Co-reporter:John M. Ovian, Christopher B. Kelly, Vincent A. Pistritto, and Nicholas E. Leadbeater
Organic Letters March 17, 2017 Volume 19(Issue 6) pp:
Publication Date(Web):March 1, 2017
DOI:10.1021/acs.orglett.7b00060
An operationally simple, robust, metal-free approach to the synthesis of N-acyl azoles from both alcohols and aldehydes is described. Oxidative amidation is facilitated by a commercially available organic oxidant (4-acetamido-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate) and proceeds under very mild conditions for an array of structurally diverse substrates. Tandem reactions of these activated amides, such as transamidation and esterification, enable further elaboration. Also, the spent oxidant can be recovered and used to regenerate the oxoammonium salt.
Co-reporter:Jyoti Nandi;John M. Ovian;Christopher B. Kelly
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 39) pp:8295-8301
Publication Date(Web):2017/10/11
DOI:10.1039/C7OB02243C
We present a new paradigm for nitroxyl-mediated processes via the merger of oxoammonium cation-mediated oxidation with visible-light photoredox catalysis. The integration of these two forms of catalysis has been realised for the oxidative amidation of aldehydes, furnishing N-acylated heterocycles. Extension of this process to the oxidative amidation of alcohols via the intermediacy of an aldehyde was successfully pursued, thus proffering a general oxidation platform. The activated amides synthesised here are excellent synthetic handles for acylation.
Co-reporter:Shelli A. Miller;James M. Bobbitt
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 13) pp:2817-2822
Publication Date(Web):2017/03/28
DOI:10.1039/C7OB00039A
A systematic study of the oxidation of a range of terminal diols is reported, employing the oxoammonium salt 4-acetamido-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate (4-NHAc-TEMPO+ BF4−) as the oxidant. For substrates bearing a hydrocarbon chain of seven carbon atoms or more, the sole product is the dialdehyde. A series of post-oxidation reactions have been performed showing that the product mixture resulting from the oxidation step can be taken on directly to a subsequent transformation. For diols containing four to six carbon atoms, the lactone product is the major product upon oxidation. In the case of 1,2-ethanediol and 1,3-propanediol, when using a 1 : 0.5 stoichiometric ratio of substrate to oxidant, the corresponding monoaldehyde is formed which reacts rapidly with further diol to yield the acetal product. This is of particular synthetic value given both the difficulty of their preparation using other approaches and also their potential application in further reaction chemistry.
Co-reporter:Jacob. J. Loman, Emma R. Carnaghan, Trevor A. Hamlin, John M. Ovian, Christopher B. Kelly, Michael A. Mercadante and Nicholas E. Leadbeater
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 16) pp:3883-3888
Publication Date(Web):18 Mar 2016
DOI:10.1039/C6OB00347H
The propensity of oxoammonium cations to facilitate the oxidative ring-opening of cyclic ethers to their corresponding distal hydroxy ketones is investigated. The reaction has been evaluated using experimental and computational methods to gain deeper insight into trends in reactivity.
Co-reporter:Monaem Balti, Shelli A. Miller, Mohamed Lotfi Efrit and Nicholas E. Leadbeater
RSC Advances 2016 vol. 6(Issue 76) pp:72165-72169
Publication Date(Web):22 Jul 2016
DOI:10.1039/C6RA15488C
A method for the preparation of 4-aryl and 5-aryl substituted thiazole-2(3H)-thiones is described. Flow processing is employed as a tool, and supported acids and bases used to facilitate both the synthetic strategy and product isolation. The methodology is applicable to a range of substrates.
Co-reporter:Monaem Balti, Mohamed Lotfi Efrit, Nicholas E. Leadbeater
Tetrahedron Letters 2016 Volume 57(Issue 16) pp:1804-1806
Publication Date(Web):20 April 2016
DOI:10.1016/j.tetlet.2016.03.037
•A Wittig reaction is used to prepare aromatic-functionalized vinyl ethers.•Hydrogenation of the vinyl ethers is performed using continuous-flow processing.•The reactions are combined for the synthesis of a biologically-relevant compound.A methodology is reported for the preparation of various aromatic-functionalized vinyl ethers using a Wittig approach. The subsequent hydrogenation of the vinyl ether products to their saturated analogs is also performed and is streamlined by employing continuous-flow processing.
Co-reporter:Kathryn A. Alexander, Emily A. Paulhus, Gillian M.L. Lazarus, Nicholas E. Leadbeater
Journal of Organometallic Chemistry 2016 Volume 812() pp:74-80
Publication Date(Web):15 June 2016
DOI:10.1016/j.jorganchem.2015.09.018
•A ruthenium complex proves active for metathesis of biorenewable feedstocks.•Microwave heating and continuous-flow processing are used as tools for performing the reactions.•For the ring-closing metathesis reactions, transition from batch to flow processing for scale-up of the reaction is possible.Tricyclohexylphosphine[4,5-dimethyl-1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene][2-thienylmethylene]ruthenium(II) dichloride proves active for the ring-closing metathesis of linalool and citronellene, the self-metathesis of eugenol, and to some extent the ethenolysis of methyl oleate. Microwave heating and continuous-flow processing have been used as tools for performing the reactions. For the ring-closing metathesis reactions, transition from batch to flow processing for scale-up of the reaction is possible but it proves problematic in the case of cross-metathesis.A ruthenium complex proves active for the ring-closing metathesis of linalool and citronellene, the self-metathesis of eugenol, and to some extent the ethenolysis of methyl oleate. Microwave heating and continuous-flow processing have been used as tools for performing the reactions.
Co-reporter:Fabrizio Politano;Elba I. Buján
Chemistry of Heterocyclic Compounds 2016 Volume 52( Issue 11) pp:952-957
Publication Date(Web):2016 November
DOI:10.1007/s10593-017-1992-1
A continuous flow process for the synthesis of nitrobenzimidazole N-oxides from 2,6-dinitrochlorobenzene and amines or amino acids is reported. The process, performed in a two-step sequence, is faster than previously reported batch processes and avoids some of the isolation and purification steps.
Co-reporter:Christopher B. Kelly, John M. Ovian, Robin M. Cywar, Taylor R. Gosselin, Rebecca J. Wiles and Nicholas E. Leadbeater
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 14) pp:4255-4259
Publication Date(Web):25 Feb 2015
DOI:10.1039/C5OB00270B
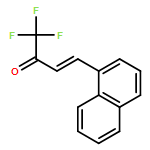
A method to oxidatively cleave allyl ethers to their corresponding aldehydes mediated by an oxoammonium salt is described. Using a biphasic solvent system and mild heating, cleavage proceeds readily, furnishing a variety of α,β-unsaturated aldehydes and ketones.
Co-reporter:Shelli A. Miller and Nicholas E. Leadbeater
RSC Advances 2015 vol. 5(Issue 113) pp:93248-93251
Publication Date(Web):20 Oct 2015
DOI:10.1039/C5RA21394K
A simple, solvent-free methodology is reported for the direct conversion of esters to amides using lithium hydroxide as a catalyst. The approach allows for the preparation of a range of amide products as well as being applicable to the ring-opening of a representative lactone.
Co-reporter:Trevor A. Hamlin, Christopher B. Kelly, John M. Ovian, Rebecca J. Wiles, Leon J. Tilley, and Nicholas E. Leadbeater
The Journal of Organic Chemistry 2015 Volume 80(Issue 16) pp:8150-8167
Publication Date(Web):July 13, 2015
DOI:10.1021/acs.joc.5b01240
A range of oxoammonium salt-based oxidation reactions have been explored computationally using density functional theory (DFT), and the results have been correlated with experimentally derived trends in reactivity. Mechanistically, most reactions involve a formal hydride transfer from an activated C–H bond to the oxygen atom of the oxoammonium cation. Several new potential modes of reactivity have been uncovered and validated experimentally.
Co-reporter:Lauren M. Stencel and Nicholas E. Leadbeater
New Journal of Chemistry 2014 vol. 38(Issue 1) pp:242-247
Publication Date(Web):29 Oct 2013
DOI:10.1039/C3NJ00784G
The results of an evaluation of the iChemExplorer for the study of bio-catalysed processes are reported. The iChemExplorer comprises of a specially-designed sample tray and a control unit, the former of which replaces a traditional tray in an HPLC autosampler assembly. It can be heated and reaction mixtures can be agitated. The system has been used to study the trypsin digestion of insulin chain B, cytochrome c and bovine serum albumin as well as the lipase-catalysed transesterification reaction between ethyl benzoate and 1-butanol.
Co-reporter:Trevor A. Hamlin, Gillian M. L. Lazarus, Christopher B. Kelly, and Nicholas E. Leadbeater
Organic Process Research & Development 2014 Volume 18(Issue 10) pp:1253-1258
Publication Date(Web):August 6, 2014
DOI:10.1021/op500190j
A continuous-flow approach to the synthesis of 3,3,3-trifluoromethylpropenes involving Grignard addition of (trimethylsilyl)methylmagnesium chloride to a trifluoromethyl ketone followed by dehydrative desilylation of the α-trifluoromethyl-β-hydroxysilyl alcohol using trimethylsilyl trifluoromethanesulfonate is reported. An inline aqueous/organic extraction and a concomitant solvent switch were key to the success of the methodology. Transition from batch to continuous flow conditions allows for higher yields, shorter reaction times, and facile scale out.
Co-reporter:Trevor A. Hamlin, Christopher B. Kelly, Robin M. Cywar, and Nicholas E. Leadbeater
The Journal of Organic Chemistry 2014 Volume 79(Issue 3) pp:1145-1155
Publication Date(Web):January 10, 2014
DOI:10.1021/jo402577n
An operationally simple, inexpensive, and rapid route for the olefination of a wide array of trifluoromethyl ketones to yield 3,3,3-trifluoromethylpropenes is reported. Using a Peterson olefination approach, the reaction gives good to excellent yields of the alkene products and can be performed without purification of the β-hydroxysilyl intermediate. The reaction can be extended to other perfluoroalkyl substituents and is easily scaled up. The alkenes prepared can be readily transformed into a variety of other perfluoroalkyl-containing compounds.
Co-reporter:Christopher B. Kelly, Michael A. Mercadante and Nicholas E. Leadbeater
Chemical Communications 2013 vol. 49(Issue 95) pp:11133-11148
Publication Date(Web):14 Oct 2013
DOI:10.1039/C3CC46266H
Trifluoromethyl ketones (TFMKs) are exceedingly valuable synthetic targets in their own right and as synthons in the construction of fluorinated pharmacons. This Feature Article provides an overview of the properties of TFMKs, an in-depth discussion of the methods available for their synthesis, and two illustrative examples of their application as key intermediates in medicinal chemistry.
Co-reporter:Christopher B. Kelly, Michael A. Mercadante, Rebecca J. Wiles, and Nicholas E. Leadbeater
Organic Letters 2013 Volume 15(Issue 9) pp:2222-2225
Publication Date(Web):April 24, 2013
DOI:10.1021/ol400785d
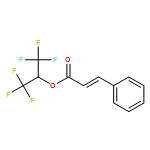
A simple, high yielding, rapid route for the oxidative esterification of a wide range aldehydes to hexafluoroisopropyl (HFIP) esters using the oxoammonium salt 4-acetylamino-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate (1a) is reported. These esters can be readily transformed into a variety of other functional groups. The spent oxidant (1b) can be recovered and conveniently reoxidized to regenerate the oxoammonium salt, 1a.
