Co-reporter:Shengtao Xu, Hong Yao, Mei Hu, Dahong Li, Zheying Zhu, Weijia Xie, Hequan Yao, Liang Wu, Zhe-Sheng Chen, and Jinyi Xu
Journal of Natural Products September 22, 2017 Volume 80(Issue 9) pp:2391-2391
Publication Date(Web):September 13, 2017
DOI:10.1021/acs.jnatprod.7b00057
Structurally unique 6,7-seco-ent-kaurenes, which are widely distributed in the genus Isodon, have attracted considerable attention because of their antitumor activities. Previously, a convenient conversion of commercially available oridonin (1) to 6,7-seco-ent-kaurenes was developed. Herein, several novel spiro-lactone-type ent-kaurene derivatives bearing various substituents at the C-1 and C-14 positions were further designed and synthesized from the natural product oridonin. Moreover, a number of seven-membered C-ring-expanded 6,7-seco-ent-kaurenes were also identified for the first time. It was observed that most of the spiro-lactone-type ent-kaurenes tested markedly inhibited the proliferation of cancer cells, with an IC50 value as low as 0.55 μM. An investigation on its mechanism of action showed that the representative compound 7b affected the cell cycle and induced apoptosis at a low micromolar level in MCF-7 human breast cancer cells. Furthermore, compound 7b inhibited liver tumor growth in an in vivo mouse model and exhibited no observable toxic effects. Collectively, the results warrant further preclinical investigations of these spiro-lactone-type ent-kaurenes as potential novel anticancer agents.
Co-reporter:Jichao Chen, Ruifan Wang, Tianyu Wang, Qilong Ding, Aliahmad Khalil, Shengtao Xu, Aijun Lin, Hequan Yao, Weijia Xie, Zheying Zhu, and Jinyi Xu
ACS Medicinal Chemistry Letters April 13, 2017 Volume 8(Issue 4) pp:443-443
Publication Date(Web):March 13, 2017
DOI:10.1021/acsmedchemlett.7b00035
A series of novel β-elemene dimer derivatives were synthesized and evaluated for their antioxidant activities. The results indicated that most of the target compounds showed more potent cytoprotective effects than positive control vitamin E. In particular, dimer D5 exhibited the strongest antioxidant activity, which was significantly superior to the active compound D1 obtained in our previous study. Besides, D5 did not produce obvious cytotoxicity in normal human umbilical vein endothelial cells (HUVECs) and increased the viability of HUVECs injured by H2O2 in a concentration-dependent manner. Further studies suggested that the cytoprotective action of D5 might be mediated, at least in part, by increasing the intracellular superoxide dismutase activity and nitric oxide secretion as well as decreasing the intracellular malonyldialdehyde content and lactate dehydrogenase release. Furthermore, D5 observably inhibited ROS generation and prevented H2O2-induced apoptosis in HUVECs possibly via inhibiting the activation of the MAPK signaling pathway.Keywords: antioxidant activity; apoptosis; dimer; MAPK pathway; β-Elemene;
Co-reporter:Shengtao Xu, Hong Yao, Shanshan Luo, Yun-Kai Zhang, Dong-Hua Yang, Dahong Li, Guangyu Wang, Mei Hu, Yangyi Qiu, Xiaoming Wu, Hequan YaoWeijia Xie, Zhe-Sheng Chen, Jinyi Xu
Journal of Medicinal Chemistry 2017 Volume 60(Issue 4) pp:
Publication Date(Web):February 6, 2017
DOI:10.1021/acs.jmedchem.6b01652
The cytotoxicity of the natural ent-kaurene diterpenoid, oridonin, has been extensively studied. However, the application of oridonin for cancer therapy was hampered primarily by its moderate potency. In this study, a series of oridonin A-ring modified analogues, and their derivatives bearing various substituents on 14-OH position, were designed, synthesized, and evaluated for anticancer efficacy. Some of the derivatives were significantly more potent than oridonin against both drug-sensitive and drug-resistant cancer cells. The most potent compound, 13p, was 200-fold more efficacious than oridonin in MCF-7 cancer cells. Furthermore, 13p induced apoptosis and cell cycle arrest at the G2/M phase. A decrease in mitochondrial membrane potential and an increase in Bax/Bcl-2 ratio, accompanied by activated caspase-3 cleavage, were observed in MCF-7 cells after treatment with 13p, suggesting that the mitochondrial pathway was involved in the 13p-mediated apoptosis. Moreover, 13p significantly inhibited tumor growth in mouse xenograft models and had no observable toxic effect.
Co-reporter:Jichao Chen, Tianyu Wang, Shengtao Xu, Aijun Lin, Hequan Yao, Weijia Xie, Zheying Zhu, Jinyi Xu
Fitoterapia 2017 Volume 120(Volume 120) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.fitote.2017.05.002
A series of novel β-elemene isopropanolamine derivatives were synthesized and evaluated for their antitumor activity. The results indicated that all of the compounds showed stronger antiproliferative activities than β-elemene as well as improved aqueous solubility. In particular dimer 6q showed the strongest cytotoxicity against four tumor cell lines (SGC-7901, HeLa, U87 and A549) with IC50 values ranging from 4.37 to 10.20 μM. Moreover, combination of 6q with cisplatin exhibited a synergistic effect on these cell lines with IC50 values ranging from 1.21 to 2.94 μM, and reversed the resistance of A549/DPP cells with an IC50 value of 2.52 μM. The mechanism study revealed that 6q caused cell cycle arrest at the G2 phase and induced apoptosis of SGC-7901 cells through a mitochondrial-dependent apoptotic pathway. Further in vivo study in H22 liver cancer xenograft mouse model validated the antitumor activity of 6q with a tumor inhibitory ratio (TIR) of 60.3%, which was higher than that of β-elemene (TIR, 49.1%) at a dose of 60 mg/kg. Altogether, the potent antitumor activity of 6q in vitro and in vivo warranted further preclinical investigation for potential anticancer chemotherapy.Download high-res image (85KB)Download full-size image
Co-reporter:Jichao Chen, Tianyu Wang, Shengtao Xu, Pengfei Zhang, Aijun Lin, Liang Wu, Hequan Yao, Weijia Xie, Zheying Zhu, Jinyi Xu
European Journal of Medicinal Chemistry 2017 Volume 135(Volume 135) pp:
Publication Date(Web):28 July 2017
DOI:10.1016/j.ejmech.2017.04.045
•Thirteen novel furoxan-based NO-donating β-elemene hybrids were synthesized.•All of the compounds exhibited significantly improved antitumor activity in vitro.•The activity of 11a in U87 cells was markedly diminished by an NO scavenger.•11a induced cell apoptosis by preventing the activation of the PI3K/Akt pathway.•11a effectively inhibited liver tumor growth in an in vivo mouse model.A series of novel furoxan-based NO-donating β-elemene hybrids were designed and synthesized to improve the anticancer efficacy of natural β-elemene. The bioassay results indicated that all of the target compounds exhibited significantly improved antiproliferative activities against three cancer cell lines (SGC-7901, HeLa and U87) compared to parent compound β-elemene. Interestingly, these compounds displayed excellent sensitivity to U87 cells with IC50 values ranging from 173 to 2 nM. Moreover, most compounds produced high levels of NO in vitro, and the antitumor activity of 11a in U87 cells was markedly attenuated by an NO scavenger (hemoglobin or carboxy-PTIO). Further mechanism studies revealed that 11a caused the G2 phase arrest of the cell cycle and induced apoptosis of U87 cells by preventing the activation of the PI3K/Akt pathway. Moreover, 11a significantly suppressed the tumor growth in H22 liver cancer xenograft mouse model with a tumor inhibitory ratio (TIR) of 64.8%, which was superior to that of β-elemene (TIR, 49.6%) at the same dose of 60 mg/kg. Together, the remarkable biological profiles of these novel NO-donating β-elemene derivatives may make them promising candidates for the intervention of human cancers.Download high-res image (185KB)Download full-size image
Co-reporter:Zhaoliang Li, Yanchun Meng, Shengtao Xu, Wang Shen, Zhaoqing Meng, Zhenzhong Wang, Gang Ding, Wenzhe Huang, Wei Xiao, Jinyi Xu
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 10(Issue 10) pp:
Publication Date(Web):15 May 2017
DOI:10.1016/j.bmc.2017.03.052
In search of novel anti-influenza agents with higher potency, a series of acylguanidine oseltamivir carboxylate analogues were synthesized and evaluated against influenza viruses (H1N1 and H3N2) in vitro. The representative compounds with strong inhibitory activities (IC50 <40 nM) against neuraminidase (NA) were further tested against the NA from oseltamivir-resistant strain (H259Y). Among them, compounds 9 and 17 were potent NA inhibitors that exhibited a 5 and 11-fold increase in activity comparing with oseltamivir carboxylate (2, OC) against the H259Y mutant, respectively. Furthermore, the effect against influenza virus H259Y mutant (H1N1) replication and cytotoxicity assays indicated that compounds 9 and 17 exhibited a 20 and 6-fold increase than the parent compound 2, and had no obvious cytotoxicity in vitro. Moreover, the molecular docking studies revealed that the docking modes of compounds 9 and 17 were different from that of oseltamivir, and the new hydrogen bonds and hydrophobic interaction were formed in this case. This work provided unique insights in the discovery of potent inhibitors against NAs from wild-type and oseltamivir-resistant strains.Download high-res image (157KB)Download full-size image
Co-reporter:Jichao Chen;Tianyu Wang;Yanpeng Liu;Tong Wang;Aijun Lin;Hequan Yao
Organic Chemistry Frontiers 2017 vol. 4(Issue 4) pp:622-626
Publication Date(Web):2017/03/28
DOI:10.1039/C6QO00765A
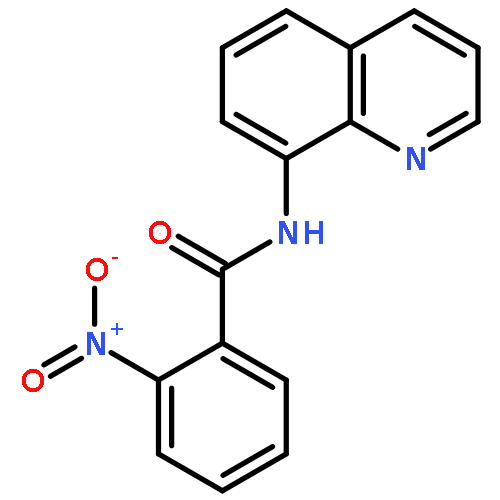
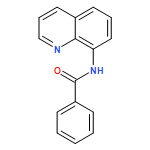
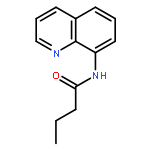
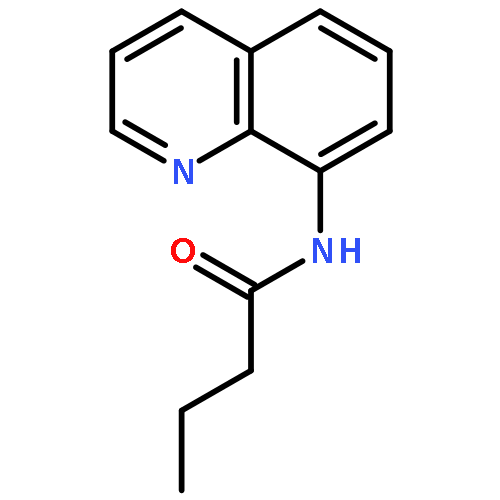
An efficient and convenient method of C5-selective halogenation of quinoline derivatives was developed. The reaction proceeds smoothly in water with readily available N-halosuccinimides (NCS, NBS and NIS) as the halogenation reagents. This method features metal-free catalysis, no additional oxidants and additives, broad substrate scope and short reaction time.
Co-reporter:Jichao Chen;Tianyu Wang;Tong Wang;Aijun Lin;Hequan Yao
Organic Chemistry Frontiers 2017 vol. 4(Issue 1) pp:130-134
Publication Date(Web):2016/12/20
DOI:10.1039/C6QO00590J
Copper-catalyzed direct C5-position thio/selenocyanation of quinolines using commercially available, inexpensive KSCN/SeCN as the thio/selenocyanation reagent was developed, which had good tolerance toward various aliphatic or aromatic 8-aminoquinoline derivatives. Importantly, the synthetic application of this protocol led to some useful compounds.
Co-reporter:Shengtao Xu, Hong Yao, Lingling Pei, Mei Hu, Dahong Li, Yangyi Qiu, Guangyu Wang, Liang Wu, Hequan Yao, Zheying Zhu, Jinyi Xu
