death. The drugs form Pt lesions with nuclear DNA, leading to the arrest of key cellular functions and triggering a variety of cellular responses. DNA probes containing Pt−DNA conjugates are important tools for studying the molecular mechanisms of these processes. In order to facilitate investigation of specific Pt−DNA lesion processing within live cells, we devised a strategy for constructing plasmids containing a single site-specific Pt−DNA adduct. The method involves the use of nicking restriction enzymes to create closely spaced tandem gaps on the plasmid followed by removal of the intervening doubly nicked DNA strand to form a short single-stranded gap. Synthetic platinated oligonucleotides were incorporated into the gapped plasmid construct to generate a covalently closed circular platinated plasmid in good yield. We discuss the application of this methodology to prepare plasmids containing a platinum 1,2-d(G*pG*) or 1,3-d(G*pTpG*) intrastrand cross-link, two notable adducts formed by the three clinically approved drugs.
Co-reporter:Ryan C. Todd and Stephen J. Lippard
Metallomics 2009 vol. 1(Issue 4) pp:280-291
Publication Date(Web):26 May 2009
DOI:10.1039/B907567D
Cisplatin, carboplatin, and oxaliplatin are three FDA-approved members of the platinum anticancer drug family. These compounds induce apoptosis in tumor cells by binding to nuclear DNA, forming a variety of structural adducts and triggering cellular responses, one of which is the inhibition of transcription. In this report we present (i) a detailed review of the structural investigations of various Pt–DNA adducts and the effects of these lesions on global DNA geometry; (ii) research detailing inhibition of cellular transcription by Pt–DNA adducts; and (iii) a mechanistic analysis of how DNA structural distortions induced by platinum damage may inhibit RNA synthesis in vivo. A thorough understanding of the molecular mechanism of action of platinum antitumor agents will aid in the development of new compounds in the family.
Co-reporter:Katherine S. Lovejoy and Stephen J. Lippard
Dalton Transactions 2009 (Issue 48) pp:10651-10659
Publication Date(Web):01 Oct 2009
DOI:10.1039/B913896J
The five platinum anticancer compounds currently in clinical use conform to structure–activity relationships formulated (M. J. Cleare and J. D. Hoeschele, Bioinorg. Chem., 1973, 2, 187–210) shortly after the discovery that cis-diamminedichloroplatinum(II), cisplatin, has antitumor activity in mice. These compounds are neutral platinum(II) species with two am(m)ine ligands or one bidentate chelating diamine and two additional ligands that can be replaced by water through aquation reactions. The resulting cations ultimately form bifunctional adducts on DNA. Information about the chemistry of these platinum compounds and correlations of their structures with anticancer activity have provided guidance for the design of novel anticancer drug candidates based on the proposed mechanisms of action. This article discusses advances in the synthesis and evaluation of such non-traditional platinum compounds, including cationic and tumor-targeting constructs.
Co-reporter:Guangyu Zhu and Stephen J. Lippard
Biochemistry 2009 Volume 48(Issue 22) pp:
Publication Date(Web):April 13, 2009
DOI:10.1021/bi900389b
The DNA-binding inorganic compound cisplatin is one of the most successful anticancer drugs. The detailed mechanism by which cells recognize and process cisplatin−DNA damage is of great interest. Although the family of proteins that bind cisplatin 1,2- and 1,3-intrastrand cross-links has been identified, much less is known about cellular protein interactions with cisplatin interstrand cross-links (ICLs). In order to address this question, a photoreactive analogue of cisplatin, PtBP6, was used to construct a DNA duplex containing a site-specific platinum ICL. This DNA probe was characterized and used in photo-cross-linking experiments to separate and identify nuclear proteins that bind to the ICL by peptide mass fingerprint analysis. Several such proteins were discovered, including PARP-1, hMutSβ, DNA ligase III, XRCC1, and PNK. The photo-cross-linking approach was independently validated by an electrophoretic mobility shift assay demonstrating hMutSβ binding to a cisplatin ICL. Proteins that recognize the platinum ICL were also identified in cisplatin-resistant cells, cells halted at various phases of the cell cycle, and in different carcinoma cells. Nuclear proteins that bind to the platinum ICL differ from those binding to intrastrand cross-links, indicating different mechanisms for disruption of cellular functions.
Co-reporter:Simone Friedle
European Journal of Inorganic Chemistry 2009 Volume 2009( Issue 36) pp:5506-5515
Publication Date(Web):
DOI:10.1002/ejic.200900821
Abstract
In this study, diiron(II) complexes were synthesized as small molecule mimics of the reduced active sites in the hydroxylase components of bacterial multicomponent monooxygenases (BMMs). Tethered aromatic substrates were introduced in the form of 2-phenoxypyridines, incorporating hydroxy and methoxy functionalities into windmill-type diiron(II) compounds [Fe2(μ-O2CArR)2(O2CArR)2(L)2] (1–4), where–O2CArR is a sterically encumbering carboxylate, 2,6-bis(4-fluorophenyl)-, or 2,6-bis(p-tolyl)benzoate (R = 4-FPh or Tol, respectively). The inability of 1–4 to hydroxylate the aromatic substrates was ascertained. Upon reaction with dioxygen, compounds 2 and 3 (L = 2-(m-MeOPhO)Py, 2-(p-MeOPhO)Py, respectively) decompose by a known bimolecular pathway to form mixed-valent diiron(II,III) species at low temperature. Use of 2-(pyridin-2-yloxy)phenol as the ligand L resulted in a doubly bridged diiron complex 4 and an unprecedented phenoxide-bridged triiron(II) complex 5 under slightly modified reaction conditions. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Christine E. Tinberg and Stephen J. Lippard
Biochemistry 2009 Volume 48(Issue 51) pp:
Publication Date(Web):November 19, 2009
DOI:10.1021/bi901672n
Stopped-flow kinetic investigations of soluble methane monooxygenase (sMMO) from M. capsulatus (Bath) have clarified discrepancies that exist in the literature regarding several aspects of catalysis by this enzyme. The development of thorough kinetic analytical techniques has led to the discovery of two novel oxygenated iron species that accumulate in addition to the well-established intermediates Hperoxo and Q. The first intermediate, P*, is a precursor to Hperoxo and was identified when the reaction of reduced MMOH and MMOB with O2 was carried out in the presence of ≥540 μM methane to suppress the dominating absorbance signal due to Q. The optical properties of P* are similar to those of Hperoxo, with ε420 = 3500 M−1 cm−1 and ε720 = 1250 M−1 cm−1. These values are suggestive of a peroxo-to-iron(III) charge-transfer transition and resemble those of peroxodiiron(III) intermediates characterized in other carboxylate-bridged diiron proteins and synthetic model complexes. The second identified intermediate, Q*, forms on the pathway of Q decay when reactions are performed in the absence of hydrocarbon substrate. Q* does not react with methane, forms independently of buffer composition, and displays a unique shoulder at 455 nm in its optical spectrum. Studies conducted at different pH values reveal that rate constants corresponding to P* decay/Hperoxo formation and Hperoxo decay/Q formation are both significantly retarded at high pH and indicate that both events require proton transfer. The processes exhibit normal kinetic solvent isotope effects (KSIEs) of 2.0 and 1.8, respectively, when the reactions are performed in D2O. Mechanisms are proposed to account for the observations of these novel intermediates and the proton dependencies of P* to Hperoxo and Hperoxo to Q conversion.
Co-reporter:Shanta Dhar
PNAS 2009 Volume 106 (Issue 52 ) pp:22199-22204
Publication Date(Web):2009-12-29
DOI:10.1073/pnas.0912276106
The unique glycolytic metabolism of most solid tumors, known as the Warburg effect, is associated with resistance to apoptosis
that enables cancer cells to survive. Dichloroacetate (DCA) is an anticancer agent that can reverse the Warburg effect by
inhibiting a key enzyme in cancer cells, pyruvate dehydrogenase kinase (PDK), that is required for the process. DCA is currently
not approved for cancer treatment in the USA. Here, we present the synthesis, characterization, and anticancer properties
of c,t,c-[Pt(NH3)2(O2CHCl2)2Cl2], mitaplatin, in which two DCA units are appended to the axial positions of a six-coordinate Pt(IV) center. The negative
intracellular redox potential reduces the platinum to release cisplatin, a Pt(II) compound, and two equivalents of DCA. By
a unique mechanism, mitaplatin thereby attacks both nuclear DNA with cisplatin and mitochondria with DCA selectively in cancer
cells. The cytotoxicity of mitaplatin in a variety of cancer cell lines equals or exceeds that of all known Pt(IV) compounds
and is comparable to that of cisplatin. Mitaplatin alters the mitochondrial membrane potential gradient (Δψm) of cancer cells, promoting apoptosis by releasing cytochrome c and translocating apoptosis inducing factor from mitochondria to the nucleus. Cisplatin formed upon cellular reduction of
mitaplatin enters the nucleus and targets DNA to form 1,2-intrastrand d(GpG) cross-links characteristic of its own potency
as an anticancer drug. These properties of mitaplatin are manifest in its ability to selectively kill cancer cells cocultured
with normal fibroblasts and to partially overcome cisplatin resistance.
Co-reporter:Evan R. Guggenheim Dr.;Dong Xu Dr.;Christiana X. Zhang Dr.;Pamela V. Chang
ChemBioChem 2009 Volume 10( Issue 1) pp:141-157
Publication Date(Web):
DOI:10.1002/cbic.200800471
Abstract
The activity of the anticancer drug cisplatin is a consequence of its ability to bind DNA. Platinum adducts bend and unwind the DNA duplex, creating recognition sites for nuclear proteins. Following DNA damage recognition, the lesions will either be repaired, facilitating cell viability, or if repair is unsuccessful and the Pt adduct interrupts vital cellular functions, apoptosis will follow. With the use of the benzophenone-modified cisplatin analogue Pt-BP6, 25 bp DNA duplexes containing either a 1,2-d(G*pG*) intrastrand or a 1,3-d(G*pTpG*) intrastrand crosslink were synthesized, where the asterisks designate platinated nucleobases. Proteins having affinity for these platinated DNAs were photocrosslinked and identified in cervical, testicular, pancreatic and bone cancer-cell nuclear extracts. Proteins identified in this manner include the DNA repair factors RPA1, Ku70, Ku80, Msh2, DNA ligase III, PARP-1, and DNA–PKcs, as well as HMG-domain proteins HMGB1, HMGB2, HMGB3, and UBF1. The latter strongly associate with the 1,2-d(G*pG*) adduct and weakly or not at all with the 1,3-d(G*pTpG*) adduct. The nucleotide excision repair protein RPA1 was photocrosslinked only by the probe containing a 1,3-d(G*pTpG*) intrastrand crosslink. The affinity of PARP-1 for platinum-modified DNA was established using this type of probe for the first time. To ensure that the proteins were not photocrosslinked because of an affinity for DNA ends, a 90-base dumbbell probe modified with Pt-BP6 was investigated. Photocrosslinking experiments with this longer probe revealed the same proteins, as well as some additional proteins involved in chromatin remodeling, transcription, or repair. These findings reveal a more complete list of proteins involved in the early steps of the mechanism of action of the cisplatin and its close analogue carboplatin than previously was available.