Co-reporter:Trevor A. Hamlin;Christopher B. Kelly
European Journal of Organic Chemistry 2013 Volume 2013( Issue 18) pp:3658-3661
Publication Date(Web):
DOI:10.1002/ejoc.201300392
Abstract
A novel dehydrogenation reaction of perfluoroalkyl ketones by the oxoammonium salt 4-acetylamino-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate (4-NHAc-TEMPO+BF4–, Bobbitt's salt, 1) is described. The reaction proceeds under mildly basic conditions and appears to be unique to perfluoroalkyl ketones. A proposed mechanism for this unusual transformation is given. The byproduct of the reaction, 4-acetylamino-2,2,6,6-tetramethyl-1-piperidinyloxy (1a), can easily be recovered and used to regenerate the oxoammonium salt.
Co-reporter:DiAndra M. Rudzinski, Christopher B. Kelly and Nicholas E. Leadbeater
Chemical Communications 2012 vol. 48(Issue 77) pp:9610-9612
Publication Date(Web):09 Aug 2012
DOI:10.1039/C2CC35037H
A novel route to access trifluoromethylketones (TFMKs) from Weinreb amides is reported. This represents the first documented case of the Ruppert–Prakash reagent (TMS–CF3) reacting in a constructive manner with an amide and enables synthesis of TMFKs without risk of over-trifluoromethylation.
Co-reporter:Michael A. Mercadante, Christopher B. Kelly, Christopher (Xiang) Lee, and Nicholas E. Leadbeater
Organic Process Research & Development 2012 Volume 16(Issue 5) pp:1064-1068
Publication Date(Web):April 4, 2012
DOI:10.1021/op300019w
A continuous-flow approach to the hydrogenation of alkenes utilizing Wilkinson’s catalyst is reported. The approach relies on a newly developed coil design in which it is possible to load gas and heat the reaction mixture simultaneously. The hydrogenation of various substrates has been performed successfully on small scale and can be scaled up substantially.
Co-reporter:Christopher B. Kelly, Michael A. Mercadante, Trevor A. Hamlin, Madison H. Fletcher, and Nicholas E. Leadbeater
The Journal of Organic Chemistry 2012 Volume 77(Issue 18) pp:8131-8141
Publication Date(Web):August 29, 2012
DOI:10.1021/jo301477s
A simple, mild method for the oxidation of α-trifluoromethyl alcohols to trifluoromethyl ketones (TFMKs) using the oxoammonium salt 4-acetylamino-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate (1) is described. Under basic conditions, oxidation proceeds rapidly and affords good to excellent yields of TFMKs, without concomitant formation of the hydrate. The byproduct of the oxidation, 4-acetylamino-2,2,6,6-tetramethyl-1-piperidinyloxy (1c), is easily recovered and can be conveniently reoxidized to regenerate the oxoammonium salt.
Co-reporter:Michael A. Mercadante and Nicholas E. Leadbeater
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 19) pp:6575-6578
Publication Date(Web):17 Aug 2011
DOI:10.1039/C1OB05808H
A prototype tube-in-tube reactor in which it is possible to load gas and heat simultaneously has been used in a continuous-flow approach to alkoxycarbonylation reactions of aryl iodides. In the stainless steel coil, liquid flows on the outside of a gas-permeable membrane. The coil can be heated and the temperature can be measured accurately via a probe touching the outer steel surface. A range of aryl iodides can be transformed to the corresponding esters in excellent conversion by reaction at 120 °C using 0.5 mol% palladium acetate as the catalyst with no additional ligand required. Small-scale optimization and substrate screening runs were followed by scale-up.
Co-reporter:Christopher B. Kelly, Christopher (Xiang) Lee, Michael A. Mercadante, and Nicholas E. Leadbeater
Organic Process Research & Development 2011 Volume 15(Issue 3) pp:717-720
Publication Date(Web):April 13, 2011
DOI:10.1021/op200037n
Using a continuous-flow approach, it is possible to perform alkoxycarbonylation reactions of aryl iodides. Optimized reactor design allows for adequate mixing of gaseous and liquid reagents. Reactions are performed at rates of around 3 mL/min and at concentrations of 1 M, allowing for significant volumes to be processed per unit time. Palladium acetate (0.5 mol %) is used as the catalyst without the need for an additional ligand.
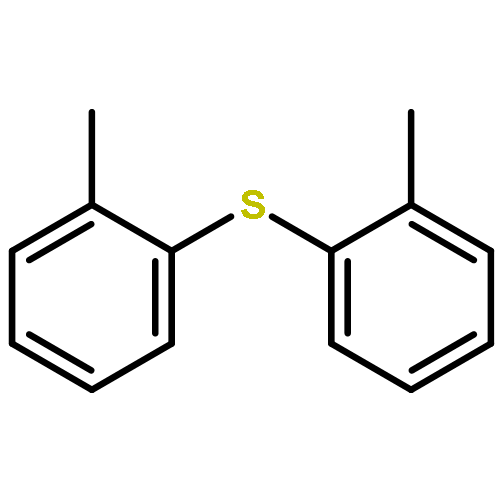
Co-reporter:Christopher B. Kelly, Christopher (Xiang) Lee, Nicholas E. Leadbeater
Tetrahedron Letters 2011 Volume 52(Issue 36) pp:4587-4589
Publication Date(Web):7 September 2011
DOI:10.1016/j.tetlet.2011.06.107
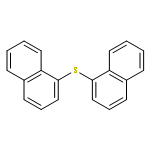
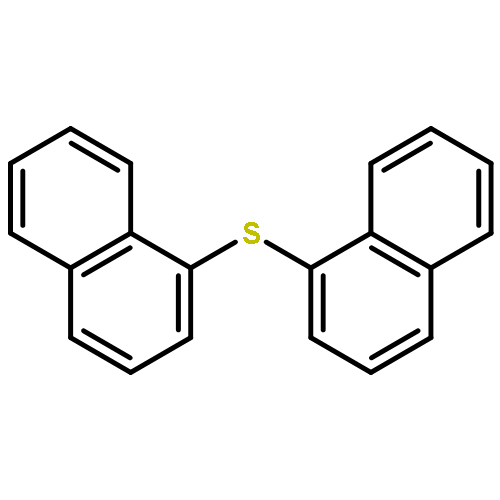
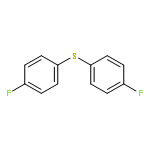
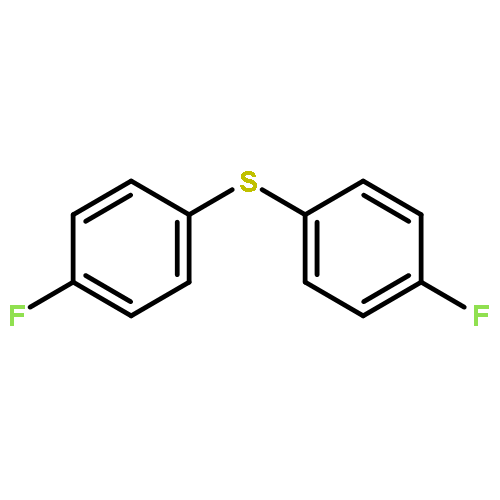
A method for direct preparation of diaryl sulfides from aryl iodides using potassium thiocyanate as a sulfur-transfer agent is reported. A catalyst system comprising of a simple copper salt, tetrabutylammonium bromide as a phase-transfer agent and water as the solvent is used. Microwave heating at 200 °C for 60 min allows for the conversion of a range of aryl iodides to the corresponding diaryl sulfides.
Co-reporter:Elizabeth A. Pedrick, Nicholas E. Leadbeater
Inorganic Chemistry Communications 2011 Volume 14(Issue 3) pp:481-483
Publication Date(Web):March 2011
DOI:10.1016/j.inoche.2011.01.005
Microwave heating has been used for the small-scale preparation of cisplatin, [cis-PtCl2(NH3)2], in isomerically pure form without concomitant formation of Magnus’ salt, [Pt(NH3)4][PtCl4]. In scaling up the reaction to the gram level, continuous-flow processing was employed.Microwave heating is used for fast, easy preparation of cisplatin. For scale up, continuous‐flow processing is employed.Research Highlights►Microwave heating used for the small‐scale synthesis of cis‐platin, [cis‐PtCl2(NH3)2]. ►Continuous‐flow processing used for scale‐up. ►Product is isomerically pure. ►Product is formed in short reaction times. ►No formation of Magnus’ salt [Pt(NH3)4][PtCl4] when using optimized conditions.
Co-reporter:Christopher B. Kelly, Christopher (Xiang) Lee, Nicholas E. Leadbeater
Tetrahedron Letters 2011 Volume 52(Issue 2) pp:263-265
Publication Date(Web):12 January 2011
DOI:10.1016/j.tetlet.2010.11.027
A simple adaptation allows batch protocols developed using microwave heating that involve formation of solid organic products to be scaled up using conventionally-heated flow chemistry with minimal or no re-optimization or modification. The product stream is intercepted with a flow of a suitable organic solvent upon exiting the heated zone, this solubilizing the product and allowing it to pass through the back-pressure regulator without aggregation of particulate material.
Co-reporter:Nicholas E. Leadbeater
Chemical Communications 2010 vol. 46(Issue 36) pp:6693-6695
Publication Date(Web):18 Aug 2010
DOI:10.1039/C0CC01921F
An IR probe has been interfaced with a scientific microwave unit, this allowing for real-time in situ monitoring of microwave-assisted reactions using IR spectroscopy. A number of organic transformations have been probed and the apparatus shown to work effectively as a tool for qualitative studies.
Co-reporter:Jason R. Schmink, Chad M. Kormos, William G. Devine and Nicholas E. Leadbeater
Organic Process Research & Development 2010 Volume 14(Issue 1) pp:205-214
Publication Date(Web):January 15, 2010
DOI:10.1021/op900287j
A new batch microwave reactor has been evaluated in the context of palladium-mediated transformations, condensation reactions, nucleophilic aromatic substitution reactions, and alkylations. Importantly, a linear scaling approach was taken, no changes being made to the protocol when moving from the small, developmental scale to larger scales. In some cases reactions were scaled over 18,000-fold when moving from small (0.1−1 mmol) to large (1−18 mol) runs.
Co-reporter:Arshad Mehmood;William G. Devine
Topics in Catalysis 2010 Volume 53( Issue 15-18) pp:1073-1080
Publication Date(Web):2010 September
DOI:10.1007/s11244-010-9535-3
We present a new methodology using copper(I) oxide as a catalyst in conjunction with cesium carbonate as a base for the preparation of diaryl and aryl–alkyl ethers. A range of aryl iodides, phenols and primary alcohols can be used as substrates and reactions are performed using microwave heating. We also present the use of Cu2Fe(CN)6 as both catalyst and cyanating agent for the direct conversion of aryl iodides to nitriles.
Co-reporter:Catherine DeBlase, Nicholas E. Leadbeater
Tetrahedron 2010 66(5) pp: 1098-1101
Publication Date(Web):
DOI:10.1016/j.tet.2009.11.016
Co-reporter:Jason R. Schmink and Nicholas E. Leadbeater
Organic Letters 2009 Volume 11(Issue 12) pp:2575-2578
Publication Date(Web):May 20, 2009
DOI:10.1021/ol900874z
A novel route to the synthesis of diarylmethanes via a Pd-catalyzed α-arylation of benzyl ketones is reported. By harnessing the inherent reactivity of enolates, it is possible to circumvent the need for a transmetalating reagent such as boron for the coupling. Additionally, the two phenyl rings of the intermediate are exploited to stabilize the high-energy carbanionic leaving group in a straightforward synthesis.
Co-reporter:Jason R. Schmink and Nicholas E. Leadbeater
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 18) pp:3842-3846
Publication Date(Web):22 Jul 2009
DOI:10.1039/B910591C
The use of in situRaman spectroscopy is reported as a tool for probing the effects of microwave irradiation on molecules. Our results show no evidence for localized superheating, an often-cited specific microwave effect. While the microwave energy may interact with the polar molecules more so than with non-polar ones, the conversion of electromagnetic energy into kinetic energy is slower than conversion of kinetic energy into thermal energy. As a result, more polar molecules are not at a temperature greater than that of the bulk.
Co-reporter:Lauren M. Stencel, Chad M. Kormos, Keri B. Avery and Nicholas E. Leadbeater
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 11) pp:2452-2457
Publication Date(Web):21 Apr 2009
DOI:10.1039/B902112D
The use of two silicon carbide plates is reported for the preparation of three libraries of organic molecules using microwave heating. In addition, a preliminary study has been carried out, showing that one of the plates can also be used in a proteomics setting. Both the 24-position and 48-position plates heated evenly when irradiated with microwave energy. The 48-position plate was used to prepare a library of N-aryl functionalized β-amino esters via an aza-Michael reaction between anilines and Michael acceptors. The 24-position plate was used to prepare a library of biaryls via a Suzuki coupling methodology and a library of 1,4-dihydropyridines via a Hantzsch synthesis. The 48-position plate was also used to perform the proteolytic digestion of insulin chain B by trypsin.
Co-reporter:Mauro Iannelli, Fabio Bergamelli, Chad M. Kormos, Stefano Paravisi and Nicholas E. Leadbeater
Organic Process Research & Development 2009 Volume 13(Issue 3) pp:634-637
Publication Date(Web):February 13, 2009
DOI:10.1021/op800296d
The ethoxycarbonylation of iodobenzene was performed on the 1 mol scale in batch mode using microwave heating. The reaction was performed using both an excess and a near stoichiometric loading of carbon monoxide, comparable yields being obtained. Six different alkoxycarbonylation reactions were then performed simultaneously on the 50 mmol scale using a near-stoichiometric loading of carbon monoxide with excellent conversions in each case.
Co-reporter:Krista A. Shoemaker, Nicholas E. Leadbeater
Inorganic Chemistry Communications 2009 Volume 12(Issue 5) pp:341-342
Publication Date(Web):May 2009
DOI:10.1016/j.inoche.2009.02.015
