Co-reporter:Eric J. Alexy, Scott C. Virgil, Michael D. Bartberger, and Brian M. Stoltz
Organic Letters October 6, 2017 Volume 19(Issue 19) pp:
Publication Date(Web):September 13, 2017
DOI:10.1021/acs.orglett.7b02354
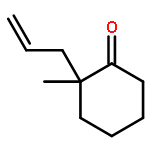
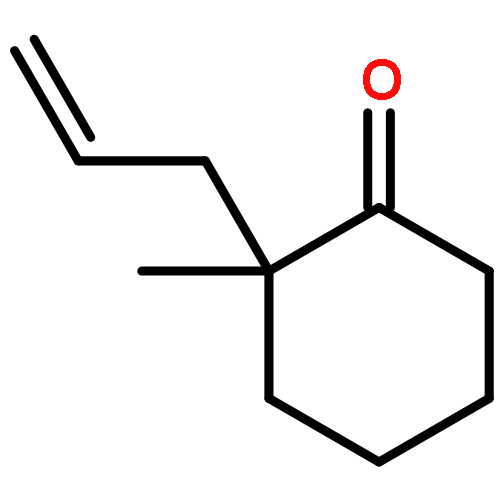
A catalytic, enantioselective decarboxylative allylic alkylation of 4-thiopyranones is reported. The α-quaternary 4-thiopyranones produced are challenging to access by standard enolate alkylation owing to facile ring-opening β-sulfur elimination. In addition, reduction of the carbon–sulfur bonds provides access to elusive acyclic α-quaternary ketones. The alkylated products are obtained in up to 92% yield and 94% enantiomeric excess.
Co-reporter:Marc Liniger, Yiyang Liu, and Brian M. Stoltz
Journal of the American Chemical Society October 4, 2017 Volume 139(Issue 39) pp:13944-13944
Publication Date(Web):September 16, 2017
DOI:10.1021/jacs.7b08496
How can you use a ruthenium isomerization catalyst twice? A ruthenium-catalyzed sequence for the formal two-carbon scission of allyl groups to carboxylic acids has been developed. The reaction includes an initial isomerization step using commercially available ruthenium catalysts followed by in situ transformation of the complex to a metal-oxo species, which is capable of catalyzing subsequent oxidation reactions. The method enables enantioselective syntheses of challenging α-tri- and tetrasubstituted α-amino acids including an expedient total synthesis of the antiepileptic drug levetiracetam.
Co-reporter:Wen-Bo Liu, David P. Schuman, Yun-Fang Yang, Anton A. Toutov, Yong Liang, Hendrik F. T. Klare, Nasri Nesnas, Martin Oestreich, Donna G. Blackmond, Scott C. Virgil, Shibdas Banerjee, Richard N. Zare, Robert H. Grubbs, K. N. Houk, and Brian M. Stoltz
Journal of the American Chemical Society May 24, 2017 Volume 139(Issue 20) pp:6867-6867
Publication Date(Web):April 12, 2017
DOI:10.1021/jacs.6b13031
We recently reported a new method for the direct dehydrogenative C–H silylation of heteroaromatics utilizing Earth-abundant potassium tert-butoxide. Herein we report a systematic experimental and computational mechanistic investigation of this transformation. Our experimental results are consistent with a radical chain mechanism. A trialkylsilyl radical may be initially generated by homolytic cleavage of a weakened Si–H bond of a hypercoordinated silicon species as detected by IR, or by traces of oxygen which can generate a reactive peroxide by reaction with [KOt-Bu]4 as indicated by density functional theory (DFT) calculations. Radical clock and kinetic isotope experiments support a mechanism in which the C–Si bond is formed through silyl radical addition to the heterocycle followed by subsequent β-hydrogen scission. DFT calculations reveal a reasonable energy profile for a radical mechanism and support the experimentally observed regioselectivity. The silylation reaction is shown to be reversible, with an equilibrium favoring products due to the generation of H2 gas. In situ NMR experiments with deuterated substrates show that H2 is formed by a cross-dehydrogenative mechanism. The stereochemical course at the silicon center was investigated utilizing a 2H-labeled silolane probe; complete scrambling at the silicon center was observed, consistent with a number of possible radical intermediates or hypercoordinate silicates.
Co-reporter:Robert A. Craig II and Brian M. Stoltz
Chemical Reviews June 28, 2017 Volume 117(Issue 12) pp:7878-7878
Publication Date(Web):May 18, 2017
DOI:10.1021/acs.chemrev.7b00083
The polycyclic furanobutenolide-derived cembranoid and norcembranoid natural products are a family of congested, stereochemically complex, and extensively oxygenated polycyclic diterpenes and norditerpenes. Although the elegant architectures and biological activity profiles of these natural products have captured the attention of chemists since the isolation of the first members of the family in the 1990s, the de novo synthesis of only a single polycyclic furanobutenolide-derived cembranoid and norcembranoid has been accomplished. This article begins with a brief discussion of the proposed biosyntheses and biosynthetic connections among the polycyclic furanobutenolide-derived cembranoids and norcembranoids and then provides a comprehensive review of the synthetic efforts toward each member of the natural product family, including biomimetic, semisynthetic, and de novo synthetic strategies. This body of knowledge has been gathered to provide insight into the reactivity and constraints of these compact and highly oxygenated polycyclic structures, as well as to offer guidance for future synthetic endeavors.
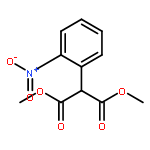
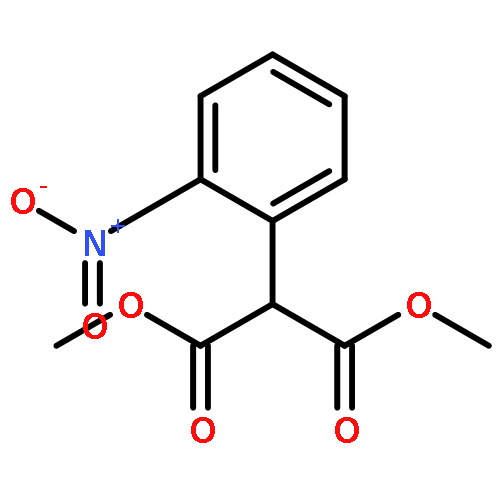
Co-reporter:Pavel Starkov, Jared T. Moore, Douglas C. Duquette, Brian M. Stoltz, and Ilan Marek
Journal of the American Chemical Society July 19, 2017 Volume 139(Issue 28) pp:9615-9615
Publication Date(Web):June 19, 2017
DOI:10.1021/jacs.7b04086
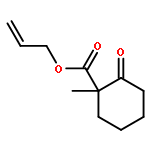
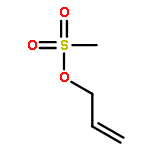
We report a divergent and modular protocol for the preparation of acyclic molecular frameworks containing newly created quaternary carbon stereocenters. Central to this approach is a sequence composed of a (1) regioselective and -retentive preparation of allyloxycarbonyl-trapped fully substituted stereodefined amide enolates and of a (2) enantioselective palladium-catalyzed decarboxylative allylic alkylation reaction using a novel bisphosphine ligand.
Co-reporter:Hosea Nelson;Qingyu Sun;Tony Ly;Brian M. Stoltz;Ryan R. Julian
Journal of Proteome Research February 6, 2009 Volume 8(Issue 2) pp:958-966
Publication Date(Web):2017-2-22
DOI:10.1021/pr800592t
A crown ether based, photolabile radical precursor which forms noncovalent complexes with peptides has been prepared. The peptide/precursor complexes can be electrosprayed, isolated in an ion trap, and then subjected to laser photolysis and collision induced dissociation to generate hydrogen deficient peptide radicals. It is demonstrated that these peptide radicals behave very differently from the hydrogen rich peptide radicals generated by electron capture methods. In fact, it is shown that side chain chemistry dictates both the occurrence and relative abundance of backbone fragments that are observed. Fragmentation at aromatic residues occurs preferentially over most other amino acids. The origin of this selectivity relates to the mechanism by which backbone dissociation is initiated. The first step is abstraction of a β-hydrogen from the side chain, followed by beta-elimination to yield primarily a-type fragment ions. Calculations reveal that those side chains which can easily lose a β-hydrogen correlate well with experimentally favored sites for backbone fragmentation. In addition, radical mediated side chain losses from the parent peptide are frequently observed. Eleven amino acids exhibit unique mass losses from side chains which positively identify that particular amino acid as part of the parent peptide. Therefore, side chain losses allow one to unambiguously narrow the possible sequences for a parent peptide, which when combined with predictable backbone fragmentation should lead to greatly increased confidence in peptide identification.Keywords: 18-crown-6 ether; direct dissociation; iodine; photodissociation; proteomics; ultraviolet;
Co-reporter:Jennifer L. Roizen, Amanda C. Jones, Russell C. Smith, Scott C. Virgil, and Brian M. Stoltz
The Journal of Organic Chemistry December 15, 2017 Volume 82(Issue 24) pp:13051-13051
Publication Date(Web):November 7, 2017
DOI:10.1021/acs.joc.7b02030
Recently, we reported a convergent cyclopropanation–Cope approach to the core of ineleganolide, which was the first disclosed synthesis of the core of the norditerpene natural product ineleganolide. In this complementary work, a model system for the core of ineleganolide has been prepared through a series of tandem cyclopropanation–Cope and translactonization–Cope rearrangements. Work with this model system has enriched our understanding of the cyclopropanation–Cope rearrangement sequence. Additionally, research into this model system has driven the development of tandem translactonization–Cope rearrangements.
Co-reporter:J. Caleb Hethcox, Samantha E. Shockley, and Brian M. Stoltz
Organic Letters April 7, 2017 Volume 19(Issue 7) pp:
Publication Date(Web):March 14, 2017
DOI:10.1021/acs.orglett.7b00449
The first enantioselective iridium-catalyzed allylic alkylation reaction of a masked acyl cyanide (MAC) reagent has been developed. The transformation allows for the use of an umpoled synthon, which serves as a carbon monoxide equivalent. The reaction proceeds with good yield and excellent selectivity up to gram scale for a wide range of substituted allylic electrophiles, delivering products amenable to the synthesis of highly desirable, enantioenriched vinylated α-aryl carbonyl derivatives.
Co-reporter:Hendrik F. T. KlareAlexander F. G. Goldberg, Douglas C. Duquette, Brian M. Stoltz
Organic Letters 2017 Volume 19(Issue 5) pp:
Publication Date(Web):February 14, 2017
DOI:10.1021/acs.orglett.6b03789
An oxidative sequence for the conversion of oxindoles to structurally distinct heterocyclic scaffolds and aniline derivatives is disclosed by the combination of a copper-catalyzed C–H peroxidation and subsequent base-mediated fragmentation reaction. In contrast to classic enzymatic (i.e., kynurenine pathway) and biomimetic methods (i.e., Witkop–Winterfeldt oxidation) for oxidative indole cleavage, this protocol allows for the incorporation of external nucleophiles. The new transformation displays broad functional group tolerance and is applicable to tryptophan derivatives, opening potential new avenues for postsynthetic modification of polypeptides, bioconjugation, and unnatural amino acid synthesis.
Co-reporter:Samantha E. Shockley, J. Caleb Hethcox, Brian M. Stoltz
Tetrahedron Letters 2017 Volume 58, Issue 34(Issue 34) pp:
Publication Date(Web):23 August 2017
DOI:10.1016/j.tetlet.2017.07.022
•Rapid access to enantioenriched spirocycles possessing a 1,4-dicarbonyl moiety spanning an all-carbon quaternary stereogenic spirocenter was achieved using a masked bromomethyl vinyl ketone reagent.•The developed protocol entails an enantioselective palladium-catalyzed allylic alkylation reaction followed by a one-pot unmasking/RCM sequence that provides access to the spirocyclic compounds in good yields and selectivities.Rapid access to enantioenriched spirocycles possessing a 1,4-dicarbonyl moiety spanning an all-carbon quaternary stereogenic spirocenter was achieved using a masked bromomethyl vinyl ketone reagent. The developed protocol entails an enantioselective palladium-catalyzed allylic alkylation reaction followed by a one-pot unmasking/RCM sequence that provides access to the spirocyclic compounds in good yields and selectivities.Download high-res image (70KB)Download full-size image
Co-reporter:Dr. Beau P. Pritchett;Dr. Etienne J. Donckele; Dr. Brian M. Stoltz
Angewandte Chemie International Edition 2017 Volume 56(Issue 41) pp:12624-12627
Publication Date(Web):2017/10/02
DOI:10.1002/anie.201707304
AbstractEnantioselective Pd-catalyzed allylic alkylations of dihydropyrido[1,2-a]indolone (DHPI) substrates were used to construct the C20-quaternary stereocenters of multiple monoterpene indole alkaloids. Stereodivergent Pictet–Spengler and Bischler–Napieralski cyclization/reduction cascades furnish the cis- and trans-fused azadecalin subunits present in Aspidosperma and Kopsia alkaloids, respectively, en route to highly efficient syntheses of (+)-limaspermidine and (+)-kopsihainanine A.
Co-reporter:Samantha E. Shockley;Dr. J. Caleb Hethcox; Dr. Brian M. Stoltz
Angewandte Chemie 2017 Volume 129(Issue 38) pp:11703-11706
Publication Date(Web):2017/09/11
DOI:10.1002/ange.201707015
AbstractThe first highly enantioselective iridium-catalyzed allylic alkylation that provides access to products bearing an allylic all-carbon quaternary stereogenic center has been developed. The reaction utilizes a masked acyl cyanide (MAC) reagent, which enables the one-pot preparation of α-quaternary carboxylic acids, esters, and amides with a high degree of enantioselectivity. The utility of these products is further explored through a series of diverse product transformations.
Co-reporter:Vikram BhatEric R. Welin, Xuelei GuoBrian M. Stoltz
Chemical Reviews 2017 Volume 117(Issue 5) pp:
Publication Date(Web):February 6, 2017
DOI:10.1021/acs.chemrev.6b00731
Stereoconvergent catalysis is an important subset of asymmetric synthesis that encompasses stereoablative transformations, dynamic kinetic resolutions, and dynamic kinetic asymmetric transformations. Initially, only enzymes were known to catalyze dynamic kinetic processes, but recently various synthetic catalysts have been developed. This Review summarizes major advances in nonenzymatic, transition-metal-promoted dynamic asymmetric transformations reported between 2005 and 2015.
Co-reporter:Wen-Bo Liu; Noriko Okamoto; Eric J. Alexy; Allen Y. Hong; Kristy Tran;Brian M. Stoltz
Journal of the American Chemical Society 2016 Volume 138(Issue 16) pp:5234-5237
Publication Date(Web):April 7, 2016
DOI:10.1021/jacs.6b02153
A catalytic, enantioselective γ-alkylation of α,β-unsaturated malonates and ketoesters is reported. This strategy entails a highly regio- and enantioselective iridium-catalyzed α-alkylation of an extended enolate, and a subsequent translocation of chirality to the γ-position via a Cope rearrangement.
Co-reporter:Masaki Hayashi; Shoshana Bachman; Satoshi Hashimoto; Chad C. Eichman;Brian M. Stoltz
Journal of the American Chemical Society 2016 Volume 138(Issue 29) pp:8997-9000
Publication Date(Web):July 3, 2016
DOI:10.1021/jacs.6b02120
A new strategy for catalytic enantioselective C-acylation to generate α-quaternary-substituted lactams is reported. Ni-catalyzed three-component coupling of lactam enolates, benzonitriles, and aryl halides produces β-imino lactams that then afford β-keto lactams by acid hydrolysis. Use of a readily available Mandyphos-type ligand and addition of LiBr enable the construction of quaternary stereocenters on α-substituted lactams to form β-keto lactams in up to 94% ee.
Co-reporter:Anton A. Toutov, Kerry N. Betz, David P. Schuman, Wen-Bo Liu, Alexey FedorovBrian M. Stoltz, Robert H. Grubbs
Journal of the American Chemical Society 2016 Volume 139(Issue 4) pp:1668-1674
Publication Date(Web):December 27, 2016
DOI:10.1021/jacs.6b12114
Disclosed is a mild, scalable, and chemoselective catalytic cross-dehydrogenative C–H bond functionalization protocol for the construction of C(sp)–Si bonds in a single step. The scope of the alkyne and hydrosilane partners is substantial, providing an entry point into various organosilane building blocks and additionally enabling the discovery of a number of novel synthetic strategies. Remarkably, the optimal catalysts are NaOH and KOH.
Co-reporter:Kelly E. Kim, Jiaming Li, Robert H. Grubbs, and Brian M. Stoltz
Journal of the American Chemical Society 2016 Volume 138(Issue 40) pp:13179-13182
Publication Date(Web):September 27, 2016
DOI:10.1021/jacs.6b08788
A new strategy for the functionalization of sterically hindered terminal olefins is reported. Alkenes bearing quaternary carbons at the allylic or homoallylic position are readily oxidized to the corresponding aldehydes by palladium/copper/nitrite catalysis. A broad range of functional groups including esters, nitriles, silyl ethers, vinylogous esters, ketones, lactones, and β-ketoesters are tolerated under the reaction conditions. The crude aldehyde products can be transformed further, enabling direct conversion of hindered terminal alkenes to various other synthetically useful functional groups, resulting in formal anti-Markovnikov hydroamination, among other transformations.
Co-reporter:J. Caleb Hethcox, Samantha E. Shockley, and Brian M. Stoltz
ACS Catalysis 2016 Volume 6(Issue 9) pp:6207
Publication Date(Web):August 17, 2016
DOI:10.1021/acscatal.6b01886
Co-reporter:Kelly E. Kim and Brian M. Stoltz
Organic Letters 2016 Volume 18(Issue 21) pp:5720-5723
Publication Date(Web):October 20, 2016
DOI:10.1021/acs.orglett.6b02962
An improved synthesis of the cyanthiwigin natural product core enabled by new catalytic technology is reported. The key double catalytic enantioselective alkylation has been reoptimized using a recently developed protocol employing low loadings of palladium catalyst, thereby facilitating large-scale production of the tricyclic cyanthiwigin framework. Additionally, preparation of the penultimate aldehyde intermediate is expedited through the application of anti-Markovnikov Tsuji–Wacker oxidation.
Co-reporter:Jiaming Li, Robert H. Grubbs, and Brian M. Stoltz
Organic Letters 2016 Volume 18(Issue 21) pp:5449-5451
Publication Date(Web):October 18, 2016
DOI:10.1021/acs.orglett.6b02722
A mild aerobic intramolecular aminoacetoxylation method for the synthesis of pyrrolidine and indoline derivatives was achieved using molecular oxygen as the oxidant. A catalytic NOx species acts as an electron transfer mediator to access a high-valent palladium intermediate as the presumed active oxidant.
Co-reporter:Austin C. Wright, Christopher K. Haley, Guillaume Lapointe, and Brian M. Stoltz
Organic Letters 2016 Volume 18(Issue 12) pp:2793-2795
Publication Date(Web):June 7, 2016
DOI:10.1021/acs.orglett.6b00994
An insertion of arenes into both imides and anhydrides via reactive aryne intermediates is presented. The reaction is performed under exceptionally mild conditions, and the corresponding ketoamide products are amenable to derivatization to deliver a variety of synthetically useful motifs such as quinolones, indoles, and ketoanilines.
Co-reporter:Jeffrey T. Bagdanoff;Douglas C. Behenna;Jennifer L. Stockdill ;Brian M. Stoltz
European Journal of Organic Chemistry 2016 Volume 2016( Issue 12) pp:2101-2104
Publication Date(Web):
DOI:10.1002/ejoc.201600223
Abstract
The enantioselective synthesis of both caprolactam and enone synthons for the DEFG ring system of zoanthenol are described. The evolution of this approach proceeds first through a synthesis using the chiral pool as a starting point. Challenges in protecting-group strategy led to the modification of this approach beginning with (±)-glycidol. Ultimately, an efficient approach was developed by employing an asymmetric hetero-Diels–Alder reaction. The caprolactam building block can be converted by an interesting selective Grignard addition into the corresponding enone synthon. Addition of a model alkyne provides support for the late-stage addition of a hindered alkyne to the caprolactam building block.
Co-reporter:Nicholas R. O'Connor;John L. Wood;Brian M. Stoltz
Israel Journal of Chemistry 2016 Volume 56( Issue 6-7) pp:431-444
Publication Date(Web):
DOI:10.1002/ijch.201500089
Abstract
Donor−acceptor cyclopropanes are convenient precursors to reactive and versatile 1,3-dipoles, and have found application in the synthesis of a variety of carbo- and heterocyclic scaffolds. This perspective review details our laboratory's use of donor−acceptor cyclopropanes as intermediates toward the total synthesis of various natural products. We also discuss our work in the development of novel cycloadditions and rearrangements of donor−acceptor cyclopropanes and aziridines, as well as an example of an aryne insertion proceeding via fragmentation of a transient donor−acceptor cyclobutane.
Co-reporter:Seo-Jung Han, Brian M. Stoltz
Tetrahedron Letters 2016 Volume 57(Issue 21) pp:2233-2235
Publication Date(Web):25 May 2016
DOI:10.1016/j.tetlet.2016.04.022
•A mild approach toward α-hydroxyethyl α,β-unsaturated δ-lactams is described.•The stereochemistry at the δ-position of the lactam was formed by Ellman’s protocol.•The α,β-unsaturated δ-lactam was smoothly constructed by ring-closing metathesis.•A Baylis–Hillman reaction afforded the substitution at the α-position of the lactam.A straightforward approach toward enantioenriched α-substituted α,β-unsaturated δ-lactams is described. Although a considerable number of approaches toward α,β-unsaturated δ-lactams have been reported, there are relatively few examples of enantioenriched α,δ-disubstituted α,β-unsaturated δ-lactams formation. The δ-stereocenter was formed by addition of allylmagnesium bromide to an N-tert-butylsulfinyl imine. The α,β-unsaturated δ-lactam was furnished by ring-closing metathesis. Although Baylis–Hillman chemistry failed on this cyclic compound, introduction of the hydroxyethyl group prior to ring-closing metathesis was successful. A Baylis–Hillman reaction was used to introduce the substituent at the α-position of the α,β-unsaturated lactam.
Co-reporter:Seo-Jung Han;Ryohei Doi ;Dr. Brian M. Stoltz
Angewandte Chemie International Edition 2016 Volume 55( Issue 26) pp:7437-7440
Publication Date(Web):
DOI:10.1002/anie.201601991
Abstract
An efficient and exceptionally mild intramolecular nickel-catalyzed carbon–oxygen bond-forming reaction between vinyl halides and primary, secondary, and tertiary alcohols has been achieved. Zinc powder was found to be an essential additive for obtaining high catalyst turnover and yields. This operationally simple method allows direct access to cyclic vinyl ethers in high yields in a single step.
Co-reporter:Benzi I. Estipona, Beau P. Pritchett, Robert A. Craig II, Brian M. Stoltz
Tetrahedron 2016 Volume 72(Issue 26) pp:3707-3712
Publication Date(Web):30 June 2016
DOI:10.1016/j.tet.2016.02.059
A catalytic enantioselective synthesis of (+)-eucomic acid is reported. A palladium-catalyzed asymmetric allylic alkylation is employed to access the chiral tetrasubstituted α-hydroxyacid moiety found in the natural product. The protecting group strategy was investigated, and a protecting group manipulation was made without any appreciable deleterious effects in the allylic alkylation reaction. Non-natural (+)-eucomic acid is synthesized in a longest linear sequence of 13 steps.
Co-reporter:Seo-Jung Han;Ryohei Doi ;Dr. Brian M. Stoltz
Angewandte Chemie 2016 Volume 128( Issue 26) pp:7563-7566
Publication Date(Web):
DOI:10.1002/ange.201601991
Abstract
An efficient and exceptionally mild intramolecular nickel-catalyzed carbon–oxygen bond-forming reaction between vinyl halides and primary, secondary, and tertiary alcohols has been achieved. Zinc powder was found to be an essential additive for obtaining high catalyst turnover and yields. This operationally simple method allows direct access to cyclic vinyl ethers in high yields in a single step.
Co-reporter:Yiyang Liu, Seo-Jung Han, Wen-Bo Liu, and Brian M. Stoltz
Accounts of Chemical Research 2015 Volume 48(Issue 3) pp:740
Publication Date(Web):February 25, 2015
DOI:10.1021/ar5004658