Co-reporter:Elizabeth M. Nolan and Stephen J. Lippard
Chemical Reviews 2008 Volume 108(Issue 9) pp:3443
Publication Date(Web):July 25, 2008
DOI:10.1021/cr068000q
Co-reporter:Erwin Reisner
European Journal of Organic Chemistry 2008 Volume 2008( Issue 1) pp:156-163
Publication Date(Web):
DOI:10.1002/ejoc.200700816
Abstract
We report an efficient convergent synthesis of a new type of C-clamp ligand with a 1,2-diethynylarene scaffold involving a chelate host capable of binding a guest molecule in its endo-dicarboxylate pocket. The chemistry involves a combination of palladium-catalyzed Sonogashira, Heck, and Suzuki cross-coupling reactions. The compounds 2,3-bis[2-(2′-carboxybiphenyl-4-yl)ethynyl]triptycene and 4,5-bis[2-(2′-carboxybiphenyl-4-yl)ethynyl]veratrole and their 2′-carboxy-m-terphenyl-4-yl analogues were designed as dinucleating ligands to assemble carboxylate-bridged transition-metal complexes with a windmill geometry. The X-ray crystal structure of one such C-clamp compound containing co-crystallized water molecules reveals strong hydrogen bonds of the aqua guest to the endo-oriented carboxylic acid entities of the C-clamp host. In addition, two syn-N-donor ligands were prepared as a synthetic scaffold to mimic the geometric arrangement of N-donor atoms in carboxylate-bridged dinuclear proteins. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Jeremy J. Kodanko, Stephen J. Lippard
Inorganica Chimica Acta 2008 Volume 361(Issue 4) pp:894-900
Publication Date(Web):3 March 2008
DOI:10.1016/j.ica.2007.02.029
The synthesis and characterization of the diiron(II) complex [Fe2(μ-OTf)2(PIC2DET)2](BARF)2 (2), where PIC2DET is a 2,3-diethynyltriptycene-linked dipicolinic methyl ester ligand, are described. The dication in 2, [Fe2(μ-OTf)2(PIC2DET)2]2+, contains two symmetry-equivalent iron atoms with octahedral coordination geometries. Each metal ion has a N2O4 atom donor set that includes four atoms from two picolinic ester N, O chelate rings, as well as two oxygen atoms from the bridging trifluoromethanesulfonate groups. The {Fe2(μ-OTf)2} core of 2 is stabilized by two PIC2DET ligands that bind the two metal ions in a head-to-head fashion, leading to an Fe⋯Fe distance of 5.173(1) Å. Molar conductivity data for 2 are consistent with [Fe2(μ-OTf)2(PIC2DET)2]2+ retaining its identity in acetone solutions, where it behaves as a 2:1 electrolyte. 1H NMR spectroscopic, and solution (d6-acetone) and solid-state magnetic susceptibility, data all indicate that the iron atoms of 2 are high-spin (S = 2). A fit of the magnetic data (2–300 K) to a spin-only isotropic exchange Hamiltonian H = −2JS1·S2 is consistent with weak antiferromagnetic coupling between the two iron atoms with J ∼ −0.99(2) cm−1 and g = 2.10(1).The diiron(II) complex [Fe2(μ-OTf)2(PIC2DET)2](BARF)2, where PIC2DET is a 2,3-diethynyltriptycene-linked dipicolinic methyl ester ligand, has been synthesized and characterized. X-ray crystallographic data reveal a new mode of coordination for the syn N-donor ligand PIC2DET. Molar conductivity, 1H NMR spectroscopic, and magnetic susceptibility data are consistent with a complex containing two high spin iron atoms that retains its structure in solution.
Co-reporter:Katherine S. Lovejoy;Ryan C. Todd;Shuzhong Zhang;Michael S. McCormick;J. Alejandro D'Aquino;Joyce T. Reardon;Aziz Sancar;Kathleen M. Giacomini
PNAS 2008 Volume 105 (Issue 26 ) pp:8902-8907
Publication Date(Web):2008-07-01
DOI:10.1073/pnas.0803441105
We have identified unique chemical and biological properties of a cationic monofunctional platinum(II) complex, cis-diammine(pyridine)chloroplatinum(II), cis-[Pt(NH3)2(py)Cl]+ or cDPCP, a coordination compound previously identified to have significant anticancer activity in a mouse tumor model. This
compound is an excellent substrate for organic cation transporters 1 and 2, also designated SLC22A1 and SLC22A2, respectively.
These transporters are abundantly expressed in human colorectal cancers, where they mediate uptake of oxaliplatin, cis-[Pt(DACH)(oxalate)] (DACH = trans-R,R-1,2-diaminocyclohexane), an FDA-approved first-line therapy for colorectal cancer. Unlike oxaliplatin, however, cDPCP binds
DNA monofunctionally, as revealed by an x-ray crystal structure of cis-{Pt(NH3)2(py)}2+ bound to the N7 atom of a single guanosine residue in a DNA dodecamer duplex. Although the quaternary structure resembles
that of B-form DNA, there is a base-pair step to the 5′ side of the Pt adduct with abnormally large shift and slide values,
features characteristic of cisplatin intrastrand cross-links. cDPCP effectively blocks transcription from DNA templates carrying
adducts of the complex, unlike DNA lesions of other monofunctional platinum(II) compounds like {Pt(dien)}2+. cDPCP–DNA adducts are removed by the nucleotide excision repair apparatus, albeit much less efficiently than bifunctional
platinum–DNA intrastrand cross-links. These exceptional characteristics indicate that cDPCP and related complexes merit consideration
as therapeutic options for treating colorectal and other cancers bearing appropriate cation transporters.
Co-reporter:Shanta Dhar;Robert Langer;Frank X. Gu;Omid C. Farokhzad
PNAS 2008 Volume 105 (Issue 45 ) pp:17356-17361
Publication Date(Web):2008-11-11
DOI:10.1073/pnas.0809154105
Cisplatin is used to treat a variety of tumors, but dose limiting toxicities or intrinsic and acquired resistance limit its
application in many types of cancer including prostate. We report a unique strategy to deliver cisplatin to prostate cancer
cells by constructing Pt(IV)-encapsulated prostate-specific membrane antigen (PSMA) targeted nanoparticles (NPs) of poly(D,L-lactic-co-glycolic acid) (PLGA)-poly(ethylene glycol) (PEG)-functionalized controlled release polymers. By using PLGA-b-PEG nanoparticles with PSMA targeting aptamers (Apt) on the surface as a vehicle for the platinum(IV) compound c,t,c-[Pt(NH3)2(O2CCH2CH2CH2CH2CH3)2Cl2] (1), a lethal dose of cisplatin was delivered specifically to prostate cancer cells. PSMA aptamer targeted delivery of Pt(IV)
cargos to PSMA+ LNCaP prostate cancer cells by endocytosis of the nanoparticle vehicles was demonstrated using fluorescence microscopy by
colocalization of green fluorescent labeled cholesterol-encapsulated NPs and early endosome marker EEA-1. The choice of linear
hexyl chains in 1 was the result of a systematic study to optimize encapsulation and controlled release from the polymer without
compromising either feature. Release of cisplatin from the polymeric nanoparticles after reduction of 1 and formation of cisplatin
1,2-intrastrand d(GpG) cross-links on nuclear DNA was confirmed by using a monoclonal antibody for the adduct. A comparison
between the cytotoxic activities of Pt(IV)-encapsulated PLGA-b-PEG NPs with the PSMA aptamer on the surface (Pt-NP-Apt), cisplatin, and the nontargeted Pt(IV)-encapsulated NPs (Pt-NP)
against human prostate PSMA-overexpressing LNCaP and PSMA- PC3 cancer cells revealed significant differences. The effectiveness of PSMA targeted Pt-NP-Apt nanoparticles against the
PSMA+ LNCaP cells is approximately an order of magnitude greater than that of free cisplatin.
Co-reporter:Ivan Gusarov;Konstantin Shatalin;Yelena Shatalina;Lindsey E. McQuade;Evgeny Nudler;Ekaterina Avetissova
PNAS 2008 Volume 105 (Issue 3 ) pp:1009-1013
Publication Date(Web):2008-01-22
DOI:10.1073/pnas.0710950105
Phagocytes generate nitric oxide (NO) and other reactive oxygen and nitrogen species in large quantities to combat infecting
bacteria. Here, we report the surprising observation that in vivo survival of a notorious pathogen—Bacillus anthracis—critically depends on its own NO-synthase (bNOS) activity. Anthrax spores (Sterne strain) deficient in bNOS lose their virulence
in an A/J mouse model of systemic infection and exhibit severely compromised survival when germinating within macrophages.
The mechanism underlying bNOS-dependent resistance to macrophage killing relies on NO-mediated activation of bacterial catalase
and suppression of the damaging Fenton reaction. Our results demonstrate that pathogenic bacteria use their own NO as a key
defense against the immune oxidative burst, thereby establishing bNOS as an essential virulence factor. Thus, bNOS represents
an attractive antimicrobial target for treatment of anthrax and other infectious diseases.
Co-reporter:Stephen J. Lippard
Journal of Inorganic Biochemistry 2007 Volume 101(11–12) pp:1546-1547
Publication Date(Web):November 2007
DOI:10.1016/j.jinorgbio.2007.08.001
Co-reporter:Katherine S. Lovejoy;Xiao-an Zhang;Alan Jasanoff
PNAS 2007 Volume 104 (Issue 26 ) pp:10780-10785
Publication Date(Web):2007-06-26
DOI:10.1073/pnas.0702393104
We report a molecular platform for dual-function fluorescence/MRI sensing of mobile zinc. Zinc-selective binding units were
strategically attached to a water-soluble porphyrin template. The synthetic strategy for achieving the designed target ligand
is flexible and convenient, and the key intermediates can be applied as general building blocks for the construction of other
metal sensors based on a similar mechanism. The metal-free form, (DPA-C2)2-TPPS3 (1), where DPA is dipicolylamine and TPPS3 is 5-phenyl-10,15,20-tris(4-sulfonatophenyl)porphine, is an excellent fluorescent sensor for zinc. It has certain superior
physical properties compared with earlier-generation zinc sensors including emission in the red and near-IR regions [λem = 645 nm (s) and 715 nm (m)], with a large Stokes shift of >230 nm. The fluorescence intensity of 1 increases by >10-fold
upon zinc binding. The fluorescence “turn-on” is highly selective for zinc versus other divalent metal ions and is relatively
pH-insensitive within the biologically relevant pH window. The manganese derivative, [(DPA-C2)2-TPPS3Mn(III)] (2), switches the function of the molecule to generate an MRI contrast agent. In the presence of zinc, the relaxivity of 2 in
aqueous solution is significantly altered, which makes it a promising zinc MRI sensor. Both metal-free and Mn(III)-inserted
forms are efficiently taken up by live cells, and the intracellular zinc can be imaged by either fluorescence or MR, respectively.
We anticipate that in vivo applications of the probes will facilitate a deeper understanding of the physiological roles of zinc and allow detection
of abnormal zinc homeostasis for pathological diagnoses.
Co-reporter:Mi Hee Lim Dr.;Chaoyuan Kuang
ChemBioChem 2006 Volume 7(Issue 10) pp:
Publication Date(Web):21 JUN 2006
DOI:10.1002/cbic.200600042
The cobalt complexes [Co(Ds-AMP)2] (1) and [Co(Ds-AQ)2] (2), where Ds-AMP and Ds-AQ are the conjugate bases of dansyl aminomethylpyridine (Ds-HAMP) and dansyl aminoquinoline (Ds-HAQ), respectively, were synthesized in two steps as fluorescence-based nitric oxide (NO) sensors and characterized by X-ray crystallography. The fluorescence of the two complexes was significantly quenched in CH3CN or CH3OH compared to that of the free Ds-HAMP or Ds-HAQ ligands. Addition of NO to a CH3CN solution of 1 or 2 enhanced the integrated fluorescence emission by factors of 2.1(±0.3) or 3.6(±0.4) within 35 or 20 min, respectively. Introduction of NO to methanolic solutions of the complexes similarly increased the fluorescence by 1.4(±0.1) for 1 or 6.5(±1.4) for 2 within 1 h. These studies demonstrate that 1 and 2 can monitor the presence of NO with turn-on emission and that their fluorescence responses are more rapid than those of previously reported cobalt systems in coordinating solvents such as CH3CN and CH3OH. 1H NMR and IR spectroscopic data revealed the formation of a {Co(NO)2}10 cobalt–dinitrosyl adduct, with concomitant dissociation of one ligand from the cobalt center, as the metal-containing product of the NO reactions, a result indicating NO-induced ligand release to be the cause of the fluorescence increase.