A fast and easy approach to the synthesis of Zeise’s salt, KPtCl3(C2H4), is reported using microwave heating. The reaction is complete after 15 min at 130 °C using K2PtCl4 as starting material, a 1:1:1 ratio of water:ethanol:concentrated HCl as solvent, and a loading of 50 psi of ethene.A fast and easy approach to the synthesis of Zeise’s salt, KPtCl3(C2H4), is reported using microwave heating. The reaction is complete after 15 min at 130 °C using K2PtCl4 as starting material, a 1:1:1 ratio of water:ethanol:concentrated HCl as solvent, and a loading of 50 psi of ethene.
Co-reporter:Keri B. Avery, William G. Devine, Chad M. Kormos, Nicholas E. Leadbeater
Tetrahedron Letters 2009 50(24) pp: 2851-2853
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.03.140
Co-reporter:Michelle L. Dean, Jason R. Schmink, Nicholas E. Leadbeater and Christian Brückner
Dalton Transactions 2008 (Issue 10) pp:1341-1345
Publication Date(Web):15 Jan 2008
DOI:10.1039/B716181F
The scope and limitations of microwave heating as a tool for insertion of Group 10 metals into meso-tetraphenyl-porphyrin, -porpholactone, and -2,3-dihydroxychlorin derivatives are discussed. In some cases it is possible to reduce reaction times dramatically while obtaining good yields of the metallated products while in others new issues arise relating to the metal salt used as well as acceleration not only of the metallation reaction but also of byproduct formation.
Co-reporter:Nicholas E. Leadbeater and M. Rashid Khan
Energy & Fuels 2008 Volume 22(Issue 3) pp:1836-1839
Publication Date(Web):April 1, 2008
DOI:10.1021/ef7007198
We have probed the effect of microwave irradiation on desulfurization of crude oil and dibenzothiophene. A range of catalysts have been screened. We find that it is possible to perform hydrodesulfurization reactions using microwave heating in conjunction with iron powder as a catalyst. The effect of hydrogen pressure, reaction temperature, reaction time, and catalyst source have been studied. The effectiveness of catalyst- and hydrogen-free desulfurization has been probed briefly. Approaches included microwave-promoted cracking, high-temperature water chemistry, and the use of metal hydrides but, with the conditions screened in this study, these proved not to be effective. We believe that our results here build on work in the scientific and patent literature suggesting that microwave irradiation can be a useful tool for effecting desulfurization chemistry.
Co-reporter:Matthew D. Bowman, Jason R. Schmink, Cynthia M. McGowan, Chad M. Kormos and Nicholas E. Leadbeater
Organic Process Research & Development 2008 Volume 12(Issue 6) pp:1078-1088
Publication Date(Web):September 19, 2008
DOI:10.1021/op8001239
A range of synthetic transformations have been scaled up successfully using a sealed-vessel multimode microwave unit. These include metal-catalyzed couplings, synthesis of heterocycles, reactions under an atmosphere of reactive gas and two-step one-pot procedures. Also, observations have been made along the way that are of use to chemists addressing scale-up of microwave-promoted reactions.
Co-reporter:Nicholas E. Leadbeater, T. Michael Barnard and Lauren M. Stencel
Energy & Fuels 2008 Volume 22(Issue 3) pp:2005-2008
Publication Date(Web):March 26, 2008
DOI:10.1021/ef700748t
The batch and continuous-flow preparation of biodiesel derived from vegetable oil and 1-butanol using a commercially available scientific microwave apparatus is reported. The methodology allows for the reaction to be run under atmospheric conditions and, in continuous-flow mode, can be performed at flow rates up to 2.3 L/min using a 4 L reaction vessel. It can be utilized with new or used vegetable oil with 1-butanol and a 1:6 molar ratio of oil to alcohol. Sulfuric acid or potassium hydroxide can be used as the catalyst.
Co-reporter:Matthew D. Bowman, Jennifer L. Holcomb, Chad M. Kormos, Nicholas E. Leadbeater and Victoria A. Williams
Organic Process Research & Development 2008 Volume 12(Issue 1) pp:41-57
Publication Date(Web):November 10, 2007
DOI:10.1021/op700187w
In this report, we look at a range of classes of reaction involving microwave heating and show how different processing techniques can be used to address scale-up needs. We look at both batch and continuous-flow processing. We have shown that when using batch methodologies working using an open reaction vessel offers operational advantages while still giving good yields of desired products. In cases where open-vessel conditions are not amenable or where particularly volatile or toxic reagents are used, parallel sealed vessels can offer an alternative approach. For continuous-flow processing, homogeneity of the reaction mixture is key. When the mixture is homogeneous, it is possible to move from small-scale sealed-vessel conditions to the continuous-flow apparatus without any modification of reaction conditions or loss in product yield. When either the starting materials or the product mixture contains particulate matter, continuous processing can prove a challenge, but reoptimization of reaction conditions as well as reduction of the concentration may allow these difficulties to be overcome.
Co-reporter:Nicholas E. Leadbeater and Krista M. Shoemaker
Organometallics 2008 Volume 27(Issue 6) pp:1254-1258
Publication Date(Web):February 22, 2008
DOI:10.1021/om701099p
By using a gas-loading accessory, we show that it is possible to perform reactions involving gases and prepare organometallic complexes easily in good yields using microwave heating. The complexes Ru3(CO)12, H4Ru4(CO)12, and H2Os3(CO)10 are prepared. Ligand substitution reactions of Ru3(CO)12 are also studied and, in the case of the reaction with triphenylphosphine, the reaction is monitored in real time by means of a digital camera interfaced with the microwave unit.
Co-reporter:JasonR. Schmink;JenniferL. Holcomb ;NicholasE. Leadbeater Dr.
Chemistry - A European Journal 2008 Volume 14( Issue 32) pp:9943-9950
Publication Date(Web):
DOI:10.1002/chem.200801158
Abstract
Raman spectroscopy has been used as an in situ tool to obtain kinetic data for an organic transformation. The model reaction studied was the synthesis of 3-acetylcoumarin from the condensation between salicylaldehyde and ethyl acetoacetate with piperidine as a catalyst. The study shows that precise kinetic data can be obtained quickly and reproducibly, allowing for the facile determination of both overall reaction order and reaction order with respect to each component of the reaction. Additionally, Arrhenius parameters such as activation energy for a reaction can be readily obtained. In conjunction with computational modeling, this data-rich analysis technique also allows for in-depth probing of mechanistic aspects of reactions. Microwave heating proves to be an ideal tool for aiding in kinetic studies. It offers reproducible noncontact heating as well as precise temperature monitoring and data recording.
Co-reporter:Kristen M. Amore
Macromolecular Rapid Communications 2007 Volume 28(Issue 4) pp:473-477
Publication Date(Web):12 FEB 2007
DOI:10.1002/marc.200600751
Microwave heating is becoming a widely accepted tool for synthetic chemists. While many reactions have been performed on a small scale using microwave heating, few have been further developed into larger-scale syntheses. Here, a microwave-promoted esterification reaction protocol is presented. The apparatus used allows for the removal of water generated during the course of the reaction and as a result the process can be driven toward completion. Reactions have been run on scales up to 3 mol.
Co-reporter:Chad M. Kormos and Nicholas E. Leadbeater
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 1) pp:65-68
Publication Date(Web):14 Nov 2006
DOI:10.1039/B614025D
The microwave-promoted alkoxycarbonylation of aryl iodides using reaction vessels pre-pressurized with carbon monoxide is reported. Reactions are performed using 0.1 mol% palladium acetate as catalyst, DBU as base and are complete within 20–30 min. A range of aryl iodide substrates can be converted to the corresponding esters using this methodology. Primary and secondary alcohols work well whereas a tertiary alcohol substrate proves less reactive. The potential for scale-up of the reaction has also been explored.
Co-reporter:Nicholas E. Leadbeater and Rebecca J. Smith
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 17) pp:2770-2774
Publication Date(Web):25 Jul 2007
DOI:10.1039/B707692D
We report the use of in situ Raman spectroscopy as a probe for the effect of power on microwave-promoted Suzuki coupling reactions. We find that increased initial microwave power leads to greater acceleration of the reaction but that the product yield obtained is essentially independent of initial microwave power. The application of simultaneous cooling lengthens the reaction time but does not alter the relative rates of the Suzuki coupling and deboronation processes. Performing the reaction at an initial microwave power of 5 W leads to an improvement in product yield.
Co-reporter:Nicholas E. Leadbeater, Lauren M. Stencel and Eric C. Wood
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 7) pp:1052-1055
Publication Date(Web):16 Feb 2007
DOI:10.1039/B617544A
The lipase-catalysed transesterification reaction of methyl acetoacetate in toluene as a solvent has been studied using carefully controlled conditions. Results suggest that microwave heating does not have a noticeable effect on reaction rate or product conversion.
Co-reporter:Jonathan M. Collins and Nicholas E. Leadbeater
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 8) pp:1141-1150
Publication Date(Web):26 Feb 2007
DOI:10.1039/B617084F
As the range of techniques for microwave heating has expanded, so have the areas in which it can have a profound impact. Two emerging areas are the application of microwave heating for the synthesis of peptides, peptoids, oligopeptides and carbohydrates and in the field of proteomics.
Co-reporter:Nicholas E. Leadbeater, Rebecca J. Smith and T. Michael Barnard
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 5) pp:822-825
Publication Date(Web):30 Jan 2007
DOI:10.1039/B615597A
Microwave-promoted esterification reactions have been monitored using in situ Raman spectroscopy. Having optimised a reaction on a 23 mmol scale, it was transferred to a larger reaction vessel and scaled up to 0.26 mol, again with Raman monitoring. With conditions in hand, an automated stop-flow apparatus was used to prepare 5.7 moles of product.
Co-reporter:Thomas M. Barnard and Nicholas E. Leadbeater
Chemical Communications 2006 (Issue 34) pp:3615-3616
Publication Date(Web):01 Aug 2006
DOI:10.1039/B608793K
An apparatus has been developed for real-time monitoring of organometallic reactions under microwave irradiation using in situ Raman spectroscopy and its application for monitoring ligand substitution reactions of Mo(CO)6 demonstrated.
Co-reporter:Arshad Mehmood, Nicholas E. Leadbeater
Catalysis Communications (25 October 2010) Volume 12(Issue 1) pp:64-66
Publication Date(Web):25 October 2010
DOI:10.1016/j.catcom.2010.07.011
A method for direct preparation of phenols from aryl halides using microwave heating is reported. A catalyst system comprising a simple copper salt and a diamine ligand is used, together with tripotassium phosphate as base and water as the solvent. Heating at 180 °C for 30 min allows for the conversion of a range of aryl bromides and iodides to the corresponding phenols. Aryl chlorides prove less reactive.Download full-size imageResearch Highlights►Conversion of aryl halides to phenols. ►Use of water as a solvent. ►Copper catalysis. ►Use of microwave heating as a tool for catalysis. ►Heating at 180 °C for 30 min allows for the conversion of a range of aryl bromides and iodides to the corresponding phenols.
