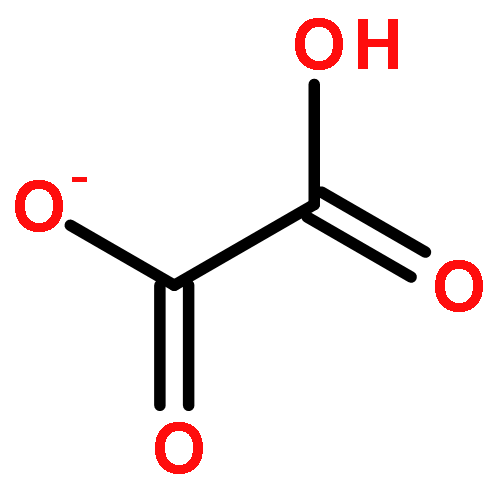
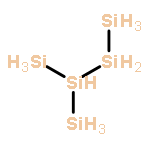
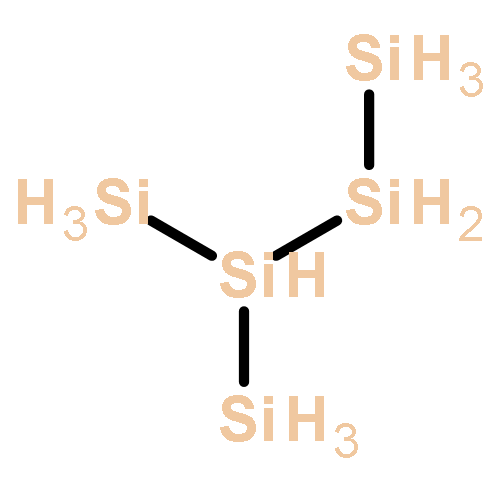
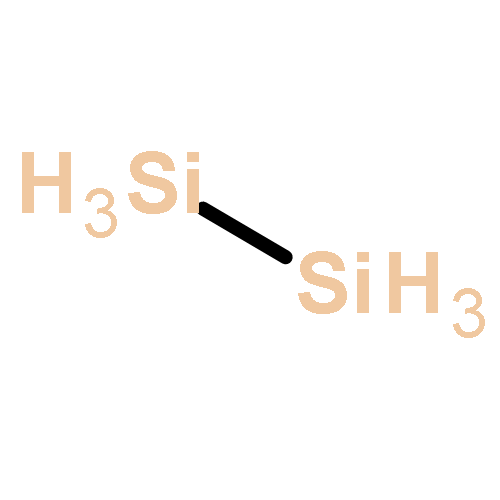
Co-reporter:Zoltan Varga, Pragya Verma, and Donald G. Truhlar
Journal of the American Chemical Society September 13, 2017 Volume 139(Issue 36) pp:12569-12569
Publication Date(Web):August 15, 2017
DOI:10.1021/jacs.7b06107
Excited spin states are important for reactivity, catalysis, and magnetic applications. This work examines the relative energies of the spin states of O atom, Fe2+ ion, and FeF2 and characterizes their excited spin states. Both single-configuration and multireference methods are used to establish the character of the lowest singlet excited state of all three systems and the lowest triplet excited state of Fe2+ and FeF2. We find that the conventional representation of the orbital occupancies is incorrect in that the states have more unpaired electrons than the minimum number required by their total electron spin quantum number. In particular, we find that, for a given spin state, an electronic configuration with more than 2S unpaired electrons is more stable than the configuration with 2S unpaired electrons (where S is the spin of the system). For instance, triplet FeF2 with four unpaired electrons is lower in energy than triplet FeF2 with two unpaired electrons. Such highly open-shell configurations are labeled as hyper open-shell electronic configurations in this work and are compared to ordinary open-shell or closed-shell electronic configurations. The hyper open-shell states considered in this work are especially interesting because, unlike typical biradicals and polyradicals, the unpaired electrons are all on the same center. This work shows that the conventional perspective on spin-state energetics that usually assumes ordinary open shells for single-centered radicals needs modification to take into account, whenever possible, hyper open-shell configurations as well.
Co-reporter:Wei-Guang Liu and Donald G. Truhlar
Chemistry of Materials October 10, 2017 Volume 29(Issue 19) pp:8073-8073
Publication Date(Web):September 8, 2017
DOI:10.1021/acs.chemmater.7b01624
NU-1000 is a metal–organic framework (MOF) with Zr6(μ3–OH)4(μ3–O)4(OH)4(OH2)4 nodes (called Zr6 nodes) and tetratopic 1,3,6,8-tetrakis(p-benzoate)pyrene (TBAPy) linkers. NU-1000 has pores of 31 Å diameter, and it has smaller pores connected perpendicularly to these large pores; this nanoporous structure makes it suitable as a catalyst or a support for catalysis. However, as ordinarily synthesized, NU-1000 has disordered Zr6 nodes populated in its large pores, and this introduces nonuniformity into the crystal and complicates structural characterization. We have examined possible MOF morphologies composed of Zr6 nodes and TBAPy linkers, and we propose that the extra Zr6 nodes in the large pores of NU-1000 are structurally similar to those of an NU-901 phase, which differs from NU-1000 in the relative orientation of Zr6 nodes and the linker conformations. This polymorphism comes from two rotamers of the TBAPy linkers; in NU-1000 the benzoates at the 1 and 3 positions and the 6 and 8 positions are disrotatorily situated, whereas in NU-901 these benzoates are conrotatorily situated. Substituting carbon-2 and carbon-7 of pyrene with groups that are unevenly disposed with respect to the pyrene plane should stabilize the rotamer that can form NU-1000 and therefore stabilize NU-1000 itself. We confirmed this by applying the PBE-D2 and M06-L exchange-correlation functionals in periodic electronic structure calculations to compare the energies of NU-1000 and NU-901 with different substituents on the linkers. In NU-1000, the benzene rings of the benzoates coordinating to the Zr6 nodes form pocketlike structures. In unsubstituted NU-1000 the pocket in the small pore is smaller and attracts precursors of the atomic layer deposition (ALD) step more strongly because of shorter benzene–precursor distances and more favorable dispersion-like interactions, leading to selective metal deposition in the small pores. We examined the pocket sizes and the adsorption energy of Zn(Et)2 (the ALD precursor) on the Zr6 node in large and small pores of substituted NU-1000, and found that bulkier substituents not only compress the pocket in the large pore, but also have shorter distances to the ALD precursor in the large pore. Both factors contribute to the more favorable dispersion interaction and lead to less tendency to deposit metals in the small pore.
Co-reporter:Lili Xing, Junwei Lucas Bao, Zhandong Wang, Feng Zhang, and Donald G. Truhlar
Journal of the American Chemical Society November 8, 2017 Volume 139(Issue 44) pp:15821-15821
Publication Date(Web):October 12, 2017
DOI:10.1021/jacs.7b08297
Oxygenates with carbonyl and hydroperoxy functional groups are important intermediates that are generated during the autoxidation of organic compounds in the atmosphere and during the autoignition of transport fuels. In the troposphere, the degradation of carbonyl hydroperoxides leads to low-vapor-pressure polyfunctional species that may precipitate in clouds and fog droplets or to the formation of secondary organic aerosols (SOAs). In combustion, the fate of carbonyl hydroperoxides is important for the performance of advanced combustion engines, especially for autoignition. A key fate of the carbonyl hydroperoxides is reaction with OH radicals, for which kinetics data are experimentally unavailable. Here, we study 4-hydroperoxy-2-pentanone (CH3C(═O)CH2CH(OOH)CH3) as a model compound to clarify the kinetics of OH reactions with carbonyl hydroperoxides, in particular H atom abstraction and OH addition reactions. With a combination of electronic structure calculations, we determine previously missing thermochemical data, and with multipath variational transition state theory (MP-VTST), a multidimensional tunneling (MT) approximation, multiple-structure anharmonicity, and torsional potential anharmonicity, we obtained much more accurate rate constants than the ones that can computed by conventional single-structure harmonic transition state theory (TST) and than the empirically estimated rate constants that are currently used in atmospheric and combustion modeling. The roles of various factors in determining the rates are elucidated. The pressure-dependent rate constants for the addition reaction are computed using system-specific quantum RRK theory. The calculated temperature range is 298–2400 K, and the pressure range is 0.01–100 atm. The accurate thermodynamic and kinetics data determined in this work are indispensable in the global modeling of SOAs in atmospheric science and in the detailed understanding and prediction of ignition properties of hydrocarbons and alternative fuels.
Co-reporter:Junwei Lucas Bao, Ying Wang, Xiao He, Laura Gagliardi, and Donald G. Truhlar
The Journal of Physical Chemistry Letters November 16, 2017 Volume 8(Issue 22) pp:5616-5616
Publication Date(Web):November 1, 2017
DOI:10.1021/acs.jpclett.7b02705
Delocalization error has been singled out by Yang and co-workers as the dominant error in Kohn–Sham density functional theory (KS-DFT) with conventional approximate functionals. In this Letter, by computing the vertical first ionization energy for well separated He clusters, we show that multiconfiguration pair-density functional theory (MC-PDFT) is free from delocalization error. To put MC-PDFT in perspective, we also compare it with some Kohn–Sham density functionals, including both traditional and modern functionals. Whereas large delocalization errors are almost universal in KS-DFT (the only exception being the very recent corrected functionals of Yang and co-workers), delocalization error is removed by MC-PDFT, which bodes well for its future as a step forward from KS-DFT.
Co-reporter:Liam Wilbraham, Pragya Verma, Donald G. Truhlar, Laura Gagliardi, and Ilaria Ciofini
The Journal of Physical Chemistry Letters May 4, 2017 Volume 8(Issue 9) pp:2026-2026
Publication Date(Web):April 4, 2017
DOI:10.1021/acs.jpclett.7b00570
The spin-state orderings in nine Fe(II) and Fe(III) complexes with ligands of diverse ligand-field strength were investigated with multiconfiguration pair-density functional theory (MC-PDFT). The performance of this method was compared to that of complete active space second-order perturbation theory (CASPT2) and Kohn–Sham density functional theory. We also investigated the dependence of CASPT2 and MC-PDFT results on the size of the active-space. MC-PDFT reproduces the CASPT2 spin-state ordering, the dependence on the ligand field strength, and the dependence on active space at a computational cost that is significantly reduced as compared to CASPT2.
Co-reporter:Yinan Shu, Kelsey A. Parker, and Donald G. Truhlar
The Journal of Physical Chemistry Letters May 18, 2017 Volume 8(Issue 10) pp:2107-2107
Publication Date(Web):April 18, 2017
DOI:10.1021/acs.jpclett.7b00594
Time-dependent Kohn–Sham density functional theory has been used successfully to compute vertical excitation energies, especially for large molecular systems. However, the lack of double excitation character in the excited amplitudes produced by linear response in the adiabatic approximation holds it back from broader applications in photochemistry; for example, it shows (3N – 7)-dimensional conical intersection seams (where N is the number of atoms) between ground and excited states, although the correct dimensionality is 3N – 8. In this letter, we present a new, conceptually simple, easy-to-implement, and easy-to-use way to employ time-dependent Kohn–Sham density functional theory that has global accuracy comparable with the conventional single-functional version and that recovers the double cone topology of the potential energy surfaces at S1/S0 conical intersection seams. The new method is called the dual-functional Tamm–Dancoff approximation (DF-TDA).
Co-reporter:Junwei Lucas Bao, Lili Xing, and Donald G. Truhlar
Journal of Chemical Theory and Computation June 13, 2017 Volume 13(Issue 6) pp:2511-2511
Publication Date(Web):May 3, 2017
DOI:10.1021/acs.jctc.7b00232
For molecules with multiple torsions, an accurate evaluation of the molecular partition function requires consideration of multiple structures and their torsional-potential anharmonicity. We previously developed a method called MS-T for this problem, and it requires an exhaustive conformational search with frequency calculations for all the distinguishable conformers; this can become expensive for molecules with a large number of torsions (and hence a large number of structures) if it is carried out with high-level methods. In the present work, we propose a cost-effective method to approximate the MS-T partition function when there are a large number of structures, and we test it on a transition state that has eight torsions. This new method is a dual-level method that combines an exhaustive conformer search carried out by a low-level electronic structure method (for instance, AM1, which is very inexpensive) and selected calculations with a higher-level electronic structure method (for example, density functional theory with a functional that is suitable for conformational analysis and thermochemistry). To provide a severe test of the new method, we consider a transition state structure that has 8 torsional degrees of freedom; this transition state structure is formed along one of the reaction pathways of the hydrogen abstraction reaction (at carbon-1) of ketohydroperoxide (KHP; its IUPAC name is 4-hydroperoxy-2-pentanone) by OH radical. We find that our proposed dual-level method is able to significantly reduce the computational cost for computing MS-T partition functions for this test case with a large number of torsions and with a large number of conformers because we carry out high-level calculations for only a fraction of the distinguishable conformers found by the low-level method. In the example studied here, the dual-level method with 40 high-level optimizations (1.8% of the number of optimizations in a coarse-grained full search and 0.13% of the number of optimizations in a fine-grained full search) reproduces the full calculation of the high-level partition function within a factor of 1.0 to 2.0 from 200 to 1000 K. The error in the dual-level method can be further reduced to factors of 0.6 to 1.1 over the whole temperature interval from 200 to 2400 K by optimizing 128 structures (5.9% of the number of optimizations in a fine-grained full search and 0.41% of the number of optimizations in a fine-grained full search). These factor-of-two or better errors are small compared to errors up to a factor of 1.0 × 103 if one neglects multistructural effects for the case under study.
Co-reporter:Shaohong L. Li and Donald G. Truhlar
Journal of Chemical Theory and Computation June 13, 2017 Volume 13(Issue 6) pp:2823-2823
Publication Date(Web):May 10, 2017
DOI:10.1021/acs.jctc.7b00325
Band shape is an essential ingredient in the simulation of electronic absorption spectra. The excitation of multiple series of vibrational levels during an electronic excitation is a main contributor to band shapes. Here we present two simple models based on the Franck–Condon displaced-harmonic-oscillator model. The models are both derived from the time-dependent formulation of electronic spectroscopy. They assume that the transition dipoles do not depend on geometry and that the potential energy surfaces are locally quadratic; one model is second order in time and is called LQ2, and the other is third order in time and is called LQ3. These models are suitable for simulating the unresolved vibronic band shapes of electronic spectra that involve many vibrational modes. The models are straightforward and can be easily applied to simulate absorption spectra that are composed of many electronic transitions. As compared to carrying out molecular dynamics simulations, they require relatively few electronic structure calculations, and the additional cost for constructing the spectra is negligible. Therefore, the models are suitable for simulating the spectra of complex systems such as transition-metal complexes.
Co-reporter:Ying Wang, Xianwei Wang, Donald G. Truhlar, and Xiao He
Journal of Chemical Theory and Computation December 12, 2017 Volume 13(Issue 12) pp:6068-6068
Publication Date(Web):November 7, 2017
DOI:10.1021/acs.jctc.7b00865
The development of better approximations to the exact exchange-correlation functional is essential to the accuracy of density functionals. A recent study suggested that functionals with few parameters provide more accurate electron densities than recently developed many-parameter functionals for light closed-shell atomic systems. In this study, we calculated electron densities, their gradients, and Laplacians of Ne, Ne6+, and Ne8+ using 19 electronic structure methods, and we compared them to the CCSD reference results. Two basis sets, namely, aug-cc-pωCV5Z and aug-cc-pV5Z, are utilized in the calculations. We found that the choice of basis set has a significant impact on the errors and rankings of some of the selected methods. The errors of electron densities, their gradients, and Laplacians calculated with the aug-cc-pV5Z basis set are substantially reduced, especially for Minnesota density functionals, as compared to the results using the aug-cc-pωCV5Z basis set (a larger basis set utilized in earlier work (Medvedev et al. Science 2017, 355, 49–52)). The rankings of the M06 suite of functionals among the 19 methods are greatly improved with the aug-cc-pV5Z basis set. In addition, the performances of the HSE06, BMK, MN12-L, and MN12-SX functionals are also improved with the aug-cc-pV5Z basis set. The M06 suite of functionals is capable of providing accurate electron densities, gradients, and Laplacians using the aug-cc-pV5Z basis set, and thus it is suitable for a wide range of applications in chemistry and physics.
Co-reporter:Luis Simón-Carballido, Junwei Lucas Bao, Tiago Vinicius Alves, Rubén Meana-Pañeda, Donald G. Truhlar, and Antonio Fernández-Ramos
Journal of Chemical Theory and Computation August 8, 2017 Volume 13(Issue 8) pp:3478-3478
Publication Date(Web):June 30, 2017
DOI:10.1021/acs.jctc.7b00451
In this work we present the extended two-dimensional torsion (E2DT) method and use it to analyze the performance of several methods that incorporate torsional anharmonicity more approximately for calculating rotational–vibrational partition functions. Twenty molecules having two hindered rotors were studied for temperatures between 100 and 2500 K. These molecules present several kinds of situations; they include molecules with nearly separable rotors, molecules in which the reduced moments of inertia change substantially with the internal rotation, and molecules presenting compound rotation. Partition functions obtained by the rigid-rotor harmonic oscillator approximation, a method involving global separability of torsions and the multistructural methods without explicit potential coupling [MS-T(U)] and with explicit potential coupling [MS-T(C)] of torsions, are compared to those obtained with a quantized version – called the extended two-dimensional torsion (E2DT) method – of the extended hindered rotor approximation of Vansteenkiste et al. (Vansteenkiste et al. J. Chem. Phys. 2006, 124, 044314). In the E2DT method, quantum effects due to the torsional modes were incorporated by the two-dimensional nonseparable method, which is a method that is based on the solution of the torsional Schrödinger equation and that includes full coupling in both the kinetic and potential energy. By comparing other methods to the E2DT method and to experimental thermochemical data, this study concludes that the harmonic approximation yields very poor results at high temperatures; the global separation of torsions from the rest of the degrees of freedom is not justified even when an accurate method to treat the torsions is employed; it is confirmed that methods based on less complete potential energy coupling of torsions, such as MS-T(U), are not accurate when dealing with rotors with different barrier heights, and more complete inclusion of torsional coupling to the method in MS-T(C) improves substantially the results in such a way that it could be used in cases where the E2DT method is unaffordable.
Co-reporter:Dr. Xiaoli Dong;Hongchuan Yu;Yuanyuan Ma;Junwei Lucas Bao; Donald G. Truhlar; Yonggang Wang; Yongyao Xia
Chemistry - A European Journal 2017 Volume 23(Issue 11) pp:2560-2565
Publication Date(Web):2017/02/21
DOI:10.1002/chem.201700063
AbstractRechargeable batteries with organic electrodes are preferred to those with transition-metal-containing electrodes for their environmental friendliness, and resource availability, but all such batteries reported to date are based on organic electrolytes, which raise concerns of safety and performance. Here an aqueous-electrolyte all-organic rechargeable battery is reported, with a maximum operating voltage of 2.1 V, in which polytriphenylamine (PTPAn) and 1,4,5,8-naphthalenetetracarboxylic dianhydride (NTCDA)-derived polyimide (PNTCDA) serve as cathode and anode material, respectively. A key feature of the design is use of a “water-in-salt” electrolyte to bind “free” water; this impedes the side reaction of water oxidation, thereby enabling excellent reversibility in aqueous solution. The battery can deliver a maximum energy density of 52.8 Wh kg−1, which is close to most of the all-organic batteries with organic electrolytes. The battery exhibits a supercapacitor-like high power of 32 000 W kg−1 and a long cycle life (700 cycles with capacity retention of 85 %), due to the kinetics not being limited by ion diffusion at either electrode.
Co-reporter:Junwei Lucas Bao
Chemical Society Reviews 2017 vol. 46(Issue 24) pp:7548-7596
Publication Date(Web):2017/12/11
DOI:10.1039/C7CS00602K
This article reviews the fundamentals of variational transition state theory (VTST), its recent theoretical development, and some modern applications. The theoretical methods reviewed here include multidimensional quantum mechanical tunneling, multistructural VTST (MS-VTST), multi-path VTST (MP-VTST), both reaction-path VTST (RP-VTST) and variable reaction coordinate VTST (VRC-VTST), system-specific quantum Rice–Ramsperger–Kassel theory (SS-QRRK) for predicting pressure-dependent rate constants, and VTST in the solid phase, liquid phase, and enzymes. We also provide some perspectives regarding the general applicability of VTST.
Co-reporter:Pragya Verma
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 20) pp:12898-12912
Publication Date(Web):2017/05/24
DOI:10.1039/C7CP01576C
Dipole moments are the first moment of electron density and are fundamental quantities that are often available from experiments. An exchange–correlation functional that leads to an accurate representation of the charge distribution of a molecule should accurately predict the dipole moments of the molecule. It is well known that Kohn–Sham density functional theory (DFT) is more accurate for the energetics of single-reference systems than for the energetics of multi-reference ones, but there has been less study of charge distributions. In this work, we benchmark 48 density functionals chosen with various combinations of ingredients, against accurate experimental data for dipole moments of 78 molecules, in particular 55 single-reference molecules and 23 multi-reference ones. We chose both organic and inorganic molecules, and within the category of inorganic molecules there are both main-group and transition-metal-containing molecules, with some of them being multi-reference. As one would expect, the multi-reference molecules are not as well described by single-reference DFT, and the functionals tested in this work do show larger mean unsigned errors (MUEs) for the 23 multi-reference molecules than the single-reference ones. Five of the 78 molecules have relatively large experimental error bars and were therefore not included in calculating the overall MUEs. For the 73 molecules not excluded, we find that three of the hybrid functionals, B97-1, PBE0, and TPSSh (each with less than or equal to 25% Hartree–Fock (HF) exchange), the range-separated hybrid functional, HSE06 (with HF exchange decreasing from 25% to 0 as interelectronic distance increases), and the hybrid functional, PW6B95 (with 28% HF exchange) are the best performing functionals with each yielding an MUE of 0.18 D. Perhaps the most significant finding of this study is that there exists great similarity among the success rate of various functionals in predicting dipole moments. In particular, of 39 functionals designed as general-purpose functionals and that do not have a global value of 100% HF exchange, the average MUE is 0.23 D, with a standard deviation of only 0.04 D. Among gradient approximations, which are especially interesting because of their speed and portability, the best overall performance is by PBE, HCTH/407, OLYP, OreLYP, and GAM, each with MUE of 0.22 D.
Co-reporter:Pragya Verma;Zoltan Varga;Johannes E. M. N. Klein;Christopher J. Cramer;Lawrence Que, Jr.
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 20) pp:13049-13069
Publication Date(Web):2017/05/24
DOI:10.1039/C7CP01263B
Our ability to understand and simulate the reactions catalyzed by iron depends strongly on our ability to predict the relative energetics of spin states. In this work, we studied the electronic structures of Fe2+ ion, gaseous FeO and 14 iron complexes using Kohn–Sham density functional theory with particular focus on determining the ground spin state of these species as well as the magnitudes of relevant spin-state energy splittings. The 14 iron complexes investigated in this work have hexacoordinate geometries of which seven are Fe(II), five are Fe(III) and two are Fe(IV) complexes. These are calculated using 20 exchange–correlation functionals. In particular, we use a local spin density approximation (LSDA) – GVWN5, four generalized gradient approximations (GGAs) – BLYP, PBE, OPBE and OLYP, two non-separable gradient approximations (NGAs) – GAM and N12, two meta-GGAs – M06-L and M11-L, a meta-NGA – MN15-L, five hybrid GGAs – B3LYP, B3LYP*, PBE0, B97-3 and SOGGA11-X, four hybrid meta-GGAs – M06, PW6B95, MPW1B95 and M08-SO and a hybrid meta-NGA – MN15. The density functional results are compared to reference data, which include experimental results as well as the results of diffusion Monte Carlo (DMC) calculations and ligand field theory estimates from the literature. For the Fe2+ ion, all functionals except M11-L correctly predict the ground spin state to be quintet. However, quantitatively, most of the functionals are not close to the experimentally determined spin-state splitting energies. For FeO all functionals predict quintet to be the ground spin state. For the 14 iron complexes, the hybrid functionals B3LYP, MPW1B95 and MN15 correctly predict the ground spin state of 13 out of 14 complexes and PW6B95 gets all the 14 complexes right. The local functionals, OPBE, OLYP and M06-L, predict the correct ground spin state for 12 out of 14 complexes. Two of the tested functionals are not recommended to be used for this type of study, in particular M08-SO and M11-L, because M08-SO systematically overstabilizes the high spin state, and M11-L systematically overstabilizes the low spin state.
Co-reporter:Jie J. Bao;Laura Gagliardi
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 44) pp:30089-30096
Publication Date(Web):2017/11/15
DOI:10.1039/C7CP05156E
Multiconfiguration pair-density functional theory (MC-PDFT) is a post multiconfiguration self-consistent field (MCSCF) method with similar performance to complete active space second-order perturbation theory (CASPT2) but with greater computational efficiency. Cyano radical (CN) is a molecule whose spectrum is well established from experiments and whose excitation energies have been used as a testing ground for theoretical methods to treat excited states of open-shell systems, which are harder and much less studied than excitation energies of closed-shell singlets. In the present work, we studied the adiabatic excitation energies of CN with MC-PDFT. Then we compared this multireference (MR) method to some single-reference (SR) methods, including time-dependent density functional theory (TDDFT) and completely renormalized equation-of-motion coupled-cluster theory with singles, doubles and noniterative triples [CR-EOM-CCSD(T)]; we also compared to some other MR methods, including configuration interaction singles and doubles (MR-CISD) and multistate CASPT2 (MS-CASPT2). Through a comparison between SR and MR methods, we achieved a better appreciation of the need to use MR methods to accurately describe higher excited states, and we found that among the MR methods, MC-PDFT stands out for its accuracy for the first four states out of the five doublet states studied this paper; this shows its efficiency for calculating doublet excited states.
Co-reporter:Junwei Lucas Bao;Xin Zhang;Xuefei Xu
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 8) pp:5839-5854
Publication Date(Web):2017/02/23
DOI:10.1039/C6CP08896A
Accurately predicting bond length and bond dissociation energy for bimetallic diatomic molecules that involve metal–metal multiple bonds is a great challenge for electronic structure theory, in part because many of these molecules have inherently multi-configuration wave functions, a characteristic that is variously labeled as strong correlation or multireference character. Although various popular density functionals are widely used in studying metal–metal bonding in catalysis, their accuracy can be questioned, and it is important to see both how well and how poorly a functional can perform. Here we test 50 Kohn–Sham exchange–correlation density functionals for selected 3d and 4d hetero- and homonuclear bimetallic diatomic molecules against experimental bond lengths and bond energies. We found that for the majority of the density functionals, the mean unsigned error in predicting the bond length is larger than 0.08 Å, and for the bond energy, half of the functionals give a mean unsigned error larger than 20 kcal mol−1. This indicates that such highly multireference bimetallic systems are challenging for KS-DFT. However, some exchange–correlation functionals perform significantly better than average for both bond energies and bond lengths, in particular, BLYP, M06-L, N12-SX, OreLYP, RPBE, and revPBE, and are recommended for both kinds of calculations. Other functionals that perform relatively well for bond lengths include MGGA_MS0, MOHLYP, OLYP, PBE, and SOGGA11, and other functionals that perform relatively well for bond energies include GAM, M05, M06, MN15, and τ-HCTHhyb. Although some of these functionals (M05, M06, MN15, N12-SX, and τ-HCTHhyb) contain a nonzero percentage of Hartree–Fock exchange, a broader conclusion is that Hartree–Fock exchange brings in a static correlation error and usually tends to make the results, especially the bond lengths, less accurate. We find some significant differences between all-electron calculations and calculations with effective core potentials. For analysis, the article also presents CASSCF calculations of the percentage contributions of the dominant configurations, and the paper compares orbitals and configurations obtained in DFT calculations to those in CASSCF calculations. The equilibrium bond distance of Rh2 is not available from experiments, and we predict it to be 2.22 Å. The bond energy of VCr is not available from experiments, and we predict it to be 52.9 kcal mol−1.
Co-reporter:Pragya VermaDonald G. Truhlar
The Journal of Physical Chemistry Letters 2017 Volume 8(Issue 2) pp:
Publication Date(Web):December 29, 2016
DOI:10.1021/acs.jpclett.6b02757
Local exchange–correlation functionals have low cost and convenient portability but are known to seriously underestimate semiconductor band gaps and the energies of molecular Rydberg states. Here we present a new local approximation to the exchange–correlation functional called HLE16 that gives good performance for semiconductor band gaps and molecular excitation energies and is competitive with hybrid functionals. By the simultaneous increase of the local exchange and decrease of the local correlation, electronic excitation energies were improved without excessively degrading the ground-state solid-state cohesive energies, molecular bond energies, or chemical reaction barrier heights, although the new functional is not recommended for optimizing lattice constants or molecular bond lengths. The new functional can be useful as-is for calculations on semiconductors or excited states where it is essential to control the cost, and it can also be useful in establishing a starting point for developing even better new functionals that perform well for excited states.
Co-reporter:Rubén Meana-Pañeda, Xuefei Xu, He Ma, and Donald G. Truhlar
The Journal of Physical Chemistry A 2017 Volume 121(Issue 8) pp:
Publication Date(Web):January 31, 2017
DOI:10.1021/acs.jpca.6b10600
Rate constants and the product branching ratio for hydrogen abstraction from CH3OH by O(3P) were computed with multistructural variational transition-state theory including microcanonically optimized multidimensional tunneling. Benchmark calculations of the forward and reverse classical barrier heights and the reaction energetics have been carried out by using coupled cluster theory and multireference calculations to select the most reliable density functional method for direct dynamics computations of the rate constants. The dynamics calculations included the anharmonicity of the zero-point energies and partition functions, with specific-reaction-parameter scaling factors for reactants and transition states, and multistructural torsional anharmonicity was included for the torsion around the C–O bond in methanol and in the transition states. The resulting rate constants are presented over a wider range than they are available from experiment, but in the temperature range where experiments are available, they agree well with experimental values, which is encouraging for their reliability over the wider temperature range and for future computations of oxygen atom reaction rates. In contrast to a previous computational prediction, the branching ratio predicted by the present work shows that the formation of CH2OH + OH is the dominant channel over the whole range of temperature from 250 to 2000 K.
Co-reporter:Kaining DuanmuDonald G. Truhlar
Journal of Chemical Theory and Computation 2017 Volume 13(Issue 2) pp:
Publication Date(Web):December 16, 2016
DOI:10.1021/acs.jctc.6b01156
The quantitative prediction of adsorption energies of radicals and molecules on surfaces is essential for the design and understanding of heterogeneous catalytic processes. A recent paper by Wellendorff et al. collected an experimental database of 39 reaction energies involving adsorption energies on transition metal surfaces that can be used as benchmarks for testing quantum mechanical electronic structure methods, and we compared the experimental data to Kohn–Sham density functional calculations with six exchange–correlation functionals. In this paper, we rearranged the data into two categories: open-shell radical adsorption reactions and closed-shell molecular adsorption reactions. We recalculated the adsorption energies with PBE, and we also calculated them with three functionals, M06-L, GAM, and MN15-L, that were not studied in the Wellendorff et al. paper; then we compared our results to the benchmark data. Of the nine functionals that have been compared to the databases, we find that BEEF-vdW, GAM, and RPBE perform best for the open-shell radical adsorption reactions, and MN15-L performs best for the closed-shell molecular adsorption, followed by BEEF-vdW and M06-L.
Co-reporter:Laura GagliardiDonald G. Truhlar, Giovanni Li ManniRebecca K. Carlson, Chad E. Hoyer, Junwei Lucas Bao
Accounts of Chemical Research 2017 Volume 50(Issue 1) pp:
Publication Date(Web):December 21, 2016
DOI:10.1021/acs.accounts.6b00471
ConspectusThe electronic energy of a system provides the Born–Oppenheimer potential energy for internuclear motion and thus determines molecular structure and spectra, bond energies, conformational energies, reaction barrier heights, and vibrational frequencies. The development of more efficient and more accurate ways to calculate the electronic energy of systems with inherently multiconfigurational electronic structure is essential for many applications, including transition metal and actinide chemistry, systems with partially broken bonds, many transition states, and most electronically excited states. Inherently multiconfigurational systems are called strongly correlated systems or multireference systems, where the latter name refers to the need for using more than one (“multiple”) configuration state function to provide a good zero-order reference wave function.This Account describes multiconfiguration pair-density functional theory (MC-PDFT), which was developed as a way to combine the advantages of wave function theory (WFT) and density functional theory (DFT) to provide a better treatment of strongly correlated systems. First we review background material: the widely used Kohn–Sham DFT (which uses only a single Slater determinant as reference wave function), multiconfiguration WFT methods that treat inherently multiconfigurational systems based on an active space, and previous attempts to combine multiconfiguration WFT with DFT. Then we review the formulation of MC-PDFT. It is a generalization of Kohn–Sham DFT in that the electron kinetic energy and classical electrostatic energy are calculated from a reference wave function, while the rest of the energy is obtained from a density functional. However, there are two main differences with respent to Kohn-Sham DFT: (i) The reference wave function is multiconfigurational rather than being a single Slater determinant. (ii) The density functional is a function of the total density and the on-top pair density rather than being a function of the spin-up and spin-down densities. In work carried out so far, the multiconfigurational wave function is a multiconfiguration self-consistent-field wave function. The new formulation has the advantage that the reference wave function has the correct spatial and spin symmetry and can describe bond dissociation (of both single and multiple bonds) and electronic excitations in a formally and physically correct way. We then review the formulation of density functionals in terms of the on-top pair density. Finally we review successful applications of the theory to bond energies and bond dissociation potential energy curves of main-group and transition metal bonds, to barrier heights (including pericyclic reactions), to proton affinities, to the hydrogen bond energy of water dimer, to ground- and excited-state charge transfer, to valence and Rydberg excitations of molecules, and to singlet–triplet splittings of radicals.We find that that MC-PDFT can give accurate results not only with complete-active-space multiconfiguration wave functions but also with generalized-active-space multiconfiguration wave functions, which are practical for larger numbers of active electrons and active orbitals than are complete-active-space wave functions. The separated-pair approximation, which is a special case of generalized active space self-consistent-field theory, is especially promising. MC-PDFT, because it requires much less computer time and storage than pure WFT methods, has the potential to open larger and more complex strongly correlated systems to accurate simulation.
Co-reporter:Junwei Lucas BaoSamuel O. Odoh, Laura GagliardiDonald G. Truhlar
Journal of Chemical Theory and Computation 2017 Volume 13(Issue 2) pp:
Publication Date(Web):December 21, 2016
DOI:10.1021/acs.jctc.6b01102
We study the performance of multiconfiguration pair-density functional theory (MC-PDFT) and multireference perturbation theory for the computation of the bond dissociation energies in 12 transition-metal-containing diatomic molecules and three small transition-metal-containing polyatomic molecules and in two transition-metal dimers. The first step is a multiconfiguration self-consistent-field calculation, for which two choices must be made: (i) the active space and (ii) its partition into subspaces, if the generalized active space formulation is used. In the present work, the active space is chosen systematically by using three correlated-participating-orbitals (CPO) schemes, and the partition is chosen by using the separated-pair (SP) approximation. Our calculations show that MC-PDFT generally has similar accuracy to CASPT2, and the active-space dependence of MC-PDFT is not very great for transition-metal–ligand bond dissociation energies. We also find that the SP approximation works very well, and in particular SP with the fully translated BLYP functional SP-ftBLYP is more accurate than CASPT2. SP greatly reduces the number of configuration state functions relative to CASSCF. For the cases of FeO and NiO with extended-CPO active space, for which complete active space calculations are unaffordable, SP calculations are not only affordable but also of satisfactory accuracy. All of the MC-PDFT results are significantly better than the corresponding results with broken-symmetry spin-unrestricted Kohn–Sham density functional theory. Finally we test a perturbation theory method based on the SP reference and find that it performs slightly worse than CASPT2 calculations, and for most cases of the nominal-CPO active space, the approximate SP perturbation theory calculations are less accurate than the much less expensive SP-PDFT calculations.
Co-reporter:Soumen Ghosh;Christopher J. Cramer;Laura Gagliardi
Chemical Science (2010-Present) 2017 vol. 8(Issue 4) pp:2741-2750
Publication Date(Web):2017/03/28
DOI:10.1039/C6SC05036K
Predicting ground- and excited-state properties of open-shell organic molecules by electronic structure theory can be challenging because an accurate treatment has to correctly describe both static and dynamic electron correlation. Strongly correlated systems, i.e., systems with near-degeneracy correlation effects, are particularly troublesome. Multiconfigurational wave function methods based on an active space are adequate in principle, but it is impractical to capture most of the dynamic correlation in these methods for systems characterized by many active electrons. We recently developed a new method called multiconfiguration pair-density functional theory (MC-PDFT), that combines the advantages of wave function theory and density functional theory to provide a more practical treatment of strongly correlated systems. Here we present calculations of the singlet–triplet gaps in oligoacenes ranging from naphthalene to dodecacene. Calculations were performed for unprecedently large orbitally optimized active spaces of 50 electrons in 50 orbitals, and we test a range of active spaces and active space partitions, including four kinds of frontier orbital partitions. We show that MC-PDFT can predict the singlet–triplet splittings for oligoacenes consistent with the best available and much more expensive methods, and indeed MC-PDFT may constitute the benchmark against which those other models should be compared, given the absence of experimental data.
Co-reporter:Shouyi Yuan;Junwei Lucas Bao;Lina Wang;Yongyao Xia;Yonggang Wang
Advanced Energy Materials 2016 Volume 6( Issue 5) pp:
Publication Date(Web):
DOI:10.1002/aenm.201501733
Co-reporter:Bo Long, Junwei Lucas Bao, and Donald G. Truhlar
Journal of the American Chemical Society 2016 Volume 138(Issue 43) pp:14409-14422
Publication Date(Web):September 29, 2016
DOI:10.1021/jacs.6b08655
Criegee intermediates are produced in the ozonolysis of unsaturated hydrocarbons in the troposphere, and understanding their fate is a prerequisite to modeling climate-controlling atmospheric aerosol formation. Although some experimental and theoretical rate data are available, they are incomplete and partially inconsistent, and they do not cover the tropospheric temperature range. Here, we report quantum chemical rate constants for the reactions of stabilized formaldehyde oxide (CH2OO) and acetaldehyde oxide (syn-CH3CHOO and anti-CH3CHOO) with H2O and for their unimolecular reactions. Our results are obtained by combining post-CCSD(T) electronic structure benchmarks, validated density functional theory potential energy surfaces, and multipath variational transition state theory with multidimensional tunneling, coupled-torsions anharmonicity, and high-frequency anharmonicity. We consider two different types of reaction mechanisms for the bimolecular reactions, namely, (i) addition-coupled hydrogen transfer and (ii) double hydrogen atom transfer (DHAT). First, we show that the MN15-L exchange-correlation functional has kJ/mol accuracy for the CH2OO + H2O and syn-CH3CHOO + H2O reactions. Then we show that, due to tunneling, the DHAT mechanism is especially important in the syn-CH3CHOO + H2O reaction. We show that the dominant pathways for reactions of Criegee intermediates depend on altitude. The results we obtain eliminate the discrepancy between experiment and theory under those conditions where experimental results are available, and we make predictions for the full range of temperatures and pressures encountered in the troposphere and stratosphere. The present results are an important cog in clarifying the atmospheric fate and oxidation processes of Criegee intermediates, and they also show how theoretical methods can provide reliable rate data for complex atmospheric processes.
Co-reporter:Junwei Lucas Bao; Jingjing Zheng
Journal of the American Chemical Society 2016 Volume 138(Issue 8) pp:2690-2704
Publication Date(Web):February 3, 2016
DOI:10.1021/jacs.5b11938
Pressure-dependent reactions are ubiquitous in combustion and atmospheric chemistry. We employ a new calibration procedure for quantum Rice–Ramsperger–Kassel (QRRK) unimolecular rate theory within a chemical activation mechanism to calculate the pressure-falloff effect of a radical association with an aromatic ring. The new theoretical framework is applied to the reaction of H with toluene, which is a prototypical reaction in the combustion chemistry of aromatic hydrocarbons present in most fuels. Both the hydrogen abstraction reactions and the hydrogen addition reactions are calculated. Our system-specific (SS) QRRK approach is adjusted with SS parameters to agree with multistructural canonical variational transition state theory with multidimensional tunneling (MS-CVT/SCT) at the high-pressure limit. The new method avoids the need for the usual empirical estimations of the QRRK parameters, and it eliminates the need for variational transition state theory calculations as a function of energy, although in this first application we do validate the falloff curves by comparing SS-QRRK results without tunneling to multistructural microcanonical variational transition state theory (MS-μVT) rate constants without tunneling. At low temperatures, the two approaches agree well with each other, but at high temperatures, SS-QRRK tends to overestimate falloff slightly. We also show that the variational effect is important in computing the energy-resolved rate constants. Multiple-structure anharmonicity, torsional–potential anharmonicity, and high-frequency-mode vibrational anharmonicity are all included in the rate computations, and torsional anharmonicity effects on the density of states are investigated. Branching fractions, which are both temperature- and pressure-dependent (and for which only limited data is available from experiment), are predicted as a function of pressure.
Co-reporter:Haoyu S. Yu, Xiao He, Shaohong L. Li and Donald G. Truhlar
Chemical Science 2016 vol. 7(Issue 8) pp:5032-5051
Publication Date(Web):06 Apr 2016
DOI:10.1039/C6SC00705H
Kohn–Sham density functionals are widely used; however, no currently available exchange–correlation functional can predict all chemical properties with chemical accuracy. Here we report a new functional, called MN15, that has broader accuracy than any previously available one. The properties considered in the parameterization include bond energies, atomization energies, ionization potentials, electron affinities, proton affinities, reaction barrier heights, noncovalent interactions, hydrocarbon thermochemistry, isomerization energies, electronic excitation energies, absolute atomic energies, and molecular structures. When compared with 82 other density functionals that have been defined in the literature, MN15 gives the second smallest mean unsigned error (MUE) for 54 data on inherently multiconfigurational systems, the smallest MUE for 313 single-reference chemical data, and the smallest MUE on 87 noncovalent data, with MUEs for these three categories of 4.75, 1.85, and 0.25 kcal mol−1, respectively, as compared to the average MUEs of the other 82 functionals of 14.0, 4.63, and 1.98 kcal mol−1. The MUE for 17 absolute atomic energies is 7.4 kcal mol−1 as compared to an average MUE of the other 82 functionals of 34.6 kcal mol−1. We further tested MN15 for 10 transition-metal coordination energies, the entire S66x8 database of noncovalent interactions, 21 transition-metal reaction barrier heights, 69 electronic excitation energies of organic molecules, 31 semiconductor band gaps, seven transition-metal dimer bond lengths, and 193 bond lengths of 47 organic molecules. The MN15 functional not only performs very well for our training set, which has 481 pieces of data, but also performs very well for our test set, which has 823 data that are not in our training set. The test set includes both ground-state properties and molecular excitation energies. For the latter MN15 achieves simultaneous accuracy for both valence and Rydberg electronic excitations when used with linear-response time-dependent density functional theory, with an MUE of less than 0.3 eV for both types of excitations.
Co-reporter:Rachel C. Klet, Timothy C. Wang, Laura E. Fernandez, Donald G. Truhlar, Joseph T. Hupp, and Omar K. Farha
Chemistry of Materials 2016 Volume 28(Issue 4) pp:1213
Publication Date(Web):February 10, 2016
DOI:10.1021/acs.chemmater.5b04887
The combination (AIM-ME) of atomic layer deposition in metal–organic frameworks (MOFs) and metal exchange (ME) is introduced as a technique to install dispersed metal atoms into the mesoporous MOF, NU-1000. Zn-AIM, which contains four Zn atoms per Zr6 node, has been synthesized through AIM and further characterized through density functional calculations to provide insight into the possible structure. Zn-AIM was then subjected to modification via transmetalation to yield uniform porous materials that present nonstructural Cu, Co, or Ni atoms.
Co-reporter:Haoyu S. Yu, Xiao He, Shaohong L. Li and Donald G. Truhlar
Chemical Science 2016 vol. 7(Issue 9) pp:6278-6279
Publication Date(Web):12 Jul 2016
DOI:10.1039/C6SC90044E
Correction for ‘MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions’ by Haoyu S. Yu et al., Chem. Sci., 2016, DOI: 10.1039/c6sc00705h.
Co-reporter:Shuping Huang, Benjamin E. Wilson, William H. Smyrl, Donald G. Truhlar, and Andreas Stein
Chemistry of Materials 2016 Volume 28(Issue 3) pp:746
Publication Date(Web):January 11, 2016
DOI:10.1021/acs.chemmater.5b03554
Lithium-ion batteries (LIBs) are promising devices for high capacity, rechargeable electrical energy storage; however, LIBs are currently limited by the low specific capacity of the cathode compared to the anode. In previous work, our group demonstrated the viability of a novel cathode material, Li8ZrO6 (LZO), through computational and experimental results. Here we report a general synthesis for transition-metal-doped LZO, and we study the effects of doping on electrochemical delithiation and relithiation. A synthesis using transition-metal (M) doped ZrO2 nanoparticle/carbon black composites as precursors produces doped M-LZO with grain sizes between 35 and 67 nm. The materials were tested as electrode materials. Specific capacities of the doped materials depend on the transition metal and on the Li:Zr ratio used in the synthesis, but they are generally higher than in similarly prepared undoped LZO. In this set of cathode materials, Fe3+-doped LZO/C composites showed the highest specific capacities, with an initial discharge capacity higher than two Li ions per formula unit, a specific capacity of 175 mAh/g maintained after 140 cycles, and a specific capacity greater than 80 mAh/g at a rate of 5C. The effects of doping were also investigated by density functional calculations of dopant locations, band gaps, and delithiation energies. We found that all of the dopants that we studied are more favorably located in Li ion sites than in Zr ion sites. The calculated doping effects on structural parameters agree well with experiments. We also found that doping with any of these ions leads to smaller band gaps. Electronic structure calculations with the HSE06 exchange-correlation functional show that deintercalation after doping with Ce3+, Cu2+, or Co2+ at a Li site decreases the attainable cell voltage, whereas Fe3+ doping at a Li site increases it. Because of the large polarization and high carbon content of the M-LZO/C composite electrodes, further materials optimization will be needed before they become practical for LIBs.
Co-reporter:Joshua Borycz, Joachim Paier, Pragya Verma, Lucy E. Darago, Dianne J. Xiao, Donald G. Truhlar, Jeffrey R. Long, and Laura Gagliardi
Inorganic Chemistry 2016 Volume 55(Issue 10) pp:4924
Publication Date(Web):May 2, 2016
DOI:10.1021/acs.inorgchem.6b00467
We report electronic, vibrational, and magnetic properties, together with their structural dependences, for the metal–organic framework Fe2(dobdc) (dobdc4– = 2,5-dioxido-1,4-benzenedicarboxylate) and its derivatives, Fe2(O)2(dobdc) and Fe2(OH)2(dobdc)—species arising in the previously proposed mechanism for the oxidation of ethane to ethanol using N2O as an oxidant. Magnetic susceptibility measurements reported for Fe2(dobdc) in an earlier study and reported in the current study for FeII0.26[FeIII(OH)]1.74(dobdc)(DMF)0.15(THF)0.22, which is more simply referred to as Fe2(OH)2(dobdc), were used to confirm the computational results. Theory was also compared to experiment for infrared spectra and powder X-ray diffraction structures. Structural and magnetic properties were computed by using Kohn–Sham density functional theory both with periodic boundary conditions and with cluster models. In addition, we studied the effects of different treatments of the exchange interactions on the magnetic coupling parameters by comparing several approaches to the exchange-correlation functional: generalized gradient approximation (GGA), GGA with empirical Coulomb and exchange integrals for 3d electrons (GGA+U), nonseparable gradient approximation (NGA) with empirical Coulomb and exchange integrals for 3d electrons (NGA+U), hybrid GGA, meta-GGA, and hybrid meta-GGA. We found the coupling between the metal centers along a chain to be ferromagnetic in the case of Fe2(dobdc) and antiferromagnetic in the cases of Fe2(O)2(dobdc) and Fe2(OH)2(dobdc). The shift in magnetic coupling behavior correlates with the changing electronic structure of the framework, which derives from both structural and electronic changes that occur upon metal oxidation and addition of the charge-balancing oxo and hydroxo ligands.