Co-reporter:Dong Wang
&
Stephen J. Lippard
Nature Reviews Drug Discovery 2005 4(4) pp:307
Publication Date(Web):
DOI:10.1038/nrd1691
Cisplatin, carboplatin and oxaliplatin are platinum-based drugs that are widely used in cancer chemotherapy. Platinum–DNA adducts, which are formed following uptake of the drug into the nucleus of cells, activate several cellular processes that mediate the cytotoxicity of these platinum drugs. This review focuses on recently discovered cellular pathways that are activated in response to cisplatin, including those involved in regulating drug uptake, the signalling of DNA damage, cell-cycle checkpoints and arrest, DNA repair and cell death. Such knowledge of the cellular processing of cisplatin adducts with DNA provides valuable clues for the rational design of more efficient platinum-based drugs as well as the development of new therapeutic strategies.
Co-reporter:Elizabeth M. Nolan and Stephen J. Lippard
Journal of Materials Chemistry A 2005 vol. 15(Issue 27-28) pp:2778-2783
Publication Date(Web):04 May 2005
DOI:10.1039/B501615K
The synthesis and photophysical characterization of Mercury Sensor 4, MS4, an aniline-derivatized seminaphthofluorescein-based dye that contains a pyridyl-amine-thioether ligand analogous to that employed in the previously reported Zinspy (ZS) Zn(II) sensor family (Nolan and Lippard, Inorg. Chem. 2004, 43, 8310–8317) are reported. Sensor MS4 provides single-excitation, dual-emission ratiometric detection of Hg(II) in aqueous solution. A ∼4-fold ratiometric change (λ624/λ524) is observed upon introduction of Hg(II) to an aqueous chloride-containing solution of MS4 at pH 8. In this milieu, MS4 shows selectivity for Hg(II) over a background of environmentally relevant alkali and alkaline earth metals, a number of divalent first-row transition metals, and its Group 12 congeners Zn(II) and Cd(II).
Co-reporter:Andrew J. Danford;Dong Wang;Qun Wang;Thomas D. Tullius
PNAS 2005 102 (35 ) pp:12311-12316
Publication Date(Web):2005-08-30
DOI:10.1073/pnas.0506025102
We constructed two site-specifically modified nucleosomes containing an intrastrand cis-{Pt(NH3)2}2+ 1,3-d(GpTpG) cross-link, similar to one formed by the anticancer drugs carboplatin and cisplatin on DNA, and investigated
their structures by hydroxyl radical footprinting and exonuclease III digestion. Hydroxyl radical footprinting demonstrated
that the presence of the platinum cross-link selects out a specific rotational setting of DNA on the histone octamer core
in each of two reconstituted nucleosomes in which the platinum positions differ by half a DNA helical turn. The {Pt(NH3)2}2+ cross-link is situated in a structurally similar location, with the undamaged strand projecting outward, forcing the DNA
to adopt opposite rotational settings in its wrapping around the histone octamer in the two nucleosomes. Enzymatic digestion
by exonuclease III of the nucleosome substrates revealed that the platinum cross-link affects the translational positioning
of the DNA, forcing it into an asymmetric arrangement with respect to the core histone proteins. We suggest that these phasing
phenomena may be central to the recognition and processing of platinum-DNA adducts in cancer cells treated with these drugs
and possibly may be common to other DNA damaging events.
Co-reporter:Ryan C. Todd, Stephen J. Lippard
Journal of Inorganic Biochemistry (September 2010) Volume 104(Issue 9) pp:902-908
Publication Date(Web):1 September 2010
DOI:10.1016/j.jinorgbio.2010.04.005
We report the 1.77-Å resolution X-ray crystal structure of a dodecamer DNA duplex with the sequence 5′-CCTCTGGTCTCC-3′ that has been modified to contain a single engineered 1,2-cis-{Pt(NH3)2}2+-d(GpG) cross-link, the major DNA adduct of cisplatin. These data represent a significant improvement in resolution over the previously published 2.6-Å structure. The ammine ligands in this structure are clearly resolved, leading to improved visualization of the cross-link geometry with respect to both the platinum center and to the nucleobases, which adopt a higher energy conformation. Also better resolved are the deoxyribose sugar puckers, which allow us to re-examine the global structure of platinum-modified DNA. Another new feature of this model is the location of four octahedral [Mg(H2O)6]2+ ions associated with bases in the DNA major groove and the identification of 124 ordered water molecules that participate in hydrogen-bonding interactions with either the nucleic acid or the diammineplatinum(II) moiety.The structure of a cisplatin-modified DNA dodecamer duplex containing a 1,2-cis-{Pt(NH3)2}2+-d(GpG) cross-link has been determined at 1.77-Å resolution. Improved electron density reveals new features not seen in the originally published 2.6 Å structure.Download full-size image
Co-reporter:Nora Graf, Stephen J. Lippard
Advanced Drug Delivery Reviews (August 2012) Volume 64(Issue 11) pp:993-1004
Publication Date(Web):1 August 2012
DOI:10.1016/j.addr.2012.01.007
This review provides an overview of metal-based anticancer drugs and drug candidates. In particular, we focus on metal complexes that can be activated in the reducing environment of cancer cells, thus serving as prodrugs. There are many reports of Pt and Ru complexes as redox-activatable drug candidates, but other d-block elements with variable oxidation states have a similar potential to serve as prodrugs in this manner. In this context are compounds based on Fe, Co, or Cu chemistry, which are also covered. A trend in the field of medicinal inorganic chemistry has been toward molecularly targeted, metal-based drugs obtained by functionalizing complexes with biologically active ligands. Another recent activity is the use of nanomaterials for drug delivery, exploiting passive targeting of tumors with nano-sized constructs made from Au, Fe, carbon, or organic polymers. Although complexes of all of the above mentioned metals will be described, this review focuses primarily on Pt compounds, including constructs containing nanomaterials.Download high-res image (848KB)Download full-size image
Co-reporter:Enhui Pan, Xiao-an Zhang, Zhen Huang, Artur Krezel, ... James O. McNamara
Neuron (22 September 2011) Volume 71(Issue 6) pp:1116-1126
Publication Date(Web):22 September 2011
DOI:10.1016/j.neuron.2011.07.019
The presence of zinc in glutamatergic synaptic vesicles of excitatory neurons of mammalian cerebral cortex suggests that zinc might regulate plasticity of synapses formed by these neurons. Long-term potentiation (LTP) is a form of synaptic plasticity that may underlie learning and memory. We tested the hypothesis that zinc within vesicles of mossy fibers (mf) contributes to mf-LTP, a classical form of presynaptic LTP. We synthesized an extracellular zinc chelator with selectivity and kinetic properties suitable for study of the large transient of zinc in the synaptic cleft induced by mf stimulation. We found that vesicular zinc is required for presynaptic mf-LTP. Unexpectedly, vesicular zinc also inhibits a form of postsynaptic mf-LTP. Because the mf-CA3 synapse provides a major source of excitatory input to the hippocampus, regulating its efficacy by these dual actions, vesicular zinc is critical to proper function of hippocampal circuitry in health and disease.Highlights► A zinc chelator, ZX1, inhibits presynaptic mf-LTP in WT mice ► Postsynaptic mf-LTP is evident in ZnT3−/− mice lacking synaptic vesicular zinc ► Dialysis of CA3 pyramid with BAPTA inhibits mf-LTP in ZnT3−/− but not WT mice ► A zinc chelator, ZX1, unmasks postsynaptic mf-LTP in rim1α mutant mice
Co-reporter:Simone Friedle, Jeremy J. Kodanko, Kyrstin L. Fornace, Stephen J. Lippard
Journal of Molecular Structure (12 November 2008) Volume 890(Issues 1–3) pp:
Publication Date(Web):12 November 2008
DOI:10.1016/j.molstruc.2008.05.030
The synthesis and characterization of diiron(II) complexes supported by 9-triptycenecarboxylate ligands (−O2CTrp) is described. The interlocking nature of the triptycenecarboxylates facilitates formation of quadruply bridged diiron(II) complexes of the type [Fe2(μ-O2CTrp)4(L)2] (L = THF, pyridine or imidazole) with a paddlewheel geometry. A systematic lengthening of the Fe–Fe distance occurs with the increase in steric bulk of the neutral donor L, resulting in values of up to 3 Å without disassembly of the paddlewheel structure. Reactions with an excess of water do not lead to decomposition of the diiron(II) core, indicating that these quadruply bridged complexes are of exceptional stability. The red-colored complexes [Fe2(μ-O2CTrp)4(4-AcPy)2] (10) and [Fe2(μ-O2CTrp)4(4-CNPy)2] (11) exhibit solvent-dependent thermochromism in coordinating solvents that was studied by variable temperature UV–vis spectroscopy. Reaction of [Fe2(μ-O2CTrp)4(THF)2] with N,N,N′,N′-tetramethylethylenediamine (TMEDA), tetra-n-butyl ammonium thiocyanate, or excess 2-methylimidazole resulted in the formation of mononuclear complexes [Fe(O2CTrp)2(TMEDA)] (13), (n-Bu4N)2[Fe(O2CTrp)2(SCN)2] (14), and [Fe(O2CTrp)2(2-MeIm)2] (15) having an O4/N2 coordination sphere composition.
Co-reporter:Shanta Dhar ; Weston L. Daniel ; David A. Giljohann ; Chad A. Mirkin
Journal of the American Chemical Society () pp:
Publication Date(Web):September 24, 2009
DOI:10.1021/ja9071282
Amine-functionalized polyvalent oligonucleotide gold nanoparticles (DNA-Au NPs) were derivatized with a cisplatin prodrug, and the resulting DNA-Au NP conjugates were used to internalize multiple platinum centers. A platinum(IV) complex, c,c,t-[Pt(NH3)2Cl2(OH)(O2CCH2CH2CO2H)], was tethered to the surface of DNA-Au NPs through amide linkages. The platinum-tethered gold nanoparticles were taken into several cancer cells. The drop in intracellular pH facilitated reductive release of cisplatin from the prodrug, which then formed 1,2-d(GpG) intrastrand cross-links in the cell nuclei, as confirmed by an antibody specific for this adduct. The cytotoxicity of the platinum(IV) complex increases significantly in several cancer cell lines when the complex is attached to the surface of the DNA-Au NPs and in some instances exceeds that of cisplatin.
Co-reporter:Timothy C. Johnstone, Sarah M. Alexander, Justin J. Wilson and Stephen J. Lippard
Dalton Transactions 2015 - vol. 44(Issue 1) pp:NaN129-129
Publication Date(Web):2014/10/20
DOI:10.1039/C4DT02627F
A series of Pt(IV) prodrugs has been obtained by oxidative halogenation of either cisplatin or carboplatin. Iodobenzene dichloride is a general reagent that cleanly provides prodrugs bearing axial chlorides without the need to prepare intervening Pt(IV) intermediates or handle chlorine gas. Elemental bromine and iodine afford Pt(IV) compounds as well, although in the case of the iodine-mediated oxidation of carboplatin, an amido-bridged Pt(IV) side product also formed. A detailed analysis of the changes in spectroscopic and structural parameters induced by varying the axial halide is presented. A number of recurring motifs are observed in the solid state structures of these compounds.
Co-reporter:Robert J. Radford, Wen Chyan and Stephen J. Lippard
Chemical Science (2010-Present) 2013 - vol. 4(Issue 8) pp:NaN3084-3084
Publication Date(Web):2013/05/29
DOI:10.1039/C3SC50974E
Combining fluorescent zinc sensors with the facile syntheses and biological targeting capabilities of peptides, we created green- and blue-emitting probes that (i) are readily prepared on the solid-phase, (ii) retain the photophysical and zinc-binding properties of the parent sensor, and (iii) can be directed to the extracellular side of plasma membranes in live cells for detection of mobile zinc.
