Co-reporter:Atanas K. Tomov, James D. Nobbs, Juan J. Chirinos, Prabhjot K. Saini, Robert Malinowski, Sarah K. Y. Ho, Craig T. Young, David S. McGuinness, Andrew J. P. White, Mark R. J. Elsegood, and George J. P. Britovsek
Organometallics February 13, 2017 Volume 36(Issue 3) pp:
Publication Date(Web):November 16, 2016
DOI:10.1021/acs.organomet.6b00671
The catalytic oligomerization of ethylene with chromium-based complexes containing bis(benzimidazolemethyl)amine (BIMA) ligands results in alternating distributions of linear α-olefins (LAOs). Extremely high activities are obtained (>100 000 g mmol–1 h–1 bar–1) with N-alkyl-substituted BIMA ligands, whereas bulky groups on the central nitrogen or alternative central donors result in much lower activities. Variations in the ligand backbone, as well as methylation of the benzimidazole units, lead to reduction in activity. The alternating LAO distributions have been mathematically analyzed using second-order recurrence relations. The shape of the distributions is affected by ethylene pressure (1–4 bar) and by the cocatalyst to some degree. On the basis of the results and analysis presented herein, we propose that the alternating behavior originates from the ability of these chromium BIMA catalysts to undergo single as well as double ethylene insertion reactions. A minor second distribution (<5 wt %) of 2-ethyl-1-alkenes is obtained under certain conditions, resulting from incorporation of 1-butene. DFT studies (M06L) and experimental observations regarding the reaction between AlMe3 and the N-methyl BIMA ligand 2 have shown that deprotonation of the benzimidazole N–H units can occur, which suggests a change in coordination of the BIMA ligand under oligomerization conditions.
Co-reporter:George J. P. Britovsek, David S. McGuinness, Tanita S. Wierenga, and Craig T. Young
ACS Catalysis 2015 Volume 5(Issue 7) pp:4152
Publication Date(Web):May 28, 2015
DOI:10.1021/acscatal.5b00989
The mechanism of ethylene trimerization and tetramerization with a chromium–diphosphinoamine (Cr–PNP) catalyst system has been studied with combined experimental and theoretical methods. Of the total product output, 1-octene, cyclopentanes, n-alkanes, and higher (C10+) olefins are formed with a fractional (∼1.4) order response to ethylene concentration, whereas 1-hexene formation is approximately first-order in ethylene. Theoretical studies suggest a mechanism involving a cationic monometallic catalyst in Cr(I) and Cr(III) formal oxidation states. A key feature of the developed model is the occurrence of a double-coordination mechanism in which a bis(ethylene) chromacyclopentane intermediate is responsible for 1-octene formation as well as the other coproducts that have a greater than first-order response to ethylene. In contrast, 1-hexene is formed primarily from a mono(ethylene) chromacyclopentane intermediate. The selectivity of catalysis is governed by the competition between single- and double-coordination pathways. The mechanistic model developed displays excellent correlation with experimental observations and is able to fully explain the formation of all products generated with this catalyst.Keywords: catalysis; chromium; oligomerization; reaction mechanism; tetramerization; trimerization
Co-reporter:George J. P. Britovsek, Robert Malinowski, David S. McGuinness, James D. Nobbs, Atanas K. Tomov, Andrew W. Wadsley, and Craig T. Young
ACS Catalysis 2015 Volume 5(Issue 11) pp:6922
Publication Date(Web):October 7, 2015
DOI:10.1021/acscatal.5b02203
The oligomerization of ethylene produces α-olefin distributions ranging from Schulz–Flory distributions to alternating and selective oligomer distributions that can be mathematically analyzed and characterized by recurrence relations.Keywords: catalysis; distributions; ethylene; oligomerization; recurrence; tetramerization; trimerization; α-olefins
Co-reporter:Allan R. Petersen ; Russell A. Taylor ; Inmaculada Vicente-Hernández ; Philip R. Mallender ; Harriet Olley ; Andrew J. P. White
Journal of the American Chemical Society 2014 Volume 136(Issue 40) pp:14089-14099
Publication Date(Web):September 8, 2014
DOI:10.1021/ja5055143
Platinum(II) and palladium(II) complexes [M(CH3)(L)]SbF6 with substituted terpyridine ligands L undergo light-driven oxygen insertion reactions into metal methyl bonds resulting in methylperoxo complexes [M(OOCH3)(L)]SbF6. The oxygen insertion reactions occur readily for complexes with methyl ligands that are activated due to steric interaction with substituents (NH2, NHMe or CH3) at the 6,6″-positions on the terpyridine ligand. All complexes exhibit attractive intermolecular π···π or M···M interactions in the solid state and in solution, which lead to excited triplet dinuclear M–M complexes upon irradiation. A mechanism is proposed whereby a dinuclear intermediate is generated upon irradiation that has a weakened M–C bond in the excited state, resulting in the observed oxygen insertion reactions.
Co-reporter:Michaela Grau, Francesco Rigodanza, Andrew J. P. White, Antonio Sorarù, Mauro Carraro, Marcella Bonchio and George J. P. Britovsek
Chemical Communications 2014 vol. 50(Issue 35) pp:4607-4609
Publication Date(Web):12 Mar 2014
DOI:10.1039/C4CC00758A
A bio-inspired manganese(II) complex with a linear pentadentate ligand framework containing soft sulfur donors and an alternating NSNSN binding motif displays excellent dual CAT/SOD-like antioxidant activity with high turnover efficiency and good operation stability in an aqueous environment.
Co-reporter:Michaela Grau, Andrew Kyriacou, Fernando Cabedo Martinez, Irene M. de Wispelaere, Andrew J. P. White and George J. P. Britovsek
Dalton Transactions 2014 vol. 43(Issue 45) pp:17108-17119
Publication Date(Web):01 Oct 2014
DOI:10.1039/C4DT02067G
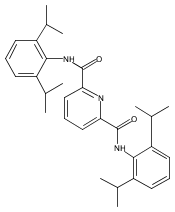
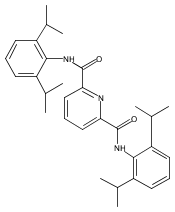
A series of potentially tetradentate and pentadentate ligands modelled on BPMEN has been prepared and their iron(II) bis(triflate) complexes have been isolated and characterised by spectroscopic and crystallographic techniques (BPMEN = N,N′-bis(pyridylmethyl)ethylenediamine). Changes to the BPMEN ligand have invariably led to complexes with different coordination modes or geometries and with inferior catalytic efficiencies for the oxidation of cyclohexane with H2O2. The reaction of an iron(II) complex containing a pentadentate BPMEN-type ligand with O2 has resulted in ligand degradation via oxidative N-dealkylation and the isolation of a bis(hydroxo)-bridged dinuclear iron(III) complex with a picolinate-type ligand.
Co-reporter:Allan R. Petersen, Russell A. Taylor, Inmaculada Vicente-Hernández, Jasmin Heinzer, Andrew J. P. White, and George J. P. Britovsek
Organometallics 2014 Volume 33(Issue 6) pp:1453-1461
Publication Date(Web):March 13, 2014
DOI:10.1021/om500048t
Square-planar palladium(II) and platinum(II) methyl complexes with terpyridine and 6,6″-diamino terpyridine ligands undergo methyl exchange reactions upon exposure to light. The half-life of the methyl exchange reactions correlates with the relative Pd–C and Pt–C bond strengths. No exchange between methyl and phenyl groups is observed, probably due to the stronger Pt–C bond in the platinum phenyl complex. A mechanism is proposed whereby a dinuclear intermediate is generated upon irradiation that has a weakened M–C bond in the excited state, resulting in the observed methyl exchange reactions.
Co-reporter:Christopher J. Whiteoak, James D. Nobbs, Evgeny Kiryushchenkov, Sandro Pagano, Andrew J.P. White, and George J.P. Britovsek
Inorganic Chemistry 2013 Volume 52(Issue 12) pp:7000-7009
Publication Date(Web):May 23, 2013
DOI:10.1021/ic4005196
Tri(pyridylmethyl)phosphine (TPPh), the remarkably elusive congener of tri(pyridylmethyl)amine (TPA), has been prepared, as well as the relative tri(N-methyl-pyridylamino)phosphine (TPAMP). The coordination properties of these new ligands have been evaluated for chromium(III), iron(II), and ruthenium(II) complexes and compared with the related TPA complexes. In all cases, a different coordination behavior has been observed whereby TPPh and TPAMP always act as tridentate ligands. A chromium(III) complex [Cr(TPPh)Cl3] has been prepared, which has shown low ethylene oligomerization activity. Octahedral low spin iron(II) complexes [Fe(TPPh)2]2+ and [Fe(TPAMP)2]2+ were obtained with two ligands bound to the metal center. Ruthenium(II) chloro complexes of TPA and TPPh undergo ligand exchange reactions in acetonitrile, and the ruthenium(II) complex [Ru(MeCN)2(TPA)]2+ can be oxidized by m-CPBA in acetonitrile to give a transient ruthenium(IV) oxo complex [Ru(O)(MeCN)(TPA)]2+. Attempts to generate high valent ruthenium(IV) oxo TPPh or TPAMP complexes could not be achieved, probably due to insufficient stabilization by these strong field ligands.
Co-reporter:Michaela Grau ; Jason England ; Rafael Torres Martin de Rosales ; Henry S. Rzepa ; Andrew J. P. White
Inorganic Chemistry 2013 Volume 52(Issue 20) pp:11867-11874
Publication Date(Web):October 10, 2013
DOI:10.1021/ic401416h
Octahedral, tetrahedral, and square planar geometries are the most often encountered coordination geometries for transition metal complexes. In certain cases, coordination equilibria can exist between different geometries, such as between six- and four-coordinate geometries in nickel(II) complexes, which were discovered half a century ago. Here, we present the first examples of a seven-five coordination equilibrium. Extensive spectroscopic studies in solution have provided evidence for a dynamic equilibrium between two iron(II) complexes, one with a seven-coordinate pentagonal bipyramidal geometry and one with a five-coordinate trigonal bipyramidal geometry.
Co-reporter:Emma Wong, Jonathan Jeck, Michaela Grau, Andrew J. P. White and George J. P. Britovsek
Catalysis Science & Technology 2013 vol. 3(Issue 4) pp:1116-1122
Publication Date(Web):11 Jan 2013
DOI:10.1039/C3CY20823K
The pentadentate ligand bis(pyridylmethyl)(bipyridylmethyl)amine BPAbipy 1 and the iron(II) complex [Fe(1)(CH3CN)](ClO4)2 have been prepared. The strong field ligand 1 stabilises the low spin iron(II) complex, resulting in a high redox potential of 1.01 V for the Fe(II)/Fe(III) redox couple (vs. SCE). High valent iron complexes are destabilised by strong field ligands and the reaction of [Fe(1)(CH3CN)](ClO4)2 with PhIO is believed to result in a short-lived iron(IV) oxo complex, which is rapidly reduced to the oxo-bridged dinuclear iron(III) complex [(Fe(1))2(μ-O)](ClO4)4. The catalytic properties of the iron(II) complex [Fe(1)(CH3CN)](ClO4)2 for the oxidation of cyclohexane with various amounts of H2O2 has been evaluated. Moderate catalytic activities, comparable to other iron(II) complexes featuring pentadentate ligands but significantly lower than the tetradentate BPMEN ligand, have been observed.
Co-reporter:Dr. Timur Coskun;Dr. Christopher M. Conifer;Laura C. Stevenson ;Dr. George J. P. Britovsek
Chemistry - A European Journal 2013 Volume 19( Issue 21) pp:6840-6844
Publication Date(Web):
DOI:10.1002/chem.201203069
Abstract
Glycerol is converted to a mixture of butyric and isobutyric acid by rhodium- or iridium-catalysed carbonylation using HI as the co-catalyst. The initial reaction of glycerol with HI results in several intermediates that lead to isopropyl iodide, which upon carbonylation forms butyric and isobutyric acid. At low HI concentration, the intermediate allyl iodide undergoes carbonylation to give vinyl acetic acid and crotonic acid. Higher polyols CnHn+2(OH)n are carbonylated to the corresponding Cn+1 mono-carboxylic acids.
Co-reporter:Theo M. Smit, Atanas K. Tomov, George J. P. Britovsek, Vernon C. Gibson, Andrew J. P. White and David J. Williams
Catalysis Science & Technology 2012 vol. 2(Issue 3) pp:643-655
Publication Date(Web):19 Dec 2011
DOI:10.1039/C2CY00448H
The synthesis, characterization and ethylene polymerisation behaviour of a series of first row transition metal complexes of the general formula LMXn (M = Fe, Co, Mn, n = 2, X = Cl; M = V, Cr, Ti, n = 3, X = Cl; M = Ni, n = 2, X = Br) with bis(imino)pyridine ligands L are reported, whereby the ligands contain heteroatom substituents DRm at the imine carbon (D = O, S, m = 1, R = Me, Ph, 2,6-Me2C6H3 or D = N, m = 2, R = Me, Ph). Only the O- and S-substituted complexes show catalytic activity for the polymerisation of ethylene, upon activation with methylaluminoxane (MAO). The Mn- and Ni-based catalysts were found to be inactive under these conditions. The S-substituted complexes are generally more active than the O-substituted complexes and the catalytic activities increase with the size of the substituents. Iron-, vanadium- and chromium-based catalysts give highly active catalyst systems, which in some cases are more active than the well-known ketimine catalysts. All catalysts produce highly linear polyethylene with molecular weights affected by M, D and R. O-substituted catalyst systems are generally less active and produce lower molecular weight polyethylene compared to S-substituted systems.
Co-reporter:James D. Nobbs, Atanas K. Tomov, Renan Cariou, Vernon C. Gibson, Andrew J. P. White and George J. P. Britovsek
Dalton Transactions 2012 vol. 41(Issue 19) pp:5949-5964
Publication Date(Web):29 Feb 2012
DOI:10.1039/C2DT30324H
A series of bis(thiazolinyl)- and bis(thiazolyl)pyridine Thio-Pybox ligands and their metal complexes of chromium(III), iron(II), cobalt(II) and nickel(II) has been prepared, as well as a nickel(II) complex containing a monoanionic bis(thiazolinyl)phenyl Thio-Phebox ligand. These new metal complexes have been characterised and used as catalysts, in combination with the co-catalyst MAO, for the polymerisation of ethylene and for the polymerisation of butadiene. In the case of ethylene polymerisation, the Thio-Pybox and Thio-Phebox metal complexes have shown relatively low polymerisation activities, much lower compared to the related bis(imino)pyridine complexes of the same metals. In the polymerisation of butadiene, several Thio-Pybox cobalt(II) complexes show very high activities, significantly higher than the other metal complexes with the same ligand. It is the metal, rather than the ligand, that appears to have the most profound effect on the catalytic activity in butadiene polymerisation, unlike in the polymerisation of ethylene, where bis(imino)pyridine ligands provide highly active catalysts for a range of 1st row transition metals.
Co-reporter:Samuel S. Karpiniec, David S. McGuinness, George J. P. Britovsek, Tanita S. Wierenga and Jim Patel
Chemical Communications 2011 vol. 47(Issue 24) pp:6945-6947
Publication Date(Web):20 May 2011
DOI:10.1039/C1CC12169C
A catalyst system composed of a 2,6-bis(arylimino)pyridineiron(II) dichloride complex and methylaluminoxane is found to be extremely active for acetylene polymerisation. The formation of poly(acetylene) gels and surface films occurs at very low catalyst concentrations, around three orders of magnitude lower than traditional catalyst systems.
Co-reporter:Christopher M. Conifer, Russell A. Taylor, David J. Law, Glenn J. Sunley, Andrew J. P. White and George J. P. Britovsek
Dalton Transactions 2011 vol. 40(Issue 5) pp:1031-1033
Publication Date(Web):22 Dec 2010
DOI:10.1039/C0DT01526A
The first square planar rhodium(I) complexes containing the 6,6′-dihydroxy-2,2′-bipyridine ligand have been prepared. The complexes form molecular wires in the solid state and are active catalysts for the carbonylation of methyl acetate.
