Co-reporter:Joshua K. G. Karlsson, Owen J. Woodford, Roza Al-Aqar, and Anthony Harriman
The Journal of Physical Chemistry A November 16, 2017 Volume 121(Issue 45) pp:8569-8569
Publication Date(Web):October 19, 2017
DOI:10.1021/acs.jpca.7b06440
Erythrosine, a popular food dye, undergoes fast O2-sensitive bleaching in water when subjected to visible light illumination. In dilute solution, erythrosine undergoes photobleaching via first-order kinetics, where the rate of bleaching depends critically on the rate of photon absorption and on the concentration of dissolved oxygen. Kinetic studies indicate that this inherent bleaching is augmented by self-catalysis at higher concentrations of erythrosine and on long exposure times. Under the conditions used, bleaching occurs by way of geminate attack of singlet molecular oxygen on the chromophore. Despite the complexity of the overall photobleaching process, the rate constants associated with both inherent and self-catalytic bleaching reactions follow Arrhenius-type behavior, allowing the activation parameters to be resolved. Bleaching remains reasonably efficient in the solid state, especially if the sample is damp, and provides a convenient means by which to construct a simple chemical actinometer.
Co-reporter:Patrycja Stachelek, Abdulrahman A. Alsimaree, Rua B. Alnoman, Anthony Harriman, and Julian G. Knight
The Journal of Physical Chemistry A March 16, 2017 Volume 121(Issue 10) pp:2096-2096
Publication Date(Web):February 28, 2017
DOI:10.1021/acs.jpca.6b11131
A small series of boron dipyrromethene (BODIPY) dyes has been synthesized whereby the boron atom is constrained in a five-membered ring formed from either o-dihydroxypyridine or o-aminophenol. In the latter case, the amino group has been converted into the corresponding amide derivative so as to curtail the possibility for light-induced charge transfer from strap to BODIPY. These compounds are weakly emissive in fluid solution but cleavage of the strap, by treatment with a photoacid generator, restores strong fluorescence. Surprisingly, the same compounds remain weakly fluorescent in a rigid glass at 80 K where light-induced charge transfer is most unlikely. In fluid solution, the fluorescence quantum yield increases with increasing temperature due to a thermally activated step but does not correlate with the thermodynamics for intramolecular charge transfer. It is proposed that the strap causes rupture of the potential energy surface for the excited state, creating traps that provide new routes by which the wave packet can return to the ground state. Access to the trap from the excited state is reversible, leading to the delayed emission. Analysis of the temperature dependent emission intensities allows estimation of the kinetic parameters associated with entering and leaving the trap.
Co-reporter:Dumitru Sirbu, Andrew C. Benniston, and Anthony Harriman
Organic Letters April 7, 2017 Volume 19(Issue 7) pp:
Publication Date(Web):March 20, 2017
DOI:10.1021/acs.orglett.7b00435
Tin(IV) catalysis allows isolation of a boron dipyrromethene derivative bearing a solitary strap around the boron center. The conditions favor internal cyclization without contamination by side products and provide high yields of product in good purity. A phenolate-based strap imposes chirality and causes geometrical distortion of the dipyrrin. Relatively strong fluorescence is observed for single crystals, evaporated films, and adsorbed layers. Single-crystal absorption and emission spectra resemble those observed from solution with contributions from a dimer.
Co-reporter:Patrycja Stachelek and Anthony Harriman
The Journal of Physical Chemistry A 2016 Volume 120(Issue 41) pp:8104-8113
Publication Date(Web):September 23, 2016
DOI:10.1021/acs.jpca.6b08284
A small series of closely spaced, bichromophoric boron dipyrromethene (BODIPY) derivatives has been examined by optical spectroscopy and compared to the corresponding mononuclear dyes. The compounds vary according to the site of attachment and also by the nature of alkyl or aryl substituents incorporated into the dipyrrin backbone. Excitonic coupling splits the lowest-energy absorption transition in each case, but to highly variable degrees. There are also marked changes in the fluorescence quantum yields across the series but much less variation in the excited-state lifetimes. After comparing different models, it is concluded that the ideal dipole approximation gives a crude qualitative representation of the observed splitting of the absorption transition, but the extended dipole approach is not applicable to these systems. Agreement is substantially improved by employing a model that takes into account the dihedral angle between the planes of the two dipyrrin units. The large variation in radiative rate constants, and those for the accompanying nonradiative processes, is accountable in terms of electronic coupling and/or intensity borrowing between the two excitonic states. In all cases, the dihedral angle between the two BODIPY units plays a key role.
Co-reporter:Joshua K. G. Karlsson and Anthony Harriman
The Journal of Physical Chemistry A 2016 Volume 120(Issue 16) pp:2537-2546
Publication Date(Web):April 5, 2016
DOI:10.1021/acs.jpca.6b01278
The optical properties are compared for two boron dipyrromethene (BODIPY) dyes that differ by virtue of the substituent at the meso-site, namely, aza-N versus C-methine atoms. Both compounds are equipped with aryl rings at the 3- and 5-positions of the dipyrrin backbone, which help to extend the degree of π-delocalization. The aza-BODIPY dye absorbs and fluoresces at much lower energy than does the conventional BODIPY dye, with red shifts of about 100 nm being observed in fluid solution, but with comparable fluorescence yield and lifetime. Hydrogen bonding donors, such as alcohols, attach to the aza-N atom and promote nonradiative decay without affecting the properties of the conventional dye. Triplet formation is ineffective in the absence of a spin-orbit coupler. Quantum chemical calculations indicate that the electronegative aza-N atom lowers the energy of the LUMO while having little effect on the corresponding HOMO energy. The resultant decrease in the HOMO–LUMO energy gap is primarily responsible for the red shift. The HOMO–LUMO energy gap is also affected by the dihedral angle subtended by the aryl rings, but this is insensitive to the geometry around the central 6-membered ring. The aza-N atom, by virtue of restricting spatial overlap between the HOMO and LUMO, decreases the energy gap between excited-singlet and -triplet states.
Co-reporter:Anthony Harriman, Patrycja Stachelek, Alexandra Sutter and Raymond Ziessel
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 39) pp:26175-26182
Publication Date(Web):28 Aug 2015
DOI:10.1039/C5CP03932K
An extended molecular array, comprising three distinct types of chromophores and two additional redox-active subunits, that harvests photons over most of the visible spectral range has been synthesized and characterised. The array exhibits a rich variety of electrochemical waves when examined by cyclic voltammetry but assignment can be made on the basis of control compounds and molecular orbital calculations. Stepwise electronic energy transfer occurs along the molecular axis, corresponding to a gradient of excitation energies, to populate the lowest-energy excited state of the ultimate acceptor. The latter species, which absorbs and emits in the far-red region, enters into light-induced charge transfer with a terminal amine group. The array is relatively stable under illumination with white light but degrades slowly via a series of well-defined steps, the first of which is autocatalytic. One of the main attributes of this system is the capability to harvest an unusually high fraction of sunlight while providing protection against exposure to UV light.
Co-reporter:Sébastien Azizi, Gilles Ulrich, Maud Guglielmino, Stéphane le Calvé, Jerry P. Hagon, Anthony Harriman, and Raymond Ziessel
The Journal of Physical Chemistry A 2015 Volume 119(Issue 1) pp:39-49
Publication Date(Web):December 4, 2014
DOI:10.1021/jp5078246
It is noted that, for a small series of 3,5-diacetyl-1,4-dihydrolutidine (DDL) derivatives and the corresponding Hantzsch esters, the presence of methyl groups at the 2,6-positions serves to extinguish fluorescence in solution but not in the solid state. Emission is weakly activated and affected by changes in solvent polarity. The latter situation arises because the optical transition involves intramolecular charge transfer. Calculations, both semiempirical and DFT, indicate that, in all cases, rotation of the carbonyl function is facile and that the dihydropyridine ring is planar. These calculations also indicate that the 2,6-methyl groups do not affect the generic structure of the molecule. It is proposed that illumination increases the molecular dipole moment and pushes electron density toward the carbonyl oxygen atom. Proton transfer can now occur from one of the methyl groups, leading to formation of a relatively low-energy, neutral intermediate, followed by a second proton transfer step that forms the enol. Reaction profiles computed for the ground-state species indicate that this route is highly favored relative to hydrogen transfer from the 4-position. The barriers for light-induced proton transfer are greatly reduced relative to the ground-state process but such large-scale structural transformations are hindered in the solid state. A rigid analogue that cannot form an enol is highly emissive in solution, supporting the conclusion that proton transfer is in competition to fluorescence in solution.
Co-reporter:Gordon J. Hedley, Arvydas Ruseckas, Andrew C. Benniston, Anthony Harriman, and Ifor D. W. Samuel
The Journal of Physical Chemistry A 2015 Volume 119(Issue 51) pp:12665-12671
Publication Date(Web):November 28, 2015
DOI:10.1021/acs.jpca.5b08640
Electronic energy transfer (EET) from a donor to an acceptor is an important mechanism that controls the light harvesting efficiency in a wide variety of systems, including artificial and natural photosynthesis and contemporary photovoltaic technologies. The detailed mechanism of EET at short distances or large angles between the donor and acceptor is poorly understood. Here the influence of the orientation between the donor and acceptor on EET is explored using a molecule with two nearly perpendicular chromophores. Very fast EET with a time constant of 120 fs is observed, which is at least 40 times faster than the time predicted by Coulombic coupling calculations. Depolarization of the emission signal indicates that the transition dipole rotates through ca. 64°, indicating the near orthogonal nature of the EET event. The rate of EET is found to be similar to structural relaxation rates in the photoexcited oligothiophene donor alone, which suggests that this initial relaxation brings the dyad to a conical intersection where the excitation jumps to the acceptor.
Co-reporter:Dr. Mohammed A. H. Alamiry; Dr. Anthony Harriman;Dr. Alexre Haefele;Dr. Raymond Ziessel
ChemPhysChem 2015 Volume 16( Issue 9) pp:1867-1872
Publication Date(Web):
DOI:10.1002/cphc.201500150
Abstract
The target artificial light-harvesting antenna, comprising 21 discrete chromophores arranged in a logical order, undergoes photochemical bleaching when dispersed in a thin plastic film. The lowest-energy component, which has an absorption maximum at 660 nm, bleaches through first-order kinetics at a relatively fast rate. The other components bleach more slowly, in part, because their excited-state lifetimes are rendered relatively short by virtue of fast intramolecular electronic energy transfer to the terminal acceptor. Two of the dyes, these being close to the terminal acceptor but interconnected through a reversible energy-transfer step, bleach by way of an autocatalytic step. Loss of the terminal acceptor, thereby switching off the energy-transfer route, escalates the rate of bleaching of these ancillary dyes. The opposite terminal, formed by a series of eight pyrene-based chromophores, does not bleach to any significant degree. Confirmation of the various bleaching steps is obtained by examination of an antenna lacking the terminal acceptor, where the autocatalytic route does not exist and bleaching is very slow.
Co-reporter:Dr. Mohammed A. H. Alamiry; Dr. Anthony Harriman;Dr. Alexre Haefele;Dr. Raymond Ziessel
ChemPhysChem 2015 Volume 16( Issue 9) pp:
Publication Date(Web):
DOI:10.1002/cphc.201500402
Abstract
The front cover artwork is provided by the groups of Tony Harriman (Newcastle University) and Raymond Ziessel (ECPM-Strasbourg). The image shows the processes following illumination of an artificial light harvester that slowly fades under continuous exposure to white light. Read the full text of the article at 10.1002/cphc.201500150.
Co-reporter:Dr. Mohammed A. H. Alamiry; Dr. Anthony Harriman;Dr. Alexre Haefele;Dr. Raymond Ziessel
ChemPhysChem 2015 Volume 16( Issue 9) pp:
Publication Date(Web):
DOI:10.1002/cphc.201590046
Co-reporter:Adela Nano, Pascal Retailleau, Jerry P. Hagon, Anthony Harriman and Raymond Ziessel
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 21) pp:10187-10198
Publication Date(Web):24 Mar 2014
DOI:10.1039/C3CP55021D
A new type of fluorescent pH indicator has been developed whereby two dissimilar amino-styryl units are attached to a boron dipyrromethene (Bodipy) dye. The photophysical properties of this hybrid dye, and its simpler counterparts bearing only a single amino-styryl residue, depend on the polarity of the surrounding medium. Of the two terminal amines, DFT (B3LYP/6-31G**) calculations and spectroscopic measurements support the notion that julolidine is oxidised and protonated under milder conditions than is N,N-dimethylaniline. For the hybrid dye, similar DFT calculations carried out for the mono-protonated analogues indicate that the julolidine residue is the stronger base while the resultant conjugate acid is the weaker one. Absorption and fluorescence spectroscopic titrations show that protonation of the hybrid dye occurs in two well-resolved steps, whereby addition of the first proton introduces a thermodynamic barrier for entry of the second. In the hybrid dye, the pKA values for the respective conjugate acids differ markedly from those derived for the mono-amino-styryl dyes and display negative co-operativity. Effectively, this means that electronic interactions running along the molecular backbone make it more difficult, relative to the individual dyes, to protonate both amino sites. As such, this dye operates as a probe over an unusually wide pH range.
Co-reporter:Anthony Harriman
European Journal of Inorganic Chemistry 2014 Volume 2014( Issue 4) pp:573-580
Publication Date(Web):
DOI:10.1002/ejic.201301540
Abstract
This essay is based on research leading to the identification of catalysts capable of the selective oxidation of water to molecular oxygen. The real need for such materials relates to the large-scale photochemical dissociation of water into its constituent elements, as opposed, for example, to the electrochemical decomposition of water at macroscopic electrodes. Combining the catalyst with the essential components needed for efficacious photochemistry brings special challenges, as does the ultimate need to scale up the system by a massive amount. Nature has developed a highly successful process for O2 evolution under ambient illumination that makes use of a cubane tetramanganese cluster having a closely associated calcium cation in attendance. This catalyst is surprisingly delicate and it is debatable as to whether we could adapt such a system for use with artificial photosystems. Historically the latter have used colloidal metal oxides, and consideration is given here as to which materials might offer the most promising catalytic performance. Moving towards heterogeneous systems has more practical meaning, but the same materials come to mind. Recent attention to cobalt-based catalysts is highlighted as a possible breakthrough that might lead to interesting electrochemical systems when combined with wind turbines. Molecular catalysts provide interesting opportunities for photochemical O2 evolution but suffer from problems of scale-up. This field has witnessed the most important progress over the past decade or so but still needs urgent attention if advanced materials are to be identified in a timely manner. Finally, consideration is given to the actual status of the field in specific terms of developing an effective artificial photosynthetic apparatus. Moving progressively from using sacrificial redox agents as a simple means to isolate the oxidative photochemical cycle towards full water cleavage will stimulate the development of demonstration models suitable for public display. The reward should be increased investment.
Co-reporter:Effat Bahaidarah, Anthony Harriman, Patrycja Stachelek, Sandra Rihn, Elodie Heyer and Raymond Ziessel
Photochemical & Photobiological Sciences 2014 vol. 13(Issue 10) pp:1397-1401
Publication Date(Web):28 Jul 2014
DOI:10.1039/C4PP00204K
The ability of an unconstrained boron dipyrromethene dye to report on changes in local viscosity is improved by appending a single aryl ring at the lower rim of the dipyrrin core. Recovering the symmetry by attaching an identical aryl ring on the opposite side of the lower rim greatly diminishes the sensory activity, as does blocking rotation of the meso-aryl group. On the basis of viscosity- and temperature-dependence studies, together with quantum chemical calculations, it is proposed that a single aryl ring at the 3-position extends the molecular surface area that undergoes structural distortion during internal rotation. The substitution pattern at the lower rim also affects the harmonic frequencies at the bottom of the potential well and at the top of the barrier. These effects can be correlated with the separation of the H1,H7 hydrogen atoms.
Co-reporter:Adela Nano;Dr. Raymond Ziessel;Patrycja Stachelek;Dr. Mohammed A. H. Alamiry; Dr. Anthony Harriman
ChemPhysChem 2014 Volume 15( Issue 1) pp:177-186
Publication Date(Web):
DOI:10.1002/cphc.201300805
Abstract
The photophysical properties of a prototypic donor–acceptor dyad, featuring a conventional boron dipyrromethene (Bodipy) dye linked to a dicyanovinyl unit through a meso-phenylene ring, have been recorded in weakly polar solvents. The absorption spectrum remains unperturbed relative to that of the parent Bodipy dye but the fluorescence is extensively quenched. At room temperature, the emission spectrum comprises roughly equal contributions from the regular π, π* excited-singlet state and from an exciplex formed by partial charge transfer from Bodipy to the dicyanovinyl residue. This mixture moves progressively in favor of the locally excited π, π* state on cooling and the exciplex is no longer seen in frozen media; the overall emission quantum yield changes dramatically near the freezing point of the solvent. The exciplex, which has a lifetime of approximately 1 ns at room temperature, can also be seen by transient absorption spectroscopy, in which it decays to form the locally excited triplet state. Under applied pressure (P<170 MPa), formation of the exciplex is somewhat hindered by restricted rotation around the semirigid linkage and again the emission profile shifts in favor of the π, π* excited state. At higher pressure (170<P<550 MPa), the molecule undergoes reversible distortion that has a small effect on the yield of π, π* emission but severely quenches exciplex fluorescence. In the limiting case, this high-pressure effect decreases the molar volume of the solute by approximately 25 cm3 and opens a new channel for nonradiative deactivation of the excited-state manifold.