The ever-present demand for drugs with better efficacy and fewer side effects continually motivates scientists to explore the vast chemical space. Traditionally, medicinal chemists have focused much attention on achiral or so-called “flat” molecules. More recently, attention has shifted toward molecules with stereogenic centers since their three-dimensional structures represent a much larger fraction of the chemical space and have a number of superior properties compared with flat aromatic compounds. Quaternary stereocenters, in particular, add greatly to the three-dimensionality and novelty of the molecule. Nevertheless, synthetic challenges in building quaternary stereocenters have largely prevented their implementation in drug discovery. The lack of effective and broadly general methods for enantioselective formation of quaternary stereocenters in simple molecular scaffolds has prompted us to investigate new chemistry and develop innovative tools and solutions.In this Account, we describe three approaches to constructing quaternary stereocenters: nucleophilic substitution of 3-halooxindoles, conjugate addition of boronic acids to cyclic enones, and allylic alkylation of enolates. In the first approach, malonic ester nucleophiles attack electrophilic 3-halooxindoles, mediated by a copper(II)-bisoxazoline catalyst. A variety of oxindoles containing a benzylic quaternary stereocenter can be accessed through this method. However, it is only applicable to the specialized 3,3-disubstituted oxindole system. To access benzylic quaternary stereocenters in a more general context, we turned our attention to the enantioselective conjugate addition of carbon nucleophiles to α,β-unsaturated carbonyl acceptors. We discovered that in the presence of catalytic palladium-pyridinooxazoline complex, arylboronic acids add smoothly to β-substituted cyclic enones to furnish ketones with a β-benzylic quaternary stereocenter in high yields and enantioselectivities. The reaction is compatible with a wide range of arylboronic acids, β-substituents, and ring sizes.Aside from benzylic quaternary stereocenters, a more challenging motif is a quaternary stereocenter not adjacent to an aromatic group. Such centers represent more general structures in chemical space but are more difficult to form by asymmetric catalysis. To address this greater challenge, and motivated by the greater reward, we entered the field of palladium-catalyzed asymmetric allylic alkylation of prochiral enolate nucleophiles about a decade ago. On the basis of Tsuji’s work, which solved the issue of positional selectivity for unsymmetrical ketones, we discovered that the phosphinooxazoline ligand effectively rendered this reaction enantioselective. Extensive investigations since then have revealed that the reaction exhibits broad scope and accepts a range of substrate classes, each with its unique advantage in synthetic applications. A diverse array of carbonyl compounds bearing α-quaternary stereocenters are obtained in excellent yields and enantioselectivities, and more possibilities have yet to be explored. As an alternative to palladium catalysis, we also studied iridium-catalyzed asymmetric allylic alkylations that generate vicinal quaternary and tertiary stereocenters in a single transformation. Overall, these methods provide access to small molecule building blocks with a single quaternary stereocenter, can be applied to various molecular scaffolds, and tolerate a wide range of functional groups. We envision that the chemistry reported in this Account will be increasingly useful in drug discovery and design.
Co-reporter:Yoshitaka Numajiri; Beau P. Pritchett; Koji Chiyoda;Brian M. Stoltz
Journal of the American Chemical Society 2015 Volume 137(Issue 3) pp:1040-1043
Publication Date(Web):January 11, 2015
DOI:10.1021/ja512124c
A catalytic enantioselective method for the synthesis of α-quaternary Mannich-type products is reported. The two-step sequence of (1) Mannich reaction followed by (2) decarboxylative enantioselective allylic alkylation serves as a novel strategy to in effect access asymmetric Mannich-type products of “thermodynamic” enolates of substrates possessing additional enolizable positions and acidic protons. Palladium-catalyzed decarboxylative allylic alkylation enables the enantioselective synthesis of five-, six-, and seven-membered ketone, lactam, and other heterocyclic systems. The mild reaction conditions are notable given the acidic free N–H groups and high functional group tolerance in each of the substrates. The utility of this method is highlighted in the first total synthesis of (+)-sibirinine.
Co-reporter:Marc Liniger; David G. VanderVelde; Michael K. Takase; Mona Shahgholi;Brian M. Stoltz
Journal of the American Chemical Society 2015 Volume 138(Issue 3) pp:969-974
Publication Date(Web):December 29, 2015
DOI:10.1021/jacs.5b11750
Derivatives of the fully twisted bicyclic amide 7-hypoquinuclidone are synthesized using a Schmidt–Aubé reaction. Their structures were unambiguously confirmed by X-ray diffraction analysis and extensive spectroscopic characterization. Furthermore, the stability and chemical reactivity of these anti-Bredt amides are investigated. 7-Hypoquinuclidonium tetrafluoroborate is shown to decompose to a unique nitrogen bound amide–BF3 complex of 7-hypoquinuclidone under anhydrous conditions and to react instantaneously with water making it one of the most reactive amides known to date.
Co-reporter:Magnus Bergner, Douglas C. Duquette, Linda Chio, and Brian M. Stoltz
Organic Letters 2015 Volume 17(Issue 12) pp:3008-3010
Publication Date(Web):June 10, 2015
DOI:10.1021/acs.orglett.5b01292
A highly efficient regio- and stereoselective total synthesis of (±)-grandifloracin via a tandem dearomative epoxidation/spontaneous Diels–Alder cyclodimerization from salicylic acid in only four steps is reported. The synthetic route allows for late-stage diversification of the core structure to give ready access to analogues of this promising agent against pancreatic cancer.
Co-reporter:Yoshitaka Numajiri, Gonzalo Jiménez-Osés, Bo Wang, K. N. Houk, and Brian M. Stoltz
Organic Letters 2015 Volume 17(Issue 5) pp:1082-1085
Publication Date(Web):February 25, 2015
DOI:10.1021/ol503425t
The enantioselective synthesis of α-disubstituted N-heterocyclic carbonyl compounds has been accomplished using palladium-catalyzed allylic alkylation. These catalytic conditions enable access to various heterocycles, such as morpholinone, thiomorpholinone, oxazolidin-4-one, 1,2-oxazepan-3-one, 1,3-oxazinan-4-one, and structurally related lactams, all bearing fully substituted α-positions. Broad functional group tolerance was explored at the α-position in the morpholinone series. We demonstrate the utility of this method by performing various transformations on our useful products to readily access a number of enantioenriched compounds.
Co-reporter:Alexer N. Marziale;Douglas C. Duquette;Robert A. Craig II;Kelly E. Kim;Marc Liniger;Yoshitaka Numajiri ;Brian M. Stoltz
Advanced Synthesis & Catalysis 2015 Volume 357( Issue 10) pp:2238-2245
Publication Date(Web):
DOI:10.1002/adsc.201500253
Co-reporter:Samantha E. Shockley, Jeffrey C. Holder, and Brian M. Stoltz
Organic Process Research & Development 2015 Volume 19(Issue 8) pp:974-981
Publication Date(Web):July 2, 2015
DOI:10.1021/acs.oprd.5b00169
This account describes our laboratory’s efforts in the development of a palladium-catalyzed asymmetric conjugate addition of arylboronic acids to cyclic conjugate acceptors. Specifically, we highlight the study of this transformation in the following areas: (a) construction of all-carbon quaternary stereocenters, (b) elucidation of the reaction mechanism, (c) addition to heterocyclic acceptors to generate tertiary stereocenters, and (d) application in the synthesis of natural products.
Co-reporter:Robert A. Craig II, Brian M. Stoltz
Tetrahedron Letters 2015 Volume 56(Issue 32) pp:4670-4673
Publication Date(Web):5 August 2015
DOI:10.1016/j.tetlet.2015.06.039
The synthesis of the novel electronically modified phosphinooxazoline (PHOX) ligand, (R)-5,5-dimethyl-(p-CF3)3-i-PrPHOX, is described. The utility of this PHOX ligand is explored in both enantio- and diastereoselective palladium-catalyzed allylic alkylations. These investigations prove (R)-5,5-dimethyl-(p-CF3)3-i-PrPHOX to be an effective and cost-efficient alternative to electronically modified PHOX ligands derived from the prohibitively expensive (R)-t-leucine.
Co-reporter:Alexander F.G. Goldberg, Robert A. Craig II, Nicholas R. O’Connor, Brian M. Stoltz
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:2983-2990
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.09.016
A series of highly substituted vinylcyclopropanes were prepared and examined as reaction partners in a palladium-catalyzed (3+2) cycloaddition with nitrostyrenes. Described herein are our efforts to synthesize an elusive 1,1-divinylcyclopropane by several distinct approaches, and to apply surrogates of this fragment toward the synthesis of the Melodinus alkaloids.
Co-reporter:Yusuke Kita, Yoshitaka Numajiri, Noriko Okamoto, Brian M. Stoltz
Tetrahedron 2015 Volume 71(Issue 37) pp:6349-6353
Publication Date(Web):16 September 2015
DOI:10.1016/j.tet.2015.05.092
The palladium-catalyzed decarboxylative allylic alkylation of enol carbonates derived from lactams and ketones is described. Employing these substrates with an electronically tuned Pd catalyst system trisubstituted chiral centers are produced. These stereocenters have been previously challenging to achieve using Pd complex/chiral P–N ligand systems.
Co-reporter:Katerina M. Korch;Dr. Christian Eidamshaus;Dr. Douglas C. Behenna;Dr. Sangkil Nam;Dr. David Horne;Dr. Brian M. Stoltz
Angewandte Chemie 2015 Volume 127( Issue 1) pp:181-185
Publication Date(Web):
DOI:10.1002/ange.201408609
Abstract
The asymmetric palladium-catalyzed decarboxylative allylic alkylation of differentially N-protected piperazin-2-ones allows the synthesis of a variety of highly enantioenriched tertiary piperazine-2-ones. Deprotection and reduction affords the corresponding tertiary piperazines, which can be employed for the synthesis of medicinally important analogues. The introduction of these chiral tertiary piperazines resulted in imatinib analogues which exhibited comparable antiproliferative activity to that of their corresponding imatinib counterparts.
Co-reporter:Dr. Xiangyou Xing;Nicholas R. O'Connor;Dr. Brian M. Stoltz
Angewandte Chemie International Edition 2015 Volume 54( Issue 38) pp:11186-11190
Publication Date(Web):
DOI:10.1002/anie.201504007
Abstract
The use of Oxone and a palladium(II) catalyst enables the efficient allylic CH oxidation of sterically hindered α-quaternary lactams which are unreactive under known conditions for similar transformations. This simple, safe, and effective system for CH activation allows for unusual tunable selectivity between a two-electron oxidation to the allylic acetates and a four-electron oxidation to the corresponding enals, with the dominant product depending on the presence or absence of water. The versatile synthetic utility of both the allylic acetate and enal products accessible through this methodology is also demonstrated.
Co-reporter:Seo-Jung Han, Florian Vogt, Jeremy A. May, Shyam Krishnan, Michele Gatti, Scott C. Virgil, and Brian M. Stoltz
The Journal of Organic Chemistry 2015 Volume 80(Issue 1) pp:528-547
Publication Date(Web):November 17, 2014
DOI:10.1021/jo502534g
Expedient synthetic approaches to the highly functionalized polycyclic alkaloids communesin F and perophoramidine are described using a unified approach featuring a key decarboxylative allylic alkylation to access a crucial and highly congested 3,3-disubstituted oxindole. Described are two distinct, stereoselective alkylations that produce structures in divergent diastereomeric series possessing the critical vicinal all-carbon quaternary centers needed for each synthesis. Synthetic studies toward these challenging core structures have revealed a number of unanticipated modes of reactivity inherent to these complex alkaloid scaffolds. Additionally, several novel and interesting intermediates en route to the target natural products, such as an intriguing propellane hexacyclic oxindole encountered in the communesin F sequence, are disclosed. Indeed, such unanticipated structures may prove to be convenient strategic intermediates in future syntheses.
Co-reporter:Chung Whan Lee, Seo-Jung Han, Scott C. Virgil, Brian M. Stoltz
Tetrahedron 2015 Volume 71(Issue 22) pp:3666-3670
Publication Date(Web):3 June 2015
DOI:10.1016/j.tet.2014.10.065
An improved method for the asymmetric alkylation of 3-bromooxindoles with α-arylated malonate esters is described. The asymmetric alkylation demonstrated was achieved up to 70% ee utilizing a copper(II) bis(phosphine) complex.
Co-reporter:Jeffrey C. Holder, Emmett D. Goodman, Kotaro Kikushima, Michele Gatti, Alexander N. Marziale, Brian M. Stoltz
Tetrahedron 2015 Volume 71(Issue 35) pp:5781-5792
Publication Date(Web):2 September 2015
DOI:10.1016/j.tet.2014.11.048
The development and optimization of a palladium-catalyzed asymmetric conjugate addition of arylboronic acids to cyclic enone conjugate acceptors is described. These reactions employ air-stable and readily-available reagents in an operationally simple and robust transformation that yields β-quaternary ketones in high yields and enantioselectivities. Notably, the reaction itself is highly tolerant of atmospheric oxygen and moisture and therefore does not require the use of dry or deoxygenated solvents, specially purified reagents, or an inert atmosphere. The ring size and β-substituent of the enone are highly variable, and a wide variety of β-quaternary ketones can be synthesized. More recently, the use of NH4PF6 has further expanded the substrate scope to include heteroatom-containing arylboronic acids and β-acyl enone substrates.
Co-reporter:Dr. Xiangyou Xing;Nicholas R. O'Connor;Dr. Brian M. Stoltz
Angewandte Chemie 2015 Volume 127( Issue 38) pp:11338-11342
Publication Date(Web):
DOI:10.1002/ange.201504007
Abstract
The use of Oxone and a palladium(II) catalyst enables the efficient allylic CH oxidation of sterically hindered α-quaternary lactams which are unreactive under known conditions for similar transformations. This simple, safe, and effective system for CH activation allows for unusual tunable selectivity between a two-electron oxidation to the allylic acetates and a four-electron oxidation to the corresponding enals, with the dominant product depending on the presence or absence of water. The versatile synthetic utility of both the allylic acetate and enal products accessible through this methodology is also demonstrated.
Co-reporter:Katerina M. Korch;Dr. Christian Eidamshaus;Dr. Douglas C. Behenna;Dr. Sangkil Nam;Dr. David Horne;Dr. Brian M. Stoltz
Angewandte Chemie International Edition 2015 Volume 54( Issue 1) pp:179-183
Publication Date(Web):
DOI:10.1002/anie.201408609
Abstract
The asymmetric palladium-catalyzed decarboxylative allylic alkylation of differentially N-protected piperazin-2-ones allows the synthesis of a variety of highly enantioenriched tertiary piperazine-2-ones. Deprotection and reduction affords the corresponding tertiary piperazines, which can be employed for the synthesis of medicinally important analogues. The introduction of these chiral tertiary piperazines resulted in imatinib analogues which exhibited comparable antiproliferative activity to that of their corresponding imatinib counterparts.
Co-reporter:Seo-Jung Han, Florian Vogt, Shyam Krishnan, Jeremy A. May, Michele Gatti, Scott C. Virgil, and Brian M. Stoltz
Organic Letters 2014 Volume 16(Issue 12) pp:3316-3319
Publication Date(Web):June 9, 2014
DOI:10.1021/ol5013263
An efficient, unified, and stereodivergent approach toward communesin F and perophoramidine was examined. The C(3) all-carbon quaternary center of an oxindole was smoothly constructed by base-promoted indolone-malonate alkylation chemistry. The complementary relative stereochemistry of the crucial vicinal quaternary centers found in communesin F and perophoramidine was selectively installed by substrate-controlled decarboxylative allylic alkylations.
Co-reporter:Samantha E. Shockley, Jeffrey C. Holder, and Brian M. Stoltz
Organic Letters 2014 Volume 16(Issue 24) pp:6362-6365
Publication Date(Web):December 9, 2014
DOI:10.1021/ol5031537
A catalytic, enantioselective formal synthesis of (+)-dichroanone and (+)-taiwaniaquinone H is reported. The all-carbon quaternary stereocenter was constructed by asymmetric conjugate addition catalyzed by a palladium(II) (S)-tert-butylpyridinooxazoline complex. The unexpected formation of a [3.2.1] bicyclic intermediate required the identification of a new route. Analysis of the Hammett constants for para-substituted arenes enabled the rational design of a highly enantioselective conjugate addition substrate that led to the completion of the formal synthesis.
Co-reporter:Corey M. Reeves, Douglas C. Behenna, and Brian M. Stoltz
Organic Letters 2014 Volume 16(Issue 9) pp:2314-2317
Publication Date(Web):April 11, 2014
DOI:10.1021/ol500355z
The development of (trimethylsilyl)ethyl ester protected enolates is reported. The application of this class of compounds in palladium-catalyzed asymmetric allylic alkylation is explored, yielding a variety of α-quaternary six- and seven-membered ketones and lactams. Independent coupling partner synthesis engenders enhanced allyl substrate scope relative to traditional β-ketoester substrates; highly functionalized α-quaternary ketones generated by the union of (trimethylsilyl)ethyl β-ketoesters and sensitive allylic alkylation coupling partners serve to demonstrate the utility of this method for complex fragment coupling.
Co-reporter:Yiyang Liu;Kelly E. Kim;Myles B. Herbert;Alexey Fedorov;Robert H. Grubbs;Brian M. Stoltz
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 1) pp:130-136
Publication Date(Web):
DOI:10.1002/adsc.201301109
Co-reporter:Seo-Jung Han, Gabriel Fernando de Melo, Brian M. Stoltz
Tetrahedron Letters 2014 Volume 55(Issue 47) pp:6467-6469
Publication Date(Web):19 November 2014
DOI:10.1016/j.tetlet.2014.10.006
A mild and efficient o- and p-nitrobenzyl cleavage protocol was developed. o- and p-nitrobenzyl groups were easily removed from a variety of substrates using 20% aqueous NaOH in methanol at 75 °C, presumably via oxidation at the benzylic position by oxygen dissolved in the solution. These easily introducible and removable nitrobenzyl groups can serve as valuable protecting groups for the synthesis of multifunctional, complex molecules.
Co-reporter:Allen Y. Hong ; Brian M. Stoltz
Angewandte Chemie International Edition 2014 Volume 53( Issue 21) pp:5248-5260
Publication Date(Web):
DOI:10.1002/anie.201309494
Abstract
Presilphiperfolanols constitute a family of biosynthetically important sesquiterpenes which can rearrange to diverse sesquiterpenoid skeletons. While the origin of these natural products can be traced to simple linear terpene precursors, the details of the enzymatic cyclization mechanism that forms the stereochemically dense tricyclic skeleton has required extensive biochemical, computational, and synthetic investigation. Parallel efforts to prepare the unique and intriguing structures of these compounds by total synthesis have also inspired novel strategies, thus resulting in four synthetic approaches and two completed syntheses. While the biosynthesis and chemical synthesis studies performed to date have provided much insight into the role and properties of these molecules, emerging questions regarding the biosynthesis of newer members of the family and subtle details of rearrangement mechanisms have yet to be explored.
Co-reporter:Allen Y. Hong ; Brian M. Stoltz
Angewandte Chemie 2014 Volume 126( Issue 21) pp:5350-5362
Publication Date(Web):
DOI:10.1002/ange.201309494
Abstract
Presilphiperfolanole bilden eine biosynthetisch bedeutende Klasse von Sesquiterpenen, die sich zu verschiedenen weiteren Sesquiterpen-Grundgerüsten umlagern können. Ihr Ursprung lässt sich auf einfache lineare Terpenvorstufen zurückverfolgen. Durch umfassende biochemische, theoretische und synthetische Arbeiten gelang es, ein mechanistisches Verständnis für die enzymatische Cyclisierung zu gewinnen. Um die ungewöhnliche und bemerkenswerte Grundstruktur der Presilphiperfolanole synthetisch zu erhalten, wurde ebenfalls intensiv an Totalsynthesen dieser Naturstoffe gearbeitet. Die dabei entwickelten Strategien mündeten in zwei Syntheseansätzen sowie zwei vollständigen Totalsynthesen. Während biosynthetisch und durch chemische Synthese schon viel zur Aufklärung der Moleküle und ihrer Mechanismen beigetragen wurde, tauchen Fragen zur Biosynthese von neueren Presilphiperfolanolen auf. Auch die Umlagerungsmechanismen sind im Detail noch unklar.
Co-reporter: Ama C. Jones; Jeremy A. May; Richmond Sarpong; Brian M. Stoltz
Angewandte Chemie International Edition 2014 Volume 53( Issue 10) pp:2556-2591
Publication Date(Web):
DOI:10.1002/anie.201302572
Abstract
Catalysis and synthesis are intimately linked in modern organic chemistry. The synthesis of complex molecules is an ever evolving area of science. In many regards, the inherent beauty associated with a synthetic sequence can be linked to a certain combination of the creativity with which a sequence is designed and the overall efficiency with which the ultimate process is performed. In synthesis, as in other endeavors, beauty is very much in the eyes of the beholder.1 It is with this in mind that we will attempt to review an area of synthesis that has fascinated us and that we find extraordinarily beautiful, namely the combination of catalysis and sigmatropic rearrangements in consecutive and cascade sequences.
Co-reporter:Robert A. Craig II;Nicholas R. O'Connor;Dr. Alexer F. G. Goldberg ; Brian M. Stoltz
Chemistry - A European Journal 2014 Volume 20( Issue 16) pp:4806-4813
Publication Date(Web):
DOI:10.1002/chem.201303699
Abstract
Alkyl and aryl isothiocyanates and carbodiimides are effective substrates in (3+2) cycloadditions with N-sulfonyl-2-substituted aziridines and 2-phenylaziridine for the synthesis of iminothiazolidines and iminoimidazolidines. Additionally, the stereoselective (3+2) cycloaddition of N-H- and N-sulfonylaziridines with isothiocyanates can be accomplished, allowing for the synthesis of highly enantioenriched iminothiazolidines. Evidence for an intimate ion-pair mechanism is presented herein in the context of these chemo-, regio-, and diastereoselective transformations. The demonstrated ability to remove the sulfonyl group from the heterocyclic products displays the utility of these compounds for further derivatization and application.
Co-reporter:Sandy Ma, Corey M. Reeves, Robert A. Craig II, Brian M. Stoltz
Tetrahedron 2014 70(27–28) pp: 4208-4212
Publication Date(Web):
DOI:10.1016/j.tet.2014.03.042
Co-reporter:Jonny R. Gordon, Hosea M. Nelson, Scott C. Virgil, and Brian M. Stoltz
The Journal of Organic Chemistry 2014 Volume 79(Issue 20) pp:9740-9747
Publication Date(Web):September 22, 2014
DOI:10.1021/jo501924u