Co-reporter:Junwei Lucas Bao, Andrew Sand, Laura Gagliardi, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 9) pp:4274-4283
Publication Date(Web):July 20, 2016
DOI:10.1021/acs.jctc.6b00569
Predicting the singlet–triplet splittings of divalent radicals is a challenging task for electronic structure theory. In the present work, we investigate the performance of multiconfiguration pair-density functional theory (MC-PDFT) for computing the singlet–triplet splitting for small main-group divalent radicals for which accurate experimental data are available. In order to define theoretical model chemistries that can be assessed consistently, we define three correlated participating orbitals (CPO) schemes (nominal, moderate, and extended, abbreviated as nom, mod, and ext) to define the constitution of complete active spaces, and we test them systematically. Broken-symmetry Kohn–Sham DFT calculations have also been carried out for comparison. We found that the extended CPO-PDFT scheme with translated on-top pair-density functionals have smaller mean unsigned errors than weighted-average broken-symmetry Kohn–Sham DFT with the corresponding exchange-correlation functional. The accuracy of the translated Perdew–Burke–Ernzerhof (tPBE) on-top pair-density functionals with ext-CPO active space is even better than some of the more accurately parametrized exchange-correlation density functionals that we tested; this is very encouraging for MC-PDFT theory.
Co-reporter:Haoyu S. Yu, Xiao He, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 3) pp:1280-1293
Publication Date(Web):January 1, 2016
DOI:10.1021/acs.jctc.5b01082
Kohn–Sham density functional theory is widely used for applications of electronic structure theory in chemistry, materials science, and condensed-matter physics, but the accuracy depends on the quality of the exchange-correlation functional. Here, we present a new local exchange-correlation functional called MN15-L that predicts accurate results for a broad range of molecular and solid-state properties including main-group bond energies, transition metal bond energies, reaction barrier heights, noncovalent interactions, atomic excitation energies, ionization potentials, electron affinities, total atomic energies, hydrocarbon thermochemistry, and lattice constants of solids. The MN15-L functional has the same mathematical form as a previous meta-nonseparable gradient approximation exchange-correlation functional, MN12-L, but it is improved because we optimized it against a larger database, designated 2015A, and included smoothness restraints; the optimization has a much better representation of transition metals. The mean unsigned error on 422 chemical energies is 2.32 kcal/mol, which is the best among all tested functionals, with or without nonlocal exchange. The MN15-L functional also provides good results for test sets that are outside the training set. A key issue is that the functional is local (no nonlocal exchange or nonlocal correlation), which makes it relatively economical for treating large and complex systems and solids. Another key advantage is that medium-range correlation energy is built in so that one does not need to add damped dispersion by molecular mechanics in order to predict accurate noncovalent binding energies. We believe that the MN15-L functional should be useful for a wide variety of applications in chemistry, physics, materials science, and molecular biology.
Co-reporter:Junwei Lucas Bao, Xin Zhang and Donald G. Truhlar
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 25) pp:16659-16670
Publication Date(Web):01 Jun 2016
DOI:10.1039/C6CP02765B
Understanding the falloff in rate constants of gas-phase unimolecular reaction rate constants as the pressure is lowered is a fundamental problem in chemical kinetics, with practical importance for combustion, atmospheric chemistry, and essentially all gas-phase reaction mechanisms. In the present work, we use our recently developed system-specific quantum RRK theory, calibrated by canonical variational transition state theory with small-curvature tunneling, combined with the Lindemann–Hinshelwood mechanism, to model the dissociation reaction of fluoroform (CHF3), which provides a definitive test for falloff modeling. Our predicted pressure-dependent thermal rate constants are in excellent agreement with experimental values over a wide range of pressures and temperatures. The present validation of our methodology, which is able to include variational transition state effects, multidimensional tunneling based on the directly calculated potential energy surface along the tunneling path, and torsional and other vibrational anharmonicity, together with state-of-the-art reaction-path-based direct dynamics calculations, is important because the method is less empirical than models routinely used for generating full mechanisms, while also being simpler in key respects than full master equation treatments and the full reduced falloff curve and modified strong collision methods of Troe.
Co-reporter:Junwei Lucas Bao and Donald G. Truhlar
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 15) pp:10097-10108
Publication Date(Web):11 Mar 2016
DOI:10.1039/C6CP00816J
The growth of anionic silicon hydride clusters is a critically important process in nanodusty plasmas. In the current study, we focus on the formation of homologs of silylene (Sin+1H2n+2−, n = 3, 4) and silyl (SinH2n+1−, n = 4, 5) anions via anion–neutral reaction pathways. Species like silyl or silylene anions and their related elementary reactions, which are involved in the formation of silicon hydride clusters, were not used in developing exchange–correlation (xc) density functionals (i.e., they were not included in the training set of semiempirical density functionals); therefore, we explored the accuracy of various widely used xc density functionals based on reaction energies and barrier heights. Among the 21 density functionals we tested, M06-2X has the best performance for a hybrid functional, and MN15-L has the best performance for a local functional. Thermal rate constants of the elementary reactions involved in the reaction mechanism are calculated using M06-2X and multistructural canonical variational transition state theory with the small-curvature tunneling approximation (MS-CVT/SCT). The pressure dependence of unimolecular isomerization reactions is treated with system-specific quantum RRK theory (SS-QRRK) and the Lindemann–Hinshelwood mechanism.
Co-reporter:Junwei Lucas Bao, Pattrawan Sripa and Donald G. Truhlar
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 2) pp:1032-1041
Publication Date(Web):23 Nov 2015
DOI:10.1039/C5CP05780A
Multi-path variational transition state theory (MP-VTST) provides a conformationally complete framework for calculating gas-phase rate constants. For reactions in which the transition state has distinguishable torsional minima (which include most reactions), there are multiple possible reaction paths. In principle MP-VTST includes the contributions from all the reaction paths, and it should explicitly treat the variational and tunneling effects of each path, but in practice one may need to truncate the number of paths included in MP-VTST calculations in order to achieve a balance between computational cost and accuracy. In this work, we present calculations including all paths for two prototype combustion reactions, namely the two hydrogen abstraction reactions from tert-butanol by HO2 radical. For both reactions we included all the reaction paths. Since abstraction at C has 46 paths, it provided a good opportunity to carry out a case study in which we investigated the errors introduced by truncating the number of paths. For the reaction studied, we found that the variational and multidimensional tunneling transmission coefficients are very different for different reaction paths, which provides new evidence that MP-VTST is necessary for treating path-dependent variational effects and multidimensional tunneling. We found that tunneling transmission coefficients can be much larger for higher-energy paths than for lower-energy ones. Interestingly, the simple hypothesis that higher barriers are narrower does not explain this finding in the present case; we found instead that the effect is due to higher-energy barriers having the possibility of tunneling at energies farther below the barrier top. We also show that a previously applied criterion for judging convergence with respect to the number of paths may not be reliable at low temperature.
Co-reporter:Chad E. Hoyer; Soumen Ghosh; Donald G. Truhlar;Laura Gagliardi
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 3) pp:586-591
Publication Date(Web):January 21, 2016
DOI:10.1021/acs.jpclett.5b02773
A correct description of electronically excited states is critical to the interpretation of visible–ultraviolet spectra, photochemical reactions, and excited-state charge-transfer processes in chemical systems. We have recently proposed a theory called multiconfiguration pair-density functional theory (MC-PDFT), which is based on a combination of multiconfiguration wave function theory and a new kind of density functional called an on-top density functional. Here, we show that MC-PDFT with a first-generation on-top density functional performs as well as CASPT2 for an organic chemistry database including valence, Rydberg, and charge-transfer excitations. The results are very encouraging for practical applications.
Co-reporter:Xin Zhang;Junwei Lucas Bao
PNAS 2016 Volume 113 (Issue 48 ) pp:13606-13611
Publication Date(Web):2016-11-29
DOI:10.1073/pnas.1616208113
Bond dissociation is a fundamental chemical reaction, and the first principles modeling of the kinetics of dissociation reactions
with a monotonically increasing potential energy along the dissociation coordinate presents a challenge not only for modern
electronic structure methods but also for kinetics theory. In this work, we use multifaceted variable-reaction-coordinate
variational transition-state theory (VRC-VTST) to compute the high-pressure limit dissociation rate constant of tetrafluoroethylene
(C2F4), in which the potential energies are computed by direct dynamics with the M08-HX exchange correlation functional. To treat
the pressure dependence of the unimolecular rate constants, we use the recently developed system-specific quantum Rice–Ramsperger–Kassel
theory. The calculations are carried out by direct dynamics using an exchange correlation functional validated against calculations
that go beyond coupled-cluster theory with single, double, and triple excitations. Our computed dissociation rate constants
agree well with the recent experimental measurements.
Co-reporter:Haoyu S. Yu ; Donald G. Truhlar
Angewandte Chemie 2016 Volume 128( Issue 31) pp:9150-9152
Publication Date(Web):
DOI:10.1002/ange.201604670
Abstract
In a recent paper, Wang et al. found an iridium-containing compound with a formal oxidation state of 9.[5] This is the highest oxidation state ever found in a stable compound. To learn if this is the highest chemical oxidation state possible, Kohn–Sham density functional theory was used to study various compounds, including PdO42+, PtO42+, PtO3F22+, PtO4OH+, PtO5, and PtO4SH+, in which the metal has an oxidation state of 10. It was found that PtO42+ has a metastable state that is kinetically stable with a barrier height for decomposition of 31 kcal mol−1 and a calculated lifetime of 0.9 years. All other compounds studied would readily decompose to lower oxidation states.
Co-reporter:Bo Wang, Sijie Luo, and Donald G. Truhlar
The Journal of Physical Chemistry B 2016 Volume 120(Issue 8) pp:1437-1439
Publication Date(Web):June 5, 2015
DOI:10.1021/acs.jpcb.5b03356
Theoretical studies on the electrode materials in lithium-ion batteries provide information on the structural changes during the charging and discharging processes. In the present study, we tested the M06-L and N12 exchange-correlation functionals on some well-studied lithium-containing materials. These functionals, which have already shown good performance for a variety of databases, outperform the widely used PBE functional for reproducing the experimental structures and averaged intercalation potentials. It is especially noteworthy that the M06-L functional gives voltages as accurate as those provided by the Heyd–Scuseria–Ernzerhof (HSE06) hybrid functional, but with less computational cost.
Co-reporter:James M. Lownsbury
The Journal of Physical Chemistry C 2016 Volume 120(Issue 30) pp:16850-16862
Publication Date(Web):July 4, 2016
DOI:10.1021/acs.jpcc.6b05707
The nature and energy of the reactions between calcium vapor and the internal surfaces of the metal–organic framework (MOF) NU-1000 have been studied by adsorption microcalorimetry, low energy He+ ion scattering spectroscopy (LEIS), X-ray photoelectron spectroscopy (XPS), and Kohn–Sham density functional theory (DFT). NU-1000 is one of the most stable MOFs with transition-metal-oxide nodes, and thus it is of interest as a potential catalyst or catalytic support when modified with other metals. The reaction heats of Ca with NU-1000 are high below 2 monolayers (ML) Ca coverage (570–366 kJ/mol), attributed (based on DFT) to Ca reacting first with free benzoic acid functionalities or water impurities, then with H2O and OH groups on the Zr6 nodes to produce Ca(OH)2 clusters. With higher Ca doses, the heat of Ca reaction decreases asymptotically to the sublimation enthalpy of bulk Ca (178 kJ/mol), attributed to the formation of Ca(solid) nanoparticles on the external surface, which only occurs after all of the H2O and OH groups are titrated deeply enough (∼20 nm) such that slow Ca diffusion prevents further reaction.
Co-reporter:Pragya Verma
The Journal of Physical Chemistry C 2016 Volume 120(Issue 18) pp:9933-9948
Publication Date(Web):April 7, 2016
DOI:10.1021/acs.jpcc.6b03240
Metal–organic frameworks, which are a special case of coordination polymers, form a class of materials with numerous applications due to their high porosities and large internal surface areas and in some cases also due to paramagnetic metal ions. A family of such materials of general formula M2(dobdc) (where M is a divalent metal ion, and dobdc4– is 2,5-dioxido-1,4-benzenedicarboxylate) has attracted considerable attention for gas separation, catalysis, and magnetism. In this work, we explore the magnetic properties of a member of this family, Fe2(dobdc), both in the activated form (bare MOF after solvent removal) and when hydrocarbons are bound to the open coordination sites of the metal. We report quantum mechanical electronic structure calculations using both cluster models and periodic models, and we compare our results to previously reported theoretical studies. We find that hydrocarbon adsorption only mildly affects the isotropic couplings but that the isotropic magnetic couplings obtained with hybrid exchange–correlation functionals for the cluster models were found to be in good agreement with the experimental values, and the ones obtained using local exchange–correlation functionals with empirical Coulomb and exchange integrals for the periodic models were also found to agree well with experiments. Furthermore, local density functionals with empirical Coulomb and exchange integrals for periodic models are found to give good agreement with the experimental result that the adsorption of ethylene changes the magnetic ordering. We also used second-order n-electron valence state perturbation theory and contracted spin–orbit configuration interaction to study the role of adsorption on the crystal-field splitting of the quintet manifold and on the single-ion anisotropy of the iron center.
Co-reporter:Xiaoyu Li, Xuefei Xu, Xiaoqing You, and Donald G. Truhlar
The Journal of Physical Chemistry A 2016 Volume 120(Issue 23) pp:4025-4036
Publication Date(Web):May 18, 2016
DOI:10.1021/acs.jpca.6b02600
It is important to determine an appropriate computational method for obtaining accurate thermochemical properties of large biodiesel molecules such as methyl linolenate. In this study, we use Kohn–Sham density functional theory (DFT) and coupled cluster theory to calculate bond dissociation enthalpies (BDEs) of seven fragment molecules of methyl linolenate, in particular, propene, methyl formate, cis-3-hexene, 1,4-pentadiene, 1-pentene, butane, and methyl butanoate. The results are compared to BDEs obtained from experiments and to Oyeyemi et al.’s multireference averaged coupled pair functional (MRACPF2) calculations. We found that with extrapolation to the complete basis set (CBS) limit, the BDEs derived from coupled cluster calculations with single, double, and triple excitations (CCSDT) and from CCSDT with a perturbative treatment of connected quadruple excitations, CCSDT(2)Q/CBS, are closer to the available experimental values than those obtained by MRACPF2 for propene and methyl formate. The CCSDT/CBS calculations were chosen as the reference for validating the DFT methods. Among the density functionals, we found that M08-HX has the best performance with a mean unsigned deviation (MUD) from CCSDT/CBS of only 1.0 kcal/mol, whereas the much more expensive MRACPF2 has an MUD of 1.1 kcal/mol. We then used the most successfully validated density functionals to calculate the BDEs of methyl linolenate and compared the results with the MRACPF2 BDEs. The present study identifies several Kohn–Sham exchange-correlation functionals that should be useful for modeling ester combustion, especially the M08-HX, M06-2X, M05-2X, M08-SO, and MPWB1K global-hybrid meta functionals, the M11 and MN12-SX range-separated-hybrid meta functionals, the ωB97 range-separated hybrid gradient approximation functional, and the SOGGA11-X global-hybrid gradient approximation functional.
Co-reporter:Haoyu S. Yu ; Donald G. Truhlar
Angewandte Chemie International Edition 2016 Volume 55( Issue 31) pp:9004-9006
Publication Date(Web):
DOI:10.1002/anie.201604670
Abstract
In a recent paper, Wang et al. found an iridium-containing compound with a formal oxidation state of 9.[5] This is the highest oxidation state ever found in a stable compound. To learn if this is the highest chemical oxidation state possible, Kohn–Sham density functional theory was used to study various compounds, including PdO42+, PtO42+, PtO3F22+, PtO4OH+, PtO5, and PtO4SH+, in which the metal has an oxidation state of 10. It was found that PtO42+ has a metastable state that is kinetically stable with a barrier height for decomposition of 31 kcal mol−1 and a calculated lifetime of 0.9 years. All other compounds studied would readily decompose to lower oxidation states.
Co-reporter:Scott T. Akin, Vicente Zamudio-Bayer, Kaining Duanmu, Georg Leistner, Konstantin Hirsch, Christine Bülow, Arkadiusz Ławicki, Akira Terasaki, Bernd von Issendorff, Donald G. Truhlar, J. Tobias Lau, and Michael A. Duncan
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 22) pp:4568-4575
Publication Date(Web):October 25, 2016
DOI:10.1021/acs.jpclett.6b01839
Cobalt–benzene cluster ions of the form Co3(bz)n+ (n = 0–3) were produced in the gas phase, mass-selected, and cooled in a cryogenic ion trap held at 3–4 K. To explore ligand effects on cluster magnetic moments, these species were investigated with X-ray absorption spectroscopy (XAS) and X-ray magnetic circular dichroism (XMCD) spectroscopy. XMCD spectra yield both the spin and orbital angular momenta of these clusters. Co3+ has a spin magnetic moment of μS = 6 μB and an orbital magnetic moment of μL = 3 μB. Co3(bz)+ and Co3(bz)2+ complexes were found to have spin and orbital magnetic moments identical to the values for ligand-free Co3+. However, coordination of the third benzene to form Co3(bz)3+ completely quenches the high spin state of the system. Density functional theory calculations elucidate the spin states of the Co3(bz)n+ species as a function of the number of attached benzene ligands, explaining the transition from septet to singlet for n = 0 → 3.
Co-reporter:Kaining Duanmu, Joachim Friedrich, and Donald G. Truhlar
The Journal of Physical Chemistry C 2016 Volume 120(Issue 45) pp:26110-26118
Publication Date(Web):October 7, 2016
DOI:10.1021/acs.jpcc.6b08371
The major obstacle that prevents reliable electronic structure studies of nanoparticles is the rapid increasing computational cost for benchmark calculations using coupled-cluster methods. We show that a CCSD(T) scheme with an MP2/CBS correction can reproduce accurate cohesive energies for magnesium clusters, and this scheme is much less computationally demanding than other reliable methods, so it is applied to Mgn with n up to 19, which enters the realm of nanoparticles. (The diameters of all Mg clusters n ≥ 11 are >1 nm). With the extended benchmark data, we validate exchange–correlation functionals into the nanoparticle regime and use the two best-validated functionals to calculate the enthalpy of formation of Mg28, with a diameter of 1.30 nm. We also calculated the enthalpy of formation of all Mg clusters and nanoparticles from Mg2 to Mg19. This kind of reliable thermodynamic data on size-selected metal nanoparticles has been hard to come by, either experimentally or theoretically, but it is badly needed to support applications in catalysis, electrochemistry, and other technologies.
Co-reporter:Annia Galano, Leonardo Muñoz-Rugeles, Juan Raul Alvarez-Idaboy, Junwei Lucas Bao, and Donald G. Truhlar
The Journal of Physical Chemistry A 2016 Volume 120(Issue 27) pp:4634-4642
Publication Date(Web):September 17, 2015
DOI:10.1021/acs.jpca.5b07662
An assessment of multireference character in transition states is considered to be an important component in establishing the expected reliability of various electronic structure methods. In the present work, the multireference characters of the transition states and the forming and breaking of bonds for a large set of hydrogen abstraction reactions from phenolic compounds by peroxyl radicals have been analyzed using the T1, M, B1, and GB1 diagnostics. The extent of multireference character depends on the system and on the conditions under which the reaction takes place, and some systematic trends are observed. In particular, the multireference character is found to be reduced by solvation, the size of the phenolic compound, and deprotonation in aqueous solution. However, the deviations of calculated rate constants from experimental ones are not correlated with the extent of multireference character. The performance of single-determinant density functional theory was investigated for the kinetics of these reactions by comparing calculated rate constants to experimental data; the results from these analyses showed that the M05 functional performs well for the task at hand.
Co-reporter:Shuping Huang
The Journal of Physical Chemistry C 2016 Volume 120(Issue 18) pp:9637-9649
Publication Date(Web):April 15, 2016
DOI:10.1021/acs.jpcc.6b02077
Doped Li8ZrO6 (LZO) is a pseudolayered material under consideration for lithium-ion battery cathodes and solid electrolyte coatings. The effects of doping LZO with Ce, Ti, Mg, Nb, and Y on structure, band gaps, conductivity, and activation energy for ion migration are investigated both experimentally and by quantum mechanical calculations. Optical band gaps decrease for all doped materials compared to undoped LZO. While all dopants reduce the electronic conductivity at room temperature slightly, doping with Mg or Nb increases ionic conductivity by an order of magnitude. Introducing a high loading of Nb into LZO decreases the activation energy for Li-ion diffusion in the 22–120 °C range. Calculations on lithium-ion diffusion in LZO show that it occurs by a polaron–vacancy complex mechanism. The energy barrier is lowest for the lithium hopping in a zigzag fashion between tetrahedral voids within adjacent layers. The diffusion barrier is reduced as the number of Li vacancies increases during battery charging. We calculated surface energies for 10 surfaces, and we find that the most stable surface is the (001) surface with the tetrahedral Li layer being exposed. The delithiation energy on the (001) surface was found to be slightly higher than that in the bulk. The Li-ion diffusion barriers from the surface to the bulk were also calculated on the (001) surface, and the diffusion energy barrier across the (001) surface was found to be smaller than the energy barrier along the (001) direction in the bulk, and also lower than the barrier for the lowest-energy path in the bulk (which is a hop between tetrahedral voids in adjacent layers as shown in the related graphic). These characterizations of surface and doping effects will assist future materials design.
Co-reporter:Kaining Duanmu
The Journal of Physical Chemistry C 2016 Volume 120(Issue 24) pp:13275-13286
Publication Date(Web):May 27, 2016
DOI:10.1021/acs.jpcc.6b03080
Equilibrium geometries, binding energies, adiabatic ionization potentials, and adiabatic electron affinities for neutral and singly charged magnesium clusters, Mgn0, ± 1, n = 1–7, have been computed using 39 exchange-correlation (XC) functionals in Kohn–Sham density functional theory and several coupled-cluster methods with single, double, and triple excitations, including CCSD(T) for all species, CCSD(2)T and CR-CC(2,3) for species with n = 1–3, and CCSDt, CC(t;3), and CCSDT for species with n = 1 and 2. We have used augmented polarized–valence and polarized–core–valence correlation-consistent basis sets. We have found that the geometry and binding energy of the weakly bound Mg2 dimer requires a robust treatment of connected triple excitations, represented in this work by the CR-CC(2,3), CC(t;3), and full CCSDT methods, which are more accurate than the popular quasi-perturbative CCSD(T) approximation, but CCSD(T) is sufficiently accurate to be applied to other Mg clusters. We have also demonstrated that for all Mg clusters examined in this study, hybrid XC functionals generally have higher accuracy than local ones, with PW6B95, SOGGA11-X, M11, and PWB6K being the most accurate, both for the geometries and for the binding energies, ionization potentials, and electron-detachment energies.
Co-reporter:Laura Masgrau and Donald G. Truhlar
Accounts of Chemical Research 2015 Volume 48(Issue 2) pp:431
Publication Date(Web):December 24, 2014
DOI:10.1021/ar500319e
The active site of an enzyme is surrounded by a fluctuating environment of protein and solvent conformational states, and a realistic calculation of chemical reaction rates and kinetic isotope effects of enzyme-catalyzed reactions must take account of this environmental diversity. Ensemble-averaged variational transition state theory with multidimensional tunneling (EA-VTST/MT) was developed as a way to carry out such calculations. This theory incorporates ensemble averaging, quantized vibrational energies, energy, tunneling, and recrossing of transition state dividing surfaces in a systematic way. It has been applied successfully to a number of hydrogen-, proton-, and hydride-transfer reactions. The theory also exposes the set of effects that should be considered in reliable rate constants calculations.We first review the basic theory and the steps in the calculation. A key role is played by the generalized free energy of activation profile, which is obtained by quantizing the classical potential of mean force as a function of a reaction coordinate because the one-way flux through the transition state dividing surface can be written in terms of the generalized free energy of activation. A recrossing transmission coefficient accounts for the difference between the one-way flux through the chosen transition state dividing surface and the net flux, and a tunneling transmission coefficient converts classical motion along the reaction coordinate to quantum mechanical motion. The tunneling calculation is multidimensional, accounting for the change in vibrational frequencies along the tunneling path and shortening of the tunneling path with respect to the minimum energy path (MEP), as promoted by reaction-path curvature. The generalized free energy of activation and the transmission coefficients both involve averaging over an ensemble of reaction paths and conformations, and this includes the coupling of protein motions to the rearrangement of chemical bonds in a statistical mechanically correct way. The standard deviations of the transmissions coefficients provide information on the diversity of the distribution of reaction paths, barriers, and protein conformations along the members of an ensemble of reaction paths passing through the transition state.We first illustrate the theory by discussing the application to both wild-type and mutant Escherichia coli dihydrofolate reductase and hyperthermophilic Thermotoga maritima dihydrofolate reductase (DHFR); DHFR is of special interest because the protein conformational changes have been widely studied. Then we present shorter discussions of several other applications of EA-VTST/MT to transfer of protons, hydrogen atoms, and hydride ions and their deuterated analogs. Systems discussed include hydride transfer in alcohol dehydrogenase, xylose isomerase, and thymidylate synthase, proton transfer in methylamine dehydrogenase, hydrogen atom transfer in methylmalonyl-CoA mutase, and nucleophilic substitution in haloalkane dehalogenase and two-dimensional potentials of mean force for potentially coupled proton and hydride transfer in the β-oxidation of butyryl-coenzyme A catalyzed by short-chain acyl-CoA dehydrogenase and in the pyruvate to lactate transformation catalyzed by lactate dehydrogenase.
Co-reporter:Pragya Verma; Konstantinos D. Vogiatzis; Nora Planas; Joshua Borycz; Dianne J. Xiao; Jeffrey R. Long; Laura Gagliardi
Journal of the American Chemical Society 2015 Volume 137(Issue 17) pp:5770-5781
Publication Date(Web):April 17, 2015
DOI:10.1021/jacs.5b00382
The catalytic properties of the metal–organic framework Fe2(dobdc), containing open Fe(II) sites, include hydroxylation of phenol by pure Fe2(dobdc) and hydroxylation of ethane by its magnesium-diluted analogue, Fe0.1Mg1.9(dobdc). In earlier work, the latter reaction was proposed to occur through a redox mechanism involving the generation of an iron(IV)–oxo species, which is an intermediate that is also observed or postulated (depending on the case) in some heme and nonheme enzymes and their model complexes. In the present work, we present a detailed mechanism by which the catalytic material, Fe0.1Mg1.9(dobdc), activates the strong C–H bonds of ethane. Kohn–Sham density functional and multireference wave function calculations have been performed to characterize the electronic structure of key species. We show that the catalytic nonheme-Fe hydroxylation of the strong C–H bond of ethane proceeds by a quintet single-state σ-attack pathway after the formation of highly reactive iron–oxo intermediate. The mechanistic pathway involves three key transition states, with the highest activation barrier for the transfer of oxygen from N2O to the Fe(II) center. The uncatalyzed reaction, where nitrous oxide directly oxidizes ethane to ethanol is found to have an activation barrier of 280 kJ/mol, in contrast to 82 kJ/mol for the slowest step in the iron(IV)–oxo catalytic mechanism. The energetics of the C–H bond activation steps of ethane and methane are also compared. Dehydrogenation and dissociation pathways that can compete with the formation of ethanol were shown to involve higher barriers than the hydroxylation pathway.
Co-reporter:Shuping Huang; Benjamin E. Wilson; Bo Wang; Yuan Fang; Keegan Buffington; Andreas Stein
Journal of the American Chemical Society 2015 Volume 137(Issue 34) pp:10992-11003
Publication Date(Web):August 11, 2015
DOI:10.1021/jacs.5b04690
We study—experimentally and theoretically—the energetics, structural changes, and charge flows during the charging and discharging processes for a new high-capacity cathode material, Li8ZrO6 (LZO), which we study both pure and yttrium-doped. We quantum mechanically calculated the stable delithiated configurations, the delithiation energy, the charge flow during delithiation, and the stability of the delithiated materials. We find that Li atoms are easier to extract from tetrahedral sites than octahedral ones. We calculate a large average voltage of 4.04 eV vs Li/Li+ for delithiation of the first Li atom in a primitive cell, which is confirmed by galvanostatic charge/discharge cycling data. Energy calculations indicate that topotactic delithiation is kinetically favored over decomposition into Li, ZrO2, and O2 during the charging process, although the thermodynamic energy of the topotactic reaction is less favorable. When one or two lithium atoms are extracted from a primitive cell of LZO, its volume and structure change little, whereas extraction of the third lithium greatly distorts the layered structure. The Li6ZrO6 and Li5ZrO6 delithiation products can be thermodynamically metastable to release of O2. Experimentally, materials with sufficiently small particle size for efficient delithiation and relithiation were achieved within an yttrium-doped LZO/carbon composite cathode that exhibited an initial discharge capacity of at least 200 mAh/g over the first 10 cycles, with 142 mAh/g maintained after 60 cycles. Computations predict that during the charging process, the oxygen ion near the Li vacancy is oxidized for both pure LZO and yttrium-doped LZO, which leads to a small-polaron hole.
Co-reporter:Xuefei Xu; Jingjing Zheng
Journal of the American Chemical Society 2015 Volume 137(Issue 25) pp:8026-8029
Publication Date(Web):June 16, 2015
DOI:10.1021/jacs.5b04845
Free energy calculations for eight enol isomers of malonaldehyde (MA) and simulation of the ultraviolet (UV) absorption spectrum in both the gas phase and water (pH = 3, where the molecule exists in neutral undeprotonated form) show that in water the two s-trans nonchelated enol conformers of MA become thermodynamically more stable than the internally hydrogen-bonded (“chelated enol”) conformer (CE). The pure CE conformer in water has a slightly red-shifted UV spectrum with respect to that in the gas phase, but the blue-shifted spectrum observed in water at pH 3 is dominated by solvent-stabilized conformations that have negligible populations in the gas phase. Density functional calculations with the solvation model based on density (SMD) and an ensemble-averaged vertical excitation model explain the experimental observations in detail.
Co-reporter:Junwei Lucas Bao, Rubén Meana-Pañeda and Donald G. Truhlar
Chemical Science 2015 vol. 6(Issue 10) pp:5866-5881
Publication Date(Web):16 Jun 2015
DOI:10.1039/C5SC01848J
The goal of the present work is modeling the kinetics of a key reaction involved in the combustion of the biofuel 2-butanol. To accomplish this we extended multi-path variational transition state theory (MP-VTST) with the small curvature tunneling (SCT) approximation to include multistructural anharmonicity factors for molecules with chiral carbons. We use the resulting theory to predict the site-dependent rate constants of the hydrogen abstraction from 2-butanol by hydroperoxyl radical. The generalized transmission coefficients were averaged over the four lowest-energy reaction paths. The computed forward reaction rate constants indicate that hydrogen abstraction from the C-2 site has the largest contribution to the overall reaction from 200 K to 2400 K, with a contribution ranging from 99.9988% at 200 K to 88.9% at 800 K to 21.2% at 3000 K, while hydrogen abstraction from the oxygen site makes the lowest contribution at all temperatures, ranging from 2.5 × 10−9% at 200 K to 0.65% at 800 K to 18% at 3000 K. This work highlights the importance of including the multiple-structure and torsional potential anharmonicity in the computation of the thermal rate constants. We also analyzed the role played by the hydrogen bond at the transition state, and we illustrated the risks of (a) considering only the lowest-energy conformations in the calculations of the rate constants or (b) ignoring the nonlinear temperature dependence of the activation energies. A hydrogen bond at the transition state can lower the enthalpy of activation, but raise the free energy of activation. We find an energy of activation that increases from 11 kcal mol−1 at 200 K to more than 36 kcal mol−1 at high temperature for this radical reaction with a biofuel molecule.
Co-reporter:Junwei Lucas Bao, Haoyu S. Yu, Kaining Duanmu, Maxim A. Makeev, Xuefei Xu, and Donald G. Truhlar
ACS Catalysis 2015 Volume 5(Issue 4) pp:2070
Publication Date(Web):February 2, 2015
DOI:10.1021/cs501675t
Metal clusters have broad applicability in catalysis due to their unique reactivity and chemical selectivity, and density functional theory has become an important method for understanding catalysis and attempting to design better catalysts. In the present paper, a main focus is on the correlation part of the exchange-correlation functional, and we tested the reliability of the Kohn–Sham density functional theory with local correlation functionals and with the nonlocal random phase approximation (RPA) correlation functional for the water splitting reaction on monatomic Fe(0) and, by implication, for transition-metal-catalyzed reactions more generally. We computed four barrier heights and six energies of reaction in the catalytic mechanism. If the results are judged by deviation from CCSD(T) calculations, it is found that many modern exchange-correlation (xc) functionals (about half of the functionals tested) with local correlation perform better than those using RPA nonlocal correlation; for example, the PWB6K, B97-3, ωB97X-D, MPW1K, M06-2X, and M05-2X hybrid xc functionals with local correlation have overall mean unsigned deviations of 1.9 kcal/mol or less from the CCSD(T) results, in comparison to a mean unsigned deviation of 3.5 kcal/mol for EXX-RPA@PBE. We also find significant differences between the predictions for catalysis at the Fe(100) surface. This work provides guidance and challenges for future theoretical investigations of transition-metal catalysis.Keywords: catalysis; density functional theory; electron correlation; exchange-correlation functionals; random phase approximation; transition metal; water splitting
Co-reporter:Zoltan Varga
Inorganic Chemistry 2015 Volume 54(Issue 17) pp:8552-8559
Publication Date(Web):August 14, 2015
DOI:10.1021/acs.inorgchem.5b01223
Cyclobutanetetrone, (CO)4, has a triplet ground state. Here we predict, based on electronic structure calculations, that the B2N2O4 molecule also has a triplet ground state and is therefore paramagnetic; the structure is an analogue of (CO)4 in which the carbon ring is replaced by a (BN)2 ring. Similar to (CO)4, the triplet ground-state structure of B2N2O4 is also thermodynamically unstable. Besides analysis of the molecular orbitals, we found that the partial atomic charges are good indicators for predicting magnetic ground states.
Co-reporter:Haoyu Yu and Donald G. Truhlar
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 7) pp:2968-2983
Publication Date(Web):May 26, 2015
DOI:10.1021/acs.jctc.5b00083
Although many transition metal complexes are known to have high multireference character, the multireference character of main-group closed-shell singlet diatomic molecules like BeF, CaO, and MgO has been less studied. However, many group-1 and group-2 diatomic molecules do have multireference character, and they provide informative systems for studying multireference character because they are simpler than transition metal compounds. The goal of the present work is to understand these multireference systems better so that, ultimately, we can apply what we learn to more complicated multireference systems and to the design of new exchange-correlation functionals for treating multireference systems more adequately. Fourteen main-group diatomic molecules and one triatomic molecule (including radicals, cations, and anions, as well as neutral closed-shell species) have been studied for this article. Eight of these molecules contain a group-1 element, and six contain a group-2 element. Seven of these molecules are multireference systems, and eight of them are single-reference systems. Fifty-three exchange-correlation functionals of 11 types [local spin-density approximation (LSDA), generalized gradient approximation (GGA), nonseparable gradient approximation (NGA), global-hybrid GGA, meta-GGA, meta-NGA, global-hybrid meta GGA, range-separated hybrid GGA, range-separated hybrid meta-GGA, range-separated hybrid meta-NGA, and DFT augmented with molecular mechanics damped dispersion (DFT-D)] and the Hartree–Fock method have been applied to calculate the bond distance, bond dissociation energy (BDE), and dipole moment of these molecules. All of the calculations are converged to a stable solution by allowing the symmetry of the Slater determinant to be broken. A reliable functional should not only predict an accurate BDE but also predict accurate components of the BDE, so each bond dissociation energy has been decomposed into ionization potential (IP) of the electropositive element, electron affinity of the electronegative bonding partner (EA), atomic excitation energy (EE) to prepare the valence states of the interacting partners, and interaction energy (IE) of the valence-prepared states. Adding Hartree–Fock exchange helps to obtain better results for atomic excitation energy, and this leads to improvements in getting the right answer for the right reason. The following functionals are singled out for reasonably good performance on all three of bond distance, BDE, and dipole moment: B97-1, B97-3, MPW1B95, M05, M06, M06-2X, M08-SO, N12-SX, O3LYP, TPSS, τ-HCTHhyb, and GAM; all but two (TPSS and GAM) of these functionals are hybrid functionals.
Co-reporter:Shaohong L. Li and Donald G. Truhlar
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 7) pp:3123-3130
Publication Date(Web):May 22, 2015
DOI:10.1021/acs.jctc.5b00369
Time-dependent density functional theory (TDDFT) with conventional local and hybrid functionals such as the local and hybrid generalized gradient approximations (GGA) seriously underestimates the excitation energies of Rydberg states, which limits its usefulness for applications such as spectroscopy and photochemistry. We present here a scheme that modifies the exchange-enhancement factor to improve GGA functionals for Rydberg excitations within the TDDFT framework while retaining their accuracy for valence excitations and for the thermochemical energetics calculated by ground-state density functional theory. The scheme is applied to a popular hybrid GGA functional and tested on data sets of valence and Rydberg excitations and atomization energies, and the results are encouraging. The scheme is simple and flexible. It can be used to correct existing functionals, and it can also be used as a strategy for the development of new functionals.
Co-reporter:Xuefei Xu, Wenjing Zhang, Mingsheng Tang, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 5) pp:2036-2052
Publication Date(Web):March 19, 2015
DOI:10.1021/acs.jctc.5b00081
Coupled-cluster (CC) methods have been extensively used as the high-level approach in quantum electronic structure theory to predict various properties of molecules when experimental results are unavailable. It is often assumed that CC methods, if they include at least up to connected-triple-excitation quasiperturbative corrections to a full treatment of single and double excitations (in particular, CCSD(T)), and a very large basis set, are more accurate than Kohn–Sham (KS) density functional theory (DFT). In the present work, we tested and compared the performance of standard CC and KS methods on bond energy calculations of 20 3d transition metal-containing diatomic molecules against the most reliable experimental data available, as collected in a database called 3dMLBE20. It is found that, although the CCSD(T) and higher levels CC methods have mean unsigned deviations from experiment that are smaller than most exchange-correlation functionals for metal–ligand bond energies of transition metals, the improvement is less than one standard deviation of the mean unsigned deviation. Furthermore, on average, almost half of the 42 exchange-correlation functionals that we tested are closer to experiment than CCSD(T) with the same extended basis set for the same molecule. The results show that, when both relativistic and core–valence correlation effects are considered, even the very high-level (expensive) CC method with single, double, triple, and perturbative quadruple cluster operators, namely, CCSDT(2)Q, averaged over 20 bond energies, gives a mean unsigned deviation (MUD(20) = 4.7 kcal/mol when one correlates only valence, 3p, and 3s electrons of transition metals and only valence electrons of ligands, or 4.6 kcal/mol when one correlates all core electrons except for 1s shells of transition metals, S, and Cl); and that is similar to some good xc functionals (e.g., B97-1 (MUD(20) = 4.5 kcal/mol) and PW6B95 (MUD(20) = 4.9 kcal/mol)) when the same basis set is used. We found that, for both coupled cluster calculations and KS calculations, the T1 diagnostics correlate the errors better than either the M diagnostics or the B1 DFT-based diagnostics. The potential use of practical standard CC methods as a benchmark theory is further confounded by the finding that CC and DFT methods usually have different signs of the error. We conclude that the available experimental data do not provide a justification for using conventional single-reference CC theory calculations to validate or test xc functionals for systems involving 3d transition metals.
Co-reporter:Rebecca K. Carlson, Giovanni Li Manni, Andrew L. Sonnenberger, Donald G. Truhlar, and Laura Gagliardi
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 1) pp:82-90
Publication Date(Web):December 8, 2014
DOI:10.1021/ct5008235
Kohn–Sham density functional theory, resting on the representation of the electronic density and kinetic energy by a single Slater determinant, has revolutionized chemistry, but for open-shell systems, the Kohn–Sham Slater determinant has the wrong symmetry properties as compared to an accurate wave function. We have recently proposed a theory, called multiconfiguration pair-density functional theory (MC-PDFT), in which the electronic kinetic energy and classical Coulomb energy are calculated from a multiconfiguration wave function with the correct symmetry properties, and the rest of the energy is calculated from a density functional, called the on-top density functional, that depends on the density and the on-top pair density calculated from this wave function. We also proposed a simple way to approximate the on-top density functional by translation of Kohn–Sham exchange-correlation functionals. The method is much less expensive than other post-SCF methods for calculating the dynamical correlation energy starting with a multiconfiguration self-consistent-field wave function as the reference wave function, and initial tests of the theory were quite encouraging. Here, we provide a broader test of the theory by applying it to bond energies of main-group molecules and transition metal complexes, barrier heights and reaction energies for diverse chemical reactions, proton affinities, and the water dimerization energy. Averaged over 56 data points, the mean unsigned error is 3.2 kcal/mol for MC-PDFT, as compared to 6.9 kcal/mol for Kohn–Sham theory with a comparable density functional. MC-PDFT is more accurate on average than complete active space second-order perturbation theory (CASPT2) for main-group small-molecule bond energies, alkyl bond dissociation energies, transition-metal–ligand bond energies, proton affinities, and the water dimerization energy.
Co-reporter:Rebecca K. Carlson, Donald G. Truhlar, and Laura Gagliardi
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 9) pp:4077-4085
Publication Date(Web):July 24, 2015
DOI:10.1021/acs.jctc.5b00609
We extend the on-top density functional of multiconfiguration pair-density functional theory (MC-PDFT) to include the gradient of the on-top density as well as the gradient of the density. We find that the theory is reasonably stable to this extension; furthermore, it provides improved accuracy for molecules containing transition metals. We illustrate the extended on-top density functionals by applying them to Cr2, Cu2, Ag2, Os2, and Re2Cl82– as well as to our previous database of 56 data for bond dissociation energies, barrier heights, reaction energies, proton affinities, and the water dimer. The performance of MC-PDFT is comparable to or better than that of CASPT2.
Co-reporter:Soumen Ghosh, Andrew L. Sonnenberger, Chad E. Hoyer, Donald G. Truhlar, and Laura Gagliardi
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 8) pp:3643-3649
Publication Date(Web):July 9, 2015
DOI:10.1021/acs.jctc.5b00456
The correct description of charge transfer in ground and excited states is very important for molecular interactions, photochemistry, electrochemistry, and charge transport, but it is very challenging for Kohn–Sham (KS) density functional theory (DFT). KS-DFT exchange-correlation functionals without nonlocal exchange fail to describe both ground- and excited-state charge transfer properly. We have recently proposed a theory called multiconfiguration pair-density functional theory (MC-PDFT), which is based on a combination of multiconfiguration wave function theory with a new type of density functional called an on-top density functional. Here we have used MC-PDFT to study challenging ground- and excited-state charge-transfer processes by using on-top density functionals obtained by translating KS exchange-correlation functionals. For ground-state charge transfer, MC-PDFT performs better than either the PBE exchange-correlation functional or CASPT2 wave function theory. For excited-state charge transfer, MC-PDFT (unlike KS-DFT) shows qualitatively correct behavior at long-range with great improvement in predicted excitation energies.
Co-reporter:Shaohong L. Li, Xuefei Xu and Donald G. Truhlar
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 31) pp:20093-20099
Publication Date(Web):05 Jun 2015
DOI:10.1039/C5CP02461G
Three singlet states, namely a closed-shell ground state and two excited states with 1ππ* and 1nσ* character, have been suggested to be responsible for the radiationless decay or photochemical reaction of photoexcited thioanisole. The correct interpretation of the electronic spectrum is critical for understanding the character of these low-lying excited states, but the experimental spectrum is yet to be fully interpreted. In the work reported here, we investigated the nature of those three states and a fourth singlet state of thioanisole using electronic structure calculations by multireference perturbation theory, by completely-renormalized equation-of-motion coupled cluster theory with single and double excitations and noniterative inclusion of connected triples (CR-EOM-CCSD(T)), and by linear-response time-dependent density functional theory (TDDFT). We clarified the assignment of the electronic spectrum by simulating it using a normal-mode sampling approach combined with TDDFT in the Tamm–Dancoff approximation (TDA). The understanding of the electronic states and of the accuracy of the electronic structure methods lays the foundation of our future work of constructing potential energy surfaces.
Co-reporter:Junwei Lucas Bao, Prasenjit Seal and Donald G. Truhlar
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 24) pp:15928-15935
Publication Date(Web):18 May 2015
DOI:10.1039/C5CP01979F
The growth of nanodusty particles, which is critical in plasma chemistry, physics, and engineering. The aim of the present work is to understand the detailed reaction mechanisms of early steps in this growth. The polymerization of neutral silane with the silylene or silyl anion, which eliminates molecular hydrogen with the formation of their higher homologues, governs the silicon hydride clustering in nanodusty plasma chemistry. The detailed mechanisms of these important polymerization reactions in terms of elementary reactions have not been proposed yet. In the present work, we investigated the initial steps of these polymerization reactions, i.e., the SiH4 + Si2H4−/Si2H5− reactions, and we propose a three-step mechanism, which is also applicable to the following polymerization steps. CM5 charges of all the silicon-containing species were computed in order to analyze the character of the species in the proposed reaction mechanisms. We also calculated thermal rate constant of each step using multi-structural canonical variational transition state theory (MS-CVT) with the small-curvature tunneling (SCT) approximation, based on the minimum energy path computed using M08-HX/MG3S electronic structure method.
Co-reporter:Haoyu S. Yu, Wenjing Zhang, Pragya Verma, Xiao He and Donald G. Truhlar
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 18) pp:12146-12160
Publication Date(Web):09 Apr 2015
DOI:10.1039/C5CP01425E
The goal of this work is to develop a gradient approximation to the exchange–correlation functional of Kohn–Sham density functional theory for treating molecular problems with a special emphasis on the prediction of quantities important for homogeneous catalysis and other molecular energetics. Our training and validation of exchange–correlation functionals is organized in terms of databases and subdatabases. The key properties required for homogeneous catalysis are main group bond energies (database MGBE137), transition metal bond energies (database TMBE32), reaction barrier heights (database BH76), and molecular structures (database MS10). We also consider 26 other databases, most of which are subdatabases of a newly extended broad database called Database 2015, which is presented in the present article and in its ESI. Based on the mathematical form of a nonseparable gradient approximation (NGA), as first employed in the N12 functional, we design a new functional by using Database 2015 and by adding smoothness constraints to the optimization of the functional. The resulting functional is called the gradient approximation for molecules, or GAM. The GAM functional gives better results for MGBE137, TMBE32, and BH76 than any available generalized gradient approximation (GGA) or than N12. The GAM functional also gives reasonable results for MS10 with an MUE of 0.018 Å. The GAM functional provides good results both within the training sets and outside the training sets. The convergence tests and the smooth curves of exchange–correlation enhancement factor as a function of the reduced density gradient show that the GAM functional is a smooth functional that should not lead to extra expense or instability in optimizations. NGAs, like GGAs, have the advantage over meta-GGAs and hybrid GGAs of respectively smaller grid-size requirements for integrations and lower costs for extended systems. These computational advantages combined with the relatively high accuracy for all the key properties needed for molecular catalysis make the GAM functional very promising for future applications.
