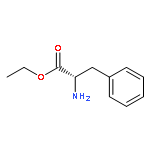
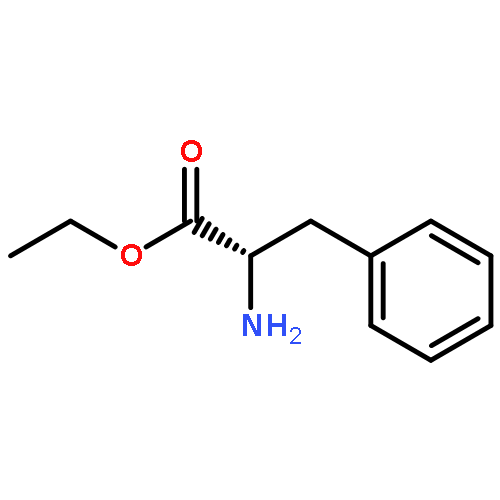
Co-reporter:Gang He, Yingsheng Zhao, Shuyu Zhang, Chengxi Lu, and Gong Chen
Journal of the American Chemical Society January 11, 2012 Volume 134(Issue 1) pp:3-6
Publication Date(Web):December 21, 2011
DOI:10.1021/ja210660g
Efficient methods have been developed to synthesize azetidine, pyrrolidine, and indoline compounds via palladium-catalyzed intramolecular amination of C–H bonds at the γ and δ positions of picolinamide (PA) protected amine substrates. These methods feature relatively a low catalyst loading, use of inexpensive reagents, and convenient operating conditions. Their selectivities are predictable. These methods highlight the use of unactivated C–H bond, especially the C(sp3)–H bond of methyl groups, as functional groups in organic synthesis.
Co-reporter:Guo-Xing Li;Cristian A. Morales-Rivera;Fang Gao;Yaxin Wang;Gang He;Peng Liu
Chemical Science (2010-Present) 2017 vol. 8(Issue 10) pp:7180-7185
Publication Date(Web):2017/09/25
DOI:10.1039/C7SC02773G
We report a unified photoredox-catalysis strategy for both hydroxylation and amidation of tertiary and benzylic C–H bonds. Use of hydroxyl perfluorobenziodoxole (PFBl–OH) oxidant is critical for efficient tertiary C–H functionalization, likely due to the enhanced electrophilicity of the benziodoxole radical. Benzylic methylene C–H bonds can be hydroxylated or amidated using unmodified hydroxyl benziodoxole oxidant Bl–OH under similar conditions. An ionic mechanism involving nucleophilic trapping of a carbocation intermediate by H2O or CH3CN cosolvent is presented.
Co-reporter:Gang He, Bo Wang, William A. Nack, and Gong Chen
Accounts of Chemical Research 2016 Volume 49(Issue 4) pp:635
Publication Date(Web):March 25, 2016
DOI:10.1021/acs.accounts.6b00022
α-Amino acids (αAA) are one of the most useful chiral building blocks for synthesis. There are numerous general strategies that have commonly been used for αAA synthesis, many of which employ de novo synthesis focused on enantioselective bond construction around the Cα center and others that consider conversion of existing αAA precursors carrying suitable functional groups on side chains (e.g., serine and aspartic acid). Despite significant advances in synthetic methodology, the efficient synthesis of enantiopure αAAs carrying complex side chains, as seen in numerous peptide natural products, remains challenging. Complementary to these “conventional” strategies, a strategy based on the selective functionalization of side chain C–H bonds, particularly sp3 hybridized C–H bonds, of various readily available αAA precursors may provide a more straightforward and broadly applicable means for the synthesis and transformation of αAAs. However, many hurdles related to the low reactivity of C(sp3)–H bonds and the difficulty of controlling selectivity must be overcome to realize the potential of C–H functionalization chemistry in this synthetic application. Over the past few years, we have carried out a systematic investigation of palladium-catalyzed bidentate auxiliary-directed C–H functionalization reactions for αAA substrates. Our strategies utilize two different types of amide-linked auxiliary groups, attached at the N or C terminus of αAA substrates, to exert complementary regio- and stereocontrol on C–H functionalization reactions through palladacycle intermediates. A variety of αAA precursors can undergo multiple modes of C(sp3)–H functionalization, including arylation, alkenylation, alkynylation, alkylation, alkoxylation, and intramolecular aminations, at the β, γ, and even δ positions to form new αAA products with diverse structures. In addition to transforming αAAs at previously unreachable positions, these palladium-catalyzed C–H functionalization strategies enable new retrosynthetic logic for the synthesis of many basic αAAs from a common alanine precursor. This approach reduces the synthetic difficulty for many αAAs by bypassing the requirement for stereocontrol at Cα and relies on straightforward and convergent single-bond coupling transformations at the β-methyl position of alanine to access a wide range of β-monosubstituted αAAs. Moreover, these β-monosubstituted αAAs can undergo further C–H functionalization at the β-methylene position to generate various β-branched αAAs in a stereoselective and programmable fashion. These new strategies offer readily applicable methods for synthesis of challenging αAAs and may facilitate the efficient total synthesis of complex peptide natural products.
Co-reporter:Bo Wang; Yunpeng Liu; Rui Jiao; Yiqing Feng; Qiong Li; Chen Chen; Long Liu; Gang He
Journal of the American Chemical Society 2016 Volume 138(Issue 11) pp:3926-3932
Publication Date(Web):February 25, 2016
DOI:10.1021/jacs.6b01384
The mannopeptimycins are a class of glycopeptide natural products with unusual structures and potent antibiotic activity against a range of Gram-positive multidrug-resistant bacteria. Their cyclic hexapeptide core features a pair of unprecedented β-hydroxyenduracididines (l- and d-βhEnd), an O-glycosylated d-Tyr carrying an α-linked dimannose, and a β-methylated Phe residue. The d-βhEnd unit also carries an α-linked mannopyranose at the most hindered N of its cyclic guanidine ring. Herein, we report the first total synthesis of mannopeptimycin α and β with fully elaborated N- and O-linked sugars. Critically, a gold-catalyzed N-glycosylation of a d-βhEnd substrate with a mannosyl ortho-alkynylbenzoate donor enabled the synthesis of the most challenging N-Man-d-βhEnd unit with excellent efficiency and stereoselectivity. The l-βMePhe unit was prepared using a Pd-catalyzed C–H arylation method. The l-βhEnd, d-Tyr(di-Man), and l-βMePhe units were prepared in gram quantities. A convergent assembly of the cyclic peptide scaffold and a single global hydrogenolysis deprotection operation provided mannopeptimycin α and β.