The total syntheses of basiliolide C and previously unreported epi-basiliolide C are achieved by an Ireland–Claisen/Diels–Alder cascade. The development of a palladium catalyzed cross-coupling of methoxy alkynyl zinc reagents allows for the protecting-group-free syntheses of transtaganolides C and D. Syntheses of transtaganolides C and D are accomplished in a single operation to generate three rings, two all-carbon quaternary centers, and four tertiary stereocenters from a monocyclic, achiral precursor.
Co-reporter:Jeffrey C. Holder ; Lufeng Zou ; Alexander N. Marziale ; Peng Liu ; Yu Lan ; Michele Gatti ; Kotaro Kikushima ; K. N. Houk ;Brian M. Stoltz
Journal of the American Chemical Society 2013 Volume 135(Issue 40) pp:14996-15007
Publication Date(Web):September 12, 2013
DOI:10.1021/ja401713g
Enantioselective conjugate additions of arylboronic acids to β-substituted cyclic enones have been previously reported from our laboratories. Air- and moisture-tolerant conditions were achieved with a catalyst derived in situ from palladium(II) trifluoroacetate and the chiral ligand (S)-t-BuPyOx. We now report a combined experimental and computational investigation on the mechanism, the nature of the active catalyst, the origins of the enantioselectivity, and the stereoelectronic effects of the ligand and the substrates of this transformation. Enantioselectivity is controlled primarily by steric repulsions between the t-Bu group of the chiral ligand and the α-methylene hydrogens of the enone substrate in the enantiodetermining carbopalladation step. Computations indicate that the reaction occurs via formation of a cationic arylpalladium(II) species, and subsequent carbopalladation of the enone olefin forms the key carbon–carbon bond. Studies of nonlinear effects and stoichiometric and catalytic reactions of isolated (PyOx)Pd(Ph)I complexes show that a monomeric arylpalladium–ligand complex is the active species in the selectivity-determining step. The addition of water and ammonium hexafluorophosphate synergistically increases the rate of the reaction, corroborating the hypothesis that a cationic palladium species is involved in the reaction pathway. These additives also allow the reaction to be performed at 40 °C and facilitate an expanded substrate scope.
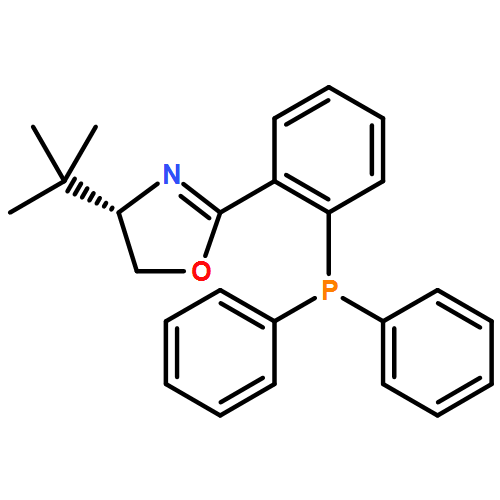
Co-reporter:Vikram Bhat ; Su Wang ; Brian M. Stoltz ;Scott C. Virgil
Journal of the American Chemical Society 2013 Volume 135(Issue 45) pp:16829-16832
Publication Date(Web):October 23, 2013
DOI:10.1021/ja409383f
A palladium-catalyzed, atroposelective C–P coupling process has been developed for the asymmetric synthesis of QUINAP and its derivatives in high enantiomeric excess. Bromide, triflate (OTf) and 4-methanesulfonylbenzenesulfonate (OSs) precursors were studied, leading in the case of the triflate to a novel dynamic kinetic resolution involving isomerization of an arylpalladium intermediate. The operationally simple methods described in this communication afford these important ligands in good to high yields and selectivity using low catalyst loading (≤3 mol % Pd).
Co-reporter:Wen-Bo Liu ; Corey M. Reeves ;Brian M. Stoltz
Journal of the American Chemical Society 2013 Volume 135(Issue 46) pp:17298-17301
Publication Date(Web):October 28, 2013
DOI:10.1021/ja4097829
The first regio-, diastereo-, and enantioselective allylic alkylation of acyclic β-ketoesters to form vicinal tertiary and all-carbon quaternary stereocenters is reported. Critical to the successful development of this method was the employment of iridium catalysis in concert with N-aryl-phosphoramidite ligands. Broad functional group tolerance is observed at the keto-, ester-, and α-positions of the nucleophile. Various transformations demonstrating the utility of this method for rapidly accessing complex enantioenriched compounds are reported.
Co-reporter:Wen-Bo Liu ; Corey M. Reeves ; Scott C. Virgil ;Brian M. Stoltz
Journal of the American Chemical Society 2013 Volume 135(Issue 29) pp:10626-10629
Publication Date(Web):July 7, 2013
DOI:10.1021/ja4052075
Highly congested vicinal stereocenters comprised of tertiary and all-carbon quaternary centers were generated via Ir-catalyzed asymmetric allylic alkylation of β-ketoesters. These catalytic reactions proceed in excellent yields with a broad scope on either reaction partner and with outstanding regio-, diastereo-, and enantiocontrol. Implementation of a subsequent Pd-catalyzed alkylation affords dialkylated products with pinpoint stereochemical control of both chiral centers.
Co-reporter:Grant M. Shibuya, John A. Enquist Jr., and Brian M. Stoltz
Organic Letters 2013 Volume 15(Issue 13) pp:3480-3483
Publication Date(Web):June 26, 2013
DOI:10.1021/ol401514s
A catalytic enantioselective double allylic alkylation reaction has been employed in the synthesis of the core of the gagunin diterpenoids. Enantioenriched material was advanced in 11 steps to afford the core of the highly oxygenated target, which includes two all-carbon quaternary stereocenters.
Co-reporter:Allen Y. Hong ;Brian M. Stoltz
European Journal of Organic Chemistry 2013 Volume 2013( Issue 14) pp:2745-2759
Publication Date(Web):
DOI:10.1002/ejoc.201201761
Abstract
All-carbon quaternary stereocenters have posed significant challenges in the synthesis of complex natural products. These important structural motifs have inspired the development of broadly applicable palladium-catalyzed asymmetric allylic alkylation reactions of unstabilized non-biased enolates for the synthesis of enantioenriched α-quaternary products. This microreview outlines key considerations in the application of palladium-catalyzed asymmetric allylic alkylation reactions and presents recent total syntheses of complex natural products that have employed these powerful transformations for the direct, catalytic, enantioselective construction of all-carbon quaternary stereocenters.
Co-reporter:Corey M. Reeves;Christian Eidamshaus;Jimin Kim ; Brian M. Stoltz
Angewandte Chemie 2013 Volume 125( Issue 26) pp:6850-6853
Publication Date(Web):
DOI:10.1002/ange.201301815
Co-reporter:Hosea M. Nelson;Jonny R. Gordon;Dr. Scott C. Virgil ;Dr. Brian M. Stoltz
Angewandte Chemie 2013 Volume 125( Issue 26) pp:6831-6835
Publication Date(Web):
DOI:10.1002/ange.201301212
Co-reporter:Christopher K. Haley, Christopher D. Gilmore, Brian M. Stoltz
Tetrahedron 2013 69(27–28) pp: 5732-5736
Publication Date(Web):
DOI:10.1016/j.tet.2013.03.085
Co-reporter:Nathan B. Bennett ; Brian M. Stoltz
Chemistry - A European Journal 2013 Volume 19( Issue 52) pp:17745-17750
Publication Date(Web):
DOI:10.1002/chem.201302353
Abstract
Access to the bicyclo[5.3.0]decane core found in the daucane and sphenolobane terpenoids via a key enone intermediate enables the enantioselective total syntheses of daucene, daucenal, epoxydaucenal B, and 14-para-anisoyloxydauc-4,8-diene. Central aspects include a catalytic asymmetric alkylation followed by a ring contraction and ring-closing metathesis to generate the five- and seven-membered rings, respectively.
Co-reporter:Jeffrey C. Holder;Dr. Alexer N. Marziale;Dr. Michele Gatti;Bin Mao ; Brian M. Stoltz
Chemistry - A European Journal 2013 Volume 19( Issue 1) pp:74-77
Publication Date(Web):
DOI:10.1002/chem.201203643
Co-reporter:Nathan B. Bennett;Douglas C. Duquette;Dr. Jimin Kim;Dr. Wen-Bo Liu;Dr. Alexer N. Marziale;Dr. Douglas C. Behenna;Dr. Scott C. Virgil ; Brian M. Stoltz
Chemistry - A European Journal 2013 Volume 19( Issue 14) pp:4414-4418
Publication Date(Web):
DOI:10.1002/chem.201300030
Co-reporter:Michael E. Meyer, John H. Phillips, Eric M. Ferreira, Brian M. Stoltz
Tetrahedron 2013 69(36) pp: 7627-7635
Publication Date(Web):
DOI:10.1016/j.tet.2013.02.034
Co-reporter:Corey M. Reeves;Christian Eidamshaus;Jimin Kim ; Brian M. Stoltz
Angewandte Chemie International Edition 2013 Volume 52( Issue 26) pp:6718-6721
Publication Date(Web):
DOI:10.1002/anie.201301815
Co-reporter:Hosea M. Nelson;Jonny R. Gordon;Dr. Scott C. Virgil ;Dr. Brian M. Stoltz
Angewandte Chemie International Edition 2013 Volume 52( Issue 26) pp:6699-6703
Publication Date(Web):
DOI:10.1002/anie.201301212
Co-reporter:Pamela M. Tadross and Brian M. Stoltz
Chemical Reviews 2012 Volume 112(Issue 6) pp:3550
Publication Date(Web):March 23, 2012
DOI:10.1021/cr200478h
Co-reporter:John A. Keith ; Douglas C. Behenna ; Nathaniel Sherden ; Justin T. Mohr ; Sandy Ma ; Smaranda C. Marinescu ; Robert J. Nielsen ; Jonas Oxgaard ; Brian M. Stoltz ;William A. Goddard ; III
Journal of the American Chemical Society 2012 Volume 134(Issue 46) pp:19050-19060
Publication Date(Web):October 28, 2012
DOI:10.1021/ja306860n
We use first principles quantum mechanics (density functional theory) to report a detailed reaction mechanism of the asymmetric Tsuji allylation involving prochiral nucleophiles and nonprochiral allyl fragments, which is consistent with experimental findings. The observed enantioselectivity is best explained with an inner-sphere mechanism involving the formation of a 5-coordinate Pd species that undergoes a ligand rearrangement, which is selective with regard to the prochiral faces of the intermediate enolate. Subsequent reductive elimination generates the product and a Pd0 complex. The reductive elimination occurs via an unconventional seven-centered transition state that contrasts dramatically with the standard three-centered C–C reductive elimination mechanism. Although limitations in the present theory prevent the conclusive identification of the enantioselective step, we note that three different computational schemes using different levels of theory all find that inner-sphere pathways are lower in energy than outer-sphere pathways. This result qualitatively contrasts with established allylation reaction mechanisms involving prochiral nucleophiles and prochiral allyl fragments. Energetic profiles of all reaction pathways are presented in detail.
Co-reporter:Robert A. Craig II, Jennifer L. Roizen, Russell C. Smith, Amanda C. Jones, and Brian M. Stoltz
Organic Letters 2012 Volume 14(Issue 22) pp:5716-5719
Publication Date(Web):October 26, 2012
DOI:10.1021/ol3027297
A brief, enantioselective synthesis of a hydroxymethyl-cis-1,3-cyclopentenediol building block is presented. This scaffold allows access to the cis-1,3-cyclopentanediol fragments found in a variety of biologically active natural and non-natural products. This rapid and efficient synthesis is highlighted by the utilization of the palladium-catalyzed enantioselective allylic alkylation of dioxanone substrates to prepare tertiary alcohols.
Co-reporter:Alexander F. G. Goldberg, Nicholas R. O’Connor, Robert A. Craig II, and Brian M. Stoltz
Organic Letters 2012 Volume 14(Issue 20) pp:5314-5317
Publication Date(Web):October 9, 2012
DOI:10.1021/ol302494n
Isocyanates, isothiocyanates, and carbodiimides are effective substrates in (3 + 2) cycloadditions with donor–acceptor cyclopropanes for the synthesis of five-membered heterocycles. These reactions exhibit a broad substrate scope, high yields, and well-defined chemoselectivity. Discussed herein are the implications of Lewis acid choice on the stereochemical outcome and the reaction mechanism.
Co-reporter:Nathan B. Bennett, Allen Y. Hong, Andrew M. Harned and Brian M. Stoltz
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 1) pp:56-59
Publication Date(Web):01 Sep 2011
DOI:10.1039/C1OB06189E
A general method for the synthesis of β-substituted and unsubstituted cycloheptenones bearing enantioenriched all-carbon γ-quaternary stereocenters is reported. Hydride or organometallic addition to a seven-membered ring vinylogous ester followed by finely tuned quenching parameters achieves elimination to the corresponding cycloheptenone. The resulting enones are elaborated to bi- and tricyclic compounds with potential for the preparation of non-natural analogs and whose structures are embedded in a number of cycloheptanoid natural products.
Co-reporter:Jimin Kim, Brian M. Stoltz
Tetrahedron Letters 2012 Volume 53(Issue 37) pp:4994-4996
Publication Date(Web):12 September 2012
DOI:10.1016/j.tetlet.2012.07.026
Intermolecular insertion of benzyne into the C–N bond of a β-lactam is described. This σ-insertion is followed by ring expansion that produces dihydroquinolinone, which rapidly reacts with an additional benzyne unit to afford an acridone through intramolecular C–C bond formation to the carbonyl group and rapid elimination of ethylene.
Co-reporter:Allen Y. Hong ; Brian M. Stoltz
Angewandte Chemie International Edition 2012 Volume 51( Issue 38) pp:9674-9678
Publication Date(Web):
DOI:10.1002/anie.201205276
Co-reporter:Allen Y. Hong ; Brian M. Stoltz
Angewandte Chemie 2012 Volume 124( Issue 38) pp:9812-9816
Publication Date(Web):
DOI:10.1002/ange.201205276
Co-reporter:Kotaro Kikushima ; Jeffrey C. Holder ; Michele Gatti ;Brian M. Stoltz
Journal of the American Chemical Society 2011 Volume 133(Issue 18) pp:6902-6905
Publication Date(Web):April 15, 2011
DOI:10.1021/ja200664x
The first enantioselective Pd-catalyzed construction of all-carbon quaternary stereocenters via 1,4-addition of arylboronic acids to β-substituted cyclic enones is reported. Reaction of a wide range of arylboronic acids and cyclic enones using a catalyst prepared from Pd(OCOCF3)2 and a chiral pyridinooxazoline ligand yields enantioenriched products bearing benzylic stereocenters. Notably, this transformation is tolerant to air and moisture, providing a practical and operationally simple method of synthesizing enantioenriched all-carbon quaternary stereocenters.
Co-reporter:Herschel Mukherjee, Nolan T. McDougal, Scott C. Virgil, and Brian M. Stoltz
Organic Letters 2011 Volume 13(Issue 5) pp:825-827
Publication Date(Web):January 27, 2011
DOI:10.1021/ol102669z
A concise asymmetric, formal synthesis of (+)-hamigeran B is reported. A Pd-catalyzed, decarboxylative allylic alkylation, employing a trifluoromethylated derivative of t-BuPHOX, is utilized as the enantioselective step to form the critical quaternary carbon center in excellent yield and enantioselectivity. The product is converted in three steps to a late-stage intermediate previously used in the synthesis of hamigeran B.
Co-reporter:Alexander F. G. Goldberg and Brian M. Stoltz
Organic Letters 2011 Volume 13(Issue 16) pp:4474-4476
Publication Date(Web):July 25, 2011
DOI:10.1021/ol2017615
A palladium-catalyzed (3 + 2) cycloaddition of a vinylcyclopropane and a β-nitrostyrene is employed to rapidly assemble the cyclopentane core of the Melodinus alkaloids. The ABCD ring system of the natural product family is prepared in six steps from commercially available materials.
Co-reporter:Pamela M. Tadross, Pradeep Bugga and Brian M. Stoltz
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 15) pp:5354-5357
Publication Date(Web):26 May 2011
DOI:10.1039/C1OB05725A
The tetracyclic core of the integrastatin natural products has been prepared in a convergent and rapid manner. Our strategy relies upon a palladium(II)-catalyzed oxidative cyclization to form the central [3.3.1]-dioxabicycle of the natural product core. Overall, the core has been completed in only 4 linear steps from known compounds.
Co-reporter:Douglas C. Behenna, Shyam Krishnan, Brian M. Stoltz
Tetrahedron Letters 2011 Volume 52(Issue 17) pp:2152-2154
Publication Date(Web):27 April 2011
DOI:10.1016/j.tetlet.2010.11.074
We confirm our previous assignment of the absolute configuration of (−)-aurantioclavine as 7R by crystallographically characterizing an advanced 3-bromoindole intermediate reported in our previous synthesis. This analysis also provides additional support for our model of enantioinduction in the palladium(II)-catalyzed oxidative kinetic resolution of secondary alcohols.
Co-reporter:Allen Y. Hong, Nathan B. Bennett, Michael R. Krout, Thomas Jensen, Andrew M. Harned, Brian M. Stoltz
Tetrahedron 2011 67(52) pp: 10234-10248
Publication Date(Web):
DOI:10.1016/j.tet.2011.10.031
Co-reporter:Kimberly S. Petersen, Brian M. Stoltz
Tetrahedron 2011 67(24) pp: 4352-4357
Publication Date(Web):
DOI:10.1016/j.tet.2011.04.046
Co-reporter:Allen Y. Hong;Dr. Michael R. Krout;Dr. Thomas Jensen;Nathan B. Bennett; Andrew M. Harned ; Brian M. Stoltz
Angewandte Chemie International Edition 2011 Volume 50( Issue 12) pp:2756-2760
Publication Date(Web):
DOI:10.1002/anie.201007814
Co-reporter:Dr. Joshua J. Day;Ryan M. McFadden;Dr. Scott C. Virgil;Helene Kolding;Jennifer L. Alleva ; Brian M. Stoltz
Angewandte Chemie 2011 Volume 123( Issue 30) pp:6946-6950
Publication Date(Web):
DOI:10.1002/ange.201101842
Co-reporter:Kevin M. Allan;Christopher D. Gilmore ; Brian M. Stoltz
Angewandte Chemie 2011 Volume 123( Issue 19) pp:4580-4583
Publication Date(Web):
DOI:10.1002/ange.201100911
Co-reporter:John A. Enquist Jr.;Dr. Scott C. Virgil ; Brian M. Stoltz
Chemistry - A European Journal 2011 Volume 17( Issue 36) pp:9957-9969
Publication Date(Web):
DOI:10.1002/chem.201100425
Abstract
A concise and versatile approach toward the preparation of the cyanthiwigin family of cyathane natural products is described. By leveraging a unique double asymmetric catalytic alkylation procedure it is possible to quickly establish two of the most critical stereocenters of the cyanthiwigin framework with high levels of selectivity and expediency. The synthetic route additionally employs both a tandem ring-closing cross-metathesis reaction, and an aldehyde-olefin radical cyclization process, in order to rapidly arrive at the tricyclic cyathane core of the cyanthiwigin molecules. From this unifying intermediate, the preparations of cyanthiwigins B, F, and G are attained swiftly and without the need for protecting groups.
Co-reporter:Dr. Douglas C. Behenna;Dr. Justin T. Mohr;Dr. Nathaniel H. Sherden;Smara C. Marinescu; Andrew M. Harned;Dr. Kousuke Tani;Dr. Masaki Seto;Dr. Sy Ma;Dr. Zoltán Novák;Dr. Michael R. Krout;Dr. Ryan M. McFadden;Dr. Jennifer L. Roizen;Dr. John A. Enquist Jr.;Dr. David E. White;Samantha R. Levine;Krastina V. Petrova;Dr. Akihiko Iwashita;Dr. Scott C. Virgil ; Brian M. Stoltz
Chemistry - A European Journal 2011 Volume 17( Issue 50) pp:14199-14223
Publication Date(Web):
DOI:10.1002/chem.201003383
Abstract
α-Quaternary ketones are accessed through novel enantioselective alkylations of allyl and propargyl electrophiles by unstabilized prochiral enolate nucleophiles in the presence of palladium complexes with various phosphinooxazoline (PHOX) ligands. Excellent yields and high enantiomeric excesses are obtained from three classes of enolate precursor: enol carbonates, enol silanes, and racemic β-ketoesters. Each of these substrate classes functions with nearly identical efficiency in terms of yield and enantioselectivity. Catalyst discovery and development, the optimization of reaction conditions, the exploration of reaction scope, and applications in target-directed synthesis are reported. Experimental observations suggest that these alkylation reactions occur through an unusual inner-sphere mechanism involving binding of the prochiral enolate nucleophile directly to the palladium center.
Co-reporter:Kevin M. Allan;Christopher D. Gilmore ; Brian M. Stoltz
Angewandte Chemie International Edition 2011 Volume 50( Issue 19) pp:4488-4491
Publication Date(Web):
DOI:10.1002/anie.201100911
Co-reporter:Dr. Joshua J. Day;Ryan M. McFadden;Dr. Scott C. Virgil;Helene Kolding;Jennifer L. Alleva ; Brian M. Stoltz
Angewandte Chemie International Edition 2011 Volume 50( Issue 30) pp:6814-6818
Publication Date(Web):
DOI:10.1002/anie.201101842
Co-reporter:Hosea M. Nelson;Kei Murakami;Dr. Scott C. Virgil ;Dr. Brian M. Stoltz
Angewandte Chemie International Edition 2011 Volume 50( Issue 16) pp:3688-3691
Publication Date(Web):
DOI:10.1002/anie.201008003
Co-reporter:Hosea M. Nelson;Kei Murakami;Dr. Scott C. Virgil ;Dr. Brian M. Stoltz
Angewandte Chemie 2011 Volume 123( Issue 16) pp:3772-3775
Publication Date(Web):
DOI:10.1002/ange.201008003
Co-reporter:Allen Y. Hong;Dr. Michael R. Krout;Dr. Thomas Jensen;Nathan B. Bennett; Andrew M. Harned ; Brian M. Stoltz
Angewandte Chemie 2011 Volume 123( Issue 12) pp:2808-2812
Publication Date(Web):
DOI:10.1002/ange.201007814
Co-reporter:Pamela M. Tadross, Christopher D. Gilmore, Pradeep Bugga, Scott C. Virgil and Brian M. Stoltz
Organic Letters 2010 Volume 12(Issue 6) pp:1224-1227
Publication Date(Web):February 18, 2010
DOI:10.1021/ol1000796
The fully regioselective reactivity of four new highly substituted silyl aryl triflate aryne precursors in aryne acyl-alkylation, acyl-alkylation/condensation, and heteroannulation reactions is reported. The application of these more complex arynes provides access to diverse natural product scaffolds and obviates late-stage functionalization of aromatic rings.
Co-reporter:Pamela M. Tadross, Scott C. Virgil and Brian M. Stoltz
Organic Letters 2010 Volume 12(Issue 7) pp:1612-1614
Publication Date(Web):March 2, 2010
DOI:10.1021/ol100335y
A general approach for the synthesis of benzannulated macrolactone natural products utilizing an aryne acyl-alkylation reaction is described. Toward this end, the total syntheses of the natural products (−)-curvularin, curvulin, and (−)-diplodialide C are reported. Furthermore, the aryne insertion technology has enabled the rapid conversion of simple diplodialide natural products to curvularin, thereby connecting these two biosynthetically distinct classes of compounds via synthetic methods.
Co-reporter:Corinne Baumgartner, Sandy Ma, Qi Liu and Brian M. Stoltz
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 13) pp:2915-2917
Publication Date(Web):24 May 2010
DOI:10.1039/C004275G
Our efforts toward the construction of the carbocylic core of cortistatin A via an enyne-ene metathesis are disclosed. Interestingly, an attempted SN2 inversion of a secondary mesylate in our five-membered D-ring piece gave a product with retention of stereochemistry.
Co-reporter:Nolan T. McDougal, Jan Streuff, Herschel Mukherjee, Scott C. Virgil, Brian M. Stoltz
Tetrahedron Letters 2010 Volume 51(Issue 42) pp:5550-5554
Publication Date(Web):20 October 2010
DOI:10.1016/j.tetlet.2010.08.039
Herein an efficient and direct copper-catalyzed coupling of oxazoline-containing aryl bromides with electron-deficient secondary phosphine oxides is reported. The resulting tertiary phosphine oxides can be reduced to prepare a range of PHOX ligands. The presented strategy is a useful alternative to known methods for constructing PHOX derivatives.
Co-reporter:David E. White, Ian C. Stewart, Brinton A. Seashore-Ludlow, Robert H. Grubbs, Brian M. Stoltz
Tetrahedron 2010 66(26) pp: 4668-4686
Publication Date(Web):
DOI:10.1016/j.tet.2010.04.128
Co-reporter:John A. Enquist, Jr. and Brian M. Stoltz
Natural Product Reports 2009 vol. 26(Issue 5) pp:661-680
Publication Date(Web):16 Mar 2009
DOI:10.1039/B811227B
Covering: 2000 to 2008. Previous review: D. L. Wright and C. R. Whitehead, Org. Prep. Proced. Int., 2000, 32, 309–330
Co-reporter:Kevin M. Allan, Boram D. Hong and Brian M. Stoltz
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 23) pp:4960-4964
Publication Date(Web):06 Oct 2009
DOI:10.1039/B913336D
A convenient method is disclosed for the synthesis of both 3-hydroxyisoquinolines and 2-hydroxy-1,4-naphthoquinones from β-ketoesters using a one-pot aryne acyl-alkylation/condensation procedure. When performed in conjunction with a one-step method for the synthesis of the β-ketoester substrates, this method provides a new route to these polyaromatic structures in only two steps from commercially available carboxylic acid starting materials. The utility of this approach is demonstrated in the synthesis of the atropisomeric P,N-ligand, QUINAP.