Co-reporter:Michael A. Mercadante, Christopher B. Kelly, Trevor A. Hamlin, Kayla R. Delle Chiaie, Michael D. Drago, Katherine K. Duffy, Megan T. Dumas, Diana C. Fager, Bryanna L. C. Glod, Katherine E. Hansen, Cameron R. Hill, Rebecca M. Leising, Catherine L. Lynes, Allyson E. MacInnis, Madeline R. McGohey, Stephanie A. Murray, Marc C. Piquette, Shaina L. Roy, Ryan M. Smith, Katherine R. Sullivan, Bao H. Truong, Kristina M. Vailonis, Vitaliy Gorbatyuk, Nicholas E. Leadbeater and Leon J. Tilley
Chemical Science (2010-Present) 2014 - vol. 5(Issue 10) pp:NaN3994-3994
Publication Date(Web):2014/07/03
DOI:10.1039/C4SC01732C
Placement of an electron-withdrawing trifluoromethyl group (–CF3) at a putative cationic centre enhances γ-silyl neighbouring-group participation (NGP). In stark contrast to previously studied γ-silyl-substituted systems, the preferred reaction pathway is 1,3-γ-silyl elimination, giving ring closure over solvent substitution or alkene formation. The scope of this cyclopropanation reaction is explored for numerous cyclic and acyclic examples, proving this method to be a viable approach to preparing CF3-substituted cyclopropanes and bicyclic systems, both containing quaternary centres. Rate-constants, kinetic isotope effects, and quantum mechanical calculations provided evidence for this enhancement and further elaborated the disparity in the reaction outcome between these systems and previously studied γ-silyl systems.
Co-reporter:Shelli A. Miller, James M. Bobbitt and Nicholas E. Leadbeater
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 13) pp:NaN2822-2822
Publication Date(Web):2017/03/03
DOI:10.1039/C7OB00039A
A systematic study of the oxidation of a range of terminal diols is reported, employing the oxoammonium salt 4-acetamido-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate (4-NHAc-TEMPO+ BF4−) as the oxidant. For substrates bearing a hydrocarbon chain of seven carbon atoms or more, the sole product is the dialdehyde. A series of post-oxidation reactions have been performed showing that the product mixture resulting from the oxidation step can be taken on directly to a subsequent transformation. For diols containing four to six carbon atoms, the lactone product is the major product upon oxidation. In the case of 1,2-ethanediol and 1,3-propanediol, when using a 1:0.5 stoichiometric ratio of substrate to oxidant, the corresponding monoaldehyde is formed which reacts rapidly with further diol to yield the acetal product. This is of particular synthetic value given both the difficulty of their preparation using other approaches and also their potential application in further reaction chemistry.
Co-reporter:Christopher B. Kelly, John M. Ovian, Robin M. Cywar, Taylor R. Gosselin, Rebecca J. Wiles and Nicholas E. Leadbeater
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 14) pp:NaN4259-4259
Publication Date(Web):2015/02/25
DOI:10.1039/C5OB00270B
A method to oxidatively cleave allyl ethers to their corresponding aldehydes mediated by an oxoammonium salt is described. Using a biphasic solvent system and mild heating, cleavage proceeds readily, furnishing a variety of α,β-unsaturated aldehydes and ketones.
Co-reporter:Michelle L. Dean, Jason R. Schmink, Nicholas E. Leadbeater and Christian Brückner
Dalton Transactions 2008(Issue 10) pp:NaN1345-1345
Publication Date(Web):2008/01/15
DOI:10.1039/B716181F
The scope and limitations of microwave heating as a tool for insertion of Group 10 metals into meso-tetraphenyl-porphyrin, -porpholactone, and -2,3-dihydroxychlorin derivatives are discussed. In some cases it is possible to reduce reaction times dramatically while obtaining good yields of the metallated products while in others new issues arise relating to the metal salt used as well as acceleration not only of the metallation reaction but also of byproduct formation.
Co-reporter:Jacob. J. Loman, Emma R. Carnaghan, Trevor A. Hamlin, John M. Ovian, Christopher B. Kelly, Michael A. Mercadante and Nicholas E. Leadbeater
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 16) pp:NaN3888-3888
Publication Date(Web):2016/03/18
DOI:10.1039/C6OB00347H
The propensity of oxoammonium cations to facilitate the oxidative ring-opening of cyclic ethers to their corresponding distal hydroxy ketones is investigated. The reaction has been evaluated using experimental and computational methods to gain deeper insight into trends in reactivity.
Co-reporter:Michael A. Mercadante and Nicholas E. Leadbeater
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 19) pp:NaN6578-6578
Publication Date(Web):2011/08/17
DOI:10.1039/C1OB05808H
A prototype tube-in-tube reactor in which it is possible to load gas and heat simultaneously has been used in a continuous-flow approach to alkoxycarbonylation reactions of aryl iodides. In the stainless steel coil, liquid flows on the outside of a gas-permeable membrane. The coil can be heated and the temperature can be measured accurately via a probe touching the outer steel surface. A range of aryl iodides can be transformed to the corresponding esters in excellent conversion by reaction at 120 °C using 0.5 mol% palladium acetate as the catalyst with no additional ligand required. Small-scale optimization and substrate screening runs were followed by scale-up.
Co-reporter:DiAndra M. Rudzinski, Christopher B. Kelly and Nicholas E. Leadbeater
Chemical Communications 2012 - vol. 48(Issue 77) pp:NaN9612-9612
Publication Date(Web):2012/08/09
DOI:10.1039/C2CC35037H
A novel route to access trifluoromethylketones (TFMKs) from Weinreb amides is reported. This represents the first documented case of the Ruppert–Prakash reagent (TMS–CF3) reacting in a constructive manner with an amide and enables synthesis of TMFKs without risk of over-trifluoromethylation.
Co-reporter:Christopher B. Kelly, Michael A. Mercadante and Nicholas E. Leadbeater
Chemical Communications 2013 - vol. 49(Issue 95) pp:NaN11148-11148
Publication Date(Web):2013/10/14
DOI:10.1039/C3CC46266H
Trifluoromethyl ketones (TFMKs) are exceedingly valuable synthetic targets in their own right and as synthons in the construction of fluorinated pharmacons. This Feature Article provides an overview of the properties of TFMKs, an in-depth discussion of the methods available for their synthesis, and two illustrative examples of their application as key intermediates in medicinal chemistry.
Co-reporter:Chad M. Kormos and Nicholas E. Leadbeater
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 1) pp:NaN68-68
Publication Date(Web):2006/11/14
DOI:10.1039/B614025D
The microwave-promoted alkoxycarbonylation of aryl iodides using reaction vessels pre-pressurized with carbon monoxide is reported. Reactions are performed using 0.1 mol% palladium acetate as catalyst, DBU as base and are complete within 20–30 min. A range of aryl iodide substrates can be converted to the corresponding esters using this methodology. Primary and secondary alcohols work well whereas a tertiary alcohol substrate proves less reactive. The potential for scale-up of the reaction has also been explored.
Co-reporter:Jason R. Schmink and Nicholas E. Leadbeater
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 18) pp:NaN3846-3846
Publication Date(Web):2009/07/22
DOI:10.1039/B910591C
The use of in situRaman spectroscopy is reported as a tool for probing the effects of microwave irradiation on molecules. Our results show no evidence for localized superheating, an often-cited specific microwave effect. While the microwave energy may interact with the polar molecules more so than with non-polar ones, the conversion of electromagnetic energy into kinetic energy is slower than conversion of kinetic energy into thermal energy. As a result, more polar molecules are not at a temperature greater than that of the bulk.
Co-reporter:Nicholas E. Leadbeater, Rebecca J. Smith and T. Michael Barnard
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 5) pp:NaN825-825
Publication Date(Web):2007/01/30
DOI:10.1039/B615597A
Microwave-promoted esterification reactions have been monitored using in situ Raman spectroscopy. Having optimised a reaction on a 23 mmol scale, it was transferred to a larger reaction vessel and scaled up to 0.26 mol, again with Raman monitoring. With conditions in hand, an automated stop-flow apparatus was used to prepare 5.7 moles of product.
Co-reporter:Nicholas E. Leadbeater, Lauren M. Stencel and Eric C. Wood
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 7) pp:NaN1055-1055
Publication Date(Web):2007/02/16
DOI:10.1039/B617544A
The lipase-catalysed transesterification reaction of methyl acetoacetate in toluene as a solvent has been studied using carefully controlled conditions. Results suggest that microwave heating does not have a noticeable effect on reaction rate or product conversion.
Co-reporter:Nicholas E. Leadbeater and Rebecca J. Smith
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 17) pp:NaN2774-2774
Publication Date(Web):2007/07/25
DOI:10.1039/B707692D
We report the use of in situ Raman spectroscopy as a probe for the effect of power on microwave-promoted Suzuki coupling reactions. We find that increased initial microwave power leads to greater acceleration of the reaction but that the product yield obtained is essentially independent of initial microwave power. The application of simultaneous cooling lengthens the reaction time but does not alter the relative rates of the Suzuki coupling and deboronation processes. Performing the reaction at an initial microwave power of 5 W leads to an improvement in product yield.
Co-reporter:Jonathan M. Collins and Nicholas E. Leadbeater
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 8) pp:NaN1150-1150
Publication Date(Web):2007/02/26
DOI:10.1039/B617084F
As the range of techniques for microwave heating has expanded, so have the areas in which it can have a profound impact. Two emerging areas are the application of microwave heating for the synthesis of peptides, peptoids, oligopeptides and carbohydrates and in the field of proteomics.
Co-reporter:Nicholas E. Leadbeater
Chemical Communications 2010 - vol. 46(Issue 36) pp:NaN6695-6695
Publication Date(Web):2010/08/18
DOI:10.1039/C0CC01921F
An IR probe has been interfaced with a scientific microwave unit, this allowing for real-time in situ monitoring of microwave-assisted reactions using IR spectroscopy. A number of organic transformations have been probed and the apparatus shown to work effectively as a tool for qualitative studies.
Co-reporter:Lauren M. Stencel, Chad M. Kormos, Keri B. Avery and Nicholas E. Leadbeater
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 11) pp:NaN2457-2457
Publication Date(Web):2009/04/21
DOI:10.1039/B902112D
The use of two silicon carbide plates is reported for the preparation of three libraries of organic molecules using microwave heating. In addition, a preliminary study has been carried out, showing that one of the plates can also be used in a proteomics setting. Both the 24-position and 48-position plates heated evenly when irradiated with microwave energy. The 48-position plate was used to prepare a library of N-aryl functionalized β-amino esters via an aza-Michael reaction between anilines and Michael acceptors. The 24-position plate was used to prepare a library of biaryls via a Suzuki coupling methodology and a library of 1,4-dihydropyridines via a Hantzsch synthesis. The 48-position plate was also used to perform the proteolytic digestion of insulin chain B by trypsin.
.jpg)







