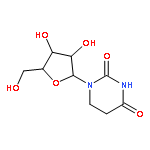
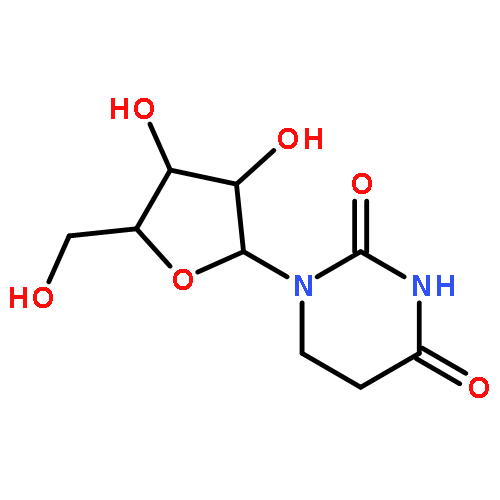
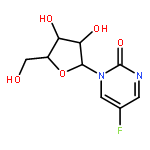
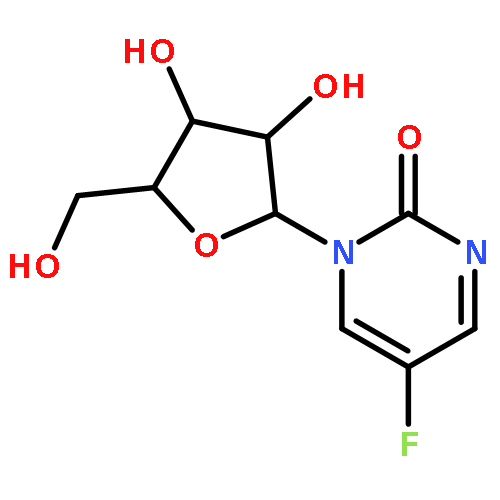
Co-reporter:Bridget C. Larman, Elizabeth A. Dethoff, and Kevin M. Weeks
Biochemistry April 25, 2017 Volume 56(Issue 16) pp:2175-2175
Publication Date(Web):March 23, 2017
DOI:10.1021/acs.biochem.6b01166
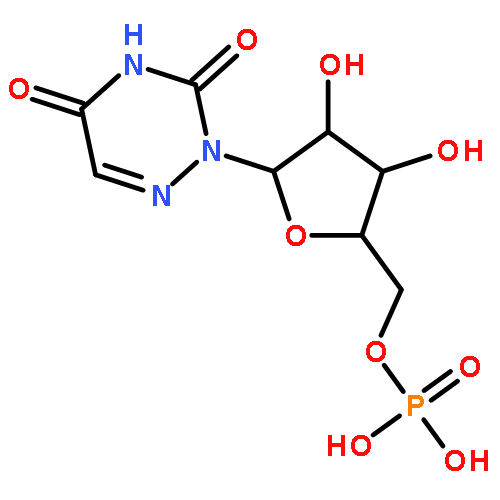
The RNA genomes of viruses likely undergo multiple functionally important conformational changes during their replication cycles, changes that are poorly understood at present. We used two complementary in-solution RNA structure probing strategies (SHAPE-MaP and RING-MaP) to examine the structure of the RNA genome of satellite tobacco mosaic virus inside authentic virions and in a capsid-free state. Both RNA states feature similar three-domain architectures in which each major replicative function—translation, capsid coding, and genome synthesis—fall into distinct domains. There are, however, large conformational differences between the in-virion and capsid-free states, primarily in one arm of the central T domain. These data support a model in which the packaged capsid-bound RNA is constrained in a local high-energy conformation by the native capsid shell. The removal of the viral capsid then allows the RNA genome to relax into a more thermodynamically stable conformation. The RNA architecture of the central T domain thus likely changes during capsid assembly and disassembly and may play a role in genome packaging.
Co-reporter:Matthew J. Smola;Thomas W. Christy;Cindo O. Nicholson;Jack D. Keene;Kaoru Inoue;Matthew Friedersdorf;David M. Lee;J. Mauro Calabrese
PNAS 2016 Volume 113 (Issue 37 ) pp:10322-10327
Publication Date(Web):2016-09-13
DOI:10.1073/pnas.1600008113
The 18-kb Xist long noncoding RNA (lncRNA) is essential for X-chromosome inactivation during female eutherian mammalian development. Global
structural architecture, cell-induced conformational changes, and protein–RNA interactions within Xist are poorly understood. We used selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP)
to examine these features of Xist at single-nucleotide resolution both in living cells and ex vivo. The Xist RNA forms complex well-defined secondary structure domains and the cellular environment strongly modulates the RNA structure,
via motifs spanning one-half of all Xist nucleotides. The Xist RNA structure modulates protein interactions in cells via multiple mechanisms. For example, repeat-containing elements adopt
accessible and dynamic structures that function as landing pads for protein cofactors. Structured RNA motifs create interaction
domains for specific proteins and also sequester other motifs, such that only a subset of potential binding sites forms stable
interactions. This work creates a broad quantitative framework for understanding structure–function interrelationships for
Xist and other lncRNAs in cells.
Co-reporter:Jillian Tyrrell, Kevin M. Weeks, and Gary J. Pielak
Biochemistry 2015 Volume 54(Issue 42) pp:6447-6453
Publication Date(Web):October 2, 2015
DOI:10.1021/acs.biochem.5b00767
There are large differences between the cellular environment and the conditions widely used to study RNA in vitro. SHAPE RNA structure probing in Escherichia coli cells has shown that the cellular environment stabilizes both long-range and local tertiary interactions in the adenine riboswitch aptamer domain. Synthetic crowding agents are widely used to understand the forces that stabilize RNA structure and in efforts to recapitulate the cellular environment under simplified experimental conditions. Here, we studied the structure and ligand binding ability of the adenine riboswitch in the presence of the macromolecular crowding agent, polyethylene glycol (PEG). Ethylene glycol and low-molecular mass PEGs destabilized RNA structure and caused the riboswitch to sample secondary structures different from those observed in simple buffered solutions or in cells. In the presence of larger PEGs, longer-range loop–loop interactions were more similar to those in cells than in buffer alone, consistent with prior work showing that larger PEGs stabilize compact RNA states. Ligand affinity was weakened by low-molecular mass PEGs but increased with high-molecular mass PEGs, indicating that PEG cosolvents exert complex chemical and steric effects on RNA structure. Regardless of polymer size, however, nucleotide-resolution structural characteristics observed in cells were not recapitulated in PEG solutions. Our results reveal that the cellular environment is difficult to recapitulate in vitro; mimicking the cellular state will likely require a combination of crowding agents and other chemical species.
Co-reporter:Matthew J. Smola, J. Mauro Calabrese, and Kevin M. Weeks
Biochemistry 2015 Volume 54(Issue 46) pp:6867-6875
Publication Date(Web):November 6, 2015
DOI:10.1021/acs.biochem.5b00977
SHAPE-MaP is unique among RNA structure probing strategies in that it both measures flexibility at single-nucleotide resolution and quantifies the uncertainties in these measurements. We report a straightforward analytical framework that incorporates these uncertainties to allow detection of RNA structural differences between any two states, and we use it here to detect RNA–protein interactions in healthy mouse trophoblast stem cells. We validate this approach by analysis of three model cytoplasmic and nuclear ribonucleoprotein complexes, in 2 min in-cell probing experiments. In contrast, data produced by alternative in-cell SHAPE probing methods correlate poorly (r = 0.2) with those generated by SHAPE-MaP and do not yield accurate signals for RNA–protein interactions. We then examine RNA–protein and RNA–substrate interactions in the RNase MRP complex and, by comparing in-cell interaction sites with disease-associated mutations, characterize these noncoding mutations in terms of molecular phenotype. Together, these results reveal that SHAPE-MaP can define true interaction sites and infer RNA functions under native cellular conditions with limited preexisting knowledge of the proteins or RNAs involved.
Co-reporter:Kevin M. Weeks
Biopolymers 2015 Volume 103( Issue 8) pp:438-448
Publication Date(Web):
DOI:10.1002/bip.22601
ABSTRACT
Profound insights regarding nucleic acid structure and function can be gleaned from very simple, direct, and chemistry-based strategies. Our approach strives to incorporate the elegant physical insights that Don Crothers instilled in those who trained in his laboratory. Don emphasized the advantages of focusing on direct and concise experiments even when the final objective was to understand something complex—potentially including the large-scale architectures of the genomes of RNA viruses and the transcriptomes of cells. Here, the author reviews the intellectual path, and a few detours, that led to the development of the SHAPE-MaP and RING-MaP technologies for interrogating RNA structure and function at large scales. The author also argues that greater attention to creating direct, less inferential experiments will convert “omics” investigations into lasting and definitive contributions to our understanding of biological function. © 2014 Wiley Periodicals, Inc. Biopolymers 103: 438–448, 2015.
Co-reporter:Michael Golden;Daisuke Yamane;David M. Mauger;Darren P. Martin;Stanley M. Lemon;Sara Williford
PNAS 2015 Volume 112 (Issue 12 ) pp:3692-3697
Publication Date(Web):2015-03-24
DOI:10.1073/pnas.1416266112
Hepatitis C virus (HCV) infects over 170 million people worldwide and is a leading cause of liver disease and cancer. The
virus has a 9,650-nt, single-stranded, messenger-sense RNA genome that is infectious as an independent entity. The RNA genome
has evolved in response to complex selection pressures, including the need to maintain structures that facilitate replication
and to avoid clearance by cell-intrinsic immune processes. Here we used high-throughput, single-nucleotide resolution information
to generate and functionally test data-driven structural models for three diverse HCV RNA genomes. We identified, de novo,
multiple regions of conserved RNA structure, including all previously characterized cis-acting regulatory elements and also multiple novel structures required for optimal viral fitness. Well-defined RNA structures
in the central regions of HCV genomes appear to facilitate persistent infection by masking the genome from RNase L and double-stranded
RNA-induced innate immune sensors. This work shows how structure-first comparative analysis of entire genomes of a pathogenic
RNA virus enables comprehensive and concise identification of regulatory elements and emphasizes the extensive interrelationships
among RNA genome structure, viral biology, and innate immune responses.
Co-reporter:Jennifer L. McGinnis;Qi Liu;Aishwarya Devaraj;Sean P. McClory;Christopher A. Lavender;Kurt Fredrick
PNAS 2015 Volume 112 (Issue 8 ) pp:2425-2430
Publication Date(Web):2015-02-24
DOI:10.1073/pnas.1411514112
It was shown decades ago that purified 30S ribosome subunits readily interconvert between “active” and “inactive” conformations
in a switch that involves changes in the functionally important neck and decoding regions. However, the physiological significance
of this conformational change had remained unknown. In exponentially growing Escherichia coli cells, RNA SHAPE probing revealed that 16S rRNA largely adopts the inactive conformation in stably assembled, mature 30S
subunits and the active conformation in translating (70S) ribosomes. Inactive 30S subunits bind mRNA as efficiently as active
subunits but initiate translation more slowly. Mutations that inhibited interconversion between states compromised translation
in vivo. Binding by the small antibiotic paromomycin induced the inactive-to-active conversion, consistent with a low-energy
barrier between the two states. Despite the small energetic barrier between states, but consistent with slow translation initiation
and a functional role in vivo, interconversion involved large-scale changes in structure in the neck region that likely propagate
across the 30S body via helix 44. These findings suggest the inactive state is a biologically relevant alternate conformation
that regulates ribosome function as a conformational switch.
Co-reporter:Jennifer L. McGinnis and Kevin M. Weeks
Biochemistry 2014 Volume 53(Issue 19) pp:
Publication Date(Web):May 12, 2014
DOI:10.1021/bi500198b