Co-reporter:Christopher M. Conifer;David J. Law;Glenn J. Sunley;Anthony Haynes;John R. Wells;Andrew J. P. White
European Journal of Inorganic Chemistry 2011 Volume 2011( Issue 23) pp:3511-3522
Publication Date(Web):
DOI:10.1002/ejic.201100423
Abstract
A series of cationic cis-dicarbonylrhodium(I) complexes [Rh(L)(CO)2]SbF6 has been prepared containing 2,2′-bipyridine ligands with proximate H-bonding substituents R in the 6- and 6′-position (R = OH, NH2, COOEt and PO(OEt)2). The solid-state structures have been determined by X-ray crystallography for the complexes where R = OH, NH2 and COOEt. The molecular structures have revealed metal–metal and π–π interactions between the square-planar complexes resulting in the formation of molecular chains in the solid state. IR studies on the reaction of [Rh(bipy)(CO)2]SbF6 with MeI have shown the formation of a neutral complex [RhI(CO)(bipy)], which undergoes oxidative addition of MeI much faster than the cationic complex [Rh(bipy)(CO)2]+. Although all complexes show good activities for the carbonylation of methyl acetate, this is believed to be due to their instability and the formation of [RhI2(CO)2]– under the reaction conditions.
Co-reporter:Christopher M. Conifer, David J. Law, Glenn J. Sunley, Andrew J. P. White, and George J. P. Britovsek
Organometallics 2011 Volume 30(Issue 15) pp:4060-4066
Publication Date(Web):July 18, 2011
DOI:10.1021/om200341t
The application of Lewis acids and Lewis acid-functionalized ligands as activators for methyl acetate in the rhodium-catalyzed carbonylation of methyl acetate to acetic anhydride has been investigated. The reaction of methyl acetate with B(C6F5)3 results in the formation of the adduct [MeOAc·B(C6F5)3]. The combination of this adduct with [Rh(OAc)(CO)2]2 results in a transfer of the Lewis acid to the rhodium complex, rather than an anticipated oxidative addition reaction. In a second approach, novel Lewis acid-functionalized rhodium(I) complexes [Rh(CO)Cl(BPP)], [Rh(CO)2(BPP)]SbF6, and [Rh(MeCN)2(BPP)]SbF6 (BPP = PhB(C6H4PPh2)2) have been prepared. The lack of reactivity of [Rh(CO)2(BPP)]SbF6 toward MeOAc has shown that the rhodium–boron interaction is too strong for activation of methyl acetate, and no carbonylation activity was observed under the conditions used.
Co-reporter:Christopher J. Whiteoak, Rafael Torres Martin de Rosales, Andrew J. P. White, and George J. P. Britovsek
Inorganic Chemistry 2010 Volume 49(Issue 23) pp:11106-11117
Publication Date(Web):November 9, 2010
DOI:10.1021/ic1016998
Tetradentate bis(aminophenolate) ligands H2salanX and H2bapenX (where X refers to the para-phenolate substituent = H, Me, F, Cl) react with [Fe{N(SiMe3)2}2] to form iron(II) complexes, which in the presence of suitable donor ligands L (L = pyridine or THF) can be isolated as the complexes [Fe(salanX)(L)2] and [Fe(bapenX)(L)2]. In the absence of donor ligands, either mononuclear complexes, for example, [Fe(salantBu,tBu)], or dinuclear complexes of the type [Fe(salanX)]2 are obtained. The dynamic coordination behavior in solution of the complexes [Fe(salanF)(L)2] and [Fe(bapenF)(L)2] has been investigated by VT 1H and 19F NMR spectroscopy, which has revealed equilibria between isomers with different ligand coordination topologies cis-α, cis-β and trans. Exposure of the iron(II) salanX complexes to O2 results in the formation of oxo-bridged iron(III) complexes of the type [{Fe(salanX)}2(μ-O)] or [{Fe(salanX)(L)}2(μ-O)]. The lack of catalytic activity of the iron(II) salan and bapen complexes in the oxidation of cyclohexane with H2O2 as the oxidant is attributed to the rapid formation of stable and catalytically inactive oxo-bridged iron(III) complexes.
Co-reporter:Renan Cariou, Juan J. Chirinos, Vernon C. Gibson, Grant Jacobsen, Atanas K. Tomov, George J. P. Britovsek and Andrew J. P. White
Dalton Transactions 2010 vol. 39(Issue 38) pp:9039-9045
Publication Date(Web):20 Aug 2010
DOI:10.1039/C0DT00402B
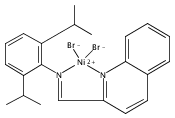
A series of bis(benzimidazole)-based cobalt(II) dichloride complexes containing a range of different central donors has been synthesized and characterized. The nature of the central donor affects the binding of the ligand to the cobalt centre and determines the coordination geometry of the metal complexes. All complexes have been shown to catalyse the polymerization of butadiene, in combination with MAO as the co-catalyst, to give cis-1,4-polybutadiene with high selectivity. The nature of the central donor has a marked influence on the polymerization activity of the catalysts, but does not affect the polymer microstructure. The addition of PPh3 generally increases the polymerization activity of these cobalt catalysts and results in predominantly (60–70%) 1,2-vinyl-polybutadiene.
Co-reporter:Christopher J. Whiteoak, George J. P. Britovsek, Vernon C. Gibson and Andrew J. P. White
Dalton Transactions 2009 (Issue 13) pp:2337-2344
Publication Date(Web):17 Feb 2009
DOI:10.1039/B820754B
A series of molybdenum(VI) cis-dioxo complexes containing tetradentate salan ligands with different para-substitutions on the phenoxy group have been prepared. These complexes catalyse the oxygen atom transfer reaction between dimethylsulfoxide and triphenylphosphine. During oxo transfer catalysis, the complexes are resistant to formation of catalytically inactive oxo-bridged dimeric Mo(V) complexes. Electronic effects influence the rate of the oxo transfer reaction and the fastest rates are achieved when the para-phenoxy substituent is an electron withdrawing nitro substituent. Hammett correlations have shown that the rate-determining step involves nucleophillic attack of the phosphine on one of the oxo ligands. Electrochemical measurements have shown that all complexes containing tertiary amine ligands exhibit quasi-reversible behaviour and that the para-substituent has a considerable effect on the half potentials (E1/2). A linear correlation between the E1/2 values and the Hammett σp parameter is observed.
Co-reporter:Jason England, Reema Gondhia, Laura Bigorra-Lopez, Allan R. Petersen, Andrew J. P. White and George J. P. Britovsek
Dalton Transactions 2009 (Issue 27) pp:5319-5334
Publication Date(Web):19 May 2009
DOI:10.1039/B901390C
A series of non-heme iron(II) bis(triflate) complexes containing linear and tripodal tetradentate ligands has been prepared. Electron withdrawing and electron donating substituents in the para position of the pyridine ligands as well as the effect of pyrazineversuspyridine and sulfur or oxygen donors instead of nitrogen donors have been investigated. The electronic effects induced by these substituents influence the strength of the ligand field. UV-vis spectroscopy and magnetic susceptibility studies have been used to quantify these effects and VT 1H and 19F NMR spectroscopy as well as X-ray diffraction have been used to elucidate structural and geometrical aspects of these complexes. The catalytic properties of the iron(II) complexes as catalysts for the oxidation of cyclohexane with hydrogen peroxide have been evaluated. In the strongly oxidising environment required to oxidise alkanes, catalyst stability determines the overall catalytic efficiency of a given catalyst, which can be related to the ligand field strength and the basicity of the ligand and its propensity to undergo oxidation.
Co-reporter:RussellA. Taylor;DavidJ. Law;GlennJ. Sunley;AndrewJ.P. White;GeorgeJ.P. Britovsek Dr.
Angewandte Chemie 2009 Volume 121( Issue 32) pp:6014-6017
Publication Date(Web):
DOI:10.1002/ange.200806187
Co-reporter:Atanas K. Tomov, Vernon C. Gibson, George J. P. Britovsek, Richard J. Long, Martin van Meurs, David J. Jones, Kilian P. Tellmann and Juan J. Chirinos
Organometallics 2009 Volume 28(Issue 24) pp:7033-7040
Publication Date(Web):December 4, 2009
DOI:10.1021/om900792x
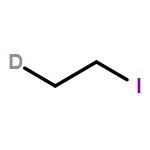
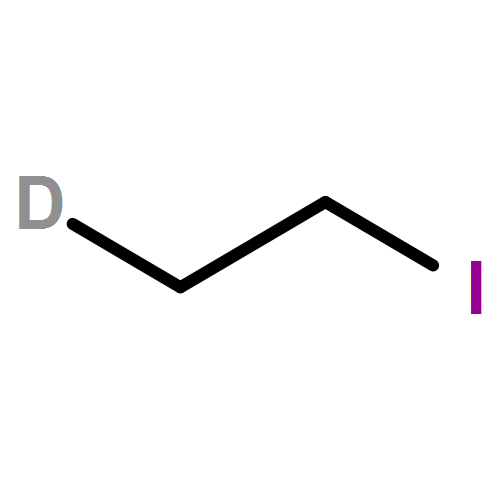
A series of co-oligomerization and copolymerization reactions of C2H4/C2D4 (1:1) mixtures have been carried out using various transition metal catalysts based on Cr, Co, and Fe in combination with MAO. The oligomeric α-olefin products have been analyzed by GC and GC/MS, and the experimental results have been compared with the theoretical mass spectra derived from mathematical models. Solid polymer samples have been analyzed by 13C{1H} and 13C DEPT-135 NMR spectroscopy. C2H4/C2D4 co-oligomerization can be used as a method to differentiate between a metallacyclic or a Cossee-type chain growth mechanism in oligomerization systems. In the case of a metallacyclic mechanism, no H/D scrambling is observed, whereas for a Cossee-type mechanism, similar rates of chain propagation and chain termination (β-H elimination) result in rapid H/D scrambling of the C2H4/C2D4 feed. This method is therefore limited to oligomerization systems and cannot be applied in polymerization systems, where the rate of chain propagation is much faster than the rate of chain termination.
Co-reporter:RussellA. Taylor;DavidJ. Law;GlennJ. Sunley;AndrewJ.P. White;GeorgeJ.P. Britovsek Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 32) pp:5900-5903
Publication Date(Web):
DOI:10.1002/anie.200806187
Co-reporter:Russell A. Taylor, David J. Law, Glenn J. Sunley, Andrew J. P. White and George J. P. Britovsek
Chemical Communications 2008 (Issue 24) pp:2800-2802
Publication Date(Web):11 Apr 2008
DOI:10.1039/B803370F
The use of ligands with proximate hydrogen bonding substituents in the oxidation of platinum(II) dimethyl complexes with H2O2 leads to the exclusive formation of an unusual cis-dihydroxo platinum(IV) complex, which can dehydrate to form a trinuclear metalla-azacrown complex.
Co-reporter:Jason Engl;CatherineR. Davies;Maria Banaru;Andrew J.P. White ;George J.P. Britovsek
Advanced Synthesis & Catalysis 2008 Volume 350( Issue 6) pp:883-897
Publication Date(Web):
DOI:10.1002/adsc.200700462
Abstract
A series of iron(II) bis(triflate) complexes [Fe(L)(OTf)2] containing linear tetradentate bis(pyridylmethyl)diamine ligands with a range of ligand backbones has been prepared. The backbone of the ligand series has been varied from a two-carbon linkage [ethylene (1), 4,5-dichlorophenylene (2) and cyclohexyl (3)] to a three-carbon [propyl (4)) and a four-carbon linkage (butyl (5)]. The coordination geometries of these complexes have been investigated in the solid state by X-ray crystallography and in solution by 1H and 19F NMR spectroscopy. Due to the labile nature of high-spin iron(II) complexes in solution, dynamic equilibria of complexes with different coordination geometries (cis-α, cis-β and trans) are observed with ligands 2–5. In these cases, the geometry observed in the solid state does not necessarily represent the only or even the major geometry present in solution. The ligand field strength in the various complexes has been investigated by variable temperature magnetic moment measurements and UV-vis spectroscopy. The strongest ligand field is observed with the most rigid ligands 1 and 2, which generate complexes [Fe(L)(OTf)2] with a cis-α coordination geometry and the corresponding complexes [Fe(L)(CH3CN)2]2+ display spin crossover behaviour. The catalytic properties of the complexes for the oxidation of cyclohexane, using hydrogen peroxide as the oxidant, have been investigated. An increased flexibility in the ligand results in a weaker ligand field, which increases the lability of the complexes. The activity and selectivity of the catalysts appear to be related to the strength of the ligand field and the stability of the catalyst in the oxidising environment.
Co-reporter:Sondra L. Hellstrom, Juri Ugolotti, George J. P. Britovsek, Tim S. Jones and Andrew J. P. White
New Journal of Chemistry 2008 vol. 32(Issue 8) pp:1379-1387
Publication Date(Web):17 Mar 2008
DOI:10.1039/B712837A
A series of boron quinolinate compounds with different degrees of fluorination {Ph2BQ 1, (4-F-C6H4)2BQ 2 and (C6F5)2BQ 3 (where Q is 8-quinolinate)} have been prepared and their electronic and luminescent behaviour has been investigated in a variety of organic light emitting device structures. Cyclic voltammetry studies have shown a decrease in ionization potential with the degree of fluorination. Electroluminescence (EL) measurements have shown increasingly red-shifted exciplex emission, originating from the different boron compounds interacting with the hole transporting layer. In layered devices, the boron compounds 1–3 are inferior in their EL performance compared to aluminium tris(8-quinolinoate) (AlQ3). However, when the boron compounds 1, 2 or 3 are doped into a 4,4′-bis(carbazol-9-yl)diphenyl (CBP) host, emission solely attributable to 1–3 is observed. In such devices, the boron compounds 1 and 2 outperform AlQ3 as an emitter at low to moderate current densities.
Co-reporter:Juri Ugolotti, Sondra Hellstrom, George J. P. Britovsek, Tim S. Jones, Patricia Hunt and Andrew J. P. White
Dalton Transactions 2007 (Issue 14) pp:1425-1432
Publication Date(Web):28 Feb 2007
DOI:10.1039/B700317J
The reaction of 8-hydroxyquinoline (HQ) with B(C6F5)3 leads to the formation of the zwitterionic compound (C6F5)3BQH (1), involving a proton migration from O to N. Compound 1 can be converted thermally to (C6F5)2BQ (2), which can also be prepared from (C6F5)2BCl and HQ. The reaction of HQ with (C6F5)B(OC6F5)2 generates initially (C6F5)(OC6F5)BQ (3), which easily hydrolyses to give the diboron compound ((C6F5)BQ)2O (4). Compounds 1, 2 and 4 have been fully characterised, including X-ray analysis. The spectroscopic properties of these compounds, including photoluminescence (PL) have been investigated and compared with the non-fluorinated luminescent boron compound (C6H5)2BQ and also with AlQ3. The changes in luminescent behaviour upon fluorination of these boron quinolinate compounds have been rationalised using computational studies.
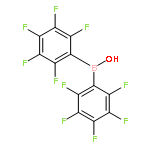
Co-reporter:George J. P. Britovsek, Juri Ugolotti, Patricia Hunt and Andrew J. P. White
Chemical Communications 2006 (Issue 12) pp:1295-1297
Publication Date(Web):10 Feb 2006
DOI:10.1039/B516262A
The combination of Lewis and Brønsted acidity as well as Lewis basicity in (C6F5)2BOH results in a remarkable reactivity towards organonitriles to give novel heterocyclic compounds containing a BOBOCN six-membered ring.
Co-reporter:George J. P. Britovsek, Jason England and Andrew J. P. White
Dalton Transactions 2006 (Issue 11) pp:1399-1408
Publication Date(Web):20 Jan 2006
DOI:10.1039/B513886H
A series of manganese(II), iron(II) and cobalt(II) bis(triflate) complexes containing linear tetradentate bis(imine) and bis(amine) ligands with a biphenyl bridge have been synthesized. The twist in the ligand backbone due to the biphenyl unit leads in the case of the bis(imine) ligands (1 and 2) containing sp2 hybridised N donors, to a distorted cis-α coordination geometry, whereas in the case of the biphenyl- and biphenylether-bridged bis(amine) ligands (7–9 and 12), a trans coordination geometry is observed. The catalytic properties of the complexes for the oxidation of cyclohexane, using H2O2 as the oxidant, have been evaluated. Only the iron complexes show any catalytic activity under the conditions used, but the low conversions and selectivies observed indicate that these catalysts lead predominantly to free radical auto-oxidation.
Co-reporter:George J. P. Britovsek, Jason England, Stefan K. Spitzmesser, Andrew J. P. White and David J. Williams