Co-reporter:Robert J. Radford, Wen Chyan and Stephen J. Lippard
Chemical Science (2010-Present) 2014 - vol. 5(Issue 11) pp:NaN4516-4516
Publication Date(Web):2014/08/12
DOI:10.1039/C4SC01280A
Fluorescein-based sensors are the most widely applied class of zinc probes but display adventitious localization in live cells. We present here a peptide-based localization strategy that affords precision in targeting of fluorescein-based zinc sensors. By appending the zinc-selective, reaction-based probe Zinpyr-1 diacetate (DA-ZP1) to the N-terminus of two different targeting peptides we achieve programmable localization and avoid unwanted sequestration within acidic vesicles. Furthermore, this approach can be generalized to other fluorescein-based sensors. When appended to a mitochondrial targeting peptide, the esterase-activated profluorophore 2′,7′-dichlorofluorescein diacetate can be used effectively at concentrations four-times lower than previously reported for analogous, non-acetylated derivatives. These results demonstrate on-resin or in-solution esterification of fluorescein to be an effective strategy to facilitate peptide-based targeting in live cells.
Co-reporter:Yao-Rong Zheng, Kogularamanan Suntharalingam, Timothy C. Johnstone and Stephen J. Lippard
Chemical Science (2010-Present) 2015 - vol. 6(Issue 2) pp:NaN1193-1193
Publication Date(Web):2014/11/24
DOI:10.1039/C4SC01892C
This report presents a novel strategy that facilitates delivery of multiple, specific payloads of Pt(IV) prodrugs using a well-defined supramolecular system. This delivery system comprises a hexanuclear Pt(II) cage that can host four Pt(IV) prodrug guest molecules. Relying on host–guest interactions between adamantyl units tethered to the Pt(IV) molecules and the cage, four prodrugs could be encapsulated within one cage. This host–guest complex, exhibiting a diameter of about 3 nm, has been characterized by detailed NMR spectroscopic measurements. Owing to the high positive charge, this nanostructure exhibits high cellular uptake. Upon entering cells and reacting with biological reductants such as ascorbic acid, the host–guest complex releases cisplatin, which leads to cell cycle arrest and apoptosis. The fully assembled complex displays cytotoxicity comparable to that of cisplatin against a panel of human cancer cell lines, whereas the cage or the Pt(IV) guest alone exhibit lower cytotoxicity. These findings indicate the potential of utilising well-defined supramolecular constructs for the delivery of prodrug molecules.
Co-reporter:Andrei Loas, Robert J. Radford, Alexandria Deliz Liang and Stephen J. Lippard
Chemical Science (2010-Present) 2015 - vol. 6(Issue 7) pp:NaN4140-4140
Publication Date(Web):2015/05/19
DOI:10.1039/C5SC00880H
We describe a modular, synthetically facile solid-phase approach aimed at separating the fluorescent reporter and binding unit of small-molecule metal-based sensors. The first representatives contain a lysine backbone functionalized with a tetramethylrhodamine fluorophore, and they operate by modulating the oxidation state of a copper ion ligated to an [N4] (cyclam) or an [N2O] (quinoline-phenolate) moiety. We demonstrate the selectivity of their Cu(II) complexes for sensing nitroxyl (HNO) and thiols (RSH), respectively, and investigate the mechanism responsible for the observed reactivity in each case. The two lysine conjugates are cell permeable in the active, Cu(II)-bound forms and retain their analyte selectivity intracellularly, even in the presence of interfering species such as nitric oxide, nitrosothiols, and hydrogen sulfide. Moreover, we apply the new probes to discriminate between distinct levels of intracellular HNO and RSH generated upon stimulation of live HeLa cells with ascorbate and hydrogen sulfide, respectively. The successful implementation of the lysine-based sensors to gain insight into biosynthetic pathways validates the method as a versatile tool for producing libraries of analogues with minimal synthetic effort.
Co-reporter:Pablo Rivera-Fuentes, Alexandra T. Wrobel, Melissa L. Zastrow, Mustafa Khan, John Georgiou, Thomas T. Luyben, John C. Roder, Kenichi Okamoto and Stephen J. Lippard
Chemical Science (2010-Present) 2015 - vol. 6(Issue 3) pp:NaN1948-1948
Publication Date(Web):2015/01/23
DOI:10.1039/C4SC03388D
Imaging mobile zinc in acidic environments remains challenging because most small-molecule optical probes display pH-dependent fluorescence. Here we report a reaction-based sensor that detects mobile zinc unambiguously at low pH. The sensor responds reversibly and with a large dynamic range to exogenously applied Zn2+ in lysosomes of HeLa cells, endogenous Zn2+ in insulin granules of MIN6 cells, and zinc-rich mossy fiber boutons in hippocampal tissue from mice. This long-wavelength probe is compatible with the green-fluorescent protein, enabling multicolor imaging, and facilitates visualization of mossy fiber boutons at depths of >100 μm, as demonstrated by studies in live tissue employing two-photon microscopy.
Co-reporter:Youngmin You, Elisa Tomat, Kevin Hwang, Tatjana Atanasijevic, Wonwoo Nam, Alan P. Jasanoff and Stephen J. Lippard
Chemical Communications 2010 - vol. 46(Issue 23) pp:NaN4141-4141
Publication Date(Web):2010/05/10
DOI:10.1039/C0CC00179A
A paramagnetic manganese complex of a fluorescein-based probe affords a dual-modality zinc sensor featuring an improved fluorescence dynamic range and an MRI readout.
Co-reporter:Loi H. Do, Hongxin Wang, Christine E. Tinberg, Eric Dowty, Yoshitaka Yoda, Stephen P. Cramer and Stephen J. Lippard
Chemical Communications 2011 - vol. 47(Issue 39) pp:NaN10947-10947
Publication Date(Web):2011/09/06
DOI:10.1039/C1CC13836G
The vibrational spectrum of an η1,η1-1,2-peroxodiiron(III) complex was measured by nuclear resonance vibrational spectroscopy and fit using an empirical force field analysis. Isotopic 18O2 labelling studies revealed a feature involving motion of the {Fe2(O2)}4+ core that was not previously observed by resonance Raman spectroscopy.
Co-reporter:Michael D. Pluth and Stephen J. Lippard
Chemical Communications 2012 - vol. 48(Issue 98) pp:NaN11983-11983
Publication Date(Web):2012/10/26
DOI:10.1039/C2CC37221E
Nitric oxide binds reversibly to the Fe(III) complex of a well-developed tetra-amido macrocyclic ligand. Reaction with NO results in formation of a species consistent with an S = 1 {Fe–NO}6 ground state as characterized by UV-vis, IR, EPR, and Mössbauer spectroscopy. The resultant nitrosyl is labile and dissociates readily upon purging with N2, thus providing a rare example of reversible NO binding to non-heme iron.
Co-reporter:Kogularamanan Suntharalingam, Ying Song and Stephen J. Lippard
Chemical Communications 2014 - vol. 50(Issue 19) pp:NaN2468-2468
Publication Date(Web):2013/12/11
DOI:10.1039/C3CC48740G
We report two platinum(IV) complexes conjugated with a vitamin E analog, α-tocopherol succinate (α-TOS). One of the conjugates displays the activity of both cisplatin and α-TOS in cancer cells, causing damage to DNA and mitochondria simultaneously. Accordingly, it serves as a promising dual-targeting anticancer agent.
Co-reporter:Wee Han Ang and Stephen J. Lippard
Chemical Communications 2009(Issue 39) pp:NaN5822-5822
Publication Date(Web):2009/09/08
DOI:10.1039/B914843D
Replacement of a single dA nucleotide positioned at a programmed site in a DNA plasmid with its 7-deaza-analog is described together with its complete resistance to restriction enzymatic cleavage.
Co-reporter:Katherine S. Lovejoy and Stephen J. Lippard
Dalton Transactions 2009(Issue 48) pp:NaN10659-10659
Publication Date(Web):2009/10/01
DOI:10.1039/B913896J
The five platinum anticancer compounds currently in clinical use conform to structure–activity relationships formulated (M. J. Cleare and J. D. Hoeschele, Bioinorg. Chem., 1973, 2, 187–210) shortly after the discovery that cis-diamminedichloroplatinum(II), cisplatin, has antitumor activity in mice. These compounds are neutral platinum(II) species with two am(m)ine ligands or one bidentate chelating diamine and two additional ligands that can be replaced by water through aquation reactions. The resulting cations ultimately form bifunctional adducts on DNA. Information about the chemistry of these platinum compounds and correlations of their structures with anticancer activity have provided guidance for the design of novel anticancer drug candidates based on the proposed mechanisms of action. This article discusses advances in the synthesis and evaluation of such non-traditional platinum compounds, including cationic and tumor-targeting constructs.
Co-reporter:Yang Li, Justin J. Wilson, Loi H. Do, Ulf-Peter Apfel and Stephen J. Lippard
Dalton Transactions 2012 - vol. 41(Issue 31) pp:NaN9275-9275
Publication Date(Web):2012/06/29
DOI:10.1039/C2DT31260C
A triptycene-based bis(benzoxazole) diacid ligand H22L2Ph4Ph4 bearing sterically encumbering groups was synthesized. Treatment of H22L2Ph4Ph4 with Fe(OTf)3 afforded a C2-symmetric trinuclear iron(III) complex, [NaFe3(L2Ph4Ph4Ph4)2(μ3-O)(μ-O2CCPh3)2(H2O)3](OTf)2 (8). The triiron core of this complex adopts the well known “basic iron acetate” structure where the heteroleptic carboxylates, comprising two Ph3CCO2− and two (L2Ph4Ph4Ph4)2− ligands, donate the six carboxylate bridges. The (L2Ph4Ph4Ph4)2− ligand undergoes only minor conformational changes upon formation of the complex.
Co-reporter:Mikael A. Minier and Stephen J. Lippard
Dalton Transactions 2015 - vol. 44(Issue 41) pp:NaN18121-18121
Publication Date(Web):2015/09/29
DOI:10.1039/C5DT02138C
A series of asymmetrically carboxylate-bridged diiron(II) complexes featuring fluorine atoms as NMR spectroscopic probes, [Fe2(PIM)(Ar4F-PhCO2)2] (10), [Fe2(F2PIM)(ArTolCO2)2] (11), and [Fe2(F2PIM)(Ar4F-PhCO2)2] (12), were prepared and characterized by X-ray crystallography, Mössbauer spectroscopy, and VT 19F NMR spectroscopy. These complexes are part of a rare family of syn N-donor diiron(II) compounds, [Fe2(X2PIM)(RCO2)2], that are structurally very similar to the active site of the hydroxylase enzyme component of reduced methane monooxygenase (MMOHred). Solution characterization of these complexes demonstrates that they undergo intramolecular carboxylate rearrangements, or carboxylate shifts, a dynamic feature relevant to the reactivity of the diiron centers in bacterial multicomponent monooxygenases.
Co-reporter:Jennifer M. Hope, Justin J. Wilson and Stephen J. Lippard
Dalton Transactions 2013 - vol. 42(Issue 9) pp:NaN3180-3180
Publication Date(Web):2012/11/06
DOI:10.1039/C2DT32462H
A platinum(II) complex of a monoanionic, tetradentate β-diketiminate (BDI) ligand with pendant quinoline arms, BDIQQH, is reported. The complex, [Pt(BDIQQ)]Cl, is emissive in DMSO, but non-emissive in aqueous buffer. Upon binding DNA in buffer, however, a 150-fold turn-on in emission intensity occurs. Dynamic light scattering and 1H NMR spectroscopy indicate that [Pt(BDIQQ)]Cl forms non-emissive aggregates in aqueous solution; DNA-binding disperses the aggregates leading to the large emission turn-on response. The cytotoxic activity of the complex, measured in two cancer cell lines, is comparable to or better than that of the established anticancer drug cisplatin.
.jpg)
![Dibenz[cd,g]indazol-6(2H)-one,5-[(3-aminopropyl)amino]-7,10-dihydroxy-2-[2-[(2-hydroxyethyl)amino]ethyl]-,hydrochloride (1:1)](http://img.cochemist.com/ccimg/118300/118201-50-6.png)
![Dibenz[cd,g]indazol-6(2H)-one,5-[(3-aminopropyl)amino]-7,10-dihydroxy-2-[2-[(2-hydroxyethyl)amino]ethyl]-,hydrochloride (1:1)](http://img.cochemist.com/ccimg/118300/118201-50-6_b.png)