In cells, RNAs likely adopt numerous intermediate conformations prior to formation of functional RNA–protein complexes. We used single-nucleotide resolution selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) to probe the structure of Escherichia coli 16S rRNA in healthy growing bacteria. SHAPE-directed modeling indicated that the predominant steady-state RNA conformational ensemble in dividing cells had a base-paired structure different from that expected on the basis of comparative sequence analysis and high-resolution studies of the 30S ribosomal subunit. We identified the major cause of these differences by stopping ongoing in-cell transcription (in essence, an in-cell RNA structure pulse–chase experiment) which caused the RNA to chase into a structure that closely resembled the expected one. Most helices that formed alternate RNA conformations under growth conditions interact directly with tertiary-binding ribosomal proteins and form a C-shape that surrounds the mRNA channel and decoding site. These in-cell experiments lead to a model in which ribosome assembly factors function as molecular struts to preorganize this intermediate and emphasize that the final stages of ribonucleoprotein assembly involve extensive protein-facilitated RNA conformational changes.
Co-reporter:Justin T. Low, Pablo Garcia-Miranda, Kathryn D. Mouzakis, Robert J. Gorelick, Samuel E. Butcher, and Kevin M. Weeks
Biochemistry 2014 Volume 53(Issue 26) pp:4282-4291
Publication Date(Web):June 13, 2014
DOI:10.1021/bi5004926
The HIV-1 ribosomal frameshift element is highly structured, regulates translation of all virally encoded enzymes, and is a promising therapeutic target. The prior model for this motif contains two helices separated by a three-nucleotide bulge. Modifications to this model were suggested by SHAPE chemical probing of an entire HIV-1 RNA genome. Novel features of the SHAPE-directed model include alternate helical conformations and a larger, more complex structure. These structural elements also support the presence of a secondary frameshift site within the frameshift domain. Here, we use oligonucleotide-directed structure perturbation, probing in the presence of formamide, and in-virion experiments to examine these models. Our data support a model in which the frameshift domain is anchored by a stable helix outside the conventional domain. Less stable helices within the domain can switch from the SHAPE-predicted to the two-helix conformation. Translational frameshifting assays with frameshift domain mutants support a functional role for the interactions predicted by and specific to the SHAPE-directed model. These results reveal that the HIV-1 frameshift domain is a complex, dynamic structure and underscore the importance of analyzing folding in the context of full-length RNAs.
Co-reporter:Philip J. Homan, Arpit Tandon, Greggory M. Rice, Feng Ding, Nikolay V. Dokholyan, and Kevin M. Weeks
Biochemistry 2014 Volume 53(Issue 43) pp:
Publication Date(Web):October 23, 2014
DOI:10.1021/bi501218g
We introduce a melded chemical and computational approach for probing and modeling higher-order intramolecular tertiary interactions in RNA. 2′-Hydroxyl molecular interference (HMX) identifies nucleotides in highly packed regions of an RNA by exploiting the ability of bulky adducts at the 2′-hydroxyl position to disrupt overall RNA structure. HMX was found to be exceptionally selective for quantitative detection of higher-order and tertiary interactions. When incorporated as experimental constraints in discrete molecular dynamics simulations, HMX information yielded accurate three-dimensional models, emphasizing the power of molecular interference to guide RNA tertiary structure analysis and fold refinement. In the case of a large, multidomain RNA, the Tetrahymena group I intron, HMX identified multiple distinct sets of tertiary structure interaction groups in a single, concise experiment.
Co-reporter:Philip J. Homan;Christopher A. Lavender;Olcay Kursun;Xiyuan Ge;Steven Busan;Nikolay V. Dokholyan;Oleg V. Favorov
PNAS 2014 Volume 111 (Issue 38 ) pp:13858-13863
Publication Date(Web):2014-09-23
DOI:10.1073/pnas.1407306111
Complex higher-order RNA structures play critical roles in all facets of gene expression; however, the through-space interaction
networks that define tertiary structures and govern sampling of multiple conformations are poorly understood. Here we describe
single-molecule RNA structure analysis in which multiple sites of chemical modification are identified in single RNA strands
by massively parallel sequencing and then analyzed for correlated and clustered interactions. The strategy thus identifies
RNA interaction groups by mutational profiling (RING-MaP) and makes possible two expansive applications. First, we identify
through-space interactions, create 3D models for RNAs spanning 80–265 nucleotides, and characterize broad classes of intramolecular
interactions that stabilize RNA. Second, we distinguish distinct conformations in solution ensembles and reveal previously
undetected hidden states and large-scale structural reconfigurations that occur in unfolded RNAs relative to native states.
RING-MaP single-molecule nucleic acid structure interrogation enables concise and facile analysis of the global architectures
and multiple conformations that govern function in RNA.
Co-reporter:Jillian Tyrrell, Jennifer L. McGinnis, Kevin M. Weeks, and Gary J. Pielak
Biochemistry 2013 Volume 52(Issue 48) pp:
Publication Date(Web):November 11, 2013
DOI:10.1021/bi401207q
There are large differences between the intracellular environment and the conditions widely used to study RNA structure and function in vitro. To assess the effects of the crowded cellular environment on RNA, we examined the structure and ligand binding function of the adenine riboswitch aptamer domain in healthy, growing Escherichia coli cells at single-nucleotide resolution on the minute time scale using SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension). The ligand-bound aptamer structure is essentially the same in cells and in buffer at 1 mM Mg2+, the approximate Mg2+ concentration we measured in cells. In contrast, the in-cell conformation of the ligand-free aptamer is much more similar to the fully folded ligand-bound state. Even adding high Mg2+ concentrations to the buffer used for in vitro analyses did not yield the conformation observed for the free aptamer in cells. The cellular environment thus stabilizes the aptamer significantly more than does Mg2+ alone. Our results show that the intracellular environment has a large effect on RNA structure that ultimately favors highly organized conformations.
Co-reporter:Christopher W. Leonard, Christine E. Hajdin, Fethullah Karabiber, David H. Mathews, Oleg V. Favorov, Nikolay V. Dokholyan, and Kevin M. Weeks
Biochemistry 2013 Volume 52(Issue 4) pp:
Publication Date(Web):January 14, 2013
DOI:10.1021/bi300755u
Accurate RNA structure modeling is an important, incompletely solved, challenge. Single-nucleotide resolution SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension) yields an experimental measurement of local nucleotide flexibility that can be incorporated as pseudo-free energy change constraints to direct secondary structure predictions. Prior work from our laboratory has emphasized both the overall accuracy of this approach and the need for nuanced interpretation of modeled structures. Recent studies by Das and colleagues [Kladwang, W., et al. (2011) Biochemistry 50, 8049; Nat. Chem. 3, 954], focused on analyzing six small RNAs, yielded poorer RNA secondary structure predictions than expected on the basis of prior benchmarking efforts. To understand the features that led to these divergent results, we re-examined four RNAs yielding the poorest results in this recent work: tRNAPhe, the adenine and cyclic-di-GMP riboswitches, and 5S rRNA. Most of the errors reported by Das and colleagues reflected nonstandard experiment and data processing choices, and selective scoring rules. For two RNAs, tRNAPhe and the adenine riboswitch, secondary structure predictions are nearly perfect if no experimental information is included but were rendered inaccurate by the SHAPE data of Das and colleagues. When best practices were used, single-sequence SHAPE-directed secondary structure modeling recovered ∼93% of individual base pairs and >90% of helices in the four RNAs, essentially indistinguishable from the results of the mutate-and-map approach with the exception of a single helix in the 5S rRNA. The field of experimentally directed RNA secondary structure prediction is entering a phase focused on the most difficult prediction challenges. We outline five constructive principles for guiding this field forward.
Co-reporter:Eva J. Archer, Mark A. Simpson, Nicholas J. Watts, Rory O’Kane, Bangchen Wang, Dorothy A. Erie, Alex McPherson, and Kevin M. Weeks
Biochemistry 2013 Volume 52(Issue 18) pp:
Publication Date(Web):April 25, 2013
DOI:10.1021/bi4001535
We have developed a model for the secondary structure of the 1058-nucleotide plus-strand RNA genome of the icosahedral satellite tobacco mosaic virus (STMV) using nucleotide-resolution SHAPE chemical probing of the viral RNA isolated from virions and within the virion, perturbation of interactions distant in the primary sequence, and atomic force microscopy. These data are consistent with long-range base pairing interactions and a three-domain genome architecture. The compact domains of the STMV RNA have dimensions of 10–45 nm. Each of the three domains corresponds to a specific functional component of the virus: The central domain corresponds to the coding sequence of the single (capsid) protein encoded by the virus, whereas the 5′ and 3′ untranslated domains span signals essential for translation and replication, respectively. This three-domain architecture is compatible with interactions between the capsid protein and short RNA helices previously visualized by crystallography. STMV is among the simplest of the icosahedral viruses but, nonetheless, has an RNA genome with a complex higher-order structure that likely reflects high information content and an evolutionary relationship between RNA domain structure and essential replicative functions.
Co-reporter:Steven Busan and Kevin M. Weeks
Biochemistry 2013 Volume 52(Issue 46) pp:
Publication Date(Web):November 7, 2013
DOI:10.1021/bi401129r