Dalton Transactions 2005 (Issue 5) pp:945-955
Publication Date(Web):04 Feb 2005
DOI:10.1039/B414813D
A series of Fe(II), Mn(II), Co(II) and Ru(II) complexes containing bis(imino)pyridine or bis(amino)pyridine ligands and weakly coordinating triflate (OTf−) or non-coordinating SbF6− anions have been prepared. The complexes have been fully characterized including several solid-state structure analyses. Two unusual mono-chelate six-coordinate bis(imino)pyridine Fe(II) and Mn(II) complexes have been observed. The catalytic properties of the complexes for the oxidation of cyclohexane with H2O2 have been evaluated. Only the Fe(II) complexes have shown catalytic activity, which is mainly due to Fenton-type free radical auto-oxidation.
Co-reporter:Samuel S. Karpiniec, David S. McGuinness, George J.P. Britovsek, Noel W. Davies, Jim Patel
Catalysis Today (15 December 2011) Volume 178(Issue 1) pp:64-71
Publication Date(Web):15 December 2011
DOI:10.1016/j.cattod.2011.07.016
Bis(imino)pyridine complexes of Co and Fe, and diimine complexes of Ni and Pd, have been tested as catalysts for acetylene oligomerisation and polymerisation. While Co, Ni and Pd complexes displayed low activity, an Fe complex led to an extremely active system for acetylene polymerisation after activation with methylaluminoxane (MAO). The product output could be shifted towards short-chain oligomers by addition of the chain transfer reagent ZnEt2, leading to polyene oligomeric chains bound to Zn. Complex branched and cyclic higher oligomers were partially identified, and some possible mechanisms for their formation proposed. The Fe system did not successfully catalyse copolymerisation of acetylene and ethylene, although there was evidence of limited acetylene incorporation into ethylene oligomers.Graphical abstractDownload high-res image (84KB)Download full-size imageHighlights► Imine complexes of Co, Fe, Ni and Pd evaluated for acetylene polym/oligomerisation. ► Fe catalyst is highly active. ► Polyacetylene formed after activation with methylaluminoxane. ► Acetylene oligomers formed in presence of chain transfer agent, ZnEt2. ► Fe catalyst does not copolymerise acetylene with ethylene.
Co-reporter:Samuel S. Karpiniec, David S. McGuinness, George J. P. Britovsek, Tanita S. Wierenga and Jim Patel
Chemical Communications 2011 - vol. 47(Issue 24) pp:NaN6947-6947
Publication Date(Web):2011/05/20
DOI:10.1039/C1CC12169C
A catalyst system composed of a 2,6-bis(arylimino)pyridineiron(II) dichloride complex and methylaluminoxane is found to be extremely active for acetylene polymerisation. The formation of poly(acetylene) gels and surface films occurs at very low catalyst concentrations, around three orders of magnitude lower than traditional catalyst systems.
Co-reporter:Michaela Grau, Andrew Kyriacou, Fernando Cabedo Martinez, Irene M. de Wispelaere, Andrew J. P. White and George J. P. Britovsek
Dalton Transactions 2014 - vol. 43(Issue 45) pp:NaN17119-17119
Publication Date(Web):2014/10/01
DOI:10.1039/C4DT02067G
A series of potentially tetradentate and pentadentate ligands modelled on BPMEN has been prepared and their iron(II) bis(triflate) complexes have been isolated and characterised by spectroscopic and crystallographic techniques (BPMEN = N,N′-bis(pyridylmethyl)ethylenediamine). Changes to the BPMEN ligand have invariably led to complexes with different coordination modes or geometries and with inferior catalytic efficiencies for the oxidation of cyclohexane with H2O2. The reaction of an iron(II) complex containing a pentadentate BPMEN-type ligand with O2 has resulted in ligand degradation via oxidative N-dealkylation and the isolation of a bis(hydroxo)-bridged dinuclear iron(III) complex with a picolinate-type ligand.
Co-reporter:Michaela Grau, Francesco Rigodanza, Andrew J. P. White, Antonio Sorarù, Mauro Carraro, Marcella Bonchio and George J. P. Britovsek
Chemical Communications 2014 - vol. 50(Issue 35) pp:NaN4609-4609
Publication Date(Web):2014/03/12
DOI:10.1039/C4CC00758A
A bio-inspired manganese(II) complex with a linear pentadentate ligand framework containing soft sulfur donors and an alternating NSNSN binding motif displays excellent dual CAT/SOD-like antioxidant activity with high turnover efficiency and good operation stability in an aqueous environment.
Co-reporter:Theo M. Smit, Atanas K. Tomov, George J. P. Britovsek, Vernon C. Gibson, Andrew J. P. White and David J. Williams
Catalysis Science & Technology (2011-Present) 2012 - vol. 2(Issue 3) pp:NaN655-655
Publication Date(Web):2011/12/19
DOI:10.1039/C2CY00448H
The synthesis, characterization and ethylene polymerisation behaviour of a series of first row transition metal complexes of the general formula LMXn (M = Fe, Co, Mn, n = 2, X = Cl; M = V, Cr, Ti, n = 3, X = Cl; M = Ni, n = 2, X = Br) with bis(imino)pyridine ligands L are reported, whereby the ligands contain heteroatom substituents DRm at the imine carbon (D = O, S, m = 1, R = Me, Ph, 2,6-Me2C6H3 or D = N, m = 2, R = Me, Ph). Only the O- and S-substituted complexes show catalytic activity for the polymerisation of ethylene, upon activation with methylaluminoxane (MAO). The Mn- and Ni-based catalysts were found to be inactive under these conditions. The S-substituted complexes are generally more active than the O-substituted complexes and the catalytic activities increase with the size of the substituents. Iron-, vanadium- and chromium-based catalysts give highly active catalyst systems, which in some cases are more active than the well-known ketimine catalysts. All catalysts produce highly linear polyethylene with molecular weights affected by M, D and R. O-substituted catalyst systems are generally less active and produce lower molecular weight polyethylene compared to S-substituted systems.
Co-reporter:Russell A. Taylor, David J. Law, Glenn J. Sunley, Andrew J. P. White and George J. P. Britovsek
Chemical Communications 2008(Issue 24) pp:NaN2802-2802
Publication Date(Web):2008/04/11
DOI:10.1039/B803370F
The use of ligands with proximate hydrogen bonding substituents in the oxidation of platinum(II) dimethyl complexes with H2O2 leads to the exclusive formation of an unusual cis-dihydroxo platinum(IV) complex, which can dehydrate to form a trinuclear metalla-azacrown complex.
Co-reporter:Jason England, Reema Gondhia, Laura Bigorra-Lopez, Allan R. Petersen, Andrew J. P. White and George J. P. Britovsek
Dalton Transactions 2009(Issue 27) pp:NaN5334-5334
Publication Date(Web):2009/05/19
DOI:10.1039/B901390C
A series of non-heme iron(II) bis(triflate) complexes containing linear and tripodal tetradentate ligands has been prepared. Electron withdrawing and electron donating substituents in the para position of the pyridine ligands as well as the effect of pyrazineversuspyridine and sulfur or oxygen donors instead of nitrogen donors have been investigated. The electronic effects induced by these substituents influence the strength of the ligand field. UV-vis spectroscopy and magnetic susceptibility studies have been used to quantify these effects and VT 1H and 19F NMR spectroscopy as well as X-ray diffraction have been used to elucidate structural and geometrical aspects of these complexes. The catalytic properties of the iron(II) complexes as catalysts for the oxidation of cyclohexane with hydrogen peroxide have been evaluated. In the strongly oxidising environment required to oxidise alkanes, catalyst stability determines the overall catalytic efficiency of a given catalyst, which can be related to the ligand field strength and the basicity of the ligand and its propensity to undergo oxidation.
Co-reporter:Christopher J. Whiteoak, George J. P. Britovsek, Vernon C. Gibson and Andrew J. P. White
Dalton Transactions 2009(Issue 13) pp:NaN2344-2344
Publication Date(Web):2009/02/17
DOI:10.1039/B820754B
A series of molybdenum(VI) cis-dioxo complexes containing tetradentate salan ligands with different para-substitutions on the phenoxy group have been prepared. These complexes catalyse the oxygen atom transfer reaction between dimethylsulfoxide and triphenylphosphine. During oxo transfer catalysis, the complexes are resistant to formation of catalytically inactive oxo-bridged dimeric Mo(V) complexes. Electronic effects influence the rate of the oxo transfer reaction and the fastest rates are achieved when the para-phenoxy substituent is an electron withdrawing nitro substituent. Hammett correlations have shown that the rate-determining step involves nucleophillic attack of the phosphine on one of the oxo ligands. Electrochemical measurements have shown that all complexes containing tertiary amine ligands exhibit quasi-reversible behaviour and that the para-substituent has a considerable effect on the half potentials (E1/2). A linear correlation between the E1/2 values and the Hammett σp parameter is observed.
Co-reporter:Juri Ugolotti, Sondra Hellstrom, George J. P. Britovsek, Tim S. Jones, Patricia Hunt and Andrew J. P. White
Dalton Transactions 2007(Issue 14) pp:NaN1432-1432
Publication Date(Web):2007/02/28
DOI:10.1039/B700317J
The reaction of 8-hydroxyquinoline (HQ) with B(C6F5)3 leads to the formation of the zwitterionic compound (C6F5)3BQH (1), involving a proton migration from O to N. Compound 1 can be converted thermally to (C6F5)2BQ (2), which can also be prepared from (C6F5)2BCl and HQ. The reaction of HQ with (C6F5)B(OC6F5)2 generates initially (C6F5)(OC6F5)BQ (3), which easily hydrolyses to give the diboron compound ((C6F5)BQ)2O (4). Compounds 1, 2 and 4 have been fully characterised, including X-ray analysis. The spectroscopic properties of these compounds, including photoluminescence (PL) have been investigated and compared with the non-fluorinated luminescent boron compound (C6H5)2BQ and also with AlQ3. The changes in luminescent behaviour upon fluorination of these boron quinolinate compounds have been rationalised using computational studies.
Co-reporter:George J. P. Britovsek, David S. McGuinness and Atanas K. Tomov
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 23) pp:NaN8241-8241
Publication Date(Web):2016/10/28
DOI:10.1039/C6CY02112C
The mechanism of ethylene trimerisation and tetramerisation with chromium–diphosphinoamine (Cr–PNP) catalysts has been studied by experimental and theoretical (DFT) methods. The effects of a pendant ether donor (ortho-methoxyaryl ligand substitution) and of anion coordination to the active species have been studied. In the former case, coordination of the ether donor to chromium favours 1-hexene by suppressing formation of the bis(ethylene) chromacyclopentane intermediate which is postulated to be the major route to 1-octene. The effect of anion coordination is similar and as the coordination strength increases, displacement of the anion by a second ethylene ligand becomes more difficult, again favouring trimerisation over tetramerisation. Hence, the experimentally observed effects of pendant donor coordination and changes in anion coordination strength can be rationalised.
Co-reporter:Emma Wong, Jonathan Jeck, Michaela Grau, Andrew J. P. White and George J. P. Britovsek
Catalysis Science & Technology (2011-Present) 2013 - vol. 3(Issue 4) pp:NaN1122-1122
Publication Date(Web):2013/01/11
DOI:10.1039/C3CY20823K
The pentadentate ligand bis(pyridylmethyl)(bipyridylmethyl)amine BPAbipy 1 and the iron(II) complex [Fe(1)(CH3CN)](ClO4)2 have been prepared. The strong field ligand 1 stabilises the low spin iron(II) complex, resulting in a high redox potential of 1.01 V for the Fe(II)/Fe(III) redox couple (vs. SCE). High valent iron complexes are destabilised by strong field ligands and the reaction of [Fe(1)(CH3CN)](ClO4)2 with PhIO is believed to result in a short-lived iron(IV) oxo complex, which is rapidly reduced to the oxo-bridged dinuclear iron(III) complex [(Fe(1))2(μ-O)](ClO4)4. The catalytic properties of the iron(II) complex [Fe(1)(CH3CN)](ClO4)2 for the oxidation of cyclohexane with various amounts of H2O2 has been evaluated. Moderate catalytic activities, comparable to other iron(II) complexes featuring pentadentate ligands but significantly lower than the tetradentate BPMEN ligand, have been observed.
Co-reporter:James D. Nobbs, Atanas K. Tomov, Renan Cariou, Vernon C. Gibson, Andrew J. P. White and George J. P. Britovsek
Dalton Transactions 2012 - vol. 41(Issue 19) pp:NaN5964-5964
Publication Date(Web):2012/02/29
DOI:10.1039/C2DT30324H
A series of bis(thiazolinyl)- and bis(thiazolyl)pyridine Thio-Pybox ligands and their metal complexes of chromium(III), iron(II), cobalt(II) and nickel(II) has been prepared, as well as a nickel(II) complex containing a monoanionic bis(thiazolinyl)phenyl Thio-Phebox ligand. These new metal complexes have been characterised and used as catalysts, in combination with the co-catalyst MAO, for the polymerisation of ethylene and for the polymerisation of butadiene. In the case of ethylene polymerisation, the Thio-Pybox and Thio-Phebox metal complexes have shown relatively low polymerisation activities, much lower compared to the related bis(imino)pyridine complexes of the same metals. In the polymerisation of butadiene, several Thio-Pybox cobalt(II) complexes show very high activities, significantly higher than the other metal complexes with the same ligand. It is the metal, rather than the ligand, that appears to have the most profound effect on the catalytic activity in butadiene polymerisation, unlike in the polymerisation of ethylene, where bis(imino)pyridine ligands provide highly active catalysts for a range of 1st row transition metals.
Co-reporter:Christopher M. Conifer, Russell A. Taylor, David J. Law, Glenn J. Sunley, Andrew J. P. White and George J. P. Britovsek
Dalton Transactions 2011 - vol. 40(Issue 5) pp:NaN1033-1033
Publication Date(Web):2010/12/22
DOI:10.1039/C0DT01526A
The first square planar rhodium(I) complexes containing the 6,6′-dihydroxy-2,2′-bipyridine ligand have been prepared. The complexes form molecular wires in the solid state and are active catalysts for the carbonylation of methyl acetate.
Co-reporter:Renan Cariou, Juan J. Chirinos, Vernon C. Gibson, Grant Jacobsen, Atanas K. Tomov, George J. P. Britovsek and Andrew J. P. White
Dalton Transactions 2010 - vol. 39(Issue 38) pp:NaN9045-9045
Publication Date(Web):2010/08/20
DOI:10.1039/C0DT00402B
A series of bis(benzimidazole)-based cobalt(II) dichloride complexes containing a range of different central donors has been synthesized and characterized. The nature of the central donor affects the binding of the ligand to the cobalt centre and determines the coordination geometry of the metal complexes. All complexes have been shown to catalyse the polymerization of butadiene, in combination with MAO as the co-catalyst, to give cis-1,4-polybutadiene with high selectivity. The nature of the central donor has a marked influence on the polymerization activity of the catalysts, but does not affect the polymer microstructure. The addition of PPh3 generally increases the polymerization activity of these cobalt catalysts and results in predominantly (60–70%) 1,2-vinyl-polybutadiene.
Co-reporter:Allan R. Petersen, Andrew J. P. White and George J. P. Britovsek
Dalton Transactions 2016 - vol. 45(Issue 37) pp:NaN14523-14523
Publication Date(Web):2016/05/30
DOI:10.1039/C6DT01691J
The 6,6′′-diaminoterpyridine palladium(II) methylperoxo complex eliminates methyl hydroperoxide and reacts with acetone to form a novel hemi-aminal palladium complex, whereas the analogous platinum(II) complex generates formaldehyde and a platinum(II) hydroxo complex.