Co-reporter:NathanielH. Sherden;DouglasC. Behenna Dr.;ScottC. Virgil Dr. ;BrianM. Stoltz
Angewandte Chemie 2009 Volume 121( Issue 37) pp:6972-6975
Publication Date(Web):
DOI:10.1002/ange.200902575
Co-reporter:Hosea M. Nelson, Brian M. Stoltz
Tetrahedron Letters 2009 50(15) pp: 1699-1701
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.01.108
Co-reporter:Jennifer L. Stockdill, Douglas C. Behenna, Andrew McClory, Brian M. Stoltz
Tetrahedron 2009 65(33) pp: 6571-6575
Publication Date(Web):
DOI:10.1016/j.tet.2009.05.023
Co-reporter:Jennifer L. Stockdill, Douglas C. Behenna, Brian M. Stoltz
Tetrahedron Letters 2009 50(26) pp: 3182-3184
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.01.121
Co-reporter:Sy Ma;Xiaoqing Han Dr.;Shyam Krishnan Dr.;ScottC. Virgil Dr.;BrianM. Stoltz
Angewandte Chemie International Edition 2009 Volume 48( Issue 43) pp:8037-8041
Publication Date(Web):
DOI:10.1002/anie.200902943
Co-reporter:NathanielH. Sherden;DouglasC. Behenna Dr.;ScottC. Virgil Dr. ;BrianM. Stoltz
Angewandte Chemie International Edition 2009 Volume 48( Issue 37) pp:6840-6843
Publication Date(Web):
DOI:10.1002/anie.200902575
Co-reporter:DavidC. Ebner Dr.;JeffreyT. Bagdanoff Dr.;EricM. Ferreira Dr.;RyanM. McFadden Dr.;DanielD. Caspi Dr.;RaissaM. Trend Dr. ;BrianM. Stoltz
Chemistry - A European Journal 2009 Volume 15( Issue 47) pp:12978-12992
Publication Date(Web):
DOI:10.1002/chem.200902172
Abstract
The first palladium-catalyzed enantioselective oxidation of secondary alcohols has been developed, utilizing the readily available diamine (−)-sparteine as a chiral ligand and molecular oxygen as the stoichiometric oxidant. Mechanistic insights regarding the role of the base and hydrogen-bond donors have resulted in several improvements to the original system. Namely, addition of cesium carbonate and tert-butyl alcohol greatly enhances reaction rates, promoting rapid resolutions. The use of chloroform as solvent allows the use of ambient air as the terminal oxidant at 23 °C, resulting in enhanced catalyst selectivity. These improved reaction conditions have permitted the successful kinetic resolution of benzylic, allylic, and cyclopropyl secondary alcohols to high enantiomeric excess with good-to-excellent selectivity factors. This catalyst system has also been applied to the desymmetrization of meso-diols, providing high yields of enantioenriched hydroxyketones.
Co-reporter:Sy Ma;Xiaoqing Han Dr.;Shyam Krishnan Dr.;ScottC. Virgil Dr.;BrianM. Stoltz
Angewandte Chemie 2009 Volume 121( Issue 43) pp:8181-8185
Publication Date(Web):
DOI:10.1002/ange.200902943
Co-reporter:DouglasC. Behenna Dr.;JenniferL. Stockdill ;BrianM. Stoltz Dr.
Angewandte Chemie 2008 Volume 120( Issue 13) pp:2400-2421
Publication Date(Web):
DOI:10.1002/ange.200703172
Abstract
Marine Naturstoffe spielen schon lange eine wichtige Rolle in der Naturstoffchemie und bei der Wirkstoffentwicklung. Die Artenvielfalt und das facettenreiche Wechselspiel im Lebensraum Meer spiegeln sich in den Eigenschaften der Substanzen wider, die aus marinen Lebensformen isoliert werden: Sie zeigen unwahrscheinlich vielseitige Molekülstrukturen und biologische Aktivitäten. Die Naturstoffe aus den Polypen der marinen Zoanthidea bilden darin keine Ausnahme. Die Zoanthamin-Alkaloide, deren erste Vertreter vor über zwanzig Jahren isoliert wurden, erregten besonderes Interesse als Syntheseziele, weil sie sich durch eine große Bandbreite an biologischen Aktivitäten auszeichnen. Hier fassen wir die wichtigsten Beiträge zur Isolierung und strukturellen Charakterisierung der natürlichen Zoanthamine zusammen und führen Studien zu ihrer biologischen Aktivität und Totalsynthese an.
Co-reporter:Masaki Seto Dr.;JenniferL. Roizen ;BrianM. Stoltz
Angewandte Chemie 2008 Volume 120( Issue 36) pp:6979-6982
Publication Date(Web):
DOI:10.1002/ange.200801424
Co-reporter:DouglasC. Behenna Dr.;JenniferL. Stockdill ;BrianM. Stoltz Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 13) pp:2365-2386
Publication Date(Web):
DOI:10.1002/anie.200703172
Abstract
Marine natural products have long played an important role in natural products chemistry and drug discovery. Mirroring the rich variety and complicated interactions of the marine environment, the substances isolated from sea creatures tend to be incredibly diverse in both molecular structure and biological activity. The natural products isolated from the polyps of marine zoanthids are no exception. The zoanthamine alkaloids, the first of which were isolated over 20 years ago, are of particular interest to the synthetic community because they feature a novel structural framework and exhibit a broad range of biological activities. In this Review, we summarize the major contributions to understanding the zoanthamine natural products with regard to their isolation and structure determination, as well as studies on their biological activity and total synthesis.
Co-reporter:DavidC. Ebner;RaissaM. Trend Dr.;Cédric Genet;MatthewJ. McGrath Dr.;Peter O'Brien ;BrianM. Stoltz
Angewandte Chemie International Edition 2008 Volume 47( Issue 34) pp:6367-6370
Publication Date(Web):
DOI:10.1002/anie.200801865
Co-reporter:Masaki Seto Dr.;JenniferL. Roizen ;BrianM. Stoltz
Angewandte Chemie International Edition 2008 Volume 47( Issue 36) pp:6873-6876
Publication Date(Web):
DOI:10.1002/anie.200801424
Co-reporter:John A. Enquist Jr
&
Brian M. Stoltz
Nature 2008 453(7199) pp:1228
Publication Date(Web):2008-06-26
DOI:10.1038/nature07046
Double catalytic enantioselective transformations are powerful synthetic methods that can facilitate the construction of stereochemically complex molecules in a single operation1, 2. In addition to generating two or more stereocentres in a single reaction, multiple asymmetric reactions also impart increased enantiomeric excess to the final product in comparison with the analogous single transformation3, 4, 5, 6. Furthermore, multiple asymmetric operations have the potential to independently construct several stereocentres at remote points within the same molecular scaffold, rather than relying on pre-existing chiral centres that are proximal to the reactive site1. Despite the inherent benefits of multiple catalytic enantioselective reactions, their application to natural product total synthesis remains largely underutilized2. Here we report the use of a double stereoablative7 enantioselective alkylation reaction in a concise synthesis of the marine diterpenoid (-)-cyanthiwigin F (ref. 8). By employing a technique for independent, selective formation of two stereocentres in a single stereoconvergent operation, we demonstrate that a complicated mixture of racemic and meso diastereomers may be smoothly converted to a synthetically useful intermediate with exceptional enantiomeric excess. The stereochemical information generated by means of this catalytic transformation facilitates the easy and rapid completion of the total synthesis of this marine natural product.
Co-reporter:Justin T. Mohr, David C. Ebner and Brian M. Stoltz
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 22) pp:3571-3576
Publication Date(Web):01 Oct 2007
DOI:10.1039/B711159M
Approaches to the preparation of enantioenriched materials via catalytic methods that destroy stereogenic elements of a molecule are discussed. Although these processes often decrease overall molecular complexity, there are several notable advantages including material recycling, enantiodivergence and convergence, and increased substrate scope. Examples are accompanied by discussion of the critical design elements required for the success of these methods.
Co-reporter:Justin T. Mohr;Brian M. Stoltz
Chemistry – An Asian Journal 2007 Volume 2(Issue 12) pp:1476-1491
Publication Date(Web):12 OCT 2007
DOI:10.1002/asia.200700183
The family of allylation reactions developed by Tsuji in the 1980s are capable of generating tertiary and quaternary carbon stereocenters from several synthetic precursors. Despite the utility of these transformations, they have seen little use in the synthesis of natural products. Recently, the power of these reactions was significantly enhanced by the development of enantioselective versions of these transformations. Applications of these methods to the enantioselective syntheses of natural products and pharmaceutical compounds highlight the importance of these developments.
Co-reporter:Douglas C. Behenna;Jennifer L. Stockdill;Brian M. Stoltz
Angewandte Chemie 2007 Volume 119(Issue 22) pp:
Publication Date(Web):19 APR 2007
DOI:10.1002/ange.200700430
Eine heiße Cyclisierung! Wenn die racemische Cyclisierungsvorstufe in reiner Trifluoressigsäure erhitzt wird, liefert eine ungewöhnliche Cyclisierung vom Friedel-Crafts-Typ das carbocyclische Gerüst des marinen Alkaloids Zoanthenol mit zwei all-C-substituierten quartären Zentren. Eine katalytische asymmetrische Alkylierung öffnet den Zugang zu einem enantioselektiven Syntheseweg. TBS=tert-Butyldimethylsilyl.
Co-reporter:Douglas C. Behenna;Jennifer L. Stockdill;Brian M. Stoltz
Angewandte Chemie International Edition 2007 Volume 46(Issue 22) pp:
Publication Date(Web):19 APR 2007
DOI:10.1002/anie.200700430
A smokin' hot cyclization! When the racemic cyclization precursor is heated in neat trifluoroacetic acid, an unusual Friedel–Crafts-type cyclization forms the carbocyclic core of the marine alkaloid zoanthenol containing two all-carbon-substituted quaternary centers. Catalytic asymmetric alkylation allows entry into an enantioselective route.
Co-reporter:Michael E. Meyer, Eric M. Ferreira and Brian M. Stoltz
Chemical Communications 2006 (Issue 12) pp:1316-1318
Publication Date(Web):15 Feb 2006
DOI:10.1039/B517719G
The formation of various α-diazo acetoacetic esters can be obtained in a single transformation with good to excellent yields using readily available 2-diazoacetoacetic acid.
Co-reporter:Neil K. Garg and Brian M. Stoltz
Chemical Communications 2006 (Issue 36) pp:3769-3779
Publication Date(Web):18 Jul 2006
DOI:10.1039/B605929E
This review describes recent developments from our laboratory involving the synthesis of the structurally complex, pyrazinone-containing dragmacidin alkaloids.
Co-reporter:Kousuke Tani
and Brian M. Stoltz
Nature 2006 441(7094) pp:731
Publication Date(Web):
DOI:10.1038/nature04842
Co-reporter:Neil K. Garg, Carolyn C. Woodroofe, Christopher J. Lacenere, Stephen R. Quake and Brian M. Stoltz
Chemical Communications 2005 (Issue 36) pp:4551-4553
Publication Date(Web):12 Aug 2005
DOI:10.1039/B505737J
A mild heterogeneous, ligand-free protocol for Sonogashira and Heck couplings has been developed and used to access several biologically important deoxynucleoside derivatives in a facile manner.
Co-reporter:Justin T. Mohr;Douglas C. Behenna;Andrew M. Harned Dr.;Brian M. Stoltz
Angewandte Chemie 2005 Volume 117(Issue 42) pp:
Publication Date(Web):5 OCT 2005
DOI:10.1002/ange.200502018
Stereochemische Alchemie! Aus racemischen Allyl-β-ketoestern lassen sich regiokontrolliert Enolate erzeugen, wobei derselbe Katalysator sowohl den Abbau (C-C-Bindungsbruch) als auch den selektiven Aufbau eines Stereozentrums (C-C-Verknüpfung) vermittelt. Auf diese Art wurden mehrere quartäre Kohlenstoffstereozentren in einer einzigen Kaskadenreaktion gebildet (siehe Schema).
Co-reporter:Justin T. Mohr, Douglas C. Behenna, Andrew M. Harned,Brian M. Stoltz
Angewandte Chemie International Edition 2005 44(42) pp:6924-6927
Publication Date(Web):
DOI:10.1002/anie.200502018
Co-reporter:Daniel D. Caspi;David C. Ebner;Jeffrey T. Bagdanoff;Brian M. Stoltz
Advanced Synthesis & Catalysis 2004 Volume 346(Issue 2-3) pp:
Publication Date(Web):29 MAR 2004
DOI:10.1002/adsc.200303188
The palladium-catalyzed aerobic oxidative kinetic resolution of key pharmaceutical building blocks is described. Substrates investigated are relevant to the enantioselective preparation of Prozac®, Singulair®, and the promising hNK-1 receptor antagonist from Merck. The latter provides the most selective aerobic oxidative kinetic resolution yet described.
Co-reporter:Haiming Zhang Dr.;Eric M. Ferreira;Brian M. Stoltz
Angewandte Chemie International Edition 2004 Volume 43(Issue 45) pp:
Publication Date(Web):17 NOV 2004
DOI:10.1002/anie.200461294
No extra functionalization step is required for palladium(II)-catalyzed oxidative carbocyclizations like that shown, which provide highly substituted benzofuran and dihydrobenzofuran derivatives by net dehydrogenation. The mechanism is similar to that of Heck cyclizations. Products containing quaternary carbon stereocenters can be obtained in diastereomerically pure form.
Co-reporter:Jeffrey T. Bagdanoff;Brian M. Stoltz
Angewandte Chemie International Edition 2004 Volume 43(Issue 3) pp:
Publication Date(Web):29 DEC 2003
DOI:10.1002/anie.200352444
Air is enough to mediate the highly enantioselective oxidation of secondary alcohols at room temperature with palladium(II) and sparteine in nonflammable solvents. Examination of the role of solvents capable of hydrogen bonding and their ability to solvate halide anions led to a novel set of conditions for the highly enantioselective oxidative kinetic resolution of secondary alcohols.
Co-reporter:Jeffrey T. Bagdanoff;Brian M. Stoltz
Angewandte Chemie 2004 Volume 116(Issue 3) pp:
Publication Date(Web):29 DEC 2003
DOI:10.1002/ange.200352444
Luft reicht aus, um die hoch enantioselektive Oxidation sekundärer Alkohole mit Palladium(II) und Spartein bei Raumtemperatur in nichtentflammbaren Solventien zu vermitteln. Die Untersuchung von Wasserstoffbrücken bildenden Lösungsmitteln und ihrer Fähigkeit, Halogenide zu solvatisieren, führte zu einem neuartigen Protokoll für die hoch enantioselektive oxidative kinetische Racematspaltung von sekundären Alkoholen.
Co-reporter:Haiming Zhang Dr.;Eric M. Ferreira;Brian M. Stoltz
Angewandte Chemie 2004 Volume 116(Issue 45) pp:
Publication Date(Web):17 NOV 2004
DOI:10.1002/ange.200461294
Ohne separaten Funktionalisierungsschritt gelingen Palladium(II)-katalysierte oxidative Carbocyclisierungen wie die gezeigte, die durch Nettodehydrierung hoch substituierte Benzo- und Dihydrobenzofurane liefern. Der Mechanismus ähnelt dem von Heck-Cyclisierungen; mit der Reaktion lassen sich Produkte mit quartären Kohlenstoffstereozentren in diastereomerenreiner Form erhalten.
Co-reporter:Ryan R. Julian, Jeremy A. May, Brian M. Stoltz, J.L. Beauchamp
International Journal of Mass Spectrometry 2003 Volume 228(2–3) pp:851-864
Publication Date(Web):15 August 2003
DOI:10.1016/S1387-3806(03)00243-4
Biomimetic reagents capable of selectively forming non-covalent complexes and initiating intermolecular reactions with peptides in the gas phase are presented. In the present work, 18-crown-6 ether (18C6) is utilized to bind specifically to various protonated primary amines, including the protonated side chain of lysine. The use of multiple crown ethers is shown to be an efficient method for enhancing the binding energy, which is a critical factor influencing the success of these reagents. The binding energy must exceed any reaction barriers to the desired chemistry, otherwise simple dissociation of the complex occurs. Two reagents containing acidic and transition metal binding functionalities, respectively, designed to selectively cleave peptide bonds, are synthesized and tested experimentally. A third class of reagent designed to covalently attach to peptides utilizing carbene insertion chemistry is also presented. The results demonstrate that combining the recognition and binding powers of 18C6 with an easily activated diazo group allows for the efficient generation of a highly reactive carbene within a non-covalent complex. Intermolecular insertion reactions initiated by the carbene can transform these non-covalent complexes into covalently bound molecules. Electrospray ionization mass spectrometry and density functional theory (DFT) are utilized to evaluate these intermolecular insertion reactions. The results from experiments with several small molecules and peptides are presented. These diazo-based reagents prove to be highly versatile molecules capable of binding to, and with appropriate activation, becoming covalently attached to virtually any molecule that contains a primary amine. For this reason, we have dubbed them “molecular mousetraps.”
Co-reporter:Raissa M. Trend;Yeeman K. Ramtohul Dr.;Eric M. Ferreira;Brian M. Stoltz
Angewandte Chemie 2003 Volume 115(Issue 25) pp:
Publication Date(Web):24 JUN 2003
DOI:10.1002/ange.200351196
Der Ring schließt sich: Eine Vielzahl oxidativer Nucleophil-Alken-Cyclisierungen verläuft in unpolaren Lösungsmitteln unter einfachen aeroben Bedingungen mit ausgezeichneten Ausbeuten. Als Nucleophile können Carbonsäuren, Amide, primäre Alkohole und Phenole eingesetzt werden. Mit einem Pd-Spartein-Katalysatorsystem (siehe Bild) gelingen einfache Phenol-Alken-Cyclisierungen mit Enantioselektivitäten bis zu 90 % ee.
Co-reporter:Raissa M. Trend;Yeeman K. Ramtohul Dr.;Eric M. Ferreira;Brian M. Stoltz
Angewandte Chemie International Edition 2003 Volume 42(Issue 25) pp:
Publication Date(Web):24 JUN 2003
DOI:10.1002/anie.200351196
A variety of Pd-catalyzed oxidative nucleophile/alkene cyclizations proceeds in excellent yield under simple aerobic conditions in nonpolar media (Pd catalyst, pyridine, and O2 in toluene). Nucleophiles for these cyclizations include phenols, carboxylic acids, amides, and primary alcohols. Additionally, enantioselective catalysis is feasible with a Pd-sparteine system (see picture). Enantioselectivities of up to 90 % ee are observed for simple phenol/alkene cyclizations.
Co-reporter:Ryan R. Julian;Jeremy A. May;Brian M. Stoltz ;J. L. Beauchamp
Angewandte Chemie International Edition 2003 Volume 42(Issue 9) pp:
Publication Date(Web):26 FEB 2003
DOI:10.1002/anie.200390258
The covalent linking of noncovalently bound host–guest complexes in the gas phase is achieved by the formation of a highly reactive carbene from the host molecule, which is generated by collision-activated dissociation. Possible mechanisms for the resulting intermolecular reactions are explored by theory and experiment.
Co-reporter:Ryan R. Julian;Jeremy A. May;Brian M. Stoltz ;J. L. Beauchamp
Angewandte Chemie International Edition 2003 Volume 42(Issue 9) pp:
Publication Date(Web):26 FEB 2003
DOI:10.1002/anie.200390276
Co-reporter:Robert A. CraigII; Steven A. Loskot; Justin T. Mohr; Douglas C. Behenna; Andrew M. Harned;Brian M. Stoltz
Organic Letters () pp:
Publication Date(Web):October 26, 2015
DOI:10.1021/acs.orglett.5b02376
The first general method for the enantioselective construction of all-carbon quaternary centers on cyclopentanones by enantioselective palladium-catalyzed decarboxylative allylic alkylation is described. Employing the electronically modified (S)-(p-CF3)3-t-BuPHOX ligand, α-quaternary cyclopentanones were isolated in yields up to >99% with ee’s up to 94%. Additionally, in order to facilitate large-scale application of this method, a low catalyst loading protocol was employed, using as little as 0.15 mol % Pd, furnishing the product without any loss in ee.
Co-reporter:Kevin M. Allan, Boram D. Hong and Brian M. Stoltz
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 23) pp:NaN4964-4964
Publication Date(Web):2009/10/06
DOI:10.1039/B913336D
A convenient method is disclosed for the synthesis of both 3-hydroxyisoquinolines and 2-hydroxy-1,4-naphthoquinones from β-ketoesters using a one-pot aryne acyl-alkylation/condensation procedure. When performed in conjunction with a one-step method for the synthesis of the β-ketoester substrates, this method provides a new route to these polyaromatic structures in only two steps from commercially available carboxylic acid starting materials. The utility of this approach is demonstrated in the synthesis of the atropisomeric P,N-ligand, QUINAP.
Co-reporter:Pamela M. Tadross, Pradeep Bugga and Brian M. Stoltz
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 15) pp:NaN5357-5357
Publication Date(Web):2011/05/26
DOI:10.1039/C1OB05725A
The tetracyclic core of the integrastatin natural products has been prepared in a convergent and rapid manner. Our strategy relies upon a palladium(II)-catalyzed oxidative cyclization to form the central [3.3.1]-dioxabicycle of the natural product core. Overall, the core has been completed in only 4 linear steps from known compounds.
Co-reporter:Zining Li, Qian Geng, Zhe Lv, Beau P. Pritchett, Katsuaki Baba, Yoshitaka Numajiri, Brian M. Stoltz and Guangxin Liang
Inorganic Chemistry Frontiers 2015 - vol. 2(Issue 3) pp:NaN240-240
Publication Date(Web):2015/01/20
DOI:10.1039/C4QO00312H
Selective syntheses of leuconolam, leuconoxine, and mersicarpine alkaloids bearing distinctive core structures were achieved through Staudinger reactions using a common intermediate. In the key cyclization step, water functioned like a switch to control which core structure to produce. The chemistry allowed for selective syntheses of the group of alkaloids from a simple intermediate through straightforward chemical operations.
Co-reporter:Nathan B. Bennett, Allen Y. Hong, Andrew M. Harned and Brian M. Stoltz
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 1) pp:NaN59-59
Publication Date(Web):2011/09/01
DOI:10.1039/C1OB06189E
A general method for the synthesis of β-substituted and unsubstituted cycloheptenones bearing enantioenriched all-carbon γ-quaternary stereocenters is reported. Hydride or organometallic addition to a seven-membered ring vinylogous ester followed by finely tuned quenching parameters achieves elimination to the corresponding cycloheptenone. The resulting enones are elaborated to bi- and tricyclic compounds with potential for the preparation of non-natural analogs and whose structures are embedded in a number of cycloheptanoid natural products.
Co-reporter:Corinne Baumgartner, Sandy Ma, Qi Liu and Brian M. Stoltz
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 13) pp:NaN2917-2917
Publication Date(Web):2010/05/24
DOI:10.1039/C004275G
Our efforts toward the construction of the carbocylic core of cortistatin A via an enyne-ene metathesis are disclosed. Interestingly, an attempted SN2 inversion of a secondary mesylate in our five-membered D-ring piece gave a product with retention of stereochemistry.
Co-reporter:Justin T. Mohr, David C. Ebner and Brian M. Stoltz
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 22) pp:NaN3576-3576
Publication Date(Web):2007/10/01
DOI:10.1039/B711159M
Approaches to the preparation of enantioenriched materials via catalytic methods that destroy stereogenic elements of a molecule are discussed. Although these processes often decrease overall molecular complexity, there are several notable advantages including material recycling, enantiodivergence and convergence, and increased substrate scope. Examples are accompanied by discussion of the critical design elements required for the success of these methods.