Co-reporter:Raymond Ziessel ; Gilles Ulrich ; Alexandre Haefele
Journal of the American Chemical Society 2013 Volume 135(Issue 30) pp:11330-11344
Publication Date(Web):July 3, 2013
DOI:10.1021/ja4049306
An artificial light-harvesting array, comprising 21 discrete chromophores arranged in a rational manner, has been synthesized and characterized fully. The design strategy follows a convergent approach that leads to a molecular-scale funnel, having an effective chromophore concentration of 0.6 M condensed into ca. 55 nm3, able to direct the excitation energy to a focal point. A cascade of electronic energy-transfer steps occurs from the rim to the focal point, with the rate slowing down as the exciton moves toward its ultimate target. Situated midway along each branch of the V-shaped array, two chromophoric relays differ only slightly in terms of their excitation energies, and this situation facilitates reverse energy transfer. Thus, the excitation energy becomes spread around the array, a situation reminiscent of a giant holding pattern for the photon that can sample many different chromophores before being trapped by the terminal acceptor. At high photon flux under conditions of relatively slow off-load to a device, such as a solar cell, electronic energy transfer encounters one or more barriers that hinder forward progress of the exciton and thereby delays arrival of the second photon. Preliminary studies have addressed the ability of the array to function as a sensitizer for amorphous silicon solar cells.
Co-reporter:Delphine Hablot, Raymond Ziessel, Mohammed A. H. Alamiry, Effat Bahraidah and Anthony Harriman
Chemical Science 2013 vol. 4(Issue 1) pp:444-453
Publication Date(Web):17 Oct 2012
DOI:10.1039/C2SC21505E
A series of molecular dyads has been synthesized and fully characterised. These linear, donor–spacer–acceptor compounds comprise terminal dyes selected to exhibit intramolecular electronic energy transfer (EET) along the molecular axis. The spacer is built by accretion of ethynylene–carborane units that give centre-to-centre separation distances of 38, 57, 76, 96, and 115 Å respectively along the series. The probability of one-way EET between terminals depends on the length of the spacer but also on temperature and applied pressure. Throughout the series, the derived EET parameters are well explained in terms of through-space interactions but the probability of EET is higher than predicted for the fully extended conformation except in a glassy matrix at low temperature. The implication is that these spacers contract under ambient conditions, with the extent of longitudinal contraction increasing under pressure but decreasing as the temperature is lowered. Longer bridges are more susceptible to such distortion, which is considered to resemble a concertina effect caused by out-of-plane bending of individual subunits. The dynamics of EET can be used to estimate the strain energy associated with molecular contraction, the amount of work done in effecting the structural change and the Young's modulus for the bridge.
Co-reporter:Dr. Anthony Harriman;Dr. Mohammed A. H. Alamiry;Dr. Jerry P. Hagon;Delphine Hablot;Dr. Raymond Ziessel
Angewandte Chemie International Edition 2013 Volume 52( Issue 26) pp:6611-6615
Publication Date(Web):
DOI:10.1002/anie.201302081
Co-reporter:Dr. Anthony Harriman;Dr. Mohammed A. H. Alamiry;Dr. Jerry P. Hagon;Delphine Hablot;Dr. Raymond Ziessel
Angewandte Chemie 2013 Volume 125( Issue 26) pp:6743-6747
Publication Date(Web):
DOI:10.1002/ange.201302081
Co-reporter:Adela Nano;Dr. Raymond Ziessel;Patrycya Stachelek;Dr. Anthony Harriman
Chemistry - A European Journal 2013 Volume 19( Issue 40) pp:13528-13537
Publication Date(Web):
DOI:10.1002/chem.201301045
Abstract
A small series of donor–acceptor molecular dyads has been synthesized and fully characterized. In each case, the acceptor is a dicyanovinyl unit and the donor is a boron dipyrromethene (BODIPY) dye equipped with a single styryl arm bearing a terminal amino group. In the absence of the acceptor, the BODIPY-based dyes are strongly fluorescent in the far-red region and the relaxed excited-singlet states possess significant charge-transfer character. As such, the emission maxima depend on both the solvent polarity and temperature. With the corresponding push–pull molecules, there is a low-energy charge-transfer state that can be observed by both absorption and emission spectroscopy. Here, charge-recombination fluorescence is weak and decays over a few hundred picoseconds or so to recover the ground state. Overall, these results permit evaluation of the factors affecting the probability of charge-recombination fluorescence in push–pull dyes. The photophysical studies are supported by cyclic voltammetry and DFT calculations.
Co-reporter:Mohammed A. H. Alamiry, Jerry P. Hagon, Anthony Harriman, Thomas Bura and Raymond Ziessel
Chemical Science 2012 vol. 3(Issue 4) pp:1041-1048
Publication Date(Web):12 Dec 2011
DOI:10.1039/C2SC00948J
This work examines the electronic energy-transfer (EET) processes inherent to a molecular dyad in which aryl polycycles attached to a boron dipyrromethene (Bodipy) dye act as ancillary light harvesters for near-UV photons. The solvent, being methyltetrahydrofuran, is compressed under applied pressure to such an extent that, over the accessible pressure range, there is a 25% decrease in molar volume. This effect serves to increase the effective concentration of the solute and increases fluorescence from Bodipy when this chromophore is excited directly. Illumination into the aryl polycycles, namely pyrene and perylene derivatives, leads to rapid intramolecular EET to Bodipy but fluorescence from these units is partially restored under high pressure. The argument is made that applied pressure restricts torsional motions around the linkages and imposes a near orthogonal geometry for transition dipole moment vectors on the reactants. In turn, this pressure-induced conformational restriction switches off Förster-type EET within the system, leaving the electron-exchange contribution. For the target dyad, the Förster component is ca. 5% for pyrene and ca. 25% for perylene. Such contributions are not inconsistent with calculations made on the basis of Förster theory but modelling is rendered difficult by the absence of accurate information about the nature of the conformational motion. Two possibilities have been considered. In the first case, the appendages remain stiff but pressure reduces the extent of displacement from the lowest-energy position. The results can be accounted for in a quantitative sense on the basis of small deviations from the lowest-energy conformation; the actual amount of displacement needed to explain the pressure effect depends on the method used to compute the Förster rates and ranges from ca. 4° for the ideal dipole approximation to only 0.5° for the extended dipole method. Secondly, pressure is assumed to bend each appendage into a banana-like shape. Again, the full effect of applied pressure can be accounted for by way of minor curvature of the linkage.
Co-reporter:Mohammed A. H. Alamiry, Andrew C. Benniston, Graeme Copley and Anthony Harriman
RSC Advances 2012 vol. 2(Issue 5) pp:1936-1941
Publication Date(Web):06 Jan 2012
DOI:10.1039/C2RA00848C
The viscosity of 1,2-dichloroethane increases steadily with increasing pressure, as does the density, refractive index and polarizability of this solvent. The pressure dependence for each of these properties can be monitored by a combination of absorption and fluorescence spectroscopy carried out in the presence of a fluorescent molecular rotor that responds to changes in the local environment. At 20 °C, dichloroethane freezes under an applied pressure of ca. 370 MPa, causing sudden extinction of the fluorescence of the molecular rotor due to the opaque nature of the frozen solvent. However, this same emission is enhanced dramatically if a small amount of inert polymer is present in the solution. The behaviour is interpreted in terms of the polymeric solute promoting establishment of a glassy matrix with reasonably good optical transparency for emission spectroscopy.
Co-reporter:Mohammed A. H. Alamiry, Effat Bahaidarah, Anthony Harriman, Thomas Bura and Raymond Ziessel
RSC Advances 2012 vol. 2(Issue 26) pp:9851-9859
Publication Date(Web):22 Aug 2012
DOI:10.1039/C2RA20786A
Sterically unhindered boron dipyrromethene dyes bearing aryl hydrocarbons at the meso position can function as fluorescent probes for monitoring changes in rheology of the surrounding environment. The key aspect of such behaviour relates to the ease of rotation of the aryl ring, which is set in part by frictional forces with nearby solvent molecules. For the target dye under consideration here, gyration of the meso-phenylene ring shows a pronounced temperature dependence but only a modest sensitivity towards applied pressure. Changing the specific viscosity of the solvent by adding a linear polymer has but a small effect on the fluorescence yield of the dye under ambient conditions and thereby indicates that there is little contact between dye and polymer. Under pressure in the presence of polymer, the fluorescence yield increases dramatically and allows design of an effective fluorescence-based pressure sensor. The simplest explanation of this phenomenon has the polymer wrapping around the dye under pressure and curtailing the rotary action. In addition, it has to be considered that the inert polymer renders the chloroform solvent more susceptible to a pressure-induced increase in density by minimising electrostatic repulsion between chlorine lone pairs. In this respect, the polymer acts as a lubricant for compression of chloroform under pressure.
Co-reporter:Mohammed A. H. Alamiry, Andrew C. Benniston, and Anthony Harriman
The Journal of Physical Chemistry B 2012 Volume 116(Issue 1) pp:253-260
Publication Date(Web):November 22, 2011
DOI:10.1021/jp2096955
The target dye, which is a derivative of Merocyanine 540 bearing a naphthoxazole headgroup, persists as a monomer in ethanol solution but dimerizes in water under ambient conditions. Analysis of the absorption spectrum indicates that the dimer has an oblique geometry with the two molecules being held at an angle of ca. 55°. Applying high pressure to the system forces the two molecules into closer contact, resulting in a decreased partial molar volume of 3.1 cm3. One molecule of the monomeric dye enters a neutral micelle formed from Triton X-100, where it is highly fluorescent and free of exciton coupling. The result of applied pressure on these latter systems depends on the concentration of surfactant. Above the critical micelle concentration (CMC), applied pressure has little effect other than to increase the viscosity inside the micelle. At very low surfactant concentration, applied pressure forces monomeric dye into the dimeric form, as observed in the absence of Triton X-100. It is notable, however, that the pressure effect on the dimerization constant is exaggerated in the presence of surfactant. At intermediate surfactant concentrations, applied pressure leads to a marked change in the CMC. In particular, applied pressure reduces the partial molar volume of the micelle by ca. 7.9 cm3 and induces micelle formation at relatively low concentration of surfactant. For example, the CMC falls from ca. 250 μM at atmospheric pressure to only 50 μM at 460 MPa.
Co-reporter:Loïc Le Pleux, Yann Pellegrin, Errol Blart, Fabrice Odobel, and Anthony Harriman
The Journal of Physical Chemistry A 2011 Volume 115(Issue 20) pp:5069-5080
Publication Date(Web):May 2, 2011
DOI:10.1021/jp2012182
A series of multiporphyrin clusters has been synthesized and characterized in which there exists a logical gradient for either energy or electron transfer between the porphyrins. A central free-base porphyrin (FbP), for example, is equipped with peripheral zinc(II) porphyrins (ZnP) which act as ancillary light harvesters and transfer excitation energy to the FbP under visible light illumination. Additional energy-transfer steps occur at the triplet level, and the series is expanded by including magnesium(II) porphyrins and/or tin(IV) porphyrins as chromophores. Light-induced electron transfer is made possible by incorporating a gold(III) porphyrin (AuP+) into the array. Although interesting by themselves, these clusters serve as control compounds by which to understand the photophysical processes occurring within a three-stage dendrimer comprising an AuP+ core, a second layer formed from four FbP units, and an outer layer containing 12 ZnP residues. Here, illumination into a peripheral ZnP leads to highly efficient electronic energy transfer to FbP, followed by charge transfer to the central AuP+. Charge recombination within the resultant charge-shift state is intercepted by secondary hole transfer to the ZnP, which occurs with a quantum yield of around 20%. The final charge-shift state survives for some microseconds in fluid solution at room temperature.
Co-reporter:Delphine Hablot;Dr. Anthony Harriman;Dr. Raymond Ziessel
Angewandte Chemie 2011 Volume 123( Issue 34) pp:7979-7982
Publication Date(Web):
DOI:10.1002/ange.201102065
Co-reporter:Delphine Hablot;Dr. Anthony Harriman;Dr. Raymond Ziessel
Angewandte Chemie International Edition 2011 Volume 50( Issue 34) pp:7833-7836
Publication Date(Web):
DOI:10.1002/anie.201102065
Co-reporter:Andrew C. Benniston, ;Victoria L. Whittle ;Mischa Zelzer
European Journal of Organic Chemistry 2010 Volume 2010( Issue 3) pp:523-530
Publication Date(Web):
DOI:10.1002/ejoc.200901135
Abstract
In order to examine how changes in the size of the rotary group affect the efficacy of molecular probes for monitoring changes in local viscosity, a boron dipyrromethene dyebearing a meso-phenanthrene unit has been synthesized and fully characterized. 19F NMR spectroscopy, together with molecular modelling, indicates that the bulky phenanthryl unit cannot rotate completely around the connecting C–C linkage but can oscillate over a reasonably large dihedral angle. This situation is to be contrasted with the corresponding dye having a meso-phenylene ring. The latter dye functions as a molecular probe for changes in viscosity of the surrounding solvent but remains essentially insensitive to changes in the polarity of the solvent. The opposite situation is found for the phenanthryl derivative, where a charge-transfer state lies at higher energy than the emissive π,π* excited state but can be accessed thermally. The results are considered in terms of energy-level diagrams taking into account rotational freedom. Photophysical properties are reported for both dyes in a range of solvents, and temperature-dependent studies are described.
Co-reporter:Soumyaditya Mula, Kristopher Elliott, Anthony Harriman, and Raymond Ziessel
The Journal of Physical Chemistry A 2010 Volume 114(Issue 39) pp:10515-10522
Publication Date(Web):September 13, 2010
DOI:10.1021/jp106626v
We have designed and synthesized a series of modular, dual-color dyes comprising a conventional boron dipyrromethene (Bodipy) dye, as a yellow emitter, and a Bodipy dye possessing extended conjugation that functions as a red emitter. A flexible tether of variable length, built from ethylene glycol residues, connects the terminal dyes. A critical design element of this type of dyad relates to a secondary amine linkage interposed between the conventional Bodipy and the tether. Cyclic voltammetry shows both Bodipy dyes to be electroactive and indicates that the secondary amine is quite easily oxidized. The ensuing fluorescence quenching is best explained in terms of the rapid formation of an intermediate charge-transfer state. In fact, exciplex-type emission is observed in weakly polar solvents and over a critical temperature range. In the dual-color dyes, direct excitation of the yellow emitter results in the appearance of red fluorescence, indicating that the exciplex is likely involved in the energy-transfer event, and provides for a virtual Stokes shift of 5000 cm−1. Replacing the red emitter with a higher energy absorber (namely, pyrene) facilitates the collection of near-UV light and extends the virtual Stokes shift to 8000 cm−1. Modulation of the efficacy of intramolecular energy transfer is achieved by preorganization of the connector in the presence of certain cations. This latter behavior, which is fully reversible, corresponds to an artificial allosteric effect.
Co-reporter:Dr. Raymond Ziessel;Sra Rihn;Dr. Anthony Harriman
Chemistry - A European Journal 2010 Volume 16( Issue 39) pp:11942-11953
Publication Date(Web):
DOI:10.1002/chem.201001142
Abstract
Synthetic strategies have been devised that allow the rational design and isolation of highly coloured boron dipyrromethene (BODIPY) dyes that absorb across much of the visible region. Each dye has an aryl polycycle (usually pyrene or perylene) connected to the central BODIPY core through a conjugated tether at the 3,5-positions. Both mono- and difunctionalised derivatives are accessible, in certain cases containing both pyrene and perylene residues. For all new compounds, the photophysical properties have been recorded in solution at ambient temperature and in a glassy matrix at 77 K. The presence of the aryl polycycle(s) affects the absorption and emission maxima of the BODIPY nucleus, thereby confirming that these units are coupled electronically. Indeed, the band maxima and oscillator strengths depend on the conjugation length of the entire molecule, whereas there is no sign of fluorescence from the polycycle. As a consequence, the radiative rate constant tends to increase with each added appendage. The nature of the linkage (styryl, ethenyl, or ethynyl) also exerts an effect on the photophysical properties and, in particular, the absorption spectrum is perturbed in the region of the aryl polycycle. The perylene-containing BODIPY derivatives absorb over a wide spectral range and emit in the far-red region in almost quantitative yield. A notable exception to this generic behaviour is provided by the anthracenyl derivative, which exhibits charge-transfer absorption and emission spectra in weakly polar media at ambient temperature. Regular BODIPY-like behaviour is restored in a glassy matrix at 77 K. Overall, these new dyes represent an important addition to the range of strongly absorbing and emitting reagents that could be used as solar concentrators.
Co-reporter:Anthony Harriman ; Laura J. Mallon ; Kristopher J. Elliot ; Alexandre Haefele ; Gilles Ulrich ;Raymond Ziessel
Journal of the American Chemical Society 2009 Volume 131(Issue 37) pp:13375-13386