The length of the CAG-repeat region in the huntingtin mRNA is predictive of Huntington’s disease. Structural studies of CAG-repeat-containing RNAs suggest that these sequences form simple hairpin structures; however, in the context of the full-length huntingtin mRNA, CAG repeats may form complex structures that could be targeted for therapeutic intervention. We examined the structures of transcripts spanning the first exon of the huntingtin mRNA with both healthy and disease-prone repeat lengths. In transcripts with 17–70 repeats, the CAG sequences base paired extensively with nucleotides in the 5′ UTR and with conserved downstream sequences including a CCG-repeat region. In huntingtin transcripts with healthy numbers of repeats, the previously observed CAG hairpin was either absent or short. In contrast, in transcripts with disease-associated numbers of repeats, a CAG hairpin was present and extended from a three-helix junction. Our findings demonstrate the profound importance of sequence context in RNA folding and identify specific structural differences between healthy and disease-inducing huntingtin alleles that may be targets for therapeutic intervention.
Co-reporter:Jacob K. Grohman;Robert J. Gorelick;Colin R. Lickwar;Jason D. Lieb;Brian D. Bower;Brent M. Znosko
Science 2013 Volume 340(Issue 6129) pp:190-195
Publication Date(Web):12 Apr 2013
DOI:10.1126/science.1230715
Simply Folding
RNA chaperones simplify what would otherwise be complex and slow RNA folding events. Grohman et al. (p. 190, published online 7 March) show that the Moloney murine leukemia virus (MuLV) nucleocapsid (NC) protein, which chaperones MuLV RNA dimerization promotes MuLV RNA folding by binding to exposed guanosine bases and destabilizing strong guanosine interactions. With base-pairs being rendered roughly of the same energy, RNA assembly pathways are simplified, promoting proper folding.
Co-reporter:Christine E. Hajdin;Stanislav Bellaousov;Wayne Huggins;Christopher W. Leonard;David H. Mathews
PNAS 2013 Volume 110 (Issue 14 ) pp:5498-5503
Publication Date(Web):2013-04-02
DOI:10.1073/pnas.1219988110
A pseudoknot forms in an RNA when nucleotides in a loop pair with a region outside the helices that close the loop. Pseudoknots
occur relatively rarely in RNA but are highly overrepresented in functionally critical motifs in large catalytic RNAs, in
riboswitches, and in regulatory elements of viruses. Pseudoknots are usually excluded from RNA structure prediction algorithms.
When included, these pairings are difficult to model accurately, especially in large RNAs, because allowing this structure
dramatically increases the number of possible incorrect folds and because it is difficult to search the fold space for an
optimal structure. We have developed a concise secondary structure modeling approach that combines SHAPE (selective 2′-hydroxyl
acylation analyzed by primer extension) experimental chemical probing information and a simple, but robust, energy model for
the entropic cost of single pseudoknot formation. Structures are predicted with iterative refinement, using a dynamic programming
algorithm. This melded experimental and thermodynamic energy function predicted the secondary structures and the pseudoknots
for a set of 21 challenging RNAs of known structure ranging in size from 34 to 530 nt. On average, 93% of known base pairs
were predicted, and all pseudoknots in well-folded RNAs were identified.
Co-reporter:Jennifer L. McGinnis ; Jack A. Dunkle ; Jamie H. D. Cate
Journal of the American Chemical Society 2012 Volume 134(Issue 15) pp:6617-6624
Publication Date(Web):April 5, 2012
DOI:10.1021/ja2104075
The biological functions of RNA are ultimately governed by the local environment at each nucleotide. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) chemistry is a powerful approach for measuring nucleotide structure and dynamics in diverse biological environments. SHAPE reagents acylate the 2′-hydroxyl group at flexible nucleotides because unconstrained nucleotides preferentially sample rare conformations that enhance the nucleophilicity of the 2′-hydroxyl. The critical corollary is that some constrained nucleotides must be poised for efficient reaction at the 2′-hydroxyl group. To identify such nucleotides, we performed SHAPE on intact crystals of the Escherichia coli ribosome, monitored the reactivity of 1490 nucleotides in 16S rRNA, and examined those nucleotides that were hyper-reactive toward SHAPE and had well-defined crystallographic conformations. Analysis of these conformations revealed that 2′-hydroxyl reactivity is broadly facilitated by general base catalysis involving multiple RNA functional groups and by two specific orientations of the bridging 3′-phosphate group. Nucleotide analog studies confirmed the contributions of these mechanisms to SHAPE reactivity. These results provide a strong mechanistic explanation for the relationship between SHAPE reactivity and local RNA dynamics and will facilitate interpretation of SHAPE information in the many technologies that make use of this chemistry.
Co-reporter:Kady-Ann Steen ; Greggory M. Rice
Journal of the American Chemical Society 2012 Volume 134(Issue 32) pp:13160-13163
Publication Date(Web):August 1, 2012
DOI:10.1021/ja304027m
Many RNA structures are composed of simple secondary structure elements linked by a few critical tertiary interactions. SHAPE chemistry has made interrogation of RNA dynamics at single-nucleotide resolution straightforward. However, de novo identification of nucleotides involved in tertiary interactions remains a challenge. Here we show that nucleotides that form noncanonical or tertiary contacts can be detected by comparing information obtained using two SHAPE reagents, N-methylisatoic anhydride (NMIA) and 1-methyl-6-nitroisatoic anhydride (1M6). Nucleotides that react preferentially with NMIA exhibit slow local nucleotide dynamics and usually adopt the less common C2′-endo ribose conformation. Experiments and first-principles calculations show that 1M6 reacts preferentially with nucleotides in which one face of the nucleobase allows an unhindered stacking interaction with the reagent. Differential SHAPE reactivities were used to detect noncanonical and tertiary interactions in four RNAs with diverse structures and to identify preformed noncanonical interactions in partially folded RNAs. Differential SHAPE reactivity analysis will enable experimentally concise, large-scale identification of tertiary structure elements and ligand binding sites in complex RNAs and in diverse biological environments.
Co-reporter:Jacob K. Grohman ; Sumith Kottegoda ; Robert J. Gorelick ; Nancy L. Allbritton
Journal of the American Chemical Society 2011 Volume 133(Issue 50) pp:20326-20334
Publication Date(Web):November 29, 2011
DOI:10.1021/ja2070945
Higher-order structure influences critical functions in nearly all noncoding and coding RNAs. Most single-nucleotide resolution RNA structure determination technologies cannot be used to analyze RNA from scarce biological samples, like viral genomes. To make quantitative RNA structure analysis applicable to a much wider array of RNA structure–function problems, we developed and applied high-sensitivity selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) to structural analysis of authentic genomic RNA of the xenotropic murine leukemia virus-related virus (XMRV). For analysis of fluorescently labeled cDNAs generated in high-sensitivity SHAPE experiments, we developed a two-color capillary electrophoresis approach with zeptomole molecular detection limits and subfemtomole sensitivity for complete SHAPE experiments involving hundreds of individual RNA structure measurements. High-sensitivity SHAPE data correlated closely (R = 0.89) with data obtained by conventional capillary electrophoresis. Using high-sensitivity SHAPE, we determined the dimeric structure of the XMRV packaging domain, examined dynamic interactions between the packaging domain RNA and viral nucleocapsid protein inside virion particles, and identified the packaging signal for this virus. Despite extensive sequence differences between XMRV and the intensively studied Moloney murine leukemia virus, architectures of the regulatory domains are similar and reveal common principles of gammaretrovirus RNA genome packaging.
Co-reporter:Kady-Ann Steen ; Arun Malhotra
Journal of the American Chemical Society 2010 Volume 132(Issue 29) pp:9940-9943
Publication Date(Web):July 2, 2010
DOI:10.1021/ja103781u
Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) is a powerful approach for characterizing RNA structure and dynamics at single-nucleotide resolution. However, SHAPE technology is limited, sometimes severely, because primer extension detection obscures structural information for ∼15 nts at the 5′ end and 40−60 nts at the 3′ end of the RNA. Moreover, detection by primer extension is more complex than the actual structure-selective chemical interrogation step. Here we quantify covalent adducts in RNA directly by adduct-inhibited exoribonuclease degradation. RNA 2′-O-adducts block processivity of a 3′→5′ exoribonuclease, RNase R, to produce fragments that terminate three nucleotides 3′ of the modification site. We analyzed the structure of the native thiamine pyrophosphate (TPP) riboswitch aptamer domain and identified large changes in local nucleotide dynamics and global RNA structure upon ligand binding. In addition to numerous changes that can be attributed to ligand recognition, we identify a single nucleotide bulge register shift, distant from the binding site, that stabilizes the ligand-bound structure. Selective 2′-hydroxyl acylation analyzed by protection from exoribonuclease (RNase-detected SHAPE) should prove broadly useful for facile structural analysis of small noncoding RNAs and for RNAs that have functionally critical structures at their 5′ and 3′ ends.
Co-reporter:Christopher A. Lavender, Feng Ding, Nikolay V. Dokholyan and Kevin M. Weeks
Biochemistry 2010 Volume 49(Issue 24) pp:
Publication Date(Web):May 27, 2010
DOI:10.1021/bi100142y
RNA function is dependent on its structure, yet three-dimensional folds for most biologically important RNAs are unknown. We develop a generic discrete molecular dynamics-based modeling system that uses long-range constraints inferred from diverse biochemical or bioinformatic analyses to create statistically significant (p < 0.01) nativelike folds for RNAs of known structure ranging from 45 to 158 nucleotides. We then predict the unknown structure of the hepatitis C virus internal ribosome entry site (IRES) pseudoknot domain. The resulting RNA model rationalizes independent solvent accessibility and cryo-electron microscopy structure information. The pseudoknot domain positions the AUG start codon near the mRNA channel and is tRNA-like, suggesting the IRES employs molecular mimicry as a functional strategy.