Co-reporter:Aleksandr V. Marenich, Christopher J. Cramer, and Donald G. Truhlar
The Journal of Physical Chemistry B 2015 Volume 119(Issue 3) pp:958-967
Publication Date(Web):August 27, 2014
DOI:10.1021/jp506293w
A physically realistic treatment of solvatochromic shifts in liquid-phase electronic absorption spectra requires a proper account for various short- and long-range equilibrium and nonequilibrium solute–solvent interactions. The present article demonstrates that such a treatment can be accomplished using a mixed discrete–continuum approach based on the two-time-scale self-consistent state-specific vertical excitation model (called VEM) for electronic excitation in solution. We apply this mixed approach in combination with time-dependent density functional theory to compute UV/vis absorption spectra in solution for the n → π* (1A2) transition for acetone in methanol and in water, the π → π* (1A1) transition for para-nitroaniline (PNA) in methanol and in water, the n → π* (1B1) transition for pyridine in water, and the n → π* (1B1) transition for pyrimidine in water. Hydrogen bonding and first-solvation-shell-specific complexation are included by means of explicit solvent molecules, and solute–solvent dispersion is included by using the solvation model with state-specific polarizability (SMSSP). Geometries of microsolvated clusters were treated in two different ways, (i) using single liquid-phase global-minimum solute–solvent clusters containing up to two explicit solvent molecules and (ii) using solute–solvent cluster snapshots derived from molecular dynamics (MD) trajectories. The calculations in water involve using VEM/TDDFT excitation energies and oscillator strengths computed over 200 MD-derived solute–solvent clusters and convoluted with Gaussian functions. We also calculate ground- and excited-state dipole moments for interpretation. We find that inclusion of explicit solvent molecules generally improves the agreement with experiment and can be recommended as a way to include the effect of hydrogen bonding in solvatochromic shifts.
Co-reporter:Chad E. Hoyer; Laura Gagliardi
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 24) pp:5015-5015
Publication Date(Web):December 7, 2015
DOI:10.1021/acs.jpclett.5b02661
Co-reporter:Kaining Duanmu
The Journal of Physical Chemistry C 2015 Volume 119(Issue 17) pp:9617-9626
Publication Date(Web):April 7, 2015
DOI:10.1021/acs.jpcc.5b01545
We report a systematic study of small silver clusters, Agn, Agn+, and Agn–, n = 1–7. We studied all possible isomers of clusters with n = 5–7. We tested 42 exchange–correlation functionals, and we assess these functionals for their accuracy in three respects: geometries (quantitative prediction of internuclear distances), structures (the nature of the lowest-energy structure, for example, whether it is planar or nonplanar), and energies. We find that the ingredients of exchange–correlation functionals are indicators of their success in predicting geometries and structures: local exchange–correlation functionals are generally better than hybrid functionals for geometries; functionals depending on kinetic energy density are the best for predicting the lowest-energy isomer correctly, especially for predicting two-dimensional to three-dimenstional transitions correctly. The accuracy for energies is less sensitive to the ingredient list. Our findings could be useful for guiding the selection of methods for computational catalyst design.
Co-reporter:Shaohong L. Li; Xuefei Xu; Chad E. Hoyer
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 17) pp:3352-3359
Publication Date(Web):August 7, 2015
DOI:10.1021/acs.jpclett.5b01609
Diabatization of potential energy surfaces is a technique that enables convenient molecular dynamics simulations of electronically nonadiabatic processes, but diabatization itself is nonunique and can be inconvenient; the best methods to achieve diabatization are still under study. Here, we present the diabatization of two electronic states of thioanisole in the S–CH3 bond stretching and C–C–S–C torsion two-dimensional nuclear coordinate space containing a conical intersection. We use two systematic methods: the (orbital-dependent) 4-fold way and the (orbital-free) Boys localization diabatization method. These very different methods yield strikingly similar diabatic potential energy surfaces that cross at geometries where the adiabatic surfaces are well separated and do not exhibit avoided crossings, and the contours of the diabatic gap and diabatic coupling are similar for the two methods. The validity of the diabatization is supported by comparing the nonadiabatic couplings calculated from the diabatic matrix elements to those calculated by direct differentiation of the adiabatic states.
Co-reporter:Jingjing Zheng, Gbenga A. Oyedepo, and Donald G. Truhlar
The Journal of Physical Chemistry A 2015 Volume 119(Issue 50) pp:12182-12192
Publication Date(Web):September 8, 2015
DOI:10.1021/acs.jpca.5b06121
The kinetics of the hydrogen abstraction from 2-butanol by hydroxyl radical have been studied using multipath variational transition-state theory with the multidimensional small curvature tunneling approximation. The rate constants for each of the five hydrogen abstraction sites (C1, C2, C3, C4, and O) and the overall reaction have been computed by direct dynamics based on M08-HX/6-311+G(2df,2p) electronic structure calculations. We show that multistructural torsional anharmonicity, anharmonicity differences of high-frequency modes between the transition structures and the reactants, and reaction-path dependence of multiple reaction paths are all important factors for determining accurate reaction rates and branching fractions for this problem. The reaction barrier heights for abstraction from various sites follow the order C2 < C3 < C4 < C1 < O, but the reactivities of the various sites do not precisely follow the inverse order of barrier heights, and the order of reactivities depends on temperature. The abstractions from C2 and C3 have the largest contribution to the total reaction rate from 200 to 2000 K.
Co-reporter:Rubén Meana-Pañeda
The Journal of Physical Chemistry C 2015 Volume 119(Issue 17) pp:9287-9301
Publication Date(Web):March 31, 2015
DOI:10.1021/acs.jpcc.5b00120
The surface chemistry of silica is strongly affected by the nature of chemically active sites (or defects) occurring on the surface. Here, we employ quantum mechanical electronic structure calculations to study an uncoordinated silicon defect, a non-bridging oxygen defect, and a peroxyl defect on the reconstructed (0001) surface of α-quartz. We characterized the spin states and energies of the defects, and calculated the reaction profiles for atomic oxygen recombination at the defects. We elucidated the diradical character by analyzing the low-lying excited states using multireference wave function methods. We show that the diradical defects consist of weakly coupled doublet radicals, and the atomic oxygen recombination can take place through a barrierless process at defects. We have delineated the recombination mechanism and computed the formation energy of the peroxyl and non-bridging oxygen defects. We found that key recombination reaction paths are barrierless. In addition, we characterize the electronically excited states that may play a role in the chemical and physical processes that occur during recombination on these surface defect sites.
Co-reporter:Prasenjit Seal
The Journal of Physical Chemistry C 2015 Volume 119(Issue 18) pp:10085-10101
Publication Date(Web):April 13, 2015
DOI:10.1021/acs.jpcc.5b00923
The present work demonstrates the importance of entropic effects on the thermodynamics of branched open chain silicon hydride clusters (SinHm and SinHm–). These clusters are important constituents in nanodusty silane plasmas. We include all categories of such single-bonded species for n = 5 and 6, namely silyl radicals, silyl anions, silylenes and silylene anions, and silanes. We calculated the statistical mechanical partition functions by employing Kohn–Sham density functional theory with the multistructural method for torsional anharmonicity to estimate thermodynamic quantities, in particular Gibbs free energy, enthalpy, entropy, and heat capacity. For each species we included contributions from all conformational structures of all possible isomers, and we calculated the thermodynamic propeties in three ways, namely by using the multistructural quasiharmonic approximation, the multistructural method with uncoupled torsional potential anharmonicity, and the multistructural method with coupled torsional potential anharmonicity. Our results show that the entropic effects are large and are primarily due to multistructural effects, although torsional potential anharmonicities are not negligible. We find that the multiple-structure effect, which is always greater than unity, is not only very large (as large as a factor of 592) but also very isomer-dependent, so that free energy differences between isomers can be greatly affected. The torsional potential effect is relatively smaller on average but certainly not negligible; it varies from a factor of 0.2 to 1.4.
Co-reporter:Pragya Verma
The Journal of Physical Chemistry C 2015 Volume 119(Issue 51) pp:28499-28511
Publication Date(Web):November 30, 2015
DOI:10.1021/acs.jpcc.5b10382
The presence of open metal sites along with a high porosity makes the Fe2(dobdc) metal–organic framework, also called Fe-MOF-74, particularly well suited for separating gaseous mixtures. For instance, since Fe2(dobdc) adsorbs O2 more strongly than N2, it can, in principle, be used to separate O2 from air [Bloch et al. J. Am. Chem. Soc. 2011, 133, 14814]. In the present work, we investigate the reversible differential adsorption of N2 and O2 on Fe2(dobdc) with Kohn–Sham density functional theory applied to an 88-atom cluster model of the MOF. The cluster is chosen such that it is large enough to allow an accurate description of the most important contributions to the binding enthalpies and small enough to perform high-level quantum mechanical calculations. For the quantum mechanical calculations, we use well-validated exchange–correlation functionals to study the ground-state structures of the Fe–N2 and Fe–O2 interacting systems. The calculations agree with experiment in that O2 binds more strongly than N2, and they reveal that the ground-state structure of the Fe–O2 subsystem has the dioxygen unit in a triplet spin state ferromagnetically coupled to the high-spin state (quintet state) of the iron center. Charge Model 5 (CM5) calculations have been performed to determine the partial atomic charges on the adsorbate molecules and the iron atom, and they show that charge transfer from the open iron(II) site is more important in the case of O2 than in the case of N2. Furthermore, bond orders, vibrational frequencies, and orbital energies were calculated to rationalize the stronger binding of O2 compared to N2 on Fe2(dobdc).
Co-reporter:Chad E. Hoyer; Laura Gagliardi
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 21) pp:4184-4188
Publication Date(Web):October 1, 2015
DOI:10.1021/acs.jpclett.5b01888
Time-dependent Kohn–Sham density functional theory (TD-KS-DFT) is useful for calculating electronic excitation spectra of large systems, but the low-energy spectra are often complicated by artificially lowered higher-energy states. This affects even the lowest energy excited states. Here, by calculating the lowest energy spin-conserving excited state for atoms from H to K and for formaldehyde, we show that this problem does not occur in multiconfiguration pair-density functional theory (MC-PDFT). We use the tPBE on-top density functional, which is a translation of the PBE exchange-correlation functional. We compare to a robust multireference method, namely, complete active space second-order perturbation theory (CASPT2), and to TD-KS-DFT with two popular exchange-correlation functionals, PBE and PBE0. We find for atoms that the mean unsigned error (MUE) of MC-PDFT with the tPBE functional improves from 0.42 to 0.40 eV with a double set of diffuse functions, whereas the MUEs for PBE and PBE0 drastically increase from 0.74 to 2.49 eV and from 0.45 to 1.47 eV, respectively.
Co-reporter:Bo Wang, Ke R. Yang, Xuefei Xu, Miho Isegawa, Hannah R. Leverentz, and Donald G. Truhlar
Accounts of Chemical Research 2014 Volume 47(Issue 9) pp:2731
Publication Date(Web):May 19, 2014
DOI:10.1021/ar500068a
The development of more efficient and more accurate ways to represent reactive potential energy surfaces is a requirement for extending the simulation of large systems to more complex systems, longer-time dynamical processes, and more complete statistical mechanical sampling. One way to treat large systems is by direct dynamics fragment methods. Another way is by fitting system-specific analytic potential energy functions with methods adapted to large systems. Here we consider both approaches.First we consider three fragment methods that allow a given monomer to appear in more than one fragment. The first two approaches are the electrostatically embedded many-body (EE-MB) expansion and the electrostatically embedded many-body expansion of the correlation energy (EE-MB-CE), which we have shown to yield quite accurate results even when one restricts the calculations to include only electrostatically embedded dimers. The third fragment method is the electrostatically embedded molecular tailoring approach (EE-MTA), which is more flexible than EE-MB and EE-MB-CE. We show that electrostatic embedding greatly improves the accuracy of these approaches compared with the original unembedded approaches.Quantum mechanical fragment methods share with combined quantum mechanical/molecular mechanical (QM/MM) methods the need to treat a quantum mechanical fragment in the presence of the rest of the system, which is especially challenging for those parts of the rest of the system that are close to the boundary of the quantum mechanical fragment. This is a delicate matter even for fragments that are not covalently bonded to the rest of the system, but it becomes even more difficult when the boundary of the quantum mechanical fragment cuts a bond. We have developed a suite of methods for more realistically treating interactions across such boundaries. These methods include redistributing and balancing the external partial atomic charges and the use of tuned fluorine atoms for capping dangling bonds, and we have shown that they can greatly improve the accuracy.Finally we present a new approach that goes beyond QM/MM by combining the convenience of molecular mechanics with the accuracy of fitting a potential function to electronic structure calculations on a specific system. To make the latter practical for systems with a large number of degrees of freedom, we developed a method to interpolate between local internal-coordinate fits to the potential energy. A key issue for the application to large systems is that rather than assigning the atoms or monomers to fragments, we assign the internal coordinates to reaction, secondary, and tertiary sets. Thus, we make a partition in coordinate space rather than atom space. Fits to the local dependence of the potential energy on tertiary coordinates are arrayed along a preselected reaction coordinate at a sequence of geometries called anchor points; the potential energy function is called an anchor points reactive potential.Electrostatically embedded fragment methods and the anchor points reactive potential, because they are based on treating an entire system by quantum mechanical electronic structure methods but are affordable for large and complex systems, have the potential to open new areas for accurate simulations where combined QM/MM methods are inadequate.
Co-reporter:Xuefei Xu ; Jingjing Zheng ; Ke R. Yang
Journal of the American Chemical Society 2014 Volume 136(Issue 46) pp:16378-16386
Publication Date(Web):October 27, 2014
DOI:10.1021/ja509016a
We report multistate trajectory simulations, including coherence, decoherence, and multidimensional tunneling, of phenol photodissociation dynamics. The calculations are based on full-dimensional anchor-points reactive potential surfaces and state couplings fit to electronic structure calculations including dynamical correlation with an augmented correlation-consistent polarized valence double-ζ basis set. The calculations successfully reproduce the experimentally observed bimodal character of the total kinetic energy release spectra and confirm the interpretation of the most recent experiments that the photodissociation process is dominated by tunneling. Analysis of the trajectories uncovers an unexpected dissociation pathway for one quantum excitation of the O–H stretching mode of the S1 state, namely, tunneling in a coherent mixture of states starting in a smaller ROH (∼0.9–1.0 Å) region than has previously been invoked. The simulations also show that most trajectories do not pass close to the S1–S2 conical intersection (they have a minimum gap greater than 0.6 eV), they provide statistics on the out-of-plane angles at the locations of the minimum energy adiabatic gap, and they reveal information about which vibrational modes are most highly activated in the products.
Co-reporter:Jingjing Zheng ; Rubén Meana-Pañeda
Journal of the American Chemical Society 2014 Volume 136(Issue 13) pp:5150-5160
Publication Date(Web):March 20, 2014
DOI:10.1021/ja5011288
Isobutanol is a prototype biofuel, and sorting out the mechanism of its combustion is an important objective where theoretical modeling can provide information that is unavailable and not easily obtained by experiment. In the present work the rate constants and branching ratios for the hydrogen abstraction reactions from isobutanol by hydroxyl radical have been calculated using multi-path variational transition-state theory with small-curvature tunneling. We use hybrid degeneracy-corrected vibrational perturbation theory to show that it is critical to consider the anharmonicity difference of high-frequency modes between reactants and transition states. To obtain accurate rate constants, we must apply different scaling factors to the calculated harmonic vibrational frequencies at the reactants and at the transition states. The factors determining the reaction rate constants have been analyzed in detail, including variational effects, tunneling contributions, the effect of multiple reaction paths on transmission coefficients, and anharmonicities of low- and high-frequency vibrational modes. The analysis quantifies the uncertainties in the rate calculations. A key result of the paper is a prediction for the site dependence of hydrogen abstraction from isobutanol by hydroxyl radical. This is very hard to measure experimentally, although it is critical for combustion mechanism modeling. The present prediction differs considerably from previous theoretical work.
Co-reporter:Prasenjit Seal
Journal of the American Chemical Society 2014 Volume 136(Issue 7) pp:2786-2799
Publication Date(Web):January 16, 2014
DOI:10.1021/ja410498d
Determination of the thermodynamic properties of reactor constituents is the first step in designing control strategies for plasma-mediated deposition processes and is also a key fundamental issue in physical chemistry. In this work, a recently proposed multistructural statistical thermodynamic method is used to show the importance of multiple structures and torsional anharmonicity in determining the thermodynamic properties of silicon hydride clusters, which are important both in plasmas and in thermally driven systems. It includes five different categories of silicon hydride clusters and radicals, including silanes, silyl radicals, and silenes. We employed a statistical mechanical approach, namely the recently developed multistructural (MS) anharmonicity method, in combination with density functional theory to calculate the partition functions, which in turn are used to estimate thermodynamic quantities, namely Gibbs free energy, enthalpy, entropy, and heat capacity, for all of the systems considered. The calculations are performed using all of the conformational structures of each molecule or radical by employing the multistructural quasiharmonic approximation (MS-QH) and also by including torsional potential anharmonicity (MS-T). For those cases where group additivity (GA) results are available, the thermodynamic quantities obtained from our MS-T calculations differ considerably due to the fact that the GA method is based on single-structure data for isomers of each stoichiometry, and hence lack multistructural effects; whereas we find that multistructural effects are very important in silicon hydride systems. Our results also indicate that the entropic effect on the thermochemistry is huge and is dominated by multistructural effects. The entropic effect of multiple structures is also expected to be important for other kinds of chain molecules, and its effect on nucleation kinetics is expected to be large.
Co-reporter:Ke R. Yang, Xuefei Xu, Jingjing Zheng and Donald G. Truhlar
Chemical Science 2014 vol. 5(Issue 12) pp:4661-4680
Publication Date(Web):2014/08/06
DOI:10.1039/C4SC01967A
We present an improved version of the anchor points reactive potential (APRP) method for potential energy surfaces; the improvement for the surfaces themselves consists of using a set of internal coordinates with better global behavior, and we also extend the method to fit the surface couplings. We use the new method to produce a 3 × 3 matrix of diabatic potential energy surfaces and couplings for the photodissociation of phenol as functions of 33 nonredundant internal coordinates. The diabatic potential matrix is based on two kinds of calculations at a sequence of anchor points along the O–H dissociation coordinate: (1) fourfold way diabatic calculations based on MC-QDPT/jul-cc-pVDZ calculations for the potential energy surfaces and diabatic couplings as functions of the O–H bond stretch, C–O–H bond angle, and C–C–O–H torsion and for the diabatic couplings as functions of the nine out-of-plane phenoxyl distortion coordinates and (2) M06-2X/jul-cc-pVDZ density functional Hessian calculations for the potentials along the 30 vibrational coordinates of the phenoxyl group. The potential energy surfaces and couplings are used to calculate and characterize adiabatic surfaces and conical intersections, and the resulting equilibrium geometries, vibrational frequencies, and vertical excitation energies are in good agreement with available reference data. We also calculate the geometries of the minimum energy conical intersections. The surfaces and couplings are used for full-dimensional tunneling calculations of the adiabatic photodissociation rate, i.e., the rate of O–H bond fission following photoexcitation. Finally we use the couplings to provide indicators of which vibrational modes are effective in promoting dissociation.
Co-reporter:Zhen Hua Li and Donald G. Truhlar
Chemical Science 2014 vol. 5(Issue 7) pp:2605-2624
Publication Date(Web):26 Feb 2014
DOI:10.1039/C4SC00052H
Metal nanoparticles have been widely used as functional materials in physics, chemistry, and biology. Understanding their unique thermodynamic properties is essential both for practical applications and from a fundamental point of view. This perspective article is an overview of recent progresses on the nanothermodynamics of metal nanoparticles and it especially highlights as examples our own studies on the structural stability, phases, phase changes, and thermodynamic functions of aluminum nanoparticles. We discuss using statistical sampling by Monte Carlo and molecular dynamics algorithms to calculate nanoparticle properties, nanophase properties, free energies, and nucleation rates, and we tried to understand the results in terms of energy landscapes by using exhaustive enumeration of the multiple structures of Al nanoparticles from all sizes up to N = 65 plus selected larger calculations.
Co-reporter:Jingjing Zheng, Xuefei Xu, Rubén Meana-Pañeda and Donald G. Truhlar
Chemical Science 2014 vol. 5(Issue 5) pp:2091-2099
Publication Date(Web):24 Jan 2014
DOI:10.1039/C3SC53290A
The classical trajectory method (also called molecular dynamics) is the most widely used method for ensemble averaging and calculating rate constants of complex dynamical systems; however it has the serious drawback of not allowing tunneling. Here, we show how to include tunneling efficiently in real-time classical trajectories by using the army ants algorithm for quantum mechanical rare event sampling and partially optimized semiclassical tunneling paths based on valence internal coordinates. Three examples, HN2 dissociation and two kinds of HCOH isomerizations, are used to illustrate the tunneling method. We show that the army ants tunneling algorithm is very efficient (even lower computational costs than calculations without tunneling) and yields physically reasonable rate constants. The new algorithm is straightforward to include in any molecular dynamics package, and it allows sampling of regions of phase space that are classically inaccessible but that may lead to different products or different energy distributions than are populated by non-tunneling processes.
Co-reporter:Bo Wang, Shaohong L. Li, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 12) pp:5640-5650
Publication Date(Web):October 30, 2014
DOI:10.1021/ct500790p
Partial atomic charges are widely used for the description of charge distributions of molecules and solids. These charges are useful to indicate the extent of charge transfer and charge flow during chemical reactions in batteries, fuel cells, and catalysts and to characterize charge distributions in capacitors, liquid-phase electrolytes, and solids and at electrochemical interfaces. However, partial atomic charges given by various charge models differ significantly, especially for systems containing metal atoms. In the present study, we have compared various charge models on both molecular systems and extended systems, including Hirshfeld, CM5, MK, ChElPG, Mulliken, MBS, NPA, DDEC, LoProp, and Bader charges. Their merits and drawbacks are compared. The CM5 charge model is found to perform well on the molecular systems, with a mean unsigned percentage deviation of only 9% for the dipole moments. We therefore formulated it for extended systems and applied it to study charge flow during the delithiation process in lithium-containing oxides used as cathodes. Our calculations show that the charges given by the CM5 charge model are reasonable and that during the delithiation process, the charge flow can occur not only on the transition metal but also on the anions. The oxygen atoms can lose a significant density of electrons, especially for deeply delithiated materials. We also discuss other methods in current use to analyze the charge transfer and charge flow in batteries, in particular the use of formal charge, spin density, and orbital occupancy. We conclude that CM5 charges provide useful information in describing charge distributions in various materials and are very promising for the study of charge transfer and charge flows in both molecules and solids.
Co-reporter:Xuefei Xu, Ke R. Yang, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 5) pp:2070-2084
Publication Date(Web):April 1, 2014
DOI:10.1021/ct500128s
Conventional time-dependent density functional theory (TDDFT) is based on a closed-shell Kohn–Sham (KS) singlet ground state with the adiabatic approximation, using either linear response (KS-LR) or the Tamm–Dancoff approximation (KS-TDA); these methods can only directly predict singly excited states. This deficiency can be overcome by using a triplet state as the reference in the KS-TDA approximation and “exciting” the singlet by a spin flip (SF) from the triplet; this is the method suggested by Krylov and co-workers, and we abbreviate this procedure as SF-KS-TDA. SF-KS-TDA can be applied either with the original collinear kernel of Krylov and co-workers or with a noncollinear kernel, as suggested by Wang and Ziegler. The SF-KS-TDA method does bring some new practical difficulties into play, but it can at least formally model doubly excited states and states with double-excitation character, so it might be more useful than conventional TDDFT (both KS-LR and KS-TDA) for photochemistry if these additional difficulties can be surmounted and if it is accurate with existing approximate exchange–correlation functionals. In the present work, we carried out calculations specifically designed to understand better the accuracy and limitations of the conventional TDDFT and SF-KS-TDA methods; we did this by studying closed-shell atoms and closed-shell monatomic cations because they provide a simple but challenging testing ground for what we might expect in studying the photochemistry of molecules with closed-shell ground states. To test their accuracy, we applied conventional KS-LR and KS-TDA and 18 versions of SF-KS-TDA (nine collinear and nine noncollinear) to the same set of vertical excitation energies (including both Rydberg and valence excitations) of Be, B+, Ne, Na+, Mg, and Al+. We did this for 10 exchange–correlation functionals of various types, both local and nonlocal. We found that the GVWN5 and M06 functionals with nonlocal kernels in spin-flip calculations can both have accuracy competitive to CASPT2 calculations. When the results were averaged over all 36 test energy differences, seven (GVWN5, M06, B3PW91, LRC-ωPBE, LRC-ωPBEh, PBE, and M06-2X) of the 10 studied density functionals had smaller mean unsigned errors for noncollinear calculations than the mean unsigned error of the best functional (M06-2X) for either conventional KS-TDA or KS-LR.
Co-reporter:Ke R. Yang, Xuefei Xu, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 3) pp:924-933
Publication Date(Web):January 20, 2014
DOI:10.1021/ct401074s
We present a new method for fitting potential energy surfaces in molecular-mechanics-like internal coordinates based on data from electronic structure calculations. The method should be applicable to chemical reactions involving either bond dissociation or isomerization and is illustrated here for bond dissociation, in particular the breaking of an O–H bond in methanol and the breaking of an N–H bond in dimethylamine. As compared to previously available systematic methods for fitting global potential energy surfaces, it extends the maximum size of the system than can be treated by at least an order of magnitude.
Co-reporter:Sijie Luo, Boris Averkiev, Ke R. Yang, Xuefei Xu, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 1) pp:102-121
Publication Date(Web):November 5, 2013
DOI:10.1021/ct400712k
The 3d-series transition metals (also called the fourth-period transition metals), Sc to Zn, are very important in industry and biology, but they provide unique challenges to computing the electronic structure of their compounds. In order to successfully describe the compounds by theory, one must be able to describe their components, in particular the constituent atoms and cations. In order to understand the ingredients required for successful computations with density functional theory, it is useful to examine the performance of various exchange–correlation functionals; we do this here for 4sN3dN′ transition-metal atoms and their cations. We analyze the results using three ways to compute the energy of the open-shell states: the direct variational method, the weighted-averaged broken symmetry (WABS) method, and a new broken-symmetry method called the reinterpreted broken symmetry (RBS) method. We find the RBS method to be comparable in accuracy with the WABS method. By examining the overall accuracy in treating 18 multiplicity-changing excitations and 10 ionization potentials with the RBS method, 10 functionals are found to have a mean-unsigned error of <5 kcal/mol, with ωB97X-D topping the list. For local density functionals, which are more practical for extended systems, the M06-L functional is the most accurate. And by combining the results with our previous studies of p-block and 4d-series elements as well as databases for alkyl bond dissociation, main-group atomization energies, and π–π noncovalent interactions, we find five functionals, namely, PW6B95, MPW1B95, M08-SO, SOGGA11-X, and MPWB1K, to be highly recommended. We also studied the performance of PW86 and C09 exchange functionals, which have drawn wide interest in recent studies due to their claimed ability to reproduce Hartree–Fock exchange at long distance. By combining them with four correlation functionals, we find the performance of the resulting functionals disappointing both for 3d transition-metal chemistry and in broader tests, and thus we do not recommend PW86 and C09 as components of generalized gradient approximations for general application.
Co-reporter:Luke Fiedler, Hannah R. Leverentz, Santhanamoorthi Nachimuthu, Joachim Friedrich, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 8) pp:3129-3139
Publication Date(Web):June 16, 2014
DOI:10.1021/ct5003169
The parametrization of the polarized molecular orbital (PMO) method, which is a neglect-of-diatomic-differential-overlap (NDDO) semiempirical method that includes polarization functions on hydrogens, is extended to include the constituents that dominate the nucleation of atmospheric aerosols, including ammonia, sulfuric acid, and water. The parametrization and validation are based mainly on CCSD(T)/CBS results for atmospheric clusters composed of sulfuric acid, dimethylamine, and ammonia and on M06-2X exchange-correlation functional calculations for other constituents of the atmospheric aerosols. The resulting model, called PMO2a, is parametrized for molecules containing any type of H, C, or O, amino or ammonium N, and S atoms bonded to O. The new method gives greatly improved electric polarization compared to any other member of the family of NDDO methods. In addition, PMO2a is shown to outperform previous NDDO methods for atomization energies and atmospheric aerosol reaction energies; therefore, its use can be recommended for realistic simulations.
Co-reporter:Haoyu Yu and Donald G. Truhlar
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 6) pp:2291-2305
Publication Date(Web):May 9, 2014
DOI:10.1021/ct5000814
In order to understand what governs the accuracy of approximate exchange–correlation functionals for intrinsically multiconfigurational systems containing metal atoms, the properties of the ground electronic state of CaO have been studied in detail. We first applied the T1, TAE(T), B1, and M diagnostics to CaO and confirmed that CaO is an intrinsically multiconfigurational system. Then, we compared the bond dissociation energies (BDEs) of CaO as calculated by 49 exchange–correlation functionals, three exchange-only functionals, and the HF method. To analyze the error in the BDEs for the various functionals, we decomposed each calculated BDE into four components, in particular the ionization potential, the electron affinity, the atomic excitation energy of the metal cation to prepare the valence state, and the interaction energy between prepared states. We found that the dominant error occurs in the calculated atomic excitation energy of the cation. Third, we compared dipole moments of CaO as calculated by the 53 methods, and we analyzed the dipole moments in terms of partial atomic charges to understand the contribution of ionic bonding and how it is affected by errors in the calculated ionization potential of the metal atom. We then analyzed the dipole moment in terms of the charge distribution among orbitals, and we found that the orbital charge distribution does not correlate well with the difference between the calculated ionization potential and electron affinity. Fourth, we examined the potential curves and internuclear distance dependence of the orbital energies of the lowest-energy CaO singlet and triplet states to analyze the near-degeneracy aspect of the correlation energy. The most important conclusion is that the error tends to be dominated by the error in the relative energies of s and d orbitals in Ca+, and the most popular density functionals predict this excitation energy poorly. Thus, even if they were to predict the BDE reasonably well, it would be due to cancellation of errors. The effect of the cation excitation energy can be understood in terms of an orbital picture, as follows. For most functionals the predicted cation excitation energy is too small, so it is too easy to delocalize charge from the oxygen 2p orbital to the Ca+ d orbital; this overestimates the covalency and explains why most functionals overestimate the bond energy.
Co-reporter:Wenjing Zhang, Donald G. Truhlar, and Mingsheng Tang
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 6) pp:2399-2409
Publication Date(Web):May 15, 2014
DOI:10.1021/ct500296a
Vanadium dimer is a notoriously difficult case for Kohn–Sham (KS) density functional theory with currently available approximations to the exchange–correlation (xc) functionals, and many approximate xc functionals yield an exceedingly large error in the calculated bond energy. In this paper, we first test the bond energies estimated by 43 xc functionals and the Hartree–Fock (HF) method. The results further confirm the large errors and show that, with the experimental bond energy being 64.2 kcal/mol, the KS calculations give predictions all over the map with errors ranging from −61.5 to +60.5 kcal/mol, and the HF method performs much worse with an error of −124.4 kcal/mol! The reason for these very large errors is examined in this article by analyzing the atomic and molecular orbital energies calculated by various xc functionals. The results show that the errors in estimates of the bond energy of vanadium dimer can primarily be related to the calculated energy gap between the 4s and 3dz2 atomic orbitals of the vanadium atom and especially to the 3dz2 orbital energy. This interesting relation between the errors in the calculated bond energy and the magnitudes of the single-particle orbital energies provides a constructive alternative to the common but more sterile explanation that it is the static correlation energy due to multicenter left–right correlation that makes the vanadium dimer and many other transition metal compounds so difficult for Kohn–Sham calculations. One of the most important factors in determining the critical atomic orbital energy is the amount of nonlocal HF exchange that is included in the xc functional, but it is still difficult to explain why different local functionals (functionals with no HF exchange) yield quite different results. We conclude that improving calculations of orbital energies of atoms may provide a route to improving the accuracy of theoretical predictions of molecular bond energies for systems containing metal atoms.
Co-reporter:Bo Wang and Donald G. Truhlar
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 10) pp:4480-4487
Publication Date(Web):August 13, 2014
DOI:10.1021/ct5005142
In a typical application of molecular mechanics (MM), the electrostatic interactions are calculated from parametrized partial atomic charges treated as point charges interacting by radial Coulomb potentials. This does not usually yield accurate electrostatic interactions at van der Waals distances, but this is compensated by additional parametrized terms, for example Lennard-Jones potentials. In the present work, we present a scheme involving radial screened Coulomb potentials that reproduces the accurate electrostatics much more accurately. The screening accounts for charge penetration of one subsystem’s charge cloud into that of another subsystem, and it is incorporated into the interaction potential in a way similar to what we proposed in a previous article (J. Chem. Theory Comput. 2010, 6, 3330) for combined quantum mechanical and molecular mechanical (QM/MM) simulations, but the screening parameters are reoptimized for MM. The optimization is carried out with electrostatic-potential-fitted partial atomic charges, but the optimized parameters should be useful with any realistic charge model. In the model we employ, the charge density of an atom is approximated as the sum of a point charge representing the nucleus and inner electrons and a smeared charge representing the outermost electrons; in particular, for all atoms except hydrogens, the smeared charge represents the two outermost electrons in the present model. We find that the charge penetration effect can cause very significant deviations from the popular point-charge model, and by comparison to electrostatic interactions calculated by symmetry-adapted perturbation theory, we find that the present results are considerably more accurate than point-charge electrostatic interactions. The mean unsigned error in electrostatics for a large and diverse data set (192 interaction energies) decreases from 9.2 to 3.3 kcal/mol, and the error in the electrostatics for 10 water dimers decreases from 1.7 to 0.5 kcal/mol. We could have decreased the average errors further, but at the cost of sometimes significantly overestimating the screening; instead we chose a more conservative (safer) parametrization that systematically underestimates the screening (which by definition means it improves over point charges) and only occasionally overestimates it. Despite this conservative choice, we find that the screened MM method is even more accurate for the electrostatics than unscreened QM/MM calculations. This new method is easy to implement in any MM program, and it can be used to develop more physical force fields for molecular simulations.
Co-reporter:Giovanni Li Manni, Rebecca K. Carlson, Sijie Luo, Dongxia Ma, Jeppe Olsen, Donald G. Truhlar, and Laura Gagliardi
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 9) pp:3669-3680
Publication Date(Web):July 21, 2014
DOI:10.1021/ct500483t
We present a new theoretical framework, called Multiconfiguration Pair-Density Functional Theory (MC-PDFT), which combines multiconfigurational wave functions with a generalization of density functional theory (DFT). A multiconfigurational self-consistent-field (MCSCF) wave function with correct spin and space symmetry is used to compute the total electronic density, its gradient, the on-top pair density, and the kinetic and Coulomb contributions to the total electronic energy. We then use a functional of the total density, its gradient, and the on-top pair density to calculate the remaining part of the energy, which we call the on-top-density-functional energy in contrast to the exchange-correlation energy of Kohn–Sham DFT. Because the on-top pair density is an element of the two-particle density matrix, this goes beyond the Hohenberg–Kohn theorem that refers only to the one-particle density. To illustrate the theory, we obtain first approximations to the required new type of density functionals by translating conventional density functionals of the spin densities using a simple prescription, and we perform post-SCF density functional calculations using the total density, density gradient, and on-top pair density from the MCSCF calculations. Double counting of dynamic correlation or exchange does not occur because the MCSCF energy is not used. The theory is illustrated by applications to the bond energies and potential energy curves of H2, N2, F2, CaO, Cr2, and NiCl and the electronic excitation energies of Be, C, N, N+, O, O+, Sc+, Mn, Co, Mo, Ru, N2, HCHO, C4H6, c-C5H6, and pyrazine. The method presented has a computational cost and scaling similar to MCSCF, but a quantitative accuracy, even with the present first approximations to the new types of density functionals, that is comparable to much more expensive multireference perturbation theory methods.
Co-reporter:Aleksandr V. Marenich, Junming Ho, Michelle L. Coote, Christopher J. Cramer and Donald G. Truhlar
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 29) pp:15068-15106
Publication Date(Web):24 Jun 2014
DOI:10.1039/C4CP01572J
This article reviews recent developments and applications in the area of computational electrochemistry. Our focus is on predicting the reduction potentials of electron transfer and other electrochemical reactions and half-reactions in both aqueous and nonaqueous solutions. Topics covered include various computational protocols that combine quantum mechanical electronic structure methods (such as density functional theory) with implicit-solvent models, explicit-solvent protocols that employ Monte Carlo or molecular dynamics simulations (for example, Car–Parrinello molecular dynamics using the grand canonical ensemble formalism), and the Marcus theory of electronic charge transfer. We also review computational approaches based on empirical relationships between molecular and electronic structure and electron transfer reactivity. The scope of the implicit-solvent protocols is emphasized, and the present status of the theory and future directions are outlined.
Co-reporter:Jingjing Zheng, Rubén Meana-Pañeda, and Donald G. Truhlar
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 11) pp:2039-2043
Publication Date(Web):May 15, 2014
DOI:10.1021/jz500653m
For electronically nonadiabatic processes in all but the simplest systems, the most practical multidimensional simulation method is a semiclassical approximation in which a trajectory or the center of a wave packet follows a classical path governed by an effective potential energy function. Here, we show how such simulations can be made more realistic by including tunneling by the army ants tunneling method. We illustrate the theory by calculations with model potential energy surfaces; one model study is in the adiabatic limit, and the other one has nonadiabatic transitions between two electronic states during the tunneling event. The army ants tunneling algorithm is used to efficiently sample tunneling events in the trajectories in both cases. This work makes it possible to simulate complex nonadiabatic chemical processes by efficiently including the important quantum effect of tunneling.Keywords: electronically nonadiabatic transition; excited state; mean-field methods; molecular dynamics; nonadiabatic trajectory; photobiology; photochemistry;
Co-reporter:Sijie Luo, Collin J. Dibble, Michael A. Duncan, and Donald. G. Truhlar
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 15) pp:2528-2532
Publication Date(Web):July 9, 2014
DOI:10.1021/jz501167s
We studied the Co4O4 subnanocluster and its MeCN-coated species using density functional theory, and we found that the Co4O4 core presents distinctive structures in bare and ligand-coated species. We propose a possible ligand-mediated ring → cube transformation mechanism during the ligand-coating process of the Co4O4 core due to the stronger binding energies of the MeCN ligands to the 3D distorted cube structure than to the 2D ring and ladder structures; theory indicates that three ligands are sufficient to stabilize the cube structure. Both ring and cube structures are ferromagnetic. Our finding is potentially useful for understanding the catalysis mechanism of Co4O4 species, which have important applications in solar energy conversion and water splitting; these catalysis reactions usually involve frequent addition and subtraction of various ligands and thus possibly involve core rearrangement processes similar to our findings.Keywords: catalysis; cluster structure; magnetism; metal oxides; nanoparticles;
Co-reporter:Shaohong L. Li, Aleksandr V. Marenich, Xuefei Xu, and Donald G. Truhlar
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 2) pp:322-328
Publication Date(Web):December 26, 2013
DOI:10.1021/jz402549p
Linear response (LR) Kohn–Sham (KS) time-dependent density functional theory (TDDFT), or KS-LR, has been widely used to study electronically excited states of molecules and is the method of choice for large and complex systems. The Tamm–Dancoff approximation to TDDFT (TDDFT-TDA or KS-TDA) gives results similar to KS-LR and alleviates the instability problem of TDDFT near state intersections. However, KS-LR and KS-TDA share a debilitating feature; conical intersections of the reference state and a response state occur in F – 1 instead of the correct F – 2 dimensions, where F is the number of internal degrees of freedom. Here, we propose a new method, named the configuration interaction-corrected Tamm–Dancoff approximation (CIC-TDA), that eliminates this problem. It calculates the coupling between the reference state and an intersecting response state by interpreting the KS reference-state Slater determinant and linear response as if they were wave functions. Both formal analysis and test results show that CIC-TDA gives similar results to KS-TDA far from a conical intersection, but the intersection occurs with the correct dimensionality. We anticipate that this will allow more realistic application of TDDFT to photochemistry.Keywords: configuration interaction-corrected; Kohn−Sham; linear response; Tamm−Dancoff; time-dependent density functional theory;
Co-reporter:Joachim Friedrich, Haoyu Yu, Hannah R. Leverentz, Peng Bai, J. Ilja Siepmann, and Donald G. Truhlar
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 4) pp:666-670
Publication Date(Web):January 30, 2014
DOI:10.1021/jz500079e
It is important to test methods for simulating water, but small water clusters for which benchmarks are available are not very representative of the bulk. Here we present benchmark calculations, in particular CCSD(T) calculations at the complete basis set limit, for water 26-mers drawn from Monte Carlo simulations of bulk water. These clusters are large enough that each water molecule participates in 2.5 hydrogen bonds on average. The electrostatically embedded three-body approximation with CCSD(T) embedded dimers and trimers reproduces the relative binding energies of eight clusters with a mean unsigned error (MUE, kcal per mole of water molecules) of only 0.009 and 0.015 kcal for relative and absolute binding energies, respectively. Using only embedded dimers (electrostatically embedded pairwise approximation) raises these MUEs to 0.038 and 0.070 kcal, and computing the energies with the M11 exchange-correlation functional, which is very economical, yields errors of only 0.029 and 0.042 kcal.Keywords: CCSD(T); correlation energy; density functional theory; explicitly correlated; fragment-based electronic structure methods; many-body expansion; overlapping fragments;
Co-reporter:Kaining Duanmu
The Journal of Physical Chemistry C 2014 Volume 118(Issue 48) pp:28069-28074
Publication Date(Web):November 12, 2014
DOI:10.1021/jp511055k
A canonical perspective on the chemical bond is the Pauling paradigm: a bond in a molecule containing only identical atoms has no ionic character. However, we show that homonuclear silver clusters have very uneven charge distributions (for example, the C2v structure of Ag4 has a larger dipole moment than formaldehyde or acetone), and we show how to predict the charge distribution from coordination numbers and Hirshfeld charges. The new charge model is validated against Kohn–Sham calculations of dipole moments with four approximations for the exchange–correlation functional. We report Kohn–Sham studies of the binding energies of CO on silver monomer and silver clusters containing 2–18 atoms. We also find that an accurate charge model is essential for understanding the site dependence of binding. In particular we find that atoms with more positive charges tend to have higher binding energies, which can be used for guidance in catalyst modeling and design. Thus, the nonuniform charge distribution of silver clusters predisposes the site preference of binding of carbon monoxide, and we conclude that nonuniform charge distributions are an important property for understanding binding of metal nanoparticles in general.
Co-reporter:Amrit Jalan ; Ionut M. Alecu ; Rubén Meana-Pañeda ; Jorge Aguilera-Iparraguirre ; Ke R. Yang ; Shamel S. Merchant ; Donald G. Truhlar ;William H. Green
Journal of the American Chemical Society 2013 Volume 135(Issue 30) pp:11100-11114
Publication Date(Web):July 17, 2013
DOI:10.1021/ja4034439
We present new reaction pathways relevant to low-temperature oxidation in gaseous and condensed phases. The new pathways originate from γ-ketohydroperoxides (KHP), which are well-known products in low-temperature oxidation and are assumed to react only via homolytic O–O dissociation in existing kinetic models. Our ab initio calculations identify new exothermic reactions of KHP forming a cyclic peroxide isomer, which decomposes via novel concerted reactions into carbonyl and carboxylic acid products. Geometries and frequencies of all stationary points are obtained using the M06-2X/MG3S DFT model chemistry, and energies are refined using RCCSD(T)-F12a/cc-pVTZ-F12 single-point calculations. Thermal rate coefficients are computed using variational transition-state theory (VTST) calculations with multidimensional tunneling contributions based on small-curvature tunneling (SCT). These are combined with multistructural partition functions (QMS–T) to obtain direct dynamics multipath (MP-VTST/SCT) gas-phase rate coefficients. For comparison with liquid-phase measurements, solvent effects are included using continuum dielectric solvation models. The predicted rate coefficients are found to be in excellent agreement with experiment when due consideration is made for acid-catalyzed isomerization. This work provides theoretical confirmation of the 30-year-old hypothesis of Korcek and co-workers that KHPs are precursors to carboxylic acid formation, resolving an open problem in the kinetics of liquid-phase autoxidation. The significance of the new pathways in atmospheric chemistry, low-temperature combustion, and oxidation of biological lipids are discussed.
Co-reporter:Kyuho Lee ; William C. Isley III; Allison L. Dzubak ; Pragya Verma ; Samuel J. Stoneburner ; Li-Chiang Lin ; Joshua D. Howe ; Eric D. Bloch ; Douglas A. Reed ; Matthew R. Hudson ; Craig M. Brown ; Jeffrey R. Long ; Jeffrey B. Neaton ; Berend Smit ; Christopher J. Cramer ; Donald G. Truhlar ;Laura Gagliardi
Journal of the American Chemical Society 2013 Volume 136(Issue 2) pp:698-704
Publication Date(Web):December 7, 2013
DOI:10.1021/ja4102979
Gas separations with porous materials are economically important and provide a unique challenge to fundamental materials design, as adsorbent properties can be altered to achieve selective gas adsorption. Metal–organic frameworks represent a rapidly expanding new class of porous adsorbents with a large range of possibilities for designing materials with desired functionalities. Given the large number of possible framework structures, quantum mechanical computations can provide useful guidance in prioritizing the synthesis of the most useful materials for a given application. Here, we show that such calculations can predict a new metal–organic framework of potential utility for separation of dinitrogen from methane, a particularly challenging separation of critical value for utilizing natural gas. An open V(II) site incorporated into a metal–organic framework can provide a material with a considerably higher enthalpy of adsorption for dinitrogen than for methane, based on strong selective back bonding with the former but not the latter.
Co-reporter:Stephen J. Klippenstein ; Vijay S. Pande
Journal of the American Chemical Society 2013 Volume 136(Issue 2) pp:528-546
Publication Date(Web):November 27, 2013
DOI:10.1021/ja408723a
This Perspective presents a personal overview of the current status of the theory of chemical kinetics and mechanisms for complex processes. We attempt to assess the status of the field for reactions in the gas phase, at gas–solid interfaces, in liquid solutions, in enzymes, and for protein folding. Some unifying concepts such as potential energy surfaces, free energy, master equations, and reaction coordinates occur in more than one area. We hope this Perspective will be useful for highlighting recent advances and for identifying important areas for future research.