![2-Methyl-1,2,3,9-tetrahydropyrrolo[2,1-b]quinazoline](http://img.cochemist.com/ccimg/881500/881404-62-2.png)
![2-Methyl-1,2,3,9-tetrahydropyrrolo[2,1-b]quinazoline](http://img.cochemist.com/ccimg/881500/881404-62-2_b.png)












![2H-1-BENZOPYRAN-2-ONE, 6-FLUORO-4-[[(4-METHYLPHENYL)SULFONYL]OXY]-](http://img.cochemist.com/ccimg/879500/879416-90-7.png)
![2H-1-BENZOPYRAN-2-ONE, 6-FLUORO-4-[[(4-METHYLPHENYL)SULFONYL]OXY]-](http://img.cochemist.com/ccimg/879500/879416-90-7_b.png)
![2H-PYRAN-2-ONE, 4-[[(4-METHYLPHENYL)SULFONYL]OXY]-](http://img.cochemist.com/ccimg/864200/864155-56-6.png)
![2H-PYRAN-2-ONE, 4-[[(4-METHYLPHENYL)SULFONYL]OXY]-](http://img.cochemist.com/ccimg/864200/864155-56-6_b.png)




![Silane, (1,1-dimethylethyl)dimethyl[1-(4-methylphenyl)ethoxy]-](http://img.cochemist.com/ccimg/774300/774244-48-3.png)
![Silane, (1,1-dimethylethyl)dimethyl[1-(4-methylphenyl)ethoxy]-](http://img.cochemist.com/ccimg/774300/774244-48-3_b.png)





















