Co-reporter:John C. K. Chu; Derek M. Dalton
Journal of the American Chemical Society 2015 Volume 137(Issue 13) pp:4445-4452
Publication Date(Web):March 30, 2015
DOI:10.1021/jacs.5b00033
We report a catalytic asymmetric synthesis of piperidines through [4 + 2] cycloaddition of 1-azadienes and nitro-alkenes. The reaction uses earth abundant Zn as catalyst and is highly diastereo- and regioselective. A novel BOPA ligand (F-BOPA) confers high reactivity and enantioselectivity in the process. The presence of ortho substitution on the arenes adjacent to the bis(oxazolines) was found to be particularly impactful, due to limiting the undesired coordination of 1-azadiene to the Lewis acid and thus allowing the reaction to be carried out at lower temperature. A series of secondary kinetic isotope effect studies using a range of ligands implicates a stepwise mechanism for the transformation, involving an initial Michael-type addition of the imine to the nitro-alkene followed by a cyclization event. The stepwise mechanism obviates the electronic requirement inherent to a concerted mechanism, explaining the successful cycloaddition between two electron-deficient partners.
Co-reporter:Wen-Zhen Zhang; John C. K. Chu; Kevin M. Oberg
Journal of the American Chemical Society 2015 Volume 137(Issue 2) pp:553-555
Publication Date(Web):January 12, 2015
DOI:10.1021/ja510348p
An enantioselective isomerization of 4-iminocrotonates catalyzed by a rhodium(I)/phosphoramidite complex is described. This reaction uses widely available amines to couple with 4-oxocrotonate to provide a convenient access to a central chiral building block in good yield and high enantioselectivity. Although the mechanism of this new transformation remains unclear, both Rh and the phosphoramidite play a central role.
Co-reporter:Todd K. Hyster, Derek M. Dalton and Tomislav Rovis
Chemical Science 2015 vol. 6(Issue 1) pp:254-258
Publication Date(Web):01 Oct 2014
DOI:10.1039/C4SC02590C
We report the regioselective synthesis of dihydroisoquinolones from aliphatic alkenes and O-pivaloyl benzhydroxamic acids mediated by a Rh(III) precatalyst bearing sterically bulky substituents. While the prototypical Cp* ligand provides product with low selectivity, sterically bulky Cpt affords product with excellent regioselectivity for a range of benzhydroxamic acids and alkenes. Crystallographic evidence offers insight as to the source of the increased regioselectivity.
Co-reporter:Todd K. Hyster and Tomislav Rovis
Chemical Communications 2015 vol. 51(Issue 26) pp:5778-5778
Publication Date(Web):09 Mar 2015
DOI:10.1039/C5CC90120K
Correction for ‘Pyridine synthesis from oximes and alkynes via rhodium(III) catalysis: Cp* and Cpt provide complementary selectivity’ by Todd K. Hyster et al., Chem. Commun., 2011, 47, 11846–11848.
Co-reporter:Claire M. Filloux
Journal of the American Chemical Society 2014 Volume 137(Issue 1) pp:508-517
Publication Date(Web):December 29, 2014
DOI:10.1021/ja511445x
Asymmetric hydroheteroarylation of alkenes represents a convenient entry to elaborated heterocyclic motifs. While chiral acids are known to mediate asymmetric addition of electron-rich heteroarenes to Michael acceptors, very few methods exploit transition metals to catalyze alkylation of heterocycles with olefins via a C–H activation, migratory insertion sequence. Herein, we describe the development of an asymmetric, intermolecular hydroheteroarylation reaction of α-substituted acrylates with benzoxazoles. The reaction provides 2-substitued benzoxazoles in moderate to excellent yields and good to excellent enantioselectivities. Notably, a series of mechanistic studies appears to contradict a pathway involving enantioselective protonation of a Rh(I)–enolate, despite the fact that such a mechanism is invoked almost unanimously in the related addition of aryl boronic acids to methacrylate derivatives. Evidence suggests instead that migratory insertion or beta-hydride elimination is enantiodetermining and that isomerization of a Rh(I)–enolate to a Rh(I)–heterobenzyl species insulates the resultant α-stereocenter from epimerization. A bulky ligand, CTH-(R)-Xylyl-P-Phos, is crucial for reactivity and enantioselectivity, as it likely discourages undesired ligation of benzoxazole substrates or intermediates to on- or off-cycle rhodium complexes and attenuates coordination-promoted product epimerization.