![[2,2'-Bipyridin]-6-amine, N,N-dimethyl-](http://img.cochemist.com/ccimg/885700/885622-35-5.png)
![[2,2'-Bipyridin]-6-amine, N,N-dimethyl-](http://img.cochemist.com/ccimg/885700/885622-35-5_b.png)




![[1,1'-Biphenyl]-2,2'-diamine, N,N'-bis(2-pyridinylmethyl)-](http://img.cochemist.com/ccimg/56300/56277-83-9.png)
![[1,1'-Biphenyl]-2,2'-diamine, N,N'-bis(2-pyridinylmethyl)-](http://img.cochemist.com/ccimg/56300/56277-83-9_b.png)
![[1,1'-Biphenyl]-2,2'-diamine, N,N'-bis[(6-methyl-2-pyridinyl)methyl]-](http://img.cochemist.com/ccimg/95100/95045-25-3.png)
![[1,1'-Biphenyl]-2,2'-diamine, N,N'-bis[(6-methyl-2-pyridinyl)methyl]-](http://img.cochemist.com/ccimg/95100/95045-25-3_b.png)
![Lithium, [2-(diphenylphosphino)phenyl]-](http://img.cochemist.com/ccimg/95100/95048-82-1.png)
![Lithium, [2-(diphenylphosphino)phenyl]-](http://img.cochemist.com/ccimg/95100/95048-82-1_b.png)
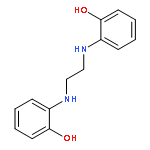
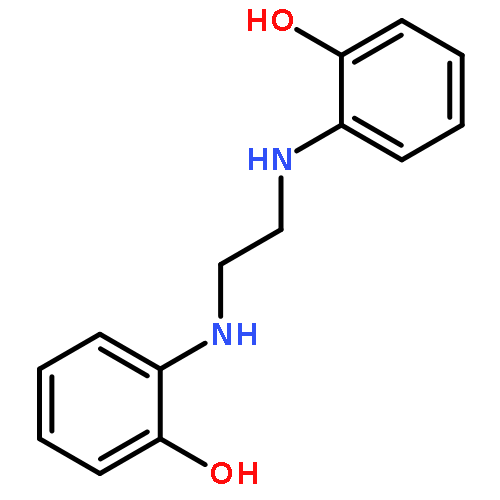
![1H-Benzimidazole, 2,2'-[oxybis(methylene)]bis-](http://img.cochemist.com/ccimg/94700/94665-42-6.png)
![1H-Benzimidazole, 2,2'-[oxybis(methylene)]bis-](http://img.cochemist.com/ccimg/94700/94665-42-6_b.png)