Co-reporter:Yaxin Wang, Guo-Xing Li, Guohui Yang, Gang He and Gong Chen
Chemical Science 2016 vol. 7(Issue 4) pp:2679-2683
Publication Date(Web):11 Jan 2016
DOI:10.1039/C5SC04169D
A highly tunable radical-mediated reaction system for the functionalization of tertiary aliphatic C–H bonds was developed. Reactions of various substrates with the Zhdankin azidoiodane reagent 1, Ru(bpy)3Cl2, and visible light irradiation at room temperature gave C–H azidated or halogenated products in an easily controllable fashion. These reactions are efficient, selective, and compatible with complex substrates. They provide a potentially valuable tool for selectively labeling tertiary C–H bonds of organic and biomolecules with tags of varied chemical and biophysical properties for comparative functional studies.
Co-reporter:Bo Wang, Daniel D. McClosky, Charles T. Anderson, Gong Chen
Carbohydrate Research 2016 Volume 433() pp:54-62
Publication Date(Web):4 October 2016
DOI:10.1016/j.carres.2016.07.012
•Design and synthesis of new click-compatible alkynyl sugar probes.•Expanding the toolbox of click-compatible alkynyl sugar probes.•Efficient and scalable synthesis strategy.Metabolic labeling based on the click chemistry between alkynyl and azido groups offers a powerful tool to study the function of carbohydrates in living systems, including plants. Herein, we describe the chemical synthesis of six alkynyl-modified sugars designed as analogs to D-glucose, D-mannose, L-rhamnose and sucrose present in plant cell walls. Among these new alkynyl probes, four of them are the 6-deoxy-alkynyl analogs of the corresponding sugars and do not possess any 6-OH groups. The other two are based on a new structural design, in which an ethynyl group is incorporated at the C-6 position of the sugar and the 6-OH group remains. The synthetic routes for both types of probes share common aldehyde intermediates, which are derived from the corresponding 6-OH precursor with other hydroxy groups protected. The overall synthesis sequence of these probes is efficient, concise, and scalable.
Co-reporter:Daniel D. McClosky, Bo Wang, Gong Chen, Charles T. Anderson
Phytochemistry 2016 Volume 123() pp:16-24
Publication Date(Web):March 2016
DOI:10.1016/j.phytochem.2016.01.007
•6-Deoxy-alkynyl glucose was synthesized as a sugar analog to study wall biosynthesis.•6dAG incorporates into growth-arrested root hairs in Arabidopsis seedlings.•6dAG incorporation is metabolism-, time-, and concentration-dependent.•Induced callose colocalizes with the site of 6dAG incorporation.Plant cell walls are dynamic structures whose polysaccharide components are rearranged and recycled during growth and morphogenesis. Covalent fluorescent tagging of these polysaccharides following a metabolic labeling approach can help elucidate these changes. Herein reported are the synthesis and seedling-incorporation of a plant polysaccharide chemical reporter, 6-deoxy-alkynyl glucose (6dAG), that is modeled on d-glucose. Whereas fucose-alkyne, a previously reported chemical reporter for pectin, incorporates diffusely throughout growing cell walls, 6dAG incorporated specifically into root hair tips. This incorporation occurs in a time- and concentration-dependent manner. 6dAG exposure both induces and colocalizes with callose deposition in this tissue, and arrests both root hair and root growth. These results show that plants can incorporate an additional alkynyl-modified sugar analog into their metabolism, and into a discrete subcellular location.Arabidopsis root epidermal cells incorporate the click-compatible sugar analog, 6-deoxy-alkynyl glucose, specifically into root hair tip cell walls where it arrests root hair growth.
Co-reporter:Bo Wang;Gang He
Science China Chemistry 2015 Volume 58( Issue 8) pp:1345-1348
Publication Date(Web):2015 August
DOI:10.1007/s11426-015-5392-z
β-Di-substituted α-amino acids (AAs) contain adjacent carbon stereogenic centers and pose considerable synthetic challenge. Complementary to the conventional synthesis strategies based on the transformation of existing functional groups, we envisioned these molecules could be quickly accessed via selective functionalization of sp3 hybridized C-H bonds on the side chains of common α-AA precursors. We report a readily applicable method to prepare β-alkynyl α-amino acids via Pd-catalyzed diastereoselective C(sp3)-H alkynylation of common α-amino acids precursors with acetylene bromide.
Co-reporter:Shu-Yu Zhang; Qiong Li; Gang He; William A. Nack
Journal of the American Chemical Society 2014 Volume 137(Issue 1) pp:531-539
Publication Date(Web):December 10, 2014
DOI:10.1021/ja511557h