European Journal of Medicinal Chemistry 2017 Volume 132(Volume 132) pp:
Publication Date(Web):26 May 2017
DOI:10.1016/j.ejmech.2017.03.055
•NQO1-targeted oridonin prodrugs possessing indolequinone moiety were prepared.•They showed relatively higher antineoplastic activities against NQO1-rich cancer cells.•The most potent compound 29h is a good substrate of NQO1.•The anti-tumor activity of 29h was verified in a liver cancer xenograft mouse model.The enzyme NQO1 is a potential target for selective cancer therapy due to its overexpression in certain hypoxic tumors. A series of prodrugs possessing a variety of cytotoxic diterpenoids (oridonin and its analogues) as the leaving groups activated by NQO1 were synthesized by functionalization of 3-(hydroxymethyl)indolequinone, which is a good substrate of NQO1. The target compounds (29a-m) exhibited relatively higher antiproliferative activities against NQO1-rich human colon carcinoma cells (HT-29) and human lung carcinoma (A549) cells (IC50 = 0.263–2.904 μM), while NQO1-defficient lung adenosquamous carcinoma cells (H596) were less sensitive to these compounds, among which, compound 29h exhibited the most potent antiproliferative activity against both A549 and HT-29 cells, with IC50 values of 0.386 and 0.263 μM, respectively. Further HPLC and docking studies demonstrated that 29h is a good substrate of NQO1. Moreover, the investigation of anticancer mechanism showed that the representative compound 29h affected cell cycle and induced NQO1 dependent apoptosis through an oxidative stress triggered mitochondria-related pathway in A549 cells. Besides, the antitumor activity of 29h was also verified in a liver cancer xenograft mouse model. Biological evaluation of these compounds concludes that there is a strong correlation between NQO1 enzyme and induction of cancer cell death. Thus, this suggests that some of the target compounds activated by NQO1 are novel prodrug candidates potential for selective anticancer therapy.Download high-res image (206KB)Download full-size image
Co-reporter:Jichao Chen, Tianyu Wang, Shengtao Xu, Aijun Lin, Hequan Yao, Weijia Xie, Zheying Zhu, Jinyi Xu
European Journal of Medicinal Chemistry 2017 Volume 132(Volume 132) pp:
Publication Date(Web):26 May 2017
DOI:10.1016/j.ejmech.2017.03.027
•Eighteen novel NO-donating protoberberine derivatives were synthesized.•Most compounds showed significantly enhanced in vitro anti-proliferative activity.•15a exhibited good selectivity between tumor cells and normal liver LO-2 cells.•The antitumor activity of 15a in HepG2 cells was diminished by an NO scavenger.•15a effectively inhibited liver tumor growth in an in vivo mouse model.A novel class of NO-donating protoberberine derivatives were synthesized and initially evaluated for their anti-hepatocellular carcinoma activities. Most of the compounds exhibited more potent activity against HepG2 cells than parent compounds berberine and palmatine. In particular, compound 15a exerted the strongest activity with an IC50 value of 1.36 μM. Moreover, most compounds released moderate levels of NO in vitro, and the antitumor activity of 15a in HepG2 cells was remarkably diminished by an NO scavenger. Interestingly, compound 15a displayed a broad-spectrum antitumor efficacy and possessed good selectivity between tumor cells (HepG2, SMMC-7721, HCT-116, HL-60) and normal liver LO-2 cells. The mechanism studies revealed that 15a blocked the G2 phase of the cell cycle and induced apoptosis of HepG2 cells by mitochondrial depolarization. Furthermore, 15a inhibited tumor growth in H22 liver cancer xenograft mouse model by 62.5% (w/w), which was significantly superior to parent compound palmatine (41.6%, w/w). Overall, the current study may provide a new approach for the discovery of novel antitumor agents.Download high-res image (207KB)Download full-size image
Co-reporter:Shengtao Xu; Shanshan Luo; Hong Yao; Hao Cai; Xiaoming Miao; Fang Wu; Dong-Hua Yang; Xiaoming Wu; Weijia Xie; Hequan Yao; Zhe-Sheng Chen
Journal of Medicinal Chemistry 2016 Volume 59(Issue 10) pp:5022-5034
Publication Date(Web):April 18, 2016
DOI:10.1021/acs.jmedchem.6b00408
Oridonin (1) is a complex ent-kaurane diterpenoid exhibiting remarkable antitumor activity. However, the detailed mechanism or cellular target that underlies this activity has not yet been identified. Herein, we report an efficient approach for exploring the anticancer mechanism of oridonin through development of the potent fluorescent analogues. A series of novel fluorescent oridonin probes linked with coumarin moieties were designed, synthesized, and characterized. Fluorescence microscopy and confocal imaging studies suggested that fluorescent oridonin probe 17d was rapidly taken up into tumor cells and the mitochondrion was the main site of its accumulation. Moreover, we confirmed that cytochrome c played an important role in oridonin induced mitochondrion-mediated apoptosis and α,β-unsaturated ketone is the active moiety of oridonin, which is crucial to its uptake, localization, and cytotoxicity. Our results provide new insights on the molecular mechanism of oridonin and would be useful for its further development into an antitumor agent.
Co-reporter:Hao Cai, Xiaojing Huang, Shengtao Xu, Hao Shen, Pengfei Zhang, Yue Huang, Jieyun Jiang, Yijun Sun, Bo Jiang, Xiaoming Wu, Hequan Yao, Jingyi Xu
European Journal of Medicinal Chemistry 2016 Volume 108() pp:89-103
Publication Date(Web):27 January 2016
DOI:10.1016/j.ejmech.2015.11.013
•Novel COX-2/5-LOX dual inhibitors were designed via pharmacophore hybrid approach.•Most compounds showed COX-2/5-LOX inhibitory and anti-proliferative activities.•The compound 15c was found to induce apoptosis and G2/M phase cell cycle arrest.•The most potent compound 22b significantly inhibited tumor growth in vivo.•The docking studies revealed possible binding mode of these compounds.Inflammation plays a key role in cancer initiation and propagation. Cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX), two important enzymes in inflammatory responses are up-regulated in various tumor types. Dual inhibition of COX-2 and 5-LOX constitutes a rational concept for the design of more efficacious anti-tumor agents with an improved safety profile. We have previously reported a series of diaryl-1,2,4-triazole derivatives as selective COX-2 inhibitors. Herein, we hybridized the diaryl-1,2,4-triazoles with caffeic acid (CA) which was reported to display 5-LOX inhibitory and anti-tumor activities, affording a novel class of COX-2/5-LOX dual inhibitors as anti-tumor drug candidates. Most of these compounds exhibited potent COX-2/5-LOX inhibitory and antiproliferative activities in vitro. And the most potent compound 22b could significantly inhibit tumor growth in vivo. Furthermore, mechanistic investigation showed that the representative compound 15c blocked cell cycle in G2 phase and induced apoptosis in human non-small cell lung cancer A549 cells in a dose-dependent manner. Our preliminary investigation results would provide new clues for the cancer theatment with COX-2/5-LOX dual inhibitors.
Co-reporter:Shengtao Xu, Guangyu Wang, Yan Lin, Yanju Zhang, Lingling Pei, Hong Yao, Mei Hu, Yangyi Qiu, Zhangjian Huang, Yihua Zhang, Jinyi Xu
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 12) pp:2795-2800
Publication Date(Web):15 June 2016
DOI:10.1016/j.bmcl.2016.04.068
Oridonin (1) is a complex ent-kaurane diterpenoid with unique antitumor profile, which is isolated from Isodon rubescens. In order to develop novel derivatives of oridonin with improved potency, a series of nitric oxide (NO)-releasing oridonin derivatives were synthesized by coupling diazeniumdiolates with oridonin and its semisynthesized analogues. Their in vitro antiproliferative activity, nitric oxide release ability, and preliminary anticancer mechanism were further evaluated. The results displayed that all the target compounds exhibited potent antiproliferative activities, with IC50 values ranging from 1.84 to 17.01 μM. Besides, it was observed that in most cases, the antiproliferative activity correlated well with levels of intracellular NO release. More interestingly, preliminary mechanism studies revealed that the most potent compound 14d induced apoptosis and arrested the cell cycle at the S phase in Bel-7402 cells, which is different from parent compound oridonin. Together, the above promising results warrant the further development of oridonin/NO hybrids as potential antitumor leads.
Co-reporter:Jingchao Liu, Junjie Fu, Wenlong Li, Yu Zou, Zhangjian Huang, Jinyi Xu, Sixun Peng, Yihua Zhang
Tetrahedron 2016 Volume 72(27–28) pp:4103-4110
Publication Date(Web):7 July 2016
DOI:10.1016/j.tet.2016.05.048
A green and efficient method for regioselective O-acylation of polyphenols has been developed. The acylation can be carried out in potassium carbonate/dimethyl sulphoxide system by utilizing the ‘inherent nucleophile’ via an intermolecular cooperative transesterification under mild condition. This method shows particular advantage in regioselective acylation of polyphenols bearing 2′,4′-dihydroxyacetophenone moiety and can be extended to the synthesis of mono or multiple acetates of polyphenols without this moiety in good yields. Compared with other reported approaches, this method is endowed with atom economy and is more environment-friendly for avoiding the use of any metal-based catalysts.
Co-reporter:Dahong Li, Tong Han, Kangtao Tian, Shuang Tang, Shengtao Xu, Xu Hu, Lei Wang, Zhanlin Li, Huiming Hua, Jinyi Xu
Bioorganic & Medicinal Chemistry Letters 2016 26(16) pp: 4191-4196
Publication Date(Web):1 September 2016
DOI:10.1016/j.bmcl.2016.07.059
Herein, we reported the cytotoxicity, NO-releasing property, and apoptosis induced ability of two series of novel nitric oxide-releasing spirolactone-type diterpenoid derivatives (10a–f and 15a–f). All the title compounds were more potent than oridonin (7) and parent compound (9 or 14) against human tumor Bel-7402, K562, MGC-803 and CaEs-17 cells. SARs were concluded based on above data. Compound 15d exhibited the strongest antiproliferative activity with the IC50 of 0.86, 1.74, 1.16 and 3.75 μM, respectively, and could produce high level (above 25 μM) of NO at the time point of 60 min. Further mechanism evaluation showed that 15d could induce S phase cell cycle arrest and apoptosis at low micromolar concentrations in Bel-7402 cells via mitochondria-related pathways. It was expected that the remarkable biological profile of the synthetic NO-releasing spirolactone-type diterpenoid analogs make them possible as promising candidates for the development of anticancer agents.