Co-reporter:Caia D. S. Duncan and Kevin M. Weeks
Biochemistry 2010 Volume 49(Issue 26) pp:
Publication Date(Web):June 9, 2010
DOI:10.1021/bi100267g
Proteins play diverse and critical roles in cellular ribonucleoproteins (RNPs) including promoting formation of and stabilizing active RNA conformations. Yet, the conformational changes required to convert large RNAs into active RNPs have proven difficult to characterize fully. Here we use high-resolution approaches to monitor both local nucleotide flexibility and solvent accessibility for nearly all nucleotides in the bI3 group I intron RNP in four assembly states: the free RNA, maturase-bound RNA, Mrs1-bound RNA, and the complete six-component holocomplex. The free RNA is misfolded relative to the secondary structure required for splicing. The maturase and Mrs1 proteins each stabilized long-range tertiary interactions, but neither protein alone induced folding into the functional secondary structure. In contrast, simultaneous binding by both proteins results in large secondary structure rearrangements in the RNA and yielded the catalytically active group I intron structure. Secondary and tertiary folding of the RNA component of the bI3 RNP are thus not independent: RNA folding is strongly nonhierarchical. These results emphasize that protein-mediated stabilization of RNA tertiary interactions functions to pull the secondary structure into an energetically disfavored, but functional, conformation and emphasize a new role for facilitator proteins in RNP assembly.
Co-reporter:Stefanie A. Mortimer, Jeffrey S. Johnson and Kevin M. Weeks
Biochemistry 2009 Volume 48(Issue 10) pp:
Publication Date(Web):February 18, 2009
DOI:10.1021/bi801939g
An important unmet experimental objective is to analyze local RNA structure in a way that is strictly governed by solvent accessibility. Essentially all chemical probes currently used to evaluate RNA (and DNA) structure via formation of stable covalent adducts employ carbon-based electrophiles, which undergo nucleophilic attack from limited spatial orientations and via highly polar transition states. Reaction by these classical electrophiles is therefore gated by both solvent accessibility and additional electrostatic factors. In contrast, silicon electrophiles react via their d-orbitals and consequently can undergo nucleophilic attack from many spatial orientations. In this work, we explore the use of silanes to react indiscriminately with RNA such that the primary factor governing reactivity is solvent accessibility. We show that N,N-(dimethylamino)dimethylchlorosilane (DMAS-Cl) reacts at the guanosine N2 position to yield a near-perfect measure (r ≥ 0.82) of solvent accessibility in an RNA with a complex tertiary structure. This silane-based chemistry represents a direct and quantitative approach for probing solvent accessibility at the base pairing face of guanosine in RNA.
Co-reporter:Tian W. Li;David H. Mathews;Katherine E. Deigan
PNAS 2009 Volume 106 (Issue 1 ) pp:97-102
Publication Date(Web):2009-01-06
DOI:10.1073/pnas.0806929106
Almost all RNAs can fold to form extensive base-paired secondary structures. Many of these structures then modulate numerous
fundamental elements of gene expression. Deducing these structure–function relationships requires that it be possible to predict
RNA secondary structures accurately. However, RNA secondary structure prediction for large RNAs, such that a single predicted
structure for a single sequence reliably represents the correct structure, has remained an unsolved problem. Here, we demonstrate
that quantitative, nucleotide-resolution information from a SHAPE experiment can be interpreted as a pseudo-free energy change
term and used to determine RNA secondary structure with high accuracy. Free energy minimization, by using SHAPE pseudo-free
energies, in conjunction with nearest neighbor parameters, predicts the secondary structure of deproteinized Escherichia coli 16S rRNA (>1,300 nt) and a set of smaller RNAs (75–155 nt) with accuracies of up to 96–100%, which are comparable to the
best accuracies achievable by comparative sequence analysis.
Co-reporter:Stefanie A. Mortimer
PNAS 2009 Volume 106 (Issue 37 ) pp:15622-15627
Publication Date(Web):2009-09-15
DOI:10.1073/pnas.0901319106
A striking and widespread observation is that higher-order folding for many RNAs is very slow, often requiring minutes. In
some cases, slow folding reflects the need to disrupt stable, but incorrect, interactions. However, a molecular explanation
for slow folding in most RNAs is unknown. The specificity domain of the Bacillus subtilis RNase P ribozyme undergoes a rate-limiting folding step on the minute time-scale. This RNA also contains a C2′-endo nucleotide
at A130 that exhibits extremely slow local conformational dynamics. This nucleotide is evolutionarily conserved and essential
for tRNA recognition by RNase P. Here we show that deleting this single nucleotide accelerates folding by an order of magnitude
even though this mutation does not change the global fold of the RNA. These results demonstrate that formation of a single
stacking interaction at a C2′-endo nucleotide comprises the rate-determining step for folding an entire 154 nucleotide RNA.
C2′-endo nucleotides exhibit slow local dynamics in structures spanning isolated helices to complex tertiary interactions.
Because the motif is both simple and ubiquitous, C2′-endo nucleotides may function as molecular timers in many RNA folding
and ligand recognition reactions.
Co-reporter:Joseph M. Watts,
Kristen K. Dang,
Robert J. Gorelick,
Christopher W. Leonard,
Julian W. Bess Jr,
Ronald Swanstrom,
Christina L. Burch
&
Kevin M. Weeks
Nature 2009 460(7256) pp:711
Publication Date(Web):2009-08-06
DOI:10.1038/nature08237
Single-stranded RNA viruses are responsible for the common cold, cancer, AIDS and other serious health threats. The genomes of these viruses form conserved secondary structures that have functional and regulatory roles, but most potential regulatory elements in viral RNA genomes remain uncharacterized. Here however, the structure of an entire HIV-1 genome at single nucleotide resolution is reported.
Co-reporter:Bin Wang, Kevin A. Wilkinson and Kevin M. Weeks
Biochemistry 2008 Volume 47(Issue 11) pp:
Publication Date(Web):February 22, 2008
DOI:10.1021/bi702372x
RNA conformation is both highly dependent on and sensitive to the presence of charged ligands. Mono- and divalent ions stabilize the native fold of RNA, whereas other polyvalent cationic ligands can act to both stabilize or disrupt native RNA structure. In this work, we analyze the effects of two ligands, Mg2+ and tobramycin, on the folding of S. cerevisiae tRNAAsp transcripts using single nucleotide resolution SHAPE chemistry. Surprisingly, reducing the Mg2+ concentration favors a structural rearrangement in which the D- and variable loops pair. The tobramycin polycation binds to loops in tRNAAsp and induces RNA unfolding in two distinct transitions: the loss of tertiary interactions between the T- and D-loops followed by complete unfolding of the D-stem. Although Mg2+ and tobramycin are relatively simple ligands, both modulate tRNAAsp folding in unanticipatedly complex ways, neither of which is consistent with simple hierarchical folding or unfolding of this RNA. Monitoring the structural consequences of ligand binding to RNA at single nucleotide resolution makes it possible to define intermediate structures that contribute to the complex energy landscapes often observed for RNA folding processes and lays the groundwork for a significantly improved understanding of the interactions between RNA and its solution environment.
Co-reporter:Caia D. S. Duncan and Kevin M. Weeks
Biochemistry 2008 Volume 47(Issue 33) pp:
Publication Date(Web):July 22, 2008
DOI:10.1021/bi800207b
Most functional RNAs require proteins to facilitate formation of their active structures. In the case of the yeast bI3 group I intron, splicing requires binding by two proteins, the intron-encoded bI3 maturase and the nuclear encoded Mrs1. Here, we use selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) chemistry coupled with analysis of point mutants to map long-range interactions in this RNA. This analysis reveals two critical features of the free RNA state. First, the catalytic intron is separated from the flanking exons via a stable anchoring helix. This anchoring helix creates an autonomous structural domain for the intron and functions to prevent misfolding with the flanking exons. Second, the thermodynamically most stable structure for the free RNA is not consistent with the catalytically active conformation as phylogenetically conserved elements form stable, non-native structures. These results highlight a fragile bI3 RNA for which binding by protein cofactors functions to promote extensive secondary structure rearrangements that are an obligatory prerequisite for forming the catalytically active tertiary structure.
Co-reporter:Costin M. Gherghe;Christopher S. Badorrek
PNAS 2006 Volume 103 (Issue 37 ) pp:13640-13645
Publication Date(Web):2006-09-12
DOI:10.1073/pnas.0606156103
Retroviruses selectively package two copies of their RNA genomes in the context of a large excess of nongenomic RNA. Specific
packaging of genomic RNA is achieved, in part, by recognizing RNAs that form a poorly understood dimeric structure at their
5′ ends. We identify, quantify the stability of, and use extensive experimental constraints to calculate a 3D model for a
tertiary structure domain that mediates specific interactions between RNA genomes in a gamma retrovirus. In an initial interaction,
two stem–loop structures from one RNA form highly stringent cross-strand loop–loop base pairs with the same structures on
a second genomic RNA. Upon subsequent folding to the final dimer state, these intergenomic RNA interactions convert to a high
affinity and compact tertiary structure, stabilized by interdigitated interactions between U-shaped RNA units. This retroviral
conformational switch model illustrates how two-step formation of an RNA tertiary structure yields a stringent molecular recognition
event at early assembly steps that can be converted to the stable RNA architecture likely packaged into nascent virions.
Co-reporter:
Nature Structural and Molecular Biology 2005 12(9) pp:779-787
Publication Date(Web):21 August 2005
DOI:10.1038/nsmb976