We report a method for the monoselective alkylation of ortho-C–H bonds of N-quinolyl benzamides with both primary and secondary alkyl halides under palladium catalysis. With promotion by NaHCO3 and (BnO)2PO2H or (PhO)2PO2H, symmetric benzamide substrates can be selectively ortho-alkylated to give either mono- or dialkylated products by simply adjusting the amount of NaHCO3 applied. The use of phosphate notably improves the alkylation yield, although it may not be directly involved in C–H palladation or the subsequent functionalization step. Kinetic isotope effect studies indicate that C–H palladation is not the rate-limiting step. Examination of the reactions of an isolated palladacycle intermediate with both cis- and trans-4-methylcyclohexyl iodides revealed surprising stereoretentive couplings of these alkyl iodides. This evidence strongly suggests that the functionalization of the palladacycle with secondary alkyl iodides proceeds via a rarely precedented concerted oxidative addition pathway.
Co-reporter:Bo Wang, William A. Nack, Gang He, Shu-Yu Zhang and Gong Chen
Chemical Science 2014 vol. 5(Issue 10) pp:3952-3957
Publication Date(Web):17 Jun 2014
DOI:10.1039/C4SC01545B
We report a highly efficient and practical protocol for palladium-catalyzed N-quinolylcarboxamide (AQ)-directed arylation of the unactivated β-C(sp3)–H bonds of alanine with aryl iodides at room temperature. For the first time, a broad range of easily accessible aryl iodides can be installed onto the β-methyl group of AQ-coupled phthaloyl alanine mono-selectively, providing both natural and unnatural aromatic α-amino acids. Access to these mono-arylated compounds enables subsequent AQ-directed diastereoselective C–H functionalization, allowing the preparation of various β-disubstituted aromatic α-amino acids in a programmable manner.
Co-reporter:Qiong Li, Shu-Yu Zhang, Gang He, Zhaoyan Ai, William A. Nack, and Gong Chen
Organic Letters 2014 Volume 16(Issue 6) pp:1764-1767
Publication Date(Web):March 4, 2014
DOI:10.1021/ol500464x
In this report, a highly efficient method for the room temperature installation of alkyl amino motifs onto the ortho position of anilines via Cu-catalyzed carboxamide-directed amination with alkylamines is described. This method offers a practical solution for the rapid synthesis of complex arylamines from simple starting materials and enables new planning strategies for the construction of arylamine-containing pharmacophores. A single electron transfer (SET)-mediated mechanism is proposed.
Co-reporter:Bo Wang, Chengxi Lu, Shu-Yu Zhang, Gang He, William A. Nack, and Gong Chen
Organic Letters 2014 Volume 16(Issue 23) pp:6260-6263
Publication Date(Web):November 20, 2014
DOI:10.1021/ol503248f
A method is reported for palladium-catalyzed N-quinolyl carboxamide-directed olefination of the unactivated C(sp3)–H bonds of phthaloyl alanine with a broad range of vinyl iodides at room temperature. This reaction represents the first example of the stereoretentive installation of multisubstituted terminal and internal olefins onto unactivated C(sp3)–H bonds. These methods enable access to a wide range of challenging β-vinyl α-amino acid products in a streamlined and controllable fashion, beginning from simple precursors.
Co-reporter:Gang He, Shu-Yu Zhang, William A. Nack, Ryan Pearson, Javon Rabb-Lynch, and Gong Chen
Organic Letters 2014 Volume 16(Issue 24) pp:6488-6491
Publication Date(Web):December 9, 2014
DOI:10.1021/ol503347d
To access the key Ile-Hpa pseudodipeptide motif in hibispeptins, a series of bidentate carboxamide-based auxiliary groups have been explored to facilitate the palladium-catalyzed arylation of unactivated γ-C(sp3)–H bonds of Ile precursor with aryl iodides. A new pyridylmethylamine-based auxiliary group PR is introduced, which permits the use of more sterically hindered ortho-substituted aryl iodide substrates and can be removed under mild conditions. Pd-catalyzed PR-directed γ-C(sp3)–H arylation enabled the first total synthesis of hibispeptin A.
Co-reporter:Qiong Li;Shu-Yu Zhang;Gang He;William A. Nack
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 7) pp:1544-1548
Publication Date(Web):
DOI:10.1002/adsc.201400121
Co-reporter:Chengxi Lu, Shu-Yu Zhang, Gang He, William A. Nack, Gong Chen
Tetrahedron 2014 70(27–28) pp: 4197-4203
Publication Date(Web):
DOI:10.1016/j.tet.2014.02.070
Co-reporter:Shu-Yu Zhang ; Gang He ; William A. Nack ; Yingsheng Zhao ; Qiong Li
Journal of the American Chemical Society 2013 Volume 135(Issue 6) pp:2124-2127
Publication Date(Web):January 27, 2013
DOI:10.1021/ja312277g
We report an efficient method for the alkylation of γ-C(sp3)–H bonds of picolinamide-protected aliphatic amine substrates with primary alkyl iodides via palladium catalysis. Ag2CO3 and dibenzyl phosphate, (BnO)2PO2H, are critical promoters of this reaction. These reactions provide a convenient and straightforward method for the preparation of high-value N-containing products from readily available amine and alkyl iodide precursors.
Co-reporter:Shu-Yu Zhang, Qiong Li, Gang He, William A. Nack, and Gong Chen
Journal of the American Chemical Society 2013 Volume 135(Issue 32) pp:12135-12141
Publication Date(Web):July 26, 2013
DOI:10.1021/ja406484v
We report a new set of reactions based on the Pd-catalyzed alkylation of methylene C(sp3)–H bonds of aliphatic quinolyl carboxamides with α-haloacetate and methyl iodide and applications in the stereoselective synthesis of various β-alkylated α-amino acids. These reactions represent the first generally applicable method for the catalytic alkylation of unconstrained and unactivated methylene C–H bonds with high synthetic relevance. When applied with simple isotope-enriched reagents, they also provide a convenient and powerful means to site-selectively incorporate isotopes into the carbon scaffolds of amino acid compounds.