Co-reporter:Hong Yao, Yu Zhou, Xia Chen, Pengfei Zhang, Jinyi Xu, and Hong Liu
The Journal of Organic Chemistry 2016 Volume 81(Issue 19) pp:8888-8899
Publication Date(Web):September 7, 2016
DOI:10.1021/acs.joc.6b01596
The ubiquitous structure of all-carbospirocyclic oxindoles makes the development of new methods for their enantioselective and stereoselective synthesis a significant ongoing challenge. Herein, we disclose a formal [4 + 2] annulation through N-heterocyclic carbene (NHC) catalysis for highly enantioselective synthesis of intriguing spirocarbocyclic oxindoles in the presence of Lewis acids. This protocol features good substrates tolerance, good yields, and excellent diastereoselectivities and enantioselectivities (up to 97% ee) under mild conditions.
Co-reporter:Hengyuan Zhang, Yiwei Wang, Peiqing Zhu, Jie Liu, Shengtao Xu, Hequan Yao, Jieyun Jiang, Wencai Ye, Xiaoming Wu, Jinyi Xu
European Journal of Medicinal Chemistry 2015 Volume 97() pp:235-244
Publication Date(Web):5 June 2015
DOI:10.1016/j.ejmech.2015.04.057
•A series of pyrazine-fused 23-hydroxybetulinic acid derivatives was synthesized.•All of the synthesized compounds showed significant anti-proliferative activity.•Compound 12a caused G1 phase arrest and apoptosis on B16 cancer cells.•Compound 12a significantly inhibited the tumor growth in vivo.Pyrazine-fused 23-hydroxybetulinic acid was synthesized by introducing a pyrazine ring between C-2 and C-3 position and further modifications were carried out by substitution of C-28 carboxyl group by ester and amide linkage to enhance the antitumor activity. The biological screening results showed that all of the derivatives exhibited more significant antiproliferative activity than the parent compound. In particular compound 12a exhibited the most potent activity with IC50 values of 3.53 μM, 4.42 μM and 5.13 μM against cell lines SF-763, B16 and Hela, respectively. In the preliminary mechanism study, 12a caused cell arrest in G1 phase and significantly induced apoptosis of B16 cells in a dose-dependent manner. Furthermore, the in vivo antitumor activity of 12a was validated (tumor inhibitory ratio of 55.6% and 62.7%, respectively) in mice with H22 liver cancer and B16 melanoma.Compound 12a caused cell arrest in G1 phase and significantly induced apoptosis of B16 cells in a dose-dependent manner.
Co-reporter:Yanchun Zhang;Yunman Li;Hequan Yao;Xiaoming Wu
Chemical Biology & Drug Design 2015 Volume 85( Issue 5) pp:541-548
Publication Date(Web):
DOI:10.1111/cbdd.12442
Two series of novel NO-releasing benzimidazole derivatives (8a–e, 9a–g) were designed and synthesized by coupling nitro ester and furoxan NO-donor moieties with benzimidazole biphenyl skeleton. The NO-releasing assay indicated that all the target compounds had different level of NO-releasing ability. Furthermore, the isolated organ assay (rat aortic strips) was used to evaluate the antagonism of Ang II-induced vasoconstriction ability. It was observed that the pA2 values of compounds 8e and 9e were better than that of lead compound 6. Moreover, the pharmacological investigation showed that the antagonism of Ang II-induced pressure response by oral administration of compound 8e was obviously superior to that of lead compound 6, and comparable to that of the positive control losartan. These results suggested that NO-releasing hybrids may provide a promising approach for the discovery of novel antihypertensive agents.
Co-reporter:Zan Li;Zhaoliang Li;Yunwei Lin;Zhaoqing Meng;Gang Ding;Liang Cao;Na Li;Wenjun Liu;Wei Xiao;Xiaoming Wu
Chemical Biology & Drug Design 2015 Volume 86( Issue 4) pp:523-530
Publication Date(Web):
DOI:10.1111/cbdd.12515
A series of novel derivatives of strictosamide were synthesized and biologically evaluated. Most of the new compounds exhibited improved activities than the parent compound strictosamide. Among them, compounds Ib and If possessed antiviral activities against influenza A virus (A/Jinan/15/90) with IC50 values of 4.12 and 12.35 μg/mL, respectively. Compound Ie possessed antiviral activity against respiratory syncytial virus (RSV) with an IC50 value of 9.58 μg/mL. Both compounds IIc and IId had moderate antiproliferative effects against five human cancer cell lines. The preliminary structure-activity relationships were also concluded. This study provides a promising new template with potential antiviral activity.
Co-reporter:Hengyuan Zhang;Fangzheng Li;Peiqing Zhu;Jie Liu;Hequan Yao;Jieyun Jiang;Wencai Ye;Xiaoming Wu
Chemical Biology & Drug Design 2015 Volume 86( Issue 4) pp:424-431
Publication Date(Web):
DOI:10.1111/cbdd.12543
A collection of isoxazole and oxadiazole substituted 23-hydroxybetulinic acid (HBA) derivatives were designed, synthesized and evaluated for their antitumor activity. Most of the newly synthesized compounds exhibited more potent antiproliferative activity than patent compound 23-hydroxybetulinic acid, especially 13e and 14a were about four- to sevenfold more potent against all tested cancer cell lines than 23-hydroxybetulinic acid. Furthermore, the in vivo antitumor activity of 13e and 14a was validated in H22 liver cancer and B16 melanoma xenograft mouse models. The structure–activity relationships of these 23-hydroxybetulinic acid derivatives were also discussed based on the present investigation.
Co-reporter:Xiao Sheng, Kai Hua, Chunyu Yang, Xiaoli Wang, Hui Ji, Jinyi Xu, Zhangjian Huang, Yihua Zhang
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 17) pp:3535-3540
Publication Date(Web):1 September 2015
DOI:10.1016/j.bmcl.2015.06.090
Fourteen hybrids (10a–g, 11a–g) of 3-n-butylphthalide (NBP) and edaravone (Eda) analogues have been designed and synthesized as potential anti-ischemic stroke agents. In vitro biological studies showed that compounds 10d and 10g exhibited more potent anti-platelet aggregation than ticlopidine (Ticlid), aspirin (ASP) and NBP. Compound 10g more significantly prevented H2O2-mediated neuronal cell (PC12) death than NBP, Eda or NBP together with Eda. Meanwhile, 10g also possessed potent radical scavenging effects on hydroxyl radical (OH) and superoxide anion radical (O2−). Our findings may provide new insights into the development of these hybrids, like 10g, for the intervention of ischemic stroke.
Co-reporter:Hengyuan Zhang, Peiqing Zhu, Jie Liu, Yan Lin, Hequan Yao, Jieyun Jiang, Wencai Ye, Xiaoming Wu, Jinyi Xu
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 3) pp:728-732
Publication Date(Web):1 February 2015
DOI:10.1016/j.bmcl.2014.11.058
A collection of pyrazole-fused 23-hydroxybetulinic acid derivatives were designed, synthesized and evaluated for their antitumor activity. Most of the newly synthesized compounds exhibited significant antiproliferative activity. Especially compound 15e displayed the most potent activity with the IC50 values of 5.58 and 6.13 μM against B16 and SF763 cancer cell lines, respectively. Furthermore, the significant in vivo antitumor activity of 15e was validated in H22 liver cancer and B16 melanoma xenograft mouse models. The structure–activity relationships of these 23-hydroxybetulinic acid derivatives were also discussed based on the present investigation.A collection of novel pyrazole-fused 23-hydroxybetulinic acid derivatives were designed, synthesized and evaluated for their antitumor activity. The results showed compound 15e as an effective chemotherapeutic agent candidate.
Co-reporter:Chaolei Wang, Zheng Wu, Hao Cai, Shengtao Xu, Jie Liu, Jieyun Jiang, Hequan Yao, Xiaoming Wu, Jinyi Xu
Bioorganic & Medicinal Chemistry Letters 2015 25(22) pp: 5212-5216
Publication Date(Web):
DOI:10.1016/j.bmcl.2015.09.063
Co-reporter:Chaolei Wang, Zheng Wu, Jia Wang, Jie Liu, Hequan Yao, Aijun Lin, Jinyi Xu
Tetrahedron 2015 Volume 71(Issue 42) pp:8172-8177
Publication Date(Web):21 October 2015
DOI:10.1016/j.tet.2015.08.028
The synthesis of 4-isochromanones via Parham-type cyclization with Weinreb amide as the internal electrophilic group, t-BuLi as the lithium reagent was described. The reaction was efficient and could be completed in one minute. The application scope of this new protocol was investigated and the desired products could be obtained in good to excellent yields. Besides, the synthetic potential of this method was further demonstrated by the synthesis of natural product (±)-XJP, which was obtained in six steps with overall yield up to 54%.
Co-reporter:Hao Cai, Yu Zhou, Dong Zhang, Jinyi Xu and Hong Liu
Chemical Communications 2014 vol. 50(Issue 94) pp:14771-14774
Publication Date(Web):03 Oct 2014
DOI:10.1039/C4CC06000H
An asymmetric cascade Mannich/cyclization reaction between 3-isothiocyanato oxindoles and sulfimides using a commercially available organocatalyst has been developed. A wide range of structurally diverse spiro[imidazolidine-4,3′-oxindole] derivatives were obtained with good yields (up to 92%) and excellent enantioselectivities (up to 99% ee).
Co-reporter:Bo Jiang, Xiaojing Huang, Hequan Yao, Jieyun Jiang, Xiaoming Wu, Siyi Jiang, Qiujuan Wang, Tao Lu and Jinyi Xu
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 13) pp:2114-2127
Publication Date(Web):14 Jan 2014
DOI:10.1039/C3OB41936C
A series of hybrids from diaryl-1,2,4-triazole and hydroxamic acid or N-hydroxyurea were synthesized and evaluated as novel anti-inflammatory agents. The biological data showed that (i) all the compounds showed dual COX-2/5-LOX inhibitory activities in vitro, and 15e showed optimal inhibitory activities (COX-2: IC50 = 0.15 μM, 5-LOX: IC50 = 0.85 μM), (ii) 15e selectively inhibited COX-2 relative to COX-1 with selectivity index (SI = 0.012) comparable to celecoxib (SI = 0.015), (iii) 15e exhibited potent anti-inflammatory activity (inhibition: 54.1%) which was comparable to the reference drug celecoxib (inhibition: 46.7%) in a xylene-induced ear edema assay, and (iv) 15e displayed promising analgesic activity in acetic acid-induced writhing response and hot-plate assay. Finally, a molecular modeling study revealed the binding interactions of 15e with COX-2 and 5-LOX. Our findings suggest that 15e may be a promising anti-inflammatory agent for further evaluation.
Co-reporter:Hengyuan Zhang, Peiqing Zhu, Jie Liu, Xue Yang, Shengtao Xu, Hequan Yao, Jieyun Jiang, Wencai Ye, Xiaoming Wu, Jinyi Xu
European Journal of Medicinal Chemistry 2014 Volume 87() pp:159-167
Publication Date(Web):24 November 2014
DOI:10.1016/j.ejmech.2014.09.058