Co-reporter:Jamie M. Neely
Journal of the American Chemical Society 2014 Volume 136(Issue 7) pp:2735-2738
Publication Date(Web):February 10, 2014
DOI:10.1021/ja412444d
α,β-Unsaturated carboxylic acids undergo Rh(III)-catalyzed decarboxylative coupling with α,β-unsaturated O-pivaloyl oximes to provide substituted pyridines in good yield. The carboxylic acid, which is removed by decarboxylation, serves as a traceless activating group, giving 5-substituted pyridines with very high levels of regioselectivity. Mechanistic studies rule out a picolinic acid intermediate, and an isolable rhodium complex sheds further light on the reaction mechanism.
Co-reporter:Tiffany Piou
Journal of the American Chemical Society 2014 Volume 136(Issue 32) pp:11292-11295
Publication Date(Web):August 5, 2014
DOI:10.1021/ja506579t
N-Enoxyphthalimides undergo a Rh(III)-catalyzed C–H activation initiated cyclopropanation of electron deficient alkenes. The reaction is proposed to proceed via a directed activation of the olefinic C–H bond followed by two migratory insertions, first across the electron-deficient alkene and then by cyclization back onto the enol moiety. A newly designed isopropylcyclopentadienyl ligand drastically improves yield and diastereoselectivity.
Co-reporter:Nicholas A. White
Journal of the American Chemical Society 2014 Volume 136(Issue 42) pp:14674-14677
Publication Date(Web):October 10, 2014
DOI:10.1021/ja5080739
A novel oxidative N-heterocyclic carbene-catalyzed reaction pathway has been discovered. Alkyl and aryl enals undergo β-hydroxylation via oxygen atom transfer from electron-deficient nitrobenzenes, followed by trapping of the resultant acyl azolium by the solvent. The proposed mechanism involves a single electron transfer event to initiate the reaction followed by radical recombination. This represents a profound mechanistic departure from the established two-electron disconnects in NHC catalysis.
Co-reporter:Benjamin L. Kohn and Tomislav Rovis
Chemical Science 2014 vol. 5(Issue 7) pp:2889-2892
Publication Date(Web):22 May 2014
DOI:10.1039/C4SC00743C
Perfluoroketones are useful products and intermediates in medicinal chemistry. Herein, cobalt-mediated fluoroacylation of 1,3-dienes is described using perfluorinated anhydrides such as TFAA. The reaction is thought to proceed through a fluoroacylcobalt reagent formed in situ. Perfluoroacylation of 1,3 dienes can also be performed to attain longer chain perfluorinated ketones.
Co-reporter:Nicholas A. White;Kerem E. Ozboya;Darrin M. Flanigan ; Tomislav Rovis
Asian Journal of Organic Chemistry 2014 Volume 3( Issue 4) pp:442-444
Publication Date(Web):
DOI:10.1002/ajoc.201402031
Abstract
A concise enantioselective synthesis of (−)-paroxetine (Paxil) and (−)-femoxetine has been achieved. Key to these syntheses is an N-heterocyclic carbene catalyzed homoenolate addition to a nitroalkene followed by in situ reduction of the nitro group to rapidly access δ-lactams.
Co-reporter:Sheng-Ying Hsieh;Benedikt Wanner;Dr. Philip Wheeler;Dr. André M. Beauchemin;Dr. Tomislav Rovis;Dr. Jeffrey W. Bode
Chemistry - A European Journal 2014 Volume 20( Issue 24) pp:7228-7231
Publication Date(Web):
DOI:10.1002/chem.201402818
Abstract
The kinetic resolution of N-heterocycles with chiral acylating agents reveals a previously unrecognized stereoelectronic effect in amine acylation. Combined with a new achiral hydroxamate, this effect makes possible the resolution of various N-heterocycles by using easily prepared reagents. A transition-state model to rationalize the stereochemical outcome of this kinetic resolution is also proposed.