![2,2-DIMETHYL-3,4-DIHYDRO-1H-PYRIDO[1,2-A]BENZIMIDAZOLE](http://img.cochemist.com/ccimg/336200/336106-30-0.png)
![2,2-DIMETHYL-3,4-DIHYDRO-1H-PYRIDO[1,2-A]BENZIMIDAZOLE](http://img.cochemist.com/ccimg/336200/336106-30-0_b.png)


![Silane, (1,1-dimethylethyl)dimethyl[[3-(4-methylphenyl)-2-propynyl]oxy]-](http://img.cochemist.com/ccimg/321600/321555-12-8.png)
![Silane, (1,1-dimethylethyl)dimethyl[[3-(4-methylphenyl)-2-propynyl]oxy]-](http://img.cochemist.com/ccimg/321600/321555-12-8_b.png)








![Silane, (1,1-dimethylethyl)[(4-nitrophenyl)methoxy]diphenyl-](http://img.cochemist.com/ccimg/161900/161803-03-8.png)
![Silane, (1,1-dimethylethyl)[(4-nitrophenyl)methoxy]diphenyl-](http://img.cochemist.com/ccimg/161900/161803-03-8_b.png)
![Silane, (1,1-dimethylethyl)dimethyl[[(2E)-5-phenyl-2-pentenyl]oxy]-](http://img.cochemist.com/ccimg/160900/160805-54-9.png)
![Silane, (1,1-dimethylethyl)dimethyl[[(2E)-5-phenyl-2-pentenyl]oxy]-](http://img.cochemist.com/ccimg/160900/160805-54-9_b.png)


![1,4-Butanediol, 1-[4-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/150100/150074-77-4.png)
![1,4-Butanediol, 1-[4-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/150100/150074-77-4_b.png)
![Silane, (1,1-dimethylethyl)dimethyl[(4-nitrophenyl)methoxy]-](http://img.cochemist.com/ccimg/135700/135605-97-9.png)
![Silane, (1,1-dimethylethyl)dimethyl[(4-nitrophenyl)methoxy]-](http://img.cochemist.com/ccimg/135700/135605-97-9_b.png)




![Silane, (1,1-dimethylethyl)[(4-methoxyphenyl)methoxy]dimethyl-](http://img.cochemist.com/ccimg/101900/101803-60-5.png)
![Silane, (1,1-dimethylethyl)[(4-methoxyphenyl)methoxy]dimethyl-](http://img.cochemist.com/ccimg/101900/101803-60-5_b.png)
![Silane, (1,1-dimethylethyl)dimethyl[(3-phenyl-2-propenyl)oxy]-, (E)-](http://img.cochemist.com/ccimg/100100/100009-29-8.png)
![Silane, (1,1-dimethylethyl)dimethyl[(3-phenyl-2-propenyl)oxy]-, (E)-](http://img.cochemist.com/ccimg/100100/100009-29-8_b.png)













![1H-1,2,3-Triazole, 1-[(2-chlorophenyl)methyl]-4-phenyl-](http://img.cochemist.com/ccimg/63800/63778-13-2.png)
![1H-1,2,3-Triazole, 1-[(2-chlorophenyl)methyl]-4-phenyl-](http://img.cochemist.com/ccimg/63800/63778-13-2_b.png)



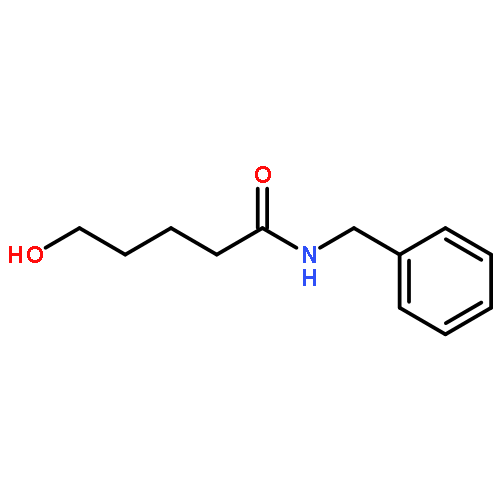


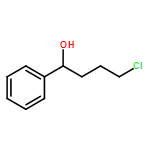
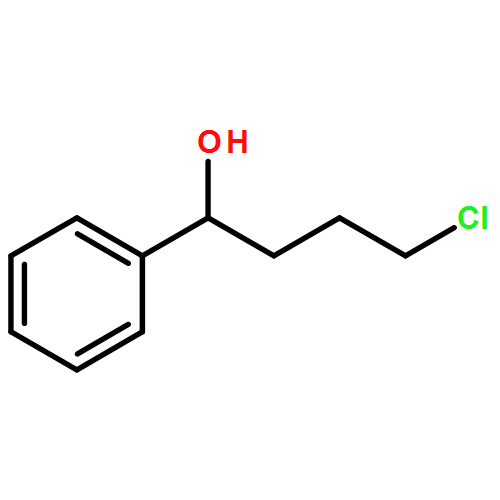


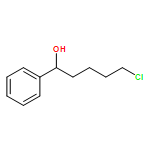
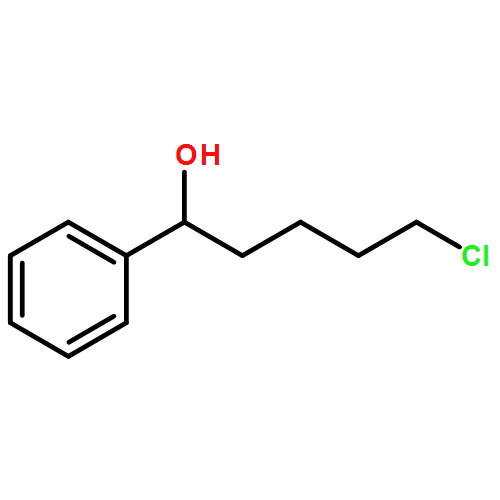




![1H-Pyrrolo[1,2-a]benzimidazole,2,3-dihydro-3-methyl-](http://img.cochemist.com/ccimg/30000/29981-98-4.png)
![1H-Pyrrolo[1,2-a]benzimidazole,2,3-dihydro-3-methyl-](http://img.cochemist.com/ccimg/30000/29981-98-4_b.png)


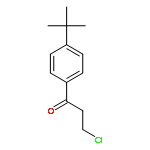

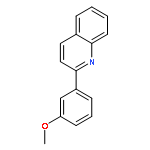
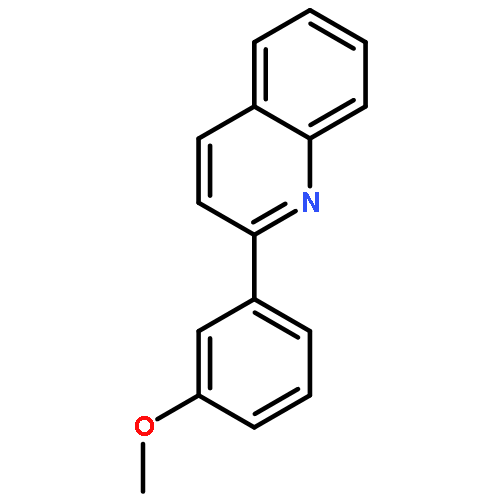





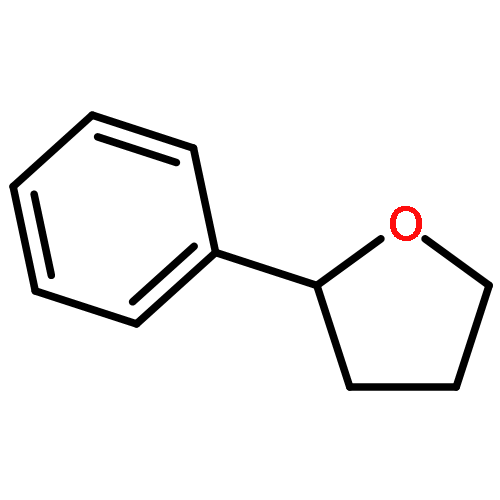





![Ethyl benzo[d][1,3]dioxole-5-carboxylate](http://img.cochemist.com/ccimg/7000/6951-08-2.png)
![Ethyl benzo[d][1,3]dioxole-5-carboxylate](http://img.cochemist.com/ccimg/7000/6951-08-2_b.png)


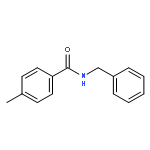










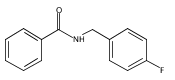
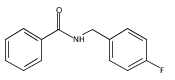
![N-[2-(BENZYLAMINO)-2-OXOETHYL]BENZAMIDE](http://img.cochemist.com/ccimg/3400/3392-91-4.png)
![N-[2-(BENZYLAMINO)-2-OXOETHYL]BENZAMIDE](http://img.cochemist.com/ccimg/3400/3392-91-4_b.png)

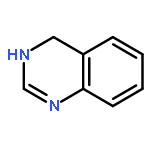
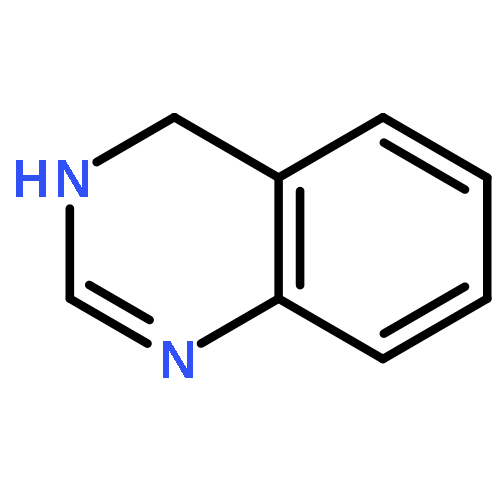
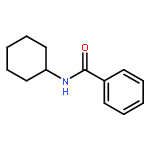
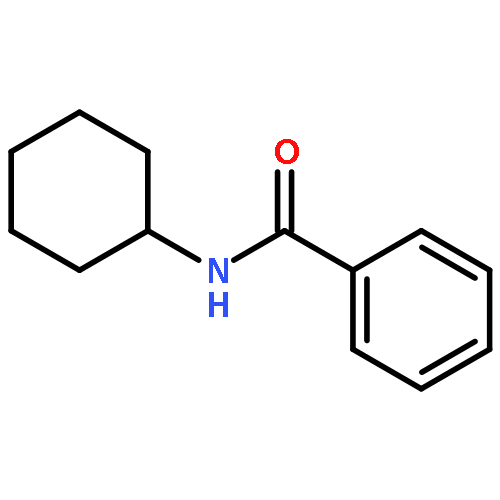