![Pyridine, 2,6-bis[[(2-pyridinylmethyl)thio]methyl]-](http://img.cochemist.com/ccimg/90800/90719-77-0.png)
![Pyridine, 2,6-bis[[(2-pyridinylmethyl)thio]methyl]-](http://img.cochemist.com/ccimg/90800/90719-77-0_b.png)
![[2,2':6',2''-Terpyridin]-6-amine](http://img.cochemist.com/ccimg/445000/444901-58-0.png)
![[2,2':6',2''-Terpyridin]-6-amine](http://img.cochemist.com/ccimg/445000/444901-58-0_b.png)
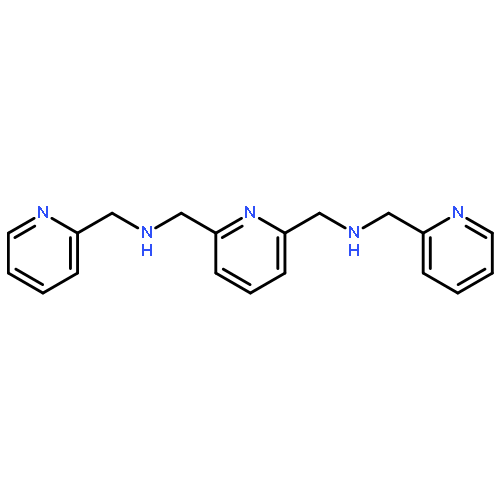