Co-reporter:Todd K. Hyster ; Kyle E. Ruhl
Journal of the American Chemical Society 2013 Volume 135(Issue 14) pp:5364-5367
Publication Date(Web):April 2, 2013
DOI:10.1021/ja402274g
The coupling of O-pivaloyl benzhydroxamic acids with donor/acceptor diazo compounds provides isoindolones in high yield. The reaction tolerates a broad range of benzhydroxamic acids and diazo compounds, including substituted 2,2,2-trifluorodiazoethanes. Mechanistic experiments suggested that C–H activation is turnover-limiting and irreversible and that insertion of the diazo compound favors electron-deficient substrates.
Co-reporter:Nicholas A. White ; Daniel A. DiRocco
Journal of the American Chemical Society 2013 Volume 135(Issue 23) pp:8504-8507
Publication Date(Web):May 28, 2013
DOI:10.1021/ja403847e
An asymmetric intermolecular reaction between enals and nitroalkenes to yield δ-nitroesters has been developed, catalyzed by a novel chiral N-heterocyclic carbene. Key to this work was the development of a catalyst that favors the δ-nitroester pathway over the established Stetter pathway. The reaction proceeds in high stereoselectivity and affords the previously unreported syn diastereomer. We also report an operationally facile two-step, one-pot procedure for the synthesis of δ-lactams.
Co-reporter:Philip Wheeler, Harit U. Vora and Tomislav Rovis
Chemical Science 2013 vol. 4(Issue 4) pp:1674-1679
Publication Date(Web):13 Feb 2013
DOI:10.1039/C3SC00089C
The scope of the NHC-redox amidation has been expanded to include a variety of α,β-unsaturated aldehydes, including α-fluoro α,β-unsaturated aldehydes which give rise to enantioenriched α-fluoroamides in good to excellent yield and enantioselectivity (up to 97% ee). Enantioenriched amines may be elaborated to either diastereomer of the product in high diastereoselectivity (up to 99:1). Functionalization of the amide products to amines and fluorohydrins is also demonstrated with retention of enantioenrichment at the fluorine stereocenter.
Co-reporter:Derek M. Dalton, Anthony K. Rappé and Tomislav Rovis
Chemical Science 2013 vol. 4(Issue 5) pp:2062-2070
Publication Date(Web):05 Mar 2013
DOI:10.1039/C3SC50271F
Perfluorinated Taddol-based phosphoramidite, CKphos, is a highly selective ligand for formation of vinylogous amide cycloadducts in the Rh(I) catalyzed [2 + 2 + 2] cycloaddition of alkenyl isocyanates and alkynes. CKphos overrides substrate bias of product selectivity in the cycloaddition, providing indolizidinones in excellent yields and enantioselectivities. The excellent selectivities are attributed to a shortened Rh–P bond and coordination of one C6F5 to rhodium via a Z-type interaction, making the phosphoramidite a bidentate L,Z-ligand on rhodium. Evidence for the shortened Rh–P and C6F5 coordination is provided by X-ray, NMR and DFT computational analyses. Additionally, an anionic cobalt complex with CKphos was synthesized and two Co–C6F5 interactions are seen. The Rh(C2H4)Cl·CKphos catalyst in the [2 + 2 + 2] cycloaddition of alkenyl isocyanates and alkynes represents a rare example of metal–C6F5 Z-type interaction affecting selectivity in transition metal catalysis.
Co-reporter:Derek M. Dalton and Tomislav Rovis
Organic Letters 2013 Volume 15(Issue 10) pp:2346-2349
Publication Date(Web):April 30, 2013
DOI:10.1021/ol400529k
The Rh(I)•CKphos catalyzed [2 + 2 + 2] cycloaddition of 1,1-disubstituted alkenyl isocyanates and alkyl alkynes selectively forms previously inaccessible vinylogous amide indolizidinone cycloadducts, establishing an aza-quaternery stereocenter with excellent enantioselectivities (up to 98% ee). This advance enables a seven step catalytic, asymmetric synthesis of the tricyclic core of the cylindricine alkaloids with excellent control of product selectivity as well as regio- and enantioselectivity.