LAGLIDADG endonucleases bind across adjacent major grooves via a saddle-shaped surface and catalyze DNA cleavage. Some LAGLIDADG proteins, called maturases, facilitate splicing by group I introns, raising the issue of how a DNA-binding protein and an RNA have evolved to function together. In this report, crystallographic analysis shows that the global architecture of the bI3 maturase is unchanged from its DNA-binding homologs; in contrast, the endonuclease active site, dispensable for splicing facilitation, is efficiently compromised by a lysine residue replacing essential catalytic groups. Biochemical experiments show that the maturase binds a peripheral RNA domain 50 Å from the splicing active site, exemplifying long-distance structural communication in a ribonucleoprotein complex. The bI3 maturase nucleic acid recognition saddle interacts at the RNA minor groove; thus, evolution from DNA to RNA function has been mediated by a switch from major to minor groove interaction.
Co-reporter:Stacy I. Chamberlin;Edward J. Merino
PNAS 2002 Volume 99 (Issue 23 ) pp:14688-14693
Publication Date(Web):2002-11-12
DOI:10.1073/pnas.212527799
The functional groups found among the RNA bases and in the phosphoribose backbone represent a limited repertoire from which
to construct a ribozyme active site. This work investigates the possibility that simple RNA phosphodiester and hydroxyl functional
groups could catalyze amide bond synthesis. Reaction of amine groups with activated esters would be catalyzed by a group that
stabilizes the partial positive charge on the amine nucleophile in the transition state. 2′-Amine substitutions adjacent to
3′-phosphodiester or 3′-hydroxyl groups react efficiently with activated esters to form 2′-amide and peptide products. In
contrast, analogs in which the 3′-phosphodiester is replaced by an uncharged phosphotriester or is constrained in a distal
conformation react at least 100-fold more slowly. Similarly, a nucleoside in which the 3′-hydroxyl group is constrained trans to the 2′-amine is also unreactive. Catalysis of synthetic reactions by RNA phosphodiester and ribose hydroxyl groups is
likely to be even greater in the context of a preorganized and solvent-excluding catalytic center. One such group is the 2′-hydroxyl
of the ribosome-bound P-site adenosine substrate, which is close to the amine nucleophile in the peptidyl synthesis reaction.
Given ubiquitous 2′-OH groups in RNA, there exists a decisive advantage for RNA over DNA in catalyzing reactions of biological
significance.
Co-reporter:Gurminder S. Bassi;Daniela M. de Oliveira;Malcolm F. White;
Proceedings of the National Academy of Sciences 2002 99(1) pp:128-133
Publication Date(Web):January 2, 2002
DOI:10.1073/pnas.012579299
Detectable splicing by the Saccharomyces
cerevisiae mitochondrial bI3 group I intron RNA in
vitro is shown to require both an intron-encoded protein, the
bI3 maturase, and the nuclear-encoded protein, Mrs1. Both proteins
bind independently to the bI3 RNA. The bI3 maturase binds as a monomer,
whereas Mrs1 is a dimer in solution that assembles as two dimers,
cooperatively, on the RNA. The active six-subunit complex has a
molecular mass of 420 kDa, splices with a
kcat of 0.3 min−1, and binds
the guanosine nucleophile with an affinity comparable to other group I
introns. The functional bI3 maturase domain is translated from within
the RNA that encodes the intron, has evolved a high-affinity
RNA-binding activity, and is a member of the LAGLIDADG family of DNA
endonucleases, but appears to have lost DNA cleavage activity. Mrs1 is
a divergent member of the RNase H fold superfamily of dimeric DNA
junction-resolving enzymes that also appears to have lost its nuclease
activity and now functions as a tetramer in RNA binding. Thus, the bI3
ribonucleoprotein is the product of a process in which a
once-catalytically active RNA now obligatorily requires two
facilitating protein cofactors, both of which are compromised in their
original functions.
Co-reporter:
Nature Structural and Molecular Biology 2001 8(6) pp:515 - 520
Publication Date(Web):
DOI:10.1038/88577
Co-reporter:
Nature Structural and Molecular Biology 2001 8(2) pp:135-140
Publication Date(Web):
DOI:10.1038/84124
Most large RNAs achieve their active, native structures only as complexes with one or more cofactor proteins. By varying the Mg2+ concentration, the catalytic core of the bI5 group I intron RNA can be manipulated into one of three states, expanded, collapsed or native, or into balanced equilibria between these states. Under near-physiological conditions, the bI5 RNA folds rapidly to a collapsed but non-native state. Hydroxyl radical footprinting demonstrates that assembly with the CBP2 protein cofactor chases the RNA from the collapsed state to the native state. In contrast, CBP2 also binds to the RNA in the expanded state to form many non-native interactions. This structural picture is reinforced by functional splicing experiments showing that RNA in an expanded state forms a non-productive, kinetically trapped complex with CBP2. Thus, rapid folding to the collapsed state functions to self-chaperone bI5 RNA folding by preventing premature interaction with its protein cofactor. This productive, self-chaperoning role for RNA collapsed states may be especially important to avert misassembly of large multi-component RNA−protein machines in the cell.
Co-reporter:
Nature Structural and Molecular Biology 2000 7(5) pp:362 - 366
Publication Date(Web):
DOI:10.1038/75125
Co-reporter:Chase A. Weidmann, Anthony M. Mustoe, Kevin M. Weeks
Trends in Biochemical Sciences (September 2016) Volume 41(Issue 9) pp:734-736
Publication Date(Web):1 September 2016
DOI:10.1016/j.tibs.2016.07.001
While a variety of powerful tools exists for analyzing RNA structure, identifying long-range and intermolecular base-pairing interactions has remained challenging. Recently, three groups introduced a high-throughput strategy that uses psoralen-mediated crosslinking to directly identify RNA–RNA duplexes in cells. Initial application of these methods highlights the preponderance of long-range structures within and between RNA molecules and their widespread structural dynamics.
Co-reporter:Justin T. Low, Kevin M. Weeks
Methods (October 2010) Volume 52(Issue 2) pp:150-158
Publication Date(Web):1 October 2010
DOI:10.1016/j.ymeth.2010.06.007
The diverse functional roles of RNA are determined by its underlying structure. Accurate and comprehensive knowledge of RNA structure would inform a broader understanding of RNA biology and facilitate exploiting RNA as a biotechnological tool and therapeutic target. Determining the pattern of base pairing, or secondary structure, of RNA is a first step in these endeavors. Advances in experimental, computational, and comparative analysis approaches for analyzing secondary structure have yielded accurate structures for many small RNAs, but only a few large (>500 nts) RNAs. In addition, most current methods for determining a secondary structure require considerable effort, analytical expertise, and technical ingenuity. In this review, we outline an efficient strategy for developing accurate secondary structure models for RNAs of arbitrary length. This approach melds structural information obtained using SHAPE chemistry with structure prediction using nearest-neighbor rules and the dynamic programming algorithm implemented in the RNAstructure program. Prediction accuracies reach ⩾95% for RNAs on the kilobase scale. This approach facilitates both development of new models and refinement of existing RNA structure models, which we illustrate using the Gag-Pol frameshift element in an HIV-1 M-group genome. Most promisingly, integrated experimental and computational refinement brings closer the ultimate goal of efficiently and accurately establishing the secondary structure for any RNA sequence.
Co-reporter:David M. Mauger, Nathan A. Siegfried, Kevin M. Weeks
FEBS Letters (17 April 2013) Volume 587(Issue 8) pp:1180-1188
Publication Date(Web):17 April 2013
DOI:10.1016/j.febslet.2013.03.002
Structured RNA elements within messenger RNA often direct or modulate the cellular production of active proteins. As reviewed here, RNA structures have been discovered that govern nearly every step in protein production: mRNA production and stability; translation initiation, elongation, and termination; protein folding; and cellular localization. Regulatory RNA elements are common within RNAs from every domain of life. This growing body of RNA-mediated mechanisms continues to reveal new ways in which mRNA structure regulates translation. We integrate examples from several different classes of RNA structure-mediated regulation to present a global perspective that suggests that the secondary and tertiary structure of RNA ultimately constitutes an additional level of the genetic code that both guides and regulates protein biosynthesis.
Co-reporter:Tuhin Subhra Maity, Kevin M. Weeks
Journal of Molecular Biology (1 June 2007) Volume 369(Issue 2) pp:512-524
Publication Date(Web):1 June 2007
DOI:10.1016/j.jmb.2007.03.032
Intermediate states play well-established roles in the folding and misfolding reactions of individual RNA and protein molecules. In contrast, the roles of transient structural intermediates in multi-component ribonucleoprotein (RNP) assembly processes and their potential for misassembly are largely unexplored. The SRP19 protein is unstructured but forms a compact core domain and two extended RNA-binding loops upon binding the signal recognition particle (SRP) RNA. The SRP54 protein subsequently binds to the fully assembled SRP19–RNA complex to form an intimate threefold interface with both SRP19 and the RNA and without significantly altering the structure of SRP19. We show, however, that the presence of SRP54 during SRP19–RNA assembly dramatically alters the folding energy landscape to create a non-native folding pathway that leads to an aberrant SRP19–RNA conformation. The misassembled complex arises from the surprising ability of SRP54 to bind rapidly to an SRP19–RNA assembly intermediate and to interfere with subsequent folding of one of the RNA binding loops at the three-way protein–RNA interface. An incorrect temporal order of assembly thus readily yields a non-native three-component ribonucleoprotein particle. We propose there may exist a general requirement to regulate the order of interaction in multi-component RNP assembly reactions by spatial or temporal compartmentalization of individual constituents in the cell.
.jpg)