Publication Date(Web):August 28, 2009
DOI:10.1021/ja9038856
A series of donor−spacer−acceptor triads has been synthesized and fully characterized. Both donor and acceptor units are built from boron dipyrromethene (BODIPY) dyes but they differ in their respective conjugation lengths, and thereby offer quite disparate optical properties. The spacer units comprise an oligomer of 1,4-phenylene-diethynylene repeat units and allow the boron−boron separation distance to be varied progressively from 18 to 38 Å. A notable feature of this series is that each subunit can be selectively excited with monochromatic light. Highly efficacious electronic energy transfer (EET) occurs from the first-excited singlet state localized on the conventional BODIPY dye to its counterpart resident on the expanded BODIPY-based nucleus, but the rate constant follows a nonlinear evolution with separation distance. Overall, the rate of EET falls by only a factor of 4-fold on moving from the shortest to the longest spacer. This shallow length dependence is a consequence of the energy gap between donor and spacer units becoming smaller as the molecular length increases. Interestingly, a simple relationship exists between the measured electronic resistance of the spacer unit and the Huang−Rhys factor determined by emission spectroscopy. Both parameters relate to the effective conjugation length. Direct illumination of the spacer unit leads to EET to both terminals, followed by EET from conventional BODIPY to the expanded version. In each case, EET to the expanded dye involves initial population of the second-singlet excited state, whereas transfer from spacer to the conventional BODIPY dye populates the S2 state for shorter lengths but the S1 state for the longer analogues. The rate of EET from spacer to conventional BODIPY dye, as measured for the corresponding molecular dyads, is extremely fast (>1011 s−1) and scales with the spectral overlap integral. The relative partitioning of EET from the spacer to each terminal is somewhat sensitive to the molecular length, with the propensity to populate the conventional BODIPY dye changing from 65% for N = 0 to 45% for N = 2. The most likely explanation for this behavior can be traced to the disparate spectral overlap integrals for the two dyes. These systems have been complemented by a molecular tetrad in which pyrene residues replace the fluorine atoms present on the conventional BODIPY-based dye. Here, rapid EET occurs from pyrene to the BODIPY dye and is followed by slower, long-range EET to the opposite terminal. Such materials are seen as highly attractive solar concentrators when dispersed in transparent plastic media and used under conditions where both inter- and intramolecular EET operate.
Co-reporter:Andrew C. Benniston ; Anthony Harriman ;Peiyi Li
Journal of the American Chemical Society 2009 Volume 132(Issue 1) pp:26-27
Publication Date(Web):December 11, 2009
DOI:10.1021/ja908865k
Two-color excitation of a molecular tetrad leads to directed electron transfer in opposite directions along the molecular axis according to which chromophore is illuminated and gives a 40 000-fold disparity in the lifetimes of the resultant charge-separated states.
Co-reporter:Kristopher J. Elliott, Anthony Harriman, Loïc Le Pleux, Yann Pellegrin, Errol Blart, Cédric R. Mayer and Fabrice Odobel
Physical Chemistry Chemical Physics 2009 vol. 11(Issue 39) pp:8767-8773
Publication Date(Web):15 Jul 2009
DOI:10.1039/B905548G
A multi-porphyrin cluster has been covalently attached to a polyoxometallate (POM) catalyst so as to form an advanced model for the photosynthetic reaction complex. This bio-inspired mimic displays efficient energy transfer from the peripheral zinc porphyrins (ZnP) to the central free-base porphyrin (FbP). The latter species participates in a light-induced electron transfer with the POM. Charge recombination is hindered by hole transfer from the FbP to one of the ZnPs. Charge accumulation occurs at the POM under illumination in the presence of a sacrificial electron donor.
Co-reporter:Andrew C. Benniston, Ben D. Allen, Anthony Harriman, Irantzu Llarena, James P. Rostron and Beverly Stewart
New Journal of Chemistry 2009 vol. 33(Issue 2) pp:417-427
Publication Date(Web):24 Nov 2008
DOI:10.1039/B814676D
Herein we describe the results of a combined theoretical and spectroscopic investigation into the design of a simple molecular system intended to act as a memory storage bank. The main operating principle revolves around the two-electron reduction of an aryl disulfide bond. Addition of the first electron leads to elongation of the S–S bond but it breaks only if there is accompanying protonation. Adding a second electron causes S–S bond cleavage, with or without protonation. The structural changes have been assessed by way of quantum chemical calculations and molecular dynamics simulations. Electrochemical studies show that the two-electron reduced product can be re-oxidised at mildly anodic potentials and the cycle can be repeated many times. Both theory and experiment point towards pronounced potential inversion whereby the second reduction potential lies at a significantly more positive potential than that for the first step. Computer simulations of the cyclic voltammograms give rise to numerical values for the reduction potentials that are in quite good agreement with the computed values and also allow determination of the electrochemical rate constants and transfer coefficients. Accurate simulation of the experimental data can be realised only if one proton accompanies the second reduction step. The possibility to design an effective molecular-scale memory device around this system is discussed briefly.
Co-reporter:Anthony Harriman ;LauraJ. Mallon;Sébastien Goeb Dr.;Gilles Ulrich Dr.;Raymond Ziessel Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 18) pp:4553-4564
Publication Date(Web):
DOI:10.1002/chem.200802477
Co-reporter:Raymond Ziessel Dr.;Pascal Retailleau Dr.;KristopherJ. Elliott Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 40) pp:10369-10374
Publication Date(Web):
DOI:10.1002/chem.200901725
Co-reporter:Raymond Ziessel Dr.;BenD. Allen;DorotaB. Rewinska Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 30) pp:7382-7393
Publication Date(Web):
DOI:10.1002/chem.200900440
Abstract
A conformationally restricted molecular dyad has been synthesized and subjected to detailed photophysical examination. The dyad comprises a borondipyrromethene (Bodipy) dye covalently linked to a buckminsterfullerene C60 residue, and is equipped with hexadecyne units at the boron centre in order to assist solubility. The linkage consists of a diphenyltolane, attached at the meso position of the Bodipy core and through an N-methylpyrrolidine ring at the C60 surface. Triplet states localised on the two terminals are essentially isoenergetic. Cyclic voltammetry indicates that light-induced electron transfer from Bodipy to C60 is thermodynamically favourable and could compete with intramolecular energy transfer in the same direction. The driving force for light-induced electron abstraction from Bodipy by the singlet excited state of C60 depends critically on the solvent polarity. Thus, in non-polar solvents, light-induced electron transfer is thermodynamically uphill, but fast excitation energy transfer occurs from Bodipy to C60 and is followed by intersystem crossing and subsequent equilibration of the two triplet excited states. Moving to a polar solvent switches on light-induced electron transfer. Now, in benzonitrile, the charge-transfer state (CTS) is positioned slightly below the triplet levels, such that charge recombination restores the ground state. However, in CH2Cl2 or methyltetrahydrofuran, the CTS is slightly higher in energy than the triplet levels, and decays, in part, to form the triplet state localized on the C60 residue. This step is highly specific and does not result in direct formation of the triplet excited state localized on the Bodipy unit. Subsequent equilibration of the two triplets takes place on a relatively slow timescale.
Co-reporter:Anthony Harriman, Kristopher J. Elliott, Mohammed A. H. Alamiry, Loïc Le Pleux, Marjorie Séverac, Yann Pellegrin, Errol Blart, Céline Fosse, Caroline Cannizzo, Cédric R. Mayer and Fabrice Odobel
The Journal of Physical Chemistry C 2009 Volume 113(Issue 14) pp:5834-5842
Publication Date(Web):2017-2-22
DOI:10.1021/jp900643m
Three photoactive, multicomponent supermolecules have been synthesized and characterized whereby a porphyrin unit is covalently linked to a Dawson-type heteropolyphosphotungstate (POM). The connection has been made via a Huisgen reaction, which gives good yields in all cases, and modified to provide linkages that vary in their degree of internal flexibility. Fluorescence from the porphyrin unit is quenched by the appended POM, for which the efficiency increases with increasing flexibility of the linker. Except for the most rigid connection, fluorescence decay profiles are nonexponential and are interpreted in terms of multiple families of conformers that differ in their ability to undergo light-induced electron transfer. The distribution of ground-state conformers was examined by high-pressure emission spectroscopy. Cyclic voltammetry and spectro-electrochemical studies provide quantitative data for the thermodynamic driving forces and spectral data for the redox products. In all cases, the first-excited singlet state resident on the porphyrin is capable of transferring an electron to the POM. The rate of electron transfer is very slow for the corresponding triplet state of the porphyrin. Photolysis of the porphyrin in the presence of triethanolamine, present as a sacrificial electron donor, leads to formation of the porphyrin π-radical anion. This latter species is able to reduce the POM, but the rate of reaction is remarkably slow. Here, bimolecular electron transfer competes effectively with the intramolecular route, confirming that the triazole linker is a poor conduit for electrons. It was not possible, under these conditions, to load the POM with more than a single electron. The one-electron reduced form of the POM transfers an electron to the singlet-excited-state of the porphyrin so as to form a relatively long-lived charge-shift state.
Co-reporter:Andrew C. Benniston, Anthony Harriman
Materials Today 2008 Volume 11(Issue 12) pp:26-34
Publication Date(Web):December 2008
DOI:10.1016/S1369-7021(08)70250-5
We raise here a series of critical issues regarding artificial photosynthesis with the intention of increasing awareness about what needs to be done to bring about a working prototype. Factors under consideration include energy and electron transfers, coupled redox reactions, repair mechanisms, and integrated photosystems.
Co-reporter:Mohammed A. H. Alamiry;Laura J. Mallon;Gilles Ulrich;Raymond Ziessel
European Journal of Organic Chemistry 2008 Volume 2008( Issue 16) pp:2774-2782
Publication Date(Web):
DOI:10.1002/ejoc.200800159
Abstract
A novel boron dipyrromethene (BODIPY) dye has been synthesized in which the F atoms, usually bound to the boron center, have been replaced with 1-ethynylperylene units and a 4-pyridine residue is attached at the meso-position. The perylene units function as photon collectors over the wavelength range from 350 to 480 nm. Despite an unfavorable spectral overlap integral, rapid energy transfer takes place from the singlet-excited state of the perylene unit to the adjacent BODIPY residue, which is itself strongly fluorescent. The mean energy-transfer time is 7 ± 2 ps at room temperature. The dominant mechanism for the energy-transfer process is Dexter-type electron exchange, with Förster-type dipole–dipole interactions accounting for less than 10 % of the total transfer probability. There are no indications for light-induced electron transfer in this system, although there is evidence for a nonradiative decay channel not normally seen for F-type BODIPY dyes. This new escape route is further exposed by the application of high pressure. The meso-pyridine group is a passive bystander until protons are added to the system. Then, protonation of the pyridine N atom leads to complete extinction of fluorescence from the BODIPY dye and slight recovery of fluorescence from the perylene units. Quenching of BODIPY-based fluorescence is due to charge-transfer to the pyridinium unit whereas the re-appearance of perylene-based emission is caused by a reduction in the Förster overlap integral upon protonation. Other cations, most notably zinc(II) ions, bind to the pyridine N-atom and induce similar effects but the resultant conjugate is weakly fluorescent.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Anthony Harriman ;Laura Mallon;Raymond Ziessel
Chemistry - A European Journal 2008 Volume 14( Issue 36) pp:
Publication Date(Web):
DOI:10.1002/chem.200890148
Co-reporter:AndrewC. Benniston Dr., ;Peiyi Li Dr.;PriteshV. Patel Dr. ;CraigA. Sams Dr.
Chemistry - A European Journal 2008 Volume 14( Issue 6) pp:1710-1717
Publication Date(Web):
DOI:10.1002/chem.200701548
Abstract
The rate constant for triplet energy transfer (kTET) has been measured in fluid solution for a series of mixed-metal Ru–Os bis(2,2′:6′,2′′-terpyridine) complexes built around a tethered biphenyl-based spacer group. The length of the tether controls the central torsion angle for the spacer, which can be varied systematically from 37 to 130°. At low temperature, but still in fluid solution, the spacer adopts the lowest-energy conformation and kTET shows a clear correlation with the torsion angle. A similar relationship holds for the inverse quantum yield for emission from the Ru–terpy donor. Triplet energy transfer is more strongly activated at higher temperature and the kinetic data require analysis in terms of two separate processes. The more weakly activated step involves electron exchange from the first-excited triplet state on the Ru–terpy donor and the size of the activation barrier matches well with that calculated from spectroscopic properties. The pre-exponential factor derived for this process correlates remarkably well with the torsion angle and there is a large disparity in electronic coupling through π and σ orbitals on the spacer. The more strongly activated step is attributed to electron exchange from an upper-lying triplet state localized on the Ru–terpy donor. Here, the pre-exponential factor is larger but shows the same dependence on the geometry of the spacer. Strangely, the difference in coupling through π and σ orbitals is much less pronounced. Despite internal flexibility around the spacer, kTET shows a marked dependence on the torsion angle computed for the lowest-energy conformation.
Co-reporter:AndrewC. Benniston Dr., ;Peiyi Li Dr.;PriteshV. Patel Dr. ;CraigA. Sams Dr.
Chemistry - A European Journal 2008 Volume 14( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/chem.200890015
Co-reporter:Anthony Harriman ;Laura Mallon;Raymond Ziessel
Chemistry - A European Journal 2008 Volume 14( Issue 36) pp:11461-11473
Publication Date(Web):
DOI:10.1002/chem.200801384
Abstract
A multicomponent cluster has been synthesised in which four disparate chromophores have been covalently linked through a logical arrangement that favours efficient photon collection and migration to a terminal emitter. The primary energy acceptor is a boron dipyrromethene (Bodipy) dye and different polycyclic aryl hydrocarbons have been substituted in place of the regular fluorine atoms attached to the boron centre. The first such unit is perylene, linked to boron through a 1,4-diethynylphenyl unit, which collects photons in the 320–490 nm region. The other photon collector is pyrene, also connected to the boron centre by a 1,4-diethynylphenyl spacer and absorbing strongly in the 280–420 nm region, which itself is equipped with an ethynylfluorene residue that absorbs in the UV region. Illumination into any of the polycyclic aryl hydrocarbons results in emission from the Bodipy unit. The rates of intramolecular electronic energy transfer have been determined from time-correlated, single-photon counting studies and compared with the rates for Coulombic interactions computed from the Förster expression. It has been necessary to allow for i) a more complex screening potential, ii) multipole–multipole coupling, iii) an extended transition dipole moment vector and iv) bridge-mediated energy transfer. The bridge-mediated energy transfer includes both modulation of the donor transition dipole vector by bridge states and Dexter-type electron exchange. The latter is a consequence of the excellent electronic coupling properties of the 1,4-diethynylphenyl spacer unit. The net result is a large antenna effect that localises the photon density at the primary acceptor without detracting from its highly favourable photophysical properties.
Une supramolécule originale comportant 4 chromophores distincts liés de façon logique sur une plateforme fluorescente permet de collecter une très vaste gamme de photons et de les faire migrer de façon directionnelle vers un chromophore qui sera le seul émetteur de lumière. L'accepteur d'énergie primaire est une sous-unité Bodipy tandis qu'un ensemble des composés polyaromatiques ont été positionné de telle manière à favoriser le transfert d'énergie. L'ensemble des accepteurs secondaires est lié de façon covalente sur l'atome de bore par l'intermédiaire de triples liaisons qui assurent une très bonne connectivité chimique et électronique. Parmis ces unités, on trouve le 1,4-diéthynylperylène qui collecte les photons dans une plage comprise entre 320 et 490 nm. Un autre collecteur de photons est le 1,6-diéthynylpyrène qui est absorbe entre 280 et 420 nm. Ces deux sous-unités sont liés au bore par un fragment 1,4-diéthynylphényle qui est indispensable pour assurer une synthèse optimale. Enfin un résidu éthynylfluorène permet d'élargir la collection de photons jusqu'à l'ultraviolet lointain et d'assurer une excellente solubilité du composé ciblé. Quelque soit la longueur d'onde d'irradiation utilisée l'ensemble des photons collectés sont transférés vers la sous-unité Bodipy qui fluorescence efficacement. Les vitesses de transfert intramoléculaire d'énergie ont été déterminées par des méthodes traditionnelles de comptage de photons et les vitesses ont été comparées aux valeurs calculées en utilisant les équations de Förster appropriées à une interaction Coulombic. Toutefois il a fallu tenir de nombreux autres facteurs comme: i) un potentiel de surface élargi; ii) de multiple interactions multipole-multipole; iii) un moment de transition dipolaire étendu; et iv) des transferts d'énergie à travers les liaisons. Ce dernier incluant à la fois la modulation du vecteur dipolaire du donneur d'énergie et d'états énergétiques localisés sur l'entretoise ainsi que du double échange d'électron de type Dexter. Ce dernier est du aux excellentes propriétés de couplage électronique au travers de l'entité diéthynylphényle. Le résultat final de ces études est un effet d'antenne étendu, une collection de photon exceptionnelle qui localise toute l'énergie excitonique sur l'accepteur final (le Bodipy) sans perturber ces propriétés optiques et de fluorescence exceptionnelles.