Co-reporter:Dr. Tyler A. Davis;Todd K. Hyster ; Tomislav Rovis
Angewandte Chemie 2013 Volume 125( Issue 52) pp:14431-14435
Publication Date(Web):
DOI:10.1002/ange.201307631
Co-reporter:Dr. Timothy J. Martin ; Tomislav Rovis
Angewandte Chemie 2013 Volume 125( Issue 20) pp:5476-5479
Publication Date(Web):
DOI:10.1002/ange.201301741
Co-reporter:Dr. Timothy J. Martin ; Tomislav Rovis
Angewandte Chemie International Edition 2013 Volume 52( Issue 20) pp:5368-5371
Publication Date(Web):
DOI:10.1002/anie.201301741
Co-reporter:Dr. Tyler A. Davis;Todd K. Hyster ; Tomislav Rovis
Angewandte Chemie International Edition 2013 Volume 52( Issue 52) pp:14181-14185
Publication Date(Web):
DOI:10.1002/anie.201307631
Co-reporter:David M. Rubush ; Michelle A. Morges ; Barbara J. Rose ; Douglas H. Thamm
Journal of the American Chemical Society 2012 Volume 134(Issue 33) pp:13554-13557
Publication Date(Web):August 7, 2012
DOI:10.1021/ja3052427
The desymmetrization of p-peroxyquinols using a Brønsted acid-catalyzed acetalization/oxa-Michael cascade was achieved in high yields and selectivities for a variety of aliphatic and aryl aldehydes. Mechanistic studies suggest that the reaction proceeds through a dynamic kinetic resolution of the peroxy hemiacetal intermediate. The resulting 1,2,4-trioxane products were derivatized and show potent cancer cell-growth inhibition.
Co-reporter:Daniel A. DiRocco ; Kevin M. Oberg
Journal of the American Chemical Society 2012 Volume 134(Issue 14) pp:6143-6145
Publication Date(Web):March 28, 2012
DOI:10.1021/ja302031v
Since Breslow’s initial report on the thiamine mode of action, the study of catalytic acyl carbanion processes has been an area of immense interest. With the advent of azolylidene catalysis, a plethora of reactivtiy has been harnessed, but the crucial nucleophilic intermediate proposed by Breslow had never been isolated or fully characterized. Herein, we report the isolation and full characterization of nitrogen analogues of the Breslow intermediate. Both stable and catalytically relevant, these species provide a model system for the study of acyl carbanion and homoenolate processes catalyzed by triazolylidene carbenes.
Co-reporter:Daniel A. DiRocco
Journal of the American Chemical Society 2012 Volume 134(Issue 19) pp:8094-8097
Publication Date(Web):May 2, 2012
DOI:10.1021/ja3030164
Cross-coupling reactions are among the most widely utilized methods for C–C bond formation; however, the requirement of preactivated starting materials still presents a major limitation. Methods that take direct advantage of the inherent reactivity of the C–H bond offer an efficient alternative to these methods, negating the requirement for substrate preactivation. In this process, two chemically distinct activation events culminate in the formation of the desired C–C bond with loss of H2 as the only byproduct. Herein we report the catalytic asymmetric α-acylation of tertiary amines with aldehydes facilitated by the combination of chiral N-heterocyclic carbene catalysis and photoredox catalysis.
Co-reporter:Jamie M. Neely
Journal of the American Chemical Society 2012 Volume 135(Issue 1) pp:66-69
Publication Date(Web):December 18, 2012
DOI:10.1021/ja3104389
α,β-Unsaturated O-pivaloyl oximes are coupled to alkenes by Rh(III) catalysis to afford substituted pyridines. The reaction with activated alkenes is exceptionally regioselective and high-yielding. Mechanistic studies suggest that heterocycle formation proceeds via reversible C–H activation, alkene insertion, and a C–N bond formation/N–O bond cleavage process.