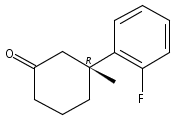



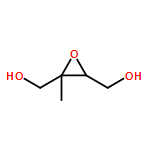
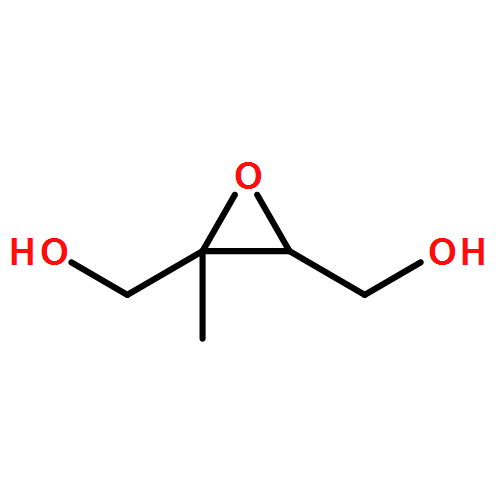

![2,2-Aziridinedicarboxylic acid, 1-[(4-methylphenyl)sulfonyl]-3-phenyl-, 2,2-dimethyl ester](/data/chemimg/3746900/1132667-77-6.png)
![2,2-Aziridinedicarboxylic acid, 1-[(4-methylphenyl)sulfonyl]-3-phenyl-, 2,2-dimethyl ester](/data/chemimg/3746900/1132667-77-6_b.png)
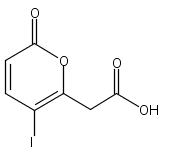