Co-reporter:William A. Nack, Gang He, Shu-Yu Zhang, Chengxi Lu, and Gong Chen
Organic Letters 2013 Volume 15(Issue 13) pp:3440-3443
Publication Date(Web):June 20, 2013
DOI:10.1021/ol4015078
A new strategy for the synthesis of tetrahydroquinolines (THQs) via the sequential functionalizations of remote C–H bonds is reported. This method uses a single picolinamide directing/protecting group to effect Pd-catalyzed γ-C(sp3)–H arylation, metal-free ε-C(sp2)–H iodination, and Cu-catalyzed intramolecular C–N cross-coupling. The overall sequence is efficient and versatile, and offers a streamlined synthesis of THQs with complex substitution patterns from readily available aryl iodide and aliphatic amine precursors.
Co-reporter:Yunpeng Liu, Jared Marshall, Qiong Li, Nicola Edwards, Gong Chen
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 8) pp:2328-2331
Publication Date(Web):15 April 2013
DOI:10.1016/j.bmcl.2013.02.068
Mannose-6-phosphate (M6P)-containing N-linked glycans are essential signaling molecules for sorting hydrolases in eukaryotic cells. Their receptors, especially the cation-independent M6P receptors (CI-MPRs), have emerged as promising protein targets for targeted drug delivery for the treatment of lysosomal storage disease and liver fibrosis. In this Letter, we describe the design and synthesis of novel bivalent mimetic ligands for CI-MPRs. We report that for the first time, a newly-discovered binding motif, GlcNAc-M6P, has been incorporated in mimetic ligands. M6P- and GlcNAc-M6P-containing building blocks, equipped with NH2 and CO2H handles, have been prepared and assembled with an ornithine linker through amide coupling reactions. Efficient global deprotection protocols have also been developed which have been showcased in the synthesis of our novel bivalent mimetic ligands.
Co-reporter:Dr. Gang He;Dr. Shu-Yu Zhang;William A. Nack;Dr. Qiong Li ;Dr. Gong Chen
Angewandte Chemie International Edition 2013 Volume 52( Issue 42) pp:11124-11128
Publication Date(Web):
DOI:10.1002/anie.201305615
Co-reporter:Dr. Gang He;Dr. Shu-Yu Zhang;William A. Nack;Dr. Qiong Li ;Dr. Gong Chen
Angewandte Chemie 2013 Volume 125( Issue 42) pp:11330-11334
Publication Date(Web):
DOI:10.1002/ange.201305615
Co-reporter:Shu-Yu Zhang ; Gang He ; Yingsheng Zhao ; Kiwan Wright ; William A. Nack
Journal of the American Chemical Society 2012 Volume 134(Issue 17) pp:7313-7316
Publication Date(Web):April 9, 2012
DOI:10.1021/ja3023972
We report the efficient synthesis of alkyl ethers by the functionalization of unactivated sp3- and sp2-hybridized C–H bonds. In the Pd(OAc)2-catalyzed, PhI(OAc)2-mediated reaction system, picolinamide-protected amine substrates undergo facile alkoxylation at the γ or δ positions with a range of alcohols, including t-BuOH, to give alkoxylated products. This method features a relatively broad substrate scope for amines and alcohols, inexpensive reagents, and convenient operating conditions. This method highlights the emerging value of unactivated C–H bonds, particularly the C(sp3)–H bond of methyl groups, as functional groups in organic synthesis.
Co-reporter:Yingsheng Zhao, Gang He, William A. Nack, and Gong Chen
Organic Letters 2012 Volume 14(Issue 12) pp:2948-2951
Publication Date(Web):June 6, 2012
DOI:10.1021/ol301214u
An efficient functionalization of ortho-C(sp2)–H bonds of picolinamide (PA)-protected benzylamine substrates with a range of vinyl iodides as well as acetylenic bromide is reported. ortho-Phenyl benzoic acid (oPBA) acts as an effective promoter in this reaction system. This method provides a practical strategy to access highly functionalized benzylamine compounds for organic synthesis.
Co-reporter:Meiqing Zheng, Yifan Yang, Ming Zhao, Xiaoyi Zhang, Jianhui Wu, Gong Chen, Li Peng, Yuji Wang, Shiqi Peng
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 5) pp:1672-1681
Publication Date(Web):May 2011
DOI:10.1016/j.ejmech.2011.02.017
Isoquinoline-3-carboxylic acid (2) was modified with amino acid benzylesters and 18 novel N-isoquinoline-3-carbonylamino acid benzylesters (3a–r) were provided. The IC50 values of 3a–r against the proliferation of HL-60 and Hela cells were less than 1 × 10−8 M and 6 × 10−7 M, respectively. On S180 mice model 100 μmol/kg of 3a–r effectively inhibited the growth of the tumors. Using MFA based Cerius2 QSAR module, two equations (r, 0.989 and 0.987) were established to correlate the structure with the in vitro and in vivo activities. The benefit of this modification was supported with both the in vitro membrane permeation test and the in vivo anti-tumor assay. The in vitro membrane permeability of N-isoquinoline-3-carbonyl-l-threonine benzylester (3n) and N-isoquinoline-3-carbonyl-l-leucine benzylester (3q) was 2.5 fold higher than that of 2, and the in vivo anti-tumor activity of 3n, q was 4.4-fold higher than that of 2.Highlights► Design, evaluation, topoisomerse I inhibition and 3D QSAR of novel N-isoquinoline-3-carbonyl-l-amino acid benzylesters were investigated as the potential intercalators. ► On S180 mouse model the oral effective dose was 1 μmol/kg. ► At the dose of 1 mmol/kg the healthy mice had no neurotoxic and acute toxic response. ► The LD50 value of N-isoquinoline-3-carbonyl-l-amino acid benzylesters is more than 540 mg/kg.