![Benzene, [(1E)-2-bromoethenyl]-](/data/chemimg/1476700/588-72-7.png)
![Benzene, [(1E)-2-bromoethenyl]-](/data/chemimg/1476700/588-72-7_b.png)
![1-bromo-4-[1-(trifluoromethyl)cyclopropyl]benzene](http://img.cochemist.com/ccimg/1227200/1227160-18-0.png)
![1-bromo-4-[1-(trifluoromethyl)cyclopropyl]benzene](http://img.cochemist.com/ccimg/1227200/1227160-18-0_b.png)

















![5-BROMO-2-[(2-METHOXYETHYL)AMINO]NICOTINONITRILE](http://img.cochemist.com/ccimg/700400/700362-32-9.png)
![5-BROMO-2-[(2-METHOXYETHYL)AMINO]NICOTINONITRILE](http://img.cochemist.com/ccimg/700400/700362-32-9_b.png)







![Spiro[4.5]dec-2-ene-6,10-dione](http://img.cochemist.com/ccimg/247100/247040-81-9.png)
![Spiro[4.5]dec-2-ene-6,10-dione](http://img.cochemist.com/ccimg/247100/247040-81-9_b.png)




























![Benzene, [3-(trifluoromethyl)-1,3-butadienyl]-, (E)-](/data/chemimg/1093300/138559-89-4.png)
![Benzene, [3-(trifluoromethyl)-1,3-butadienyl]-, (E)-](/data/chemimg/1093300/138559-89-4_b.png)









![2-Propenoic acid, 3-[4-(trifluoromethyl)phenyl]-, ethyl ester, (2E)-](http://img.cochemist.com/ccimg/128500/128408-03-7.png)
![2-Propenoic acid, 3-[4-(trifluoromethyl)phenyl]-, ethyl ester, (2E)-](http://img.cochemist.com/ccimg/128500/128408-03-7_b.png)


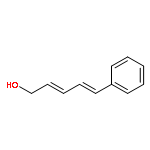
![2-Propen-1-ol, 3-[4-(trifluoromethyl)phenyl]-, (2E)-](/data/chemimg/115900/125617-18-7.png)
![2-Propen-1-ol, 3-[4-(trifluoromethyl)phenyl]-, (2E)-](/data/chemimg/115900/125617-18-7_b.png)












![Cyclohexanamine, N-[1-(2-pyridinyl)ethylidene]-](http://img.cochemist.com/ccimg/108000/107954-71-2.png)
![Cyclohexanamine, N-[1-(2-pyridinyl)ethylidene]-](http://img.cochemist.com/ccimg/108000/107954-71-2_b.png)




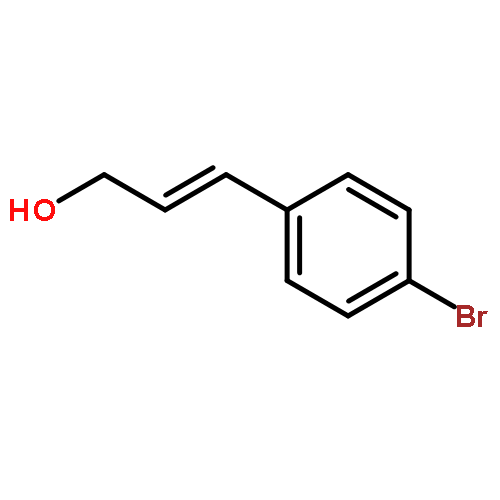








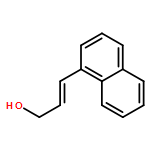
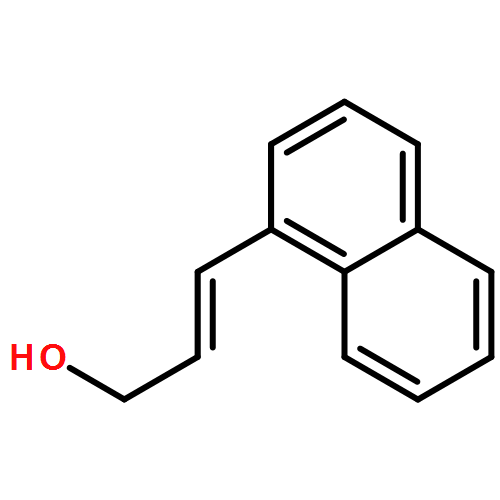
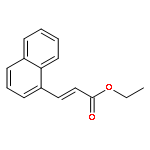

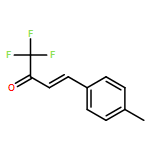



![2-Propenal, 3-[4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/95200/95123-61-8.png)
![2-Propenal, 3-[4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/95200/95123-61-8_b.png)











![Benzene, [(1E)-3-iodo-1-propenyl]-](http://img.cochemist.com/ccimg/82900/82859-39-0.png)
![Benzene, [(1E)-3-iodo-1-propenyl]-](http://img.cochemist.com/ccimg/82900/82859-39-0_b.png)



![7,12-DIHYDROXY-9-METHOXY-2,2-DIMETHYL-10-(3-METHYL-2-BUTEN-1-YL)-<WBR />2H,6H-PYRANO[3,2-B]XANTHEN-6-ONE](http://img.cochemist.com/ccimg/77000/76911-73-4.png)
![7,12-DIHYDROXY-9-METHOXY-2,2-DIMETHYL-10-(3-METHYL-2-BUTEN-1-YL)-<WBR />2H,6H-PYRANO[3,2-B]XANTHEN-6-ONE](http://img.cochemist.com/ccimg/77000/76911-73-4_b.png)


















![5-Ethylbenzo[d][1,3]dioxole](http://img.cochemist.com/ccimg/60400/60373-70-8.png)
![5-Ethylbenzo[d][1,3]dioxole](http://img.cochemist.com/ccimg/60400/60373-70-8_b.png)


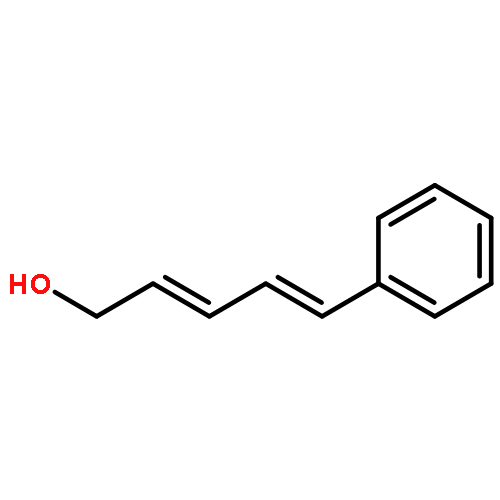


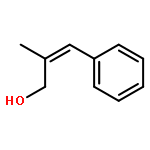
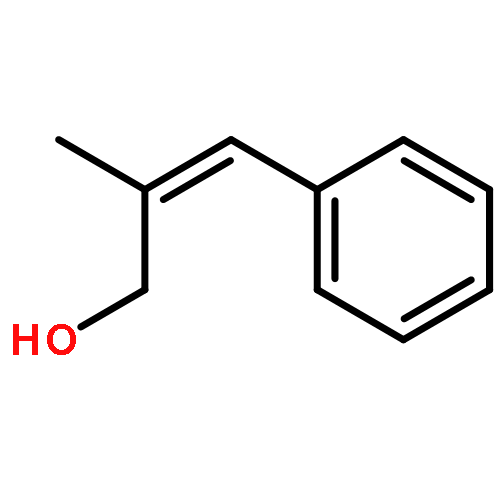

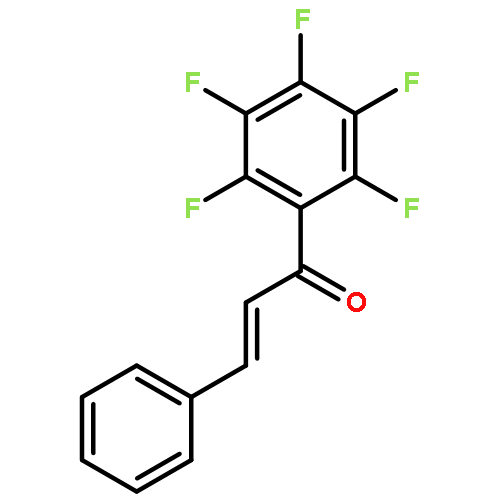


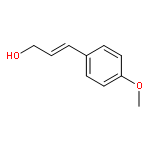
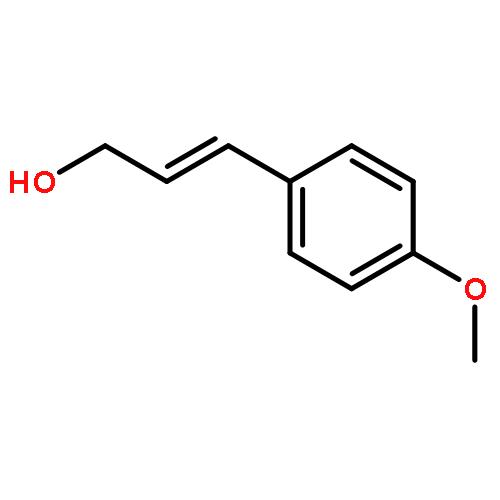


![Cyclohexanamine, N-[1-(4-methoxyphenyl)ethylidene]-](http://img.cochemist.com/ccimg/41900/41801-73-4.png)
![Cyclohexanamine, N-[1-(4-methoxyphenyl)ethylidene]-](http://img.cochemist.com/ccimg/41900/41801-73-4_b.png)
![Cyclohexanamine, N-[1-(4-methylphenyl)ethylidene]-](http://img.cochemist.com/ccimg/41900/41801-69-8.png)
![Cyclohexanamine, N-[1-(4-methylphenyl)ethylidene]-](http://img.cochemist.com/ccimg/41900/41801-69-8_b.png)