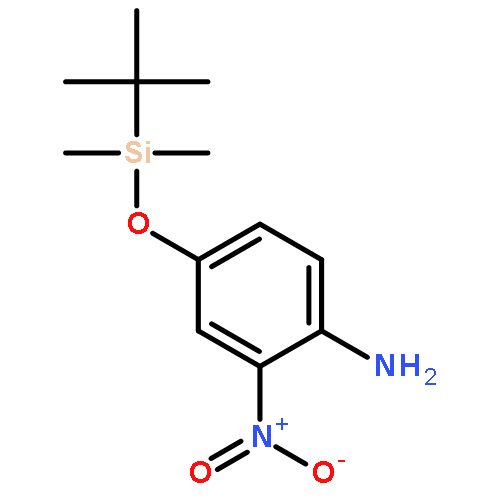








![2-hydroxy-n-[2-[(2-hydroxybenzoyl)amino]phenyl]benzamide](http://img.cochemist.com/ccimg/103600/103528-00-3.png)
![2-hydroxy-n-[2-[(2-hydroxybenzoyl)amino]phenyl]benzamide](http://img.cochemist.com/ccimg/103600/103528-00-3_b.png)
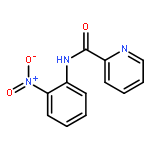
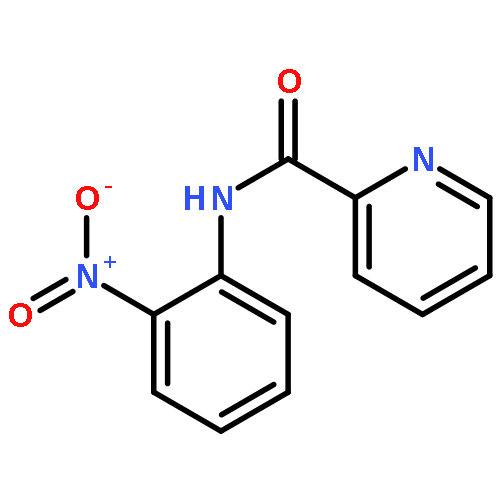
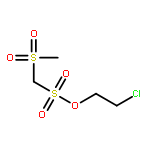
![Imidazo[5,1-d]-1,2,3,5-tetrazine-8-carboxamide,3-(2-chloroethyl)-3,4-dihydro-4-oxo-](http://img.cochemist.com/ccimg/85700/85622-95-3.png)
![Imidazo[5,1-d]-1,2,3,5-tetrazine-8-carboxamide,3-(2-chloroethyl)-3,4-dihydro-4-oxo-](http://img.cochemist.com/ccimg/85700/85622-95-3_b.png)
![9,10-ANTHRACENEDICARBOXALDEHYDE, BIS[(4,5-DIHYDRO-1H-IMIDAZOL-2-YL)HYDRAZONE], DIHYDROCHLORIDE](http://img.cochemist.com/ccimg/71500/71439-68-4.png)
![9,10-ANTHRACENEDICARBOXALDEHYDE, BIS[(4,5-DIHYDRO-1H-IMIDAZOL-2-YL)HYDRAZONE], DIHYDROCHLORIDE](http://img.cochemist.com/ccimg/71500/71439-68-4_b.png)