Co-reporter:Yunpeng Liu and Gong Chen
The Journal of Organic Chemistry 2011 Volume 76(Issue 21) pp:8682-8689
Publication Date(Web):September 28, 2011
DOI:10.1021/jo2010999
Mannose-6-phosphate (M6P) containing N-linked glycans are the essential targeting signals for hydrolases sorting in eukaryotic cells. To facilitate their structural and binding analyses, a highly efficient and convergent method has been developed to prepare complex N-linked glycans with well-defined M6P and N-acetylglucosamine (GlcNAc)-M6P motifs, a newly identified binding element for M6P receptors. The GlcNAc-M6P motif was stereoselectively installed at the late stage of the synthesis. Sequential deprotection of benzyl and acetate groups provided the fully deprotected N-glycans in excellent yield.
Co-reporter:Dr. Gang He ;Dr. Gong Chen
Angewandte Chemie 2011 Volume 123( Issue 22) pp:5298-5302
Publication Date(Web):
DOI:10.1002/ange.201100984
Co-reporter:Dr. Yunpeng Liu;Yan Mei Chan;Dr. Jianhui Wu;Dr. Chen Chen; Dr. Alan Benesi;Jing Hu; Dr. Yanming Wang; Dr. Gong Chen
ChemBioChem 2011 Volume 12( Issue 5) pp:685-690
Publication Date(Web):
DOI:10.1002/cbic.201000785
Co-reporter:Dr. Gang He ;Dr. Gong Chen
Angewandte Chemie International Edition 2011 Volume 50( Issue 22) pp:5192-5196
Publication Date(Web):
DOI:10.1002/anie.201100984
Co-reporter:Yiqing Feng, Yuji Wang, Bradley Landgraf, Shi Liu and Gong Chen
Organic Letters 2010 Volume 12(Issue 15) pp:3414-3417
Publication Date(Web):June 28, 2010
DOI:10.1021/ol101220x
A practical synthetic method for the annulation of benzo-rings by the intramolecular coupling of an aryl iodide and a methylene C−H bond is described. The palladium-catalyzed C−H functionalization is directed by an aminoquinoline carboxamide group, which can be easily installed and removed. High yields and broad substrate scope were achieved. An additive of ortho-phenyl benzoic acid, identified from a systematic screening, functions as a critical ligand for the catalytic process under mild condition, even at near room temperature.
Co-reporter:Yiqing Feng
Angewandte Chemie 2010 Volume 122( Issue 5) pp:970-973
Publication Date(Web):
DOI:10.1002/ange.200905134
Co-reporter:Yiqing Feng
Angewandte Chemie International Edition 2010 Volume 49( Issue 5) pp:958-961
Publication Date(Web):
DOI:10.1002/anie.200905134
Co-reporter:Gang He ; Yingsheng Zhao ; Shuyu Zhang ; Chengxi Lu
Journal of the American Chemical Society () pp:
Publication Date(Web):December 21, 2011
DOI:10.1021/ja210660g
Efficient methods have been developed to synthesize azetidine, pyrrolidine, and indoline compounds via palladium-catalyzed intramolecular amination of C–H bonds at the γ and δ positions of picolinamide (PA) protected amine substrates. These methods feature relatively a low catalyst loading, use of inexpensive reagents, and convenient operating conditions. Their selectivities are predictable. These methods highlight the use of unactivated C–H bond, especially the C(sp3)–H bond of methyl groups, as functional groups in organic synthesis.
Co-reporter:Yaxin Wang, Guo-Xing Li, Guohui Yang, Gang He and Gong Chen
Chemical Science (2010-Present) 2016 - vol. 7(Issue 4) pp:NaN2683-2683
Publication Date(Web):2016/01/11
DOI:10.1039/C5SC04169D
A highly tunable radical-mediated reaction system for the functionalization of tertiary aliphatic C–H bonds was developed. Reactions of various substrates with the Zhdankin azidoiodane reagent 1, Ru(bpy)3Cl2, and visible light irradiation at room temperature gave C–H azidated or halogenated products in an easily controllable fashion. These reactions are efficient, selective, and compatible with complex substrates. They provide a potentially valuable tool for selectively labeling tertiary C–H bonds of organic and biomolecules with tags of varied chemical and biophysical properties for comparative functional studies.
Co-reporter:Bo Wang, William A. Nack, Gang He, Shu-Yu Zhang and Gong Chen
Chemical Science (2010-Present) 2014 - vol. 5(Issue 10) pp:NaN3957-3957
Publication Date(Web):2014/06/17
DOI:10.1039/C4SC01545B
We report a highly efficient and practical protocol for palladium-catalyzed N-quinolylcarboxamide (AQ)-directed arylation of the unactivated β-C(sp3)–H bonds of alanine with aryl iodides at room temperature. For the first time, a broad range of easily accessible aryl iodides can be installed onto the β-methyl group of AQ-coupled phthaloyl alanine mono-selectively, providing both natural and unnatural aromatic α-amino acids. Access to these mono-arylated compounds enables subsequent AQ-directed diastereoselective C–H functionalization, allowing the preparation of various β-disubstituted aromatic α-amino acids in a programmable manner.
Co-reporter:B. Wang, X. Wu, R. Jiao, S.-Y. Zhang, W. A. Nack, G. He and G. Chen
Inorganic Chemistry Frontiers 2015 - vol. 2(Issue 10) pp:NaN1321-1321
Publication Date(Web):2015/07/23
DOI:10.1039/C5QO00112A
An efficient protocol for the synthesis of β-alkyl α-amino acids via palladium-catalyzed β-C(sp3)–H alkylation of aminoquinoline-coupled phthaloyl alanine is developed. The new TFA-promoted reaction conditions provide excellent C–H alkylation reactivity with alkyl iodides bearing moderately electron-withdrawing groups at room temperature. A range of β-alkyl α-amino acid products, including some difficult to access by other means, can be quickly prepared from readily available primary alkyl iodides and alanine precursors.