Co-reporter:Harit U. Vora;Philip Wheeler
Advanced Synthesis & Catalysis 2012 Volume 354( Issue 9) pp:1617-1639
Publication Date(Web):
DOI:10.1002/adsc.201200031
Abstract
N-heterocyclic carbenes (NHC) are well known for their role in catalyzing benzoin and Stetter reactions: the generation of acyl anion equivalents from simple aldehydes to react with a variety of electrophiles. However, when an aldehyde bearing a leaving group or unsaturation adjacent to the acyl anion equivalent is subjected to an NHC, a new avenue of reactivity is unlocked, leading to a number of novel transformations which can generate highly complex products from simple starting materials, many of which are assembled through unconventional bond disconnections. The field of these new reactions – those utilizing α-reducible aldehydes to access previously unexplored catalytic intermediates – has expanded rapidly in the past eight years. This review aims to provide the reader with a historical perspective on the underlying discoveries that led to the current state of the art, a mechanistic description of these reactions, and a summary of the recent advances in this area.
Co-reporter:Daniel A. DiRocco ; Tomislav Rovis
Angewandte Chemie 2012 Volume 124( Issue 24) pp:6006-6008
Publication Date(Web):
DOI:10.1002/ange.201202442
Co-reporter:Livia Knörr;Thomas R. Ward;Todd K. Hyster
Science 2012 Volume 338(Issue 6106) pp:500-503
Publication Date(Web):26 Oct 2012
DOI:10.1126/science.1226132
Co-reporter:Dr. Xiaodan Zhao;Kyle E. Ruhl ; Tomislav Rovis
Angewandte Chemie 2012 Volume 124( Issue 49) pp:12496-12499
Publication Date(Web):
DOI:10.1002/ange.201206490
Co-reporter:Daniel A. DiRocco;Elizabeth L. Noey; K. N. Houk; Tomislav Rovis
Angewandte Chemie International Edition 2012 Volume 51( Issue 10) pp:2391-2394
Publication Date(Web):
DOI:10.1002/anie.201107597
Co-reporter:Daniel A. DiRocco ; Tomislav Rovis
Angewandte Chemie International Edition 2012 Volume 51( Issue 24) pp:5904-5906
Publication Date(Web):
DOI:10.1002/anie.201202442
Co-reporter:Dr. Xiaodan Zhao;Kyle E. Ruhl ; Tomislav Rovis
Angewandte Chemie International Edition 2012 Volume 51( Issue 49) pp:12330-12333
Publication Date(Web):
DOI:10.1002/anie.201206490
Co-reporter:Kevin M. Oberg
Journal of the American Chemical Society 2011 Volume 133(Issue 13) pp:4785-4787
Publication Date(Web):March 14, 2011
DOI:10.1021/ja200766k
A [4 + 2] cycloaddition of α,β-unsaturated imines and isocyanates catalyzed by a phosphoramidite−rhodium complex provides pyrimidinones in good yields and high enantioselectivities.
Co-reporter:Daniel A. DiRocco
Journal of the American Chemical Society 2011 Volume 133(Issue 27) pp:10402-10405
Publication Date(Web):June 16, 2011
DOI:10.1021/ja203810b
An asymmetric intermolecular Stetter reaction of enals with nitroalkenes catalyzed by chiral N-heterocyclic carbenes has been developed. The reaction rate and efficiency are profoundly impacted by the presence of catechol. The reaction proceeds with high selectivities and affords good yields of the Stetter product. Internal redox products were not observed despite of the protic conditions. The impact of catechol has been found to be general, facilitating far lower catalyst loadings than were previously achievable.
Co-reporter:Joann M. Um ; Daniel A. DiRocco ; Elizabeth L. Noey ; Tomislav Rovis ;K. N. Houk
Journal of the American Chemical Society 2011 Volume 133(Issue 29) pp:11249-11254
Publication Date(Web):June 15, 2011
DOI:10.1021/ja202444g
The asymmetric intermolecular Stetter reaction was investigated using the B3LYP and M06-2X functionals. Fluorination of a triazolium bicyclic catalyst had been found to significantly influence reaction yields and enantiomeric ratios. Computations indicate that the improved reactivity of the fluorinated catalyst is due to better electrostatic interactions between the nitroalkene and catalyst. Computational investigations of preferred conformations of the ground state catalyst and acyl anion equivalent, and the transition structures leading to both enantiomers of the products, are reported.