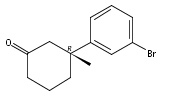




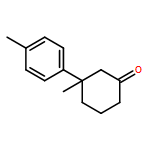
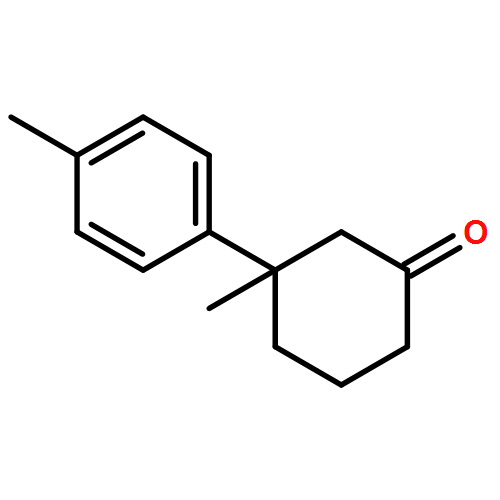
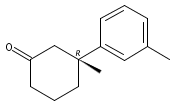

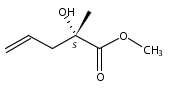






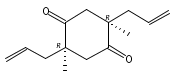
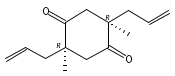
![2-(Benzo[b]thiophen-7-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane](http://img.cochemist.com/ccimg/1000200/1000160-74-6.png)
![2-(Benzo[b]thiophen-7-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane](http://img.cochemist.com/ccimg/1000200/1000160-74-6_b.png)














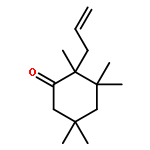
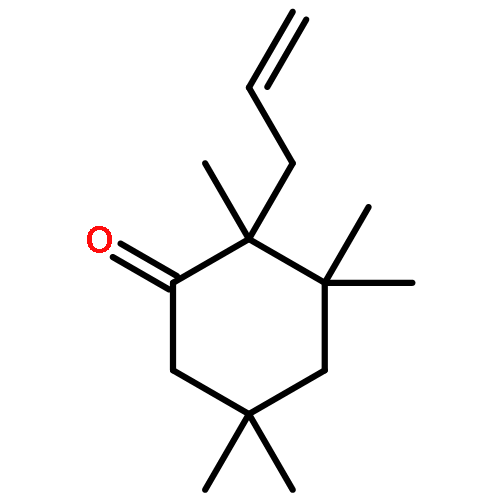

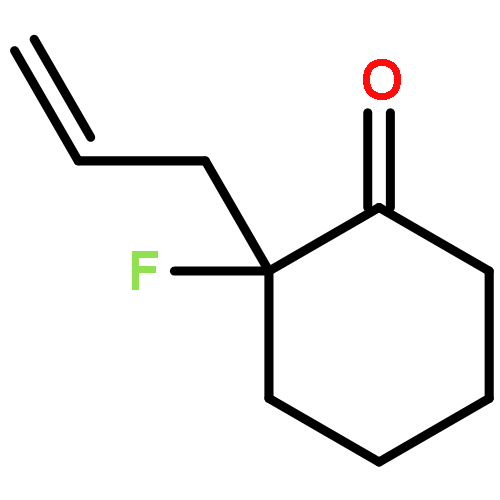

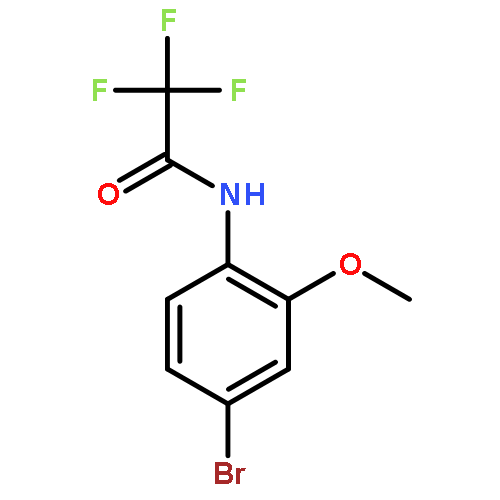




![Ferrocene,1,1'-bis[bis(4-methoxy-3,5-dimethylphenyl)phosphino]-2,2'-bis[(S)-(dimethylamino)phenylmethyl]-,(1S,1'S)- (9CI)](http://img.cochemist.com/ccimg/850000/849925-12-8.png)
![Ferrocene,1,1'-bis[bis(4-methoxy-3,5-dimethylphenyl)phosphino]-2,2'-bis[(S)-(dimethylamino)phenylmethyl]-,(1S,1'S)- (9CI)](http://img.cochemist.com/ccimg/850000/849925-12-8_b.png)

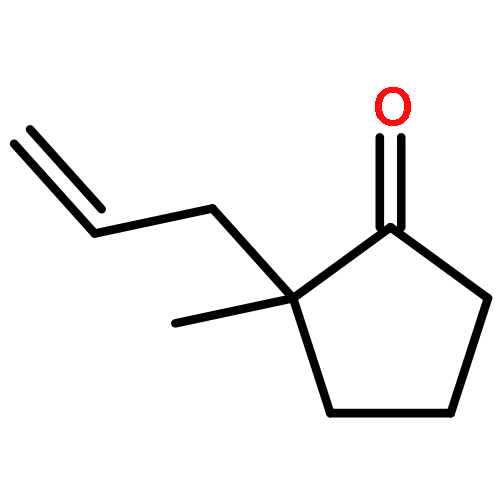
![Rhodium, bis[m-[a,a,a',a'-tetramethyl-1,3-benzenedipropanoato(2-)-kO1,kO'3:kO3,kO'1]]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/819100/819050-89-0.png)
![Rhodium, bis[m-[a,a,a',a'-tetramethyl-1,3-benzenedipropanoato(2-)-kO1,kO'3:kO3,kO'1]]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/819100/819050-89-0_b.png)


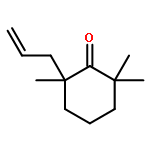

![2-[1-(4-METHYLPHENYL)SULFONYLAZIRIDIN-2-YL]PYRIDINE](http://img.cochemist.com/ccimg/797000/796975-18-3.png)
![2-[1-(4-METHYLPHENYL)SULFONYLAZIRIDIN-2-YL]PYRIDINE](http://img.cochemist.com/ccimg/797000/796975-18-3_b.png)




![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-(2,4,6-trimethylphenyl)-](http://img.cochemist.com/ccimg/727400/727379-99-9.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-(2,4,6-trimethylphenyl)-](http://img.cochemist.com/ccimg/727400/727379-99-9_b.png)





![2-Thiazolidinimine, 3-[(4-methylphenyl)sulfonyl]-N,5-diphenyl-, (2Z)-](http://img.cochemist.com/ccimg/639500/639476-90-7.png)
![2-Thiazolidinimine, 3-[(4-methylphenyl)sulfonyl]-N,5-diphenyl-, (2Z)-](http://img.cochemist.com/ccimg/639500/639476-90-7_b.png)
![1-Azatricyclo[3.3.1.13,7]decan-2-one, 3-methyl-](http://img.cochemist.com/ccimg/631900/631860-12-3.png)
![1-Azatricyclo[3.3.1.13,7]decan-2-one, 3-methyl-](http://img.cochemist.com/ccimg/631900/631860-12-3_b.png)


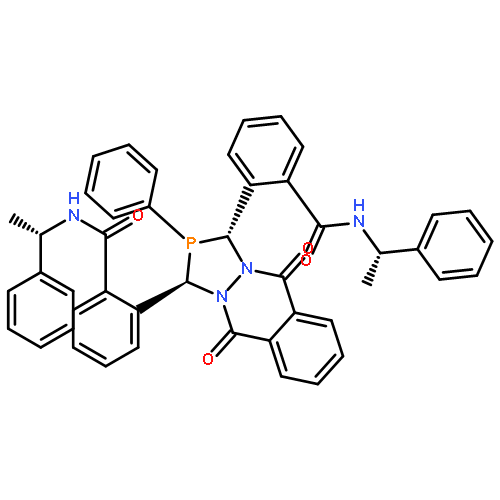
![Benzenesulfonamide, 4-methyl-N-[(1E)-2-phenyl-1-propenyl]-](http://img.cochemist.com/ccimg/602400/602302-01-2.png)
![Benzenesulfonamide, 4-methyl-N-[(1E)-2-phenyl-1-propenyl]-](http://img.cochemist.com/ccimg/602400/602302-01-2_b.png)
![BENZENESULFONAMIDE, 4-METHYL-N-[(1Z)-2-PHENYL-1-PROPENYL]-](http://img.cochemist.com/ccimg/602400/602302-00-1.png)
![BENZENESULFONAMIDE, 4-METHYL-N-[(1Z)-2-PHENYL-1-PROPENYL]-](http://img.cochemist.com/ccimg/602400/602302-00-1_b.png)


![BENZENESULFONAMIDE, N-[2-(1,2-DIHYDROXYETHYL)PHENYL]-4-METHYL-](http://img.cochemist.com/ccimg/544700/544696-62-0.png)
![BENZENESULFONAMIDE, N-[2-(1,2-DIHYDROXYETHYL)PHENYL]-4-METHYL-](http://img.cochemist.com/ccimg/544700/544696-62-0_b.png)

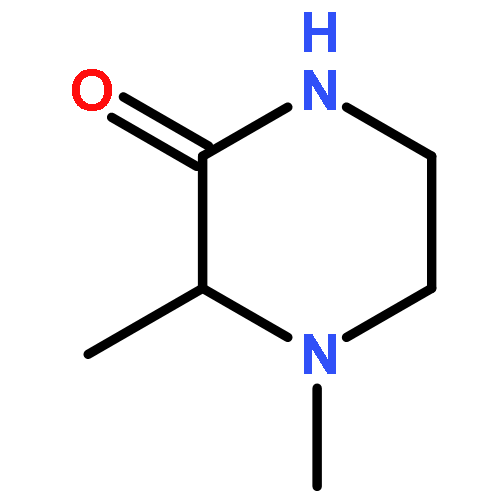




![4-HEXENAL, 6-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-4-METHYL-, (4E)-](http://img.cochemist.com/ccimg/447500/447454-31-1.png)
![4-HEXENAL, 6-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-4-METHYL-, (4E)-](http://img.cochemist.com/ccimg/447500/447454-31-1_b.png)

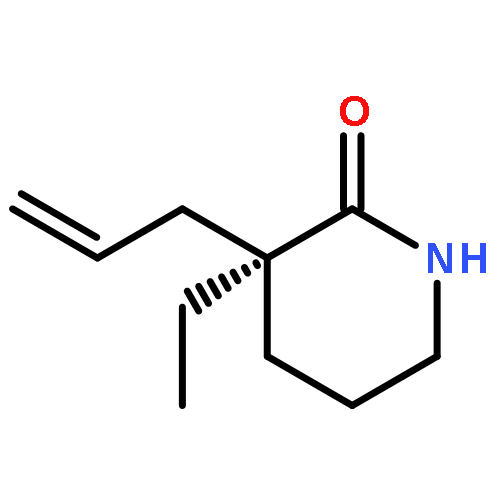
![Benzamide,N-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]-4-(1-piperazinylmethyl)-](http://img.cochemist.com/ccimg/404900/404844-02-6.png)
![Benzamide,N-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]-4-(1-piperazinylmethyl)-](http://img.cochemist.com/ccimg/404900/404844-02-6_b.png)


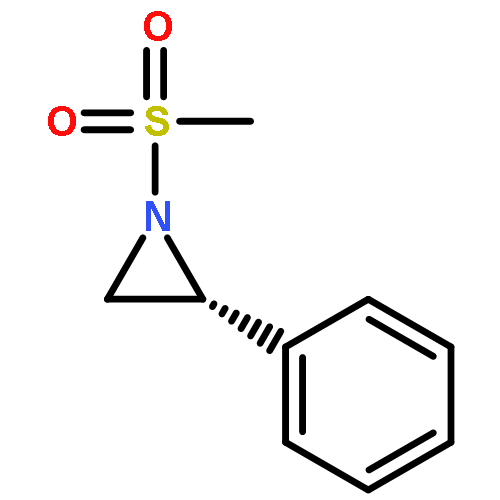
![Benzene, [(1Z)-3-bromo-2-iodo-1-propenyl]-](http://img.cochemist.com/ccimg/353500/353454-94-1.png)
![Benzene, [(1Z)-3-bromo-2-iodo-1-propenyl]-](http://img.cochemist.com/ccimg/353500/353454-94-1_b.png)






![Methyl 1-[(4-methylphenyl)sulfonyl]-4-piperidinecarboxylate](http://img.cochemist.com/ccimg/311800/311794-12-4.png)
![Methyl 1-[(4-methylphenyl)sulfonyl]-4-piperidinecarboxylate](http://img.cochemist.com/ccimg/311800/311794-12-4_b.png)
![1,2,3,4,5-cyclopentanepentayl, compd. with 1-[bis[3,5-bis(trifluoromethyl)phenyl]phosphino]-2-[(1S)-1-(dicyclohexylphosphino)ethyl]-1,2,3,4,5-cyclopentanepentayl, iron salt (1:1:1)](http://img.cochemist.com/ccimg/292700/292638-88-1.png)
![1,2,3,4,5-cyclopentanepentayl, compd. with 1-[bis[3,5-bis(trifluoromethyl)phenyl]phosphino]-2-[(1S)-1-(dicyclohexylphosphino)ethyl]-1,2,3,4,5-cyclopentanepentayl, iron salt (1:1:1)](http://img.cochemist.com/ccimg/292700/292638-88-1_b.png)


![Aziridine, 2-[4-(acetyloxy)phenyl]-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/250300/250260-26-5.png)
![Aziridine, 2-[4-(acetyloxy)phenyl]-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/250300/250260-26-5_b.png)


![2H-Pyran-2-one,6-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-5,6-dihydro-, (6R)-](http://img.cochemist.com/ccimg/232000/231963-90-9.png)
![2H-Pyran-2-one,6-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-5,6-dihydro-, (6R)-](http://img.cochemist.com/ccimg/232000/231963-90-9_b.png)





![Boronic acid, [4-[(trifluoroacetyl)amino]phenyl]-](http://img.cochemist.com/ccimg/210000/209917-42-0.png)
![Boronic acid, [4-[(trifluoroacetyl)amino]phenyl]-](http://img.cochemist.com/ccimg/210000/209917-42-0_b.png)

![Aziridine, 2-(3-chlorophenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/200900/200803-16-3.png)
![Aziridine, 2-(3-chlorophenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/200900/200803-16-3_b.png)
![Aziridine, 1-[(4-methoxyphenyl)sulfonyl]-2-phenyl-, (R)-](http://img.cochemist.com/ccimg/200600/200503-34-0.png)
![Aziridine, 1-[(4-methoxyphenyl)sulfonyl]-2-phenyl-, (R)-](http://img.cochemist.com/ccimg/200600/200503-34-0_b.png)
![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-, (2R)-](http://img.cochemist.com/ccimg/194200/194156-25-7.png)
![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-, (2R)-](http://img.cochemist.com/ccimg/194200/194156-25-7_b.png)
![1,4-Ethanonaphthalene-6,10(4H)-dione, 5,9-bis[(benzoyloxy)methyl]-1,4a,5,8a-tetrahydro-5,9-dihydroxy-, (1R,4S,4aS,5S,8aR,9S)-](/data/chemimg/3769200/193410-88-7.png)
![1,4-Ethanonaphthalene-6,10(4H)-dione, 5,9-bis[(benzoyloxy)methyl]-1,4a,5,8a-tetrahydro-5,9-dihydroxy-, (1R,4S,4aS,5S,8aR,9S)-](/data/chemimg/3769200/193410-88-7_b.png)



![Aziridine, 1-[(4-methoxyphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/181400/181306-57-0.png)
![Aziridine, 1-[(4-methoxyphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/181400/181306-57-0_b.png)
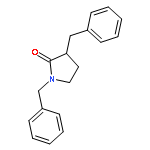
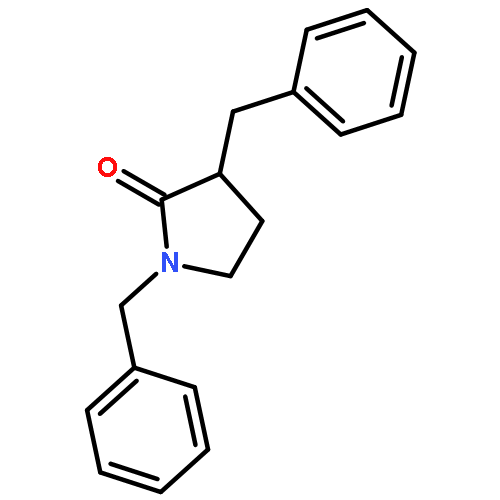
![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/175300/175222-83-0.png)
![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/175300/175222-83-0_b.png)
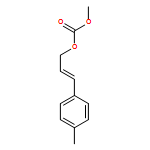
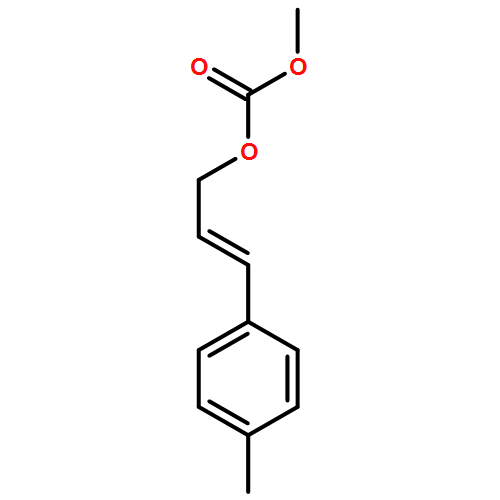
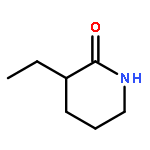
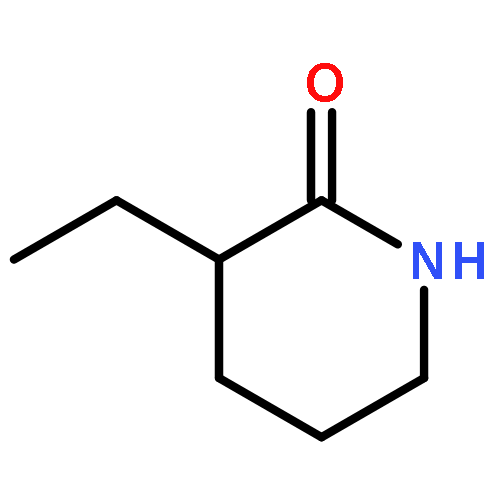
![Oxazole,4-(1,1-dimethylethyl)-2-[2-(diphenylphosphino)phenyl]-4,5-dihydro-,(4R)-](/data/chemimg/109300/164858-79-1.png)
![Oxazole,4-(1,1-dimethylethyl)-2-[2-(diphenylphosphino)phenyl]-4,5-dihydro-,(4R)-](/data/chemimg/109300/164858-79-1_b.png)
![2-BUTEN-1-OL, 4-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-2-METHYL-, (2E)-](http://img.cochemist.com/ccimg/162200/162188-27-4.png)
![2-BUTEN-1-OL, 4-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-2-METHYL-, (2E)-](http://img.cochemist.com/ccimg/162200/162188-27-4_b.png)
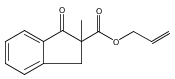
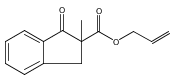

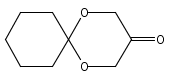
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/155800/155721-37-2.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-(4-nitrophenyl)-](http://img.cochemist.com/ccimg/155800/155721-37-2_b.png)
![Ethyl 2-methyl-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-2-carboxylate](http://img.cochemist.com/ccimg/154400/154365-33-0.png)
![Ethyl 2-methyl-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-2-carboxylate](http://img.cochemist.com/ccimg/154400/154365-33-0_b.png)
![1-(phenylmethyl)-1H-Pyrrolo[2,3-b]pyridine](http://img.cochemist.com/ccimg/153000/152955-68-5.png)
![1-(phenylmethyl)-1H-Pyrrolo[2,3-b]pyridine](http://img.cochemist.com/ccimg/153000/152955-68-5_b.png)