•A series of 3-oxo-23-hydroxybetulinic acid derivatives was synthesized.•Most of the derivatives showed improved antitumor activity than parent compound.•Compound 10e displayed significant in vitro and in vivo antitumor activity.A series of novel derivatives of 3-oxo-23-hydroxybetulinic acid was designed, synthesized, and evaluated for their antiproliferative activity against a panel of cancer cell lines (HL-60, BEL-7402, SF-763, HeLa, B16 and A375). The results indicated that majority of the derivatives exhibited more significant antitumor activity than the parent compound. In particular compound 10e showed the most potent activity with IC50 values of 5.85, 6.23 and 7.22 μM against B16, SF-763 and BEL-7402 cells, respectively. Furthermore, 10e inhibited tumor growth by 51.8% and 62.7% (w/w) in H22 and B16 xenograft mouse models, comparable to cyclophosphamide and 5-fluorouracil, respectively.Compound 10e displayed significant antitumor activity against mice bearing H22 liver cancer and B16 melanoma.
Co-reporter:Yi Bi;Cong Ma;Hengyuan Zhang;Zhiwen Zhou;Jian Yang;Zhenlei Zhang;Qingguo Meng;Peter J. Lewis
Chemical Biology & Drug Design 2014 Volume 84( Issue 4) pp:489-496
Publication Date(Web):
DOI:10.1111/cbdd.12337
Plant-derived triterpenoid saponins are involved in the plant defense system by targeting bacterial membranes. A series of ocotillol-type triterpenoid derivatives were synthesized starting from PPD, one of the main components of Panax ginseng and their antibacterial activity against several representative bacteria were evaluated. Compounds 5 and 11 exhibited excellent antibacterial activity with MIC values of 1 μg/mL against Staphylococcus aureus and 8 μg/mL and 4 μg/mL against Bacillus subtilis, respectively. Furthermore, when compounds 5 and 11 were combined with two commercial antibiotics kanamycin and chloramphenicol, they showed strong synergistic activity at sub-MIC levels against S. aureus USA300 and B. subtilis 168. Moreover, chloramphenicol turned from a bacteriostatic to a bactericidal agent when combined with compound 11 against B. subtilis 168.
Co-reporter:Shengtao Xu, Lingling Pei, Chengqian Wang, Yun-Kai Zhang, Dahong Li, Hequan Yao, Xiaoming Wu, Zhe-Sheng Chen, Yijun Sun, and Jinyi Xu
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 7) pp:797
Publication Date(Web):May 28, 2014
DOI:10.1021/ml500141f
A series of novel hybrids from natural product oridonin and nitrogen mustards were designed and synthesized to obtain more efficacious and less toxic antitumor agents. The antiproliferative evaluation showed that most conjugates were more potent than their parent compounds oridonin and clinically used nitrogen mustards against four human cancer cell lines (K562, MCF-7, Bel-7402, and MGC-803). Furthermore, the representative compounds 16a–c exhibited antiproliferative activities against the multidrug resistant cell lines (SW620/AD300 and NCI-H460/MX20). It was shown that the most effective compound 16b possesses a strong inhibitory activity with an IC50 value 21-fold lower than that of oridonin in MCF-7 cells and also exhibits selective cytotoxicity toward the cancer cells. Intriguingly, compound 16b has been demonstrated to significantly induce apoptosis and affect cell cycle progression in human hepatoma Bel-7402 cells.Keywords: antiproliferative activities; apoptosis; combination principle; drug-resistant; nitrogen mustards; Oridonin;
Co-reporter:Shengtao Xu, Dahong Li, Lingling Pei, Hong Yao, Chengqian Wang, Hao Cai, Hequan Yao, Xiaoming Wu, Jinyi Xu
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 13) pp:2811-2814
Publication Date(Web):1 July 2014
DOI:10.1016/j.bmcl.2014.04.119
In an effort to develop novel potent antitubercular drugs, thirty-one oridonin derivatives were designed and prepared. All the compounds obtained were screened for their in vitro activities against Mycobacterium phlei, Mycobacterium smegmatis and Mycobacterium marinum. Among them, thirteen compounds showed significant inhibitory activity against M. phlei with MICs less than 2 μg/mL. Compounds 2k, 8d, 10c, 10d containing trans-cinnamic acid moiety were the most potent (MIC = 0.5 μg/mL), comparable to the well-known antitubercular drug streptomycin. The preliminary structure–activity relationships (SARs) were also analyzed.Four natural derived compounds (2k, 8d, 10c, 10d) containing trans-cinnamic acid moiety exhibited potent antimycobacterial activity against M. phlei at a concentration of 0.5 μg/mL, which was comparable to that of streptomycin (MIC = 0.5 μg/mL).
Co-reporter:Jichao Chen, Wenli Duan, Renren Bai, Hequan Yao, Jing Shang, Jinyi Xu
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 15) pp:3407-3411
Publication Date(Web):1 August 2014
DOI:10.1016/j.bmcl.2014.05.078
Forty β-elemene derivatives were prepared and their antioxidant activity in H2O2-treated human umbilical vein endothelial cells (HUVECs) was first investigated. Among which, the dimer compounds 5r and 5s exhibited the most potent antioxidant activity against reactive oxygen species production. Meanwhile, 5r and 5s led to a significant increase in superoxide dismutase and nitric oxide levels and decrease in malonyldialdehyde and lactate dehydrogenase contents. Furthermore, MTT assay showed that 5r and 5s did not produce obvious cytotoxicity and had significantly cytoprotective effects against oxidative damage on HUVECs.The dimer compounds 5r and 5s exhibited potent ROS inhibitory activities in HUVECs injured by H2O2 in a dose-dependent manner, which was comparable or even superior to that of positive control Vitamin E.
Co-reporter:Wei Cong, Lei Zhao, Xiaoming Wu, Jinyi Xu, Hequan Yao
Tetrahedron 2014 70(2) pp: 312-317
Publication Date(Web):
DOI:10.1016/j.tet.2013.11.053
Co-reporter:Shengtao Xu, Yu Zhou, Jinyi Xu, Hualiang Jiang and Hong Liu
Green Chemistry 2013 vol. 15(Issue 3) pp:718-726
Publication Date(Web):17 Dec 2012
DOI:10.1039/C2GC36301A
The report describes a gold(I) complex and trifluoroacetic acid (TFA) cocatalyzed one-pot, Michael addition/intramolecular cyclization cascade reaction for the synthesis of unusual tetracyclic indoles containing a seven-membered ring in water with microwave irradiation (MW). This protocol presents an operationally simple, rapid and environmentally friendly strategy for preparing potential biologically interesting fused-indole molecular architectures from some simple starting materials.
Co-reporter:Xiaoli Wang ; Linna Wang ; Tingting Li ; Zhangjian Huang ; Yisheng Lai ; Hui Ji ; Xiaolong Wan ; Jinyi Xu ; Jide Tian ;Yihua Zhang
Journal of Medicinal Chemistry 2013 Volume 56(Issue 7) pp:3078-3089
Publication Date(Web):March 19, 2013
DOI:10.1021/jm4001693
In search of novel anti-ischemic stroke agents with higher potency than a known drug 3-n-butylphthalide (NBP), a series of hybrids ((S)- and (R)-5a–f) from optically active ring-opened NBP derivative and isosorbide were synthesized for evaluating their anti-ischemic stroke activity. Compound (S)-5e displayed the strongest activity in inhibiting the adenosine diphosphate (ADP) and arachidonic acid (AA)-induced platelet aggregation in vitro, with 10.0- and 8.4-fold more effectiveness than (S)-NBP, respectively. Furthermore, (S)-5e was stable in artificial gastrointestinal fluids and could penetrate the blood–brain barrier (BBB) with an appreciate lipid/water partition coefficient relative to (S)-NBP. More importantly, oral treatment with (S)-5e protected from acute thrombosis and inhibited the ischemia/reperfusion-related brain injury in animals. Our findings suggest that (S)-5e may be promising for further evaluation for the intervention of ischemic stroke.
Co-reporter:Dahong Li, Shengtao Xu, Hao Cai, Lingling Pei, Hengyuan Zhang, Lei Wang, Hequan Yao, Xiaoming Wu, Jieyun Jiang, Yijun Sun, Jinyi Xu
European Journal of Medicinal Chemistry 2013 Volume 64() pp:215-221
Publication Date(Web):June 2013
DOI:10.1016/j.ejmech.2013.04.012
•Promising enmein-type diterpenoid analogs were obtained from natural oridonin.•All the derivatives showed improved anti-proliferative activities.•The most promising compound 17 was selected for further mechanistic evaluation.•The influence of cell cycle progression by compound 17 was observed.•Inducing of apoptosis involves mitochondria-related caspase-dependent pathways.A series of enmein-type diterpenoid analogs (11–20) derived from natural kaurene-type diterpenoid oridonin were synthesized and biologically evaluated. All target compounds showed improved anti-proliferative activities against four human cancer cell lines compared with natural oridonin and parent compound 10. Some compounds were more potent than positive control Taxol. Furthermore, mechanistic investigation showed that the representative compound 17 affected cell cycle and induced apoptosis at low micro-molar level in human hepatoma Bel-7402 cells, via an oxidative stress triggered mitochondria-related caspase-dependent pathway.The synthetic enmein-type diterpenoid analog 17 exhibited potent anti-proliferative activities and induced apoptosis via mitochondria-related caspase-dependent pathways.
Co-reporter:Dahong Li, Hao Cai, Bowen Jiang, Guyue Liu, Yuetong Wang, Lei Wang, Hequan Yao, Xiaoming Wu, Yijun Sun, Jinyi Xu
European Journal of Medicinal Chemistry 2013 Volume 59() pp:322-328
Publication Date(Web):January 2013
DOI:10.1016/j.ejmech.2012.11.002
A series of novel spirolactone-type diterpenoid derivatives of oridonin (12a–j) were designed and synthesized. All the target compounds showed improved anti-proliferative activity against a panel of human cancer cell lines and the most effective compound 12j was more potent than positive control Taxol in K562 and Bel-7402 cells with IC50 values of 0.39 μM and 1.39 μM, respectively. The cellular mechanisms showed that compound 12j induced apoptosis at low micromolar concentrations in human hepatoma Bel-7402 cells. These results demonstrate that the spirolactone-type diterpenoid derivatives of oridonin have optimized growth inhibitory activity against cancer cells and interesting apoptosis-inducing ability.Graphical abstractThe most effective synthetic spirolactone-type diterpenoid analog 12j exhibited similar anti-proliferative activity as the positive control Taxol and induced apoptosis at low micromolar concentrations in human hepatoma Bel-7402 cells.Highlights► Spirolactone-type diterpenoids could be got from commercial available oridonin. ► A series of derivatives with improved anti-proliferative activities were synthesized. ► Compound 12j showed similar anti-proliferative activity as Taxol in vitro. ► Induction of apoptosis and influence of cell cycle by 12j were investigated. ► The structure–activity relationships of the derivatives were concluded.