![PYRIDINE, 2,6-BIS[(2-PYRIDINYLMETHOXY)METHYL]-](http://img.cochemist.com/ccimg/90800/90719-76-9.png)
![PYRIDINE, 2,6-BIS[(2-PYRIDINYLMETHOXY)METHYL]-](http://img.cochemist.com/ccimg/90800/90719-76-9_b.png)
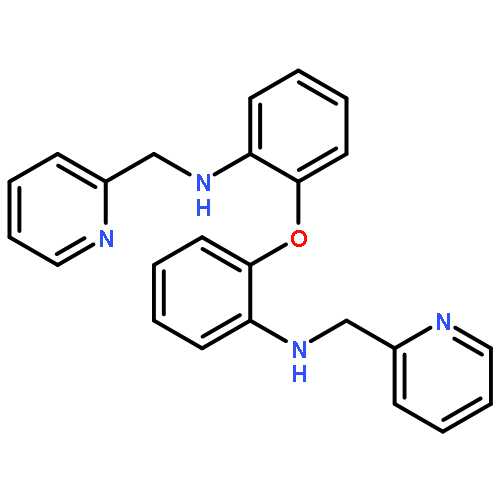


![[1,1'-BIPHENYL]-2,2'-DIAMINE, 6,6'-DIMETHYL-N,N'-BIS(2-PYRIDINYLMETHYL)-](http://img.cochemist.com/ccimg/746700/746642-51-3.png)
![[1,1'-BIPHENYL]-2,2'-DIAMINE, 6,6'-DIMETHYL-N,N'-BIS(2-PYRIDINYLMETHYL)-](http://img.cochemist.com/ccimg/746700/746642-51-3_b.png)
![Phenol, 2,2'-[1,2-ethanediylbis(iminomethylene)]bis[4-methyl-](http://img.cochemist.com/ccimg/151000/150920-59-5.png)
![Phenol, 2,2'-[1,2-ethanediylbis(iminomethylene)]bis[4-methyl-](http://img.cochemist.com/ccimg/151000/150920-59-5_b.png)
![[1,1'-BIPHENYL]-2,2'-DIAMINE, N,N'-DIMETHYL-N,N'-BIS(2-PYRIDINYLMETHYL)-](http://img.cochemist.com/ccimg/884900/884843-27-0.png)
![[1,1'-BIPHENYL]-2,2'-DIAMINE, N,N'-DIMETHYL-N,N'-BIS(2-PYRIDINYLMETHYL)-](http://img.cochemist.com/ccimg/884900/884843-27-0_b.png)
![METHANONE, [6-(PHENYLMETHYL)-1,3,5-TRIAZINE-2,4-DIYL]BIS[PHENYL-](http://img.cochemist.com/ccimg/549500/549494-66-8.png)
![METHANONE, [6-(PHENYLMETHYL)-1,3,5-TRIAZINE-2,4-DIYL]BIS[PHENYL-](http://img.cochemist.com/ccimg/549500/549494-66-8_b.png)