Co-reporter:Xiaodan Zhao ; Daniel A. DiRocco
Journal of the American Chemical Society 2011 Volume 133(Issue 32) pp:12466-12469
Publication Date(Web):July 25, 2011
DOI:10.1021/ja205714g
An efficient enantioselective approach to form trans-γ-lactams in up to 99% yield, 93% ee, and >20/1 dr using unactivated imines has been developed. The cyclohexyl-substituted azolium and the weak base sodium o-chlorobenzoate are most suitable for this transformation. Notably, the process involves cooperative catalysis by an N-heterocyclic carbene and a Brønsted acid.
Co-reporter:Kerem E. Ozboya and Tomislav Rovis
Chemical Science 2011 vol. 2(Issue 9) pp:1835-1838
Publication Date(Web):16 Jun 2011
DOI:10.1039/C1SC00175B
Herein we report an enantioselective synthesis of complex cyclopentanones using aliphatic aldehydes and activated enones. With the combination of a chiral secondary amine and a chiral triazolium catalyst, high diastereoselectivity and excellent enantioselectivity can be achieved. We present evidence of a clear cooperative effect when these two catalysts are present simultaneously in the system.
Co-reporter:Todd K. Hyster and Tomislav Rovis
Chemical Science 2011 vol. 2(Issue 8) pp:1606-1610
Publication Date(Web):16 May 2011
DOI:10.1039/C1SC00235J
We have developed a method for preparing pyridones from the coupling reaction of acrylamides and alkynes with either stoichometric Cu(OAc)2 or catalyticCu(OAc)2 and air as oxidants. In the course of these studies, it was found that a larger ligand, 1,3-di-tert-butylcyclopentadienyl (termed Cpt) results in higher degrees of regioselectivity in the alkyneinsertion event. The transformation tolerates a broad variety of alkynes and acrylamides. Furthermore, Cpt and Cp* demonstrate similar catalytic activity. This similarity allows for mechanistic studies to be undertaken which suggest a difference in mechanism between this reaction and the previously studied benzamide system.
Co-reporter:Ya Du, Todd K. Hyster and Tomislav Rovis
Chemical Communications 2011 vol. 47(Issue 44) pp:12074-12076
Publication Date(Web):17 Oct 2011
DOI:10.1039/C1CC15843K
An efficient strategy for the oxidative carbonylation of aromatic amidesvia C–H/N–H activation to form phthalimides using an Rh(III) catalyst has been developed. The reaction shows a preference for C–H bonds of electron-rich aromatic amides and tolerates a variety of functional groups.
Co-reporter:Jennifer L. Moore, Anthony P. Silvestri, Javier Read de Alaniz, Daniel A. DiRocco, and Tomislav Rovis
Organic Letters 2011 Volume 13(Issue 7) pp:1742-1745
Publication Date(Web):February 28, 2011
DOI:10.1021/ol200256a
A study on the mechanism of the asymmetric intramolecular Stetter reaction is reported. This investigation includes the determination of the rate law, kinetic isotope effects, and competition experiments. The reaction was found to be first order in aldehyde and azolium catalyst or free carbene. A primary kinetic isotope effect was found for the proton of the aldehyde. Taken together with a series of competition experiments, these results suggest that proton transfer from the tetrahedral intermediate formed upon nucleophilic attack of the carbene onto the aldehyde is the first irreversible step.
Co-reporter:Todd K. Hyster and Tomislav Rovis
Chemical Communications 2011 vol. 47(Issue 43) pp:11846-11848
Publication Date(Web):10 Oct 2011
DOI:10.1039/C1CC15248C
The synthesis of pyridines from readily available α,β-unsaturated oximes and alkynes under mild conditions and low temperatures using Rh(III) catalysis has been developed. It was found that the use of sterically different ligands allows for complementary selectivities to be achieved.