Co-reporter:Zhiwen Zhou, Cong Ma, Hengyuan Zhang, Yi Bi, Xia Chen, Hua Tian, Xiaoni Xie, Qingguo Meng, Peter John Lewis, Jinyi Xu
European Journal of Medicinal Chemistry 2013 Volume 68() pp:444-453
Publication Date(Web):October 2013
DOI:10.1016/j.ejmech.2013.07.041
•A series of ocotillol-type derivatives derived from natural PPD were obtained.•Compounds 3, 5, 16 and 24 showed potent activities against Gram-(+) bacteria.•Compounds 3 and 16 highly sensitized Gram-(+) bacteria to antibiotics KAN and CHL.•Compounds 3, 5 and 16 exhibited no cytotoxicity at their MICs.A novel class of ocotillol-type triterpenoid derivatives have been synthesized and evaluated for their in vitro antibacterial activity against several representative pathogenic bacterial strains. Compounds 20(S)-protopanaxadiol (PPD), 3, 5, 16 and 24 exhibited potent antibacterial activity against Gram-positive bacteria. Compounds 3 and 5 also displayed promising antibacterial activity against a community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA; strain USA300). Furthermore, compounds PPD, 3 and 16 combined with two commercially available antibiotics kanamycin and chloramphenicol showed strong synergistic inhibitory effects at their sub-MIC concentrations against S. aureus USA300 and Bacillus subtilis 168. Additionally, cytotoxic activity assay showed that the compounds tested did not affect cell viability of the human epithelial kidney (HEK-293) and human cervical (HeLa) cells at their MICs.Compounds 3 and 16 combined with kanamycin and chloramphenicol showed strong synergistic inhibitory effects at their sub-MIC concentrations against Staphylococcus aureus USA300 and Bacillus subtilis 168.
Co-reporter:Jie Liu, Qin Liu, Xue Yang, Shengtao Xu, Hengyuan Zhang, Renren Bai, Hequan Yao, Jieyun Jiang, Mingqin Shen, Xiaoming Wu, Jinyi Xu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 24) pp:7742-7751
Publication Date(Web):15 December 2013
DOI:10.1016/j.bmc.2013.10.017
A series of novel 1,2,4-triazole bearing 5-substituted biphenyl-2-sulfonamide derivatives were designed and synthesized to develop new angiotensin II subtype 2 (AT2) receptor agonists as novel antihypertensive candidates. It was found that 14f (IC50 = 0.4 nM) and 15e (IC50 = 5.0 nM) displayed potent AT2 receptor affinity and selectivity in binding assays. Biological evaluation in vivo suggested that 14f is obviously superior to that of reference drug losartan in RHRs, and meanwhile, 14f has no significant impact on heart rate. The interesting activities of these compounds may make them promising candidates as antihypertensive agents.A series of novel 1,2,4-triazole bearing 5-substituted biphenyl-2-sulfonamide derivatives were designed and synthesized to develop new angiotensin II subtype 2 (AT2) receptor agonists as novel antihypertensive candidates. It was found that 14f (IC50 = 0.4 nM) and 15e (IC50 = 5.0 nM) displayed potent AT2 receptor affinity and selectivity in binding assays. Biological evaluation in vivo suggested that 14f is obviously superior to that of reference drug losartan in RHRs, and meanwhile, 14f has no significant impact on heart rate. The interesting activities of these compounds may make them promising candidates as antihypertensive agents.
Co-reporter:Xiaoli Wang, Linna Wang, Zhangjian Huang, Xiao Sheng, Tingting Li, Hui Ji, Jinyi Xu, Yihua Zhang
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 7) pp:1985-1988
Publication Date(Web):1 April 2013
DOI:10.1016/j.bmcl.2013.02.035
A series of novel nitric oxide releasing derivatives of 6-amino-3-n-butylphthalide were designed, synthesized and evaluated as potential antiplatelet agents. Compound 10b significantly inhibited the adenosine diphosphate (ADP)-induced platelet aggregation in vitro, superior to 6-amino-3-n-butylphthalide, 3-n-butylphthalide (NBP) and ticlopidine. Meanwhile 10b released moderate levels of NO, which could be beneficial for improving cardiovascular and cerebral circulation. Furthermore, 10b had an enhanced aqueous solubility relative to NBP. These findings may provide new insights into the development of novel antiplatelet agents for the treatment of thrombosis-related ischemic stroke.The inhibitory activity of NO-releasing compound 10b on ADP-induced platelet aggregation was better than that of 6-amino-NBP (1), NBP and ticlopidine.
Co-reporter:Renren Bai, Xiaojing Huang, Xue Yang, Wen Hong, Yiqun Tang, Hequan Yao, Jieyun Jiang, Jie Liu, Mingqin Shen, Xiaoming Wu, Jinyi Xu
Bioorganic & Medicinal Chemistry 2013 21(9) pp: 2495-2502
Publication Date(Web):
DOI:10.1016/j.bmc.2013.02.044
Co-reporter:Dr. Dahong Li;Dr. Shengtao Xu;Hao Cai;Lingling Pei;Dr. Lei Wang; Xiaoming Wu; Hequan Yao; Jieyun Jiang;Yijun Sun; Jinyi Xu
ChemMedChem 2013 Volume 8( Issue 5) pp:812-818
Publication Date(Web):
DOI:10.1002/cmdc.201200559
Abstract
A library of promising enmein-type 14-O-diterpenoid derivatives was constructed from a commercially available kaurene-type oridonin by practical and efficient synthetic methods. These synthetic derivatives were evaluated for their antiproliferative activities against a set of four human cancer cell lines. The IC50 values are similar to or improved over those of the parent molecule and paclitaxel, the latter of which was used as a positive control. Compound 29 was further investigated for its apoptotic properties against human hepatocarcinoma Bel-7402 cells to better understand its mode of action. Moreover, compound 29 was shown to have potent antitumor activity in vivo in studies with a murine model of gastric cancer (MGC-803 mice). These results warrant further preclinical investigations of these diterpenoid-based analogues as potential novel anticancer chemotherapeutics.
Co-reporter:Lei Wang, Dahong Li, Shengtao Xu, Hao Cai, Hequan Yao, Yihua Zhang, Jieyun Jiang, Jinyi Xu
European Journal of Medicinal Chemistry 2012 Volume 52() pp:242-250
Publication Date(Web):June 2012
DOI:10.1016/j.ejmech.2012.03.024
Starting from commercial available natural product oridonin (1), a practical synthesis of ent-6,7-seco-oridonin derivatives (2, 3, 5, and 9) was accomplished and their biological activities were evaluated. The conversion of spirolactone-type diterpenoid to enmein-type was first completed. The results demonstrated that all synthesized ent-6,7-seco-oridonin derivatives could markedly inhibit the proliferation of cancer cells. Compared with Taxol, the most cytotoxic compound 5 has similar potency in A549 cell and slightly less cytotoxicity in Bel-7402 cell. Compound 5 was also more potent than parent compound oridonin in mice with MGC-803 gastric cancer in vivo. Then a series of novel 14-O-derivatives of 5 were further designed and synthesized, which showed better activity than 5 and similar activity as Taxol in vitro. The structure–activity relationships of oridonin derivatives were also discussed in the present investigations.The conversion of oridonin to spirolactone-type or enmein-type diterpenoid were accomplished, and a series of 14-O-derivatives of ent-6,7-seco-oridonin were synthesized and evaluated as novel potential anticancer agents.Highlights► The synthesis of ent-6,7-seco-oridonin derivatives were accomplished and evaluated. ► Compound 5 showed stronger anticancer activity than oridonin in vitro and in vivo. ► A series of 14-O-derivatives of 5 with improved potency were designed and obtained. ► Some 14-O-derivatives of 5 showed similar cytotoxicity as Taxol in vitro. ► The structure–activity relationships of the derivatives of oridonin were concluded.
Co-reporter:Xiaoli Wang, Qian Zhao, Xuliang Wang, Tingting Li, Yisheng Lai, Sixun Peng, Hui Ji, Jinyi Xu and Yihua Zhang
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 45) pp:9030-9040
Publication Date(Web):14 Sep 2012
DOI:10.1039/C2OB26511G
ZJM-289 is a potent racemic agent which inhibits both platelet aggregation and thrombosis superior to a known anti-ischemic stroke drug 3-n-butylphthalide (NBP). Herein, the enantiomers of ZJM-289, (S)-ZJM-289 and (R)-ZJM-289, were synthesized and evaluated for their biological activities. It was observed that the two enantiomers appeared to be almost as effective as ZJM-289 in inhibiting platelet aggregation in vitro and thrombus formation in vivo. Moreover, like ZJM-289, its enantiomers could regulate the ratio of thromboxane B2 (TXB2) and 6-keto-prostaglandin F1α, and enhanced levels of nitric oxide (NO), cAMP and cGMP, suggesting that the anti-platelet and antithrombotic activities of the enantiomers and ZJM-289 are associated with both the arachidonic acid cascade and cGMP–NO signal pathway. Furthermore, it was found that oral administration of the enantiomers and ZJM-289 for three days significantly reduced the infarct size, brain water content and neurological deficit in rats after cerebral ischemia reperfusion. Importantly, the two enantiomers equally improved blood flow in the ischemic stroke model and modulated endothelial function through releasing moderate levels of NO, which might, at least partially, contribute to their neuroprotection. Collectively, the present study demonstrates that the two enantiomers are as potent as ZJM-289 in inhibition of platelet aggregation and thrombosis and in neuroprotection, and (S)-ZJM-289 shows somewhat better effects than (R)-ZJM-289 and ZJM-289 in a few cases. These findings may provide new insights into the development of therapeutic agents like ZJM-289 for the intervention of thrombosis-related ischemic stroke.
Co-reporter:Renren Bai, Xue Yang, Yao Zhu, Zhiwen Zhou, Weijia Xie, Hequan Yao, Jieyun Jiang, Jie Liu, Mingqin Shen, Xiaoming Wu, Jinyi Xu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 23) pp:6848-6855
Publication Date(Web):1 December 2012
DOI:10.1016/j.bmc.2012.09.043
By coupling nitric oxide (NO)-donor moieties with a natural antihypertensive product (±)-7,8-dihydroxy-3-methyl-isochroman-4-one [(±)-XJP] and its analogue (±)-XJP-B, a series of novel NO-releasing isochroman-4-one derivatives were designed and synthesized. The NO-releasing assay indicated that compounds Ia, Id, IIIb and IIIe released the maximum amount of NO. The maximum reductions of blood pressure of Ia, IIIb and IIIe in SHRs were nearly 40%, which was obviously superior to that of the lead compounds and comparable to that of reference drug captopril. These results suggested that NO-donor/natural product hybrids may provide a promising approach for the discovery of novel antihypertensive agents.
Co-reporter:Renren Bai, Jie Liu, Yao Zhu, Xue Yang, Chen Yang, Lingyi Kong, Xiaobing Wang, Hengyuan Zhang, Hequan Yao, Mingqin Shen, Xiaoming Wu, Jinyi Xu