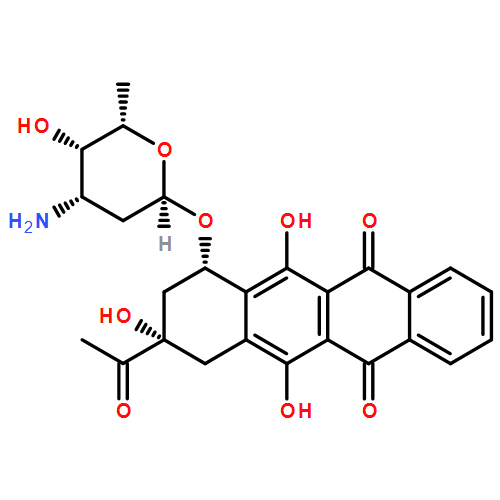
![Carbamic acid,N,N'-[2,5-bis(1-aziridinyl)-3,6-dioxo-1,4-cyclohexadiene-1,4-diyl]bis-,C,C'-diethyl ester](http://img.cochemist.com/ccimg/58000/57998-68-2.png)
![Carbamic acid,N,N'-[2,5-bis(1-aziridinyl)-3,6-dioxo-1,4-cyclohexadiene-1,4-diyl]bis-,C,C'-diethyl ester](http://img.cochemist.com/ccimg/58000/57998-68-2_b.png)
![(6R,8S,8aR)-6,7,8-tribenzyloxy-2-phenyl-4,4a,6,7,8,8a-hexahydropyrano[3,2-d][1,3]dioxine](http://img.cochemist.com/ccimg/57800/57783-66-1.png)
![(6R,8S,8aR)-6,7,8-tribenzyloxy-2-phenyl-4,4a,6,7,8,8a-hexahydropyrano[3,2-d][1,3]dioxine](http://img.cochemist.com/ccimg/57800/57783-66-1_b.png)
![D-Glucose,2-[[[(2-chloroethyl)nitrosoamino]carbonyl]amino]-2-deoxy-](http://img.cochemist.com/ccimg/54800/54749-90-5.png)
![D-Glucose,2-[[[(2-chloroethyl)nitrosoamino]carbonyl]amino]-2-deoxy-](http://img.cochemist.com/ccimg/54800/54749-90-5_b.png)
![N-[(e)-1-[(2s,4s)-4-(4-amino-5-hydroxy-6-methyloxan-2-yl)oxy-2,5,12-trihydroxy-7-methoxy-6,11-dioxo-3,4-dihydro-1h-tetracen-2-yl]ethylideneamino]benzamide](http://img.cochemist.com/ccimg/54100/54083-22-6.png)
![N-[(e)-1-[(2s,4s)-4-(4-amino-5-hydroxy-6-methyloxan-2-yl)oxy-2,5,12-trihydroxy-7-methoxy-6,11-dioxo-3,4-dihydro-1h-tetracen-2-yl]ethylideneamino]benzamide](http://img.cochemist.com/ccimg/54100/54083-22-6_b.png)


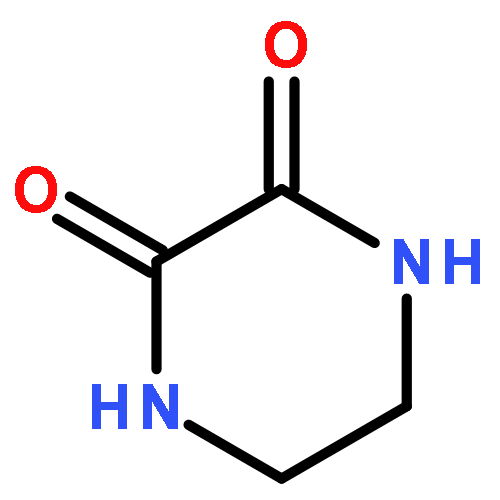

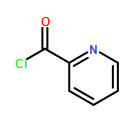


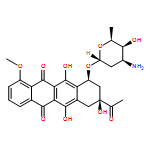
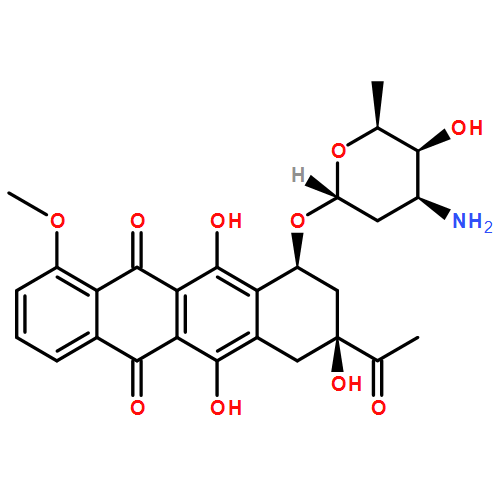


![4-(5-(Bis(2-chloroethyl)amino)-1-methyl-1H-benzo[d]imidazol-2-yl)butanoic acid](http://img.cochemist.com/ccimg/16600/16506-27-7.png)
![4-(5-(Bis(2-chloroethyl)amino)-1-methyl-1H-benzo[d]imidazol-2-yl)butanoic acid](http://img.cochemist.com/ccimg/16600/16506-27-7_b.png)
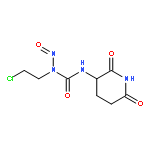
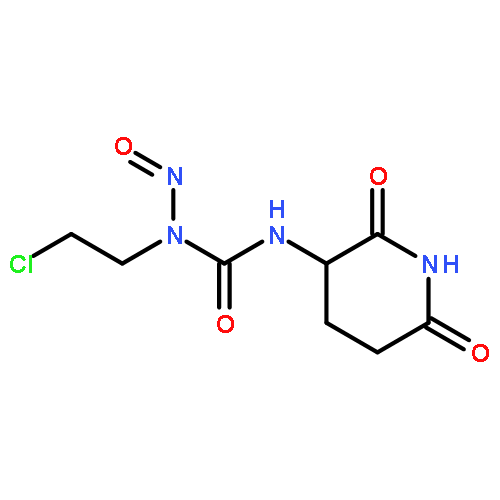
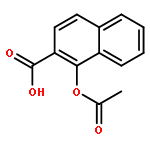
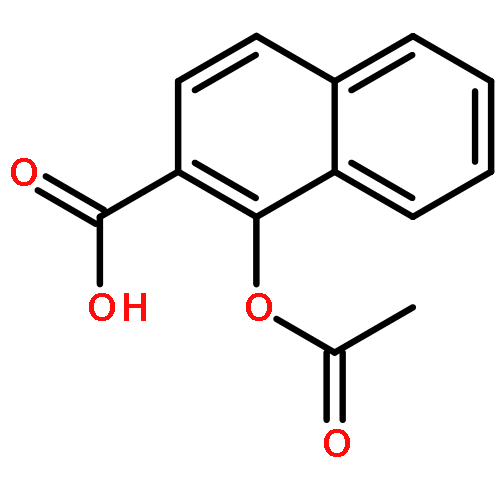






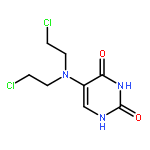
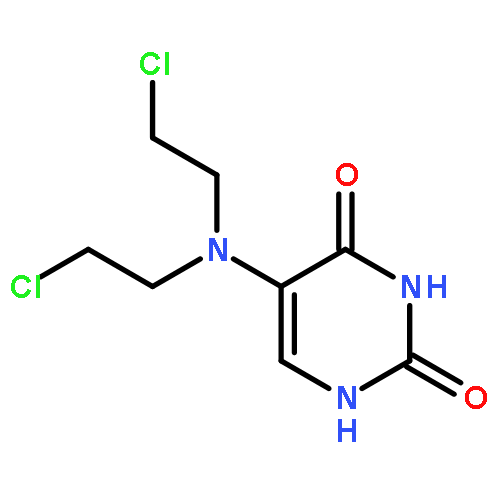





![1,4,8,11-Tetraazacyclotetradecane, 1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/128500/128495-29-4.png)
![1,4,8,11-Tetraazacyclotetradecane, 1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/128500/128495-29-4_b.png)
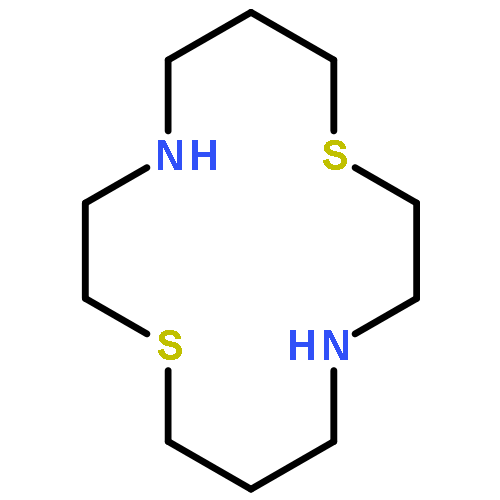







![3-Pyridinecarboxaldehyde, 6-benzo[b]thien-2-yl-](/data/chemimg/3781800/1314880-65-3.png)
![3-Pyridinecarboxaldehyde, 6-benzo[b]thien-2-yl-](/data/chemimg/3781800/1314880-65-3_b.png)