Co-reporter:Aleksandr V. Marenich, Christopher J. Cramer and Donald G. Truhlar
Chemical Science 2013 vol. 4(Issue 6) pp:2349-2356
Publication Date(Web):18 Mar 2013
DOI:10.1039/C3SC50242B
Polarizability is a key molecular property controlling induction and dispersion forces in molecules, and atomic polarizabilities in molecules are widely used elements both in qualitative schemes for understanding molecular interactions and in quantitative methods for modeling them. Unfortunately, experimental probes of local polarizability are not readily available. Here we predict the polarizability of individual atoms and functional groups in a variety of systems, and we draw both general and specific conclusions with broad consequences. We find that the polarizability of the same functional group (e.g., the carbonyl group) can differ substantially, depending on the position of this group in a molecule (e.g., in a protein). More specifically, we find that the polarizability of buried atoms and groups is screened and thereby diminished; thus the outermost atoms and functional groups (for example, those lying closer to the molecular van der Waals surface) are more polarizable than buried ones, even when acted on by the same electric field. These findings mitigate against attributing isolated system behavior to molecular fragments since their polarizability depends on their environment, and the methods used here provide a way to probe molecular polarizability with a finer grain than has previously been possible.
Co-reporter:Jingjing Zheng, Prasenjit Seal and Donald G. Truhlar
Chemical Science 2013 vol. 4(Issue 1) pp:200-212
Publication Date(Web):24 Sep 2012
DOI:10.1039/C2SC21090H
Aldehyde–radical reactions are important in atmospheric and combustion chemistry, and the reactions studied here also serve more generally to illustrate a fundamental aspect of chemical kinetics that has been relatively unexplored from a quantitative point of view, in particular the roles of multiple structures and torsional anharmonicity in determining the rate constants and branching ratios (product yields). We consider hydrogen abstraction from four carbon sites of butanal (carbonyl-C, α-C, β-C and γ-C) by hydroperoxyl radical. We employed multi-structural variational transition state theory for studying the first three channels; this uses a multi-faceted dividing surface and allows us to include the contributions of multiple structures of both reacting species and transition states. Multi-configurational Shepard interpolation (MCSI) was used to obtain the geometries and energies of the potential energy surface along the minimum-energy paths, with gradients and Hessians calculated by the M08-HX/maug-cc-pVTZ method. We find the numbers of structures obtained for the transition states are 46, 60, 72 and 76respectively for the H abstraction at the carbonyl C, the α position, the β position and the γ position. Our results show that neglecting the factors arising from multiple structures and torsional anharmonicity would lead to errors at 300, 1000 and 2400 K of factors of 8, 11 and 10 for abstraction at the carbonyl-O, 2, 11 and 25 at the α-C position, 2, 23 and 47 at the β-C position, and 0.6, 8 and 18 at the γ-C position. The errors would be even larger at high temperature for the reverse of the H abstraction at the β-C. Relative yields are changed as much as a factor of 7.0 at 200 K, a factor of 5.0 at 298 K, and a factor of 3.7 in the other direction at 2400 K. The strong dependence of the product ratios on the multi-structural anharmonicity factors shows that such factors play an important role in controlling branching ratios in reaction mechanism networks.
Co-reporter:Sijie Luo and Donald G. Truhlar
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 12) pp:5349-5355
Publication Date(Web):November 19, 2013
DOI:10.1021/ct4007508
When the spins of molecular orbitals are allowed to be aligned with different directions in space rather than being aligned collinearly, the resulting noncollinear spin orbitals add extra flexibility to variational optimization of the orbitals, and solutions obtained with collinear spin orbitals may be unstable with respect to becoming noncollinear in the expanded variational space. The goal of the present work is to explore whether and in what way the molecular orbitals of the Kohn–Sham density functional theory become noncollinear when fully optimized for multi-reference molecules, transition states, and reaction paths. (We note that a noncollinear determinant has intermediate flexibility between a collinear determinant and a linear combination of many collinear determinants with completely independent coefficients. However, the Kohn–Sham method is defined to involve the variational optimization of a single determinant, and a noncollinear determinant represents the limit of complete optimization in the Kohn–Sham scheme.) We compare the results obtained with the noncollinear Kohn–Sham (NKS) scheme to those obtained with the widely used unrestricted Kohn–Sham (UKS) scheme for two types of multi-reference systems. For the dissociation of the MnO and NiO transition metal oxides, we find UKS fails to dissociate to the ground states of neutral atoms, while NKS dissociates to the correct limit and predicts potential energy curves that vary smoothly at intermediate bond lengths. This is due to the instability of UKS solutions at large bond distances. For barrier heights of O3, BeH2, and H4, NKS is shown to stabilize the multi-reference transition states by expanding the variational space. Although the errors vary because they are closely coupled with the capability of the employed exchange–correlation functionals in treating the multi-configurational states, these findings demonstrate that results with collinear spin orbitals should be further scrutinized, and future development of exchange–correlation functionals for multi-reference systems should incorporate the flexibilities of NKS.
Co-reporter:Wenjing Zhang, Donald G. Truhlar, and Mingsheng Tang
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 9) pp:3965-3977
Publication Date(Web):July 10, 2013
DOI:10.1021/ct400418u
One of the greatest challenges for the theoretical study of transition-metal-containing compounds is the treatment of intrinsically multiconfigurational atoms and molecules, which require a multireference (MR) treatment in wave function theory. The accuracy of density functional theory for such systems is still being explored. Here, we continue that exploration by presenting the predictions of 42 exchange-correlation (xc) functionals of 11 types [local spin density approximation (LSDA), generalized gradient approximation (GGA), nonseparable gradient approximation (NGA), global-hybrid GGA, meta-GGA, meta-NGA, global-hybrid meta-GGA, range-separated hybrid GGA, range-separated hybrid meta-GGA, range-separated hybrid meta-NGA, and DFT augmented with molecular mechanics damped dispersion (DFT-D)]. DFT-D is tested both for Grimme’s DFT-D3(BJ) model with Becke-Johnson damping and for ωB97X-D, which has the empirical atom–atom dispersion parametrized by Chai and Head-Gordon. The Hartree–Fock (HF) method has also been included because it can be viewed as a functional with 100% HF exchange and no correlation. These methods are tested against a database including 70 first-transition-row (3d) transition-metal-containing molecules (19 single-reference molecules and 51 MR molecules), all of which have estimated experimental uncertainties equal to or less than 2.0 kcal/mol in the heat of formation. We analyze the accuracy in terms of the atomization energy per bond instead of the enthalpy of formation of the molecule because it allows us to test electronic energies without the possibility of cancellation of errors in electronic energies with errors in vibrational energies. All the density functional and HF wave functions have been optimized to a stable solution, in which the spatial symmetry is allowed to be broken to minimize the energy to a stable solution. We find that τ-HCTHhyb has the smallest mean unsigned error (MUE) in average bond energy, in particular 2.5 kcal/mol, for the full set of 70 molecules, and it also gives the smallest MUE for MR systems. For single-reference systems, MPW1B95 has the best performance, with an MUE of 1.6 kcal/mol. Among local functionals, which are the least expensive, the best performance (MUE = 3.4 kcal/mol) for the total database is achieved by OreLYP. It is observed that adding HF exchange does not guarantee better accuracy for GGAs or for the NGA, but inclusion of the kinetic energy densities can benefit the GGAs and NGA calculations. The metal hydrides and metal oxides are demonstrated to be the most difficult bond types to predict, and CrO3, FeH, CrO, VH, and MnS are found to be the most difficult molecules to predict. The middle transition metals (V, Cr, and Mn) lead to larger errors on average than either the early or late transition metals.
Co-reporter:Aleksandr V. Marenich, Christopher J. Cramer, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 8) pp:3649-3659
Publication Date(Web):June 25, 2013
DOI:10.1021/ct400329u
We present a new kind of treatment of the solute–solvent dispersion contribution to the free energy of solvation using a solvation model with state-specific polarizability (SMSSP). To evaluate the solute–solvent dispersion contribution, the SMSSP model utilizes only two descriptors, namely, the spherically averaged dipole polarizability of the solute molecule (either in its ground or excited electronic state) and the refractive index of the solvent. The model was parametrized over 643 ground-state solvation free energy data for 231 solutes in 14 nonpolar, non-hydrogen-bonding solvents. We show that the SMSSP model is applicable to solutes in both the ground and the excited electronic state. For example, in comparison to available experimental data, the model yields qualitatively accurate predictions of the solvatochromic shifts for a number of systems where solute–solvent dispersion is the dominant contributor to the shift.
Co-reporter:Aleksandr V. Marenich, Christopher J. Cramer, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 1) pp:609-620
Publication Date(Web):December 3, 2012
DOI:10.1021/ct300900e
We present a new self-consistent reaction-field implicit solvation model that employs the generalized Born approximation for the bulk electrostatic contribution to the free energy of solvation. The new solvation model (SM) is called SM12 (where ″12″ stands for 2012), and it is available with two sets of parameters, SM12CM5 and SM12ESP. The SM12CM5 parametrization is based on CM5 partial atomic charges, and the SM12ESP parametrization is based on charges derived from a quantum-mechanically calculated electrostatic potential (ESP) (in particular, we consider ChElPG and Merz–Kollman–Singh charges). The model was parametrized over 10 combinations of theoretical levels including the 6-31G(d) and MG3S basis sets and the B3LYP, mPW1PW, M06-L, M06, and M06-2X density functionals against 2979 reference experimental data. The reference data include 2503 solvation free energies and 144 transfer free energies of neutral solutes composed of H, C, N, O, F, Si, P, S, Cl, Br, and I in water and in 90 organic solvents as well as 332 solvation free energies of singly charged anions and cations in acetonitrile, dimethyl sulfoxide, methanol, and water. The advantages of the new solvation model over our previous generalized Born model (SM8) and all other previous generalized Born solvation models are (i) like the SMD model based on electron density distributions, it may be applied with a single set of parameters with arbitrary extended basis sets, whereas the SM8 model involves CM4 or CM4M charges that become unstable for extended basis sets, (ii) it is parametrized against a more diverse training sets than any previous solvation model, and (iii) it is defined for the entire periodic table.
Co-reporter:Bo Wang and Donald G. Truhlar
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 2) pp:1036-1042
Publication Date(Web):December 20, 2012
DOI:10.1021/ct300935m
Tuned and balanced redistributed charge schemes have been developed for modeling the electrostatic fields of bonds that are cut by a quantum mechanical–molecular mechanical boundary in combined quantum mechanical and molecular mechanical (QM/MM) methods. First, the charge is balanced by adjusting the charge on the MM boundary atom to conserve the total charge of the entire QM/MM system. In the balanced smeared redistributed charge (BSRC) scheme, the adjusted MM boundary charge is smeared with a smearing width of 1.0 Å and is distributed in equal portions to the midpoints of the bonds between the MM boundary atom and the MM atoms bonded to it; in the balanced redistributed charge-2 (BRC2) scheme, the adjusted MM boundary charge is distributed as point charges in equal portions to the MM atoms that are bonded to the MM boundary atom. The QM subsystem is capped by a fluorine atom that is tuned to reproduce the sum of partial atomic charges of the uncapped portion of the QM subsystem. The new aspect of the present study is a new way to carry out the tuning process; in particular, the CM5 charge model, rather than the Mulliken population analysis applied in previous studies, is used for tuning the capping atom that terminates the dangling bond of the QM region. The mean unsigned error (MUE) of the QM/MM deprotonation energy for a 15-system test suite of deprotonation reactions is 2.3 kcal/mol for the tuned BSRC scheme (TBSRC) and 2.4 kcal/mol for the tuned BRC2 scheme (TBRC2). As was the case for the original tuning method based on Mulliken charges, the new tuning method performs much better than using conventional hydrogen link atoms, which have an MUE on this test set of about 7 kcal/mol. However, the new scheme eliminates the need to use small basis sets, which can be problematic, and it allows one to be more consistent by tuning the parameters with whatever basis set is appropriate for applications. (Alternatively, since the tuning parameters and partial charges obtained by the new method do not depend strongly on basis set, one can continue to use available CM5-tuned parameters even when one changes the basis set.) Furthermore, we found that, as compared to Mulliken charges, the CM5 charges describe the charge distributions in test molecules better, and they reproduce the dipole moments of full quantum mechanical calculations better; therefore the new tuning procedure is more physical and should be more reliable and robust.
Co-reporter:Miho Isegawa, Luke Fiedler, Hannah R. Leverentz, Yingjie Wang, Santhanamoorthi Nachimuthu, Jiali Gao, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 1) pp:33-45
Publication Date(Web):November 13, 2012
DOI:10.1021/ct300509d
The polarized molecular orbital (PMO) method, a neglect-of-diatomic-differential-overlap (NDDO) semiempirical molecular orbital method previously parametrized for systems composed of O and H, is here extended to carbon. We modified the formalism and optimized all the parameters in the PMO Hamiltonian by using a genetic algorithm and a database containing both electrostatic and energetic properties; the new parameter set is called PMO2. The quality of the resulting predictions is compared to results obtained by previous NDDO semiempirical molecular orbital methods, both including and excluding dispersion terms. We also compare the PMO2 properties to SCC-DFTB calculations. Within the class of semiempirical molecular orbital methods, the PMO2 method is found to be especially accurate for polarizabilities, atomization energies, proton transfer energies, noncovalent complexation energies, and chemical reaction barrier heights and to have good across-the-board accuracy for a range of other properties, including dipole moments, partial atomic charges, and molecular geometries.
Co-reporter:Xuefei Xu, Ke R. Yang, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 8) pp:3612-3625
Publication Date(Web):June 13, 2013
DOI:10.1021/ct400447f
Complete-active-space self-consistent-field (CASSCF) calculations provide useful reference wave functions for configuration interaction or perturbation theory calculations of excited-state potential energy surfaces including dynamical electron correlation. However, the canonical molecular orbitals (MOs) of CASSCF calculations usually have mixed character in regions of strong interaction of two or more electronic states; therefore, they are unsuitable for diabatization using the configurational uniformity approach. Here, CASSCF diabatic MOs for phenol have been obtained by the 4-fold way, and comparison to the CASSCF canonical MOs shows that they are much smoother. Using these smooth CASSCF diabatic MOs, we performed direct diabatization calculations for the three low-lying states (1ππ, 1ππ*, and 1πσ*) and their diabatic (scalar) couplings at the dynamically correlated multiconfiguration quasidegenerate perturbation theory (MC-QDPT) level. We present calculations along the O–H stretching and C–C–O–H torsion coordinates for the nonadiabatic photodissociation of phenol to the phenoxyl radical and hydrogen atom. The seams of 1ππ*/1πσ* and 1ππ/1πσ* diabatic crossings are plotted as functions of these coordinates. We also present diabatization calculations for displacements along the out-of-plane ring distortion modes 16a and 16b of the phenyl group. The dominant coupling modes of the two conical intersections (1ππ*/1πσ* and 1ππ/1πσ*) are discussed. The present diabatization method is confirmed to be valid even for significantly distorted ring structures by diabatization calculations along a reaction path connecting the planar equilibrium geometry of phenol to its strongly distorted prefulvenic form. The present work provides insight into the mode specificity of phenol photodissociation and shows that diabatization at the MC-QDPT level employing CASSCF diabatic MOs can be a good starting point for multidimensional dynamics calculations of photochemical reactions.
Co-reporter:Jingjing Zheng and Donald G. Truhlar
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 7) pp:2875-2881
Publication Date(Web):May 28, 2013
DOI:10.1021/ct400231q
We reformulate multistructural variational transition state theory by removing the approximation of calculating torsional anharmonicity only at stationary points. The multistructural method with torsional anharmonicity is applied to calculate the reaction-path free energy of the hydrogen abstraction from the carbon-1 position in isobutanol by OH radical. The torsional potential anharmonicity along the reaction path is taken into account by a coupled torsional potential. The calculations show that it can be critical to include torsional anharmonicity in searching for canonical and microcanonical variational transition states. The harmonic-oscillator approximation fails to yield reasonable free energy curves along the reaction path.
Co-reporter:Jingjing Zheng and Donald G. Truhlar
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 3) pp:1356-1367
Publication Date(Web):January 23, 2013
DOI:10.1021/ct3010722
We present a new approximation for calculating partition functions and thermodynamic functions by the multistructural method with torsional anharmonicity (MS-T). The new approximation is based on a reference potential with torsional barriers obtained from a calculation that includes local torsional coupling. By comparing to a fully coupled classical rotational-torsional partition function evaluated as a numerical phase space integral, the method is shown to provide improved accuracy in the classical limit. Quantum effects, which are most important at low temperatures, are included based on the harmonic approximation (which can be upgraded to a quasiharmonic approximation, that is, harmonic formulas with effective frequencies). Calculations were performed for six molecules (ethanol, 1-butanol, hexane, isohexane, heptane, and isoheptane), one radical (1-pentyl radical), and the saddle point structures of a hydrogen abstraction reaction (hydroxyl plus ethanol) to illustrate the difference between the new coupled-potential MS-T approximation and the original uncoupled-potential MS-T approximation. The new method improves the agreement with experimental results of calcuated thermodynamic functions for 1-butanol, hexane, isohexane, and heptane.
Co-reporter:Miho Isegawa, Bo Wang, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 3) pp:1381-1393
Publication Date(Web):February 12, 2013
DOI:10.1021/ct300845q
We add higher-order electronic polarization effects to the molecular tailoring approach (MTA) by embedding each fragment in background charges as in combined quantum mechanical and molecular mechanical (QM/MM) methods; the resulting method considered here is called electrostatically embedded MTA (EE-MTA). We compare EE-MTA to MTA for a test peptide, Ace-(Ala)20-NMe, and we find that including background charges (embedding charges) greatly improves the performance. The fragmentation is performed on the basis of amino acids as monomers, and several sizes of fragment are tested. The fragments are capped with either hydrogen cap atoms or tuned fluorine cap atoms. The effective core potential of the tuned fluorine cap atom is optimized so as to reproduce the proton affinity for seven types of tetrapeptide, and the proton affinity calculated with the tuned cap atom shows a lower mean unsigned error than that obtained by using a hydrogen cap atom. In the application to the test peptide, these generically tuned cap atoms show better performance compared with the hydrogen cap atom for both the electronic energy and the energy difference between an α helix and a β sheet (in the latter case, 1.0% vs 2.7% when averaged over three sizes of fragments and two locations for cut bonds). Also, we compared the accuracy of several charge redistribution schemes, and we find that the results are not particularly sensitive to these choices for the Ace–(Ala)20–NMe peptide. We also illustrate the dependence on the choice of charge model for the embedding charges, including both fixed embedding charges and embedding charges that depend on conformation.
Co-reporter:Hannah R. Leverentz, Helena W. Qi, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 2) pp:995-1006
Publication Date(Web):December 11, 2012
DOI:10.1021/ct300848z
The binding energies and relative conformational energies of five configurations of the water 16-mer are computed using 61 levels of density functional (DF) theory, 12 methods combining DF theory with molecular mechanics damped dispersion (DF-MM), seven semiempirical-wave function (SWF) methods, and five methods combining SWF theory with molecular mechanics damped dispersion (SWF-MM). The accuracies of the computed energies are assessed by comparing them to recent high-level ab initio results; this assessment is more relevant to bulk water than previous tests on small clusters because a 16-mer is large enough to have water molecules that participate in more than three hydrogen bonds. We find that for water 16-mer binding energies the best DF, DF-MM, SWF, and SWF-MM methods (and their mean unsigned errors in kcal/mol) are respectively M06-2X (1.6), ωB97X-D (2.3), SCC-DFTB-γh (35.2), and PM3-D (3.2). We also mention the good performance of CAM-B3LYP (1.8), M05-2X (1.9), and TPSSLYP (3.0). In contrast, for relative energies of various water nanoparticle 16-mer structures, the best methods (and mean unsigned errors in kcal/mol), in the same order of classes of methods, are SOGGA11-X (0.3), ωB97X-D (0.2), PM6 (0.4), and PMOv1 (0.6). We also mention the good performance of LC-ωPBE-D3 (0.3) and ωB97X (0.4). When both relative and binding energies are taken into consideration, the best methods overall (out of the 85 tested) are M05-2X without molecular mechanics and ωB97X-D when molecular mechanics corrections are included; with considerably higher average errors and considerably lower cost, the best SWF or SWF-MM method is PMOv1. We use six of the best methods for binding energies of the water 16-mers to calculate the binding energies of water hexamers and water 17-mers to test whether these methods are also reliable for binding energy calculations on other types of water clusters.
Co-reporter:Rémi Maurice, Pragya Verma, Joseph M. Zadrozny, Sijie Luo, Joshua Borycz, Jeffrey R. Long, Donald G. Truhlar, and Laura Gagliardi
Inorganic Chemistry 2013 Volume 52(Issue 16) pp:9379-9389
Publication Date(Web):July 30, 2013
DOI:10.1021/ic400953e
The metal–organic framework Fe2(dobdc) (dobdc4– = 2,5-dioxido-1,4-benzenedicarboxylate), often referred to as Fe-MOF-74, possesses many interesting properties such as a high selectivity in olefin/paraffin separations. This compound contains open-shell FeII ions with open coordination sites which may have large single-ion magnetic anisotropies, as well as isotropic couplings between the nearest and next nearest neighbor magnetic sites. To complement a previous analysis of experimental data made by considering only isotropic couplings [Bloch et al. Science 2012, 335, 1606], the magnitude of the main magnetic interactions are here assessed with quantum chemical calculations performed on a finite size cluster. It is shown that the single-ion anisotropy is governed by same-spin spin–orbit interactions (i.e., weak crystal-field regime), and that this effect is not negligible compared to the nearest neighbor isotropic couplings. Additional magnetic data reveal a metamagnetic behavior at low temperature. This effect can be attributed to various microscopic interactions, and the most probable scenarios are discussed.
Co-reporter:Elbek K. Kurbanov, Hannah R. Leverentz, Donald G. Truhlar, and Elizabeth A. Amin
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 6) pp:2617-2628
Publication Date(Web):May 7, 2013
DOI:10.1021/ct4001872
In the present paper, we apply the electrostatically embedded many-body expansion of the correlation energy (EE-MB-CE) to the calculation of zinc–ligand and cadmium–ligand bond dissociation energies, and we analyze the errors due to various fragmentation schemes in a variety of neutral, positively charged, and negatively charged Zn2+ and Cd2+ coordination complexes. As a result of the analysis, we are able to present a new, simple, and unambiguous fragmentation strategy. Following this strategy, we show that both methods perform well for zinc–ligand and cadmium–ligand bond dissociation energies for all systems studied in the paper, including a model of the catalytic site of the zinc-bearing anthrax toxin lethal factor (LF), which has garnered substantial attention as a target for drug development. To draw general conclusions, we consider ten pentacoordinate and hexacoordinate zinc and cadmium containing coordination complexes, each with 10 or 15 different fragmentation schemes. By analyzing errors, we developed a prescription for the optimal fragmentation strategy. With this scheme, and using MP2 correlation energies as a test, we find that the electrostatically embedded three-body expansion of the correlation energy (EE-3B-CE) method is able to reproduce all 53 conventionally calculated bond energies with an average absolute error of only 0.59 kcal/mol. The paper also presents EE-MB-CE calculations using the CCSD(T) level of theory on an LF model system. With CCSD(T), EE-3B-CE has an average error of 0.30 kcal/mol.
Co-reporter:Ke R. Yang, Amrit Jalan, William H. Green, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 1) pp:418-431
Publication Date(Web):December 6, 2012
DOI:10.1021/ct3009528
We examine the accuracy of single-reference and multireference correlated wave function methods for predicting accurate energies and potential energy curves of biradicals. The biradicals considered are intermediate species along the bond dissociation coordinates for breaking the F–F bond in F2, the O–O bond in H2O2, and the C–C bond in CH3CH3. We apply a host of single-reference and multireference approximations in a consistent way to the same cases to provide a better assessment of their relative accuracies than was previously possible. The most accurate method studied is coupled cluster theory with all connected excitations through quadruples, CCSDTQ. Without explicit quadruple excitations, the most accurate potential energy curves are obtained by the single-reference RCCSDt method, followed, in order of decreasing accuracy, by UCCSDT, RCCSDT, UCCSDt, seven multireference methods, including perturbation theory, configuration interaction, and coupled-cluster methods (with MRCI+Q being the best and Mk-MR-CCSD the least accurate), four CCSD(T) methods, and then CCSD.
Co-reporter:Ke R. Yang, Xuefei Xu, Donald G. Truhlar
Chemical Physics Letters 2013 Volume 573() pp:84-89
Publication Date(Web):6 June 2013
DOI:10.1016/j.cplett.2013.04.036
Highlights•Simplification of fourfold-way diabatization procedure.•Improved algorithm for diabatic states for simulating photochemical dynamics.•Complete active space SCF diabatic MOs for excited states with inclusion of dynamical correlation.We propose a new scheme for the direct diabatization of MC-QDPT wave functions. Our new scheme utilizes CASSCF diabatic molecular orbitals (DMOs); this is conceptually simpler than the previous approach and can lead to smoother diabatic potentials. We validated the new diabatization scheme, in comparison to CASSCF diabatization and to the original MC-QDPT diabatization scheme, for two test cases, the dissociation of LiF and the reaction of Li + FH → LiF + H. The results with our new scheme suggest that the new scheme with CASSCF DMOs would be a good choice for nonadiabatic dynamics studies in the future.
Co-reporter:Oksana Tishchenko and Donald G. Truhlar
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 3) pp:422-425
Publication Date(Web):January 16, 2013
DOI:10.1021/jz3020259
Endohedral fullerences have great potential for a variety of techological applications. Here we consider B@C60 and show that the amount of charge transfer from the semimetal boron atom to the cage is a strong function of the radial distance of the atom from the center of the fullerene, and it is controlled by multistate conical intersections whose associated ridge of avoided crossings has the topology of a Euclidean sphere. The potential energy surfaces of B@C60 are characterized by two kinds of local minima: those with a boron atom located in the geometric center of the fullerene, and those with a boron atom bound to the fullerene inner wall. At the lowest-energy minimum, at the center, the boron atom is neutral, whereas the transition to the wall is accompanied by an electron transfer from boron to the fullerene cage. The two kinds of minima are separated by a ridge of avoided crossings that forms a surface with a nearly spherical shape. The properties of such systems may be altered by controlling the populations of the two kinds of minima, for example, by application of an external field. Such switchable atom–cage charge transfer may find applications in novel molecular devices.Keywords: conical intersection; functional molecules; molecular switching; photochemistry;
Co-reporter:Xuefei Xu, Samer Gozem, Massimo Olivucci, and Donald G. Truhlar
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 2) pp:253-258
Publication Date(Web):December 22, 2012
DOI:10.1021/jz301935x
We present a new approach to calculating potential energy surfaces for photochemical reactions by combining self-consistent-field calculations for single-reference ground and excited states with symmetry-corrected spin-flip Tamm–Dancoff approximation calculations for multireference electronic states. The method is illustrated by an application with the M05-2X exchange-correlation functional to cis–trans isomerization of the penta-2,4-dieniminium cation, which is a model (with three conjugated double bonds) of the protonated Schiff base of retinal. We find good agreement with multireference configuration interaction-plus-quadruples (MRCISD+Q) wave function calculations along three key paths in the strong-interaction region of the ground and first excited singlet states.Keywords: broken symmetry; conical intersection; electronically excited states; multireference configuration interaction; SF2 method; time-dependent density functional theory;
Co-reporter:Hannah R. Leverentz, J. Ilja Siepmann, Donald G. Truhlar, Ville Loukonen, and Hanna Vehkamäki
The Journal of Physical Chemistry A 2013 Volume 117(Issue 18) pp:3819-3825
Publication Date(Web):April 10, 2013
DOI:10.1021/jp402346u
The formation of atmospheric aerosol particles through clustering of condensable vapors is an important contributor to the overall concentration of these atmospheric particles. However, the details of the nucleation process are not yet well understood and are difficult to probe by experimental means. Computational chemistry is a powerful tool for gaining insights about the nucleation mechanism. Here, we report accurate electronic structure calculations of the potential energies of small clusters made from sulfuric acid, ammonia, and dimethylamine. We also assess and validate the accuracy of less expensive methods that might be used for the calculation of the binding energies of larger clusters for atmospheric modeling. The PW6B95-D3 density-functional-plus-molecular-mechanics calculation with the MG3S basis set stands out as yielding excellent accuracy while still being affordable for very large clusters.
Co-reporter:Steven L. Mielke, Arindam Chakraborty, and Donald G. Truhlar
The Journal of Physical Chemistry A 2013 Volume 117(Issue 32) pp:7327-7343
Publication Date(Web):April 8, 2013
DOI:10.1021/jp4011789
We present vibrational configuration interaction calculations employing the Watson Hamiltonian and a multimode expansion. Results for the lowest 36 eigenvalues of the zero total angular momentum rovibrational spectrum of methane agree with the accurate benchmarks of Wang and Carrington to within a mean unsigned deviation of 0.68, 0.033, and 0.014 cm–1 for 4-mode, 5-mode, and 6-mode representations, respectively. We note that in the case of the 5-mode results, this is a factor of 10 better agreement than for 5-mode calculations reported earlier by Wu, Huang, Carter, and Bowman for the same set of eigenvalues, which indicates that the multimode expansion is even more rapidly convergent than previously demonstrated. Our largest calculations employ a tiered approach with matrix elements treated using a variable-order multimode expansion with orders ranging from 4-mode to 7-mode; strategies for assigning matrix elements to particular multimode tiers are discussed. Improvements of 7-mode coupling over 6-mode coupling are small (averaging 0.002 cm–1 for the first 36 eigenvalues) suggesting that 7-mode coupling is sufficient to fully converge the results. A number of approximate treatments of the computationally expensive vibrational angular momentum terms are explored. The use of optimized vibrational quadratures allows rapid integration of the matrix elements, especially the vibrational angular momentum terms, which require significantly fewer quadrature points than are required to integrate the potential. We assign the lowest 243 states and compare our results to those of Wang and Carrington, who provided assignments for the same set of states. Excellent agreement is observed for most states, but our results are lower for some of the higher-energy states by as much as 20 cm–1, with the largest deviations being for the states with six quanta of excitation in the F2 bends, suggesting that the earlier results were not fully converged with respect to the basis set. We also provide corrections to several of the state assignments published previously.
Co-reporter:Helena W. Qi, Hannah R. Leverentz, and Donald G. Truhlar
The Journal of Physical Chemistry A 2013 Volume 117(Issue 21) pp:4486-4499
Publication Date(Web):April 30, 2013
DOI:10.1021/jp401463f
This work presents a new fragment method, the electrostatically embedded many-body expansion of the nonlocal energy (EE-MB-NE), and shows that it, along with the previously proposed electrostatically embedded many-body expansion of the correlation energy (EE-MB-CE), produces accurate results for large systems at the level of CCSD(T) coupled cluster theory. We primarily study water 16-mers, but we also test the EE-MB-CE method on water hexamers. We analyze the distributions of two-body and three-body terms to show why the many-body expansion of the electrostatically embedded correlation energy converges faster than the many-body expansion of the entire electrostatically embedded interaction potential. The average magnitude of the dimer contributions to the pairwise additive (PA) term of the correlation energy (which neglects cooperative effects) is only one-half of that of the average dimer contribution to the PA term of the expansion of the total energy; this explains why the mean unsigned error (MUE) of the EE-PA-CE approximation is only one-half of that of the EE-PA approximation. Similarly, the average magnitude of the trimer contributions to the three-body (3B) term of the EE-3B-CE approximation is only one-fourth of that of the EE-3B approximation, and the MUE of the EE-3B-CE approximation is one-fourth that of the EE-3B approximation. Finally, we test the efficacy of two- and three-body density functional corrections. One such density functional correction method, the new EE-PA-NE method, with the OLYP or the OHLYP density functional (where the OHLYP functional is the OptX exchange functional combined with the LYP correlation functional multiplied by 0.5), has the best performance-to-price ratio of any method whose computational cost scales as the third power of the number of monomers and is competitive in accuracy in the tests presented here with even the electrostatically embedded three-body approximation.
Co-reporter:Ruifang Li, Roberto Peverati, Miho Isegawa, and Donald G. Truhlar
The Journal of Physical Chemistry A 2013 Volume 117(Issue 1) pp:169-173
Publication Date(Web):December 14, 2012
DOI:10.1021/jp3079106
Using quantum chemical approximations to understand and predict complex transition metal chemistry, such as catalytic processes and materials properties, is an important activity in modern computational chemistry. High-level theory can sometimes provide high-precision benchmarks for systems containing transition metals, and these benchmarks can be used to understand the reliability of less expensive quantum chemical approximations that are applicable to complex systems. Here, we studied the ionization potential energy of Fe and FeC and the bond dissociation energies of FeC and FeC+ by 15 density functional approximations: M05, M06, M06-L, ωB97, ωB97X, ωB97X-D, τ-HCTHhyb, BLYP, B3LYP, M08-HX, M08-SO, SOGGA11, SOGGA11-X, M11, and M11-L. All of the functionals predict the correct spin state as the ground state of neutral iron atom, but five of them predict the wrong spin state for Fe+. In the final analysis, four functionals, namely M11-L, τ-HCTHhyb, SOGGA11, and M06-L, have small mean unsigned errors when averaged over two bond dissociation energies and two ionization potentials. In fact, the results show that M11-L gives the smallest averaged mean unsigned error, i.e., M11-L is the most reliable density functional for these iron carbide systems among those studied.
Co-reporter:Prasenjit Seal, Gbenga Oyedepo, and Donald G. Truhlar
The Journal of Physical Chemistry A 2013 Volume 117(Issue 2) pp:275-282
Publication Date(Web):December 17, 2012
DOI:10.1021/jp310910f
In the present work, we study the H atom abstraction reactions by hydroxyl radical at all five sites of 1-butanol. Multistructural variational transition state theory (MS-VTST) was employed to estimate the five thermal rate constants. MS-VTST utilizes a multifaceted dividing surface that accounts for the multiple conformational structures of the transition state, and we also include all the structures of the reactant molecule. The vibrational frequencies and minimum energy paths (MEPs) were computed using the M08-HX/MG3S electronic structure method. The required potential energy surfaces were obtained implicitly by direct dynamics employing interpolated variational transition state theory with mapping (IVTST-M) using a variational reaction path algorithm. The M08-HX/MG3S electronic model chemistry was then used to calculate multistructural torsional anharmonicity factors to complete the MS-VTST rate constant calculations. The results indicate that torsional anharmonicity is very important at higher temperatures, and neglecting it would lead to errors of 26 and 32 at 1000 and 1500 K, respectively. Our results for the sums of the site-specific rate constants agree very well with the experimental values of Hanson and co-workers at 896–1269 K and with the experimental results of Campbell et al. at 292 K, but slightly less well with the experiments of Wallington et al., Nelson et al., and Yujing and Mellouki at 253–372 K; nevertheless, the calculated rates are within a factor of 1.61 of all experimental values at all temperatures. This gives us confidence in the site-specific values, which are currently inaccessible to experiment.
Co-reporter:Tao Yu, Masahiro Higashi, Alessandro Cembran, Jiali Gao, and Donald G. Truhlar
The Journal of Physical Chemistry B 2013 Volume 117(Issue 28) pp:8422-8429
Publication Date(Web):May 31, 2013
DOI:10.1021/jp404292t
We calculate the free energy profile for the postulated hydride transfer reaction mechanism for the catalysis of lysine demethylation by lysine-specific demethylase LSD1. The potential energy surface is obtained by using combined electrostatically embedded multiconfiguration molecular mechanics (EE-MCMM) and single-configuration molecular mechanics (MM). We employ a constant valence bond coupling term to obtain analytical energies and gradients of the EE-MCMM subsystem, which contains 45 quantum mechanics (QM) atoms and which is parametrized with density functional calculations employing specific reaction parameters obtained by matching high-level wave function calculations. In the MM region, we employ the Amber ff03 and TIP3P force fields. The free energy of activation at 300 K is calculated by molecular dynamics (MD) umbrella sampling on a system with 102 090 atoms as the maximum of the free energy profile along the reaction coordinate as obtained by the weighted histogram analysis method with 17 umbrella sampling windows. This yields a free energy of activation of only 10 kcal/mol, showing that the previously postulated direct hydride transfer reaction mechanism is plausible, although we find that it is better interpreted as a concerted transfer of a hydrogen atom and an electron.
Co-reporter:Anant D. Kulkarni, Donald G. Truhlar, Sriram Goverapet Srinivasan, Adri C. T. van Duin, Paul Norman, and Thomas E. Schwartzentruber
The Journal of Physical Chemistry C 2013 Volume 117(Issue 1) pp:258-269
Publication Date(Web):December 4, 2012
DOI:10.1021/jp3086649
We consider oxygen interactions with realistic silica surfaces, including both experimentally observed nondefective surface reconstructions and experimentally observed surface defects. Nondefective models include clusters representing the site above a fully coordinated surface Si atom and bridging O atoms, and the defective models include clusters representing an under-coordinated Si defect, a nonbridging O defect, and a ring structure. Energies were obtained for the approach of atomic and molecular oxygen to these clusters in various configurations by using explicitly correlated CCSD(T)-F12b electronic structure theory and the Minnesota density functionals, which were found to be in good agreement. The Minnesota functionals were employed in binding energy calculations for all of the clusters, considering the singlet and triplet spin states for nondefective clusters and doublet and quartet spin states for defective clusters. We find that the chosen defects are energetically favorable sites for binding. The density functional energies were used to extend the empirical ReaxFFSiO potential for silica, which was previously parametrized for bulk silica polymorphs (van Duin et al. J. Phys. Chem. A, 2003, 107, 3803–3811), to model the gas–surface interactions represented by the defective and nondefective clusters presented here. Interaction energy predictions from ReaxFFSiOGSI agree very well with the density functional energies. ReaxFFSiOGSI can now be employed in reactive large-scale molecular dynamics simulations involving oxygen–silica gas–surface interactions such as oxide growth and the heterogeneous recombination of oxygen occurring on real silica surfaces.
Co-reporter:Paul Norman, Thomas E. Schwartzentruber, Hannah Leverentz, Sijie Luo, Rubén Meana-Pañeda, Yuliya Paukku, and Donald G. Truhlar
The Journal of Physical Chemistry C 2013 Volume 117(Issue 18) pp:9311-9321
Publication Date(Web):April 10, 2013
DOI:10.1021/jp4019525
In this work we use molecular dynamics (MD) simulations with the ReaxFFSiOGSI potential to model the structure of quartz and amorphous silica surfaces exposed to atomic oxygen. The ReaxFFSiOGSI potential is a reactive force field that was specifically parametrized to describe gas surface interactions in silica–oxygen systems (Kulkarni, A. D.; et al. J. Phys. Chem. C2012, 117, 258–269). We show that the ReaxFFSiOGSI potential accurately describes the experimentally measured bulk structure of quartz and amorphous silica, as well as experimentally and computationally characterized surface features for these materials. A flux boundary condition is implemented in molecular dynamics simulations to model the exposure of silica surfaces to atomic oxygen. We find that the types of defects occurring on silica surfaces under vacuum and exposed to atomic oxygen at high temperatures are in agreement with previous MD simulations and experimental measurements of silica surfaces. The ReaxFFSiOGSI potential predicts a peroxyl defect that has not been observed in previous MD simulations of silica surfaces, but has been experimentally identified. Density functional theory (DFT) calculations are used to validate the extent to which the ReaxFFSiOGSI potential predicts the structure and binding energy of the oxygen molecule on this defect. Through MD simulation and comparison with experiment, we identify the chemical surface defects that exist on real silica surfaces exposed to atomic oxygen at high temperatures and discuss their role in the catalytic recombination of atomic oxygen.
Co-reporter:Junming Ho, Jingjing Zheng, Rubén Meana-Pañeda, Donald G. Truhlar, Eun Jung Ko, G. Paul Savage, Craig M. Williams, Michelle L. Coote, and John Tsanaktsidis
The Journal of Organic Chemistry 2013 Volume 78(Issue 13) pp:6677-6687
Publication Date(Web):June 3, 2013
DOI:10.1021/jo400927y
The utility of chloroform as both a solvent and a hydrogen atom donor in Barton reductive decarboxylation of a range of carboxylic acids was recently demonstrated (Ko, E. J. et al. Org. Lett. 2011, 13, 1944). In the present work, a combination of electronic structure calculations, direct dynamics calculations, and experimental studies was carried out to investigate how chloroform acts as a hydrogen atom donor in Barton reductive decarboxylations and to determine the scope of this process. The results from this study show that hydrogen atom transfer from chloroform occurs directly under kinetic control and is aided by a combination of polar effects and quantum mechanical tunneling. Chloroform acts as an effective hydrogen atom donor for primary, secondary, and tertiary alkyl radicals, although significant chlorination was also observed with unstrained tertiary carboxylic acids.
Co-reporter:Pragya Verma, Xuefei Xu, and Donald G. Truhlar
The Journal of Physical Chemistry C 2013 Volume 117(Issue 24) pp:12648-12660
Publication Date(Web):May 22, 2013
DOI:10.1021/jp402884h
An increase in demand for energy efficient processes for the separation of a mixture of hydrocarbons drives the need for understanding metal–organic frameworks (MOFs) that can provide better noncryogenic alternatives for the fractionation of hydrocarbon mixtures. Here we study the structure and properties of a metal–organic framework, Fe-MOF-74, and its effectiveness to separate C1–C3 hydrocarbon mixtures. The binding of these hydrocarbons to the open metal sites of Fe-MOF-74 has been investigated using a meta-generalized gradient approximation density functional, M06-L, which has previously been validated for systems containing transition metals. For interpretive purposes, charge model 5 (CM5) is used to determine the partial atomic charges on the metal cations and the oxygen atoms of the ligands surrounding these metal centers. Our computations show preferential binding to the metal center of Fe-MOF-74 of unsaturated hydrocarbons over saturated ones in agreement with experimental results, and the calculated binding energies are in semiquantitative agreement with experiment. The results are analyzed in terms of various factors contributing to the binding, including structural distortion, electrostatics, damped dispersion, charge transfer, back bonding, and ligand field effects on the d orbitals. The CM5 charges are not sensitive to small differences in structure.
Co-reporter:Yan Zhao, Hou T. Ng, Roberto Peverati, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 8) pp:2824-2834
Publication Date(Web):July 9, 2012
DOI:10.1021/ct300457c
We report a database of 18 ylidic bond dissociation energies obtained by using highly accurate quantum mechanical methods, and we use it to test approximate electronic structure methods. The new benchmark database is called YBDE18 and is used to test a large number of electronic structure methods, including eight wave function methods and 98 density functional exchange-correlation functionals. Among them, we include some very recent density functionals, including the SOGGA11 GGA functional, the SOGGA11-X hybrid GGA functional, the M11-L local meta-GGA functional, and the M11 range-separated hybrid meta-GGA functional. We also consider other functionals of these classes plus a local spin density approximation, global-hybrid meta-GGAs, range-separated hybrid GGAs, doubly hybrid GGAs, and doubly hybrid meta-GGAs. We found M05-2X-D3, MPWB1K-D3, M05-2X, LC-BLYP, PBE0-D3, and MC3MPWB to be the best DFT methods for this database. Although they do not place in the top four overall, our new-generation functionals show overall competitive performances; each of the new functionals provides the smallest mean signed error within its class, while in terms of mean unsigned errors, SOGGA11 is the best GGA, and SOGGA11-X and M11-L are among the first three best functionals in their categories, global-hybrid GGA and local meta-GGA. The best local functionals are VSXC and M06-L, the best global-hybrids are M05-2X, M08-HX, M06-2X, and MPWB1K, and the best range-separated hybrids are LC-BLYP, ωB97, ωB97X, and M11.
Co-reporter:Steven L. Mielke and Donald G. Truhlar
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 5) pp:1589-1596
Publication Date(Web):April 5, 2012
DOI:10.1021/ct300098p
We present two new methods to accelerate the convergence of Feynman path integral calculations of thermodynamic partition functions. The first enhancement uses information from instantaneous normal mode (INM) calculations to decrease the number of discretized points necessary to represent the Feynman paths and is denoted the local generalized Pitzer–Gwinn (LGPG) scheme. The second enhancement, denoted harmonically guided variance reduction (HGVR), reduces the variance in Monte Carlo (MC) calculations by exploiting the correlation between the sampling error associated with the sum over paths at a particular centroid location for the accurate potential and for the INM approximation of a model potential, the latter of which can be exactly calculated. The LGPG scheme can reduce the number of quadrature points required along the paths by nearly an order of magnitude, and the HGVR scheme can reduce the number of MC samples needed to achieve a target accuracy by more than an order of magnitude. Numerical calculations are presented for H2O2, a very anharmonic system where torsional motion is important, and H2O, a system more amenable to harmonic reference treatment.
Co-reporter:Denis Jacquemin, Yan Zhao, Rosendo Valero, Carlo Adamo, Ilaria Ciofini, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 4) pp:1255-1259
Publication Date(Web):February 3, 2012
DOI:10.1021/ct200721d
We assess the accuracy of eight Minnesota density functionals (M05 through M08-SO) and two others (PBE and PBE0) for the prediction of electronic excitation energies of a family of four cyanine dyes. We find that time-dependent density functional theory (TDDFT) with the five most recent of these functionals (from M06-HF through M08-SO) is able to predict excitation energies for cyanine dyes within 0.10–0.36 eV accuracy with respect to the most accurate available Quantum Monte Carlo calculations, providing a comparable accuracy to the latest generation of CASPT2 calculations, which have errors of 0.16–0.34 eV. Therefore previous conclusions that TDDFT cannot treat cyanine dyes reasonably accurately must be revised.
Co-reporter:Xuefei Xu and Donald G. Truhlar
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 1) pp:80-90
Publication Date(Web):October 20, 2011
DOI:10.1021/ct200558j
The performance of popular Hartree–Fock-based effective core potentials in Hartree–Fock and density functional calculations of 3d transition metals has been evaluated by basis-set convergence studies for ten cases: the equilibrium bond dissociation energy (De) for dissociation of ground-state Ti2 to ground and excited atoms, the ground-state dissociation energies of FeO, Cu2, ScH, TiH, Sc2, Fe2, and TiV+, and the first excitation energy (Ex) of Ti atom. Each case is studied with 11 or 13 density functionals. For comparison, the accuracy of the all-electron def2-TZVP basis set is tested with both relativistic and nonrelativistic treatments. Convergence and accuracy are assessed by comparing to relativistic all-electron calculations with a nearly complete relativistic basis set (NCBS-DK, which denotes the cc-pV5Z-DK basis set for 3d metals and hydrogen and the ma-cc-pV5Z-DK basis set for oxygen) and to nonrelativistic all-electron calculations with a nearly complete nonrelativistic basis set (NCBS-NR, which denotes the cc-pV5Z basis set for 3d metals and hydrogen and the ma-cc-pV5Z basis set for oxygen). As compared to NCBS-DK results, all ECP calculations perform worse than def2-TZVP all-electron relativistic calculations when averaged over all 130 data (13 functionals and ten test cases). The compact effective potential (CEP) relativistic effective core potential (RECP) combined with a valence basis set developed for the many-electron Dirac–Fock (MDF10) RECP performs best in effective core potential calculations and has an average basis-set incompleteness error of 3.7 kcal/mol, which is much larger than that (0.9 kcal/mol) of def2-TZVP relativistic all-electron results. Hence, the def2-TZVP relativistic all-electron calculations are recommended for accurate DFT calculations on 3d transition metals. In addition to our general findings, we observed that all kinds of density functionals do not show the same trends. For example, when ECPs are used with hybrid functionals, which sometimes are not recommended for calculations of transition metal systems, they are found to perform better at achieving the basis-set limit than when used with local functionals and meta-GGA functionals. The most successful combination of RECP and basis set has a basis-set incompleteness error of 1.7–2.4 kcal/mol for hybrid generalized gradient approximations, which is smaller than that of nonrelativistic NCBS calculations (whose average basis-set incompleteness error for hybrid functionals is 2.7–2.9 kcal/mol). The average basis-set incompleteness error in Hartree–Fock calculations is 1.0–4.4 kcal/mol for five of the ECP basis sets but is 5.8–10.8 kcal/mol for six others.