![cis/trans-[PtCl2(SMe2)2]](http://img.cochemist.com/ccimg/17900/17836-09-8.png)
![cis/trans-[PtCl2(SMe2)2]](http://img.cochemist.com/ccimg/17900/17836-09-8_b.png)
![2-[(trimethylsilyl)methyl]-Pyridine](http://img.cochemist.com/ccimg/17900/17881-80-0.png)
![2-[(trimethylsilyl)methyl]-Pyridine](http://img.cochemist.com/ccimg/17900/17881-80-0_b.png)


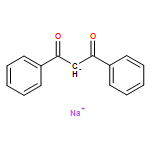
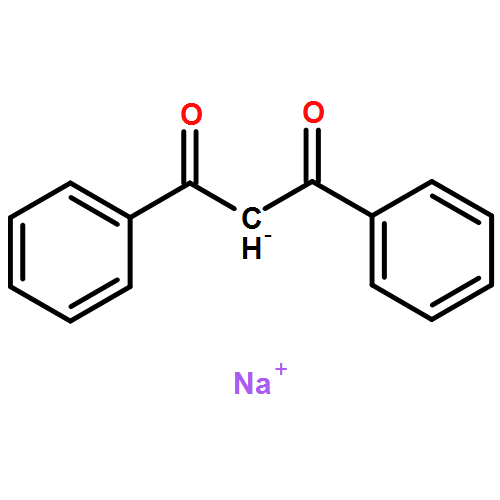
![[2,2'-Bipyridin]-6-amine](/data/chemimg/1757100/178039-84-4.png)
![[2,2'-Bipyridin]-6-amine](/data/chemimg/1757100/178039-84-4_b.png)


![Bis((1H-benzo[d]imidazol-2-yl)methyl)amine](http://img.cochemist.com/ccimg/89600/89505-04-4.png)
![Bis((1H-benzo[d]imidazol-2-yl)methyl)amine](http://img.cochemist.com/ccimg/89600/89505-04-4_b.png)
![1-cyclohexyl-4-[(4-methoxyphenyl)sulfonyl]piperazine](http://img.cochemist.com/ccimg/6000/5999-14-4.png)
![1-cyclohexyl-4-[(4-methoxyphenyl)sulfonyl]piperazine](http://img.cochemist.com/ccimg/6000/5999-14-4_b.png)




![6,6'-Dimethyl-[1,1'-biphenyl]-2,2'-diamine](/data/chemimg/410500/20261-65-8.png)
![6,6'-Dimethyl-[1,1'-biphenyl]-2,2'-diamine](/data/chemimg/410500/20261-65-8_b.png)
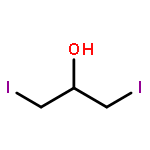

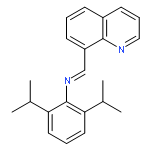
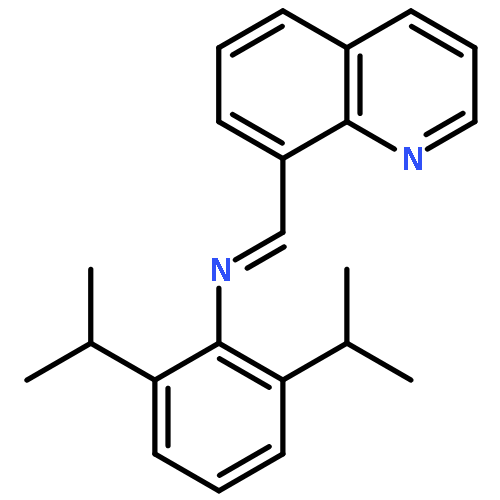
![N-[2,6-DI(PROPAN-2-YL)PHENYL]-1-QUINOLIN-2-YLMETHANIMINE](/data/chemimg/420400/220001-08-1.png)
![N-[2,6-DI(PROPAN-2-YL)PHENYL]-1-QUINOLIN-2-YLMETHANIMINE](/data/chemimg/420400/220001-08-1_b.png)
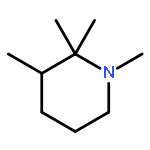
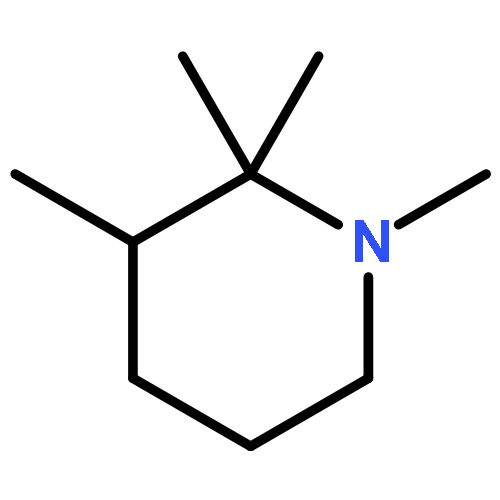