![2-Pyridinecarboxamide, N-[(3-fluorophenyl)methyl]-](http://img.cochemist.com/ccimg/867400/867350-28-5.png)
![2-Pyridinecarboxamide, N-[(3-fluorophenyl)methyl]-](http://img.cochemist.com/ccimg/867400/867350-28-5_b.png)
![2-PYRIDINECARBOXAMIDE, N-[3-(4-METHOXYPHENYL)-1-METHYLPROPYL]-](http://img.cochemist.com/ccimg/867400/867347-59-9.png)
![2-PYRIDINECARBOXAMIDE, N-[3-(4-METHOXYPHENYL)-1-METHYLPROPYL]-](http://img.cochemist.com/ccimg/867400/867347-59-9_b.png)












![2-PYRIDINECARBOXAMIDE, N-[(1R)-1-PHENYLETHYL]-](http://img.cochemist.com/ccimg/668500/668461-31-2.png)
![2-PYRIDINECARBOXAMIDE, N-[(1R)-1-PHENYLETHYL]-](http://img.cochemist.com/ccimg/668500/668461-31-2_b.png)
![Acetamide, 2,2,2-trifluoro-N-[3-(4-iodophenyl)propyl]-](http://img.cochemist.com/ccimg/643100/643093-84-9.png)
![Acetamide, 2,2,2-trifluoro-N-[3-(4-iodophenyl)propyl]-](http://img.cochemist.com/ccimg/643100/643093-84-9_b.png)





