Co-reporter:Aleksandr V. Marenich, Steven V. Jerome, Christopher J. Cramer, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 2) pp:527-541
Publication Date(Web):January 11, 2012
DOI:10.1021/ct200866d
We propose a novel approach to deriving partial atomic charges from population analysis. The new model, called Charge Model 5 (CM5), yields class IV partial atomic charges by mapping from those obtained by Hirshfeld population analysis of density functional electronic charge distributions. The CM5 model utilizes a single set of parameters derived by fitting to reference values of the gas-phase dipole moments of 614 molecular structures. An additional test set (not included in the CM5 parametrization) contained 107 singly charged ions with nonzero dipole moments, calculated from the accurate electronic charge density, with respect to the center of nuclear charges. The CM5 model is applicable to any charged or uncharged molecule composed of any element of the periodic table in the gas phase or in solution. The CM5 model predicts dipole moments for the tested molecules that are more accurate on average than those from the original Hirshfeld method or from many other popular schemes including atomic polar tensor and Löwdin, Mulliken, and natural population analyses. In addition, the CM5 charge model is essentially independent of a basis set. It can be used with larger basis sets, and thereby this model significantly improves on our previous charge models CMx (x = 1–4 or 4M) and other methods that are prone to basis set sensitivity. CM5 partial atomic charges are less conformationally dependent than those derived from electrostatic potentials. The CM5 model does not suffer from ill conditioning for buried atoms in larger molecules, as electrostatic fitting schemes sometimes do. The CM5 model can be used with any level of electronic structure theory (Hartree–Fock, post-Hartree–Fock, and other wave function correlated methods or density functional theory) as long as an accurate electronic charge distribution and a Hirshfeld analysis can be computed for that level of theory.
Co-reporter:Bo Wang and Donald G. Truhlar
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 6) pp:1989-1998
Publication Date(Web):March 30, 2012
DOI:10.1021/ct2009285
We propose a new screened charge method for calculating partial atomic charges in molecules by electrostatic potential (ESP) fitting. The model, called full density screening (FDS), is used to approximate the screening effect of full charge densities of atoms in molecules. The results are compared to the conventional ESP fitting method based on point charges and to our previously proposed outer density screening (ODS) method, in which the parameters are reoptimized for the present purpose. In ODS, the charge density of an atom is represented by the sum of a point charge and a smeared negative charge distributed in a Slater-type orbital (STO). In FDS, the charge density of an atom is taken to be the sum of the charge density of the neutral atom and a partial atomic charge (of either sign) distributed in an STO. The ζ values of the STOs used in these two models are optimized in the present study to best reproduce the electrostatic potentials. The quality of the fit to the electrostatics is improved in the screened charge methods, especially for the regions that are within one van der Waals radius of the centers of atoms. It is also found that the charges derived by fitting electrostatic potentials with screened charges are less sensitive to the positions of the fitting points than are those derived with conventional electrostatic fitting. Moreover, we found that the electrostatic-potential-fitted (ESP) charges from the screened charge methods are similar to those from the point-charge method except for molecules containing the methyl group, where we have explored the use of restraints on nonpolar H atoms. We recommend the FDS model if the only goal is ESP fitting to obtain partial atomic charges or a fit to the ESP field. However, the ODS model is more accurate for electronic embedding in combined quantum mechanical and molecular mechanical (QM/MM) modeling and is more accurate than point-charge models for ESP fitting, and it is recommended for applications involving QM/MM methods. Since the screened charges describe the electrostatic potentials more accurately than point charges, since they asymptotically act as point charges at long distances, and since the electrostatic potential in terms of the screened charges is still a sum of functions centered at the atoms, the screened-charge representation of the electrostatic potential can be used in the same way as the conventional point-charge representation to model the electrostatic interactions, but it is more realistic. For the H atom and p block elements, the error in the fit to the electrostatic potential is reduced by about a factor of 3, and the sensitivity of the derived partial atomic charges to the choice of fitting points is reduced by about a factor of 2. For s and d block elements, there are also improvements in the inner regions but not necessarily in the outer regions.
Co-reporter:Roberto Peverati and Donald G. Truhlar
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 7) pp:2310-2319
Publication Date(Web):June 5, 2012
DOI:10.1021/ct3002656
The generalized gradient approximation (GGA) has been a workhorse exchange–correlation functional for electronic structure studies of extended systems (liquid-phase reactions, solids, heterogeneous and enzymatic catalysis, biopolymers) because its dependence on only the spin-labeled electron densities and their reduced gradients makes it the most affordable choice that produces realistic results for thermochemistry. However, much recent research has focused on its poor performance for solid-state lattice constants; the results for lattice constants can be improved but only at the cost of making the energetic predictions worse. In the present article, we propose a new density functional, called N12, which may be thought of as a generalization of range-separated functionals. The N12 functional depends only on the spin-labeled electron densities and their reduced gradients, but with a new kind of nonseparable term that gives it much greater flexibility. The N12 functional is the first exchange–correlation functional depending only on the spin-labeled electron densities and their reduced gradients that simultaneously provides good accuracy for the four key energetic and structural properties of solids and molecules, namely, solid-state cohesive energies and lattice constants and molecular atomization energies and bond lengths.
Co-reporter:Jingjing Zheng, Steven L. Mielke, Kenneth L. Clarkson, Donald G. Truhlar
Computer Physics Communications 2012 Volume 183(Issue 8) pp:1803-1812
Publication Date(Web):August 2012
DOI:10.1016/j.cpc.2012.03.007
We present a Fortran program package, MSTor, which calculates partition functions and thermodynamic functions of complex molecules involving multiple torsional motions by the recently proposed MS-T method. This method interpolates between the local harmonic approximation in the low-temperature limit, and the limit of free internal rotation of all torsions at high temperature. The program can also carry out calculations in the multiple-structure local harmonic approximation. The program package also includes six utility codes that can be used as stand-alone programs to calculate reduced moment of inertia matrices by the method of Kilpatrick and Pitzer, to generate conformational structures, to calculate, either analytically or by Monte Carlo sampling, volumes for torsional subdomains defined by Voronoi tessellation of the conformational subspace, to generate template input files, and to calculate one-dimensional torsional partition functions using the torsional eigenvalue summation method.Program summaryProgram title: MSTorCatalogue identifier: AEMF_v1_0Program summary URL:http://cpc.cs.qub.ac.uk/summaries/AEMF_v1_0.htmlProgram obtainable from: CPC Program Library, Queenʼs University, Belfast, N. IrelandLicensing provisions: Standard CPC licence, http://cpc.cs.qub.ac.uk/licence/licence.htmlNo. of lines in distributed program, including test data, etc.: 77 434No. of bytes in distributed program, including test data, etc.: 3 264 737Distribution format: tar.gzProgramming language: Fortran 90, C, and PerlComputer: Itasca (HP Linux cluster, each node has two-socket, quad-core 2.8 GHz Intel Xeon X5560 “Nehalem EP” processors), Calhoun (SGI Altix XE 1300 cluster, each node containing two quad-core 2.66 GHz Intel Xeon “Clovertown”-class processors sharing 16 GB of main memory), Koronis (Altix UV 1000 server with 190 6-core Intel Xeon X7542 “Westmere” processors at 2.66 GHz), Elmo (Sun Fire X4600 Linux cluster with AMD Opteron cores), and Mac Pro (two 2.8 GHz Quad-core Intel Xeon processors)Operating system: Linux/Unix/Mac OSRAM: 2 MbytesClassification: 16.3, 16.12, 23Nature of problem: Calculation of the partition functions and thermodynamic functions (standard-state energy, enthalpy, entropy, and free energy as functions of temperatures) of complex molecules involving multiple torsional motions.Solution method: The multi-structural approximation with torsional anharmonicity (MS-T). The program also provides results for the multi-structural local harmonic approximation [1].Restrictions: There is no limit on the number of torsions that can be included in either the Voronoi calculation or the full MS-T calculation. In practice, the range of problems that can be addressed with the present method consists of all multi-torsional problems for which one can afford to calculate all the conformations and their frequencies.Unusual features: The method can be applied to transition states as well as stable molecules. The program package also includes the hull program for the calculation of Voronoi volumes and six utility codes that can be used as stand-alone programs to calculate reduced moment-of-inertia matrices by the method of Kilpatrick and Pitzer, to generate conformational structures, to calculate, either analytically or by Monte Carlo sampling, volumes for torsional subdomain defined by Voronoi tessellation of the conformational subspace, to generate template input files, and to calculate one-dimensional torsional partition functions using the torsional eigenvalue summation method.Additional comments: The program package includes a manual, installation script, and input and output files for a test suite.Running time: There are 24 test runs. The running time of the test runs on a single processor of the Itasca computer is less than 2 seconds.References:[1]J. Zheng, T. Yu, E. Papajak, I.M. Alecu, S.L. Mielke, D.G. Truhlar, Practical methods for including torsional anharmonicity in thermochemical calculations of complex molecules: The internal-coordinate multi-structural approximation, Phys. Chem. Chem. Phys. 13 (2011) 10885–10907.
Co-reporter:Roberto Peverati and Donald G. Truhlar
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 38) pp:13171-13174
Publication Date(Web):02 Aug 2012
DOI:10.1039/C2CP42025B
We report a new local exchange–correlation energy functional that has significantly improved across-the-board performance, including main-group and transition metal chemistry and solid-state physics, especially atomization energies, ionization potentials, barrier heights, noncovalent interactions, isomerization energies of large moleucles, and solid-state lattice constants and cohesive energies.
Co-reporter:Roberto Peverati and Donald G. Truhlar
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 32) pp:11363-11370
Publication Date(Web):30 May 2012
DOI:10.1039/C2CP41295K
Adiabatic time-dependent density functional theory is a powerful method for calculating electronic excitation energies of complex systems, but the quality of the results depends on the choice of approximate density functional. In this article we test two promising new density functionals, M11 and M11-L, against databases of 214 diverse electronic excitation energies, and we compare the results to those for 16 other density functionals of various kinds and to time-dependent Hartree–Fock. Charge transfer excitations are well known to be the hardest challenge for TDDFT. M11 is a long-range-corrected hybrid meta-GGA, and it shows better performance for charge transfer excitations than any of the other functionals except M06-HF, which is a specialized functional that does not do well for valence excitations. Several other long-range-corrected hybrid functionals also do well, and we especially recommend M11, ωB97X, and M06-2X for general spectroscopic applications because they do exceptionally well on ground-state properties as well as excitation energies. Local functionals are preferred for many applications to extended systems because of their significant cost advantage for large systems. M11-L is a dual-range local functional and—unlike all previous local functionals—it has good performance for Rydberg states as well as for valence states. Thus it is highly recommended for excitation energy calculations on extended systems.
Co-reporter:Xuefei Xu, Ewa Papajak, Jingjing Zheng and Donald G. Truhlar
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 12) pp:4204-4216
Publication Date(Web):16 Jan 2012
DOI:10.1039/C2CP23692C
We investigate the statistical thermodynamics and kinetics of the 1,5-hydrogen shift isomerization reaction of the 1-butoxyl radical and its reverse isomerization. The partition functions and thermodynamic functions (entropy, enthalpy, heat capacity, and Gibbs free energy) are calculated using the multi-structural torsional (MS-T) anharmonicity method including all structures for three species (reactant, product, and transition state) involved in the reaction. The calculated thermodynamic quantities have been compared to those estimated by the empirical group additivity (GA) method. The kinetics of the unimolecular isomerization reaction was investigated using multi-structural canonical variational transition state theory (MS-CVT) including both multiple-structure and torsional (MS-T) anharmonicity effects. In these calculations, multidimensional tunneling (MT) probabilities were evaluated by the small-curvature tunneling (SCT) approximation and compared to results obtained with the zero-curvature tunneling (ZCT) approximation. The high-pressure-limit rate constants for both the forward and reverse reactions are reported as calculated by MS-CVT/MT, where MT can be ZCT or SCT. Comparison with the rate constants obtained by the single-structural harmonic oscillator (SS-HO) approximation shows the importance of anharmonicity in the rate constants of these reactions, and the effect of multi-structural anharmonicity is found to be very large. Whereas the tunneling effect increases the rate constants, the MS-T anharmonicity decreases them at all temperatures. The two effects counteract each other at temperatures 385 K and 264 K for forward and reverse reactions, respectively, and tunneling dominates at lower temperatures while MS-T anharmonicity has a larger effect at higher temperatures. The multi-structural torsional anharmonicity effect reduces the final reverse reaction rate constants by a much larger factor than it does to the forward ones as a result of the existence of more low-energy structures of the product 4-hydroxy-1-butyl radical than the reactant 1-butoxyl radical. As a consequence there is also a very large effect on the equilibrium constant. The neglect of multi-structural anharmonicity will lead to large errors in the estimation of reverse reaction rate constants.
Co-reporter:Tao Yu, Jingjing Zheng and Donald G. Truhlar
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 2) pp:482-494
Publication Date(Web):25 Nov 2011
DOI:10.1039/C1CP22578B
We have employed electronic structure calculations and the recently proposed multi-structural (MS) anharmonicity method to calculate partition functions and thermodynamic quantities, in particular entropy and heat capacity, for n-heptane and isoheptane. We included all structures, of which there are 59 for n-heptane and 37 for isoheptane, and we carried out the calculations both in the local harmonic approximation and by including torsional (T) anharmonicity. In addition, ΔS°, ΔH, and ΔG° for the isomerization reaction between these two species were also calculated. It is found that all calculated thermodynamic quantities based on the MS-T approximation in the temperature range from 298 K to 1500 K agree well with experimental data from the American Petroleum Institute (API) tables or Thermodynamics Research Center (TRC) data series and with values obtained from Benson's empirical parameters fit to experiment. This demonstrates not only the high accuracy of the electronic structure calculations but also that the MS-T method can be used to include both multiple-structure anharmonicity and torsional anharmonicity in the calculation of thermodynamic properties for complex molecules that contain many torsions. It also gives us confidence that we can apply the MS-T statistical thermodynamic method to obtain thermodynamic properties (i) over a broader temperature range than that for which data are available in the API tables, TRC data series, or from empirical estimation and (ii) to the many molecules for which experimental data are not available at any temperature.
Co-reporter:Roberto Peverati and Donald G. Truhlar
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 47) pp:16187-16191
Publication Date(Web):17 Sep 2012
DOI:10.1039/C2CP42576A
We present two new exchange–correlation functionals for hybrid Kohn–Sham electronic structure calculations based on the nonseparable functional form introduced recently in the N12 and MN12-L functionals but now with the addition of screened Hartree–Fock exchange. The first functional depends on the density and the density gradient and is called N12-SX; the second functional depends on the density, the density gradient, and the kinetic energy density and is called MN12-SX. Both new functionals include a portion of the Hartree–Fock exchange at short-range, but Hartree–Fock exchange is screened at long range. The accuracies of the two new functionals are compared to those of the recent N12 and MN12-L local functionals to show the effect of adding screened exchange, are compared to the previously best available screened exchange functional, HSE06, and are compared to the best available global-hybrid generalized gradient approximation (GGA) and to a high-performance long-range-corrected meta-GGA.
Co-reporter:Donald G. Truhlar
Journal of Chemical Education 2012 Volume 89(Issue 5) pp:573-574
Publication Date(Web):March 12, 2012
DOI:10.1021/ed200565h
In contrast to statements in a recent article in this Journal, the bonding electrons in methane can be properly described in terms of localized electrons, and photoelectron spectroscopy does not indicate otherwise. There is no good reason to abandon the description of the chemical bonds of methane in terms of localized bonds formed by covalent linking of hybrid orbitals.Keywords: Covalent Bonding; First-Year Undergraduate/General; Lewis Structures; Misconceptions/Discrepant Events; MO Theory; Physical Chemistry; Quantum Chemistry; Upper-Division Undergraduate; Valence Bond Theory; VSEPR Theory;
Co-reporter:Aleksandr V. Marenich, Wendu Ding, Christopher J. Cramer, and Donald G. Truhlar
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 11) pp:1437-1442
Publication Date(Web):May 6, 2012
DOI:10.1021/jz300416r
First and second dissociation constants (pKa values) of oxalic acid, malonic acid, and adipic acid were computed by using a number of theoretical protocols based on density functional theory and using both continuum solvation models and mixed discrete–continuum solvation models. We show that fully implicit solvation models (in which the entire solvent is represented by a dielectric continuum) fail badly for dicarboxylic acids with mean unsigned errors (averaged over six pKa values) of 2.4–9.0 log units, depending on the particular implicit model used. The use of water–solute clusters and accounting for multiple conformations in solution significantly improve the performance of both generalized Born solvation models and models that solve the nonhomogeneous dielectric Poisson equation for bulk electrostatics. The four most successful models have mean unsigned errors of only 0.6–0.8 log units.Keywords: dicarboxylic acid; dielectric continuum; generalized Born; PCM; SM8; SM8AD; SMD; solvation model;
Co-reporter:Prasenjit Seal, Ewa Papajak, and Donald G. Truhlar
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 2) pp:264-271
Publication Date(Web):January 3, 2012
DOI:10.1021/jz201546e
We estimated rate constants for the hydrogen abstraction from carbon-3 of 1-butanol by hydroperoxyl radical, a critically important reaction in the combustion of biofuel. We employed the recently developed multi-structural variational transition-state theory (MS-VTST), which utilizes a multifaceted dividing surface that allows us to include the contributions of multiple structures for reacting species and transition states. First, multiconfigurational Shepard interpolation—based on molecular-mechanics-guided interpolation of electronic-structure Hessian data obtained by the M08 HX/jun-cc-pVTZ electronic model chemistry—was used to obtain the portion of the potential energy surface needed for single-structure variational transition-state theory rate constants including multidimensional tunneling; then, the M08-HX/MG3S electronic model chemistry was used to calculate multi-structural torsional anharmonicity factors to complete the MS-VTST rate constant calculations. The lowest-energy structures of the transition state have strongly bent hydrogen bonds. Our results indicate that neglect of multi-structural anharmonicity would lead to errors of factors of 0.3, 46, and 171 at 200, 1000, and 2400 K for this reaction.Keywords: barrier height; butanol; combustion chemistry; hydrogen abstraction; hydrogen bonds; multi-structural variational transition state theory; torsional anharmonicity;
Co-reporter:Oksana Tishchenko and Donald G. Truhlar
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 19) pp:2834-2839
Publication Date(Web):September 19, 2012
DOI:10.1021/jz3011817
This Letter presents benchmark results for the transition-state geometry and classical barrier height for the hydrogen atom abstraction from phenol by an organic peroxyl radical. We use multireference Møller–Plesset perturbation theory (MRMP2) based on a complete active space self-consistent field (CASSCF) wave function with a previously defined well-balanced nom-CPO+π active space and a triple-ζ one-electron basis set for the benchmark calculations, including full geometry optimization of the saddle point at the MRMP2 level. The classical barrier height for the abstraction reaction by the methylperoxyl radical is found to be 7.4 kcal/mol. A variety of density functionals are tested for their ability to reproduce the benchmark calculations for this reaction to provide guidance for selecting a reliable density functional in future calculations of larger systems involving phenolic antioxidants. The best-performing density functional is M05, and other functionals with above-average performance (for both transition-state geometry and transition-state barrier height) are B1LYP, B3LYP, B98, B97-1, and M06.Keywords: density functionals; hydrogen atom abstraction; MRMP2; phenolic antioxidant; reaction barrier height; transition-state geometry;
Co-reporter:Sijie Luo, Yan Zhao, and Donald G. Truhlar
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 20) pp:2975-2979
Publication Date(Web):September 26, 2012
DOI:10.1021/jz301182a
A notorious failing of approximate exchange–correlation functionals when applied to problems involving catalysis has been the inability of most local functionals to predict the correct adsorption site for CO on metal surfaces or to simultaneously predict accurate surface formation energies and adsorption energies for transition metals. By adding the kinetic energy density τ to the density functional, the revTPSS density functional was shown recently to achieve a balanced description of surface energies and adsorption energies. Here, we show that the older M06-L density functional, also containing τ, provides improved surface formation energies and CO adsorption energies over revTPSS for five transition metals and correctly predicted the on-top/hollow site adsorption preferences for four of the five metals, which was not achieved by most other local functionals. Because M06-L was entirely designed on the basis of atomic and molecular energies, its very good performance is a confirmation of the reasonableness of its functional form. Two GGA functionals with an expansion in the reduced gradient that is correct through second order, namely, SOGGA and SOGGA11, were also tested and found to produce the best surface formation energies of all tested GGA functionals, although they significantly overestimate the adsorption energies.Keywords: chemisorption; electronic structure theory; hollow sites versus on-top sites; meta-generalized gradient approximation;
Co-reporter:Tao Yu, Jingjing Zheng, and Donald G. Truhlar
The Journal of Physical Chemistry A 2012 Volume 116(Issue 1) pp:297-308
Publication Date(Web):November 29, 2011
DOI:10.1021/jp209146b
We propose a new formulation of variational transition state theory called multipath variational transition state theory (MP-VTST). We employ this new formulation to calculate the forward and reverse thermal rate constant of the 1,4-hydrogen shift isomerization of the 2-cyclohexylethyl radical in the gas phase. First, we find and optimize all the local-minimum-energy structures of the reaction, product, and transition state. Then, for the lowest-energy transition state structures, we calculate the reaction path by using multiconfiguration Shepard interpolation (MSCI) method to represent the potential energy surface, and, from this representation, we also calculate the ground-state vibrationally adiabatic potential energy curve, the reaction-path curvature vector, and the generalized free energy of activation profile. With this information, the path-averaged generalized transmission coefficients ⟨γ⟩ are evaluated. Then, thermal rate constant containing the multiple-structure anharmonicity and torsional anharmonicity effects is calculated using multistructural transition state theory (MS-TST). The final MP-VTST thermal rate constant is obtained by multiplying kMS-TMS-TST by ⟨γ⟩. In these calculations, the M06 density functional is utilized to compute the energy, gradient, and Hessian at the Shepard points, and the M06-2X density functional is used to obtain the structures (conformers) of the reactant, product, and the saddle point for computing the multistructural anharmonicity factors.
Co-reporter:Xuefei Xu, Tao Yu, Ewa Papajak, and Donald G. Truhlar
The Journal of Physical Chemistry A 2012 Volume 116(Issue 43) pp:10480-10487
Publication Date(Web):September 28, 2012
DOI:10.1021/jp307504p
We calculated the forward and reverse rate constants of the hydrogen abstraction reaction from carbon-2 of 2-methyl-1-propanol by hydroperoxyl radical over the temperature range 250–2400 K by using multistructural canonical variational transition state theory (MS-CVT) including both multiple-structure and torsional potential anharmonicity effects by the multistructural torsional anharmonicity (MS-T) method. In these calculations, multidimensional tunneling (MT) probabilities used to compute the tunneling transmission coefficients were evaluated by the small-curvature tunneling (SCT) approximation. Comparison with the rate constants obtained by the single-structural harmonic oscillator (SS-HO) approximation shows that multistructural anharmonicity increases the forward rate constants for all temperatures, but the reverse rate constants are reduced for temperatures lower than 430 K and increased for higher temperatures. The neglect of multistructural torsional anharmonicity would lead to errors of factors of 1.5, 8.8, and 13 at 300, 1000, and 2400 K, respectively, for the forward reaction, and would lead to errors of factors of 0.76, 3.0, and 6.0, respectively, at these temperatures for the reverse reaction.
Co-reporter:I. M. Alecu, Jingjing Zheng, Ewa Papajak, Tao Yu, and Donald G. Truhlar
The Journal of Physical Chemistry A 2012 Volume 116(Issue 50) pp:12206-12213
Publication Date(Web):November 14, 2012
DOI:10.1021/jp308460y
Multistructural canonical variational transition-state theory with small-curvature multidimensional tunneling (MS-CVT/SCT) is employed to calculate thermal rate constants for hydrogen-atom abstraction from carbon-1 of n-butanol by the hydroperoxyl radical over the temperature range 250–2000 K. The M08-SO hybrid meta-GGA density functional was validated against CCSD(T)-F12a explicitly correlated wave function calculations with the jul-cc-pVTZ basis set. It was then used to compute the properties of all stationary points and the energies and Hessians of a few nonstationary points along the reaction path, which were then used to generate a potential energy surface by the multiconfiguration Shepard interpolation (MCSI) method. The internal rotations in the transition state for this reaction (like those in the reactant alcohol) are strongly coupled to each other and generate multiple stable conformations, which make important contributions to the partition functions. It is shown that neglecting to account for the multiple-structure effects and torsional potential anharmonicity effects that arise from the torsional modes would lead to order-of-magnitude errors in the calculated rate constants at temperatures of interest in combustion.
Co-reporter:Roberto Peverati and Donald G. Truhlar
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 1) pp:117-124
Publication Date(Web):December 12, 2011
DOI:10.1021/jz201525m
Local approximations to the exchange-correlation functional are of special interest because of their cost advantages and their useful accuracy for efficient calculations on systems (such as many transition metal catalysts) with significant multiconfigurational wave function character. We present a meta-GGA exchange-correlation functional, called M11-L, that employs dual-range local exchange to provide broad accuracy for both single-configurational and multiconfigurational molecules and for solid-state lattice constants. Also notable is the high accuracy (for a local functional) for chemical reaction barrier heights. The mean unsigned error on a broad chemistry database of 338 energetic data is lower than that for any other known functional, even hybrid functionals and range-separated hybrid functionals. This success shows that the dependence of the exchange energy density on interelectronic distance is quite different at short-range and long-range, and it establishes a new standard for the limit of what can be achieved with a local exchange-correlation functional.Keywords: bond dissociation energy; density functional theory; energy of reaction; exchange- correlation functionals;
Co-reporter:Aleksr V. Marenich;Abir Majumdar;Michelle Lenz; Christopher J. Cramer; Donald G. Truhlar
Angewandte Chemie International Edition 2012 Volume 51( Issue 51) pp:12810-12814
Publication Date(Web):
DOI:10.1002/anie.201206012
Co-reporter:Santhanamoorthi Nachimuthu, Jiali Gao, Donald G. Truhlar
Chemical Physics 2012 400() pp: 8-12
Publication Date(Web):25 May 2012
DOI:10.1016/j.chemphys.2012.01.014
We present benchmark calculations of nine selected points on potential energy surfaces describing proton transfer processes in three model systems, H5O2+, CH3OH…H+…OH2, and CH3COOH…OH2. The calculated relative energies of these geometries are compared to those calculated by various wave function and density functional methods, including the polarized molecular orbital (PMO) model recently developed in our research group and other semiempirical molecular orbital methods. We found that the SCC-DFTB and PMO methods (the latter available so far only for molecules consisting of only O and H and therefore only for the first of the three model systems) give results that are, on average, within 2 kcal/mol of the benchmark results. Other semiempirical molecular orbital methods have mean unsigned errors (MUEs) of 3–8 kcal/mol, local density functionals have MUEs in the range 0.7–3.7 kcal/mol, and hybrid density functionals have MUEs of only 0.3–1.0 kcal/mol, with the best density functional performance obtained by hybrid meta-GGAs, especially M06 and PW6B95.Graphical abstractHighlights► We present benchmark calculations of energies of complexation and barriers for proton transfer to water. ► Benchmark calculations are used to test methods suitable for application to large and complex systems. ► Methods tested include hybrid meta-GGAs, M06-L, PW6B95, SOGGA11, MP2, SCC-DFTB, PMO, and NDDO.
Co-reporter:Varinia S. Bernales, Aleksandr V. Marenich, Renato Contreras, Christopher J. Cramer, and Donald G. Truhlar
The Journal of Physical Chemistry B 2012 Volume 116(Issue 30) pp:9122-9129
Publication Date(Web):June 26, 2012
DOI:10.1021/jp304365v
The quantum mechanical SMD continuum universal solvation model can be applied to predict the free energy of solvation of any solute in any solvent following specification of various macroscopic solvent parameters. For three ionic liquids where these descriptors are readily available, the SMD solvation model exhibits a mean unsigned error of 0.48 kcal/mol for 93 solvation free energies of neutral solutes and a mean unsigned error of 1.10 kcal/mol for 148 water-to-IL transfer free energies. Because the necessary solvent parameters are not always available for a given ionic liquid, we determine average values for a set of ionic liquids over which measurements have been made in order to define a generic ionic liquid solvation model, SMD-GIL. Considering 11 different ionic liquids, the SMD-GIL solvation model exhibits a mean unsigned error of 0.43 kcal/mol for 344 solvation free energies of neutral solutes and a mean unsigned error of 0.61 kcal/mol for 431 water-to-IL transfer free energies. As these errors are similar in magnitude to those typically observed when applying continuum solvation models to ordinary liquids, we conclude that the SMD universal solvation model may be applied to ionic liquids as well as ordinary liquids.
Co-reporter:Tao Yu, Jingjing Zheng and Donald G. Truhlar
Chemical Science 2011 vol. 2(Issue 11) pp:2199-2213
Publication Date(Web):10 Aug 2011
DOI:10.1039/C1SC00225B
We present a new formulation of variational transition state theory (VTST) called multi-structural VTST (MS-VTST) and the use of this to calculate the rate constant for the 1,4-hydrogen shift isomerization reaction of 1-pentyl radical and that for the reverse reaction. MS-VTST uses a multi-faceted dividing surface and provides a convenient way to include the contributions of many structures (typically conformers) of the reactant and the transition state in rate constant calculations. In this particular application, we also account for the torsional anharmonicity. We used the multi-configuration Shepard interpolation method to efficiently generate a semi-global portion of the potential energy surface from a small number of high-level electronic structure calculations using the M06 density functional in order to compute the energies and Hessians of Shepard points along a reaction path. The M06-2X density functional was used to calculate the multi-structural anharmonicity effect, including all of the structures of the reactant, product and transition state. To predict the thermal rate constant, VTST calculations were performed to obtain the canonical variational rate constant over the temperature range 200–2000 K. A transmission coefficient is calculated by the multidimensional small-curvature tunneling (SCT) approximation. The final MS-CVT/SCT thermal rate constant was determined by combining a reaction rate calculation in the single-structural harmonic oscillator approximation (including tunneling) with the multi-structural anharmonicity torsional factor. The calculated forward rate constant agrees very well with experimentally-based evaluations of the high-pressure limit for the temperature range 300–1300 K, although it is a factor of 2.5–3.0 lower than the single-structural harmonic oscillator approximation over this temperature range. We anticipate that MS-VTST will be generally useful for calculating the reaction rates of complex molecules with multiple torsions.
Co-reporter:Aleksandr V. Marenich, Christopher J. Cramer, Donald G. Truhlar, Ciro A. Guido, Benedetta Mennucci, Giovanni Scalmani and Michael J. Frisch
Chemical Science 2011 vol. 2(Issue 11) pp:2143-2161
Publication Date(Web):05 Aug 2011
DOI:10.1039/C1SC00313E
We present a unified treatment of solvatochromic shifts in liquid-phase absorption spectra, and we develop a self-consistent state-specific vertical excitation model (called VEM) for electronic excitation in solution. We discuss several other approaches to calculate vertical excitations in solution as an approximation to VEM. We illustrate these methods by presenting calculations of the solvatochromic shifts of the lowest excited states of several solutes (acetone, acrolein, coumarin 153, indolinedimethine-malononitrile, julolidine-malononitrile, methanal, methylenecyclopropene, and pyridine) in polar and nonpolar solvents (acetonitrile, cyclohexane, dimethyl sulfoxide, methanol, n-hexane, n-pentane, and water) using implicit solvation models combined with configuration interaction based on single excitations and with time-dependent density functional theory.
Co-reporter:Ruifang Li, Yan Zhao and Donald G. Truhlar
Chemical Communications 2011 vol. 47(Issue 8) pp:2357-2359
Publication Date(Web):16 Dec 2010
DOI:10.1039/C0CC02845B
Adequate polarization functions reduce the error of density functional theory (DFT) for the heat of reaction for CF4 + SiCl4 from ∼9–12 kcal mol−1 to ∼2–4 kcal mol−1, and using an improved density functional further reduces it to ∼1 kcal mol−1. This reaction was previously identified as a stumbling block for DFT, but we show that the problem with the previous calculations was not DFT but rather inadequate basis sets to account for intramolecular charge polarization.
Co-reporter:Boris B. Averkiev and Donald G. Truhlar
Catalysis Science & Technology 2011 vol. 1(Issue 8) pp:1526-1529
Publication Date(Web):12 Sep 2011
DOI:10.1039/C1CY00227A
The Gibbs energy of reaction of oxidative addition of ammonia to an iridium complex in diethyl ether was calculated by seven density functional methods, in particular B3LYP, PBE, CAM-B3LYP, M05, M06, M06-L, and ωB97X. The calculated free energies, based on geometry optimization and frequency calculations in both the gas phase and solution, were compared with the experimental result, −1.3 kcal mol−1, obtained by Hartwig and coworkers. The M06-L method gives the best result: −1.4 kcal/mol.
Co-reporter:Roberto Peverati and Donald G. Truhlar
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 12) pp:3983-3994
Publication Date(Web):October 27, 2011
DOI:10.1021/ct2006192
We present a natural cubic spline implementation of the exchange enhancement factor as a function of the reduced density gradient, and we demonstrate its performance by replicating the results of common GGA functionals. We also investigate the effect on the accuracy of various calculated properties of changing the shape of the exchange enhancement factor and an analogous factor for correlation. The properties considered are main group atomization energies, ionization potentials, electron affinities, proton affinities, alkyl bond dissociation energies, difficult hydrocarbon cases, barrier heights for chemical reactions, noncovalent interactions, atomic energies, metal bond energies, and main group bond lengths.
Co-reporter:Xuefei Xu and Donald G. Truhlar
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 9) pp:2766-2779
Publication Date(Web):June 30, 2011
DOI:10.1021/ct200234r
For molecules containing the fourth-period element arsenic, we test (i, ii) the accuracy of all-electron (AE) basis sets from the def2-xZVP and ma-xZVP series (where xZ is S, TZ, or QZ), (iii) the accuracy of the 6-311G series of AE basis sets with additional polarization and diffuse functions, and (iv) the performance of effective core potentials (ECPs). The first set of tests involves basis-set convergence studies with eleven density functionals for five cases: equilibrium dissociation energy (De) of As2, vertical ionization potential (VIP) of As2, IP of As, acid dissociation of H3AsO4, and De of FeAs. A second set of tests involves the same kinds of basis-set convergence studies for the VIP and De values of As3 and As4 clusters. Both relativistic and nonrelativistic calculations are considered, including in each case both AE calculations and calculations with ECPs. Convergence and accuracy are assessed by comparing to relativistic AE calculations with the cc-pV5Z-DK or ma-cc-pV5Z-DK basis and to nonrelativistic AE calculations with the cc-pV5Z or ma-cc-pV5Z basis. The primary objective of this study is to evaluate the abilities of ECPs with both their recommended basis sets and other basis sets to reproduce the results of all-electron relativistic calculations. The performance of the def2 and ma series basis sets is consistent with their sizes, and quadruple-ζ basis sets are the best. The def2-TZVP basis set performs better than most of the 6-311G series basis sets, which are the most commonly used basis sets in the previous studies of arsenic compounds. However, relativistic def2-TZVP calculations are not recommended. The large-core ECPs, which are the only available ECPs for arsenic in the popular Gaussian program, have average errors of 9–12 kcal/mol for the arsenic systems studied; therefore, these ECPs are not recommended. The triple-ζ small-core relativistic ECP (RECP) basis set cc-pVTZ-PP is found to have performance better than that of the def2-TZVP basis set, and it is highly recommended for arsenic-containing systems. The double-ζ RECP basis set ma-sc-SVP is recommended for large arsenic systems for which the def2-TZVP and cc-pVTZ-PP basis sets are unaffordable, if a basis-set error of ∼2 kcal/mol can be tolerated.
Co-reporter:Anant D. Kulkarni and Donald G. Truhlar
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 7) pp:2325-2332
Publication Date(Web):May 27, 2011
DOI:10.1021/ct200188n
We assess the performance of density functional theory (DFT) and Møller–Plesset second-order perturbation theory (MP2) for predicting structural parameters in Ru complexes, in particular, a Ru(IV) allyl dicationic complex with the formula [Ru(η5-Cp*)(η3-CH2CHCHC6H5)(NCCH3)2]2+ and the molecules RuO4 and Ru(C2O4)2(H2O)2–, where Cp* denotes C5Me5 and Me denotes methyl. The density functionals studied are B3LYP, B3PW91, M05, M06, M06-L, MOHLYP, MPW3LYP, PBE0, PW6B95, SOGGA, τHCTHhyb, ωB97X, and ωB97X-D, in combination with three different basis sets, namely, LANL2DZ, def2-SVP, and def2-TZVP. The theoretically computed Ru–C distances corresponding to the phenylallyl complex are especially well predicted by the SOGGA (pure DFT) and ωB97X-D (DFT plus an empirical molecular mechanics term) methods. This contrasts with an article in this Journal [Calhorda, M. J., Pregosin, P. S., and Veiros, L. F. J. Chem. Theory Comput. 2007, 3, 665−670] in which it was found that DFT cannot account for these Ru–C distances. Averaging over four Ru–C distances in the allyl complex and three unique Ru–O distances in RuO4 and Ru(C2O4)2(H2O)2–, the SOGGA and ωB97X-D methods have both a smaller mean unsigned error than MP2 and the same maximum error. The M06, PW6B95, PBE0, M06-L, and ωB97X density functionals also have a smaller or the same mean unsigned error as MP2.
Co-reporter:Ewa Papajak, Jingjing Zheng, Xuefei Xu, Hannah R. Leverentz, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 10) pp:3027-3034
Publication Date(Web):August 5, 2011
DOI:10.1021/ct200106a
We present a perspective on the use of diffuse basis functions for electronic structure calculations by density functional theory and wave function theory. We especially emphasize minimally augmented basis sets and calendar basis sets. We base our conclusions on our previous experience with commonly computed quantities, such as bond energies, barrier heights, electron affinities, noncovalent (van der Waals and hydrogen bond) interaction energies, and ionization potentials, on Stephens et al.’s results for optical rotation and on our own new calculations (presented here) of polarizabilities and of potential energy curves of van der Waals complexes. We emphasize the benefits of partial augmentation of the higher-zeta basis sets in preference to full augmentation at a lower ζ level. Benefits and limitations of the use of fully, partially, and minimally augmented basis sets are reviewed for different electronic structure methods and molecular properties. We have found that minimal augmentation is almost always enough for density functional theory (DFT) when applied to ionization potentials, electron affinities, atomization energies, barrier heights, and hydrogen-bond energies. For electric dipole polarizabilities, we find that augmentation beyond minimal has an average effect of 8% at the polarized triple-ζ level and 5% at the polarized quadruple-ζ level. The effects are larger for potential energy curves of van der Waals complexes. The effects are also larger for wave function theory (WFT). Even for WFT though, full augmentation is not needed for most purposes, and a level of augmentation between minimal and full is optimal for most problems. The calendar basis sets named after the months provide a convergent sequence of partially augmented basis sets that can be used for such calculations. The jun-cc-pV(T+d)Z basis set is very useful for MP2-F12 calculations of barrier heights and hydrogen bond strengths.
Co-reporter:Xuefei Xu, I. M. Alecu, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 6) pp:1667-1676
Publication Date(Web):April 26, 2011
DOI:10.1021/ct2001057
We introduce a new database called TSG48 containing 48 transition state geometrical data (in particular, internuclear distances in transition state structures) for 16 main group reactions. The 16 reactions are the 12 reactions in the previously published DBH24 database (which includes hydrogen transfer reactions, heavy-atom transfer reactions, nucleophilic substitution reactions, and association reactions plus one unimolecular isomerization) plus four H-transfer reactions in which a hydrogen atom is abstracted by the methyl or hydroperoxyl radical from the two different positions in methanol. The data in TSG48 include data for four reactions that have previously been treated at a very high level in the literature. These data are used to test and validate methods that are affordable for the entire test suite, and the most accurate of these methods is found to be the multilevel BMC-CCSD method. The data that constitute the TSG48 database are therefore taken to consist of these very high level calculations for the four reactions where they are available and BMC-CCSD calculations for the other 12 reactions. The TSG48 database is used to assess the performance of the eight Minnesota density functionals from the M05–M08 families and 26 other high-performance and popular density functionals for locating transition state geometries. For comparison, the MP2 and QCISD wave function methods have also been tested for transition state geometries. The MC3BB and MC3MPW doubly hybrid functionals and the M08-HX and M06-2X hybrid meta-GGAs are found to have the best performance of all of the density functionals tested. M08-HX is the most highly recommended functional due to the excellent performance for all five subsets of TSG48, as well as having a lower cost when compared to doubly hybrid functionals. The mean absolute errors in transition state internuclear distances associated with breaking and forming bonds as calculated by the B2PLYP, MP2, and B3LYP methods are respectively about 2, 3, and 5 times larger than those calculated by MC3BB and M08-HX.
Co-reporter:Luke Fiedler, Jiali Gao, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 4) pp:852-856
Publication Date(Web):March 3, 2011
DOI:10.1021/ct1006373
The objective of this paper is to examine the minimal requirements for obtaining semiquantitative polarizabilities of molecules, in order to provide a well-founded starting point for a new semiempirical molecular orbital formulation that is more suitable than presently available methods for simulating electronic polarization effects. For this purpose, we present polarizability calculations for 38 molecules with 36 basis sets, including many unconventional ones, and five semiempirical molecular orbital theories based on neglect of diatomic differential overlap. We conclude that two basis sets are particularly promising to serve as bases for semiempirical improvement, namely, STO-3G(,P), in which diffuse p functions are added to all hydrogens, and 3-(21,3,21)G, in which a minimal basis set is augmented with one extra s function on every atom. We especially recommend the former because all intra-atomic overlap integrals are zero by symmetry, which makes it a better candidate for neglect-of-differential-overlap treatments.
Co-reporter:Peng Zhang, Luke Fiedler, Hannah R. Leverentz, Donald G. Truhlar, and Jiali Gao
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 4) pp:857-867
Publication Date(Web):March 3, 2011
DOI:10.1021/ct100638g
We present a new semiempirical molecular orbital method based on neglect of diatomic differential overlap. This method differs from previous NDDO-based methods in that we include p orbitals on hydrogen atoms to provide a more realistic modeling of polarizability. As in AM1-D and PM3-D, we also include damped dispersion. The formalism is based on the original MNDO one, but in the process of parametrization we make some specific changes to some of the functional forms. The present article is a demonstration of the capability of the new approach, and it presents a successful parametrization for compounds composed only of hydrogen and oxygen atoms, including the important case of water clusters.
Co-reporter:Ewa Papajak and Donald G. Truhlar
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 1) pp:10-18
Publication Date(Web):December 9, 2010
DOI:10.1021/ct1005533
We present sets of convergent, partially augmented basis set levels corresponding to subsets of the augmented “aug-cc-pV(n+d)Z” basis sets of Dunning and co-workers. We show that for many molecular properties a basis set fully augmented with diffuse functions is computationally expensive and almost always unnecessary. On the other hand, unaugmented cc-pV(n+d)Z basis sets are insufficient for many properties that require diffuse functions. Therefore, we propose using intermediate basis sets. We developed an efficient strategy for partial augmentation, and in this article, we test it and validate it. Sequentially deleting diffuse basis functions from the “aug” basis sets yields the “jul”, “jun”, “may”, “apr”, etc. basis sets. Tests of these basis sets for Møller−Plesset second-order perturbation theory (MP2) show the advantages of using these partially augmented basis sets and allow us to recommend which basis sets offer the best accuracy for a given number of basis functions for calculations on large systems. Similar truncations in the diffuse space can be performed for the aug-cc-pVxZ, aug-cc-pCVxZ, etc. basis sets.
Co-reporter:Yan Zhao and Donald G. Truhlar
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 3) pp:669-676
Publication Date(Web):February 3, 2011
DOI:10.1021/ct1006604
The present study compares the accuracy of 30 density functionals for four databases of reaction energies studied recently by Grimme and co-workers. For 20 of the density functionals, the calculations are new, and the calculations are compared to previous work for the other 10. We present the results in detail for 11 of the functionals and as mean unsigned errors for the others. The results presented in detail are for the seven most recent Minnesota functionals (M05-2X, M06-L, M06-HF, M06, M06-2X, M08-HX, and M08-SO), three range-separated functionals (HSE, LC-ωPBE, and ωB97X-D), and one dispersion-corrected global hybrid generalized gradient approximation (B97-D); the other functionals include five dispersion-corrected functionals and their uncorrected analogs, eight high-performing functionals on a recent catalytic-energies test, and the TPSSh functional because it is of special interest to compare its performance to that of M08-SO. Three of the four databases contain a total of 21 rearrangement reaction energies and 13 diverse dissociation or association energies, and the fourth contains three dissociation reaction energies of alkali metal clusters and three dissociation reaction energies of alkali-metal-cation−benzene complexes. The results are especially promising for the Minnesota hybrid meta-GGA functionals and the ωB97X-D, B2PLYP-D, and HSE functionals.
Co-reporter:Jingjing Zheng, Tao Yu and Donald G. Truhlar
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 43) pp:19318-19324
Publication Date(Web):10 Oct 2011
DOI:10.1039/C1CP21829H
The C–H bond dissociation processes of n-hexane and isohexane involve 23 and 13 conformational structures, respectively in the parent molecules and 14–45 conformational structures in each of the seven isomeric products that we studied. Here we use the recently developed multi-structural (MS) thermodynamics method and CCSD(T)-F12a/jul-cc-pVTZ//M06-2X/6-311+G(2df,2p) potential energy surfaces to calculate the enthalpy, entropy, and heat capacity of n-hexane, isohexane, and seven of the possible radical products of dissociation of C–H bonds. We compare our calculations with the limited experimental data and with values obtained by group additivity fits used to extend the experimental data. This work shows that using the MS method involving a full set of structural isomers with density functional geometries, scaled density functional frequencies, and coupled cluster single-point energies can predict thermodynamic functions of complex molecules and bond dissociation reactions with chemical accuracy. The method should be useful to obtain thermodynamic data for complex molecules for which such data has not been measured and to obtain thermodynamic data at temperatures outside the temperature range where measurements are available.
Co-reporter:Raphael F. Ribeiro, Aleksandr V. Marenich, Christopher J. Cramer and Donald G. Truhlar
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 23) pp:10908-10922
Publication Date(Web):12 May 2011
DOI:10.1039/C0CP02784G
We present M06-2X density functional calculations of the chloroform/water partition coefficients of cytosine, thymine, uracil, adenine, and guanine and calculations of the free energies of association of selected unsubstituted and alkylated nucleotide base pairs in chloroform and water. Both hydrogen bonding and π–π stacking interactions are considered. Solvation effects are treated using the continuum solvent models SM8, SM8AD, and SMD, including geometry optimization in solution. Comparison of theoretical results with available experimental data indicates that all three of these solvation models predict the chloroform–water partition coefficients for the studied nucleobases qualitatively well, with mean unsigned errors in the range of 0.4–1.3 log units. All three models correctly predict the preference for hydrogen bonding over stacking for nucleobase pairs solvated in chloroform, and SM8, SM8AD, and SMD show similar accuracy in predicting the corresponding free energies of association. The agreement between theory and experiment for the association free energies of the dimers in water is more difficult to assess, as the relevant experimental data are indirect. Theory predicts that the stacking interaction of nucleobases in water is more favorable than hydrogen bonding for only two out of three tested hetero-dimers.