Co-reporter:Anthony Harriman, Laura J. Mallon, Sébastien Goeb and Raymond Ziessel
Physical Chemistry Chemical Physics 2007 vol. 9(Issue 38) pp:5199-5201
Publication Date(Web):25 Jul 2007
DOI:10.1039/B709358F
Despite limited spectral overlap and quite wide spatial separation, essentially quantitative electronic energy transfer occurs from peripheral Bodipy units to an expanded Bodipy core, the latter being attached via its B centre, so as to generate near-IR fluorescence.
Co-reporter:Anthony Harriman and Guillaume Izzet
Physical Chemistry Chemical Physics 2007 vol. 9(Issue 8) pp:944-948
Publication Date(Web):11 Jan 2007
DOI:10.1039/B613854N
The luminescence properties of ruthenium(II) tris(2,2′-bipyridine) have been recorded in butyronitrile solution and in a transparent KBr disk over a reasonable temperature range. In solution, spectral curve fitting routines indicate that emission arises solely from an ensemble of triplet states, each of which is of Metal-to-Ligand, Charge-Transfer (MLCT) character and of closely comparable energy. At ambient temperature, dual emission is observed for the KBr disk and interpreted in terms of luminescence from both the ensemble and the fourth MLCT triplet state that lies at slightly higher energy. Relative reorganisation energies, energies, Huang–Rhys factors and radiative rate constants have been calculated for the two emissive states. It is confirmed that the fourth MLCT triplet state possesses more singlet character than the ensemble.
Co-reporter:Raymond Ziessel;Laura J. Mallon;Gilles Ulrich;Beverly Stewart
European Journal of Organic Chemistry 2007 Volume 2007(Issue 19) pp:3191-3198
Publication Date(Web):10 MAY 2007
DOI:10.1002/ejoc.200700190
Two new boron-dipyrromethene dyes have been synthesised and their photophysical properties have been investigated by steady-state and time-resolved fluorescence spectroscopy. These dyes are equipped with ancillary 2,2′:6′,2″-terpyridine (terpy) residues, attached either at the B centre or at the meso position of the organic framework; in this latter case two ethynylated pyrene units are bound to the B atom. Under most experimental conditions, the absorption and emission spectral properties are typical of this class of dye. Complexation of zinc(II) cations to the terpy units attached at the B centre causes complete extinction of the fluorescence due to intramolecular electron transfer from the excited state dye to the resultant zinc(II) bis-terpy complex. Similar behaviour is noted when the terpy is attached at the meso site, although the rate of electron transfer is slower due to the weaker driving force. A most unusual feature of the pyrene-substituted dye is its facile self association in acetonitrile solution. This results in the reversible formation of a fluorescent associate, believed to be a symmetric dimer, that absorbs and emits to the red of the monomer. Self association is considered to be driven by a solvophobic effect in which the pyrene units seek to minimize surface contact with the surrounding solvent molecules.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2007)
Co-reporter:Andrew C. Benniston Dr. ;Peiyi Li Dr.;James P. Rostron Dr.;Ross W. Harrington Dr.;William Clegg
Chemistry - A European Journal 2007 Volume 13(Issue 28) pp:
Publication Date(Web):23 AUG 2007
DOI:10.1002/chem.200700872
A small series of N,N′-dimethyl-4,4′-bipyridinium dication derivatives (commonly known as viologens) has been synthesized and fully characterized; a short dialkoxy tether attached at the 3,3′-positions is used to alter the central dihedral angle. These angles were determined by both single-crystal X-ray diffraction and by computational studies made for the dication, radical cation, and neutral species in a solvent reservoir. The dihedral angle derived for the dication controls the first reduction potential, whereas the geometry of the resultant π-radical cation determines the magnitude of the second reduction potential. The optical absorption spectra recorded for the various species, and especially those of the radical cations, and the EPR spectral parameters of the π-radical cations also depend on the molecular geometry. In particular, the central dihedral angle influences the spin density distribution around the aromatic nucleus and, by way of comparison to the parent viologen, it has been possible to resolve the angle dependence from the inherent inductive effect of the strap. These results are considered in terms of the degree of electronic communication between the two aromatic rings, as controlled by the length of the tether.
Co-reporter:Andrew C. Benniston Dr. ;Dorota B. Rewinska;Songjie Yang Dr.;Yong-Gang Zhi Dr.
Chemistry - A European Journal 2007 Volume 13(Issue 36) pp:
Publication Date(Web):23 OCT 2007
DOI:10.1002/chem.200701235
The synthesis is described for a small series of oligomers built from (2, 3, 4 or 6) ethynyl-naphthalene repeat units and end-capped with solubilising 1,2,3-tris-dodecyloxy-benzene groups. These compounds absorb in the near-UV region and exhibit strong fluorescence in both fluid solution and a glassy matrix at 77 K. The spectral profiles are fully consistent with a structurally heterogeneous ground state becoming more planar upon excitation and with the low-temperature glass further stabilising the co-planar orientation. The absorption and fluorescence maxima move towards lower energy with increasing number of repeat units and there is a corresponding increase in the Huang–Rhys factor for the radiative process. The non-radiative rate constants also depend on molecular length and are well explained in terms of the energy-gap law. In contrast, very weak phosphorescence is observed at 77 K for which the peak maximum and lifetime remain insensitive to the number of naphthalene units. The triplet lifetimes recorded at ambient temperature are also independent of the molecular length but the triplet–triplet absorption spectra change throughout the series. These results are discussed in terms of the degree of electronic coupling between adjacent repeat units for each of the relevant excited states. During these studies it was noted that the rate of intersystem crossing to the triplet manifold is but weakly affected by heavy-atom perturbers. A non-fluorescent complex is formed between iodoethane and the molecular rod but the corresponding bimolecular process occurs at well below the diffusion-controlled limit. This behaviour is considered in terms of spin-orbit coupling between the excited states and takes account of the differing conjugation lengths.
Co-reporter:Andrew C. Benniston Dr. ;Sarah L. Howell Dr.;Craig A. Sams Dr.;Yong-Gang Zhi Dr.
Chemistry - A European Journal 2007 Volume 13(Issue 16) pp:
Publication Date(Web):7 FEB 2007
DOI:10.1002/chem.200601498
The synthesis is described for a series of five molecular dyads comprising pyrene-based terminals covalently linked through a 1,3-disubstituted phenylene spacer. The extent of through-space communication between the pyrene units is modulated by steric interactions imposed by bulky moieties attached at the 6,8-positions of each pyrene unit. For the control compound, only hydrogen atoms occupy the 6,8 positions (DP1), whereas the remaining compounds incorporate ethynylene groups terminated with either triisopropylsilyl (DP2), 1-tert-butylbenzene (DP3), 2,6-di-tert-butylbenzene (DP4) or 1-tert-butyl-3,5-dimethylbenzene (DP5) units. Each compound shows a mixture of monomer and excimer fluorescence in fluid solution at room temperature, but only monomer emission in a glassy matrix at 77 K. The ratio of monomer to excimer fluorescence depends markedly on the molecular structure; DP1 is heavily biased in favour of the excimer and DP4 is enriched with monomer fluorescence. Photophysical properties, including laser induced and delayed fluorescence data, are reported for each compound. Delayed fluorescence occurs by both intramolecular and bimolecular steps, but these events take place on different timescales. The possibility is raised for using intramolecular triplet–triplet annihilation as a means of molecular imaging.
Co-reporter:Andrew C. Benniston, Anthony Harriman, Peiyi Li, James P. Rostron and Jan W. Verhoeven
Chemical Communications 2005 (Issue 21) pp:2701-2703
Publication Date(Web):08 Apr 2005
DOI:10.1039/B501262G
Photolysis of the 9-mesityl-10-methylacridinium cation in benzonitrile forms an acridinyl radical, detected by EPR and UV–visible spectroscopy, by way of a sacrificial process.
Co-reporter:Ben D. Allen, Andrew C. Benniston, Anthony Harriman, Sarah A. Rostron and Chunfang Yu
Physical Chemistry Chemical Physics 2005 vol. 7(Issue 16) pp:3035-3040
Publication Date(Web):06 Jul 2005
DOI:10.1039/B507165H
The photophysical properties of 9-dicyanovinyljulolidine are sensitive to solvent viscosity but are little affected by changes in polarity. In fluid solution, the lifetime of the first-excited singlet state is very short and triplet state formation cannot be detected by laser flash photolysis. Decay of the excited singlet state is strongly activated and weak phosphorescence can be observed in a glassy matrix at 77 K. Temperature dependent 1H NMR studies indicate that the molecule undergoes slow internal rotation in solution, for which the activation energy has a value of ca. 35 kJ mol−1. This process is unlikely to account for the poor fluorescence quantum yield found in fluid solution. Instead, it is considered that the target compound undergoes rapid rotation around the dicyanovinyl double bond from the excited singlet state. The rate of rotation depends weakly on the viscosity of the solvent in a range of linear alcohols at room temperature. This might represent the fact that the rotor is relatively small and can pack into cavities in the solvent structure. In glycerol, the rate of rotation is more sensitive to viscosity effects but a quite complex temperature dependence is observed in ethanol. Here, the rate is almost activationless in a glassy matrix and in fluid solution at high temperature but strongly activated at intermediate temperatures.
Co-reporter:Andrew C. Benniston, Anthony Harriman and James P. Rostron
Physical Chemistry Chemical Physics 2005 vol. 7(Issue 16) pp:3041-3047
Publication Date(Web):13 Jul 2005
DOI:10.1039/B506776F
The photophysical properties of the target compound are extremely sensitive to changes in solvent polarity since the lowest-energy excited states possess considerable charge-transfer character. Excitation results in a greatly increased dipole moment, with the resultant excited singlet state retaining a lifetime of ca. 1 ns in all solvents. Radiative decay involves coupling between the lowest-energy excited singlet state and both the ground state and an upper excited singlet state. The level of coupling to the upper singlet decreases in non-polar solvents, presumably due to symmetry factors. The radiative rate constant decreases smoothly with increasing solvent polarity function as the molecule acquires an ever increasing dipolar character. Non-radiative decay includes both intersystem crossing and internal conversion, but the former process dominates in polar solvents. The excited singlet state lifetime is very weakly dependent upon temperature in the solid state. However, in polar solutions where the Stokes’ shift decreases with decreasing temperature, there is clear evidence for an activated process. This is believed to involve coupling to the upper-lying singlet excited state.
Co-reporter:Andrew C. Benniston, Anthony Harriman, Peiyi Li, Pritesh V. Patel and Craig A. Sams
Physical Chemistry Chemical Physics 2005 vol. 7(Issue 21) pp:3677-3679
Publication Date(Web):23 Sep 2005
DOI:10.1039/B512307K
The magnitude of electronic coupling between the terminal chromophores shows a precise dependence on the dihedral angle around a bridging biphenyl group.
Co-reporter:Anthony Harriman, Annabelle Mayeux, Christophe Stroh and Raymond Ziessel
Dalton Transactions 2005 (Issue 17) pp:2925-2932
Publication Date(Web):25 Jul 2005
DOI:10.1039/B506818E
A binuclear complex has been synthesized having ruthenium(II) bis(2,2′:6′,2″-terpyridine) terminals attached to a central 2,2′-bipyrimidine unit via ethynylene groups. Cyclic voltammetry indicates that the substituted terpyridine is the most easily reduced subunit and the main chromophore involves charge transfer from the metal centre to this ligand. The resultant metal-to-ligand, charge-transfer (MLCT) triplet state is weakly emissive and has a lifetime of 60 ns in deoxygenated solution at room temperature. The luminescence yield and lifetime increase with decreasing temperature in a manner that indicates the lowest-energy MLCT triplet couples to at least two higher-energy triplets. Cations can bind to the central bipyrimidine unit, forming both 1 : 1 and 1 : 2 (ligand : metal) complexes as confirmed by electrospray MS analysis. The photophysical properties depend on the number of bound cations and on the nature of the cation. In the specific case of binding zinc(II) cations, the 1 : 1 complex has a triplet lifetime of 8.0 ns while that of the 1 : 2 complex is 1.8 ns. The 1 : 1 complexes formed with Ba2+ and Mg2+ are more luminescent than is the parent compound while the 1 : 2 complexes are much less luminescent. It is shown that the coordinated cations raise the reduction potential of the central bipyrimidine unit and thereby increase the activation energy for coupling with the metal-centred state. Complexation also introduces a non-emissive intramolecular charge-transfer (ICT) state that couples to the lowest-energy MLCT triplet and provides an additional non-radiative decay route. The triplet state of the 1 : 2 complex formed with added Zn2+ cations decays preferentially via this ICT state.
Co-reporter:Anthony Harriman, Maryam Mehrabi and Bashkar G. Maiya
Photochemical & Photobiological Sciences 2005 vol. 4(Issue 1) pp:47-53
Publication Date(Web):21 Sep 2004
DOI:10.1039/B410141C
The fluorescence from a set of porphyrin–calixarene complexes is quenched upon addition of benzo-1,4-quinone (BQ) in fluid solution. In N,N-dimethylformamide solution, fluorescence quenching involves both static and dynamic interactions but there are no obvious differences between porphyrins with or without the appended calixarene. Under such conditions, the static quenching behaviour is attributed to π-complexation between the reactants and it is concluded that the calixarene cavity does not bind BQ. An additional static component is apparent in dichloromethane solution. This latter effect involves partial fluorescence quenching, for which the intramolecular rate constant can be obtained by time-resolved fluorescence spectroscopy. The derived rate constants depend on molecular structure in a manner consistent with fluorescence quenching being due to electron transfer. In all cases, however, the dominant quenching step involves diffusional contact between the porphyrin nucleus and a non-bound molecule of BQ.
Co-reporter:Andrew C. Benniston, Anthony Harriman, Francisco M. Romero and Raymond Ziessel
Dalton Transactions 2004 (Issue 8) pp:1233-1238
Publication Date(Web):15 Mar 2004
DOI:10.1039/B400933A
The photophysical properties of closely-coupled, binuclear complexes formed by connecting two ruthenium(II) tris(2,2′-bipyridine) complexes via an alkynylene group differ significantly from those of the relevant mononuclear complex. In particular, the energy of the first triplet excited state is lowered relative to the parent complex, because of the presence of the alkynylene substituent, while the triplet lifetime is prolonged, in part, because of extended electron delocalisation. We now report that the triplet lifetime is also affected by the nature of the spectator 2,2′-bipyridyl ligands. Thus, replacing the parent 2,2′-bipyridine ligands with the corresponding 4,4′-dinitro-substituted ligands serves to decrease the luminescence yield and lifetime. With the corresponding carboxylate ester, the luminescence yield and lifetime are increased. Perdeuteration of the parent 2,2′-bipyridine ligands also leads to a modest increase in the luminescence yield. Such observations are indicative of electronic coupling between the various metal-to-ligand, charge-transfer excited triplet states. Temperature dependence studies confirm that these excited states are closely spaced and thermally accessible at ambient temperature. For some of the binuclear complexes, the quantum yield for formation of the lowest-energy triplet state is significantly less than unity.
Co-reporter:Andrew C. Benniston, Vincent Grosshenny, Anthony Harriman and Raymond Ziessel
Dalton Transactions 2004 (Issue 8) pp:1227-1232
Publication Date(Web):15 Mar 2004
DOI:10.1039/B400931B
The photophysical properties of closely-coupled, binuclear complexes formed by connecting two ruthenium(II) bis(2,2′:6′,2″-terpyridine) complexes via an alkynylene group are compared to those of the parent complex. The dimers exhibit red-shifted emission maxima and prolonged triplet lifetimes in deoxygenated solution. Triplet quantum yields are much less than unity and the dimers generate singlet molecular oxygen with low quantum efficiency. Temperature dependence emission studies indicate coupling to higher-energy triplet states while cyclic voltammetry shows that the metal centres are only very weakly coupled but that extensive electron delocalisation occurs upon one-electron reduction. The radiative rate constants derived for these dimers are relatively low, because the lowest-energy metal-to-ligand, charge-transfer states possess increased triplet character. In contrast, the rate constants for nonradiative decay of the lowest-energy triplet states are kept low by extended electron delocalisation over the polytopic ligand. The poor triplet yields are a consequence of partitioning at the second triplet level.
Co-reporter:Andrew C. Benniston, Anthony Harriman, Peiyi Li and Craig A. Sams
Physical Chemistry Chemical Physics 2004 vol. 6(Issue 5) pp:875-877
Publication Date(Web):30 Jan 2004
DOI:10.1039/B316620A
On the basis of temperature-dependent emission studies made with the target binuclear compounds, it is concluded that the extent of electron delocalisation at the triplet level depends on the torsion angle imposed by the bridge.
Co-reporter:Ata Amini, Anthony Harriman and Annabelle Mayeux
Physical Chemistry Chemical Physics 2004 vol. 6(Issue 6) pp:1157-1164
Publication Date(Web):20 Feb 2004
DOI:10.1039/B313526H
The emission spectrum of ruthenium(II) bis(2,2′:6′,2″-terpyridine) has been recorded as a function of temperature over the range 77–290 K. Analysis of the spectrum allows calculation of the triplet energy and of the total reorganization energy accompanying deactivation of the metal-to-ligand, charge-transfer (MLCT) triplet state in both frozen glasses and fluid solutions. Emission quantum yields and triplet lifetimes decrease markedly with increasing temperature above 140 K and can be explained satisfactorily in terms of a 4-state model wherein the lowest-energy MLCT triplet interacts with two other triplets and with the ground state. The barrier for reaching the highest-energy triplet state is 1,700 cm−1 but, although this upper-lying triplet is usually described as being of metal-centred (MC) character, the experimental work does not help identify the nature of the interacting states. The same parameters were calculated by quantum chemical methods. It is seen that agreement between experiment and theory is rather good. Although the quantum chemical calculations indicate that the vertical energy difference between MLCT and MC triplets is about 3,000 cm−1, the latter species is characterised by a relatively large reorganization energy. When the fully optimised structures are taken into account, the energy difference between these two triplets is computed to be less than 1,300 cm−1.