![2-Cyclohexen-1-one, 5-[1-(hydroxymethyl)ethenyl]-2-methyl-, (R)-](http://img.cochemist.com/ccimg/152000/151910-94-0.png)
![2-Cyclohexen-1-one, 5-[1-(hydroxymethyl)ethenyl]-2-methyl-, (R)-](http://img.cochemist.com/ccimg/152000/151910-94-0_b.png)
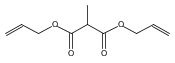

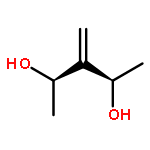
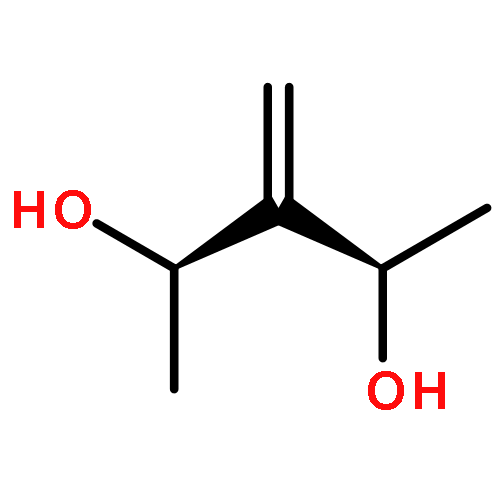
![Benzene, 1-[(1E)-3-chloro-1-propenyl]-4-fluoro-](http://img.cochemist.com/ccimg/145800/145798-29-4.png)
![Benzene, 1-[(1E)-3-chloro-1-propenyl]-4-fluoro-](http://img.cochemist.com/ccimg/145800/145798-29-4_b.png)
![2-Propenoic acid, 2-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-,methyl ester](http://img.cochemist.com/ccimg/144900/144828-45-5.png)
![2-Propenoic acid, 2-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-,methyl ester](http://img.cochemist.com/ccimg/144900/144828-45-5_b.png)

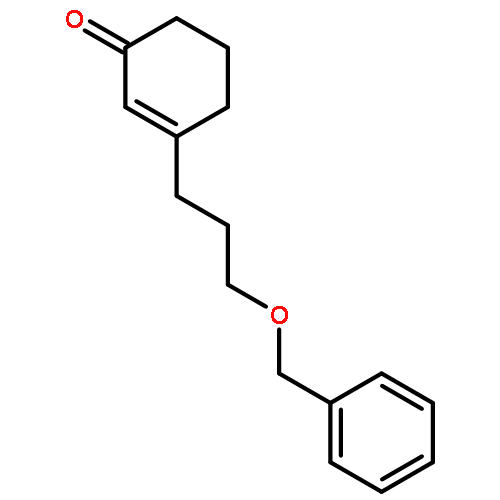

![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/137600/137595-22-3.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/137600/137595-22-3_b.png)
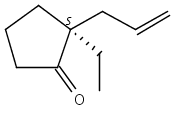



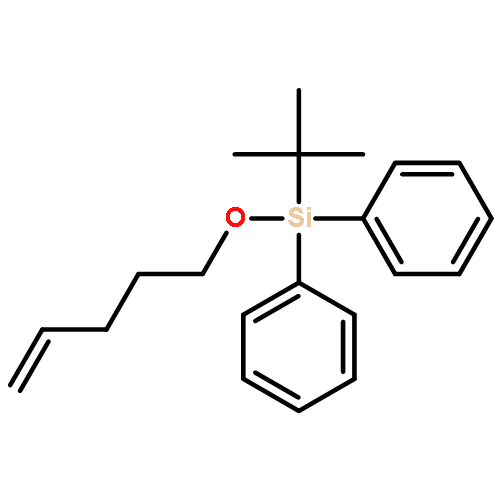
![Benzene, 1-nitro-4-[(2-phenylethoxy)methyl]-](http://img.cochemist.com/ccimg/128800/128702-29-4.png)
![Benzene, 1-nitro-4-[(2-phenylethoxy)methyl]-](http://img.cochemist.com/ccimg/128800/128702-29-4_b.png)
![2H-Pyran-2-one, 5,6-dihydro-6-[(phenylmethoxy)methyl]-, (S)-](http://img.cochemist.com/ccimg/124700/124662-66-4.png)
![2H-Pyran-2-one, 5,6-dihydro-6-[(phenylmethoxy)methyl]-, (S)-](http://img.cochemist.com/ccimg/124700/124662-66-4_b.png)

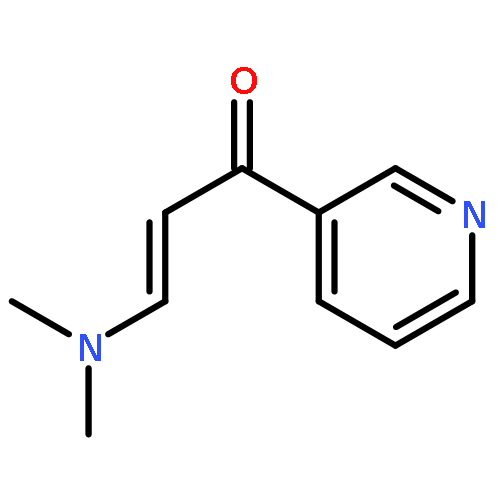
![Hexanoic acid, 6-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/122900/122846-05-3.png)
![Hexanoic acid, 6-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/122900/122846-05-3_b.png)



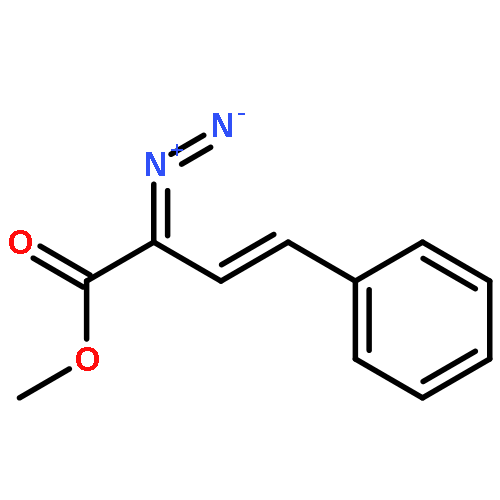

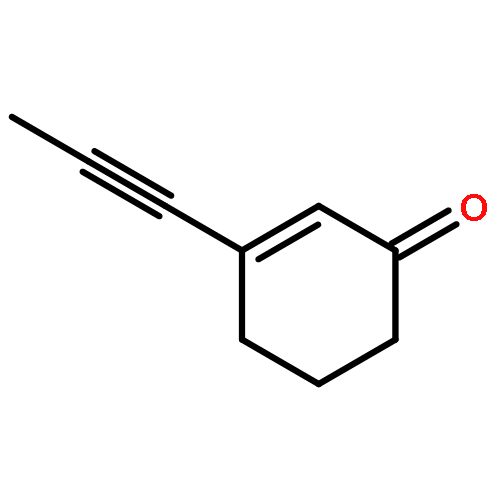


![Aziridine, 2-butyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/117000/116905-61-4.png)
![Aziridine, 2-butyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/117000/116905-61-4_b.png)
![1-Propanamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/115400/115306-75-7.png)
![1-Propanamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/115400/115306-75-7_b.png)


![Silane, (1,1-dimethylethyl)dimethyl[(3-methyl-2-butenyl)oxy]-](http://img.cochemist.com/ccimg/114800/114701-65-4.png)
![Silane, (1,1-dimethylethyl)dimethyl[(3-methyl-2-butenyl)oxy]-](http://img.cochemist.com/ccimg/114800/114701-65-4_b.png)








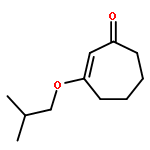


![Aziridine, 2-(4-chlorophenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/97500/97401-93-9.png)
![Aziridine, 2-(4-chlorophenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/97500/97401-93-9_b.png)
![Aziridine, 2-(4-methylphenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/97500/97401-87-1.png)
![Aziridine, 2-(4-methylphenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/97500/97401-87-1_b.png)
![Benzene, 1-[(1E)-3-chloro-1-propen-1-yl]-4-methyl-](/data/chemimg/3742100/96000-21-4.png)
![Benzene, 1-[(1E)-3-chloro-1-propen-1-yl]-4-methyl-](/data/chemimg/3742100/96000-21-4_b.png)
![Benzene, 1-[(1E)-3-chloro-1-propen-1-yl]-4-methoxy-](/data/chemimg/2074200/94607-41-7.png)
![Benzene, 1-[(1E)-3-chloro-1-propen-1-yl]-4-methoxy-](/data/chemimg/2074200/94607-41-7_b.png)




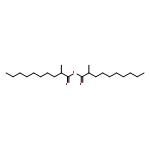
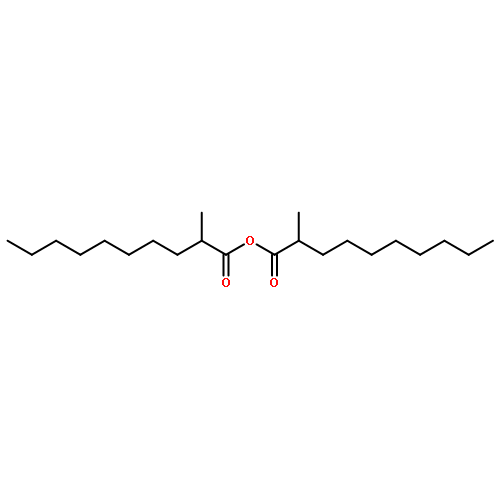
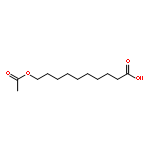
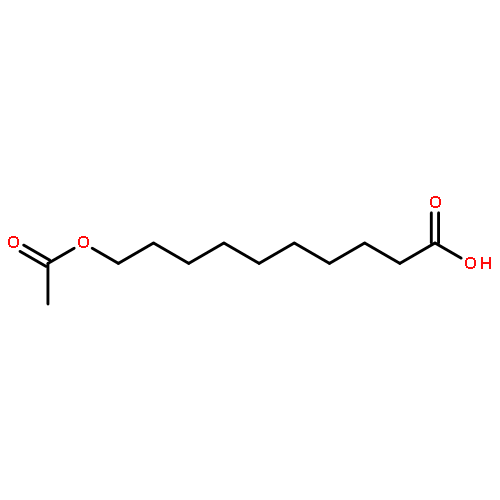
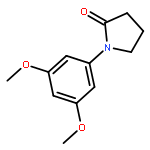
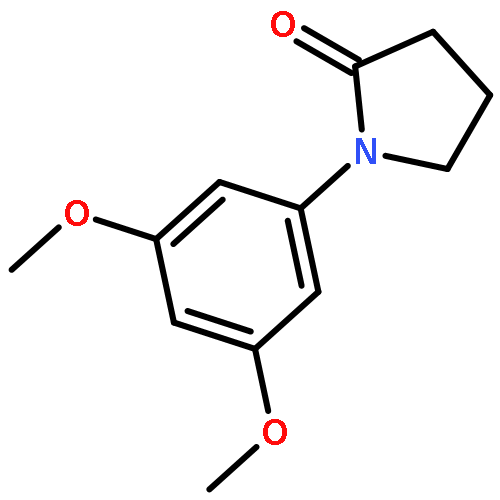


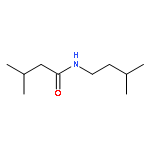



![1H-Indole, 1-[[2-(trimethylsilyl)ethoxy]methyl]-](http://img.cochemist.com/ccimg/88000/87954-27-6.png)
![1H-Indole, 1-[[2-(trimethylsilyl)ethoxy]methyl]-](http://img.cochemist.com/ccimg/88000/87954-27-6_b.png)
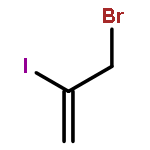
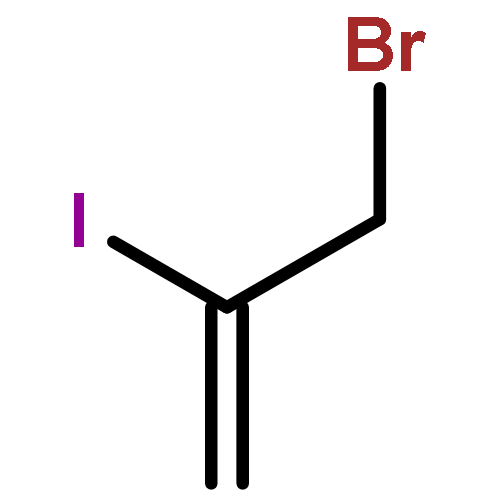
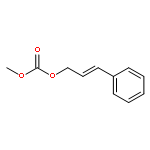
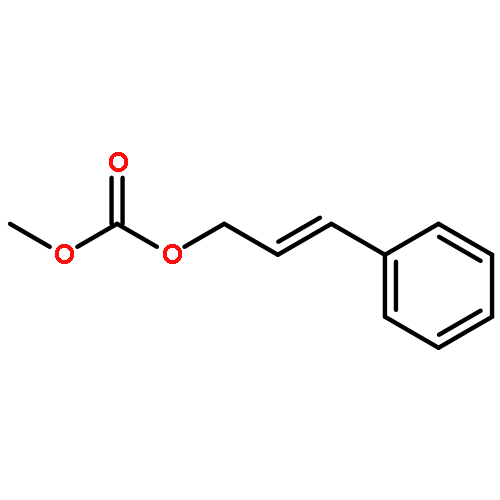

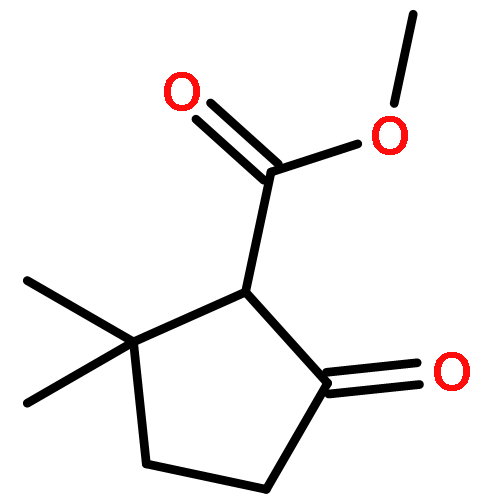



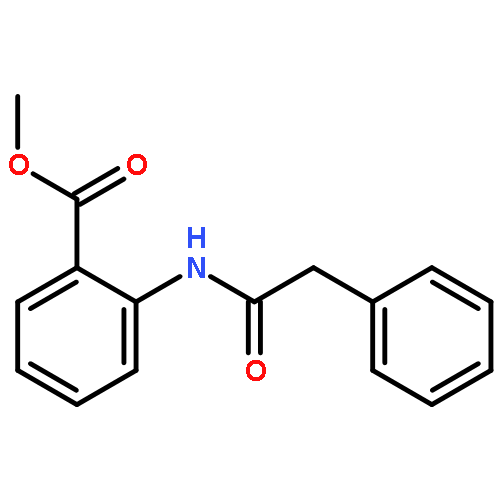



![(3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-4-(butanoyloxy)-3,3a-dihydroxy-3,6,9-trimethyl-8-{[(2Z)-2-methylbut-2-enoyl]oxy}-2-oxo-2,3,3a,4,5,6,6a,7,8,9b-decahydroazuleno[4,5-b]furan-7-yl octanoate](http://img.cochemist.com/ccimg/67600/67526-95-8.png)
![(3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-4-(butanoyloxy)-3,3a-dihydroxy-3,6,9-trimethyl-8-{[(2Z)-2-methylbut-2-enoyl]oxy}-2-oxo-2,3,3a,4,5,6,6a,7,8,9b-decahydroazuleno[4,5-b]furan-7-yl octanoate](http://img.cochemist.com/ccimg/67600/67526-95-8_b.png)






![2H-Pyran-2-one, 6-[(acetyloxy)methyl]-5,6-dihydro-, (6S)-](http://img.cochemist.com/ccimg/64000/63952-83-0.png)
![2H-Pyran-2-one, 6-[(acetyloxy)methyl]-5,6-dihydro-, (6S)-](http://img.cochemist.com/ccimg/64000/63952-83-0_b.png)




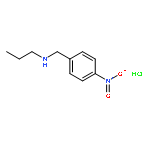
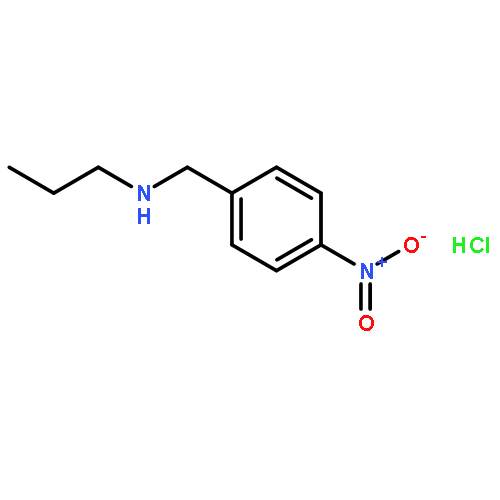
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-, (R)-](http://img.cochemist.com/ccimg/62600/62596-62-7.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-, (R)-](http://img.cochemist.com/ccimg/62600/62596-62-7_b.png)


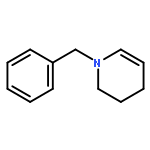
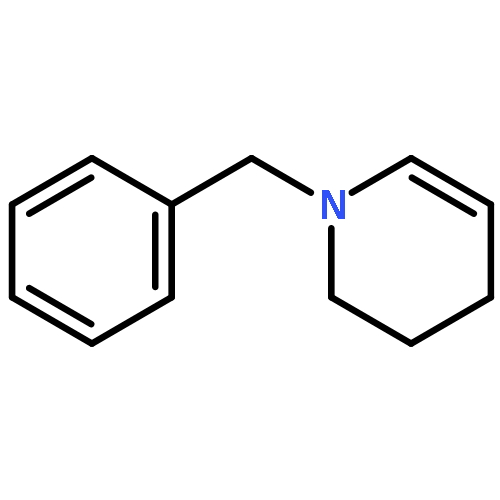




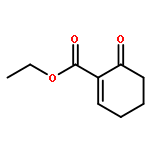



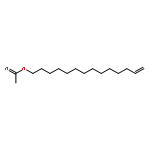

![5-(2-Chlorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine](http://img.cochemist.com/ccimg/55200/55142-85-3.png)
![5-(2-Chlorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine](http://img.cochemist.com/ccimg/55200/55142-85-3_b.png)
![CYCLOPENTANONE, 2-[(4-METHYLPHENYL)METHYL]-](http://img.cochemist.com/ccimg/54200/54158-56-4.png)
![CYCLOPENTANONE, 2-[(4-METHYLPHENYL)METHYL]-](http://img.cochemist.com/ccimg/54200/54158-56-4_b.png)


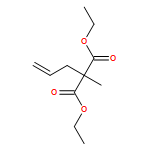
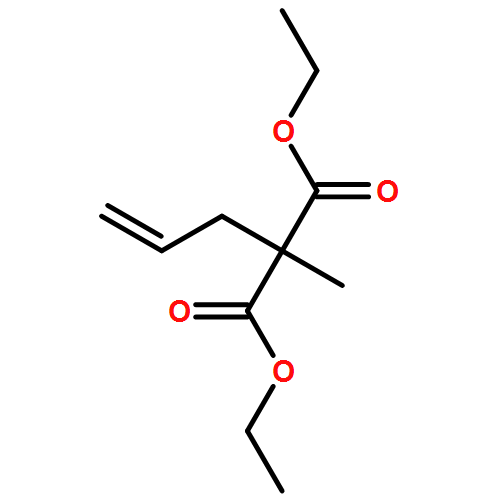
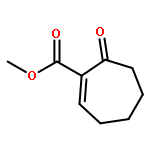