Co-reporter:Jingjing Zheng, Tao Yu, Ewa Papajak, I. M. Alecu, Steven L. Mielke and Donald G. Truhlar
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 23) pp:10885-10907
Publication Date(Web):11 May 2011
DOI:10.1039/C0CP02644A
Many methods for correcting harmonic partition functions for the presence of torsional motions employ some form of one-dimensional torsional treatment to replace the harmonic contribution of a specific normal mode. However, torsions are often strongly coupled to other degrees of freedom, especially other torsions and low-frequency bending motions, and this coupling can make assigning torsions to specific normal modes problematic. Here, we present a new class of methods, called multi-structural (MS) methods, that circumvents the need for such assignments by instead adjusting the harmonic results by torsional correction factors that are determined using internal coordinates. We present three versions of the MS method: (i) MS-AS based on including all structures (AS), i.e., all conformers generated by internal rotations; (ii) MS-ASCB based on all structures augmented with explicit conformational barrier (CB) information, i.e., including explicit calculations of all barrier heights for internal-rotation barriers between the conformers; and (iii) MS-RS based on including all conformers generated from a reference structure (RS) by independent torsions. In the MS-AS scheme, one has two options for obtaining the local periodicity parameters, one based on consideration of the nearly separable limit and one based on strongly coupled torsions. The latter involves assigning the local periodicities on the basis of Voronoi volumes. The methods are illustrated with calculations for ethanol, 1-butanol, and 1-pentyl radical as well as two one-dimensional torsional potentials. The MS-AS method is particularly interesting because it does not require any information about conformational barriers or about the paths that connect the various structures.
Co-reporter:Yan Zhao, Donald G. Truhlar
Chemical Physics Letters 2011 Volume 502(1–3) pp:1-13
Publication Date(Web):18 January 2011
DOI:10.1016/j.cplett.2010.11.060
Abstract
We discuss and review selected recent applications and validations of the Minnesota density functionals, especially the M06 family, emphasizing nanochemistry, organic, inorganic, and biological chemistry, and catalysis and highlighting the broad accuracy of these functionals as compared to previous popular functionals for thermochemistry, kinetics, and noncovalent interactions.
Co-reporter:Jeremy O. B. Tempkin, Hannah R. Leverentz, Bo Wang, and Donald G. Truhlar
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 17) pp:2141-2144
Publication Date(Web):August 9, 2011
DOI:10.1021/jz200893t
The electrostatically embedded many-body (EE-MB) method has been very successful for calculating energies of molecular clusters. Here, we introduce screened charges in the EE-MB method and evaluate the accuracy of the resulting method for calculating the binding energy for five water hexamers. The screened EE-MB method shows dramatic improvement over the unscreened method. The mean unsigned deviation of the screened EE-MB binding energies relative to unfragmented calculations on the entire cluster is 0.60 kcal/mol at the pairwise additive (PA) level of approximation and 0.24 kcal/mol at the three-body (3B) level, as compared to mean unsigned deviations of 1.32 (PA) and 0.54 (3B) kcal/mol with unscreened charges. The mean unsigned percentage deviations with screened embedding are only 1.1% (PA) and 0.5% (3B). The high accuracy obtained with the very affordable and quadratically scaling PA method is very encouraging and opens the door to more accurate simulations on complex systems.Keywords: electrostatic embedding; fragment-based methods; pairwise additive approximation; screened charges; three-body approximation;
Co-reporter:Roberto Peverati, Yan Zhao, and Donald G. Truhlar
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 16) pp:1991-1997
Publication Date(Web):June 29, 2011
DOI:10.1021/jz200616w
We present a new generalized gradient approximation (GGA) to the exchange-correlation functional of density functional theory, called SOGGA11, that has better overall performance for a broad chemical database than any previously available GGA and in addition is correct to second order (SO) in the density-gradient. It provides excellent accuracy for predicting molecular bond lengths.Keywords: bond dissociation energy; density functional theory; energy of reaction; exchange-correlation functionals;
Co-reporter:Jingjing Zheng;Xuefei Xu
Theoretical Chemistry Accounts 2011 Volume 128( Issue 3) pp:295-305
Publication Date(Web):2011 February
DOI:10.1007/s00214-010-0846-z
We propose an extension of the basis sets proposed by Ahlrichs and coworkers at Karlsruhe (these basis sets are designated as the second-generation default or “def2” basis sets in the Turbomole program). The Karlsruhe basis sets are very appealing because they constitute balanced and economical basis sets of graded quality from partially polarized double zeta to heavily polarized quadruple zeta for all elements up to radon (Z = 86). The extension consists of adding a minimal set of diffuse functions to a subset of the elements. This yields basis sets labeled minimally augmented or with “ma” as a prefix. We find that diffuse functions are not quite as important for the def2 basis sets as they are for Pople basis sets, but they are still necessary for good results on barrier heights and electron affinities. We provide assessments and validations of this extension for a variety of data sets and representative cases. We recommend the new ma-TZVP basis set for general-purpose applications of density functional theory.
Co-reporter:Roberto Peverati and Donald G. Truhlar
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 21) pp:2810-2817
Publication Date(Web):October 18, 2011
DOI:10.1021/jz201170d
The Minnesota family of exchange–correlation functionals, which consists of meta generalized gradient approximations (meta-GGAs) and global-hybrid meta-GGAs, has been successful for density functional calculations of molecular structure, properties, and thermochemistry, kinetics, noncovalent interactions, and spectroscopy. Here, we generalize the functional form by using range-separated hybrid meta-GGA exchange. We optimize a functional, called M11, with the new form against a broad database of energetic chemical properties and compare its performance to that of several other functionals, including previous Minnesota functionals. We require the percentage of Hartree–Fock exchange to be 100 at large interelectronic distance, and we find an optimum percentage of 42.8 at short range. M11 has good across-the-board performance and the smallest mean unsigned error over the whole test set of 332 data; it has especially good performance for main-group atomization energies, proton affinities, electron affinities, alkyl bond dissociation energies, barrier heights, noncovalent interaction energies, and charge-transfer electronic excitation.Keywords: bond dissociation energy; charge transfer; density functional theory; kinetic energy density;
Co-reporter:Sijie Luo, Ivan Rivalta, Victor Batista, and Donald G. Truhlar
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 20) pp:2629-2633
Publication Date(Web):September 26, 2011
DOI:10.1021/jz201077n
We employ noncollinear density functional theory to show that the low-spin state of Mn3 in a model of the oxygen-evolving complex of photosystem II avoids frustrated spin coupling by adopting a noncollinear arrangement of spins, thereby lowering the energy by 7 kcal/mol. The high-spin state also has noncollinear spins. The optimum self-consistent field solutions for this multinuclear oxomanganese complex correspond to states that cannot be described by the unrestricted Slater determinants used in Kohn–Sham collinear density functional methods. This kind of spin coupling can be important in many open-shell systems, and the conventional collinear spin interpretation of chemical bonding in such systems should be viewed with caution.Keywords: bioinorganic chemistry; bonding theory; density functional calculations; frustrated spins; magnetic state;
Co-reporter:I. M. Alecu and Donald G. Truhlar
The Journal of Physical Chemistry A 2011 Volume 115(Issue 13) pp:2811-2829
Publication Date(Web):March 15, 2011
DOI:10.1021/jp110024e
The reactions of CH3OH with the HO2 and CH3 radicals are important in the combustion of methanol and are prototypes for reactions of heavier alcohols in biofuels. The reaction energies and barrier heights for these reaction systems are computed with CCSD(T) theory extrapolated to the complete basis set limit using correlation-consistent basis sets, both augmented and unaugmented, and further refined by including a fully coupled treatment of the connected triple excitations, a second-order perturbative treatment of quadruple excitations (by CCSDT(2)Q), core−valence corrections, and scalar relativistic effects. It is shown that the M08-HX and M08-SO hybrid meta-GGA density functionals can achieve sub-kcal mol−1 agreement with the high-level ab initio results, identifying these functionals as important potential candidates for direct dynamics studies on the rates of these and homologous reaction systems.
Co-reporter:I. M. Alecu and Donald G. Truhlar
The Journal of Physical Chemistry A 2011 Volume 115(Issue 51) pp:14599-14611
Publication Date(Web):November 7, 2011
DOI:10.1021/jp209029p
Multistructural canonical variational-transition-state theory with multidimensional tunneling (MS-CVT/MT) is employed to calculate thermal rate constants for the abstraction of hydrogen atoms from both positions of methanol by the hydroperoxyl and methyl radicals over the temperature range 100–3000 K. The M08-HX hybrid meta-generalized gradient approximation density functional and M08-HX with specific reaction parameters, both with the maug-cc-pVTZ basis set, were validated in part 1 of this study (Alecu, I. M.; Truhlar, D. G. J. Phys. Chem. A2011, 115, 2811) against highly accurate CCSDT(2)Q/CBS calculations for the energetics of these reactions, and they are used here to compute the properties of all stationary points and the energies, gradients, and Hessians of nonstationary points along each considered reaction path. The internal rotations in some of the transition states are found to be highly anharmonic and strongly coupled to each other, and they generate multiple structures (conformations) whose contributions are included in the partition function. It is shown that the previous estimates for these rate constants used to build kinetic models for the combustion of methanol, some of which were based on transition state theory calculations with one-dimensional tunneling corrections and harmonic-oscillator approximations or separable one-dimensional hindered rotor treatments of torsions, are appreciably different than the ones presently calculated using MS-CVT/MT. The rate constants obtained from the best MS-CVT/MT calculations carried out in this study, in which the important effects of corner cutting due to small and large reaction path curvature are captured via a microcanonical optimized multidimensional tunneling (μOMT) treatment, are recommended for future refinement of the kinetic model for methanol combustion.
Co-reporter:Raphael F. Ribeiro, Aleksandr V. Marenich, Christopher J. Cramer, and Donald G. Truhlar
The Journal of Physical Chemistry B 2011 Volume 115(Issue 49) pp:14556-14562
Publication Date(Web):August 29, 2011
DOI:10.1021/jp205508z
We find that vibrational contributions to a solute’s free energy are in general insensitive to whether the solute vibrational frequencies are computed in the gas phase or in solution. In most cases, the difference is smaller than the intrinsic error in solvation free energies associated with the continuum approximation to solvation modeling, although care must be taken to avoid spurious results associated with limitations in the quantum-mechanical harmonic-oscillator approximation for very low-frequency molecular vibrations. We compute solute vibrational partition functions in aqueous and carbon tetrachloride solution and compare them to gas-phase molecular partition functions computed with the same level of theory and the same quasiharmonic approximation for the diverse and extensive set of molecules and ions included in the training set of the SMD continuum solvation model, and we find mean unsigned differences in vibrational contributions to the solute free energy of only about 0.2 kcal/mol. On the basis of these results and a review of the theory, we conclude, in contrast to previous work (Ho, J.; Klamt, H.; Coote, M. L. J. Phys. Chem. A 2010, 114, 13442), that using partition functions computed for molecules optimized in solution is a correct and useful approach for averaging over solute degrees of freedom when computing free energies of solutes in solution, and it is moreover recommended for cases where liquid and gas-phase solute structures differ appreciably or when stationary points present in liquid solution do not exist in the gas phase, for which we provide some examples. When gas-phase and solution-phase geometries and frequencies are similar, the use of gas-phase geometries and frequencies is a useful approximation.
Co-reporter:Hsiao-Ching Yang, Yen-Chin Huang, Yi-Kang Lan, Tien-Yau Luh, Yan Zhao, and Donald G. Truhlar
Organometallics 2011 Volume 30(Issue 15) pp:4196-4200
Publication Date(Web):July 8, 2011
DOI:10.1021/om200529m
As a long-standing puzzle, experimental observations reveal faster organophosphine dissociation in the olefin metathesis by Grubbs’s first-generation precatalyst (Gen I) than by the second-generation precatalyst (Gen II), but Gen I shows less catalytic activity. Here we show by electronic structure calculations with the M06-L density functional that carbene rotamer energetic effects are responsible for the inverse relation between organophosphine dissociation rate and catalytic activity. The carbene rotamer acts as a toggle switch, triggering the dissociative mechanism that produces the active catalyst. The slower catalyst production in Gen II as compared to Gen I is not a pure electronic effect but results from rotameric coupling to the dissociation coordinate speeding up Gen I dissociation more than Gen II dissociation. If organophosphine dissociation were to occur with fixed rotamer orientation, Gen II would be produced faster than Gen I, as originally expected. The rotameric energetics also contributes to the higher catalytic activity of the Gen II catalyst.
Co-reporter:Yongho Kim ; Jerry R. Mohrig
Journal of the American Chemical Society 2010 Volume 132(Issue 32) pp:11071-11082
Publication Date(Web):July 26, 2010
DOI:10.1021/ja101104q
Distinguishing between the concerted second-order mechanism for β-eliminations and nonconcerted mechanisms with discrete carbanion intermediates is very difficult experimentally, but the ability of quantum chemistry to find stationary points of the free-energy surface in liquid-phase solutions, even for complex reagents, provides a new tool for elucidating such mechanisms. Here we use liquid-phase density functional theory calculations to find transition states and intermediates on the free-energy surfaces of four base-initiated α,β-eliminations of acetoxy and mesyloxy esters and their analogous thioesters. The geometries, free energies, and charge distributions of these structures support a stepwise irreversible first-order elimination from a conjugate base (E1cBI) mechanism with acetoxy ester 3, acetoxy thioester 4, and mesyloxy thioester 6. However, mesyloxy ester 5, which has an excellent nucleofuge and a less-acidic proton, follows a concerted but asynchronous E2 mechanism with an E1cB-like transition state. The anti transition state is more favorable than the syn one, even for the poorer nucleofuge and more-acidic thioesters. The article includes a general scheme for describing liquid-phase reactions in terms of free-energy surfaces.
Co-reporter:Alessandro Cembran, Peng Bao, Yingjie Wang, Lingchun Song, Donald G. Truhlar and Jiali Gao
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 8) pp:2469-2476
Publication Date(Web):July 20, 2010
DOI:10.1021/ct100268p
The inclusion of exchange repulsion terms in the explicit polarization (X-Pol) model is examined by antisymmetrizing the X-Pol Hartree product wave function; this yields X-Pol with full eXchange, called X-Pol-X. When the monomers are treated by Hartree−Fock theory, this calculation can be accomplished by using the formalism of block-localized wave functions (BLW) that has been used in a variety of applications. In this case the block-localized structure in the X-Pol-X wave function allows for decomposition of the full Fock matrix of a dimension of M blocks into M smaller Fock matrices. The method is illustrated by considering two trimer structures of water clusters, and it is found that the total exchange repulsion energies in these hydrogen-bonding test cases are adequately treated and—to a good approximation—are pairwise additive. We also present a formalism to yield a simplified Fock matrix by making use of the neglect of interfragment differential overlap (NIDO) approximation, which is less severe than the neglect of diatomic differential overlap (NDDO) approximation.
Co-reporter:Aleksandr V. Marenich, Christopher J. Cramer, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 9) pp:2829-2844
Publication Date(Web):August 25, 2010
DOI:10.1021/ct100267s
Conventional polarized continuum model calculations of solvatochromic shifts on electronic excitation energies using popular quantum chemical programs (e.g., Gaussian or Turbomole) include the noninertial and inertial bulk-solvent polarization, which will be called electrostatics, but not dispersion interactions and specific effects like hydrogen bonding. For the n→π* excitation of acetone in several solvents, we estimated the nonelectrostatic contributions in two ways: (i) the vertical excitation model (VEM) of Li et al. (Int. J. Quantum Chem. 2000, 77, 264), but updated to use TD-DFT corrected linear response with SMD atomic radii, and (ii) in the case of acetone in water, ensemble averaging over supermolecule calculations with up to 12 explicit solvent molecules selected from a molecular dynamics trajectory, with the explicit solvent surrounded by a continuum solvent. The TD-DFT VEM calculations carried out with the M06 density functional for 23 solvents result in a dispersion contribution to the red of 261−356 cm−1 and a hydrogen-bonding contribution to the blue of up to 289 cm−1.
Co-reporter:I. M. Alecu, Jingjing Zheng, Yan Zhao, and Donald G. Truhlar
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 9) pp:2872-2887
Publication Date(Web):August 20, 2010
DOI:10.1021/ct100326h
Optimized scale factors for calculating vibrational harmonic and fundamental frequencies and zero-point energies have been determined for 145 electronic model chemistries, including 119 based on approximate functionals depending on occupied orbitals, 19 based on single-level wave function theory, three based on the neglect-of-diatomic-differential-overlap, two based on doubly hybrid density functional theory, and two based on multicoefficient correlation methods. Forty of the scale factors are obtained from large databases, which are also used to derive two universal scale factor ratios that can be used to interconvert between scale factors optimized for various properties, enabling the derivation of three key scale factors at the effort of optimizing only one of them. A reduced scale factor optimization model is formulated in order to further reduce the cost of optimizing scale factors, and the reduced model is illustrated by using it to obtain 105 additional scale factors. Using root-mean-square errors from the values in the large databases, we find that scaling reduces errors in zero-point energies by a factor of 2.3 and errors in fundamental vibrational frequencies by a factor of 3.0, but it reduces errors in harmonic vibrational frequencies by only a factor of 1.3. It is shown that, upon scaling, the balanced multicoefficient correlation method based on coupled cluster theory with single and double excitations (BMC−CCSD) can lead to very accurate predictions of vibrational frequencies. With a polarized, minimally augmented basis set, the density functionals with zero-point energy scale factors closest to unity are MPWLYP1M (1.009), τHCTHhyb (0.989), BB95 (1.012), BLYP (1.013), BP86 (1.014), B3LYP (0.986), MPW3LYP (0.986), and VSXC (0.986).
Co-reporter:Ewa Papajak and Donald G. Truhlar
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 3) pp:597-601
Publication Date(Web):February 16, 2010
DOI:10.1021/ct900566x
Eliminating all but the s and p diffuse functions on the non-hydrogenic atoms and all diffuse functions on the hydrogen atoms from the aug-cc-pV(x+d)Z basis sets of Dunning and co-workers, where x = D, T, Q, ..., yields the previously proposed “minimally augmented” basis sets, called maug-cc-pV(x+d)Z. Here, we present extensive and systematic tests of these basis sets for density functional calculations of chemical reaction barrier heights, hydrogen bond energies, electron affinities, ionization potentials, and atomization energies. The tests show that the maug-cc-pV(x+d)Z basis sets are as accurate as the aug-cc-pV(x+d)Z ones for density functional calculations, but the computational cost savings are a factor of about two to seven.
Co-reporter:Bo Wang and Donald G. Truhlar
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 2) pp:359-369
Publication Date(Web):January 7, 2010
DOI:10.1021/ct900366m
The combined quantum mechanical and molecular mechanical (QM/MM) method is one of the most powerful approaches for including correlation and polarization effects in simulations of large and complex systems, and the present article is concerned with the systematics of treating a QM/MM boundary that passes through a covalent bond, especially a polar covalent bond. In this study, we develop a new algorithm to treat such boundaries; the new method is called the balanced redistributed charge (balanced RC or BRC) scheme with a tuned fluorine link atom. The MM point charge on the MM boundary atom is modified to conserve the total charge of the entire system, and the modified charge is redistributed to the midpoints of the bonds between an MM boundary atom and its neighboring MM atoms. A pseudopotential is added to the fluorine link atom to reproduce the partial charge of the uncapped portion of the QM subsystem. We select proton affinities as the property used to validate the new method because the energy change associated with the addition of an entire charge (proton) to the QM system is very sensitive to the treatment of electrostatics at the boundary; we apply the new method to calculate proton affinities of 25 molecules with 13 different kinds of bonds being cut. The average proton affinity in the test set is 373 kcal/mol, and the test set provides a more challenging test than those usually used for testing QM/MM methods. For this challenging test set, common unbalanced schemes give a mean unsigned error (MUE) of 15−21 kcal/mol for H link atoms or 16−24 kcal/mol for F link atoms, much larger than the 5 kcal/mol obtained by simply omitting the MM region with either kind of link atom. Balancing the charges reduces the error to 5−7 kcal/mol for H link atoms and 4−6 kcal/mol for F link atoms. Balancing the charges and also tuning an F link atom lowers the MUE to 1.3−4 kcal/mol, with the best result for the balanced RC scheme. We conclude that properly tuning the link atom and correctly treating the point charges near the QM/MM boundary significantly improves the accuracy of the calculated proton affinities.
Co-reporter:Junjun Liu, Casey P. Kelly, Alan C. Goren, Aleksandr V. Marenich, Christopher J. Cramer, Donald G. Truhlar and Chang-Guo Zhan
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 4) pp:1109-1117
Publication Date(Web):March 4, 2010
DOI:10.1021/ct100025j
Building on the SVPE (surface and volume polarization for electrostatics) model for electrostatic contributions to the free energy of solvation with explicit consideration of both surface and volume polarization effects, on the SMx approach to including first-solvation-shell contributions, and on the linear relationship between the electric field and short-range electrostatic contributions found by Chipman, we have developed a new method for computing absolute aqueous solvation free energies by combining the SVPE method with semiempirical terms that account for effects beyond bulk electrostatics. The new method is called SMVLE, and the elements it contains are denoted by SVPE-CDSL, where SVPE denotes accounting for bulk electrostatic interactions between solute and solvent with both surface and volume contributions, CDS denotes the inclusion of solvent cavitation, changes in dispersion energy, and possible changes in local solvent structure by a semiempirical term utilizing geometry-dependent atomic surface tensions as implemented in SMx models, and L represents the local electrostatic effect derived from the outward-directed normal electric field on the cavity surface. The semiempirical CDS and L terms together represent the deviation of short-range contributions to the free energy of solvation from those accounted for by the SVPE term based on the bulk solvent dielectric constant. A solute training set containing a broad range of molecules used previously in the development of SM6 is used here for SMVLE model calibration. The aqueous solvation free energies predicted by the parametrized SMVLE model correlate exceedingly well with experimental values. The square of the correlation coefficient is 0.9949 and the slope is 1.0079. Comparison of the final SMVLE model against the earlier SMx solvation model shows that the parametrized SMVLE model not only yields good accuracy for neutrals but also significantly increases the accuracy for ions, making it the best implicit solvation model to date for aqueous solvation free energies of ions. The semiempirical terms associated with the outward-directed electric field account in a physical way for the improvement in the predictive accuracy for ions. The SMVLE method greatly decreases the need to include explicit water molecules for accurate modeling of solvation free energies of ions.
Co-reporter:Yan Zhao and Donald G. Truhlar
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 4) pp:1104-1108
Publication Date(Web):March 17, 2010
DOI:10.1021/ct100082z
We have computed stationary points on the potential energy surface for the anti-E2, syn-E2, and SN2 pathways of the reactions of F− and Cl− with CH3CH2F and CH3CH2Cl with fully self-consistent fields and Gaussian basis functions. We find large differences from previously reported [Bento, A. P.; Solà, M.; Bickelhaupt, F. M. J. Chem. Theory Comput. 2008, 4, 929] calculations with Slater-type orbitals. We revise the findings of the previous study; in particular, we find average absolute errors in kcal/mol compared to benchmark calculations of 20 stationary point energies (6 saddle points and 14 minima) of 0.9 for M06-2X, 1.2 for M08-SO, 1.4 for M06-HF, 2.0 for M06, 2.3 for B3LYP, 2.5 for OLYP, 2.7 for M06-L, and 3.5 kcal/mol for TPSS. We also compare the predictions of various density functionals for the partial atomic charges at the transition states.
Co-reporter:Bo Wang and Donald G. Truhlar
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 11) pp:3330-3342
Publication Date(Web):October 11, 2010
DOI:10.1021/ct1003862
Electrostatic effects are often the dominant component of intermolecular interactions, but they are often modeled without accounting for charge penetration effects due to the finite extent of electronic orbitals. Here, we propose a new scheme to include charge penetration effects in electrostatic modeling, and we parametrize it and illustrate it by employing the electronically embedded combined quantum mechanical and molecular mechanical (QM/MM) method. It can also be extended to other molecular modeling approximations that include electrostatic effects. The method, which is based on introduction of a single parameter for each element, is simple in concept and implementation, modest in cost, and easily incorporated into existing codes. In the new scheme, the MM atomic charge density of an atom in a molecule is represented by a screened charge rather than by a point charge. The screened charge includes a point charge for the nucleus, core electrons, and inner valence electrons, and a smeared charge for the outer valence electron density, which is distributed in a Slater-type orbital representing the outer part of the atomic charge distribution such that the resulting pairwise interactions are still analytic central potentials. We optimize the exponential parameters of the Slater-type orbitals for 10 elements, in particular H, C, N, O, F, Si, P, S, Cl, and Br, to minimize the mean unsigned error (MUE) of the QM/MM electrostatic and induction energies with respect to the Hartree−Fock electrostatic energies and the sum of induction and induction-exchange energies calculated by symmetry-adapted perturbation theory (SAPT). The resulting optimized exponential parameters are very physical, which allows one to assign parameters to all nonmetal elements (except rare gases) with atomic number less than or equal to 35. For a test set of complexes, the improved description of MM charge densities reduces the error of electrostatic interactions between QM and MM regions in the QM/MM method from 8.1 to 2.8 kcal/mol and reduces the error of induction interactions from 1.9 to 1.4 kcal/mol.
Co-reporter:Jingjing Zheng and Donald G. Truhlar
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 28) pp:7782-7793
Publication Date(Web):25 May 2010
DOI:10.1039/B927504E
Five isomerization reactions involving intramolecular hydrogen-transfer in butoxyl radicals have been studied using variational transition state theory with small curvature tunneling. A set of best estimates of barrier heights and reaction energies for these five reactions was obtained by using coupled cluster theory including single and double excitations with a quasiperturbative treatment of connected triple excitations and a basis set extrapolated to the complete basis set limit plus core–valence correlation contributions and scalar relativistic corrections. This work predicts high-pressure limiting rate constants of these five reactions over the temperature range 200–2500 K and clarifies the available experimental data from indirect measurements. This study shows the importance of performing rate calculations with proper accounting for tunneling and torsional anharmonicity. We also proposed two new models for use in fitting rate constants over wide ranges of temperature.
Co-reporter:Donald G. Truhlar
Journal of Physical Organic Chemistry 2010 Volume 23( Issue 7) pp:660-676
Publication Date(Web):
DOI:10.1002/poc.1676
Abstract
This paper is a response to an invitation to share my viewpoint by writing an opinion piece (not a review) on proton tunneling, especially from the point of view of whether it has a greater importance in enzymatic reactions than in other reactions. The paper begins with a discussion of the emergence of a conceptual framework for including tunneling in reaction rate calculations; the framework is general enough to include not only transfer of protons but also transfer of hydrogen atoms and hydride ions and their isotopes, and not only enzymatically catalyzed reactions but also nonenzymatic reactions. Then the paper discusses the special issues that arise when the reaction rate under consideration is for an enzyme-catalyzed reaction. The emphasis is on physical considerations in reaction rate calculations, not on system-specific comparison of results for different modes of reaction. It is argued that enzymatic and nonenzymatic reactions may be treated within the same basic framework except that ensemble averaging, which is not usually required for gas-phase reactions, is essential for treating enzyme reactions. Enzymes explicitly discussed include methylamine dehydrogenase, aromatic amine dehydrogenase, E. coli dihydrofolate reductase, hyperthermophilic dihydrofolate reductase, liver alcohol dehydrogenase, methylmalonyl-CoA mutase, soybean lipoxygenase, copper amine oxidase, pentaerythritol tetranitrate reductase, morphinone reductase, enolase, xylose isomerase, and 4-oxalocrotonate tautomerase. Copyright © 2010 John Wiley & Sons, Ltd.
Co-reporter:Boris B. Averkiev, Yan Zhao, Donald G. Truhlar
Journal of Molecular Catalysis A: Chemical 2010 324(1–2) pp: 80-88
Publication Date(Web):
DOI:10.1016/j.molcata.2010.03.016
Co-reporter:Michael A. North, Sudeep Bhattacharyya, and Donald G. Truhlar
The Journal of Physical Chemistry B 2010 Volume 114(Issue 46) pp:14907-14915
Publication Date(Web):October 20, 2010
DOI:10.1021/jp108024b
The energetics of electrochemical changes have been investigated for several substituted flavins with the M06-L density functional. The reduction potentials for one- and two-electron reductions of these molecules have been determined and the results are consistent with experimental findings with a mean unsigned error of only 42 mV. It is especially noteworthy that the M06-L density functional makes a significant difference in the computed free energy of the first reduction of lumiflavin, which produces a neutral semiquinone. We also investigate the effects of flavin ring substituents on the geometries, charge distributions, reduction potentials, pKa’s, ionization potentials, electron affinities, hardnesses, softnesses, electrophilic powers, and nucleophilicities.
Co-reporter:Raphael F. Ribeiro;Aleksandr V. Marenich
Journal of Computer-Aided Molecular Design 2010 Volume 24( Issue 4) pp:317-333
Publication Date(Web):2010 April
DOI:10.1007/s10822-010-9333-9
We applied the solvation models SM8, SM8AD, and SMD in combination with the Minnesota M06-2X density functional to predict vacuum-water transfer free energies (Task 1) and tautomeric ratios in aqueous solution (Task 2) for the SAMPL2 test set. The bulk-electrostatic contribution to the free energy of solvation is treated as follows: SM8 employs the generalized Born model with the Coulomb field approximation, SM8AD employs the generalized Born approximation with asymmetric descreening, and SMD solves the nonhomogeneous Poisson equation. The non-bulk-electrostatic contribution arising from short-range interactions between the solute and solvent molecules in the first solvation shell is treated as a sum of terms that are products of geometry-dependent atomic surface tensions and solvent-accessible surface areas of the individual atoms of the solute. On average, three models tested in the present work perform similarly. In particular, we achieved mean unsigned errors of 1.3 (SM8), 2.0 (SM8AD), and 2.6 kcal/mol (SMD) for the aqueous free energies of 30 out of 31 compounds with known reference data involved in Task 1 and mean unsigned errors of 2.7 (SM8), 1.8 (SM8AD), and 2.4 kcal/mol (SMD) in the free energy differences (tautomeric ratios) for 21 tautomeric pairs in aqueous solution involved in Task 2.
Co-reporter:Oksana Tishchenko;Ruifang Li
PNAS 2010 107 (45 ) pp:19139-19145
Publication Date(Web):2010-11-09
DOI:10.1073/pnas.1010287107
The present paper illustrates key features of charge transfer between calcium atoms and prototype conjugated hydrocarbons
(ethylene, benzene, and coronene) as elucidated by electronic structure calculations. One- and two-electron charge transfer
is controlled by two sequential conical intersections. The two lowest electronic states that undergo a conical intersection
have closed-shell and open-shell dominant configurations correlating with the 4s2 and 4s13d1 states of Ca, respectively. Unlike the neutral-ionic state crossing in, for example, hydrogen halides or alkali halides,
the path from separated reactants to the conical intersection region is uphill and the charge-transferred state is a biradical.
The lowest-energy adiabatic singlet state shows at least two minima along a single approach path of Ca to the π system: (i) a van der Waals complex with a doubly occupied highest molecular orbital, denoted , and a small negative charge on Ca and (ii) an open-shell singlet (biradical) at intermediate approach (Ca⋯C distance ≈2.5–2.7 Å) with molecular orbital structure ϕ1ϕ2, where ϕ2 is an orbital showing significant charge transfer form Ca to the π-system, leading to a one-electron multicentered bond.
A third minimum (iii) at shorter distances along the same path corresponding to a closed-shell state with molecular orbital structure has also been found; however, it does not necessarily represent the ground state at a given Ca⋯C distance in all three systems.
The topography of the lowest adiabatic singlet potential energy surface is due to the one- and two-electron bonding patterns
in Ca-π complexes.
Co-reporter:Jingjing Zheng, Yan Zhao and Donald G. Truhlar
Journal of Chemical Theory and Computation 2009 Volume 5(Issue 4) pp:808-821
Publication Date(Web):March 23, 2009
DOI:10.1021/ct800568m
The diverse barrier height database DBH24 is updated by using W4 and W3.2 data (Karton, A.; Tarnopolsky, A.; Lamère, J.-F.; Schatz, G. C.; Martin, J. M. L. J. Phys. Chem. A 2008, 112, 12868) to replace previous W1 values; we call the new database DBH24/08. We used the new database to assess 348 model chemistries, each consisting of a combination of a wave function theory level or a density functional approximation with a one-electron basis set. All assessments are made by simultaneous consideration of accuracy and cost. The assessment includes several electronic structure methods and basis sets that have not previously been systematically tested for barrier heights. Some conclusions drawn in our previous work (Zheng, J.; Zhao, Y.; Truhlar, D. G. J. Chem. Theory Comput. 2007, 3, 569) are still valid when using this improved database and including more model chemistries. For example, BMC-CCSD is again found to be the best method whose cost scales as N6, and its cost is an order of magnitude smaller than the N7 method with best performance-to-cost ratio, G3SX(MP3), although the mean unsigned error is only marginally higher, namely 0.70 kcal/mol vs 0.57 kcal/mol. Other conclusions are now broader in scope. For example, among single-reference N5 methods (that is, excluding MRMP2), we now conclude not only that doubly hybrid density functionals and multicoefficient extrapolated density functional methods perform better than second-order Møller−Plesset-type perturbation theory (MP2) but also that they perform better than any correlation-energy-scaled MP2 method. The most recommended hybrid density functionals, if functionals are judged only on the basis of barrier heights, are M08-SO, M06-2X, M08-HX, BB1K, BMK, PWB6K, MPW1K, BHandHLYP, and TPSS25B95. MOHLYP and HCTH are found to be the best performing local density functionals for barrier heights. The basis set cc-pVTZ+ is more efficient than aug-cc-pVTZ with similar accuracy, especially for density functional theory. The basis sets cc-pVDZ+, 6−31+G(d,p), 6−31B(d,p), 6−31B(d), MIDIY+, MIDIX+, and MIDI! are recommended for double-ζ-quality density functional calculations on large systems for their good balance between accuracy and cost, and the basis sets cc-pVTZ+, MG3S, MG3SXP, and aug-cc-pVDZ are recommended for density functional calculations when larger basis sets are affordable. The best performance of any methods tested is attained by CCSD(T)(full)/aug-cc-pCV(T+d)Z with a mean unsigned error of 0.46 kcal/mol; however, this is several orders of magnitude more expensive than M08-SO/cc-pVTZ+, which has a mean unsigned error of only 0.90 kcal/mol.
Co-reporter:Aleksandr V. Marenich, Christopher J. Cramer and Donald G. Truhlar
Journal of Chemical Theory and Computation 2009 Volume 5(Issue 9) pp:2447-2464
Publication Date(Web):August 12, 2009
DOI:10.1021/ct900312z
We present a new self-consistent reaction field continuum solvation model based on the generalized Born (GB) approximation for the bulk electrostatic contribution to the free energy of solvation. The new model improves on the earlier SM8 model by using the asymmetric descreening algorithm of Grycuk to treat dielectric descreening effects rather than the Coulomb field approximation; it will be called Solvation Model 8 with asymmetric descreening (SM8AD). The SM8AD model is applicable to any charged or uncharged solute in any solvent or liquid medium for which a few key descriptors are known, in particular dielectric constant, refractive index, bulk surface tension, and acidity and basicity parameters. It does not require the user to assign molecular mechanics types to an atom or a group; all parameters are unique and continuous functions of geometry. This model employs a single set of parameters (solvent acidity-dependent intrinsic Coulomb radii for the treatment of bulk electrostatics and solvent description-dependent atomic surface tensions coefficients for the treatment of nonelectrostatic and short-range electrostatic effects). The SM8AD model was optimized over 26 combinations of theoretical levels including various basis sets (MIDI!, 6-31G*, 6-31+G*, 6-31+G**, 6-31G**, cc-pVDZ, DZVP, 6-31B*) and electronic structure methods (M05-2X, M05, M06-2X, M06, M06-HF, M06-L, mPW1PW, mPWPW, B3LYP, HF). It may be used with confidence with any level of electronic structure theory as long as self-consistently polarized Charge Model 4 or other self-consistently polarized charges compatible with CM4 charges are used, for example, CM4M charges can be used. With M05-2X/6-31G*, the SM8AD model achieves a mean unsigned error of 0.6 kcal/mol on average over 2 560 solvation free energies of tested aqueous and nonaqueous neutral solutes and a mean unsigned error of 3.9 kcal/mol on average over 332 solvation free energies of aqueous and nonaqueous ions.
Co-reporter:Masahiro Higashi and Donald G. Truhlar
Journal of Chemical Theory and Computation 2009 Volume 5(Issue 11) pp:2925-2929
Publication Date(Web):October 16, 2009
DOI:10.1021/ct900301d
Density functional theory is a powerful and efficient method for calculating potential energy surfaces for chemical reactions, but its application to complex systems, such as reactions in enzymes, is often prohibitively expensive, even when high-level theory is applied only to a primary subsystem, such as an active site, and when the remaining system is treated by molecular mechanics. Here we show how the combination of multiconfiguration molecular mechanics with charge response kernels can speed up such calculations by three or more orders of magnitude. The resulting method, called electrostatically embedded multiconfiguration molecular mechanics, is illustrated by calculating the free energy of activation profile for the dehalogenation of 1,2-dichloroethane by haloalkane dehalogenase. This shows how hybrid density functionals or other high-level electronic structure methods can now be used efficiently in simulations that require extensive sampling, such as for calculating free energy profiles along a high-barrier reaction coordinate.
Co-reporter:Oksana Tishchenko and Donald G. Truhlar
Journal of Chemical Theory and Computation 2009 Volume 5(Issue 6) pp:1454-1461
Publication Date(Web):May 21, 2009
DOI:10.1021/ct900077g
We present a new version of the multiconfiguration molecular mechanics (MCMM) algorithm for fitting potential energy surfaces of complex reactive systems. The main improvement consists in allowing the valence bond configuration interaction matrix to be non-Hermitian, which broadens the range of geometries over which the potential energy surface can be fit accurately. A second improvement is that the new algorithm has simpler gradients and Hessians and executes faster. The performance of the new algorithm is evaluated using the example of two model reactions.
Co-reporter:Wangshen Xie, Modesto Orozco, Donald G. Truhlar and Jiali Gao
Journal of Chemical Theory and Computation 2009 Volume 5(Issue 3) pp:459-467
Publication Date(Web):February 17, 2009
DOI:10.1021/ct800239q
A recently proposed electronic structure-based force field called the explicit polarization (X-Pol) potential is used to study many-body electronic polarization effects in a protein, in particular by carrying out a molecular dynamics (MD) simulation of bovine pancreatic trypsin inhibitor (BPTI) in water with periodic boundary conditions. The primary unit cell is cubic with dimensions ∼54 × 54 × 54 Å3, and the total number of atoms in this cell is 14281. An approximate electronic wave function, consisting of 29026 basis functions for the entire system, is variationally optimized to give the minimum Born−Oppenheimer energy at every MD step; this allows the efficient evaluation of the required analytic forces for the dynamics. Intramolecular and intermolecular polarization and intramolecular charge transfer effects are examined and are found to be significant; for example, 17 out of 58 backbone carbonyls differ from neutrality on average by more than 0.1 electron, and the average charge on the six alanines varies from −0.05 to +0.09. The instantaneous excess charges vary even more widely; the backbone carbonyls have standard deviations in their fluctuating net charges from 0.03 to 0.05, and more than half of the residues have excess charges whose standard deviation exceeds 0.05. We conclude that the new-generation X-Pol force field permits the inclusion of time-dependent quantum mechanical polarization and charge transfer effects in much larger systems than was previously possible.
Co-reporter:Meiyu Zhao, Mark A. Iron, Przemysław Staszewski, Nathan E. Schultz, Rosendo Valero and Donald G. Truhlar
Journal of Chemical Theory and Computation 2009 Volume 5(Issue 3) pp:594-604
Publication Date(Web):February 10, 2009
DOI:10.1021/ct8004535
The extension of molecular mechanics to reactive systems, metals, and covalently bonded clusters with variable coordination numbers requires new functional forms beyond those popular for organic chemistry and biomolecules. Here we present a new scheme for reactive molecular mechanics, which is denoted as the valence–bond order model, for approximating reactive potential energy surfaces in large molecules, clusters, nanoparticles, solids, and other condensed-phase materials, especially those containing metals. The model is motivated by a moment approximation to tight binding molecular orbital theory, and we test how well one can approximate potential energy surfaces with a very simple functional form involving only interatomic distances with no explicit dependence on bond angles or dihedral angles. For large systems the computational requirements scale linearly with system size, and no diagonalizations or iterations are required; thus the method is well suited to large-scale simulations. The method is illustrated here by developing a force field for particles and solids composed of aluminum and hydrogen. The parameters were optimized against both interaction energies and relative interaction energies. The method performs well for pure aluminum clusters, nanoparticles, and bulk lattices and reasonably well for pure hydrogen clusters; the mean unsigned error per atom for the aluminum−hydrogen clusters is 0.1 eV/atom.
Co-reporter:Yongho Kim, Aleksandr V. Marenich, Jingjing Zheng, Kyung Hyun Kim, Magdalena Kołodziejska-Huben, Michał Rostkowski, Piotr Paneth and Donald G. Truhlar
Journal of Chemical Theory and Computation 2009 Volume 5(Issue 1) pp:59-67
Publication Date(Web):December 4, 2008
DOI:10.1021/ct800345j
The primary and secondary deuterium kinetic isotope effects as well as leaving-group fluorine kinetic isotope effects have been calculated for the base-promoted elimination of hydrogen fluoride from 4-fluoro-4-(4′-nitrophenyl)butane-2-one in 75% aqueous methanol solution. The elimination was studied for both formate and imidazole as the catalytic base; and reactant and transition state structures and vibrational frequencies have been calculated by including the base explicitly and by including the solvent by an implicit solvation model that includes both electrostatics by class IV charges and first-solvation-shell effects by atomic surface tensions. We used the M06-L density functional for all calculations. The optimized stationary points, the geometry changes along the solution-phase minimum free energy path, and the solution-phase free energy profile indicate that the elimination reaction occurs concertedly but asynchronously via an E1cb-like transition state. Reaction rates were calculated by the equilibrium solvation path method, using variational transition state theory with multidimensional tunneling. The primary deuterium kinetic isotope effects are calculated to be large: 1.67 and 5.13 for formate and imidazole, respectively. The corresponding C4-secondary deuterium kinetic isotope effects are 1.044 and 1.044, and the leaving group fluorine kinetic isotope effects are respectively 1.020 and 1.015.
Co-reporter:Ewa Papajak, Hannah R. Leverentz, Jingjing Zheng and Donald G. Truhlar
Journal of Chemical Theory and Computation 2009 Volume 5(Issue 5) pp:1197-1202
Publication Date(Web):March 31, 2009
DOI:10.1021/ct800575z
We combine the diffuse basis functions from the 6-31+G basis set of Pople and co-workers with the correlation-consistent basis sets of Dunning and co-workers. In both wave function and density functional calculations, the resulting basis sets reduce the basis set superposition error almost as much as the augmented correlation-consistent basis sets, although they are much smaller. In addition, in density functional calculations the new basis sets, called cc-pVxZ+ where x = D, T, Q, ..., or x = D+d, T+d, Q+d, ..., give very similar energetic predictions to the much larger aug-cc-pVxZ basis sets. However, energetics calculated from correlated wave function calculations are more slowly convergent with respect to the addition of diffuse functions. We also examined basis sets with the same number and type of functions as the cc-pVxZ+ sets but using the diffuse exponents of the aug-cc-pVxZ basis sets and found very similar performance to cc-pVxZ+; these basis sets are called minimally augmented cc-pVxZ, which we abbreviate as maug-cc-pVxZ.
Co-reporter:Rosendo Valero, Lingchun Song, Jiali Gao and Donald G. Truhlar
Journal of Chemical Theory and Computation 2009 Volume 5(Issue 1) pp:1-22
Publication Date(Web):December 4, 2008
DOI:10.1021/ct800318h
Co-reporter:Yan Zhao and Donald G. Truhlar
Journal of Chemical Theory and Computation 2009 Volume 5(Issue 2) pp:324-333
Publication Date(Web):January 8, 2009
DOI:10.1021/ct800386d
We present benchmark relative energetics in the catalytic cycle of a model system for Grubbs second-generation olefin metathesis catalysts. The benchmark data were determined by a composite approach based on CCSD(T) calculations, and they were used as a training set to develop a new spin-component-scaled MP2 method optimized for catalysis, which is called SCSC-MP2. The SCSC-MP2 method has improved performance for modeling Grubbs II olefin metathesis catalysts as compared to canonical MP2 or SCS-MP2. We also employed the benchmark data to test 17 WFT methods and 39 density functionals. Among the tested density functionals, M06 is the best performing functional. M06/TZQS gives an MUE of only 1.06 kcal/mol, and it is a much more affordable method than the SCSC-MP2 method or any other correlated WFT methods. The best performing meta-GGA is M06-L, and M06-L/DZQ gives an MUE of 1.77 kcal/mol. PBEh is the best performing hybrid GGA, with an MUE of 3.01 kcal/mol; however, it does not perform well for the larger, real Grubbs II catalyst. B3LYP and many other functionals containing the LYP correlation functional perform poorly, and B3LYP underestimates the stability of stationary points for the cis-pathway of the model system by a large margin. From the assessments, we recommend the M06, M06-L, and MPW1B95 functionals for modeling Grubbs II olefin metathesis catalysts. The local M06-L method is especially efficient for calculations on large systems.
Co-reporter:Christopher J. Cramer and Donald G. Truhlar
Physical Chemistry Chemical Physics 2009 vol. 11(Issue 46) pp:10757-10816
Publication Date(Web):21 Oct 2009
DOI:10.1039/B907148B
We introduce density functional theory and review recent progress in its application to transition metal chemistry. Topics covered include local, meta, hybrid, hybrid meta, and range-separated functionals, band theory, software, validation tests, and applications to spin states, magnetic exchange coupling, spectra, structure, reactivity, and catalysis, including molecules, clusters, nanoparticles, surfaces, and solids.
Co-reporter:Aleksandr V. Marenich, Christopher J. Cramer, and Donald G. Truhlar
The Journal of Physical Chemistry B 2009 Volume 113(Issue 18) pp:6378-6396
Publication Date(Web):April 14, 2009
DOI:10.1021/jp810292n
We present a new continuum solvation model based on the quantum mechanical charge density of a solute molecule interacting with a continuum description of the solvent. The model is called SMD, where the “D” stands for “density” to denote that the full solute electron density is used without defining partial atomic charges. “Continuum” denotes that the solvent is not represented explicitly but rather as a dielectric medium with surface tension at the solute−solvent boundary. SMD is a universal solvation model, where “universal” denotes its applicability to any charged or uncharged solute in any solvent or liquid medium for which a few key descriptors are known (in particular, dielectric constant, refractive index, bulk surface tension, and acidity and basicity parameters). The model separates the observable solvation free energy into two main components. The first component is the bulk electrostatic contribution arising from a self-consistent reaction field treatment that involves the solution of the nonhomogeneous Poisson equation for electrostatics in terms of the integral-equation-formalism polarizable continuum model (IEF-PCM). The cavities for the bulk electrostatic calculation are defined by superpositions of nuclear-centered spheres. The second component is called the cavity-dispersion-solvent-structure term and is the contribution arising from short-range interactions between the solute and solvent molecules in the first solvation shell. This contribution is a sum of terms that are proportional (with geometry-dependent proportionality constants called atomic surface tensions) to the solvent-accessible surface areas of the individual atoms of the solute. The SMD model has been parametrized with a training set of 2821 solvation data including 112 aqueous ionic solvation free energies, 220 solvation free energies for 166 ions in acetonitrile, methanol, and dimethyl sulfoxide, 2346 solvation free energies for 318 neutral solutes in 91 solvents (90 nonaqueous organic solvents and water), and 143 transfer free energies for 93 neutral solutes between water and 15 organic solvents. The elements present in the solutes are H, C, N, O, F, Si, P, S, Cl, and Br. The SMD model employs a single set of parameters (intrinsic atomic Coulomb radii and atomic surface tension coefficients) optimized over six electronic structure methods: M05-2X/MIDI!6D, M05-2X/6-31G*, M05-2X/6-31+G**, M05-2X/cc-pVTZ, B3LYP/6-31G*, and HF/6-31G*. Although the SMD model has been parametrized using the IEF-PCM protocol for bulk electrostatics, it may also be employed with other algorithms for solving the nonhomogeneous Poisson equation for continuum solvation calculations in which the solute is represented by its electron density in real space. This includes, for example, the conductor-like screening algorithm. With the 6-31G* basis set, the SMD model achieves mean unsigned errors of 0.6−1.0 kcal/mol in the solvation free energies of tested neutrals and mean unsigned errors of 4 kcal/mol on average for ions with either Gaussian03 or GAMESS.