Co-reporter:Andrew C. Benniston, Anthony Harriman, Donald J. Lawrie and Annabelle Mayeux
Physical Chemistry Chemical Physics 2004 vol. 6(Issue 1) pp:51-57
Publication Date(Web):02 Dec 2003
DOI:10.1039/B312286G
A tripartite supermolecule, comprising a pyrene moiety tethered to a 2,2′:6′,2″-terpyridyl ligand via a 2,5-diethynylated thiophene linker, has been synthesized. This compound is highly fluorescent in solution due to the formation of an intramolecular charge-transfer (CT) state. From consideration of the electrochemical properties, it is concluded that the CT state arises because of charge transfer from pyrene to the central thiophene-based unit. Formation of the CT state involves an increase in dipole moment of ca. 18.5 D. Phosphorescence was not observed but the intermediate population of the triplet state was confirmed by laser flash photolysis. Addition of Zn2+ cations results in a drastic decrease in the fluorescence yield while the absorption spectrum exhibits a pronounced red shift. It appears that the dipole is extended upon cation binding, with the zinc terpyridine terminal acting as the electron acceptor. Again, no phosphorescence was apparent at 77 K. Coordination of a ruthenium(II) 2,2′;6′,2″-terpyridyl metallo-fragment to the vacant terpyridine terminal causes the appearance of weak phosphorescence in fluid solution at room temperature. The emitting species has a lifetime of 2.6 μs in deoxygenated acetonitrile at 20°C. Luminescence, which shows a complex temperature dependence, is attributed to either the lowest-energy metal-to-ligand, charge-transfer triplet localised on the ruthenium(II) complex or to the intraligand CT state. In the later case, spin–orbit coupling effects induced by the ruthenium atom are responsible for promoting emission.
Co-reporter:Anthony Harriman
Angewandte Chemie 2004 Volume 116(Issue 38) pp:
Publication Date(Web):26 JUL 2004
DOI:10.1002/ange.200301762
In neuem Licht betrachtet: Nicht nur der häufig genutzte Kaskadeneffekt, sondern auch einfache geometrische Vorgaben können zur Stabilisierung von ladungsgetrennten Zuständen in künstlichen Photosynthesesystemen verwendet werden. In diesem Fall folgt auf die lichtinduzierte Ladungstrennung (CS) in einer kompakten molekularen Dyade eine außergewöhnlich langsame Ladungsrekombination (CR, siehe Bild). Der ladungsgetrennte Zustand hat eine Lebensdauer von bis zu 120 s.
Co-reporter:Anthony Harriman
Angewandte Chemie International Edition 2004 Volume 43(Issue 38) pp:
Publication Date(Web):26 JUL 2004
DOI:10.1002/anie.200301762
Shedding new light on the matter: Rather than the conventional approach of utilizing the cascade effect, charge separation can be stabilized in artificial photosynthetic systems simply by the geometry. In these latter systems, light-induced charge separation (CS) in a closely spaced molecular dyad is followed by exceptionally slow charge recombination (CR, see picture) to give a charge-separated state with a lifetime of up to 120 s.
Co-reporter:Ata Amini and Anthony Harriman
Physical Chemistry Chemical Physics 2003 vol. 5(Issue 7) pp:1344-1351
Publication Date(Web):24 Feb 2003
DOI:10.1039/B210886K
The photophysical properties of 4-cyano-(4′-methylthio)diphenylacetylene have been examined by quantum chemical methods. It is concluded that the lowest-energy, excited singlet state in a polar solvent like water corresponds to intramolecular charge transfer from the S-based donor to the benzonitrile unit. This excited state adopts a twisted geometry, in marked contrast to the planar ground state. Because of the structural change that follows from UV excitation there is a substantial Stokes shift, the magnitude of which depends on the polarity of the surrounding solvent. Indeed, formation of the twisted intramolecular charge-transfer state involves a significant increase in dipole moment. The charge transfer state decays via a combination of fluorescence, charge recombination and intersystem crossing. The calculations reveal values for the accompanying nuclear and solvent reorganisation energies and assign relative values for the electronic coupling matrix elements associated with formation of the ground and triplet states. Overall, the computed results remain in good agreement with the relevant experimental data.
Co-reporter:Ata Amini and Anthony Harriman
Physical Chemistry Chemical Physics 2003 vol. 5(Issue 20) pp:4556-4562
Publication Date(Web):08 Sep 2003
DOI:10.1039/B308059E
2-Methyl-1,3-dihydrobenz[d,e]isoquinoline (DHBIQ) undergoes rapid intramolecular charge transfer from the amino donor to a locally-excited singlet state of the naphthalene acceptor. In order to better understand the mechanism of charge transfer in this class of molecules, we have performed a series of quantum chemical calculations intended to compute the relevant electronic coupling matrix elements. The reactants are separated by σ-bonds and it appears that both through-space and through-bond electronic coupling must be taken into account. Furthermore, the ground state of DHBIQ exists as a mixture of two interconverting conformers and, surprisingly, the extent of through-space interactions is significantly greater than the corresponding through-bond interactions for both conformers. Comparison of electronic coupling within the two conformers shows that an axial conformation more strongly favours charge transfer than does the complementary equatorial geometry. An additional calculation made on a twisted conformation of DHBIQ further illustrates the effect of molecular geometry on the extent of electronic coupling, especially for through-bond interactions. Overall, the calculations agree well with experimental observations and it is stressed that the configuration interaction single and double excitation method, used with 12 molecular orbitals in the active space, has distinct advantages relative to other methods available for calculating the extent of electronic coupling in donor–connector–acceptor systems.
Co-reporter:Anthony Harriman, Abderrahim Khatyr and Raymond Ziessel
Dalton Transactions 2003 (Issue 10) pp:2061-2068
Publication Date(Web):23 Apr 2003
DOI:10.1039/B300618M
A set of ruthenium(II) poly(pyridine) complexes has been synthesized in which a central diethynylated pyrene moiety separates the 2,2′-bipyridine- and 2,2′:6′,2″-terpyridine-based terminals. The mononuclear complex, having only the 2,2′-bipyridine ligand coordinated with the metal cation, and the corresponding binuclear complex show remarkably similar luminescence properties in deoxygenated acetonitrile solution at room temperature. Two emission bands are evident in the spectrum. These bands appear to be in thermal equilibrium over the temperature range 0–60 °C but only a single emitting species is seen in a frozen glass at 77 K. The phosphorescence lifetimes are significantly longer than those associated with the parent complexes under the same experimental conditions but, unlike most other metal complex–pyrene dyads, the luminescence yield is extremely sensitive to the presence of trace amounts of molecular oxygen. The analogous compound having two ruthenium(II) tris(2,2′-bipyridine)-based terminals shows comparable behaviour. Allowing for all of the measured photophysical and electrochemical properties, it is concluded that the triplet manifold has the metal-to-ligand, charge-transfer state localised on the metal complex in equilibrium with an intramolecular charge-transfer state involving the pyrene and a coordinated poly(pyridine) group. The latter state lies at lower energy in a polar solvent and controls the photophysics. At low temperature, only the metal-to-ligand, charge-transfer triplet is observed.
Co-reporter:Anthony Harriman, Raymond Ziessel, Jean-Claude Moutet and Eric Saint-Aman
Physical Chemistry Chemical Physics 2003 vol. 5(Issue 8) pp:1593-1598
Publication Date(Web):14 Mar 2003
DOI:10.1039/B210591H
Copper(I) cations bind to a ferrocene-based ligand bearing two pendant 2,2′-bipyridine groups to form three distinct complexes possessing compositions of 1∶1, 1∶2 and 2∶2 (metal to ligand), respectively. The 2∶2 complex exists in the form of a double-stranded metallo-helicate but it is unstable in solution with respect to dissociation into the mononuclear species. Stability constants have been determined for each of the complexes using UV-visible spectrophotometric titrations. With this information, together with data derived from competitive complexation studies, electrospray mass spectrometry and electrochemistry, the mechanism by which the metallo-helicate self assembles in solution has been deduced. It is clear that the binuclear complex forms on dimerization of the 1∶1 complex, for which there is only a small thermodynamic driving force (ΔG0=−3.5 kJ mol−1).
Co-reporter:Anthony Harriman;Muriel Hissler;Abderrahim Khatyr;Raymond Ziessel
European Journal of Inorganic Chemistry 2003 Volume 2003(Issue 5) pp:
Publication Date(Web):18 FEB 2003
DOI:10.1002/ejic.200390127
Stepwise cross-coupling reactions between functionalized metallo synthons and bipyridine- or terpyridine-based building blocks afford novel photoactive, hybrid complexes bearing multiple chromophores that function separately or cooperatively according to the nature of the coordinated metal cations. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2003)
Co-reporter:Ata Amini, Anthony Harriman
Journal of Photochemistry and Photobiology C: Photochemistry Reviews 2003 Volume 4(Issue 2) pp:155-177
Publication Date(Web):31 October 2003
DOI:10.1016/S1389-5567(03)00027-3
Electron-transfer processes, and especially light-induced electron-transfer reactions, play an extremely important role in natural and artificial energy transduction. Following many decades of intensive theoretical and experimental study, it is now opportune to explore electron-transfer processes by way of modern computational chemistry. In essence, this requires the meaningful calculation of those thermodynamic parameters that combine to control the rate of electron-transfer between remote donor and acceptor species. The most important parameters are the nuclear and solvent re-organisation energies, the electronic coupling matrix element, the change in Gibbs free-energy and the activation energy change accompanying electron-transfer. Clearly, the surrounding environment has to be taken into account. Restricting attention to intramolecular electron-transfer in tripartite supermolecules of general type donor–bridge–acceptor (D–B–A), it is possible to compute each of the required thermodynamic properties from first principles. We examine here the most common quantum chemical approaches for estimation of each term and show that it is possible to arrive at a realistic estimate of the overall rate of electron-transfer. Attention is focused on readily accessible computational methodology.
Co-reporter:Anthony Harriman, Laura J. Mallon, Sébastien Goeb and Raymond Ziessel
Physical Chemistry Chemical Physics 2007 - vol. 9(Issue 38) pp:NaN5201-5201
Publication Date(Web):2007/07/25
DOI:10.1039/B709358F
Despite limited spectral overlap and quite wide spatial separation, essentially quantitative electronic energy transfer occurs from peripheral Bodipy units to an expanded Bodipy core, the latter being attached via its B centre, so as to generate near-IR fluorescence.
Co-reporter:Kristopher J. Elliott, Anthony Harriman, Loïc Le Pleux, Yann Pellegrin, Errol Blart, Cédric R. Mayer and Fabrice Odobel
Physical Chemistry Chemical Physics 2009 - vol. 11(Issue 39) pp:NaN8773-8773
Publication Date(Web):2009/07/15
DOI:10.1039/B905548G
A multi-porphyrin cluster has been covalently attached to a polyoxometallate (POM) catalyst so as to form an advanced model for the photosynthetic reaction complex. This bio-inspired mimic displays efficient energy transfer from the peripheral zinc porphyrins (ZnP) to the central free-base porphyrin (FbP). The latter species participates in a light-induced electron transfer with the POM. Charge recombination is hindered by hole transfer from the FbP to one of the ZnPs. Charge accumulation occurs at the POM under illumination in the presence of a sacrificial electron donor.
Co-reporter:Anthony Harriman and Guillaume Izzet
Physical Chemistry Chemical Physics 2007 - vol. 9(Issue 8) pp:NaN948-948
Publication Date(Web):2007/01/11
DOI:10.1039/B613854N
The luminescence properties of ruthenium(II) tris(2,2′-bipyridine) have been recorded in butyronitrile solution and in a transparent KBr disk over a reasonable temperature range. In solution, spectral curve fitting routines indicate that emission arises solely from an ensemble of triplet states, each of which is of Metal-to-Ligand, Charge-Transfer (MLCT) character and of closely comparable energy. At ambient temperature, dual emission is observed for the KBr disk and interpreted in terms of luminescence from both the ensemble and the fourth MLCT triplet state that lies at slightly higher energy. Relative reorganisation energies, energies, Huang–Rhys factors and radiative rate constants have been calculated for the two emissive states. It is confirmed that the fourth MLCT triplet state possesses more singlet character than the ensemble.
Co-reporter:Delphine Hablot, Raymond Ziessel, Mohammed A. H. Alamiry, Effat Bahraidah and Anthony Harriman
Chemical Science (2010-Present) 2013 - vol. 4(Issue 1) pp:NaN453-453
Publication Date(Web):2012/10/17
DOI:10.1039/C2SC21505E
A series of molecular dyads has been synthesized and fully characterised. These linear, donor–spacer–acceptor compounds comprise terminal dyes selected to exhibit intramolecular electronic energy transfer (EET) along the molecular axis. The spacer is built by accretion of ethynylene–carborane units that give centre-to-centre separation distances of 38, 57, 76, 96, and 115 Å respectively along the series. The probability of one-way EET between terminals depends on the length of the spacer but also on temperature and applied pressure. Throughout the series, the derived EET parameters are well explained in terms of through-space interactions but the probability of EET is higher than predicted for the fully extended conformation except in a glassy matrix at low temperature. The implication is that these spacers contract under ambient conditions, with the extent of longitudinal contraction increasing under pressure but decreasing as the temperature is lowered. Longer bridges are more susceptible to such distortion, which is considered to resemble a concertina effect caused by out-of-plane bending of individual subunits. The dynamics of EET can be used to estimate the strain energy associated with molecular contraction, the amount of work done in effecting the structural change and the Young's modulus for the bridge.
Co-reporter:Anthony Harriman, Patrycja Stachelek, Alexandra Sutter and Raymond Ziessel
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 39) pp:NaN26182-26182
Publication Date(Web):2015/08/28
DOI:10.1039/C5CP03932K
An extended molecular array, comprising three distinct types of chromophores and two additional redox-active subunits, that harvests photons over most of the visible spectral range has been synthesized and characterised. The array exhibits a rich variety of electrochemical waves when examined by cyclic voltammetry but assignment can be made on the basis of control compounds and molecular orbital calculations. Stepwise electronic energy transfer occurs along the molecular axis, corresponding to a gradient of excitation energies, to populate the lowest-energy excited state of the ultimate acceptor. The latter species, which absorbs and emits in the far-red region, enters into light-induced charge transfer with a terminal amine group. The array is relatively stable under illumination with white light but degrades slowly via a series of well-defined steps, the first of which is autocatalytic. One of the main attributes of this system is the capability to harvest an unusually high fraction of sunlight while providing protection against exposure to UV light.
Co-reporter:Mohammed A. H. Alamiry, Jerry P. Hagon, Anthony Harriman, Thomas Bura and Raymond Ziessel
Chemical Science (2010-Present) 2012 - vol. 3(Issue 4) pp:NaN1048-1048
Publication Date(Web):2011/12/12
DOI:10.1039/C2SC00948J
This work examines the electronic energy-transfer (EET) processes inherent to a molecular dyad in which aryl polycycles attached to a boron dipyrromethene (Bodipy) dye act as ancillary light harvesters for near-UV photons. The solvent, being methyltetrahydrofuran, is compressed under applied pressure to such an extent that, over the accessible pressure range, there is a 25% decrease in molar volume. This effect serves to increase the effective concentration of the solute and increases fluorescence from Bodipy when this chromophore is excited directly. Illumination into the aryl polycycles, namely pyrene and perylene derivatives, leads to rapid intramolecular EET to Bodipy but fluorescence from these units is partially restored under high pressure. The argument is made that applied pressure restricts torsional motions around the linkages and imposes a near orthogonal geometry for transition dipole moment vectors on the reactants. In turn, this pressure-induced conformational restriction switches off Förster-type EET within the system, leaving the electron-exchange contribution. For the target dyad, the Förster component is ca. 5% for pyrene and ca. 25% for perylene. Such contributions are not inconsistent with calculations made on the basis of Förster theory but modelling is rendered difficult by the absence of accurate information about the nature of the conformational motion. Two possibilities have been considered. In the first case, the appendages remain stiff but pressure reduces the extent of displacement from the lowest-energy position. The results can be accounted for in a quantitative sense on the basis of small deviations from the lowest-energy conformation; the actual amount of displacement needed to explain the pressure effect depends on the method used to compute the Förster rates and ranges from ca. 4° for the ideal dipole approximation to only 0.5° for the extended dipole method. Secondly, pressure is assumed to bend each appendage into a banana-like shape. Again, the full effect of applied pressure can be accounted for by way of minor curvature of the linkage.
Co-reporter:Adela Nano, Pascal Retailleau, Jerry P. Hagon, Anthony Harriman and Raymond Ziessel
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 21) pp:
Publication Date(Web):
DOI:10.1039/C3CP55021D