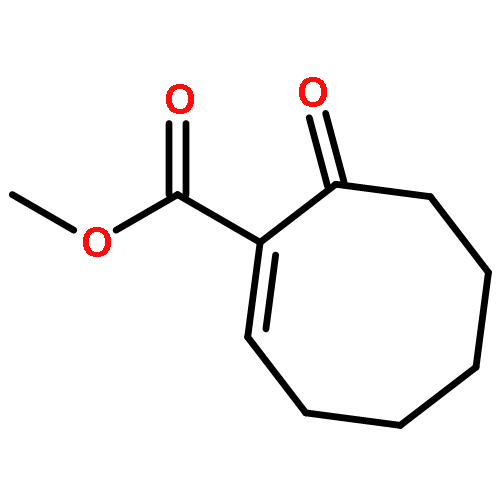
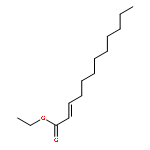
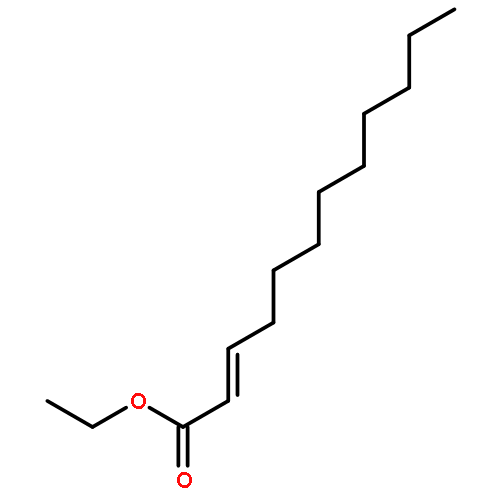








![3,4,5,6-tetrahydro-1H-Pyrrolo[4,3,2-ef][2]benzazepine](http://img.cochemist.com/ccimg/38100/38073-24-4.png)
![3,4,5,6-tetrahydro-1H-Pyrrolo[4,3,2-ef][2]benzazepine](http://img.cochemist.com/ccimg/38100/38073-24-4_b.png)
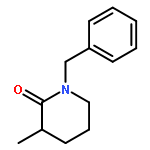
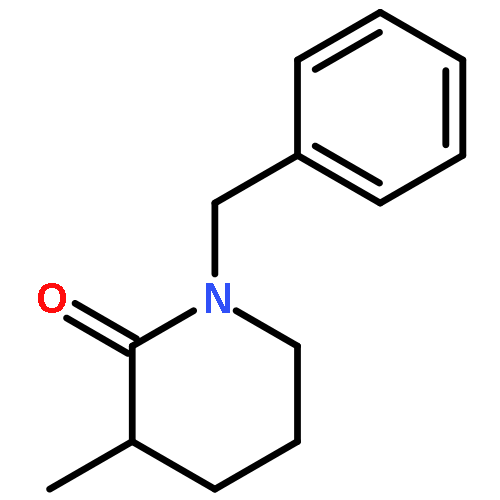

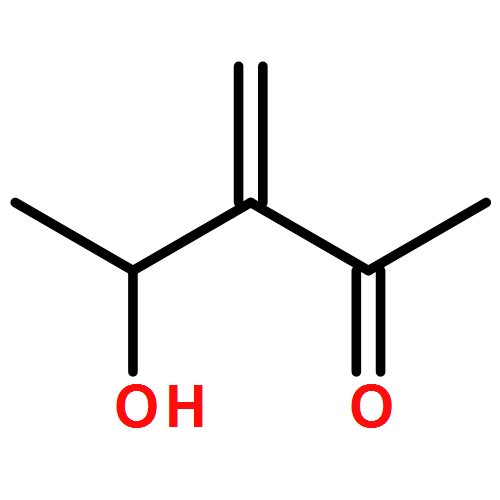
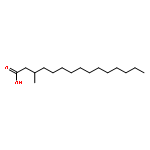
![ETHYL 6-BROMO-3-CHLORO-8-(TRIFLUOROMETHYL)IMIDAZO[1,2-A]PYRIDINE-<WBR />2-CARBOXYLATE](http://img.cochemist.com/ccimg/35200/35194-39-9.png)
![ETHYL 6-BROMO-3-CHLORO-8-(TRIFLUOROMETHYL)IMIDAZO[1,2-A]PYRIDINE-<WBR />2-CARBOXYLATE](http://img.cochemist.com/ccimg/35200/35194-39-9_b.png)



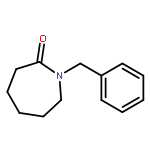
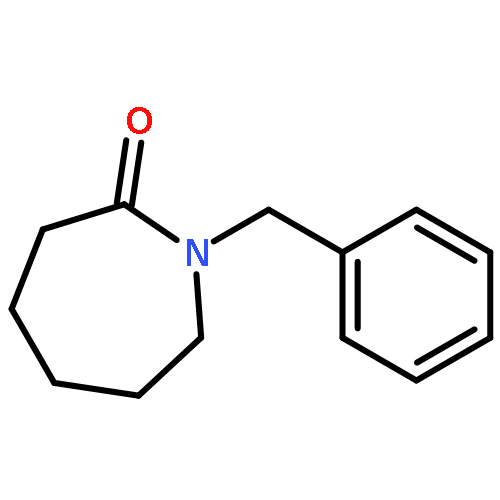
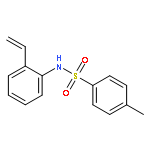
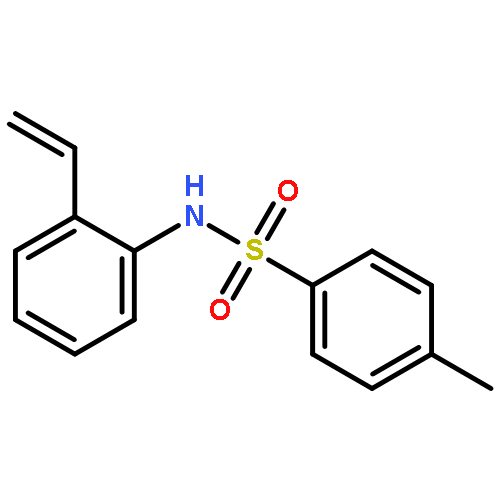


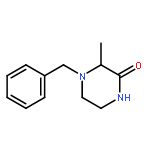
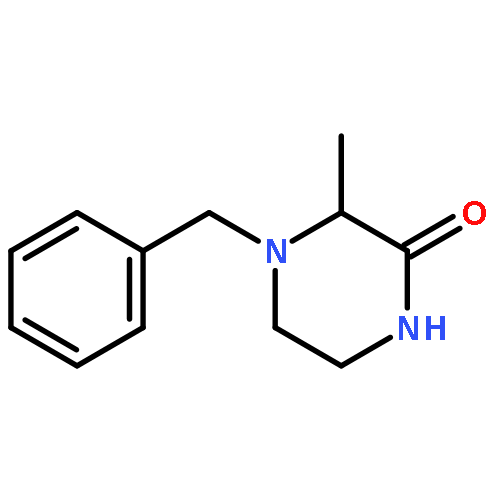






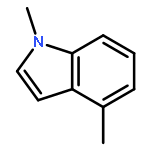
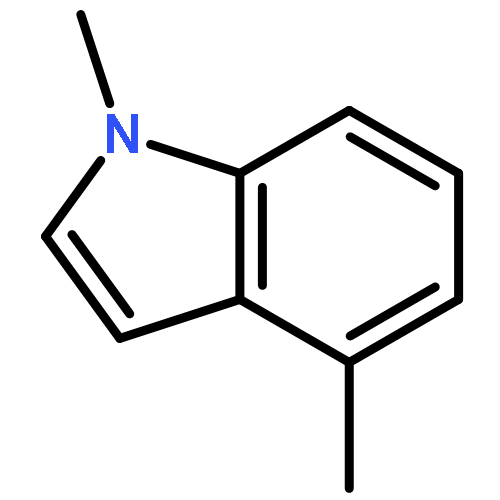




![Aziridine,1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24400/24395-14-0.png)
![Aziridine,1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24400/24395-14-0_b.png)







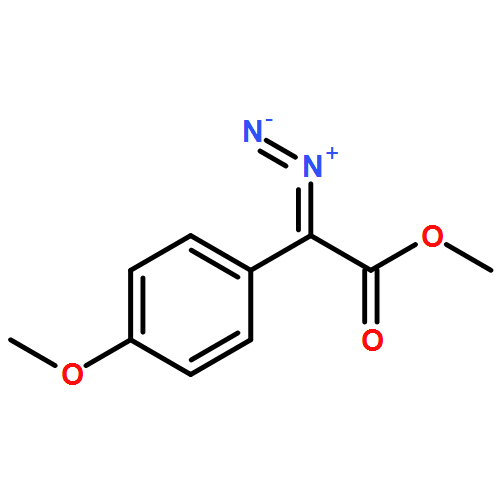


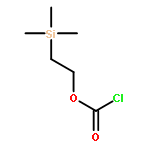

















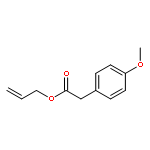
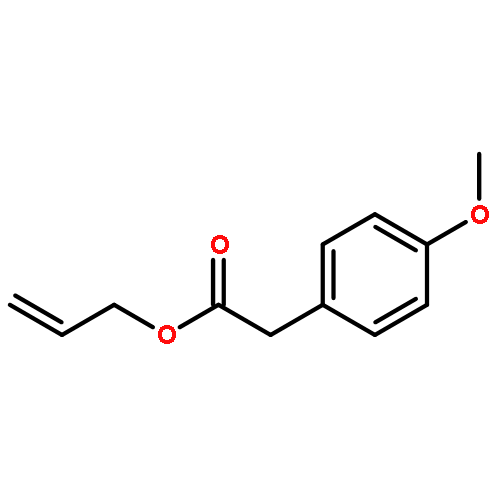
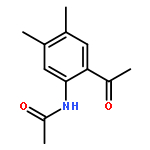
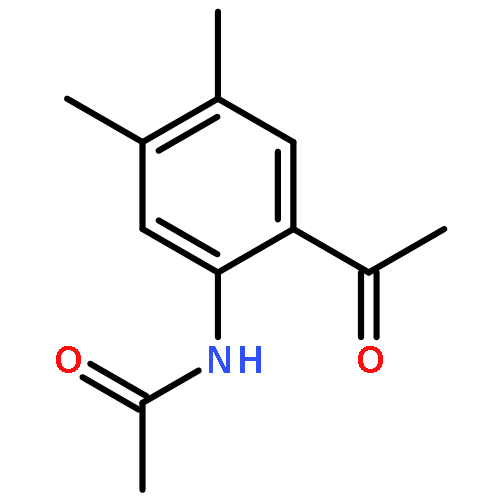





![Bicyclo[7.2.0]undec-4-ene,4,11,11-trimethyl-8-methylene-](http://img.cochemist.com/ccimg/13900/13877-93-5.png)
![Bicyclo[7.2.0]undec-4-ene,4,11,11-trimethyl-8-methylene-](http://img.cochemist.com/ccimg/13900/13877-93-5_b.png)
![Propanamide, N-[2-(1-oxopropyl)phenyl]-](http://img.cochemist.com/ccimg/13900/13837-11-1.png)
![Propanamide, N-[2-(1-oxopropyl)phenyl]-](http://img.cochemist.com/ccimg/13900/13837-11-1_b.png)
![Bicyclo[3.1.1]heptan-3-amine,2,6,6-trimethyl-, (1S,2S,3S,5R)-](http://img.cochemist.com/ccimg/13300/13293-47-5.png)
![Bicyclo[3.1.1]heptan-3-amine,2,6,6-trimethyl-, (1S,2S,3S,5R)-](http://img.cochemist.com/ccimg/13300/13293-47-5_b.png)







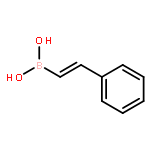
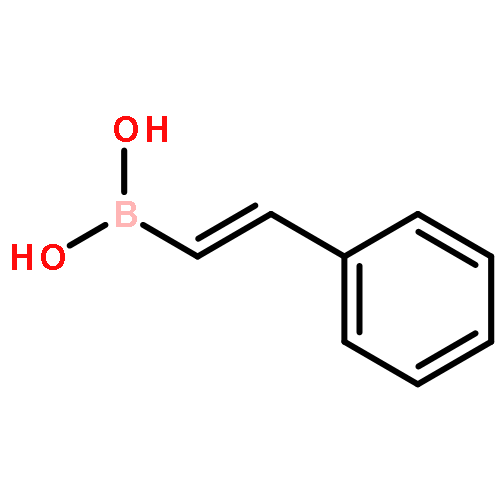
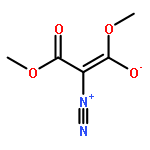
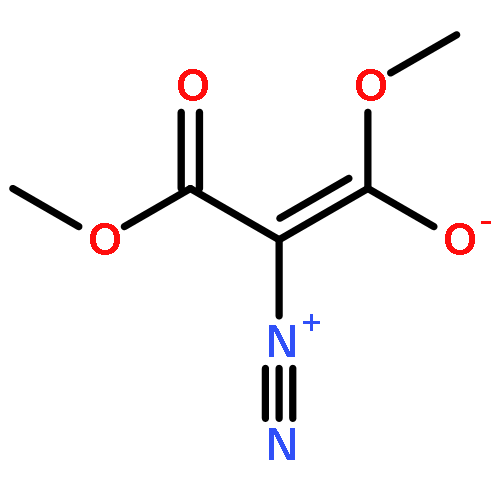
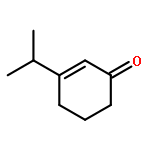
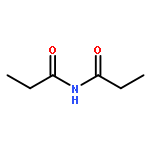
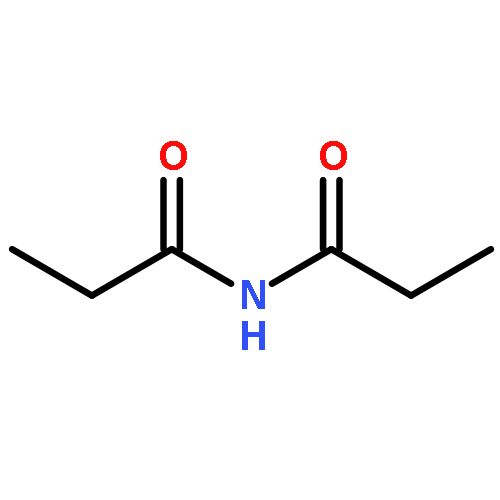






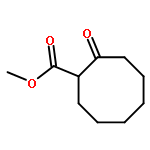
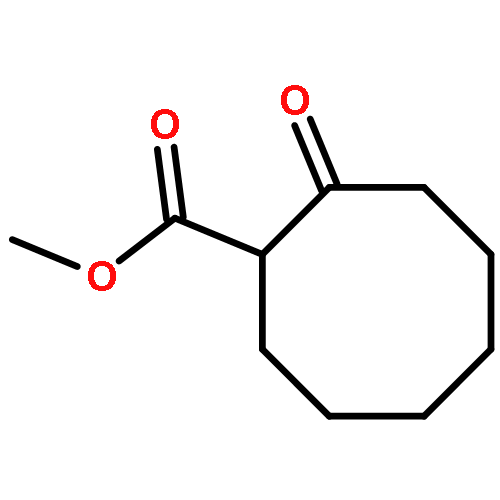















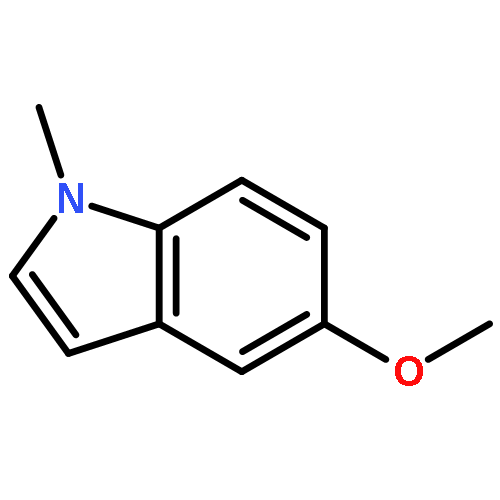
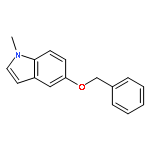
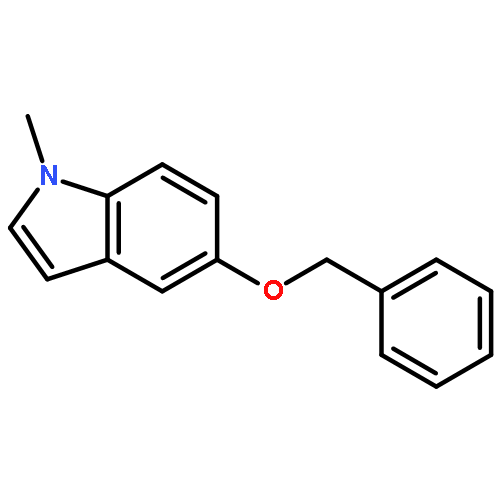
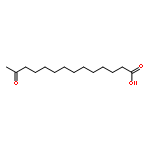
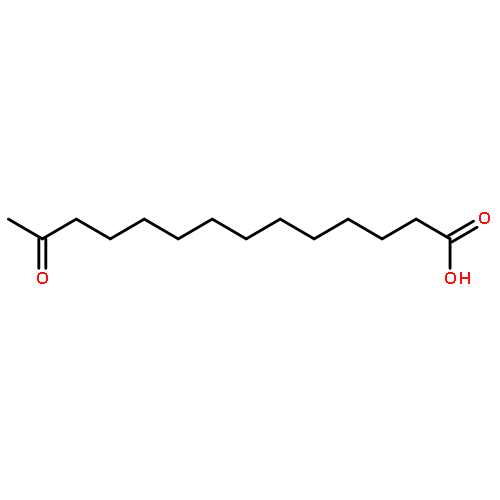



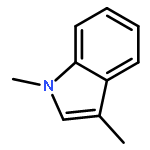
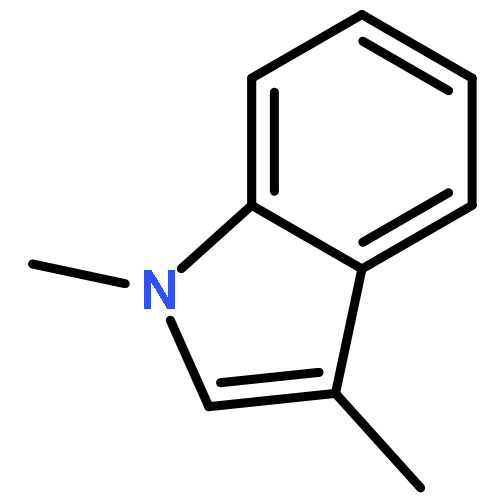




![Cyclopropa[d]naphthalene,1,1a,4,4a,5,6,7,8-octahydro-2,4a,8,8-tetramethyl-, (1aS,4aS,8aS)-](http://img.cochemist.com/ccimg/500/470-40-6.png)
![Cyclopropa[d]naphthalene,1,1a,4,4a,5,6,7,8-octahydro-2,4a,8,8-tetramethyl-, (1aS,4aS,8aS)-](http://img.cochemist.com/ccimg/500/470-40-6_b.png)