Co-reporter:Manjeera Mantina, Adam C. Chamberlin, Rosendo Valero, Christopher J. Cramer, and Donald G. Truhlar
The Journal of Physical Chemistry A 2009 Volume 113(Issue 19) pp:5806-5812
Publication Date(Web):April 21, 2009
DOI:10.1021/jp8111556
Atomic radii are not precisely defined but are nevertheless widely used parameters in modeling and understanding molecular structure and interactions. The van der Waals radii determined by Bondi from molecular crystals and data for gases are the most widely used values, but Bondi recommended radius values for only 28 of the 44 main-group elements in the periodic table. In the present Article, we present atomic radii for the other 16; these new radii were determined in a way designed to be compatible with Bondi’s scale. The method chosen is a set of two-parameter correlations of Bondi’s radii with repulsive-wall distances calculated by relativistic coupled-cluster electronic structure calculations. The newly determined radii (in Å) are Be, 1.53; B, 1.92; Al, 1.84; Ca, 2.31; Ge, 2.11; Rb, 3.03; Sr, 2.49; Sb, 2.06; Cs, 3.43; Ba, 2.68; Bi, 2.07; Po, 1.97; At, 2.02; Rn, 2.20; Fr, 3.48; and Ra, 2.83.
Co-reporter:Kelly E. Anderson, Steven L. Mielke, J. Ilja Siepmann and Donald G. Truhlar
The Journal of Physical Chemistry A 2009 Volume 113(Issue 10) pp:2053-2059
Publication Date(Web):January 27, 2009
DOI:10.1021/jp808711y
Recent work has focused attention on possible shifts in the bond angle distribution of CO2 as a consequence of intermolecular interactions in the supercritical phase. To investigate the temperature and phase dependence of the intramolecular structure of CO2, we performed Feynman path integral Monte Carlo calculations based on a spectroscopically derived analytical potential, first principles molecular dynamics simulations using Kohn−Sham density functional theory, and Monte Carlo simulations employing empirical interaction potentials. On the basis of various distributions used to characterize the intramolecular structure, we conclude that the aggregation state has a negligible influence on the intramolecular structure, in particular we find that in the classical limit the distributions are remarkably similar for the ideal gas, supercritical, and solid phases when considered at the same temperature. In contrast, an increase in the temperature from 325 to 673 K or inclusion of nuclear quantum effects leads to a significant broadening of the distributions. With respect to the first C−O bond vector, the second bond vector most prefers a collinear arrangement. However, due to the Jacobian factor the maximum in the bond angle distribution at 325 K is shifted to an angle of about 175.7° in the classical limit or to 173.0° if nuclear quantum effects are included. Nevertheless, an analysis of the temperature dependence of the constant-volume heat capacity demonstrates that carbon dioxide should be viewed as a linear molecule.
Co-reporter:Jingjing Zheng and Donald G. Truhlar
The Journal of Physical Chemistry A 2009 Volume 113(Issue 43) pp:11919-11925
Publication Date(Web):July 17, 2009
DOI:10.1021/jp903345x
The rate constants of three intramolecular hydrogen-transfer isomerization reactions, namely, 1-4 isomerization of the 1-pentyl radical and 1-4 and 1-5 isomerizations of the 1-hexyl radical, are calculated using variational transition state theory with multidimensional tunneling, in particular by using canonical variational theory (CVT, which is the version of variational transition state theory in which the transition state dividing surface is optimized for a canonical ensemble) with small-curvature tunneling (SCT) for the transmission coefficient. The required potential energy surfaces were obtained implicitly by direct dynamics employing interpolated variational transition state theory with mapping (IVTST-M) and variational transition state theory with interpolated single-point energies (VTST-ISPE). Single-level direct dynamics calculations were performed for all of the reactions by IVTST-M using M06-2X/MG3S or M08-HX/cc-pVTZ+ potential energy surfaces or both. The stationary points of 1-4 isomerization of 1-pentyl and the stationary points for the forward reactions of 1-4 and 1-5 isomerizations of 1-hexyl were also optimized by BMC-CCSD, and for all three reactions we also performed dual-level direct dynamics calculations using VTST-ISPE in which MCG3-MPW single-point energies served as the higher level. The calculated MCG3-MPW//M06-2X/MG3S rate constants agree well with experimental values for 1-4 isomerization of the 1-pentyl radical at high temperature, and this validates the accuracy of this theoretical method for 1-4 isomerization. The MCG3-MPW//M06-2X/MG3S method was therefore used to make a reliable prediction for the rata constants of 1-4 isomerization of the 1-hexyl radical for which a direct experimental measurement is not available. The calculated CVT/SCT/M08-HX/cc-pVTZ+ rate constants agree well with experimental values for 1-5 isomerization of the 1-hexyl radical, and they show that the tunneling effect for these reactions was underestimated in previous work.
Co-reporter:Yan Zhao, Oksana Tishchenko, Jeffrey R. Gour, Wei Li, Jesse J. Lutz, Piotr Piecuch, and Donald G. Truhlar
The Journal of Physical Chemistry A 2009 Volume 113(Issue 19) pp:5786-5799
Publication Date(Web):April 17, 2009
DOI:10.1021/jp811054n
The 1,3-dipolar cycloadditions of ozone to ethyne and ethene provide extreme examples of multireference singlet-state chemistry, and they are examined here to test the applicability of several approaches to thermochemical kinetics of systems with large static correlation. Four different multireference diagnostics are applied to measure the multireference characters of the reactants, products, and transition states; all diagnostics indicate significant multireference character in the reactant portion of the potential energy surfaces. We make a more complete estimation of the effect of quadruple excitations than was previously available, and we use this with CCSDT/CBS estimation of Wheeler et al. (Wheeler, S. E.; Ess, D. H.; Houk, K. N. J. Phys. Chem. A 2008, 112, 1798.) to make new best estimates of the van der Waals association energy, the barrier height, and the reaction energy to form the cycloadduct for both reactions. Comparing with these best estimates, we present comprehensive mean unsigned errors for a variety of coupled cluster, multilevel, and density functional methods. Several computational aspects of multireference reactions are considered: (i) the applicability of multilevel theory, (ii) the convergence of coupled cluster theory for reaction barrier heights, (iii) the applicability of completely renormalized coupled cluster methods to multireference systems, (iv) the treatment by density functional theory, (v) the multireference perturbation theory for multireference reactions, and (vi) the relative accuracy of scaling-type multilevel methods as compared with additive ones. It is found that scaling-type multilevel methods do not perform better than the additive-type multilevel methods. Among the 48 tested density functionals, only M05 reproduces the best estimates within their uncertainty. Multireference perturbation theory based on the complete-active-space reference wave functions constructed using a small number of reaction-specific active orbitals gives accurate forward barrier heights; however, it significantly underestimates reaction energies.
Co-reporter:Yan Zhao and Donald G. Truhlar
Accounts of Chemical Research 2008 Volume 41(Issue 2) pp:157
Publication Date(Web):January 11, 2008
DOI:10.1021/ar700111a
Although density functional theory is widely used in the computational chemistry community, the most popular density functional, B3LYP, has some serious shortcomings: (i) it is better for main-group chemistry than for transition metals; (ii) it systematically underestimates reaction barrier heights; (iii) it is inaccurate for interactions dominated by medium-range correlation energy, such as van der Waals attraction, aromatic−aromatic stacking, and alkane isomerization energies. We have developed a variety of databases for testing and designing new density functionals. We used these data to design new density functionals, called M06-class (and, earlier, M05-class) functionals, for which we enforced some fundamental exact constraints such as the uniform-electron-gas limit and the absence of self-correlation energy. Our M06-class functionals depend on spin-up and spin-down electron densities (i.e., spin densities), spin density gradients, spin kinetic energy densities, and, for nonlocal (also called hybrid) functionals, Hartree−Fock exchange. We have developed four new functionals that overcome the above-mentioned difficulties: (a) M06, a hybrid meta functional, is a functional with good accuracy “across-the-board” for transition metals, main group thermochemistry, medium-range correlation energy, and barrier heights; (b) M06-2X, another hybrid meta functional, is not good for transition metals but has excellent performance for main group chemistry, predicts accurate valence and Rydberg electronic excitation energies, and is an excellent functional for aromatic−aromatic stacking interactions; (c) M06-L is not as accurate as M06 for barrier heights but is the most accurate functional for transition metals and is the only local functional (no Hartree−Fock exchange) with better across-the-board average performance than B3LYP; this is very important because only local functionals are affordable for many demanding applications on very large systems; (d) M06-HF has good performance for valence, Rydberg, and charge transfer excited states with minimal sacrifice of ground-state accuracy. In this Account, we compared the performance of the M06-class functionals and one M05-class functional (M05-2X) to that of some popular functionals for diverse databases and their performance on several difficult cases. The tests include barrier heights, conformational energy, and the trend in bond dissociation energies of Grubbsʼ ruthenium catalysts for olefin metathesis. Based on these tests, we recommend (1) the M06-2X, BMK, and M05-2X functionals for main-group thermochemistry and kinetics, (2) M06-2X and M06 for systems where main-group thermochemistry, kinetics, and noncovalent interactions are all important, (3) M06-L and M06 for transition metal thermochemistry, (4) M06 for problems involving multireference rearrangements or reactions where both organic and transition-metal bonds are formed or broken, (5) M06-2X, M05-2X, M06-HF, M06, and M06-L for the study of noncovalent interactions, (6) M06-HF when the use of full Hartree−Fock exchange is important, for example, to avoid the error of self-interaction at long-range, (7) M06-L when a local functional is required, because a local functional has much lower cost for large systems.
Co-reporter:Christopher J. Cramer and Donald G. Truhlar
Accounts of Chemical Research 2008 Volume 41(Issue 6) pp:760
Publication Date(Web):May 31, 2008
DOI:10.1021/ar800019z
Continuum mean-field models that have been carefully designed to address the various electrostatic and nonelectrostatic interactions that develop between a molecule and a surrounding medium are particularly efficient tools for studying the effects of condensed phases on molecular structure, energetics, properties, spectra, interaction potentials, and dynamics. The SM8 model may be combined with density functional theory or Hartree–Fock theory to describe a solute’s electronic structure and its self-consistent-field polarization by a solvent. A key feature is the use of class IV charge models to obtain accurate charge distributions (either in the vapor phase or in solution), even when using small basis sets that are affordable for large systems. A second key feature is that nonelectrostatic effects due to cavity formation, dispersion interactions, and changes in solvent structure are included in terms of empirical atomic surface tensions that depend on geometry but do not require atom-type assignments by the user. Use of an analytic surface area algorithm provides very stable energy gradients that allow geometry optimization in solution. The SM8 continuum model, the culmination of a series of SMx models (x = 1–8), permits the modeling of such diverse media as aqueous and organic solvents, soils, lipid bilayers, and air–water interfaces. In addition to predicting accurate transfer free energies between gaseous and condensed phases or between two different condensed phases, SMx models have been useful for predicting the significant influence of condensed phases on processes associated with a change in molecular charge, including acid/base equilibria and oxidation/reduction processes. In this Account, we provide an overview of the algorithms associated with the computation of free energies of solvation in the SM8 model. We also compare the accuracies of the SM8 model with those of other continuum solvation models. Finally, we highlight applications of the SM8 models to compute ionic solvation free energies, oxidation and reduction potentials, and pKa values.
Co-reporter:Yan Zhao and Donald G. Truhlar
Journal of Chemical Theory and Computation 2008 Volume 4(Issue 11) pp:1849-1868
Publication Date(Web):October 15, 2008
DOI:10.1021/ct800246v
The hybrid meta density functionals M05-2X and M06-2X have been shown to provide broad accuracy for main group chemistry. In the present article we make the functional form more flexible and improve the self-interaction term in the correlation functional to improve its self-consistent-field convergence. We also explore the constraint of enforcing the exact forms of the exchange and correlation functionals through second order (SO) in the reduced density gradient. This yields two new functionals called M08-HX and M08-SO, with different exact constraints. The new functionals are optimized against 267 diverse main-group energetic data consisting of atomization energies, ionization potentials, electron affinities, proton affinities, dissociation energies, isomerization energies, barrier heights, noncovalent complexation energies, and atomic energies. Then the M08-HX, M08-SO, M05-2X, and M06-2X functionals and the popular B3LYP functional are tested against 250 data that were not part of the original training data for any of the functionals, in particular 164 main-group energetic data in 7 databases, 39 bond lengths, 38 vibrational frequencies, and 9 multiplicity-changing electronic transition energies. These tests include a variety of new challenges for complex systems, including large-molecule atomization energies, organic isomerization energies, interaction energies in uracil trimers, and bond distances in crowded molecules (in particular, cyclophanes). The M08-HX functional performs slightly better than M08-SO and M06-2X on average, significantly better than M05-2X, and much better than B3LYP for a combination of main-group thermochemistry, kinetics, noncovalent interactions, and electronic spectroscopy. More important than the slight improvement in accuracy afforded by M08-HX is the conformation that the optimization procedure works well for data outside the training set. Problems for which the accuracy is especially improved by the new M08-HX functional include large-molecule atomization energies, noncovalent interaction energies, conformational energies in aromatic peptides, barrier heights, multiplicity-changing excitation energies, and bond lengths in crowded molecules.
Co-reporter:Aleksandr V. Marenich, Christopher J. Cramer and Donald G. Truhlar
Journal of Chemical Theory and Computation 2008 Volume 4(Issue 6) pp:877-887
Publication Date(Web):May 8, 2008
DOI:10.1021/ct800029c
Co-reporter:Oksana Tishchenko, Jingjing Zheng and Donald G. Truhlar
Journal of Chemical Theory and Computation 2008 Volume 4(Issue 8) pp:1208-1219
Publication Date(Web):July 1, 2008
DOI:10.1021/ct800077r
By combining the generalized valence bond ansatz of correlated participating orbitals (CPO) with the complete-active-space prescription for selecting configurations and with the use of multireference second order perturbation theory (MRMP2) for including dynamical correlation, we define three levels of multireference (MR) theoretical model chemistries for electronic structure calculations of chemical reaction energies and barrier heights. The three levels differ in their choice of which orbitals are considered to be participating; the choices are called nominal (nom-CPO), moderate (mod-CPO), and extended (ext-CPO). Combining any of these three choices with a method for treatment of dynamical correlation energy and a one-electron basis set yields a theoretical model chemistry. Unlike the full-valence choice of active orbitals, the CPO choices lead to active spaces that contain the orbitals needed to include important static correlation effects on chemical reactions but do not increase with the size of the nonparticipating portion of the system, and hence they remain viable computational options even for many large and complex reacting systems. The accuracies of the new levels, combined with the MG3S basis set (a partially augmented, multiply polarized valence triple-ζ basis with appropriately tight d functions for 3p-block elements) and with the fully augmented correlation-consistent aug-cc-pVTZ basis set, are assessed against a previously presented database of barrier heights for diverse reaction types. We find that nom-CPO level captures the bulk of the static correlation energy, and MRMP2/nom-CPO calculations have an average error of only 1.4 kcal/mol in barrier heights, which may be compared to 5.0 kcal/mol for single-reference MP2 theory, 2.5 kcal/mol for CCSD, and 4.1 and 1.0 kcal/mol for the B3LYP and M06−2X density functionals, respectively. The accuracy of MRMP2/CPO for transition structure bond lengths and donor−acceptor distances is excellent, with a mean unsigned error of only 0.007 Å as compared to 0.018 Å for CCSD, 0.019 Å for M06−2X, and 0.039 Å for MP2 and B3LYP. We also introduce a new multireference diagnostic, called the M diagnostic, that allows one to measure the importance of static correlation in a given reagent or transition state.
Co-reporter:Anastassia Sorkin, Erin E. Dahlke and Donald G. Truhlar
Journal of Chemical Theory and Computation 2008 Volume 4(Issue 5) pp:683-688
Publication Date(Web):April 19, 2008
DOI:10.1021/ct7003462
The electrostatically embedded many-body expansion (EE-MB), at both the second and third order, that is, the electrostatically embedded pairwise additive (EE-PA) approximation and the electrostatically embedded three-body (EE-3B) approximation, are tested for mixed ammonia−water clusters. We examine tetramers, pentamers, and hexamers for three different density functionals and two levels of wave function theory, We compare the many-body results to the results of full calculations performed without many-body expansions. Because of the differing charge distributions in the two kinds of monomers, this provides a different kind of test of the usefulness of the EE-MB method than was provided by previous tests on pure water clusters. We find only small errors due to the truncation of the many-body expansion for the mixed clusters. In particular, for tests on tetramers and pentamers, the mean absolute deviations for truncation at second order are 0.36−0.98 kcal/mol (average: 0.66 kcal/mol), and the mean absolute deviations for truncation at third order are 0.04−0.28 (average: 0.16 kcal/mol). These may be compared to a spread of energies as large as 4.24 kcal/mol in the relative energies of various structures of pentamers and to deviations of up to 8.57 kcal/mol of the full calculations of relative energies from the best estimates of the relative energies. When the methods are tested on hexamers, the mean unsigned deviation per monomer remains below 0.10 kcal/mol for EE-PA and below 0.03 kcal/mol for EE-3B. Thus the additional error due to the truncation of the expansion is small compared to the accuracy needed or the other approximations involved in practical calculations. This means that the EE-MB expansion in combination with density functional theory or wave function theory for the oligomers provides a useful practical model chemistry for making electronic structure calculations and simulations more affordable by improving the scaling with respect to system size.
Co-reporter:Mark A. Iron, Andreas Heyden, Grażyna Staszewska and Donald G. Truhlar
Journal of Chemical Theory and Computation 2008 Volume 4(Issue 5) pp:804-818
Publication Date(Web):April 25, 2008
DOI:10.1021/ct700343t
We present a new electronic structure approximation called Tight Binding Configuration Interaction. It uses a tight-binding Hamiltonian to obtain orbitals that are used in a configuration interaction calculation that includes explicit charge interactions. This new method is better capable of predicting energies, ionization potentials, and fragmentation charges than the Wolfsberg−Helmholz Tight-Binding and Many-Body Tight-Binding models reported earlier (Staszewska, G.; Staszewski, P.; Schultz, N. E.; Truhlar, D. Phys. Rev. B 2005, 71, 045423). The method is illustrated for clusters and nanoparticles containing aluminum.
Co-reporter:Masahiro Higashi and Donald G. Truhlar
Journal of Chemical Theory and Computation 2008 Volume 4(Issue 5) pp:790-803
Publication Date(Web):March 26, 2008
DOI:10.1021/ct800004y
We present a new method for generating global or semiglobal potential energy surfaces in the presence of an electrostatic potential; the new method can be used to model chemical reactions in solution or in an enzyme, nanocavity, or other chemical environment. The method extends the multiconfiguration molecular mechanics method so that the energy depends on the electrostatic potential at each atomic center. The charge distribution of the system can also be calculated. We illustrate the method by applying it to the symmetric bimolecular reaction Cl– + CH3Cl′ → ClCH3 + Cl′– in aqueous solution, where the potential energy information is obtained by the combined density functional and molecular mechanical method, that is, by the combined quantum mechanical and molecular mechanical method (QM/MM) with the QM level being density functional theory. It is found that we can describe a semiglobal potential energy surface in aqueous solution with electronic structure information obtained entirely in the gas phase, including the linear and quadratic responses to variations in the electrostatic potential distribution. The semiglobal potential energy surface calculated by the present method is in good agreement with that calculated directly without any fitting.
Co-reporter:Masahiro Higashi and Donald G. Truhlar
Journal of Chemical Theory and Computation 2008 Volume 4(Issue 7) pp:1032-1039
Publication Date(Web):May 29, 2008
DOI:10.1021/ct8000816
We here combine the electrostatically embedded multiconfiguration molecular mechanics (EE−MCMM) method for generating global potential energy surfaces in the presence of an electrostatic potential with molecular mechanics (MM). The resulting EE−MCMM/MM method is illustrated by applying it to carry out a molecular dynamics simulation for the symmetric bimolecular reaction Cl− + CH3Cl′ → ClCH3 + Cl′− in aqueous solution with hybrid density functional theory as the quantum mechanical level. The potential of mean force is calculated, and the free energy barrier is found to be 25.3 kcal/mol, which is in good agreement with previous work. The advantage of the combined EE−MCMM and MM method is that the number of quantum mechanical calculations required for the active subsystem is very small compared to straight direct dynamics.
Co-reporter:Yan Zhao and Donald G. Truhlar
Physical Chemistry Chemical Physics 2008 vol. 10(Issue 19) pp:2813-2818
Publication Date(Web):18 Feb 2008
DOI:10.1039/B717744E
The geometries and binding energies of a recent buckyball tweezers (C60H28) and its supramolecular complexes are investigated using recently developed density functionals (M06-L and M06-2X) that include an accurate treatment of medium-range correlation energy. The pincer part of the tweezers, corannulene, has a strong attractive interaction with C60. However, due to the entropy penalty, the calculated gas-phase free energy of association of the C60@corannulene supramolecule is positive 3.5 kcal mol−1; and this entropy penalty explains why it is difficult to observe C60@corannulene supramolecule experimentally. By using a π-extended tetrathiafulvalene (TTF), in particular 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene (TTFAQ or C20H10S4), as the pincer part, we modeled a new buckyball tweezers. The geometries and binding energies of the new buckyball tweezers and its supramolecular complexes are also calculated. Due to fact that the attractive interaction between TTFAQ and C60 is weaker than that between corannulene and C60, the gas-phase binding free energy in the C60@C60H 32S8 supramolecular complex is smaller than that in the C60@C60H28 supramolecule. We also discuss solvent effects.
Co-reporter:Adam C. Chamberlin, David G. Levitt, Christopher J. Cramer and Donald G. Truhlar
Molecular Pharmaceutics 2008 Volume 5(Issue 6) pp:1064-1079
Publication Date(Web):October 16, 2008
DOI:10.1021/mp800059u
Olive oil partition coefficients are useful for modeling the bioavailability of drug-like compounds. We have recently developed an accurate solvation model called SM8 for aqueous and organic solvents (Marenich, A. V.; Olson, R. M.; Kelly, C. P.; Cramer, C. J.; Truhlar, D. G. J. Chem. Theory Comput. 2007, 3, 2011) and a temperature-dependent solvation model called SM8T for aqueous solution (Chamberlin, A. C.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2008, 112, 3024). Here we describe an extension of SM8T to predict air−olive oil and water−olive oil partitioning for drug-like solutes as functions of temperature. We also describe the database of experimental partition coefficients used to parametrize the model; this database includes 371 entries for 304 compounds spanning the 291−310 K temperature range.Keywords: Bioavailability; continuum model; lipid−water partitioning; membrane partitioning;
Co-reporter:Donald G. Truhlar
Theoretical Chemistry Accounts 2008 Volume 121( Issue 1-2) pp:103
Publication Date(Web):2008 September
DOI:10.1007/s00214-008-0448-1
Co-reporter:Yan Zhao
Theoretical Chemistry Accounts 2008 Volume 120( Issue 1-3) pp:215-241
Publication Date(Web):2008 May
DOI:10.1007/s00214-007-0310-x
We present two new hybrid meta exchange- correlation functionals, called M06 and M06-2X. The M06 functional is parametrized including both transition metals and nonmetals, whereas the M06-2X functional is a high-nonlocality functional with double the amount of nonlocal exchange (2X), and it is parametrized only for nonmetals.The functionals, along with the previously published M06-L local functional and the M06-HF full-Hartree–Fock functionals, constitute the M06 suite of complementary functionals. We assess these four functionals by comparing their performance to that of 12 other functionals and Hartree–Fock theory for 403 energetic data in 29 diverse databases, including ten databases for thermochemistry, four databases for kinetics, eight databases for noncovalent interactions, three databases for transition metal bonding, one database for metal atom excitation energies, and three databases for molecular excitation energies. We also illustrate the performance of these 17 methods for three databases containing 40 bond lengths and for databases containing 38 vibrational frequencies and 15 vibrational zero point energies. We recommend the M06-2X functional for applications involving main-group thermochemistry, kinetics, noncovalent interactions, and electronic excitation energies to valence and Rydberg states. We recommend the M06 functional for application in organometallic and inorganometallic chemistry and for noncovalent interactions.
Co-reporter:Peifeng Su, Wei Wu, Casey P. Kelly, Christopher J. Cramer and Donald G. Truhlar
The Journal of Physical Chemistry A 2008 Volume 112(Issue 50) pp:12761-12768
Publication Date(Web):August 1, 2008
DOI:10.1021/jp711655k
A new solvation model, called VBSM, is presented. The model combines valence bond (VB) theory with parameters determined for the SM6 solvation model (Kelly, C. P.; Cramer, C. J.; Truhlar, D. G. J. Chem. Theo. Comp. 2005, 1, 1133−1152). VBSM, like SM6, is based on the generalized Born (GB) approximation for bulk electrostatics and atomic surface tensions to account for cavitation, dispersion, and solvent structure (CDS). The solvation free energy of VBSM includes (i) a self-consistent polarization term obtained by using VB atomic charges in a GB reaction field with a VB self-consistent field procedure that minimizes the total energy of the system with respect to the valence bond orbitals and (ii) a geometry-dependent CDS term to account for deviations from bulk-electrostatic solvation. Test calculations for a few systems show that the liquid-phase partial atomic charges obtained by VBSM are in good agreement with liquid-phase charges obtained by charge model CM4 (Kelly, C. P.; Cramer, C. J.; Truhlar, D. G. J. Chem. Theo. Comp. 2005, 1, 1133−1152). Free energies of solvation are calculated for two prototype test cases, namely, for the degenerate SN2 reaction of Cl− with CH3Cl in water and for a Menshutkin reaction in water. These calculations show that the VBSM method provides a practical alternative to single-configuration self-consistent field theory for solvent effects in molecules and chemical reactions.
Co-reporter:Zhen Hua Li
The Journal of Physical Chemistry C 2008 Volume 112(Issue 30) pp:11109-11121
Publication Date(Web):July 2, 2008
DOI:10.1021/jp711349v
The association reactions of Al atoms with Aln clusters and nanoparticles and the unimolecular dissociation reactions of Aln clusters and nanoparticles have been studied using classical molecular dynamics trajectory simulations. Thermal reaction rate constants of the association rate constant with m = 1 and the dissociation rate constant with m = 1−8 have been simulated, and subsequently, the association rate constants for m > 1 can be determined indirectly. It was found that the monomer association rate constants depend weakly on temperature. For the unimolecular dissociation reactions, the rate constants depend strongly on temperature, and the temperature dependences can be fitted using the Arrhenius equation. The results indicate that the unimolecular dissociation reaction has a high activation barrier that tends to increase with particle size and furthermore that the preferred dissociation process is always monomer emission. With both the monomer association and monomer emission rate constants, the standard Gibbs free energy changes of the Alm + Aln−m ↔ Aln reactions on the ground-state potential energy surface with n = 2−20, 30, 40, 50, and 60 have been determined. These standard Gibbs free energy changes determined by the molecular dynamics trajectory simulations agree fairly well with the corresponding values determined by previous Monte Carlo equilibrium simulations. The rate constants determined in this study can be used to model the formation and growth of metal nanoparticles under a wide range of conditions from 1100 to 3300 K.
Co-reporter:Rosendo Valero and Donald G. Truhlar, Ahren W. Jasper
The Journal of Physical Chemistry A 2008 Volume 112(Issue 25) pp:5756-5769
Publication Date(Web):June 5, 2008
DOI:10.1021/jp800738b
The development of spin-coupled diabatic representations for theoretical semiclassical treatments of photodissociation dynamics is an important practical goal, and some of the assumptions required to carry this out may be validated by applications to simple systems. With this objective, we report here a study of the photodissociation dynamics of the prototypical HBr system using semiclassical trajectory methods. The valence (spin-free) potential energy curves and the permanent and transition dipole moments were computed using high-level ab initio methods and were transformed to a spin-coupled diabatic representation. The spin−orbit coupling used in the transformation was taken as that of atomic bromine at all internuclear distances. Adiabatic potential energy curves, nonadiabatic couplings and transition dipole moments were then obtained from the diabatic ones and were used in all the dynamics calculations. Nonadiabatic photodissociation probabilities were computed using three semiclassical trajectory methods, namely, coherent switching with decay of mixing (CSDM), fewest switches with time uncertainty (FSTU), and its recently developed variant with stochastic decoherence (FTSU/SD), each combined with semiclassical sampling of the initial vibrational state. The calculated branching fraction to the higher fine-structure level of the bromine atom is in good agreement with experiment and with more complete theoretical treatments. The present study, by comparing our new calculations to wave packet calculations with distance-dependent ab initio spin−orbit coupling, validates the semiclassical trajectory methods, the semiclassical initial state sample scheme, and the use of a distance-independent spin−orbit coupling for future applications to polyatomic photodissociation. Finally, using LiBr+ as a model system, it is shown that accurate spin-coupled potential curves can also be constructed for odd-electron systems using the same strategy as for HBr.
Co-reporter:Hannah R. Leverentz and Donald G. Truhlar
The Journal of Physical Chemistry A 2008 Volume 112(Issue 26) pp:6009-6016
Publication Date(Web):June 10, 2008
DOI:10.1021/jp8018364
Noncovalent interactions of a hydrogen bond donor with an aromatic π system present a challenge for density functional theory, and most density functionals do not perform well for this kind of interaction. Here we test seven recent density functionals from our research group, along with the popular B3LYP functional, for the dimer of H2S with benzene. The functionals considered include the four new meta and hybrid meta density functionals of the M06 suite, three slightly older hybrid meta functionals, and the B3LYP hybrid functional, and they were tested for their abilities to predict the dissociation energies of three conformations of the H2S−benzene dimer and to reproduce the key geometric parameters of the equilibrium conformation of this dimer. All of the functionals tested except B3LYP correctly predict which of the three conformations of the dimer is the most stable. The functionals that are best able to reproduce the geometry of the equilibrium conformation of the dimer with a polarized triple-ζ basis set are M06-L, PWB6K, and MPWB1K, each having a mean unsigned relative error across the two experimentally verifiable geometric parameters of only 8%. The success of M06-L is very encouraging because it is a local functional, which reduces the cost for large simulations. The M05-2X functional yields the most accurate binding energy of a conformation of the dimer for which a binding energy calculated at the CCSD(T) level of theory is available; M05-2X gives a binding energy for the system with a difference of merely 0.02 kcal/mol from that obtained by the CCSD(T) calculation. The M06 functional performs well in both categories by yielding a good representation of the geometry of the equilibrium structure and by giving a binding energy that is only 0.19 kcal/mol different from that calculated by CCSD(T). We conclude that the new generation of density functionals should be useful for a variety of problems in biochemistry and materials where aromatic functional groups can serve as hydrogen bond acceptors.
Co-reporter:Jingjing Zheng, Shuxia Zhang and Donald G. Truhlar
The Journal of Physical Chemistry A 2008 Volume 112(Issue 46) pp:11509-11513
Publication Date(Web):October 24, 2008
DOI:10.1021/jp806617m
Rate constants of the prototypical methyl−methyl radical association reaction are calculated on the basis of variational transition-state theory with a variable reaction coordinate and a multifaceted dividing surface. The potential energies required in the Monte Carlo integrations are evaluated directly using the M06 and M06-L density functionals. The rate constants are calculated at the canonical, microcanonical, and E,J-resolved microcanonical levels. The best prediction of rate constants is based on the potential energies calculated by the M06-L density functional; these agree with experimental data quantitatively from 300 to 1000 K. This study shows that density functional theory can be accurate enough for calculating rate constants of reactions with loose transition states, whereas previously only multireference wave function methods, which are more complicated and more expensive, had been demonstrated to be sufficiently accurate. The application of density functional theory for the loose transition states will allow larger and complicated systems to be studied efficiently.
Co-reporter:Yan Zhao and Donald G. Truhlar
The Journal of Physical Chemistry A 2008 Volume 112(Issue 30) pp:6794-6799
Publication Date(Web):July 10, 2008
DOI:10.1021/jp804583d
The performance of the M06-L density functional has been tested for four databases of NMR isotropic chemical shielding constants. Comparison with the B3LYP, BLYP, HCTH, KT1, KT2, LSDA, OPBE, OLYP, PBE, TPSS, and VSXC functionals shows that M06-L has improved performance for calculating NMR chemical shielding constants, especially for highly correlated systems. We also found that VSXC and M06-L have encouraging accuracy for calculating 13C chemical shielding constants, and both functionals perform very well for the chemical shielding constants in the o-benzyne molecule.
Co-reporter:Yan Zhao
Theoretical Chemistry Accounts 2008 Volume 119( Issue 5-6) pp:525
Publication Date(Web):2008 April
DOI:10.1007/s00214-007-0401-8
Co-reporter:Donald G. Truhlar
Theoretical Chemistry Accounts 2008 Volume 121( Issue 1-2) pp:105-106
Publication Date(Web):2008 September
DOI:10.1007/s00214-008-0449-0
Co-reporter:Antonio Fernández-Ramos;Benjamin A. Ellingson
Theoretical Chemistry Accounts 2007 Volume 118( Issue 4) pp:813-826
Publication Date(Web):2007 October
DOI:10.1007/s00214-007-0328-0
This article shows how to evaluate rotational symmetry numbers for different molecular configurations and how to apply them to transition state theory. In general, the symmetry number is given by the ratio of the reactant and transition state rotational symmetry numbers. However, special care is advised in the evaluation of symmetry numbers in the following situations: (i) if the reaction is symmetric, (ii) if reactants and/or transition states are chiral, (iii) if the reaction has multiple conformers for reactants and/or transition states and, (iv) if there is an internal rotation of part of the molecular system. All these four situations are treated systematically and analyzed in detail in the present article. We also include a large number of examples to clarify some complicated situations, and in the last section we discuss an example involving an achiral diasteroisomer.
Co-reporter:Zhen Hua Li;Rosendo Valero
Theoretical Chemistry Accounts 2007 Volume 118( Issue 1) pp:9-24
Publication Date(Web):2007 July
DOI:10.1007/s00214-006-0237-7
Diabatic potential energy surfaces are a convenient starting point for dynamics calculations of photochemical processes, and they can be calculated by the fourfold way direct diabatization scheme. Here we present an improved definition of the reference orbital for applying the fourfold way direct diabatization scheme to ammonia. The improved reference orbital is a geometry-dependent hybrid orbital that allows one to define consistent dominant configuration lists at all geometries important for photodissociation. Using diabatic energies calculated with the new reference orbital and consistent dominant configuration lists, we have refitted the analytical representations of the ground and the first electronically excited singlet-state potential energy surfaces and the diabatic coupling surface. Improved functional forms were used to reproduce the experimental dissociation energies and excitation energies, which will be important for subsequent simulations of photochemical dynamics. We find that the lowest-energy conical intersection point is at 5.16 eV, with C2v symmetry.
Co-reporter:Hai Lin
Theoretical Chemistry Accounts 2007 Volume 117( Issue 2) pp:
Publication Date(Web):2007 February
DOI:10.1007/s00214-006-0143-z
This paper briefly reviews the current status of the most popular methods for combined quantum mechanical/molecular mechanical (QM/MM) calculations, including their advantages and disadvantages. There is a special emphasis on very general link-atom methods and various ways to treat the charge near the boundary. Mechanical and electric embedding are contrasted. We consider methods applicable to gas-phase organic chemistry, liquid-phase organic and organometallic chemistry, biochemistry, and solid-state chemistry. Then we review some recent tests of QM/MM methods and summarize what we learn about QM/MM from these studies. We also discuss some available software. Finally, we present a few comments about future directions of research in this exciting area, where we focus on more intimate blends of QM with MM.
Co-reporter:Agnieszka Dybala-Defratyka;Piotr Paneth;Ruma Banerjee
PNAS 2007 Volume 104 (Issue 26 ) pp:10774-10779
Publication Date(Web):2007-06-26
DOI:10.1073/pnas.0702188104
Hydrogen transfer reactions catalyzed by coenzyme B12-dependent methylmalonyl-CoA mutase have very large kinetic isotope effects, indicating that they proceed by a highly quantal
tunneling mechanism. We explain the kinetic isotope effect by using a combined quantum mechanical/molecular mechanical potential
and semiclassical quantum dynamics calculations. Multidimensional tunneling increases the magnitude of the calculated intrinsic
hydrogen kinetic isotope effect by a factor of 3.6 from 14 to 51, in excellent agreement with experimental results. These
calculations confirm that tunneling contributions can be large enough to explain even a kinetic isotope effect >50, not because
the barrier is unusually thin but because corner-cutting tunneling decreases the distance over which the system tunnels without
a comparable increase in either the effective potential barrier or the effective mass for tunneling.
Co-reporter:Jingzhi Pu Dr.;Jiali Gao
ChemPhysChem 2005 Volume 6(Issue 9) pp:
Publication Date(Web):5 AUG 2005
DOI:10.1002/cphc.200400602
The generalized hybrid orbital (GHO) method has previously been formulated for combining molecular mechanics with various levels of quantum mechanics, in particular semiempirical neglect of diatomic differential overlap theory, ab initio Hartree–Fock theory, and self-consistent charge density functional tight-binding theory. To include electron-correlation effects accurately and efficiently in GHO calculations, we extend the GHO method to density functional theory in the generalized-gradient approximation and hybrid density functional theory (denoted by GHO-DFT and GHO-HDFT, respectively) using Gaussian-type orbitals as basis functions. In the proposed GHO-(H)DFT formalism, charge densities in auxiliary hybrid orbitals are included to calculate the total electron density. The orthonormality constraints involving the auxiliary Kohn–Sham orbitals are satisfied by carrying out the hybridization in terms of a set of Löwdin symmetrically orthogonalized atomic basis functions. Analytical gradients are formulated for GHO-(H)DFT by incorporating additional forces associated with GHO basis transformations. Scaling parameters are introduced for some of the one-electron integrals and are optimized to obtain the correct charges and geometry near the QM/MM boundary region. The GHO-(H)DFT method based on the generalized gradient approach (GGA) (BLYP and mPWPW91) and HDFT methods (B3 LYP, mPW1PW91, and MPW1 K) is tested—for geometries and atomic charges—against a set of small molecules. The following quantities are tested: 1) the CC stretch potential in ethane, 2) the torsional barrier for internal rotation around the central CC bond in n-butane, 3) proton affinities for a set of alcohols, amines, thiols, and acids, 4) the conformational energies of alanine dipeptide, and 5) the barrier height of the hydrogen-atom transfer between n-C4H10 and n-C4H9, where the reaction center is described at the MPW1 K/6–31G(d) level of theory.
Co-reporter:Mireia Garcia-Viloca;Jiali Gao;Martin Karplus
Science 2004 Vol 303(5655) pp:186-195
Publication Date(Web):09 Jan 2004
DOI:10.1126/science.1088172
Abstract
Advances in transition state theory and computer simulations are providing new insights into the sources of enzyme catalysis. Both lowering of the activation free energy and changes in the generalized transmission coefficient (recrossing of the transition state, tunneling, and nonequilibrium contributions) can play a role. A framework for understanding these effects is presented, and the contributions of the different factors, as illustrated by specific enzymes, are identified and quantified by computer simulations. The resulting understanding of enzyme catalysis is used to comment on alternative proposals of how enzymes work.
Co-reporter:Brian K. Kendrick, C. Alden Mead, Donald G. Truhlar
Chemical Physics 2002 Volume 277(Issue 1) pp:31-41
Publication Date(Web):1 March 2002
DOI:10.1016/S0301-0104(02)00281-1
Abstract
We present a new analysis of the nonadiabatic coupling terms in the coupled equations for nuclear motion wave functions when the Born–Oppenheimer (BO) representation is used for the electronic wave function. The new analysis leads to a criterion for truncating the series and neglecting terms in the coupled equations of motion. We show that in general the nonremovable part of the coupling is of the same magnitude as the removable part, except near intersections of the adiabatic states.
Co-reporter:Cristóbal Alhambra, Maria Luz Sánchez, José C. Corchado, Jiali Gao, Donald G. Truhlar
Chemical Physics Letters 2002 Volume 355(3–4) pp:388-394
Publication Date(Web):2 April 2002
DOI:10.1016/S0009-2614(02)00057-X
We report a calculation for a trideuteration kinetic isotope effect (KIE) for the proton transfer step in the oxidation of methylamine by the quinoprotein methylamine dehydrogenase (MADH). The potential field includes 11 025 atoms, and the dynamics are based on a quantum mechanical/molecular mechanical (QM/MM) dynamics simulation and ensemble-averaged canonical variational transition state theory with small-curvature multidimensional tunneling contributions. About 1% of the reaction occurs by overbarrier processes, with the rest due to tunneling. We compute a KIE of 18.3, in good accord with experiment (17.2), but the calculated KIE is reduced to 5.9 when we omit tunneling. This provides the most striking evidence yet for the contribution of tunneling processes to enzymatic reactions at physiological temperatures.
Co-reporter:Cristóbal Alhambra, Maria Luz Sánchez, José Corchado, Jiali Gao, Donald G. Truhlar
Chemical Physics Letters 2001 Volume 347(4–6) pp:512-518
Publication Date(Web):26 October 2001
DOI:10.1016/S0009-2614(01)00921-6
We report a calculation for a trideuteration kinetic isotope effect (KIE) for the proton transfer step in the oxidation of methylamine by the quinoprotein methylamine dehydrogenase (MADH). The potential field includes 11 025 atoms, and the dynamics are based on a quantum mechanical/molecular mechanical (QM/MM) direct dynamics simulation and canonical variational transition state theory with small-curvature multidimensional tunneling contributions. About 1% of the reaction occurs by overbarrier processes, with the rest due to tunneling, and the calculated KIE is reduced to 5.9 when we omit tunneling. This provides the most striking evidence yet for the contribution of tunneling processes to enzymatic reactions at physiological temperatures.
Co-reporter:Donald G. Truhlar;Amnon Kohen
PNAS 2001 Volume 98 (Issue 3 ) pp:848-851
Publication Date(Web):2001-01-30
DOI:10.1073/pnas.98.3.848
This paper draws attention to selected experiments on
enzyme-catalyzed reactions that show convex Arrhenius plots, which are
very rare, and points out that Tolman's interpretation of the
activation energy places a fundamental model-independent constraint on
any detailed explanation of these reactions. The analysis presented
here shows that in such systems, the rate coefficient as a function of
energy is not just increasing more slowly than expected, it is actually
decreasing. This interpretation of the data provides a constraint on
proposed microscopic models, i.e., it requires that any successful
model of a reaction with a convex Arrhenius plot should be consistent
with the microcanonical rate coefficient being a decreasing function of
energy. The implications and limitations of this analysis to
interpreting enzyme mechanisms are discussed. This model-independent
conclusion has broad applicability to all fields of kinetics, and we
also draw attention to an analogy with diffusion in metastable fluids
and glasses.
Co-reporter:Haoyu S. Yu, Lucas J. Fiedler, I.M. Alecu, Donald G. Truhlar
Computer Physics Communications (January 2017) Volume 210() pp:
Publication Date(Web):January 2017
DOI:10.1016/j.cpc.2016.09.004
We present a Python program, FREQ, for calculating the optimal scale factors for calculating harmonic vibrational frequencies, fundamental vibrational frequencies, and zero-point vibrational energies from electronic structure calculations. The program utilizes a previously published scale factor optimization model (Alecu et al., 2010) to efficiently obtain all three scale factors from a set of computed vibrational harmonic frequencies. In order to obtain the three scale factors, the user only needs to provide zero-point energies of 15 or 6 selected molecules. If the user has access to the Gaussian 09 or Gaussian 03 program, we provide the option for the user to run the program by entering the keywords for a certain method and basis set in the Gaussian 09 or Gaussian 03 program. Four other Python programs, input.py, input6, pbs.py, and pbs6.py, are also provided for generating Gaussian 09 or Gaussian 03 input and PBS files. The program can also be used with data from any other electronic structure package. A manual of how to use this program is included in the code package.Program summaryProgram title: FREQCatalogue identifier: AFBH_v1_0Program summary URL:http://cpc.cs.qub.ac.uk/summaries/AFBH_v1_0.htmlProgram obtainable from: CPC Program Library, Queen’s University, Belfast, N. Ireland at http://www.cpc.cs.qub.ac.uk or from Truhlar group software page at comp.chem.umn.edu/freq/Licensing provisions: GNU GPL v3No. of lines in distributed program, including test data, etc.: 3013No. of bytes in distributed program, including test data, etc.: 212537Distribution format: tar.gzProgramming language: PYTHON.Computer: Any computer with PYTHON compiler.Operating systems: Linux, Unix.Classifications: 16.3, 23.External routines: Gaussian 03 or Gaussian 09 (see “Restrictions”).Nature of problem:Optimization of property-specific scale factors for vibrational frequencies for a specific electronic model chemistry.Solution method:The method is based on minimizing the root-mean-square deviation between a set of zero-point energies derived from harmonic vibrational frequencies (either provided by the user or computed on the fly) and their experimentally determined counterparts.Restrictions:In order to compute the electronic model chemistry’s harmonic zero-point energies on the fly, the user must have access to the Gaussian 03 or Gaussian 09 program. If the electronic model chemistry’s zero-point energies are read in, no other program is required.Additional comments:After opening the FREQ.tar.gz file, the user will find a run.sh file which can be used to run all the programs to obtain the scaling factors for a user-chosen electronic structure model chemistry.Running time:Less than a second if the user provides the zero-point energies; if zero-point energies are to be computed, the running time depends on the electronic model chemistry used to compute them as well as the efficiency of the Gaussian 09 program, in our test we obtain the results within 10 min by using one node with eight processors for each Gaussian 09 input on Minnesota Supercomputing Institute’s Mesabi supercomputer.