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 20) pp:6490-6493
Publication Date(Web):15 October 2012
DOI:10.1016/j.bmcl.2012.08.040
(±)-7,8-Dihydroxy-3-methyl-isochromanone-4 [(±)-XJP] is a natural antihypertensive product contained in banana (Musa sapientum L.) peel. (−)-XJP and (+)-XJP were first obtained by chiral resolution, meanwhile circular dichroism (CD) calculations and chiral synthesis were employed to investigate the absolute configuration. The results indicated that the absolute configuration of (+)-XJP is S-configured and the absolute configuration of (−)-XJP is R-configured. Furthermore, the evaluation of antihypertensive effects in vivo proved that R-(−)-XJP was more potent than S-(+)-XJP and [(±)-XJP].
Co-reporter:Yiyun Wang, Ziyuan Li, Yue Huang, Changhua Tang, Xiaoming Wu, Jinyi Xu, Hequan Yao
Tetrahedron 2011 67(38) pp: 7406-7411
Publication Date(Web):
DOI:10.1016/j.tet.2011.07.016
Co-reporter:Bo Jiang;Yi Zeng;Meng-Jie Li;Jin-Yi Xu;Yong-Na Zhang;Qiu-Juan Wang;Ni-Yue Sun;Tao Lu;Xiao-Ming Wu
Archiv der Pharmazie 2010 Volume 343( Issue 9) pp:500-508
Publication Date(Web):
DOI:10.1002/ardp.200900227
Abstract
A series of 1,5-diaryl-1,2,4-triazole derivatives were synthesized and evaluated as cyclooxygenase-2 (COX-2) inhibitors. The results of the preliminary biological assays in vivo showed that eight compounds 5b, 6b, 6c, 7c, 8b, 8d, 9c, and 9d have potent anti-inflammatory activity (P < 0.01), while compounds 6b, 6c, and 9c exhibit marked potency. Compound 6c was then selected for further investigation. In the COX inhibition assay in vitro, compound 6c was identified as a potent and selective inhibitor of COX-2 (COX-2 IC50 = 0.37 µM; SI = 0.018), being equipotent to celecoxib (COX-2 IC50 = 0.26 µM; SI = 0.015). In a rat carrageenan-induced paw edema assay, 6c exhibited moderate anti-inflammatory activity (35% inhibition of inflammation) at 2 h after administration of 15 mg/kg as an oral dose. A docking study also revealed that compound 6c binds in the active site of COX-2 in a similar mode to that of the known selective COX-2 inhibitor SC-558.
Co-reporter:Peiqing Zhu, Yi Bi, Jinyi Xu, Zan Li, Jun Liu, Luyong Zhang, Wencai Ye, Xiaoming Wu
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 24) pp:6966-6969
Publication Date(Web):15 December 2009
DOI:10.1016/j.bmcl.2009.10.055
A series of 23-hydroxybetulinic acid derivatives were prepared and tested in vitro as a new class of inhibitors of glycogen phosphorylase (GP). Within this series of compounds, 12b (IC50 = 3.5 μM) is the most potent GPa inhibitor. The preliminary SAR results of the 23-hydroxybetulinic acid derivatives are discussed.A series of 23-hydroxybetulinic acid derivatives were prepared and evaluated as a new class of inhibitors of glycogen phosphorylase (GP), among which 12b was the most potent GPa inhibitor (IC50 = 3.5 μM).
Co-reporter:Jie Liu, Hao Ren, Jinyi Xu, Renren Bai, Qi Yan, Wenlong Huang, Xiaoming Wu, Jihua Fu, Qiujuan Wang, Qian Wu, Rong Fu
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 6) pp:1822-1824
Publication Date(Web):15 March 2009
DOI:10.1016/j.bmcl.2008.12.102
This letter describes the total synthesis, preliminary biological evaluation and mechanism studies of a novel and structurally unique isochromanone, (±)7,8-dihydroxy-3-methyl-isochromanone-4 (1), a nature product contained in banana (Musa sapientum L.) peel. The bioassay showed that compound 1 displays potent antihypertensive activity in renal hypertensive rats and further mechanism studies revealed that it is an ACE inhibitor.The total synthesis of (±)7,8-dihydroxy-3-methyl-isochromanone-4 (1) is described and this compound displays potent antihypertensive activity and moderate ACE inhibitory activity.
Co-reporter:Jinyi Xu, JingYi Yang, Qian Ran, Lei Wang, Jie Liu, Zhixuan Wang, Xiaoming Wu, Weiyi Hua, Shengtao Yuan, Luyong Zhang, Mingqin Shen, Yongfang Ding
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 16) pp:4741-4744
Publication Date(Web):15 August 2008
DOI:10.1016/j.bmcl.2008.06.097
Novel 1-O- and 14-O-derivatives of oridonin were synthesized and biologically evaluated. All of the derivatives exhibited stronger cytotoxicity against six cancer cell lines (BGC-7901, SW-480, HL-60, BEL-7402, A549, and B16) than oridonin in vitro, and some of them were more potent than oridonin and cyclophosphamide in vivo. Compounds Ib and IIg were the most potent with the IC50 values of 0.84 μM for Ib in HL-60 cell and 1.00 μM for IIg in BEL-7402 cell.1-O- and 14-O-derivatives of oridonin exhibited stronger cytotoxicity against six cancer cell lines than oridonin in vitro, compounds Ib and IIg were more potent than oridonin and cyclophosphamide in vivo.
Co-reporter:Jin Yi Xu, Yi Zeng, Qian Ran, Zhen Wei, Yi Bi, Qian Hui He, Qiu Juan Wang, Song Hu, Jing Zhang, Ming Yue Tang, Wei Yi Hua, Xiao Ming Wu
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 10) pp:2921-2926
Publication Date(Web):15 May 2007
DOI:10.1016/j.bmcl.2007.02.042
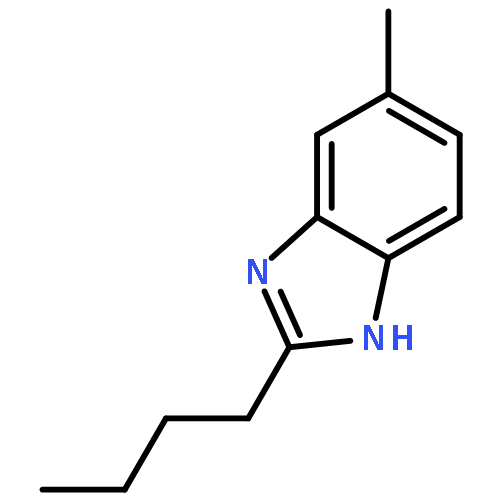
A series of 2-alkylbenzimidazoles bearing a N-phenylpyrrole moiety were synthesized and evaluated as a novel class of AT1 receptor antagonists. Among them, compounds 10a and 10g inhibited [125I] AngII-binding affinity to AT1 receptor at nanomolar level and potently inhibited the Ang II-induced pressor response by oral administration. Moreover, evaluation in spontaneously hypertensive rats showed that 10a is an orally active AT1 receptor antagonist.2-Alkylbenzimidazoles bearing a N-phenylpyrrole moiety 10a and 10g inhibited [125I] AngII-binding affinity to AT1 receptor at nanomolar level and evaluation in spontaneously hypertensive rats showed that 10a is an orally active AT1 receptor antagonist.
Co-reporter:Jichao Chen, Tianyu Wang, Tong Wang, Aijun Lin, Hequan Yao and Jinyi Xu
Inorganic Chemistry Frontiers 2017 - vol. 4(Issue 1) pp:NaN134-134
Publication Date(Web):2016/10/26
DOI:10.1039/C6QO00590J
Copper-catalyzed direct C5-position thio/selenocyanation of quinolines using commercially available, inexpensive KSCN/SeCN as the thio/selenocyanation reagent was developed, which had good tolerance toward various aliphatic or aromatic 8-aminoquinoline derivatives. Importantly, the synthetic application of this protocol led to some useful compounds.
Co-reporter:Jichao Chen, Tianyu Wang, Yanpeng Liu, Tong Wang, Aijun Lin, Hequan Yao and Jinyi Xu
Inorganic Chemistry Frontiers 2017 - vol. 4(Issue 4) pp:NaN626-626
Publication Date(Web):2017/02/07
DOI:10.1039/C6QO00765A
An efficient and convenient method of C5-selective halogenation of quinoline derivatives was developed. The reaction proceeds smoothly in water with readily available N-halosuccinimides (NCS, NBS and NIS) as the halogenation reagents. This method features metal-free catalysis, no additional oxidants and additives, broad substrate scope and short reaction time.
Co-reporter:Bo Jiang, Xiaojing Huang, Hequan Yao, Jieyun Jiang, Xiaoming Wu, Siyi Jiang, Qiujuan Wang, Tao Lu and Jinyi Xu
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 13) pp:NaN2127-2127
Publication Date(Web):2014/01/14
DOI:10.1039/C3OB41936C
A series of hybrids from diaryl-1,2,4-triazole and hydroxamic acid or N-hydroxyurea were synthesized and evaluated as novel anti-inflammatory agents. The biological data showed that (i) all the compounds showed dual COX-2/5-LOX inhibitory activities in vitro, and 15e showed optimal inhibitory activities (COX-2: IC50 = 0.15 μM, 5-LOX: IC50 = 0.85 μM), (ii) 15e selectively inhibited COX-2 relative to COX-1 with selectivity index (SI = 0.012) comparable to celecoxib (SI = 0.015), (iii) 15e exhibited potent anti-inflammatory activity (inhibition: 54.1%) which was comparable to the reference drug celecoxib (inhibition: 46.7%) in a xylene-induced ear edema assay, and (iv) 15e displayed promising analgesic activity in acetic acid-induced writhing response and hot-plate assay. Finally, a molecular modeling study revealed the binding interactions of 15e with COX-2 and 5-LOX. Our findings suggest that 15e may be a promising anti-inflammatory agent for further evaluation.
Co-reporter:Xiaoli Wang, Qian Zhao, Xuliang Wang, Tingting Li, Yisheng Lai, Sixun Peng, Hui Ji, Jinyi Xu and Yihua Zhang
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 45) pp:NaN9040-9040
Publication Date(Web):2012/09/14
DOI:10.1039/C2OB26511G
ZJM-289 is a potent racemic agent which inhibits both platelet aggregation and thrombosis superior to a known anti-ischemic stroke drug 3-n-butylphthalide (NBP). Herein, the enantiomers of ZJM-289, (S)-ZJM-289 and (R)-ZJM-289, were synthesized and evaluated for their biological activities. It was observed that the two enantiomers appeared to be almost as effective as ZJM-289 in inhibiting platelet aggregation in vitro and thrombus formation in vivo. Moreover, like ZJM-289, its enantiomers could regulate the ratio of thromboxane B2 (TXB2) and 6-keto-prostaglandin F1α, and enhanced levels of nitric oxide (NO), cAMP and cGMP, suggesting that the anti-platelet and antithrombotic activities of the enantiomers and ZJM-289 are associated with both the arachidonic acid cascade and cGMP–NO signal pathway. Furthermore, it was found that oral administration of the enantiomers and ZJM-289 for three days significantly reduced the infarct size, brain water content and neurological deficit in rats after cerebral ischemia reperfusion. Importantly, the two enantiomers equally improved blood flow in the ischemic stroke model and modulated endothelial function through releasing moderate levels of NO, which might, at least partially, contribute to their neuroprotection. Collectively, the present study demonstrates that the two enantiomers are as potent as ZJM-289 in inhibition of platelet aggregation and thrombosis and in neuroprotection, and (S)-ZJM-289 shows somewhat better effects than (R)-ZJM-289 and ZJM-289 in a few cases. These findings may provide new insights into the development of therapeutic agents like ZJM-289 for the intervention of thrombosis-related ischemic stroke.
Co-reporter:Hao Cai, Yu Zhou, Dong Zhang, Jinyi Xu and Hong Liu
Chemical Communications 2014 - vol. 50(Issue 94) pp:NaN14774-14774
Publication Date(Web):2014/10/03
DOI:10.1039/C4CC06000H
An asymmetric cascade Mannich/cyclization reaction between 3-isothiocyanato oxindoles and sulfimides using a commercially available organocatalyst has been developed. A wide range of structurally diverse spiro[imidazolidine-4,3′-oxindole] derivatives were obtained with good yields (up to 92%) and excellent enantioselectivities (up to 99% ee).

