![Pentacyclo[19.3.1.13,7.19,13.115,19]octacosa-1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-25,26,27,28-tetrol, 5,11,17,23-tetrakis(1,1-dimethylethyl)-2-[3-[3-(10,15,20-triphenyl-21H,23H-porphin-5-yl)phenyl]propyl]-](/data/chemimg/3724600/855473-58-4.png)
![Pentacyclo[19.3.1.13,7.19,13.115,19]octacosa-1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-25,26,27,28-tetrol, 5,11,17,23-tetrakis(1,1-dimethylethyl)-2-[3-[3-(10,15,20-triphenyl-21H,23H-porphin-5-yl)phenyl]propyl]-](/data/chemimg/3724600/855473-58-4_b.png)
![Pentacyclo[19.3.1.13,7.19,13.115,19]octacosa-1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-25,26,27,28-tetrol, 5,11,17,23-tetrakis(1,1-dimethylethyl)-2-[3-[2-(10,15,20-triphenyl-21H,23H-porphin-5-yl)phenoxy]propyl]-](/data/chemimg/3724600/855473-57-3.png)
![Pentacyclo[19.3.1.13,7.19,13.115,19]octacosa-1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-25,26,27,28-tetrol, 5,11,17,23-tetrakis(1,1-dimethylethyl)-2-[3-[2-(10,15,20-triphenyl-21H,23H-porphin-5-yl)phenoxy]propyl]-](/data/chemimg/3724600/855473-57-3_b.png)
![Pentacyclo[19.3.1.13,7.19,13.115,19]octacosa-1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-25,26,27,28-tetrol, 5,11,17,23-tetrakis(1,1-dimethylethyl)-2-[3-[4-(10,15,20-triphenyl-21H,23H-porphin-5-yl)phenyl]propyl]-](/data/chemimg/3724600/855473-59-5.png)
![Pentacyclo[19.3.1.13,7.19,13.115,19]octacosa-1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-25,26,27,28-tetrol, 5,11,17,23-tetrakis(1,1-dimethylethyl)-2-[3-[4-(10,15,20-triphenyl-21H,23H-porphin-5-yl)phenyl]propyl]-](/data/chemimg/3724600/855473-59-5_b.png)
![Benzenamine, 4-[10,15,20-tris[3,5-bis(1,1-dimethylethyl)phenyl]-21H,23H-porphin-5-yl]-](/data/chemimg/3724200/163811-80-1.png)
![Benzenamine, 4-[10,15,20-tris[3,5-bis(1,1-dimethylethyl)phenyl]-21H,23H-porphin-5-yl]-](/data/chemimg/3724200/163811-80-1_b.png)
![Dibenzo[q,s][1,4,7,10,13,16]hexaoxacycloeicosin, 6,7,9,10,12,13,15,16,18,19-decahydro-](/data/chemimg/3724000/41051-91-6.png)
![Dibenzo[q,s][1,4,7,10,13,16]hexaoxacycloeicosin, 6,7,9,10,12,13,15,16,18,19-decahydro-](/data/chemimg/3724000/41051-91-6_b.png)
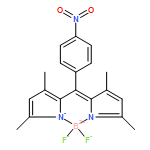