Co-reporter:Donald G. Truhlar
Archives of Biochemistry and Biophysics (15 September 2015) Volume 582() pp:10-17
Publication Date(Web):15 September 2015
DOI:10.1016/j.abb.2015.05.004
Co-reporter:Pragya Verma and Donald G. Truhlar
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 20) pp:NaN12912-12912
Publication Date(Web):2017/04/24
DOI:10.1039/C7CP01576C
Dipole moments are the first moment of electron density and are fundamental quantities that are often available from experiments. An exchange–correlation functional that leads to an accurate representation of the charge distribution of a molecule should accurately predict the dipole moments of the molecule. It is well known that Kohn–Sham density functional theory (DFT) is more accurate for the energetics of single-reference systems than for the energetics of multi-reference ones, but there has been less study of charge distributions. In this work, we benchmark 48 density functionals chosen with various combinations of ingredients, against accurate experimental data for dipole moments of 78 molecules, in particular 55 single-reference molecules and 23 multi-reference ones. We chose both organic and inorganic molecules, and within the category of inorganic molecules there are both main-group and transition-metal-containing molecules, with some of them being multi-reference. As one would expect, the multi-reference molecules are not as well described by single-reference DFT, and the functionals tested in this work do show larger mean unsigned errors (MUEs) for the 23 multi-reference molecules than the single-reference ones. Five of the 78 molecules have relatively large experimental error bars and were therefore not included in calculating the overall MUEs. For the 73 molecules not excluded, we find that three of the hybrid functionals, B97-1, PBE0, and TPSSh (each with less than or equal to 25% Hartree–Fock (HF) exchange), the range-separated hybrid functional, HSE06 (with HF exchange decreasing from 25% to 0 as interelectronic distance increases), and the hybrid functional, PW6B95 (with 28% HF exchange) are the best performing functionals with each yielding an MUE of 0.18 D. Perhaps the most significant finding of this study is that there exists great similarity among the success rate of various functionals in predicting dipole moments. In particular, of 39 functionals designed as general-purpose functionals and that do not have a global value of 100% HF exchange, the average MUE is 0.23 D, with a standard deviation of only 0.04 D. Among gradient approximations, which are especially interesting because of their speed and portability, the best overall performance is by PBE, HCTH/407, OLYP, OreLYP, and GAM, each with MUE of 0.22 D.
Co-reporter:Junwei Lucas Bao, Xin Zhang, Xuefei Xu and Donald G. Truhlar
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 8) pp:NaN5854-5854
Publication Date(Web):2017/01/30
DOI:10.1039/C6CP08896A
Accurately predicting bond length and bond dissociation energy for bimetallic diatomic molecules that involve metal–metal multiple bonds is a great challenge for electronic structure theory, in part because many of these molecules have inherently multi-configuration wave functions, a characteristic that is variously labeled as strong correlation or multireference character. Although various popular density functionals are widely used in studying metal–metal bonding in catalysis, their accuracy can be questioned, and it is important to see both how well and how poorly a functional can perform. Here we test 50 Kohn–Sham exchange–correlation density functionals for selected 3d and 4d hetero- and homonuclear bimetallic diatomic molecules against experimental bond lengths and bond energies. We found that for the majority of the density functionals, the mean unsigned error in predicting the bond length is larger than 0.08 Å, and for the bond energy, half of the functionals give a mean unsigned error larger than 20 kcal mol−1. This indicates that such highly multireference bimetallic systems are challenging for KS-DFT. However, some exchange–correlation functionals perform significantly better than average for both bond energies and bond lengths, in particular, BLYP, M06-L, N12-SX, OreLYP, RPBE, and revPBE, and are recommended for both kinds of calculations. Other functionals that perform relatively well for bond lengths include MGGA_MS0, MOHLYP, OLYP, PBE, and SOGGA11, and other functionals that perform relatively well for bond energies include GAM, M05, M06, MN15, and τ-HCTHhyb. Although some of these functionals (M05, M06, MN15, N12-SX, and τ-HCTHhyb) contain a nonzero percentage of Hartree–Fock exchange, a broader conclusion is that Hartree–Fock exchange brings in a static correlation error and usually tends to make the results, especially the bond lengths, less accurate. We find some significant differences between all-electron calculations and calculations with effective core potentials. For analysis, the article also presents CASSCF calculations of the percentage contributions of the dominant configurations, and the paper compares orbitals and configurations obtained in DFT calculations to those in CASSCF calculations. The equilibrium bond distance of Rh2 is not available from experiments, and we predict it to be 2.22 Å. The bond energy of VCr is not available from experiments, and we predict it to be 52.9 kcal mol−1.
Co-reporter:Raphael F. Ribeiro, Aleksandr V. Marenich, Christopher J. Cramer and Donald G. Truhlar
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 23) pp:NaN10922-10922
Publication Date(Web):2011/05/12
DOI:10.1039/C0CP02784G
We present M06-2X density functional calculations of the chloroform/water partition coefficients of cytosine, thymine, uracil, adenine, and guanine and calculations of the free energies of association of selected unsubstituted and alkylated nucleotide base pairs in chloroform and water. Both hydrogen bonding and π–π stacking interactions are considered. Solvation effects are treated using the continuum solvent models SM8, SM8AD, and SMD, including geometry optimization in solution. Comparison of theoretical results with available experimental data indicates that all three of these solvation models predict the chloroform–water partition coefficients for the studied nucleobases qualitatively well, with mean unsigned errors in the range of 0.4–1.3 log units. All three models correctly predict the preference for hydrogen bonding over stacking for nucleobase pairs solvated in chloroform, and SM8, SM8AD, and SMD show similar accuracy in predicting the corresponding free energies of association. The agreement between theory and experiment for the association free energies of the dimers in water is more difficult to assess, as the relevant experimental data are indirect. Theory predicts that the stacking interaction of nucleobases in water is more favorable than hydrogen bonding for only two out of three tested hetero-dimers.
Co-reporter:Jingjing Zheng and Donald G. Truhlar
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 28) pp:NaN7793-7793
Publication Date(Web):2010/05/25
DOI:10.1039/B927504E
Five isomerization reactions involving intramolecular hydrogen-transfer in butoxyl radicals have been studied using variational transition state theory with small curvature tunneling. A set of best estimates of barrier heights and reaction energies for these five reactions was obtained by using coupled cluster theory including single and double excitations with a quasiperturbative treatment of connected triple excitations and a basis set extrapolated to the complete basis set limit plus core–valence correlation contributions and scalar relativistic corrections. This work predicts high-pressure limiting rate constants of these five reactions over the temperature range 200–2500 K and clarifies the available experimental data from indirect measurements. This study shows the importance of performing rate calculations with proper accounting for tunneling and torsional anharmonicity. We also proposed two new models for use in fitting rate constants over wide ranges of temperature.
Co-reporter:Christopher J. Cramer and Donald G. Truhlar
Physical Chemistry Chemical Physics 2009 - vol. 11(Issue 46) pp:NaN10816-10816
Publication Date(Web):2009/10/21
DOI:10.1039/B907148B
We introduce density functional theory and review recent progress in its application to transition metal chemistry. Topics covered include local, meta, hybrid, hybrid meta, and range-separated functionals, band theory, software, validation tests, and applications to spin states, magnetic exchange coupling, spectra, structure, reactivity, and catalysis, including molecules, clusters, nanoparticles, surfaces, and solids.
Co-reporter:Pragya Verma, Zoltan Varga, Johannes E. M. N. Klein, Christopher J. Cramer, Lawrence Que and Donald G. Truhlar
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 20) pp:NaN13069-13069
Publication Date(Web):2017/05/02
DOI:10.1039/C7CP01263B
Our ability to understand and simulate the reactions catalyzed by iron depends strongly on our ability to predict the relative energetics of spin states. In this work, we studied the electronic structures of Fe2+ ion, gaseous FeO and 14 iron complexes using Kohn–Sham density functional theory with particular focus on determining the ground spin state of these species as well as the magnitudes of relevant spin-state energy splittings. The 14 iron complexes investigated in this work have hexacoordinate geometries of which seven are Fe(II), five are Fe(III) and two are Fe(IV) complexes. These are calculated using 20 exchange–correlation functionals. In particular, we use a local spin density approximation (LSDA) – GVWN5, four generalized gradient approximations (GGAs) – BLYP, PBE, OPBE and OLYP, two non-separable gradient approximations (NGAs) – GAM and N12, two meta-GGAs – M06-L and M11-L, a meta-NGA – MN15-L, five hybrid GGAs – B3LYP, B3LYP*, PBE0, B97-3 and SOGGA11-X, four hybrid meta-GGAs – M06, PW6B95, MPW1B95 and M08-SO and a hybrid meta-NGA – MN15. The density functional results are compared to reference data, which include experimental results as well as the results of diffusion Monte Carlo (DMC) calculations and ligand field theory estimates from the literature. For the Fe2+ ion, all functionals except M11-L correctly predict the ground spin state to be quintet. However, quantitatively, most of the functionals are not close to the experimentally determined spin-state splitting energies. For FeO all functionals predict quintet to be the ground spin state. For the 14 iron complexes, the hybrid functionals B3LYP, MPW1B95 and MN15 correctly predict the ground spin state of 13 out of 14 complexes and PW6B95 gets all the 14 complexes right. The local functionals, OPBE, OLYP and M06-L, predict the correct ground spin state for 12 out of 14 complexes. Two of the tested functionals are not recommended to be used for this type of study, in particular M08-SO and M11-L, because M08-SO systematically overstabilizes the high spin state, and M11-L systematically overstabilizes the low spin state.
Co-reporter:Tao Yu, Jingjing Zheng and Donald G. Truhlar
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 2) pp:NaN494-494
Publication Date(Web):2011/11/25
DOI:10.1039/C1CP22578B
We have employed electronic structure calculations and the recently proposed multi-structural (MS) anharmonicity method to calculate partition functions and thermodynamic quantities, in particular entropy and heat capacity, for n-heptane and isoheptane. We included all structures, of which there are 59 for n-heptane and 37 for isoheptane, and we carried out the calculations both in the local harmonic approximation and by including torsional (T) anharmonicity. In addition, ΔS°, ΔH, and ΔG° for the isomerization reaction between these two species were also calculated. It is found that all calculated thermodynamic quantities based on the MS-T approximation in the temperature range from 298 K to 1500 K agree well with experimental data from the American Petroleum Institute (API) tables or Thermodynamics Research Center (TRC) data series and with values obtained from Benson's empirical parameters fit to experiment. This demonstrates not only the high accuracy of the electronic structure calculations but also that the MS-T method can be used to include both multiple-structure anharmonicity and torsional anharmonicity in the calculation of thermodynamic properties for complex molecules that contain many torsions. It also gives us confidence that we can apply the MS-T statistical thermodynamic method to obtain thermodynamic properties (i) over a broader temperature range than that for which data are available in the API tables, TRC data series, or from empirical estimation and (ii) to the many molecules for which experimental data are not available at any temperature.
Co-reporter:Roberto Peverati and Donald G. Truhlar
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 47) pp:NaN16191-16191
Publication Date(Web):2012/09/17
DOI:10.1039/C2CP42576A
We present two new exchange–correlation functionals for hybrid Kohn–Sham electronic structure calculations based on the nonseparable functional form introduced recently in the N12 and MN12-L functionals but now with the addition of screened Hartree–Fock exchange. The first functional depends on the density and the density gradient and is called N12-SX; the second functional depends on the density, the density gradient, and the kinetic energy density and is called MN12-SX. Both new functionals include a portion of the Hartree–Fock exchange at short-range, but Hartree–Fock exchange is screened at long range. The accuracies of the two new functionals are compared to those of the recent N12 and MN12-L local functionals to show the effect of adding screened exchange, are compared to the previously best available screened exchange functional, HSE06, and are compared to the best available global-hybrid generalized gradient approximation (GGA) and to a high-performance long-range-corrected meta-GGA.
Co-reporter:Roberto Peverati and Donald G. Truhlar
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 32) pp:NaN11370-11370
Publication Date(Web):2012/05/30
DOI:10.1039/C2CP41295K
Adiabatic time-dependent density functional theory is a powerful method for calculating electronic excitation energies of complex systems, but the quality of the results depends on the choice of approximate density functional. In this article we test two promising new density functionals, M11 and M11-L, against databases of 214 diverse electronic excitation energies, and we compare the results to those for 16 other density functionals of various kinds and to time-dependent Hartree–Fock. Charge transfer excitations are well known to be the hardest challenge for TDDFT. M11 is a long-range-corrected hybrid meta-GGA, and it shows better performance for charge transfer excitations than any of the other functionals except M06-HF, which is a specialized functional that does not do well for valence excitations. Several other long-range-corrected hybrid functionals also do well, and we especially recommend M11, ωB97X, and M06-2X for general spectroscopic applications because they do exceptionally well on ground-state properties as well as excitation energies. Local functionals are preferred for many applications to extended systems because of their significant cost advantage for large systems. M11-L is a dual-range local functional and—unlike all previous local functionals—it has good performance for Rydberg states as well as for valence states. Thus it is highly recommended for excitation energy calculations on extended systems.
Co-reporter:Roberto Peverati and Donald G. Truhlar
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 38) pp:NaN13174-13174
Publication Date(Web):2012/08/02
DOI:10.1039/C2CP42025B
We report a new local exchange–correlation energy functional that has significantly improved across-the-board performance, including main-group and transition metal chemistry and solid-state physics, especially atomization energies, ionization potentials, barrier heights, noncovalent interactions, isomerization energies of large moleucles, and solid-state lattice constants and cohesive energies.
Co-reporter:Jingjing Zheng, Tao Yu, Ewa Papajak, I. M. Alecu, Steven L. Mielke and Donald G. Truhlar
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 23) pp:NaN10907-10907
Publication Date(Web):2011/05/11
DOI:10.1039/C0CP02644A
Many methods for correcting harmonic partition functions for the presence of torsional motions employ some form of one-dimensional torsional treatment to replace the harmonic contribution of a specific normal mode. However, torsions are often strongly coupled to other degrees of freedom, especially other torsions and low-frequency bending motions, and this coupling can make assigning torsions to specific normal modes problematic. Here, we present a new class of methods, called multi-structural (MS) methods, that circumvents the need for such assignments by instead adjusting the harmonic results by torsional correction factors that are determined using internal coordinates. We present three versions of the MS method: (i) MS-AS based on including all structures (AS), i.e., all conformers generated by internal rotations; (ii) MS-ASCB based on all structures augmented with explicit conformational barrier (CB) information, i.e., including explicit calculations of all barrier heights for internal-rotation barriers between the conformers; and (iii) MS-RS based on including all conformers generated from a reference structure (RS) by independent torsions. In the MS-AS scheme, one has two options for obtaining the local periodicity parameters, one based on consideration of the nearly separable limit and one based on strongly coupled torsions. The latter involves assigning the local periodicities on the basis of Voronoi volumes. The methods are illustrated with calculations for ethanol, 1-butanol, and 1-pentyl radical as well as two one-dimensional torsional potentials. The MS-AS method is particularly interesting because it does not require any information about conformational barriers or about the paths that connect the various structures.
Co-reporter:Boris B. Averkiev and Donald G. Truhlar
Catalysis Science & Technology (2011-Present) 2011 - vol. 1(Issue 8) pp:NaN1529-1529
Publication Date(Web):2011/09/12
DOI:10.1039/C1CY00227A
The Gibbs energy of reaction of oxidative addition of ammonia to an iridium complex in diethyl ether was calculated by seven density functional methods, in particular B3LYP, PBE, CAM-B3LYP, M05, M06, M06-L, and ωB97X. The calculated free energies, based on geometry optimization and frequency calculations in both the gas phase and solution, were compared with the experimental result, −1.3 kcal mol−1, obtained by Hartwig and coworkers. The M06-L method gives the best result: −1.4 kcal/mol.
Co-reporter:Haoyu S. Yu, Xiao He, Shaohong L. Li and Donald G. Truhlar
Chemical Science (2010-Present) 2016 - vol. 7(Issue 9) pp:NaN6279-6279
Publication Date(Web):2016/07/12
DOI:10.1039/C6SC90044E
Correction for ‘MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions’ by Haoyu S. Yu et al., Chem. Sci., 2016, DOI: 10.1039/c6sc00705h.
Co-reporter:Junwei Lucas Bao, Rubén Meana-Pañeda and Donald G. Truhlar
Chemical Science (2010-Present) 2015 - vol. 6(Issue 10) pp:NaN5881-5881
Publication Date(Web):2015/06/16
DOI:10.1039/C5SC01848J
The goal of the present work is modeling the kinetics of a key reaction involved in the combustion of the biofuel 2-butanol. To accomplish this we extended multi-path variational transition state theory (MP-VTST) with the small curvature tunneling (SCT) approximation to include multistructural anharmonicity factors for molecules with chiral carbons. We use the resulting theory to predict the site-dependent rate constants of the hydrogen abstraction from 2-butanol by hydroperoxyl radical. The generalized transmission coefficients were averaged over the four lowest-energy reaction paths. The computed forward reaction rate constants indicate that hydrogen abstraction from the C-2 site has the largest contribution to the overall reaction from 200 K to 2400 K, with a contribution ranging from 99.9988% at 200 K to 88.9% at 800 K to 21.2% at 3000 K, while hydrogen abstraction from the oxygen site makes the lowest contribution at all temperatures, ranging from 2.5 × 10−9% at 200 K to 0.65% at 800 K to 18% at 3000 K. This work highlights the importance of including the multiple-structure and torsional potential anharmonicity in the computation of the thermal rate constants. We also analyzed the role played by the hydrogen bond at the transition state, and we illustrated the risks of (a) considering only the lowest-energy conformations in the calculations of the rate constants or (b) ignoring the nonlinear temperature dependence of the activation energies. A hydrogen bond at the transition state can lower the enthalpy of activation, but raise the free energy of activation. We find an energy of activation that increases from 11 kcal mol−1 at 200 K to more than 36 kcal mol−1 at high temperature for this radical reaction with a biofuel molecule.
Co-reporter:Ke R. Yang, Xuefei Xu, Jingjing Zheng and Donald G. Truhlar
Chemical Science (2010-Present) 2014 - vol. 5(Issue 12) pp:NaN4680-4680
Publication Date(Web):2014/08/06
DOI:10.1039/C4SC01967A
We present an improved version of the anchor points reactive potential (APRP) method for potential energy surfaces; the improvement for the surfaces themselves consists of using a set of internal coordinates with better global behavior, and we also extend the method to fit the surface couplings. We use the new method to produce a 3 × 3 matrix of diabatic potential energy surfaces and couplings for the photodissociation of phenol as functions of 33 nonredundant internal coordinates. The diabatic potential matrix is based on two kinds of calculations at a sequence of anchor points along the O–H dissociation coordinate: (1) fourfold way diabatic calculations based on MC-QDPT/jul-cc-pVDZ calculations for the potential energy surfaces and diabatic couplings as functions of the O–H bond stretch, C–O–H bond angle, and C–C–O–H torsion and for the diabatic couplings as functions of the nine out-of-plane phenoxyl distortion coordinates and (2) M06-2X/jul-cc-pVDZ density functional Hessian calculations for the potentials along the 30 vibrational coordinates of the phenoxyl group. The potential energy surfaces and couplings are used to calculate and characterize adiabatic surfaces and conical intersections, and the resulting equilibrium geometries, vibrational frequencies, and vertical excitation energies are in good agreement with available reference data. We also calculate the geometries of the minimum energy conical intersections. The surfaces and couplings are used for full-dimensional tunneling calculations of the adiabatic photodissociation rate, i.e., the rate of O–H bond fission following photoexcitation. Finally we use the couplings to provide indicators of which vibrational modes are effective in promoting dissociation.
Co-reporter:Aleksandr V. Marenich, Christopher J. Cramer and Donald G. Truhlar
Chemical Science (2010-Present) 2013 - vol. 4(Issue 6) pp:NaN2356-2356
Publication Date(Web):2013/03/18
DOI:10.1039/C3SC50242B
Polarizability is a key molecular property controlling induction and dispersion forces in molecules, and atomic polarizabilities in molecules are widely used elements both in qualitative schemes for understanding molecular interactions and in quantitative methods for modeling them. Unfortunately, experimental probes of local polarizability are not readily available. Here we predict the polarizability of individual atoms and functional groups in a variety of systems, and we draw both general and specific conclusions with broad consequences. We find that the polarizability of the same functional group (e.g., the carbonyl group) can differ substantially, depending on the position of this group in a molecule (e.g., in a protein). More specifically, we find that the polarizability of buried atoms and groups is screened and thereby diminished; thus the outermost atoms and functional groups (for example, those lying closer to the molecular van der Waals surface) are more polarizable than buried ones, even when acted on by the same electric field. These findings mitigate against attributing isolated system behavior to molecular fragments since their polarizability depends on their environment, and the methods used here provide a way to probe molecular polarizability with a finer grain than has previously been possible.
Co-reporter:Jingjing Zheng, Xuefei Xu, Rubén Meana-Pañeda and Donald G. Truhlar
Chemical Science (2010-Present) 2014 - vol. 5(Issue 5) pp:NaN2099-2099
Publication Date(Web):2014/01/24
DOI:10.1039/C3SC53290A
The classical trajectory method (also called molecular dynamics) is the most widely used method for ensemble averaging and calculating rate constants of complex dynamical systems; however it has the serious drawback of not allowing tunneling. Here, we show how to include tunneling efficiently in real-time classical trajectories by using the army ants algorithm for quantum mechanical rare event sampling and partially optimized semiclassical tunneling paths based on valence internal coordinates. Three examples, HN2 dissociation and two kinds of HCOH isomerizations, are used to illustrate the tunneling method. We show that the army ants tunneling algorithm is very efficient (even lower computational costs than calculations without tunneling) and yields physically reasonable rate constants. The new algorithm is straightforward to include in any molecular dynamics package, and it allows sampling of regions of phase space that are classically inaccessible but that may lead to different products or different energy distributions than are populated by non-tunneling processes.
Co-reporter:Aleksandr V. Marenich, Christopher J. Cramer, Donald G. Truhlar, Ciro A. Guido, Benedetta Mennucci, Giovanni Scalmani and Michael J. Frisch
Chemical Science (2010-Present) 2011 - vol. 2(Issue 11) pp:NaN2161-2161
Publication Date(Web):2011/08/05
DOI:10.1039/C1SC00313E
We present a unified treatment of solvatochromic shifts in liquid-phase absorption spectra, and we develop a self-consistent state-specific vertical excitation model (called VEM) for electronic excitation in solution. We discuss several other approaches to calculate vertical excitations in solution as an approximation to VEM. We illustrate these methods by presenting calculations of the solvatochromic shifts of the lowest excited states of several solutes (acetone, acrolein, coumarin 153, indolinedimethine-malononitrile, julolidine-malononitrile, methanal, methylenecyclopropene, and pyridine) in polar and nonpolar solvents (acetonitrile, cyclohexane, dimethyl sulfoxide, methanol, n-hexane, n-pentane, and water) using implicit solvation models combined with configuration interaction based on single excitations and with time-dependent density functional theory.
Co-reporter:Tao Yu, Jingjing Zheng and Donald G. Truhlar
Chemical Science (2010-Present) 2011 - vol. 2(Issue 11) pp:NaN2213-2213
Publication Date(Web):2011/08/10
DOI:10.1039/C1SC00225B
We present a new formulation of variational transition state theory (VTST) called multi-structural VTST (MS-VTST) and the use of this to calculate the rate constant for the 1,4-hydrogen shift isomerization reaction of 1-pentyl radical and that for the reverse reaction. MS-VTST uses a multi-faceted dividing surface and provides a convenient way to include the contributions of many structures (typically conformers) of the reactant and the transition state in rate constant calculations. In this particular application, we also account for the torsional anharmonicity. We used the multi-configuration Shepard interpolation method to efficiently generate a semi-global portion of the potential energy surface from a small number of high-level electronic structure calculations using the M06 density functional in order to compute the energies and Hessians of Shepard points along a reaction path. The M06-2X density functional was used to calculate the multi-structural anharmonicity effect, including all of the structures of the reactant, product and transition state. To predict the thermal rate constant, VTST calculations were performed to obtain the canonical variational rate constant over the temperature range 200–2000 K. A transmission coefficient is calculated by the multidimensional small-curvature tunneling (SCT) approximation. The final MS-CVT/SCT thermal rate constant was determined by combining a reaction rate calculation in the single-structural harmonic oscillator approximation (including tunneling) with the multi-structural anharmonicity torsional factor. The calculated forward rate constant agrees very well with experimentally-based evaluations of the high-pressure limit for the temperature range 300–1300 K, although it is a factor of 2.5–3.0 lower than the single-structural harmonic oscillator approximation over this temperature range. We anticipate that MS-VTST will be generally useful for calculating the reaction rates of complex molecules with multiple torsions.
Co-reporter:Junwei Lucas Bao, Pattrawan Sripa and Donald G. Truhlar
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 2) pp:NaN1041-1041
Publication Date(Web):2015/11/23
DOI:10.1039/C5CP05780A
Multi-path variational transition state theory (MP-VTST) provides a conformationally complete framework for calculating gas-phase rate constants. For reactions in which the transition state has distinguishable torsional minima (which include most reactions), there are multiple possible reaction paths. In principle MP-VTST includes the contributions from all the reaction paths, and it should explicitly treat the variational and tunneling effects of each path, but in practice one may need to truncate the number of paths included in MP-VTST calculations in order to achieve a balance between computational cost and accuracy. In this work, we present calculations including all paths for two prototype combustion reactions, namely the two hydrogen abstraction reactions from tert-butanol by HO2 radical. For both reactions we included all the reaction paths. Since abstraction at C has 46 paths, it provided a good opportunity to carry out a case study in which we investigated the errors introduced by truncating the number of paths. For the reaction studied, we found that the variational and multidimensional tunneling transmission coefficients are very different for different reaction paths, which provides new evidence that MP-VTST is necessary for treating path-dependent variational effects and multidimensional tunneling. We found that tunneling transmission coefficients can be much larger for higher-energy paths than for lower-energy ones. Interestingly, the simple hypothesis that higher barriers are narrower does not explain this finding in the present case; we found instead that the effect is due to higher-energy barriers having the possibility of tunneling at energies farther below the barrier top. We also show that a previously applied criterion for judging convergence with respect to the number of paths may not be reliable at low temperature.
Co-reporter:Junwei Lucas Bao and Donald G. Truhlar
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 15) pp:NaN10108-10108
Publication Date(Web):2016/03/11
DOI:10.1039/C6CP00816J
The growth of anionic silicon hydride clusters is a critically important process in nanodusty plasmas. In the current study, we focus on the formation of homologs of silylene (Sin+1H2n+2−, n = 3, 4) and silyl (SinH2n+1−, n = 4, 5) anions via anion–neutral reaction pathways. Species like silyl or silylene anions and their related elementary reactions, which are involved in the formation of silicon hydride clusters, were not used in developing exchange–correlation (xc) density functionals (i.e., they were not included in the training set of semiempirical density functionals); therefore, we explored the accuracy of various widely used xc density functionals based on reaction energies and barrier heights. Among the 21 density functionals we tested, M06-2X has the best performance for a hybrid functional, and MN15-L has the best performance for a local functional. Thermal rate constants of the elementary reactions involved in the reaction mechanism are calculated using M06-2X and multistructural canonical variational transition state theory with the small-curvature tunneling approximation (MS-CVT/SCT). The pressure dependence of unimolecular isomerization reactions is treated with system-specific quantum RRK theory (SS-QRRK) and the Lindemann–Hinshelwood mechanism.
Co-reporter:Junwei Lucas Bao, Xin Zhang and Donald G. Truhlar
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 25) pp:NaN16670-16670
Publication Date(Web):2016/06/01
DOI:10.1039/C6CP02765B
Understanding the falloff in rate constants of gas-phase unimolecular reaction rate constants as the pressure is lowered is a fundamental problem in chemical kinetics, with practical importance for combustion, atmospheric chemistry, and essentially all gas-phase reaction mechanisms. In the present work, we use our recently developed system-specific quantum RRK theory, calibrated by canonical variational transition state theory with small-curvature tunneling, combined with the Lindemann–Hinshelwood mechanism, to model the dissociation reaction of fluoroform (CHF3), which provides a definitive test for falloff modeling. Our predicted pressure-dependent thermal rate constants are in excellent agreement with experimental values over a wide range of pressures and temperatures. The present validation of our methodology, which is able to include variational transition state effects, multidimensional tunneling based on the directly calculated potential energy surface along the tunneling path, and torsional and other vibrational anharmonicity, together with state-of-the-art reaction-path-based direct dynamics calculations, is important because the method is less empirical than models routinely used for generating full mechanisms, while also being simpler in key respects than full master equation treatments and the full reduced falloff curve and modified strong collision methods of Troe.
Co-reporter:Haoyu S. Yu, Wenjing Zhang, Pragya Verma, Xiao He and Donald G. Truhlar
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 18) pp:NaN12160-12160
Publication Date(Web):2015/04/09
DOI:10.1039/C5CP01425E
The goal of this work is to develop a gradient approximation to the exchange–correlation functional of Kohn–Sham density functional theory for treating molecular problems with a special emphasis on the prediction of quantities important for homogeneous catalysis and other molecular energetics. Our training and validation of exchange–correlation functionals is organized in terms of databases and subdatabases. The key properties required for homogeneous catalysis are main group bond energies (database MGBE137), transition metal bond energies (database TMBE32), reaction barrier heights (database BH76), and molecular structures (database MS10). We also consider 26 other databases, most of which are subdatabases of a newly extended broad database called Database 2015, which is presented in the present article and in its ESI. Based on the mathematical form of a nonseparable gradient approximation (NGA), as first employed in the N12 functional, we design a new functional by using Database 2015 and by adding smoothness constraints to the optimization of the functional. The resulting functional is called the gradient approximation for molecules, or GAM. The GAM functional gives better results for MGBE137, TMBE32, and BH76 than any available generalized gradient approximation (GGA) or than N12. The GAM functional also gives reasonable results for MS10 with an MUE of 0.018 Å. The GAM functional provides good results both within the training sets and outside the training sets. The convergence tests and the smooth curves of exchange–correlation enhancement factor as a function of the reduced density gradient show that the GAM functional is a smooth functional that should not lead to extra expense or instability in optimizations. NGAs, like GGAs, have the advantage over meta-GGAs and hybrid GGAs of respectively smaller grid-size requirements for integrations and lower costs for extended systems. These computational advantages combined with the relatively high accuracy for all the key properties needed for molecular catalysis make the GAM functional very promising for future applications.
Co-reporter:Shaohong L. Li, Xuefei Xu and Donald G. Truhlar
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 31) pp:NaN20099-20099
Publication Date(Web):2015/06/05
DOI:10.1039/C5CP02461G
Three singlet states, namely a closed-shell ground state and two excited states with 1ππ* and 1nσ* character, have been suggested to be responsible for the radiationless decay or photochemical reaction of photoexcited thioanisole. The correct interpretation of the electronic spectrum is critical for understanding the character of these low-lying excited states, but the experimental spectrum is yet to be fully interpreted. In the work reported here, we investigated the nature of those three states and a fourth singlet state of thioanisole using electronic structure calculations by multireference perturbation theory, by completely-renormalized equation-of-motion coupled cluster theory with single and double excitations and noniterative inclusion of connected triples (CR-EOM-CCSD(T)), and by linear-response time-dependent density functional theory (TDDFT). We clarified the assignment of the electronic spectrum by simulating it using a normal-mode sampling approach combined with TDDFT in the Tamm–Dancoff approximation (TDA). The understanding of the electronic states and of the accuracy of the electronic structure methods lays the foundation of our future work of constructing potential energy surfaces.
Co-reporter:Aleksandr V. Marenich, Junming Ho, Michelle L. Coote, Christopher J. Cramer and Donald G. Truhlar
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 29) pp:NaN15106-15106
Publication Date(Web):2014/06/24
DOI:10.1039/C4CP01572J
This article reviews recent developments and applications in the area of computational electrochemistry. Our focus is on predicting the reduction potentials of electron transfer and other electrochemical reactions and half-reactions in both aqueous and nonaqueous solutions. Topics covered include various computational protocols that combine quantum mechanical electronic structure methods (such as density functional theory) with implicit-solvent models, explicit-solvent protocols that employ Monte Carlo or molecular dynamics simulations (for example, Car–Parrinello molecular dynamics using the grand canonical ensemble formalism), and the Marcus theory of electronic charge transfer. We also review computational approaches based on empirical relationships between molecular and electronic structure and electron transfer reactivity. The scope of the implicit-solvent protocols is emphasized, and the present status of the theory and future directions are outlined.
Co-reporter:Xuefei Xu, Ewa Papajak, Jingjing Zheng and Donald G. Truhlar
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 12) pp:NaN4216-4216
Publication Date(Web):2012/01/16
DOI:10.1039/C2CP23692C
We investigate the statistical thermodynamics and kinetics of the 1,5-hydrogen shift isomerization reaction of the 1-butoxyl radical and its reverse isomerization. The partition functions and thermodynamic functions (entropy, enthalpy, heat capacity, and Gibbs free energy) are calculated using the multi-structural torsional (MS-T) anharmonicity method including all structures for three species (reactant, product, and transition state) involved in the reaction. The calculated thermodynamic quantities have been compared to those estimated by the empirical group additivity (GA) method. The kinetics of the unimolecular isomerization reaction was investigated using multi-structural canonical variational transition state theory (MS-CVT) including both multiple-structure and torsional (MS-T) anharmonicity effects. In these calculations, multidimensional tunneling (MT) probabilities were evaluated by the small-curvature tunneling (SCT) approximation and compared to results obtained with the zero-curvature tunneling (ZCT) approximation. The high-pressure-limit rate constants for both the forward and reverse reactions are reported as calculated by MS-CVT/MT, where MT can be ZCT or SCT. Comparison with the rate constants obtained by the single-structural harmonic oscillator (SS-HO) approximation shows the importance of anharmonicity in the rate constants of these reactions, and the effect of multi-structural anharmonicity is found to be very large. Whereas the tunneling effect increases the rate constants, the MS-T anharmonicity decreases them at all temperatures. The two effects counteract each other at temperatures 385 K and 264 K for forward and reverse reactions, respectively, and tunneling dominates at lower temperatures while MS-T anharmonicity has a larger effect at higher temperatures. The multi-structural torsional anharmonicity effect reduces the final reverse reaction rate constants by a much larger factor than it does to the forward ones as a result of the existence of more low-energy structures of the product 4-hydroxy-1-butyl radical than the reactant 1-butoxyl radical. As a consequence there is also a very large effect on the equilibrium constant. The neglect of multi-structural anharmonicity will lead to large errors in the estimation of reverse reaction rate constants.
Co-reporter:Soumen Ghosh, Christopher J. Cramer, Donald G. Truhlar and Laura Gagliardi
Chemical Science (2010-Present) 2017 - vol. 8(Issue 4) pp:NaN2750-2750
Publication Date(Web):2017/01/19
DOI:10.1039/C6SC05036K
Predicting ground- and excited-state properties of open-shell organic molecules by electronic structure theory can be challenging because an accurate treatment has to correctly describe both static and dynamic electron correlation. Strongly correlated systems, i.e., systems with near-degeneracy correlation effects, are particularly troublesome. Multiconfigurational wave function methods based on an active space are adequate in principle, but it is impractical to capture most of the dynamic correlation in these methods for systems characterized by many active electrons. We recently developed a new method called multiconfiguration pair-density functional theory (MC-PDFT), that combines the advantages of wave function theory and density functional theory to provide a more practical treatment of strongly correlated systems. Here we present calculations of the singlet–triplet gaps in oligoacenes ranging from naphthalene to dodecacene. Calculations were performed for unprecedently large orbitally optimized active spaces of 50 electrons in 50 orbitals, and we test a range of active spaces and active space partitions, including four kinds of frontier orbital partitions. We show that MC-PDFT can predict the singlet–triplet splittings for oligoacenes consistent with the best available and much more expensive methods, and indeed MC-PDFT may constitute the benchmark against which those other models should be compared, given the absence of experimental data.
Co-reporter:Jingjing Zheng, Prasenjit Seal and Donald G. Truhlar
Chemical Science (2010-Present) 2013 - vol. 4(Issue 1) pp:NaN212-212
Publication Date(Web):2012/09/24
DOI:10.1039/C2SC21090H
Aldehyde–radical reactions are important in atmospheric and combustion chemistry, and the reactions studied here also serve more generally to illustrate a fundamental aspect of chemical kinetics that has been relatively unexplored from a quantitative point of view, in particular the roles of multiple structures and torsional anharmonicity in determining the rate constants and branching ratios (product yields). We consider hydrogen abstraction from four carbon sites of butanal (carbonyl-C, α-C, β-C and γ-C) by hydroperoxyl radical. We employed multi-structural variational transition state theory for studying the first three channels; this uses a multi-faceted dividing surface and allows us to include the contributions of multiple structures of both reacting species and transition states. Multi-configurational Shepard interpolation (MCSI) was used to obtain the geometries and energies of the potential energy surface along the minimum-energy paths, with gradients and Hessians calculated by the M08-HX/maug-cc-pVTZ method. We find the numbers of structures obtained for the transition states are 46, 60, 72 and 76respectively for the H abstraction at the carbonyl C, the α position, the β position and the γ position. Our results show that neglecting the factors arising from multiple structures and torsional anharmonicity would lead to errors at 300, 1000 and 2400 K of factors of 8, 11 and 10 for abstraction at the carbonyl-O, 2, 11 and 25 at the α-C position, 2, 23 and 47 at the β-C position, and 0.6, 8 and 18 at the γ-C position. The errors would be even larger at high temperature for the reverse of the H abstraction at the β-C. Relative yields are changed as much as a factor of 7.0 at 200 K, a factor of 5.0 at 298 K, and a factor of 3.7 in the other direction at 2400 K. The strong dependence of the product ratios on the multi-structural anharmonicity factors shows that such factors play an important role in controlling branching ratios in reaction mechanism networks.
Co-reporter:Haoyu S. Yu, Xiao He, Shaohong L. Li and Donald G. Truhlar
Chemical Science (2010-Present) 2016 - vol. 7(Issue 8) pp:NaN5051-5051
Publication Date(Web):2016/04/06
DOI:10.1039/C6SC00705H
Kohn–Sham density functionals are widely used; however, no currently available exchange–correlation functional can predict all chemical properties with chemical accuracy. Here we report a new functional, called MN15, that has broader accuracy than any previously available one. The properties considered in the parameterization include bond energies, atomization energies, ionization potentials, electron affinities, proton affinities, reaction barrier heights, noncovalent interactions, hydrocarbon thermochemistry, isomerization energies, electronic excitation energies, absolute atomic energies, and molecular structures. When compared with 82 other density functionals that have been defined in the literature, MN15 gives the second smallest mean unsigned error (MUE) for 54 data on inherently multiconfigurational systems, the smallest MUE for 313 single-reference chemical data, and the smallest MUE on 87 noncovalent data, with MUEs for these three categories of 4.75, 1.85, and 0.25 kcal mol−1, respectively, as compared to the average MUEs of the other 82 functionals of 14.0, 4.63, and 1.98 kcal mol−1. The MUE for 17 absolute atomic energies is 7.4 kcal mol−1 as compared to an average MUE of the other 82 functionals of 34.6 kcal mol−1. We further tested MN15 for 10 transition-metal coordination energies, the entire S66x8 database of noncovalent interactions, 21 transition-metal reaction barrier heights, 69 electronic excitation energies of organic molecules, 31 semiconductor band gaps, seven transition-metal dimer bond lengths, and 193 bond lengths of 47 organic molecules. The MN15 functional not only performs very well for our training set, which has 481 pieces of data, but also performs very well for our test set, which has 823 data that are not in our training set. The test set includes both ground-state properties and molecular excitation energies. For the latter MN15 achieves simultaneous accuracy for both valence and Rydberg electronic excitations when used with linear-response time-dependent density functional theory, with an MUE of less than 0.3 eV for both types of excitations.
Co-reporter:Zhen Hua Li and Donald G. Truhlar
Chemical Science (2010-Present) 2014 - vol. 5(Issue 7) pp:NaN2624-2624
Publication Date(Web):2014/02/26
DOI:10.1039/C4SC00052H
Metal nanoparticles have been widely used as functional materials in physics, chemistry, and biology. Understanding their unique thermodynamic properties is essential both for practical applications and from a fundamental point of view. This perspective article is an overview of recent progresses on the nanothermodynamics of metal nanoparticles and it especially highlights as examples our own studies on the structural stability, phases, phase changes, and thermodynamic functions of aluminum nanoparticles. We discuss using statistical sampling by Monte Carlo and molecular dynamics algorithms to calculate nanoparticle properties, nanophase properties, free energies, and nucleation rates, and we tried to understand the results in terms of energy landscapes by using exhaustive enumeration of the multiple structures of Al nanoparticles from all sizes up to N = 65 plus selected larger calculations.
Co-reporter:Ruifang Li, Yan Zhao and Donald G. Truhlar
Chemical Communications 2011 - vol. 47(Issue 8) pp:NaN2359-2359
Publication Date(Web):2010/12/16
DOI:10.1039/C0CC02845B
Adequate polarization functions reduce the error of density functional theory (DFT) for the heat of reaction for CF4 + SiCl4 from ∼9–12 kcal mol−1 to ∼2–4 kcal mol−1, and using an improved density functional further reduces it to ∼1 kcal mol−1. This reaction was previously identified as a stumbling block for DFT, but we show that the problem with the previous calculations was not DFT but rather inadequate basis sets to account for intramolecular charge polarization.
Co-reporter:Yan Zhao and Donald G. Truhlar
Physical Chemistry Chemical Physics 2008 - vol. 10(Issue 19) pp:NaN2818-2818
Publication Date(Web):2008/02/18
DOI:10.1039/B717744E
The geometries and binding energies of a recent buckyball tweezers (C60H28) and its supramolecular complexes are investigated using recently developed density functionals (M06-L and M06-2X) that include an accurate treatment of medium-range correlation energy. The pincer part of the tweezers, corannulene, has a strong attractive interaction with C60. However, due to the entropy penalty, the calculated gas-phase free energy of association of the C60@corannulene supramolecule is positive 3.5 kcal mol−1; and this entropy penalty explains why it is difficult to observe C60@corannulene supramolecule experimentally. By using a π-extended tetrathiafulvalene (TTF), in particular 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene (TTFAQ or C20H10S4), as the pincer part, we modeled a new buckyball tweezers. The geometries and binding energies of the new buckyball tweezers and its supramolecular complexes are also calculated. Due to fact that the attractive interaction between TTFAQ and C60 is weaker than that between corannulene and C60, the gas-phase binding free energy in the C60@C60H 32S8 supramolecular complex is smaller than that in the C60@C60H28 supramolecule. We also discuss solvent effects.
Co-reporter:Jingjing Zheng, Tao Yu and Donald G. Truhlar
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 43) pp:NaN19324-19324
Publication Date(Web):2011/10/10
DOI:10.1039/C1CP21829H
The C–H bond dissociation processes of n-hexane and isohexane involve 23 and 13 conformational structures, respectively in the parent molecules and 14–45 conformational structures in each of the seven isomeric products that we studied. Here we use the recently developed multi-structural (MS) thermodynamics method and CCSD(T)-F12a/jul-cc-pVTZ//M06-2X/6-311+G(2df,2p) potential energy surfaces to calculate the enthalpy, entropy, and heat capacity of n-hexane, isohexane, and seven of the possible radical products of dissociation of C–H bonds. We compare our calculations with the limited experimental data and with values obtained by group additivity fits used to extend the experimental data. This work shows that using the MS method involving a full set of structural isomers with density functional geometries, scaled density functional frequencies, and coupled cluster single-point energies can predict thermodynamic functions of complex molecules and bond dissociation reactions with chemical accuracy. The method should be useful to obtain thermodynamic data for complex molecules for which such data has not been measured and to obtain thermodynamic data at temperatures outside the temperature range where measurements are available.
Co-reporter:Junwei Lucas Bao, Prasenjit Seal and Donald G. Truhlar
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 24) pp:NaN15935-15935
Publication Date(Web):2015/05/18
DOI:10.1039/C5CP01979F
The growth of nanodusty particles, which is critical in plasma chemistry, physics, and engineering. The aim of the present work is to understand the detailed reaction mechanisms of early steps in this growth. The polymerization of neutral silane with the silylene or silyl anion, which eliminates molecular hydrogen with the formation of their higher homologues, governs the silicon hydride clustering in nanodusty plasma chemistry. The detailed mechanisms of these important polymerization reactions in terms of elementary reactions have not been proposed yet. In the present work, we investigated the initial steps of these polymerization reactions, i.e., the SiH4 + Si2H4−/Si2H5− reactions, and we propose a three-step mechanism, which is also applicable to the following polymerization steps. CM5 charges of all the silicon-containing species were computed in order to analyze the character of the species in the proposed reaction mechanisms. We also calculated thermal rate constant of each step using multi-structural canonical variational transition state theory (MS-CVT) with the small-curvature tunneling (SCT) approximation, based on the minimum energy path computed using M08-HX/MG3S electronic structure method.
Co-reporter:Adam C. Chamberlin ; Christopher J. Cramer
The Journal of Physical Chemistry B () pp:
Publication Date(Web):June 26, 2008
DOI:10.1021/jp8028038
The SM8 quantum mechanical aqueous continuum solvation model is applied to a 17-molecule test set proposed by Nicholls et al. (J. Med. Chem. 2008, 51, 769) to predict free energies of solvation. With the M06-2X density functional, the 6-31G(d) basis set, and CM4M charge model, the root-mean-square error (RMSE) of SM8 is 1.08 kcal mol−1 for aqueous geometries and 1.14 kcal mol−1 for gas-phase geometries. These errors compare favorably with optimal explicit and continuum models reported by Nicholls et al., having RMSEs of 1.33 and 1.87 kcal mol−1, respectively. Other models examined by these workers had RMSEs of 1.5−2.6 kcal mol−1. We also explore the use of other density functionals and charge models with SM8 and the RMSE increases to 1.21 kcal mol−1 for mPW1/CM4 with gas-phase geometries, to 1.50 kcal mol−1 for M06-2X/CM4 with gas-phase geometries, and to 1.27−1.64 kcal mol−1 with three different models at B3LYP gas-phase geometries.
.gif)




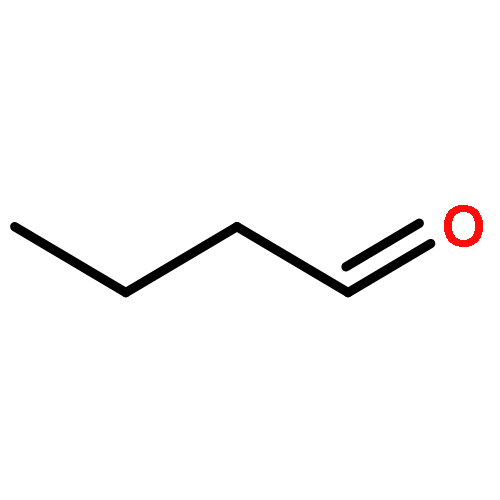



















































![N,N-DIMETHYL-2-{[2-PHENYL-8-(TRIFLUOROMETHYL)-4-QUINAZOLINYL]SULF<WBR />ANYL}ACETAMIDE](http://img.cochemist.com/ccimg/7800/7783-29-1.png)
![N,N-DIMETHYL-2-{[2-PHENYL-8-(TRIFLUOROMETHYL)-4-QUINAZOLINYL]SULF<WBR />ANYL}ACETAMIDE](http://img.cochemist.com/ccimg/7800/7783-29-1_b.png)






![ETHYL 3-AMINO-4-[(6-AMINO-4-OXO-1H-PYRIMIDIN-2-YL)SULFANYL]BUT-2-ENOATE](http://img.cochemist.com/ccimg/7300/7233-58-1.png)
![ETHYL 3-AMINO-4-[(6-AMINO-4-OXO-1H-PYRIMIDIN-2-YL)SULFANYL]BUT-2-ENOATE](http://img.cochemist.com/ccimg/7300/7233-58-1_b.png)