![1,3-Benzenediol, 5-[2-[4-(acetyloxy)phenyl]ethenyl]-, diacetate](http://img.cochemist.com/ccimg/54500/54443-64-0.png)
![1,3-Benzenediol, 5-[2-[4-(acetyloxy)phenyl]ethenyl]-, diacetate](http://img.cochemist.com/ccimg/54500/54443-64-0_b.png)

![Ethanone, 1-[2-(acetyloxy)-4-hydroxyphenyl]-](http://img.cochemist.com/ccimg/52800/52751-42-5.png)
![Ethanone, 1-[2-(acetyloxy)-4-hydroxyphenyl]-](http://img.cochemist.com/ccimg/52800/52751-42-5_b.png)

![Benzaldehyde, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/52100/52095-44-0.png)
![Benzaldehyde, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/52100/52095-44-0_b.png)




![ISOQUINOLINE, 1-[(4-CHLOROPHENYL)METHYL]-3,4-DIHYDRO-6,7-DIMETHOXY-](http://img.cochemist.com/ccimg/47300/47216-54-6.png)
![ISOQUINOLINE, 1-[(4-CHLOROPHENYL)METHYL]-3,4-DIHYDRO-6,7-DIMETHOXY-](http://img.cochemist.com/ccimg/47300/47216-54-6_b.png)