![PROPANEDIOIC ACID, [(1-METHYLETHYLIDENE)AMINO]- (9CI)](http://img.cochemist.com/ccimg/691100/691001-12-4.png)
![PROPANEDIOIC ACID, [(1-METHYLETHYLIDENE)AMINO]- (9CI)](http://img.cochemist.com/ccimg/691100/691001-12-4_b.png)
![Sulfamic acid,6,7,8,9,10,11-hexahydro-6-oxobenzo[b]cyclohepta[d]pyran-3-yl ester](http://img.cochemist.com/ccimg/288700/288628-05-7.png)
![Sulfamic acid,6,7,8,9,10,11-hexahydro-6-oxobenzo[b]cyclohepta[d]pyran-3-yl ester](http://img.cochemist.com/ccimg/288700/288628-05-7_b.png)





![2,5-Dioxabicyclo[4.1.0]heptane, 7-chloro-7-fluoro-](http://img.cochemist.com/ccimg/120300/120201-74-3.png)
![2,5-Dioxabicyclo[4.1.0]heptane, 7-chloro-7-fluoro-](http://img.cochemist.com/ccimg/120300/120201-74-3_b.png)


![Coenzyme A,S-[2-[[4-[(3-aminopropyl)amino]butyl]amino]-2-oxoethyl]- (9CI)](http://img.cochemist.com/ccimg/83900/83889-68-3.png)
![Coenzyme A,S-[2-[[4-[(3-aminopropyl)amino]butyl]amino]-2-oxoethyl]- (9CI)](http://img.cochemist.com/ccimg/83900/83889-68-3_b.png)