![Dibenzo[e,g][1,4]dioxocin-3,10-diamine, 6,7-dihydro-](http://img.cochemist.com/ccimg/563600/563539-69-5.png)
![Dibenzo[e,g][1,4]dioxocin-3,10-diamine, 6,7-dihydro-](http://img.cochemist.com/ccimg/563600/563539-69-5_b.png)
![9,12-Dioxa-2,3-diazatricyclo[11.3.1.14,8]octadeca-1(17),2,4,6,8(18),13,
15-heptaene](http://img.cochemist.com/ccimg/563600/563539-62-8.png)
![9,12-Dioxa-2,3-diazatricyclo[11.3.1.14,8]octadeca-1(17),2,4,6,8(18),13,
15-heptaene](http://img.cochemist.com/ccimg/563600/563539-62-8_b.png)






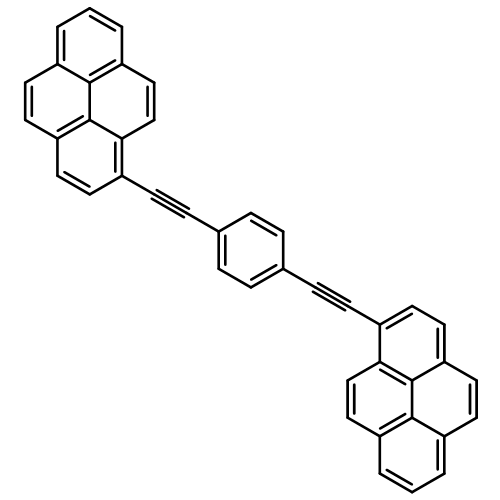








![21H,23H-Porphine, 5,10,15-tris[3,5-bis(1,1-dimethylethyl)phenyl]-20-(4-ethynylphenyl)-](/data/chemimg/3723300/1000788-85-1.png)
![21H,23H-Porphine, 5,10,15-tris[3,5-bis(1,1-dimethylethyl)phenyl]-20-(4-ethynylphenyl)-](/data/chemimg/3723300/1000788-85-1_b.png)
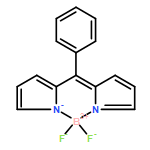






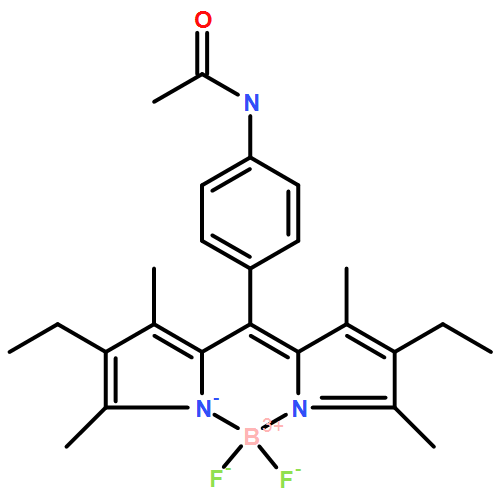


![Dibenzo[d,f][1,3]dioxepin, 3,9-diiodo-](http://img.cochemist.com/ccimg/849100/849042-53-1.png)
![Dibenzo[d,f][1,3]dioxepin, 3,9-diiodo-](http://img.cochemist.com/ccimg/849100/849042-53-1_b.png)
![9,16-Dioxa-2,3-diazatricyclo[15.3.1.14,8]docosa-1(21),2,4,6,8(22),17,1
9-heptaene](http://img.cochemist.com/ccimg/563600/563539-64-0.png)
![9,16-Dioxa-2,3-diazatricyclo[15.3.1.14,8]docosa-1(21),2,4,6,8(22),17,1
9-heptaene](http://img.cochemist.com/ccimg/563600/563539-64-0_b.png)
![9,21-Dioxa-2,3-diazatricyclo[20.3.1.14,8]heptacosa-1(26),2,4,6,8(27),2
2,24-heptaene](http://img.cochemist.com/ccimg/563600/563539-66-2.png)
![9,21-Dioxa-2,3-diazatricyclo[20.3.1.14,8]heptacosa-1(26),2,4,6,8(27),2
2,24-heptaene](http://img.cochemist.com/ccimg/563600/563539-66-2_b.png)

![Dibenzo[e,g][1,4]dioxocin-1,12-diamine, 6,7-dihydro-](http://img.cochemist.com/ccimg/563600/563539-74-2.png)
![Dibenzo[e,g][1,4]dioxocin-1,12-diamine, 6,7-dihydro-](http://img.cochemist.com/ccimg/563600/563539-74-2_b.png)
![2,2'-Bipyrimidine, 5,5'-bis[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/718700/718606-28-1.png)
![2,2'-Bipyrimidine, 5,5'-bis[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/718700/718606-28-1_b.png)
![Dibenzo[d,f][1,3]dioxepin, 3,9-diphenyl-](http://img.cochemist.com/ccimg/42500/42447-99-4.png)
![Dibenzo[d,f][1,3]dioxepin, 3,9-diphenyl-](http://img.cochemist.com/ccimg/42500/42447-99-4_b.png)
![2,2'-Bipyridine, 5-[(3,4-dibutyl-2-thienyl)ethynyl]-5'-ethynyl-](http://img.cochemist.com/ccimg/794600/794529-18-3.png)
![2,2'-Bipyridine, 5-[(3,4-dibutyl-2-thienyl)ethynyl]-5'-ethynyl-](http://img.cochemist.com/ccimg/794600/794529-18-3_b.png)
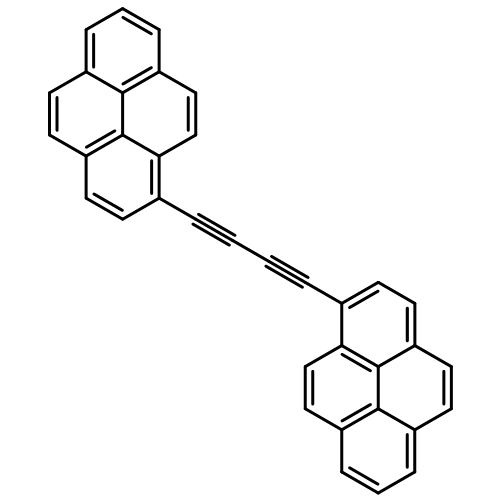
![DIPYRIDO[1,2-A:2',1'-C][1,4]DIAZOCINEDIIUM, 6,7,8,9-TETRAHYDRO-](http://img.cochemist.com/ccimg/16700/16651-68-6.png)
![DIPYRIDO[1,2-A:2',1'-C][1,4]DIAZOCINEDIIUM, 6,7,8,9-TETRAHYDRO-](http://img.cochemist.com/ccimg/16700/16651-68-6_b.png)
![[1,1'-Biphenyl]-2,2'-diol, 4,4'-diiodo-](http://img.cochemist.com/ccimg/204400/204315-81-1.png)
![[1,1'-Biphenyl]-2,2'-diol, 4,4'-diiodo-](http://img.cochemist.com/ccimg/204400/204315-81-1_b.png)
![DIBENZO[C,E][1,2]DITHIIN, 3,8-DIIODO-](http://img.cochemist.com/ccimg/916900/916899-31-5.png)
![DIBENZO[C,E][1,2]DITHIIN, 3,8-DIIODO-](http://img.cochemist.com/ccimg/916900/916899-31-5_b.png)
![1H-Benz[de]isoquinoline, 2,3-dihydro-2-methyl-](http://img.cochemist.com/ccimg/20000/19961-48-9.png)
![1H-Benz[de]isoquinoline, 2,3-dihydro-2-methyl-](http://img.cochemist.com/ccimg/20000/19961-48-9_b.png)
![2,2'-Bipyridine, 5,5'-bis[[2,5-bis(dodecyloxy)phenyl]ethynyl]-](http://img.cochemist.com/ccimg/314000/313947-68-1.png)
![2,2'-Bipyridine, 5,5'-bis[[2,5-bis(dodecyloxy)phenyl]ethynyl]-](http://img.cochemist.com/ccimg/314000/313947-68-1_b.png)
![1H-Pyrrole, 2,2'-[[3,5-bis(1,1-dimethylethyl)phenyl]methylene]bis-](http://img.cochemist.com/ccimg/181800/181762-72-1.png)
![1H-Pyrrole, 2,2'-[[3,5-bis(1,1-dimethylethyl)phenyl]methylene]bis-](http://img.cochemist.com/ccimg/181800/181762-72-1_b.png)
![Benzonitrile, 4-[[4-(methylthio)phenyl]ethynyl]-](http://img.cochemist.com/ccimg/125200/125138-98-9.png)
![Benzonitrile, 4-[[4-(methylthio)phenyl]ethynyl]-](http://img.cochemist.com/ccimg/125200/125138-98-9_b.png)
![2,2'-BIPYRIDINE, 5,5'-BIS[(3,4-DIBUTYL-5-IODO-2-THIENYL)ETHYNYL]-](http://img.cochemist.com/ccimg/794600/794529-19-4.png)
![2,2'-BIPYRIDINE, 5,5'-BIS[(3,4-DIBUTYL-5-IODO-2-THIENYL)ETHYNYL]-](http://img.cochemist.com/ccimg/794600/794529-19-4_b.png)
![BENZENE, 1,1'-[1,6-HEXANEDIYLBIS(OXY)]BIS[3-NITRO-](http://img.cochemist.com/ccimg/5300/5226-69-7.png)
![BENZENE, 1,1'-[1,6-HEXANEDIYLBIS(OXY)]BIS[3-NITRO-](http://img.cochemist.com/ccimg/5300/5226-69-7_b.png)
![BENZENE, 1,1'-[1,9-NONANEDIYLBIS(OXY)]BIS[3-NITRO-](http://img.cochemist.com/ccimg/5300/5226-70-0.png)
![BENZENE, 1,1'-[1,9-NONANEDIYLBIS(OXY)]BIS[3-NITRO-](http://img.cochemist.com/ccimg/5300/5226-70-0_b.png)
![2,2'-BIPYRIDINE, 5-[(3,4-DIBUTYL-2-THIENYL)ETHYNYL]-5'-[(TRIETHYLSILYL)ETHYNYL]-](http://img.cochemist.com/ccimg/794600/794529-21-8.png)
![2,2'-BIPYRIDINE, 5-[(3,4-DIBUTYL-2-THIENYL)ETHYNYL]-5'-[(TRIETHYLSILYL)ETHYNYL]-](http://img.cochemist.com/ccimg/794600/794529-21-8_b.png)
![BENZENE, 1,1'-[OXYBIS(2,1-ETHANEDIYLOXY-2,1-ETHANEDIYLOXY)]BIS[3-NITRO-](http://img.cochemist.com/ccimg/563600/563539-57-1.png)
![BENZENE, 1,1'-[OXYBIS(2,1-ETHANEDIYLOXY-2,1-ETHANEDIYLOXY)]BIS[3-NITRO-](http://img.cochemist.com/ccimg/563600/563539-57-1_b.png)