![3-iodo-1-[(4-methylphenyl)sulfonyl]-1H-Indole](http://img.cochemist.com/ccimg/170500/170456-80-1.png)
![3-iodo-1-[(4-methylphenyl)sulfonyl]-1H-Indole](http://img.cochemist.com/ccimg/170500/170456-80-1_b.png)


![Benzamide, N-[3-(4-iodophenyl)propyl]-](http://img.cochemist.com/ccimg/151000/150932-98-2.png)
![Benzamide, N-[3-(4-iodophenyl)propyl]-](http://img.cochemist.com/ccimg/151000/150932-98-2_b.png)




![L-Phenylalanine,N-[(1,1-dimethylethoxy)carbonyl]-b-methyl-, (bR)-](http://img.cochemist.com/ccimg/145500/145432-51-5.png)
![L-Phenylalanine,N-[(1,1-dimethylethoxy)carbonyl]-b-methyl-, (bR)-](http://img.cochemist.com/ccimg/145500/145432-51-5_b.png)


![Acetic acid, 2,2'-[(11,23-dihydroxypentacyclo[19.3.1.13,7.19,13.115,19]octacosa-1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-5,17-diyl)bis(oxy)]bis-, 1,1'-diethyl ester](/data/chemimg/110600/142267-55-8.png)
![Acetic acid, 2,2'-[(11,23-dihydroxypentacyclo[19.3.1.13,7.19,13.115,19]octacosa-1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-5,17-diyl)bis(oxy)]bis-, 1,1'-diethyl ester](/data/chemimg/110600/142267-55-8_b.png)








![Zinc, bromo[1-(trifluoromethyl)ethenyl]-](http://img.cochemist.com/ccimg/140000/139954-92-0.png)
![Zinc, bromo[1-(trifluoromethyl)ethenyl]-](http://img.cochemist.com/ccimg/140000/139954-92-0_b.png)
![Cyclohexene, 1-[1-(trifluoromethyl)ethenyl]-](http://img.cochemist.com/ccimg/138600/138559-93-0.png)
![Cyclohexene, 1-[1-(trifluoromethyl)ethenyl]-](http://img.cochemist.com/ccimg/138600/138559-93-0_b.png)
![1-(2,6-DIMETHYL-1-PIPERIDINYL)-2-({1-[2-OXO-2-(1-PYRROLIDINYL)ETHYL]-1H-BENZIMIDAZOL-2-YL}SULFANYL)ETHANONE](http://img.cochemist.com/ccimg/134900/134833-83-3.png)
![1-(2,6-DIMETHYL-1-PIPERIDINYL)-2-({1-[2-OXO-2-(1-PYRROLIDINYL)ETHYL]-1H-BENZIMIDAZOL-2-YL}SULFANYL)ETHANONE](http://img.cochemist.com/ccimg/134900/134833-83-3_b.png)






![L-Threonine, O-[(1,1-dimethylethyl)dimethylsilyl]-, methyl ester](http://img.cochemist.com/ccimg/127700/127680-34-6.png)
![L-Threonine, O-[(1,1-dimethylethyl)dimethylsilyl]-, methyl ester](http://img.cochemist.com/ccimg/127700/127680-34-6_b.png)












![Silane, [(1E)-2-(1-cyclohexen-1-yl)ethenyl]trimethyl-](http://img.cochemist.com/ccimg/116200/116103-21-0.png)
![Silane, [(1E)-2-(1-cyclohexen-1-yl)ethenyl]trimethyl-](http://img.cochemist.com/ccimg/116200/116103-21-0_b.png)




















![2-Pyridinecarboxamide, N-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/77300/77250-70-5.png)
![2-Pyridinecarboxamide, N-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/77300/77250-70-5_b.png)












![Benzene, [(1E)-2-iodo-1-methylethenyl]-](http://img.cochemist.com/ccimg/66700/66644-32-4.png)
![Benzene, [(1E)-2-iodo-1-methylethenyl]-](http://img.cochemist.com/ccimg/66700/66644-32-4_b.png)






![2-Pyridinecarboxamide, N-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/61400/61350-01-4.png)
![2-Pyridinecarboxamide, N-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/61400/61350-01-4_b.png)














![Benzene, [(1E)-2-iodoethenyl]-](http://img.cochemist.com/ccimg/42600/42599-24-6.png)
![Benzene, [(1E)-2-iodoethenyl]-](http://img.cochemist.com/ccimg/42600/42599-24-6_b.png)