![4-amino-1-[(2-hydroxyethoxy)methyl]pyrimidin-2(1H)-one](http://img.cochemist.com/ccimg/68800/68724-12-9.png)
![4-amino-1-[(2-hydroxyethoxy)methyl]pyrimidin-2(1H)-one](http://img.cochemist.com/ccimg/68800/68724-12-9_b.png)


![9-[3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]purine-6-carboxamide](http://img.cochemist.com/ccimg/65200/65134-53-4.png)
![9-[3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]purine-6-carboxamide](http://img.cochemist.com/ccimg/65200/65134-53-4_b.png)
![Ethanol, 1-[(2-methoxyethyl)thio]-](http://img.cochemist.com/ccimg/64800/64743-40-4.png)
![Ethanol, 1-[(2-methoxyethyl)thio]-](http://img.cochemist.com/ccimg/64800/64743-40-4_b.png)















![Acetamide, N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/35600/35593-64-7.png)
![Acetamide, N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/35600/35593-64-7_b.png)
![N-[(2S)-1-oxo-3-phenylpropan-2-yl]benzamide](http://img.cochemist.com/ccimg/35600/35593-57-8.png)
![N-[(2S)-1-oxo-3-phenylpropan-2-yl]benzamide](http://img.cochemist.com/ccimg/35600/35593-57-8_b.png)