![9,12,15,18,21-PENTAOXA-2,3-DIAZATRICYCLO[20.3.1.14,8]HEPTACOSA-1(26),2,4,6,8(27),22,24-HEPTAENE](http://img.cochemist.com/ccimg/563600/563539-63-9.png)
![9,12,15,18,21-PENTAOXA-2,3-DIAZATRICYCLO[20.3.1.14,8]HEPTACOSA-1(26),2,4,6,8(27),22,24-HEPTAENE](http://img.cochemist.com/ccimg/563600/563539-63-9_b.png)


![[4,4'-BIPYRIDINE]-3,3'-DIOL](http://img.cochemist.com/ccimg/90800/90770-89-1.png)
![[4,4'-BIPYRIDINE]-3,3'-DIOL](http://img.cochemist.com/ccimg/90800/90770-89-1_b.png)


![2,2':6',2''-Terpyridine, 4'-[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/187100/187026-88-6.png)
![2,2':6',2''-Terpyridine, 4'-[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/187100/187026-88-6_b.png)
![BENZENE, 1,1'-[1,11-UNDECANEDIYLBIS(OXY)]BIS[3-NITRO-](http://img.cochemist.com/ccimg/563600/563539-58-2.png)
![BENZENE, 1,1'-[1,11-UNDECANEDIYLBIS(OXY)]BIS[3-NITRO-](http://img.cochemist.com/ccimg/563600/563539-58-2_b.png)
![6H-DIBENZO[F,H][1,5]DIOXONIN, 7,8-DIHYDRO-3,11-DIIODO-](http://img.cochemist.com/ccimg/564500/564461-78-5.png)
![6H-DIBENZO[F,H][1,5]DIOXONIN, 7,8-DIHYDRO-3,11-DIIODO-](http://img.cochemist.com/ccimg/564500/564461-78-5_b.png)
![9,19-DIOXA-2,3-DIAZATRICYCLO[18.3.1.14,8]PENTACOSA-1(24),2,4,6,8(25),20,22-HEPTAENE](http://img.cochemist.com/ccimg/563600/563539-65-1.png)
![9,19-DIOXA-2,3-DIAZATRICYCLO[18.3.1.14,8]PENTACOSA-1(24),2,4,6,8(25),20,22-HEPTAENE](http://img.cochemist.com/ccimg/563600/563539-65-1_b.png)


![1H,3H-NAPHTHO[1,8-CD]PYRAN-1,3-DIONE, 6-(DIMETHYLAMINO)-](http://img.cochemist.com/ccimg/78000/77976-79-5.png)
![1H,3H-NAPHTHO[1,8-CD]PYRAN-1,3-DIONE, 6-(DIMETHYLAMINO)-](http://img.cochemist.com/ccimg/78000/77976-79-5_b.png)





![Silane, [(4-ethyl-5-iodo-2-thienyl)ethynyl]trimethyl-](http://img.cochemist.com/ccimg/676500/676446-62-1.png)
![Silane, [(4-ethyl-5-iodo-2-thienyl)ethynyl]trimethyl-](http://img.cochemist.com/ccimg/676500/676446-62-1_b.png)









![9(10H)-Acridinone, 10-[(2-methoxyethoxy)methyl]-](http://img.cochemist.com/ccimg/147700/147643-41-2.png)
![9(10H)-Acridinone, 10-[(2-methoxyethoxy)methyl]-](http://img.cochemist.com/ccimg/147700/147643-41-2_b.png)





![Methanesulfonic acid, trifluoro-, [2,2':6',2''-terpyridin]-4'-yl ester](http://img.cochemist.com/ccimg/134700/134653-69-3.png)
![Methanesulfonic acid, trifluoro-, [2,2':6',2''-terpyridin]-4'-yl ester](http://img.cochemist.com/ccimg/134700/134653-69-3_b.png)











![Propanedinitrile,2-[(2,3,6,7-tetrahydro-1H,5H-benzo[ij]quinolizin-9-yl)methylene]-](http://img.cochemist.com/ccimg/58300/58293-56-4.png)
![Propanedinitrile,2-[(2,3,6,7-tetrahydro-1H,5H-benzo[ij]quinolizin-9-yl)methylene]-](http://img.cochemist.com/ccimg/58300/58293-56-4_b.png)


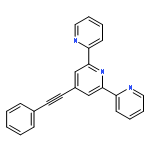
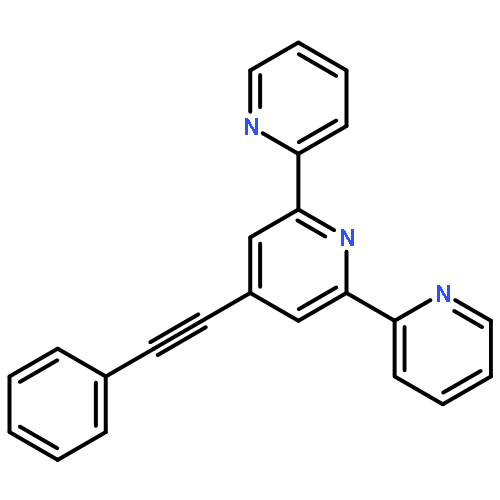
![7,8-DIHYDRO-6H-DIPYRIDO[1,2-B:1',2'-E][1,4]DIAZEPINE-5,9-DIIUM](http://img.cochemist.com/ccimg/7400/7325-63-5.png)
![7,8-DIHYDRO-6H-DIPYRIDO[1,2-B:1',2'-E][1,4]DIAZEPINE-5,9-DIIUM](http://img.cochemist.com/ccimg/7400/7325-63-5_b.png)




![2,2'-Bipyridine,5,5''-[[2,5-bis(dodecyloxy)-1,4-phenylene]di-2,1-ethynediyl]bis-](http://img.cochemist.com/ccimg/271300/271251-02-6.png)
![2,2'-Bipyridine,5,5''-[[2,5-bis(dodecyloxy)-1,4-phenylene]di-2,1-ethynediyl]bis-](http://img.cochemist.com/ccimg/271300/271251-02-6_b.png)


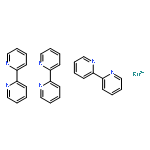
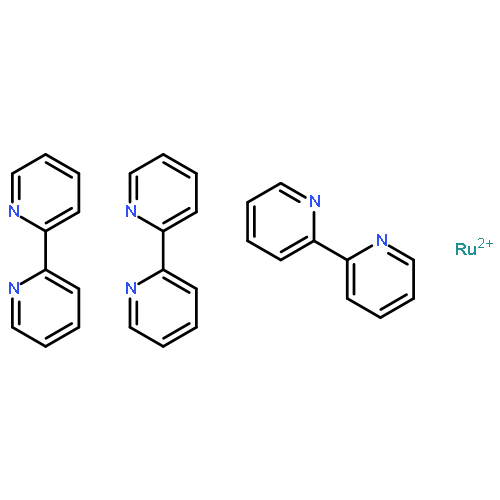













![4-[4-[BIS(1H-PYRROL-2-YL)METHYL]PHENYL]-2,6-DIPYRIDIN-2-YLPYRIDINE](http://img.cochemist.com/ccimg/799300/799292-09-4.png)
![4-[4-[BIS(1H-PYRROL-2-YL)METHYL]PHENYL]-2,6-DIPYRIDIN-2-YLPYRIDINE](http://img.cochemist.com/ccimg/799300/799292-09-4_b.png)






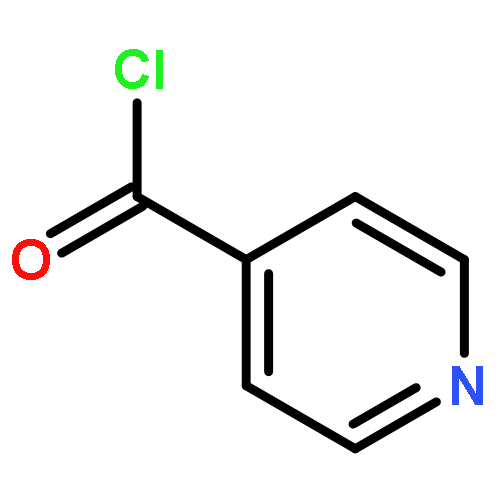
![4-([2,2':6',2''-Terpyridin]-4'-yl)benzaldehyde](http://img.cochemist.com/ccimg/138300/138253-30-2.png)
![4-([2,2':6',2''-Terpyridin]-4'-yl)benzaldehyde](http://img.cochemist.com/ccimg/138300/138253-30-2_b.png)



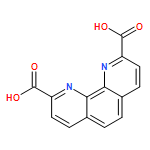

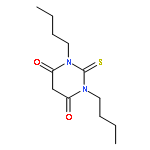
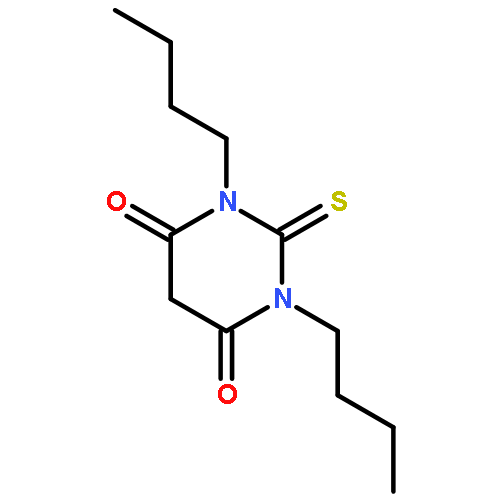
![(R)-(2,3,4,5-TETRAHYDRO-1H-BENZO[B]AZEPIN-5-YL)AMINE](http://img.cochemist.com/ccimg/400/353-41-3.png)
![(R)-(2,3,4,5-TETRAHYDRO-1H-BENZO[B]AZEPIN-5-YL)AMINE](http://img.cochemist.com/ccimg/400/353-41-3_b.png)

![21H,23H-Porphine, 5-(4-azidophenyl)-10,15,20-tris[3,5-bis(1,1-dimethylethyl)phenyl]-](/data/chemimg/147500/1141485-91-7.png)
![21H,23H-Porphine, 5-(4-azidophenyl)-10,15,20-tris[3,5-bis(1,1-dimethylethyl)phenyl]-](/data/chemimg/147500/1141485-91-7_b.png)



![1,1'-[ethane-1,2-diylbis(oxy)]bis(3-nitrobenzene)](http://img.cochemist.com/ccimg/26000/25986-11-2.png)
![1,1'-[ethane-1,2-diylbis(oxy)]bis(3-nitrobenzene)](http://img.cochemist.com/ccimg/26000/25986-11-2_b.png)











![6,7-dihydrodipyrido[1,2-b:1',2'-e]pyrazine-5,8-diium](http://img.cochemist.com/ccimg/2800/2764-72-9.png)
![6,7-dihydrodipyrido[1,2-b:1',2'-e]pyrazine-5,8-diium](http://img.cochemist.com/ccimg/2800/2764-72-9_b.png)






![1,2,3,5,6,7-Hexahydropyrido[3,2,1-ij]quinoline-9-carbaldehyde](http://img.cochemist.com/ccimg/34000/33985-71-6.png)
![1,2,3,5,6,7-Hexahydropyrido[3,2,1-ij]quinoline-9-carbaldehyde](http://img.cochemist.com/ccimg/34000/33985-71-6_b.png)
















![Boron,[2-[1-(3,5-dimethyl-2H-pyrrol-2-ylidene-kN)ethyl]-3,5-dimethyl-1H-pyrrolato-kN]difluoro-, (T-4)-](/data/chemimg/110400/121207-31-6.png)
![Boron,[2-[1-(3,5-dimethyl-2H-pyrrol-2-ylidene-kN)ethyl]-3,5-dimethyl-1H-pyrrolato-kN]difluoro-, (T-4)-](/data/chemimg/110400/121207-31-6_b.png)
![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2R)-](http://img.cochemist.com/ccimg/155900/155806-35-2.png)
![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2R)-](http://img.cochemist.com/ccimg/155900/155806-35-2_b.png)

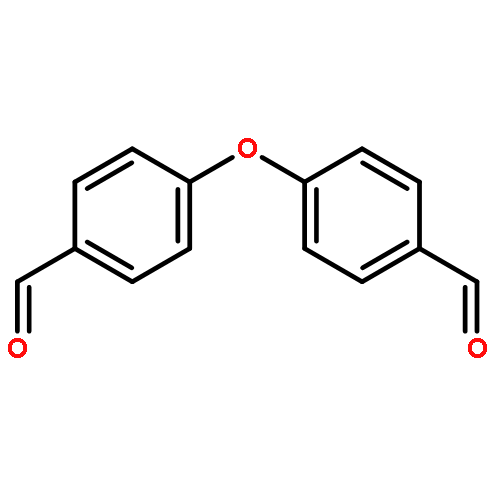












![2,2'-[(4-nitrophenyl)methylene]bis-1H-Pyrrole](http://img.cochemist.com/ccimg/143900/143859-77-2.png)
![2,2'-[(4-nitrophenyl)methylene]bis-1H-Pyrrole](http://img.cochemist.com/ccimg/143900/143859-77-2_b.png)



![[(2s,3r)-3-(methylsulfonyloxymethyl)-2-bicyclo[2.2.1]heptanyl]methyl Methanesulfonate](http://img.cochemist.com/ccimg/2500/2434-88-0.png)
![[(2s,3r)-3-(methylsulfonyloxymethyl)-2-bicyclo[2.2.1]heptanyl]methyl Methanesulfonate](http://img.cochemist.com/ccimg/2500/2434-88-0_b.png)
![Bicyclo[2.2.1]heptane-2,3-dimethanol,2,3-dimethanesulfonate, (1R,2R,3R,4S)-rel-](http://img.cochemist.com/ccimg/2500/2434-90-4.png)
![Bicyclo[2.2.1]heptane-2,3-dimethanol,2,3-dimethanesulfonate, (1R,2R,3R,4S)-rel-](http://img.cochemist.com/ccimg/2500/2434-90-4_b.png)