![2-Pyridinemethanamine, 4-methoxy-N,N-bis[(4-methoxy-3,5-dimethyl-2-pyridinyl)methyl]-3,5-dimethyl-](/data/chemimg/178100/1006899-82-6.png)
![2-Pyridinemethanamine, 4-methoxy-N,N-bis[(4-methoxy-3,5-dimethyl-2-pyridinyl)methyl]-3,5-dimethyl-](/data/chemimg/178100/1006899-82-6_b.png)




![METHYL 3-[(1,3-DIOXO-1,3-DIHYDRO-2H-ISOINDOL-2-YL)METHYL]BENZOATE](http://img.cochemist.com/ccimg/781700/781632-38-0.png)
![METHYL 3-[(1,3-DIOXO-1,3-DIHYDRO-2H-ISOINDOL-2-YL)METHYL]BENZOATE](http://img.cochemist.com/ccimg/781700/781632-38-0_b.png)





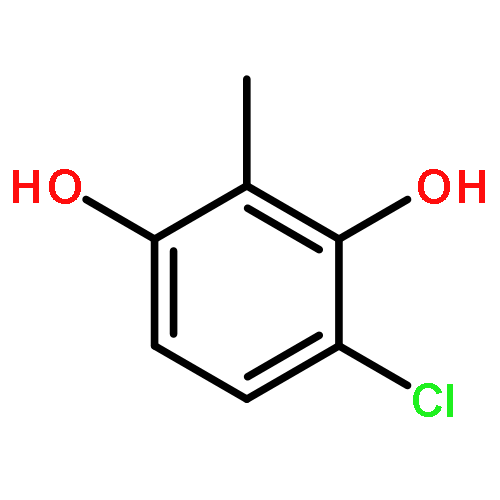



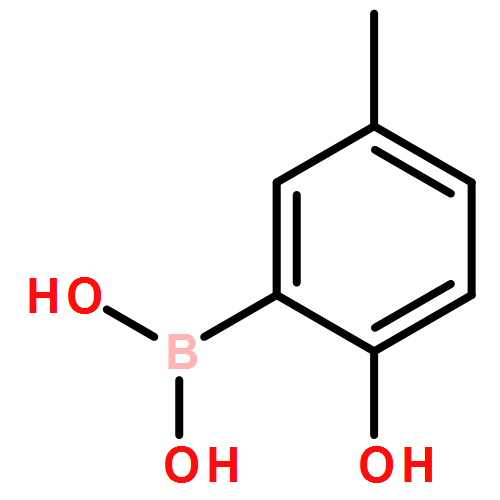






![[1,1':3',1''-Terphenyl]-2'-carboxylic acid, 5'-methyl-](http://img.cochemist.com/ccimg/171700/171669-68-4.png)
![[1,1':3',1''-Terphenyl]-2'-carboxylic acid, 5'-methyl-](http://img.cochemist.com/ccimg/171700/171669-68-4_b.png)






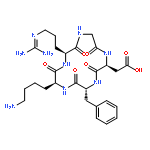
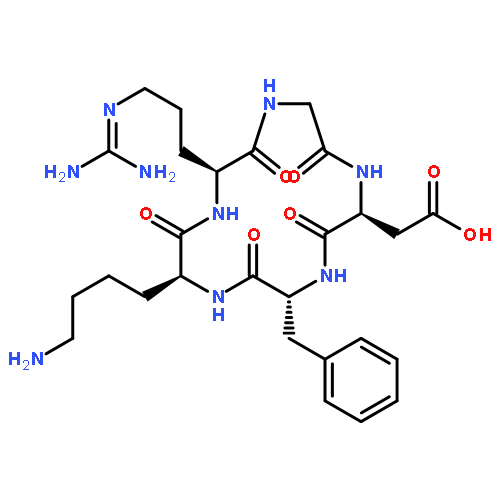
![5-[[2-(4-BROMOPHENOXY)ACETYL]CARBAMOTHIOYLAMINO]-2-CHLOROBENZOIC ACID](/data/chemimg/532100/144489-10-1.png)
![5-[[2-(4-BROMOPHENOXY)ACETYL]CARBAMOTHIOYLAMINO]-2-CHLOROBENZOIC ACID](/data/chemimg/532100/144489-10-1_b.png)
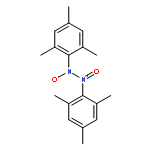

![1H-1,4,7-Triazonine, octahydro-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/129500/129422-96-4.png)
![1H-1,4,7-Triazonine, octahydro-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/129500/129422-96-4_b.png)


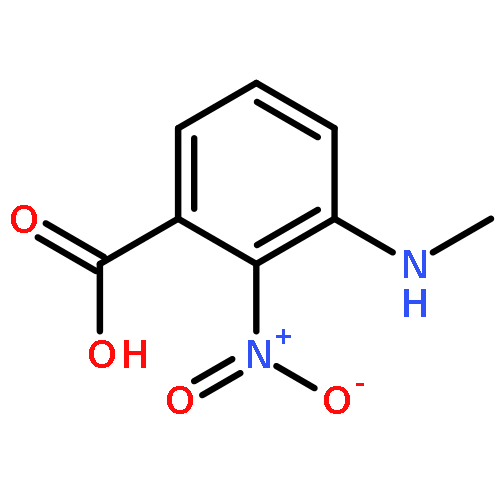
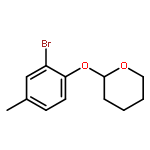

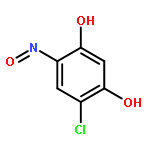
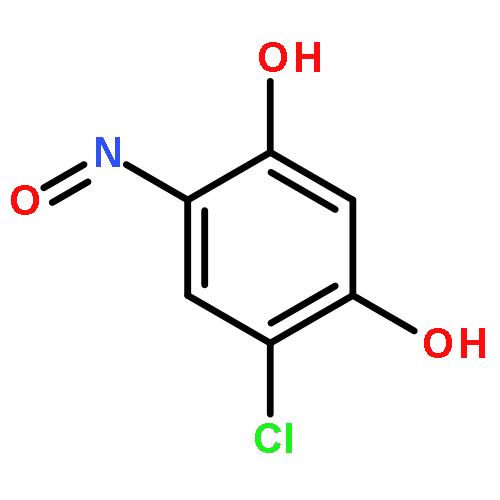


![D-Tryptophan, N-[(1,1-dimethylethoxy)carbonyl]-1-methyl-](http://img.cochemist.com/ccimg/104000/103943-63-1.png)
![D-Tryptophan, N-[(1,1-dimethylethoxy)carbonyl]-1-methyl-](http://img.cochemist.com/ccimg/104000/103943-63-1_b.png)


![1H-1,4,7-Triazonine, octahydro-1,4-bis[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/95400/95388-08-2.png)
![1H-1,4,7-Triazonine, octahydro-1,4-bis[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/95400/95388-08-2_b.png)
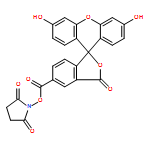
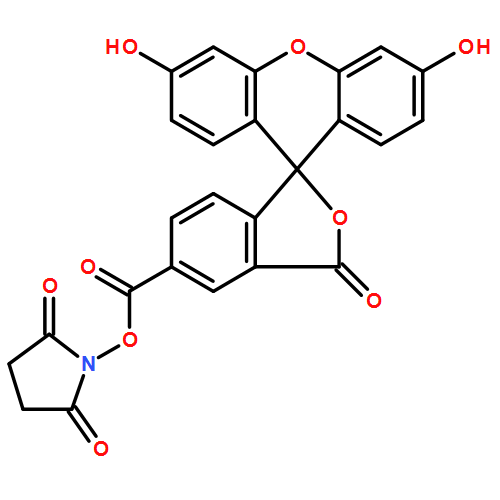








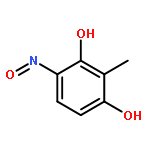
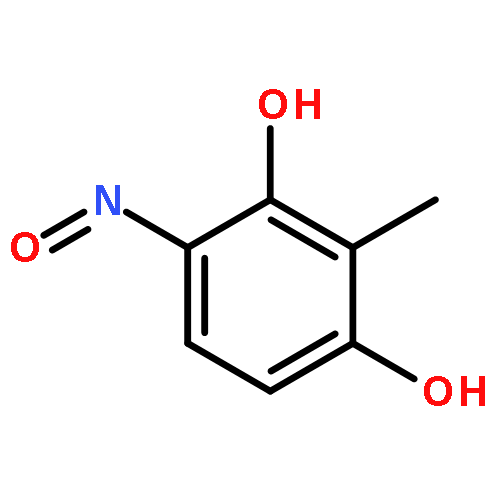


![11-oxo-2,3,5,6,7,11-Hexahydro-1H-pyrano[2,3-f]pyrido[3,2,1-ij]quinoline-10-carboxylic acid](/data/chemimg/86700/55804-65-4.png)
![11-oxo-2,3,5,6,7,11-Hexahydro-1H-pyrano[2,3-f]pyrido[3,2,1-ij]quinoline-10-carboxylic acid](/data/chemimg/86700/55804-65-4_b.png)
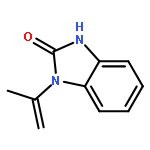
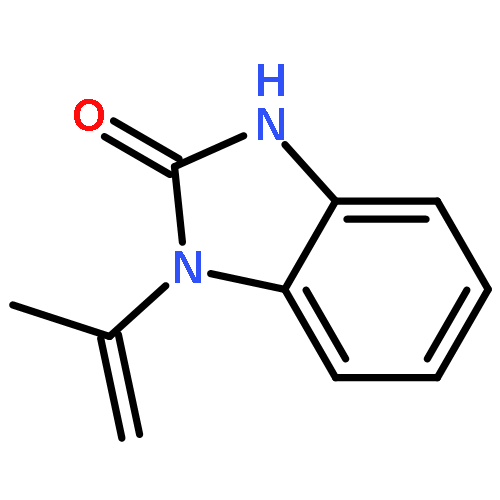
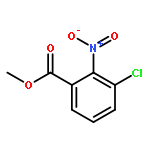
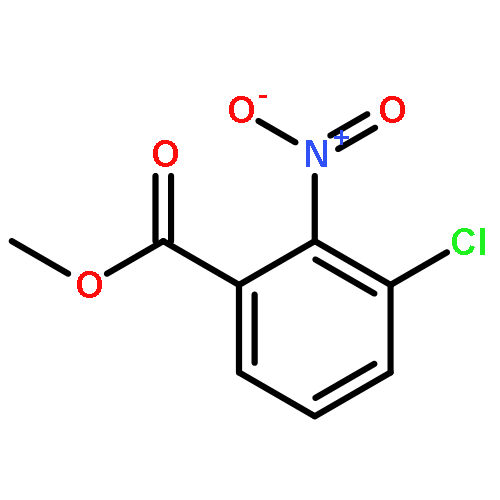