![1-[4-(3-METHYL-BUTOXY)-PHENYL]-ETHANONE](http://img.cochemist.com/ccimg/30300/30237-26-4.png)
![1-[4-(3-METHYL-BUTOXY)-PHENYL]-ETHANONE](http://img.cochemist.com/ccimg/30300/30237-26-4_b.png)
































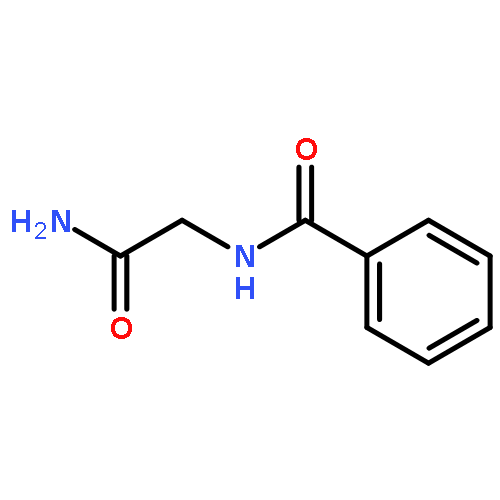

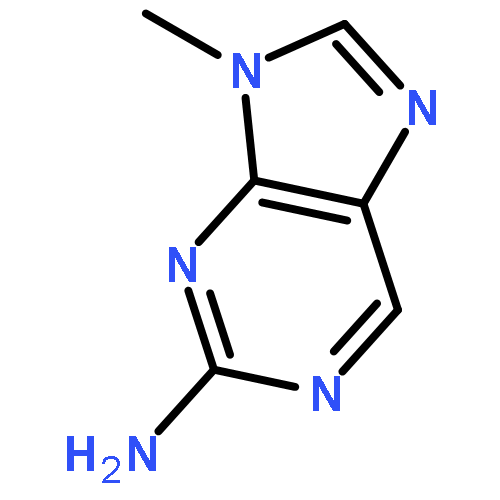


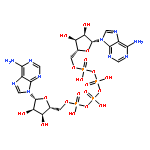
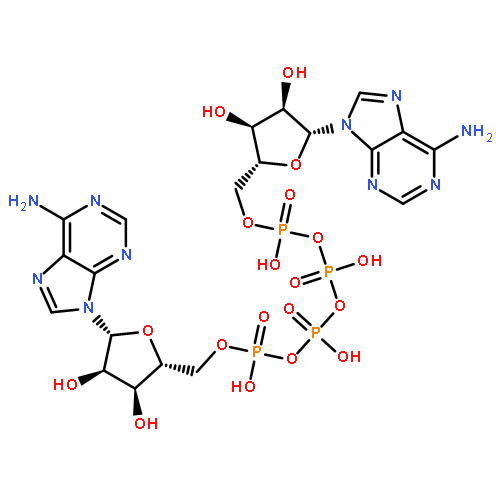











![N~2~-(2,4-dimethoxyphenyl)-N~2~-[(4-methylphenyl)sulfonyl]-N-(1-phenylethyl)glycinamide](http://img.cochemist.com/ccimg/4400/4354-95-4.png)
![N~2~-(2,4-dimethoxyphenyl)-N~2~-[(4-methylphenyl)sulfonyl]-N-(1-phenylethyl)glycinamide](http://img.cochemist.com/ccimg/4400/4354-95-4_b.png)






































![N-[2-(1H-INDOL-3-YL)ETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/1100/1016-47-3.png)
![N-[2-(1H-INDOL-3-YL)ETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/1100/1016-47-3_b.png)










































![ACETAMIDE, 2-(FORMYLAMINO)-N-[2-HYDROXY-1,1-BIS(HYDROXYMETHYL)ETHYL]-](http://img.cochemist.com/ccimg/740900/740841-26-3.png)
![ACETAMIDE, 2-(FORMYLAMINO)-N-[2-HYDROXY-1,1-BIS(HYDROXYMETHYL)ETHYL]-](http://img.cochemist.com/ccimg/740900/740841-26-3_b.png)
![Acetamide, 2-(acetylamino)-N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]-](http://img.cochemist.com/ccimg/740900/740841-25-2.png)
![Acetamide, 2-(acetylamino)-N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]-](http://img.cochemist.com/ccimg/740900/740841-25-2_b.png)
![Estra-1,3,5(10)-trien-17-one,3-[(aminosulfonyl)oxy]-](http://img.cochemist.com/ccimg/148700/148672-09-7.png)
![Estra-1,3,5(10)-trien-17-one,3-[(aminosulfonyl)oxy]-](http://img.cochemist.com/ccimg/148700/148672-09-7_b.png)


![(R)-3-((2S,4S,5R)-4-Hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-3,4,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol](http://img.cochemist.com/ccimg/54000/53910-25-1.png)
![(R)-3-((2S,4S,5R)-4-Hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-3,4,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol](http://img.cochemist.com/ccimg/54000/53910-25-1_b.png)


![Benzamide,N,N',N''-[(3S,7S,11S)-2,6,10-trioxo-1,5,9-trioxacyclododecane-3,7,11-triyl]tris[2,3-dihydroxy-](http://img.cochemist.com/ccimg/28400/28384-96-5.png)
![Benzamide,N,N',N''-[(3S,7S,11S)-2,6,10-trioxo-1,5,9-trioxacyclododecane-3,7,11-triyl]tris[2,3-dihydroxy-](http://img.cochemist.com/ccimg/28400/28384-96-5_b.png)






![[[(2R,3S,4R,5R)-5-(6-AMINOPURIN-9-YL)-3,4-DIHYDROXYOXOLAN-2-YL]METHOXY-HYDROXYPHOSPHORYL] [HYDROXY-[HYDROXY-[[(2R,3S,5R)-3-HYDROXY-5-(5-METHYL-2,4-DIOXOPYRIMIDIN-1-YL)OXOLAN-2-YL]METHOXY]PHOSPHORYL]OXYPHOSPHORYL] HYDROGEN PHOSPHATE](/data/chemimg/495700/13457-68-6.png)
![[[(2R,3S,4R,5R)-5-(6-AMINOPURIN-9-YL)-3,4-DIHYDROXYOXOLAN-2-YL]METHOXY-HYDROXYPHOSPHORYL] [HYDROXY-[HYDROXY-[[(2R,3S,5R)-3-HYDROXY-5-(5-METHYL-2,4-DIOXOPYRIMIDIN-1-YL)OXOLAN-2-YL]METHOXY]PHOSPHORYL]OXYPHOSPHORYL] HYDROGEN PHOSPHATE](/data/chemimg/495700/13457-68-6_b.png)