![Bicyclo[2.2.1]heptan-2-amine,(1R,2S,4S)-rel-](http://img.cochemist.com/ccimg/31100/31002-73-0.png)
![Bicyclo[2.2.1]heptan-2-amine,(1R,2S,4S)-rel-](http://img.cochemist.com/ccimg/31100/31002-73-0_b.png)
![9-Borabicyclo[3.3.1]nonane, 9-octyl-](http://img.cochemist.com/ccimg/30100/30089-00-0.png)
![9-Borabicyclo[3.3.1]nonane, 9-octyl-](http://img.cochemist.com/ccimg/30100/30089-00-0_b.png)





![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-L-leucyl-](http://img.cochemist.com/ccimg/26100/26055-05-0.png)
![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-L-leucyl-](http://img.cochemist.com/ccimg/26100/26055-05-0_b.png)






































![furo[3,4-b]pyridin-5(7H)-one](http://img.cochemist.com/ccimg/4800/4733-69-1.png)
![furo[3,4-b]pyridin-5(7H)-one](http://img.cochemist.com/ccimg/4800/4733-69-1_b.png)


















![Bicyclo[2.2.1]heptan-2-amine,1,7,7-trimethyl-, (1R,2S,4R)-rel-](http://img.cochemist.com/ccimg/500/464-42-6.png)
![Bicyclo[2.2.1]heptan-2-amine,1,7,7-trimethyl-, (1R,2S,4R)-rel-](http://img.cochemist.com/ccimg/500/464-42-6_b.png)




![2-PYRIDINECARBOXAMIDE, N-[(3-METHYLPHENYL)METHYL]-](http://img.cochemist.com/ccimg/867400/867350-27-4.png)
![2-PYRIDINECARBOXAMIDE, N-[(3-METHYLPHENYL)METHYL]-](http://img.cochemist.com/ccimg/867400/867350-27-4_b.png)
![2-Pyridinecarboxamide, N-[(2-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/867400/867350-26-3.png)
![2-Pyridinecarboxamide, N-[(2-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/867400/867350-26-3_b.png)
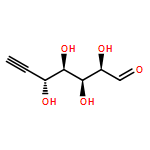









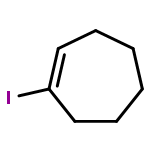


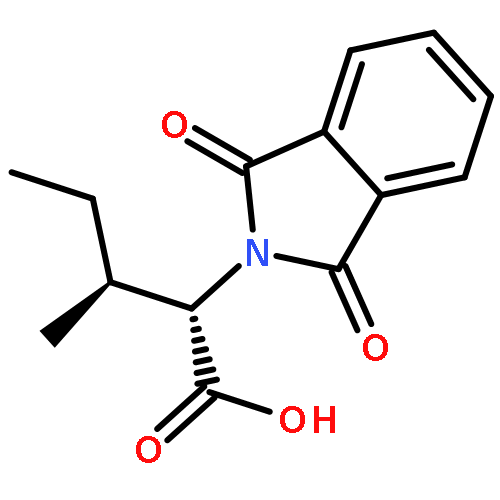




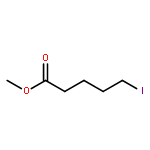



![2-PYRIDINECARBOXAMIDE, N-[(3-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/867400/867350-23-0.png)
![2-PYRIDINECARBOXAMIDE, N-[(3-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/867400/867350-23-0_b.png)
![2-PYRIDINECARBOXAMIDE, N-[(3-BROMOPHENYL)METHYL]-](http://img.cochemist.com/ccimg/867400/867347-57-7.png)
![2-PYRIDINECARBOXAMIDE, N-[(3-BROMOPHENYL)METHYL]-](http://img.cochemist.com/ccimg/867400/867347-57-7_b.png)
![2-PYRIDINECARBOXAMIDE, N-[(2-FLUOROPHENYL)METHYL]-](http://img.cochemist.com/ccimg/850200/850169-83-4.png)
![2-PYRIDINECARBOXAMIDE, N-[(2-FLUOROPHENYL)METHYL]-](http://img.cochemist.com/ccimg/850200/850169-83-4_b.png)















![2H-1,2,4-Benzothiadiazine-7-sulfonamide,3-bicyclo[2.2.1]hept-5-en-2-yl-6-chloro-3,4-dihydro-, 1,1-dioxide](http://img.cochemist.com/ccimg/2300/2259-96-3.png)
![2H-1,2,4-Benzothiadiazine-7-sulfonamide,3-bicyclo[2.2.1]hept-5-en-2-yl-6-chloro-3,4-dihydro-, 1,1-dioxide](http://img.cochemist.com/ccimg/2300/2259-96-3_b.png)
![2-PYRIDINECARBOXAMIDE, N-[(2-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/867400/867350-21-8.png)
![2-PYRIDINECARBOXAMIDE, N-[(2-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/867400/867350-21-8_b.png)