![2,5-Cyclohexadiene-1,4-dione, 3-[(2E)-3,7-dimethyl-2,6-octadien-1-yl]-5-methoxy-2-methyl-](/data/chemimg/1789400/20839-93-4.png)
![2,5-Cyclohexadiene-1,4-dione, 3-[(2E)-3,7-dimethyl-2,6-octadien-1-yl]-5-methoxy-2-methyl-](/data/chemimg/1789400/20839-93-4_b.png)
![Butanedioic acid,1-[3,4-dihydro-2,5,7,8-tetramethyl-2-(4,8,12-trimethyltridecyl)-2H-1-benzopyran-6-yl]ester](http://img.cochemist.com/ccimg/17500/17407-37-3.png)
![Butanedioic acid,1-[3,4-dihydro-2,5,7,8-tetramethyl-2-(4,8,12-trimethyltridecyl)-2H-1-benzopyran-6-yl]ester](http://img.cochemist.com/ccimg/17500/17407-37-3_b.png)


![Platinate(2-),dichloro[[N,N'-1,2-ethanediylbis[glycinato-kN]](2-)]-, dihydrogen, (SP-4-2)- (9CI)](http://img.cochemist.com/ccimg/16800/16786-97-3.png)
![Platinate(2-),dichloro[[N,N'-1,2-ethanediylbis[glycinato-kN]](2-)]-, dihydrogen, (SP-4-2)- (9CI)](http://img.cochemist.com/ccimg/16800/16786-97-3_b.png)
![Iron, chloro[5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-12)-](/data/chemimg/1024600/16456-81-8.png)
![Iron, chloro[5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-12)-](/data/chemimg/1024600/16456-81-8_b.png)


















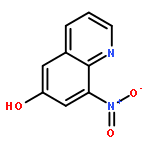
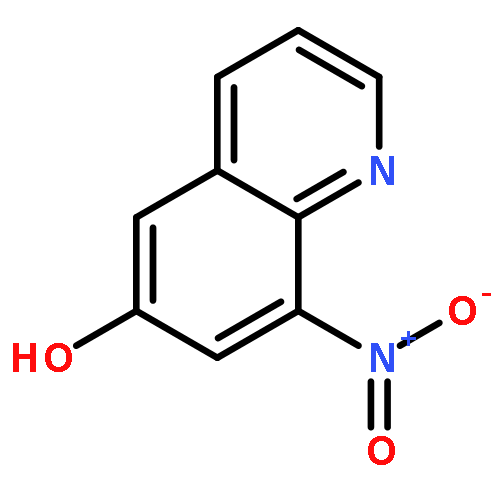


![6-({[5-(DIMETHYLAMINO)-1-NAPHTHYL]SULFONYL}AMINO)HEXANOIC ACID](http://img.cochemist.com/ccimg/3400/3353-33-1.png)
![6-({[5-(DIMETHYLAMINO)-1-NAPHTHYL]SULFONYL}AMINO)HEXANOIC ACID](http://img.cochemist.com/ccimg/3400/3353-33-1_b.png)








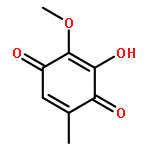
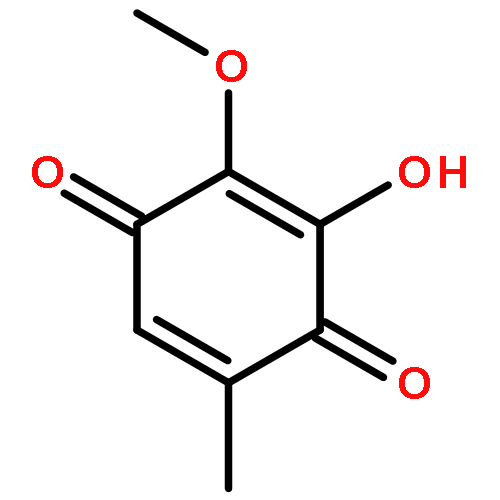
![Benzo[f]quinoline](http://img.cochemist.com/ccimg/100/85-02-9.png)
![Benzo[f]quinoline](http://img.cochemist.com/ccimg/100/85-02-9_b.png)










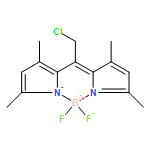
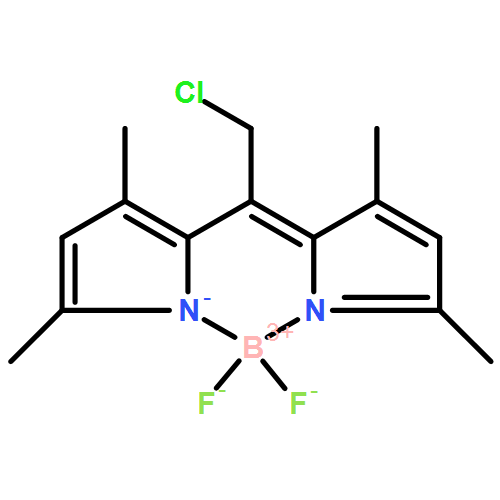


![Acetic acid,2-[[2-methyl-8-[[(4-methylphenyl)sulfonyl]amino]-6-quinolinyl]oxy]-](http://img.cochemist.com/ccimg/151700/151606-29-0.png)
![Acetic acid,2-[[2-methyl-8-[[(4-methylphenyl)sulfonyl]amino]-6-quinolinyl]oxy]-](http://img.cochemist.com/ccimg/151700/151606-29-0_b.png)
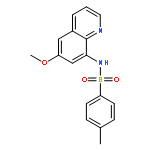
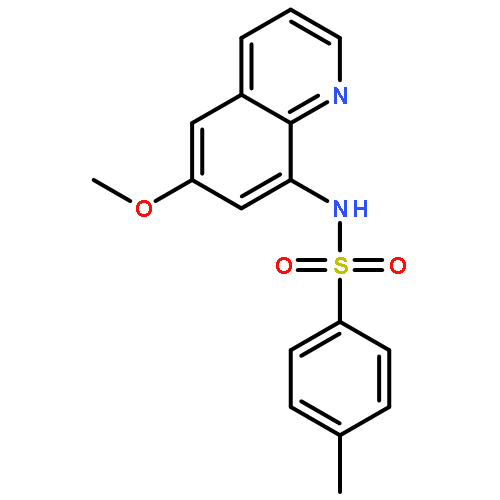
![1,3-BIS[BIS[(1-ETHYLBENZIMIDAZOL-2-YL)METHYL]AMINO]PROPAN-2-OL](http://img.cochemist.com/ccimg/79800/79724-80-4.png)
![1,3-BIS[BIS[(1-ETHYLBENZIMIDAZOL-2-YL)METHYL]AMINO]PROPAN-2-OL](http://img.cochemist.com/ccimg/79800/79724-80-4_b.png)
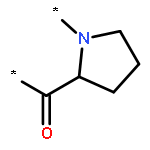
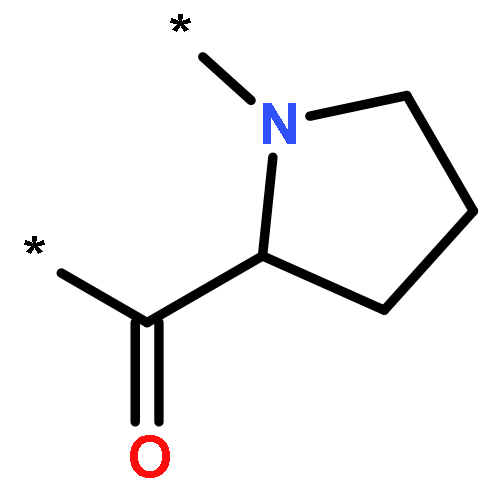















![1,1',1''-[(nitrososulfanyl)methanetriyl]tribenzene](http://img.cochemist.com/ccimg/6400/6316-86-5.png)
![1,1',1''-[(nitrososulfanyl)methanetriyl]tribenzene](http://img.cochemist.com/ccimg/6400/6316-86-5_b.png)
![Acetic acid, [(8-amino-2-methyl-6-quinolinyl)oxy]-, ethyl ester](http://img.cochemist.com/ccimg/257900/257892-18-5.png)
![Acetic acid, [(8-amino-2-methyl-6-quinolinyl)oxy]-, ethyl ester](http://img.cochemist.com/ccimg/257900/257892-18-5_b.png)
![[1,1':3',1''-Terphenyl]-2'-carboxylic acid, 4,4''-dimethyl-](http://img.cochemist.com/ccimg/155200/155186-06-4.png)
![[1,1':3',1''-Terphenyl]-2'-carboxylic acid, 4,4''-dimethyl-](http://img.cochemist.com/ccimg/155200/155186-06-4_b.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)
![2-(4,5-bis{[bis(pyridin-2-ylmethyl)ammonio]methyl}-2,7-dichloro-6-oxido-3-oxo-3H-xanthen-9-yl)benzoate](/data/chemimg/461100/288574-78-7.png)
![2-(4,5-bis{[bis(pyridin-2-ylmethyl)ammonio]methyl}-2,7-dichloro-6-oxido-3-oxo-3H-xanthen-9-yl)benzoate](/data/chemimg/461100/288574-78-7_b.png)