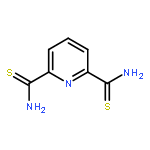
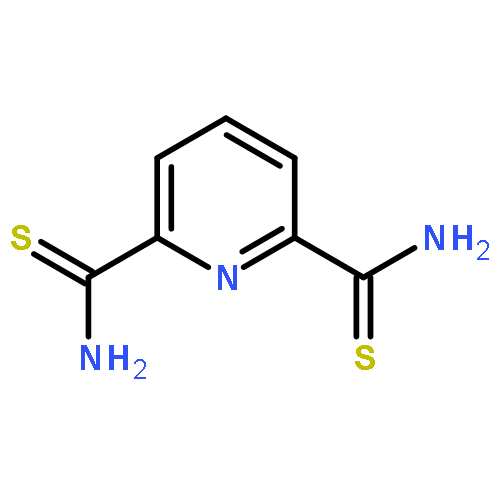
![[2,2':6',2'']TERPYRIDINE-6,6''-DICARBONITRILE](http://img.cochemist.com/ccimg/129100/129077-54-9.png)
![[2,2':6',2'']TERPYRIDINE-6,6''-DICARBONITRILE](http://img.cochemist.com/ccimg/129100/129077-54-9_b.png)
![Pyridine, 2,6-bis[(4S)-4,5-dihydro-4-(1-methylethyl)-2-thiazolyl]-](http://img.cochemist.com/ccimg/776400/776318-56-0.png)
![Pyridine, 2,6-bis[(4S)-4,5-dihydro-4-(1-methylethyl)-2-thiazolyl]-](http://img.cochemist.com/ccimg/776400/776318-56-0_b.png)
![2,6-Pyridinedicarboxamide, N,N'-bis[(1R)-2-hydroxy-1-phenylethyl]-](http://img.cochemist.com/ccimg/661500/661464-60-4.png)
![2,6-Pyridinedicarboxamide, N,N'-bis[(1R)-2-hydroxy-1-phenylethyl]-](http://img.cochemist.com/ccimg/661500/661464-60-4_b.png)












![Phenol, 2,2'-[1,2-ethanediylbis[(methylimino)methylene]]bis-](http://img.cochemist.com/ccimg/121800/121788-87-2.png)
![Phenol, 2,2'-[1,2-ethanediylbis[(methylimino)methylene]]bis-](http://img.cochemist.com/ccimg/121800/121788-87-2_b.png)


![[2,2'-Bipyridine]-6,6'-diamine](http://img.cochemist.com/ccimg/93200/93127-75-4.png)
![[2,2'-Bipyridine]-6,6'-diamine](http://img.cochemist.com/ccimg/93200/93127-75-4_b.png)






















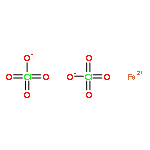
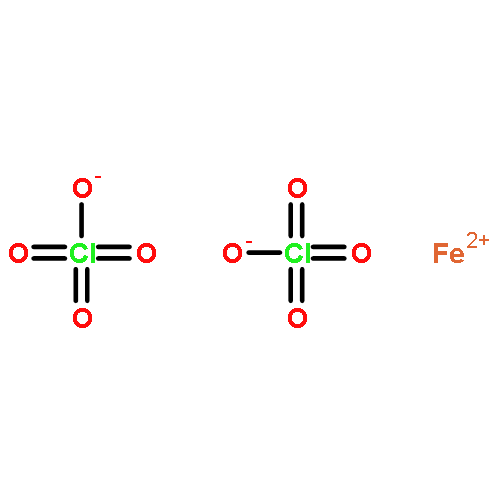


![[2,2'-Bipyridine]-6-carboxaldehyde](/data/chemimg/481100/134296-07-4.png)
![[2,2'-Bipyridine]-6-carboxaldehyde](/data/chemimg/481100/134296-07-4_b.png)


![2,6-Bis[1-(2,6-di-i-propylphenylimino)ethyl]pyridine](/data/chemimg/6300/204203-14-5.png)
![2,6-Bis[1-(2,6-di-i-propylphenylimino)ethyl]pyridine](/data/chemimg/6300/204203-14-5_b.png)




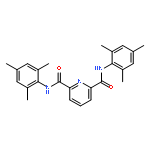
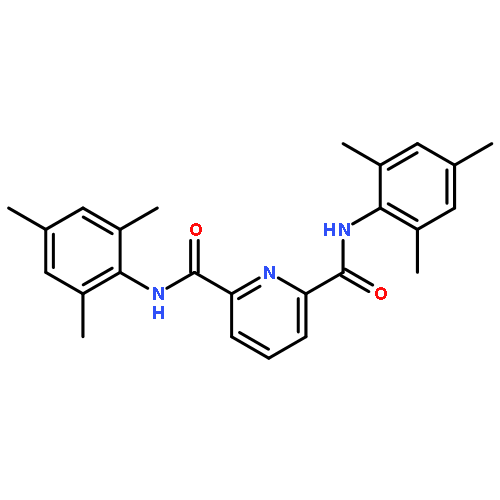
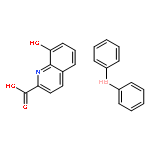





![2,6-BIS[1-(2-METHYLPHENYLIMINO)ETHYL]PYRIDINE](http://img.cochemist.com/ccimg/210600/210537-32-9.png)
![2,6-BIS[1-(2-METHYLPHENYLIMINO)ETHYL]PYRIDINE](http://img.cochemist.com/ccimg/210600/210537-32-9_b.png)
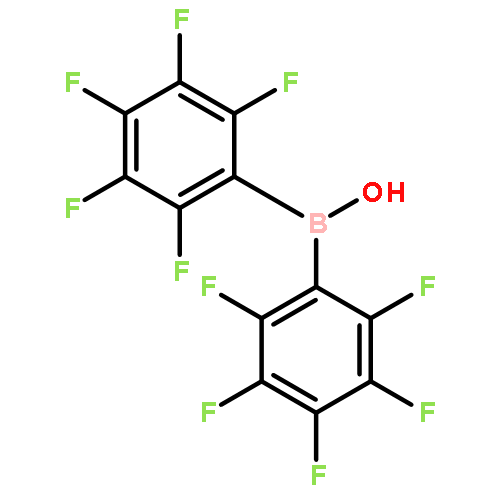
![[2,2'-Bipyridine]-6,6'-dicarboxaldehyde](/data/chemimg/70500/49669-26-3.png)
![[2,2'-Bipyridine]-6,6'-dicarboxaldehyde](/data/chemimg/70500/49669-26-3_b.png)
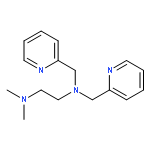
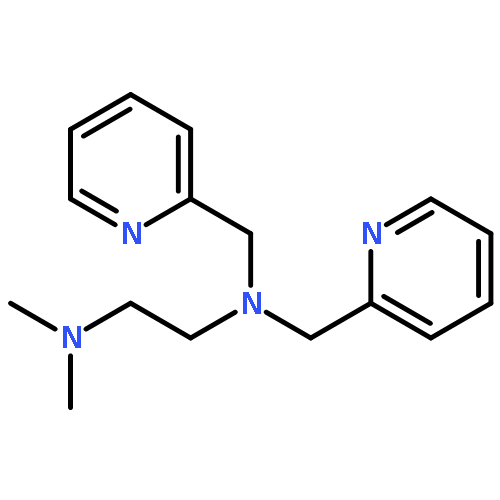


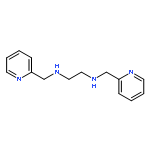

![1,2-Ethanediamine,N2-[2-(dimethylamino)ethyl]-N1,N1-dimethyl-](http://img.cochemist.com/ccimg/40600/40538-81-6.png)
![1,2-Ethanediamine,N2-[2-(dimethylamino)ethyl]-N1,N1-dimethyl-](http://img.cochemist.com/ccimg/40600/40538-81-6_b.png)
























![Ethanone, 1-[2,2'-bipyridin]-6-yl-](/data/chemimg/105500/126770-42-1.png)
![Ethanone, 1-[2,2'-bipyridin]-6-yl-](/data/chemimg/105500/126770-42-1_b.png)