![Ethanone, 1-[4-(acetyloxy)-2-hydroxyphenyl]-](http://img.cochemist.com/ccimg/42100/42059-48-3.png)
![Ethanone, 1-[4-(acetyloxy)-2-hydroxyphenyl]-](http://img.cochemist.com/ccimg/42100/42059-48-3_b.png)


















![2-Propenoic acid, 3-[4-(1,1-dimethylethyl)phenyl]-, methyl ester, (2E)-](http://img.cochemist.com/ccimg/36300/36215-20-0.png)
![2-Propenoic acid, 3-[4-(1,1-dimethylethyl)phenyl]-, methyl ester, (2E)-](http://img.cochemist.com/ccimg/36300/36215-20-0_b.png)



![L-ALANINE, 3-(P-[BIS(2-CHLOROETHYL)AMINO]PHENYL)-N-FORMYL-](http://img.cochemist.com/ccimg/35900/35849-41-3.png)
![L-ALANINE, 3-(P-[BIS(2-CHLOROETHYL)AMINO]PHENYL)-N-FORMYL-](http://img.cochemist.com/ccimg/35900/35849-41-3_b.png)

































![Oxayohimban-21-one, 19,20-didehydro-16-ethenyl-17-[(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)oxy]-, (15β,16α,17β)-](/data/chemimg/1799800/23141-26-6.png)
![Oxayohimban-21-one, 19,20-didehydro-16-ethenyl-17-[(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)oxy]-, (15β,16α,17β)-](/data/chemimg/1799800/23141-26-6_b.png)
![5H-Indolo[2,3-a]pyrano[3,4-g]quinolizin-5-one,1-ethenyl-2-(b-D-glucopyranosyloxy)-1,2,7,8,13,13b,14,14a-octahydro-,(1R,2S,13bS,14aS)-](http://img.cochemist.com/ccimg/23200/23141-25-5.png)
![5H-Indolo[2,3-a]pyrano[3,4-g]quinolizin-5-one,1-ethenyl-2-(b-D-glucopyranosyloxy)-1,2,7,8,13,13b,14,14a-octahydro-,(1R,2S,13bS,14aS)-](http://img.cochemist.com/ccimg/23200/23141-25-5_b.png)























![Benzoic acid,4-[bis(2-hydroxyethyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/15800/15716-30-0.png)
![Benzoic acid,4-[bis(2-hydroxyethyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/15800/15716-30-0_b.png)








![Benzeneethanamine, 3,4-dimethoxy-N-[2-(4-methoxyphenoxy)ethyl]-](http://img.cochemist.com/ccimg/10500/10450-25-6.png)
![Benzeneethanamine, 3,4-dimethoxy-N-[2-(4-methoxyphenoxy)ethyl]-](http://img.cochemist.com/ccimg/10500/10450-25-6_b.png)
![Benzeneethanamine, 3,4-dimethoxy-N-[2-(2-methylphenoxy)ethyl]-](http://img.cochemist.com/ccimg/10500/10414-90-1.png)
![Benzeneethanamine, 3,4-dimethoxy-N-[2-(2-methylphenoxy)ethyl]-](http://img.cochemist.com/ccimg/10500/10414-90-1_b.png)


![Isoquinoline,3,4-dihydro-6,7-dimethoxy-1-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/10300/10268-39-0.png)
![Isoquinoline,3,4-dihydro-6,7-dimethoxy-1-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/10300/10268-39-0_b.png)
![Isoquinoline, 1,2,3,4-tetrahydro-6,7-dimethoxy-1-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/10300/10268-26-5.png)
![Isoquinoline, 1,2,3,4-tetrahydro-6,7-dimethoxy-1-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/10300/10268-26-5_b.png)





















![3-BROMO-5-[2-(TRIFLUOROMETHYL)PHENYL]PYRIDINE](http://img.cochemist.com/ccimg/7100/7025-91-4.png)
![3-BROMO-5-[2-(TRIFLUOROMETHYL)PHENYL]PYRIDINE](http://img.cochemist.com/ccimg/7100/7025-91-4_b.png)





















![2-Propanone, 1-diazo-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/2800/2725-60-2.png)
![2-Propanone, 1-diazo-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/2800/2725-60-2_b.png)















![Benzoic acid,4-[bis(2-chloroethyl)amino]-](http://img.cochemist.com/ccimg/1200/1141-37-3.png)
![Benzoic acid,4-[bis(2-chloroethyl)amino]-](http://img.cochemist.com/ccimg/1200/1141-37-3_b.png)
![4H-1-Benzopyran-4-one,3,5,7-tris(acetyloxy)-2-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/1100/1064-06-8.png)
![4H-1-Benzopyran-4-one,3,5,7-tris(acetyloxy)-2-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/1100/1064-06-8_b.png)


![METHANONE, [4-HYDROXY-3,5-BIS(1-METHYLETHYL)PHENYL]PHENYL-](http://img.cochemist.com/ccimg/800/738-15-8.png)
![METHANONE, [4-HYDROXY-3,5-BIS(1-METHYLETHYL)PHENYL]PHENYL-](http://img.cochemist.com/ccimg/800/738-15-8_b.png)














![Ethanol, 2-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/1005600/901792-84-5.png)
![Ethanol, 2-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/1005600/901792-84-5_b.png)


![1-Propanol, 3-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/3151700/361541-87-9.png)
![1-Propanol, 3-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/3151700/361541-87-9_b.png)


![1-{2-[4-(Trifluoromethyl)phenyl]-1,3-thiazol-4-yl}ethan-1-one](http://img.cochemist.com/ccimg/263600/263564-37-0.png)
![1-{2-[4-(Trifluoromethyl)phenyl]-1,3-thiazol-4-yl}ethan-1-one](http://img.cochemist.com/ccimg/263600/263564-37-0_b.png)
![Ethanone, 1-[2-[4-(trifluoromethyl)phenyl]-4-thiazolyl]-, oxime](http://img.cochemist.com/ccimg/206700/206653-23-8.png)
![Ethanone, 1-[2-[4-(trifluoromethyl)phenyl]-4-thiazolyl]-, oxime](http://img.cochemist.com/ccimg/206700/206653-23-8_b.png)


![Ethyl 2-[4-(trifluoromethyl)phenyl]thiazole-4-carboxylate](http://img.cochemist.com/ccimg/175300/175204-88-3.png)
![Ethyl 2-[4-(trifluoromethyl)phenyl]thiazole-4-carboxylate](http://img.cochemist.com/ccimg/175300/175204-88-3_b.png)


![METHYL 1-[4-(BROMOMETHYL)PHENYL]PYRROLE-2-CARBOXYLATE](http://img.cochemist.com/ccimg/144100/144062-63-5.png)
![METHYL 1-[4-(BROMOMETHYL)PHENYL]PYRROLE-2-CARBOXYLATE](http://img.cochemist.com/ccimg/144100/144062-63-5_b.png)






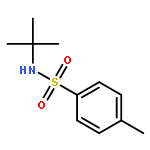
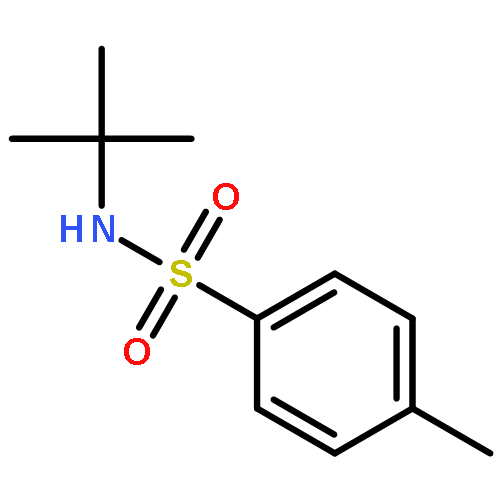
![Benzoic acid,4-[[(1,1-dimethylethyl)amino]sulfonyl]-](http://img.cochemist.com/ccimg/100000/99987-05-0.png)
![Benzoic acid,4-[[(1,1-dimethylethyl)amino]sulfonyl]-](http://img.cochemist.com/ccimg/100000/99987-05-0_b.png)

![Ethanone, 1-[2-(4-methoxyphenyl)-4-thiazolyl]-](http://img.cochemist.com/ccimg/65900/65823-91-8.png)
![Ethanone, 1-[2-(4-methoxyphenyl)-4-thiazolyl]-](http://img.cochemist.com/ccimg/65900/65823-91-8_b.png)

















![ACETATE, 2,2',2'',2'''-[(1R,2R)-1,2-CYCLOHEXANEDIYLDINITRILO]TETR<WBR />AKIS-, COPPER(2+) SALT (1:1)](http://img.cochemist.com/ccimg/300900/300800-07-1.png)
![ACETATE, 2,2',2'',2'''-[(1R,2R)-1,2-CYCLOHEXANEDIYLDINITRILO]TETR<WBR />AKIS-, COPPER(2+) SALT (1:1)](http://img.cochemist.com/ccimg/300900/300800-07-1_b.png)
![1-[4-(BROMOMETHYL)PHENYL]PYRROLE-2-CARBONITRILE](http://img.cochemist.com/ccimg/142100/142044-78-8.png)
![1-[4-(BROMOMETHYL)PHENYL]PYRROLE-2-CARBONITRILE](http://img.cochemist.com/ccimg/142100/142044-78-8_b.png)










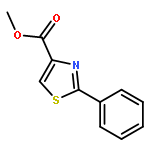
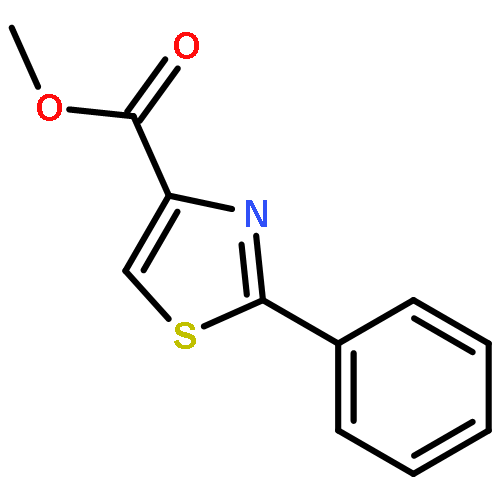
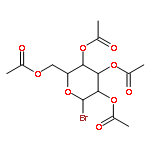

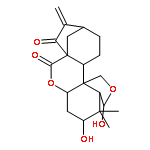



![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)



![4-OXAZOLECARBOXYLIC ACID, 2-[4-(TRIFLUOROMETHYL)PHENYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/753500/753479-58-2.png)
![4-OXAZOLECARBOXYLIC ACID, 2-[4-(TRIFLUOROMETHYL)PHENYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/753500/753479-58-2_b.png)