![Benzene, [(1E)-3-phenoxy-1-propenyl]-](http://img.cochemist.com/ccimg/37500/37464-41-8.png)
![Benzene, [(1E)-3-phenoxy-1-propenyl]-](http://img.cochemist.com/ccimg/37500/37464-41-8_b.png)
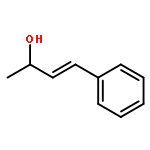
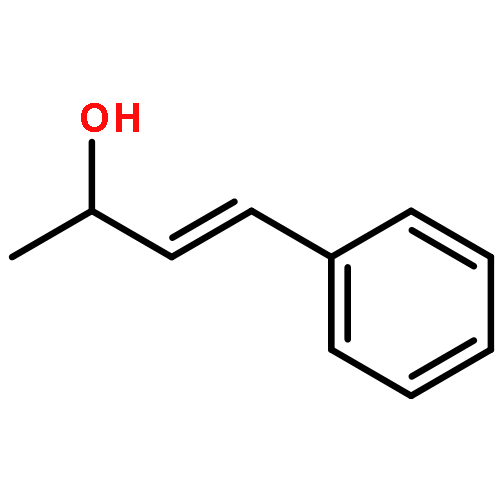
































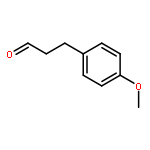
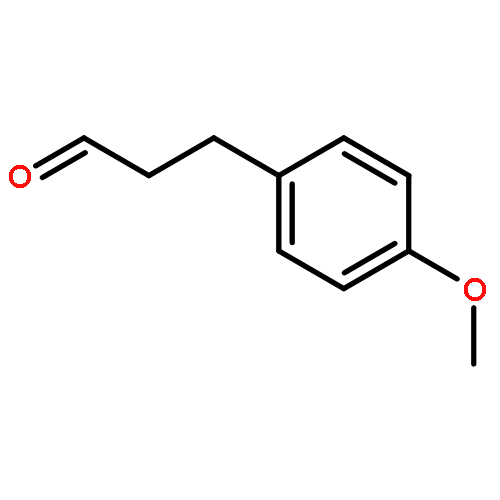

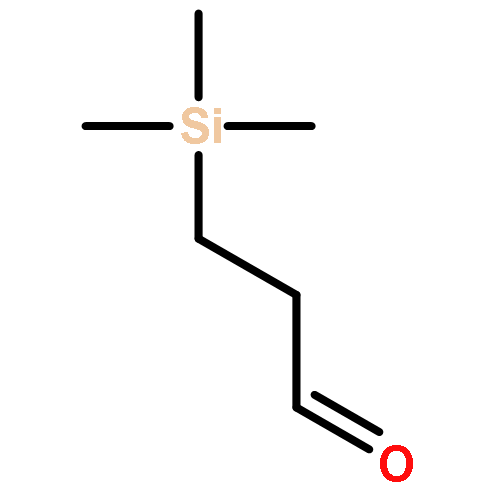




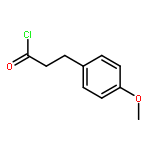
![Butanoic acid, 3-oxo-2-[(trimethylsilyl)methyl]-, ethyl ester](/data/chemimg/2152500/17906-77-3.png)
![Butanoic acid, 3-oxo-2-[(trimethylsilyl)methyl]-, ethyl ester](/data/chemimg/2152500/17906-77-3_b.png)












![Benzene, [3-(1-methylethoxy)-1-propenyl]-, (E)-](/data/chemimg/1724800/14289-95-3.png)
![Benzene, [3-(1-methylethoxy)-1-propenyl]-, (E)-](/data/chemimg/1724800/14289-95-3_b.png)






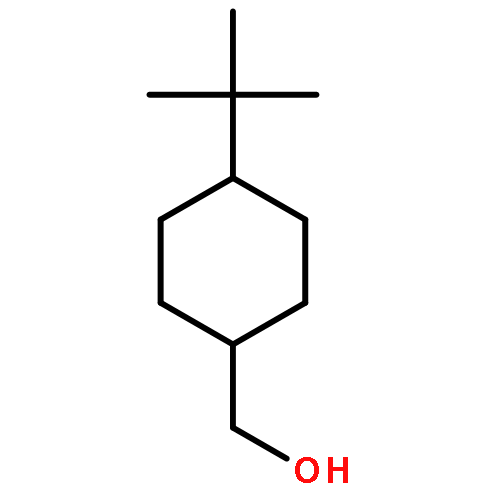
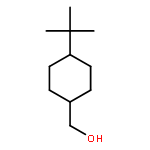
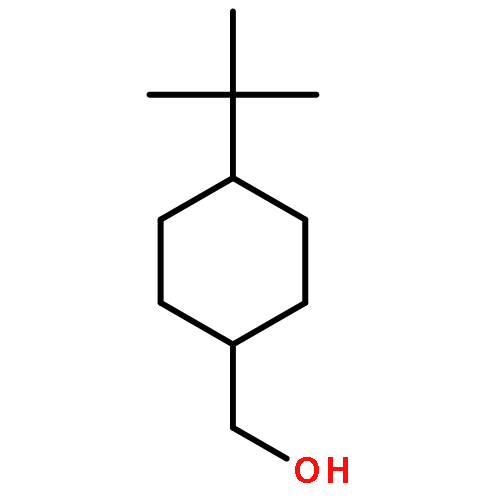
![Chromium,tricarbonyl[(1,2,3,4,5,6-h)-methylbenzene]-](http://img.cochemist.com/ccimg/12100/12083-24-8.png)
![Chromium,tricarbonyl[(1,2,3,4,5,6-h)-methylbenzene]-](http://img.cochemist.com/ccimg/12100/12083-24-8_b.png)












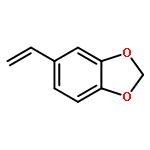






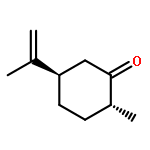
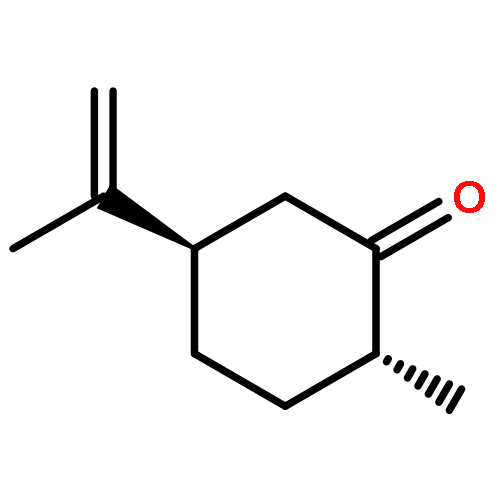


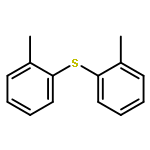
![BENZENE, [(1E)-3-ETHOXY-1-PROPENYL]-](http://img.cochemist.com/ccimg/5900/5882-84-8.png)
![BENZENE, [(1E)-3-ETHOXY-1-PROPENYL]-](http://img.cochemist.com/ccimg/5900/5882-84-8_b.png)








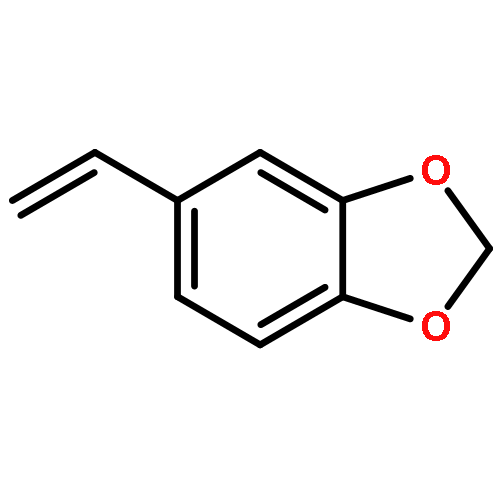
![Benzo[d][1,3]dioxole-5-carbonitrile](http://img.cochemist.com/ccimg/4500/4421-09-4.png)
![Benzo[d][1,3]dioxole-5-carbonitrile](http://img.cochemist.com/ccimg/4500/4421-09-4_b.png)


![Benzene, [(1,1-dimethylethoxy)methyl]-](http://img.cochemist.com/ccimg/3500/3459-80-1.png)
![Benzene, [(1,1-dimethylethoxy)methyl]-](http://img.cochemist.com/ccimg/3500/3459-80-1_b.png)


















































![Benzene, 1-(1,1-dimethylethyl)-4-[1-(trifluoromethyl)ethenyl]-](http://img.cochemist.com/ccimg/185200/185140-55-0.png)
![Benzene, 1-(1,1-dimethylethyl)-4-[1-(trifluoromethyl)ethenyl]-](http://img.cochemist.com/ccimg/185200/185140-55-0_b.png)


![Naphthalene, 1-[1-(trifluoromethyl)ethenyl]-](/data/chemimg/501200/136476-28-3.png)
![Naphthalene, 1-[1-(trifluoromethyl)ethenyl]-](/data/chemimg/501200/136476-28-3_b.png)










![BENZENE, 1-METHOXY-4-[1-(TRIFLUOROMETHYL)ETHENYL]-](http://img.cochemist.com/ccimg/69900/69843-08-9.png)
![BENZENE, 1-METHOXY-4-[1-(TRIFLUOROMETHYL)ETHENYL]-](http://img.cochemist.com/ccimg/69900/69843-08-9_b.png)








![Benzene, [(1E)-3-methoxy-1-propen-1-yl]-](/data/chemimg/1798100/22688-03-5.png)
![Benzene, [(1E)-3-methoxy-1-propen-1-yl]-](/data/chemimg/1798100/22688-03-5_b.png)