Co-reporter:Daniel A. DiRocco ;Dr. Tomislav Rovis
Angewandte Chemie International Edition 2011 Volume 50( Issue 35) pp:7982-7983
Publication Date(Web):
DOI:10.1002/anie.201102920
Co-reporter:Daniel A. DiRocco ;Dr. Tomislav Rovis
Angewandte Chemie 2011 Volume 123( Issue 35) pp:8130-8132
Publication Date(Web):
DOI:10.1002/ange.201102920
Co-reporter:Harit U. Vora
Journal of the American Chemical Society 2010 Volume 132(Issue 9) pp:2860-2861
Publication Date(Web):February 12, 2010
DOI:10.1021/ja910281s
Asymmetric hydration of α,α-dichloro aldehydes and α-halo enals via a NHC-catalyzed redox process to yield enantioenriched α-chloro and α-fluoro carboxylic acids is described herein. The developed reaction allows for installation of an α-deuterium to give rise to enantioenriched α-deutero α-halo acids using D2O as the deuteron source.
Co-reporter:Todd K. Hyster
Journal of the American Chemical Society 2010 Volume 132(Issue 30) pp:10565-10569
Publication Date(Web):July 13, 2010
DOI:10.1021/ja103776u
The oxidative cycloaddition of benzamides and alkynes has been developed. The reaction utilizes Rh(III) catalysts in the presence of Cu(II) oxidants, and is proposed to proceed by N−H metalation of the amide followed by ortho C−H activation. The resultant rhodacycle undergoes alkyne insertion to form isoquinolones in good yield. The reaction is tolerant of extensive substitution on the amide, alkyne, and arene, including halides, silyl ethers, and unprotected aldehydes as substituents. Unsymmetrical alkynes proceed with excellent regioselectivity, and heteroaryl carboxamides are tolerated leading to intriguing scaffolds for medicinal chemistry. A series of competition experiments shed further light on the mechanism of the transformation and reasons for selectivity.
Co-reporter:Claire M. Filloux;Stephen P. Lathrop
PNAS 2010 Volume 107 (Issue 48 ) pp:20666-20671
Publication Date(Web):2010-11-30
DOI:10.1073/pnas.1002830107
We report the development of a multicatalytic, one-pot, asymmetric Michael/Stetter reaction between salicylaldehydes and electron-deficient
alkynes. The cascade proceeds via amine-mediated Michael addition followed by an N-heterocyclic carbene-promoted intramolecular Stetter reaction. A variety of salicylaldehydes, doubly activated alkynes, and
terminal, electrophilic allenes participate in a one-step or two-step protocol to give a variety of benzofuranone products
in moderate to good yields and good to excellent enantioselectivities. The origin of enantioselectivity in the reaction is
also explored; E/Z geometry of the reaction intermediate as well as the presence of catalytic amounts of catechol additive are found to influence
reaction enantioselectivity.
Co-reporter:Stéphane Perreault and Tomislav Rovis
Chemical Society Reviews 2009 vol. 38(Issue 11) pp:3149-3159
Publication Date(Web):15 Sep 2009
DOI:10.1039/B816702H
Cycloaddition reactions are attractive strategies for the rapid formation of molecular complexity in organic synthesis, as multiple bonds are formed in a single process. To this end, several research groups have been actively involved in the development of catalytic methods to activate readily accessible π-components to achieve cycloadditions. However, the use of C–N π-components for the formation of heterocycles by these processes is less well developed. It has been previously demonstrated that the combination of different isocyanates with two alkynes yields pyridones of several types by metal-catalyzed [2 + 2 + 2] cycloadditions. The potential of this chemistry has been extended to alkenes as C–C π-components, allowing the formation of sp3-stereocenters. In this tutorial review directed towards [n + 2 + 2] cycloadditions of heterocumulenes, alkynes and alkenes, the recent advances in the catalytic asymmetric synthesis of indolizidine, quinolizidine and azocine skeletons are discussed.
Co-reporter:Daniel A. DiRocco ; Kevin M. Oberg ; Derek M. Dalton
Journal of the American Chemical Society 2009 Volume 131(Issue 31) pp:10872-10874
Publication Date(Web):July 16, 2009
DOI:10.1021/ja904375q
The catalytic asymmetric intermolecular Stetter reaction of heterocyclic aldehydes and nitroalkenes has been developed. We have identified a strong stereoelectronic effect on catalyst structure when a fluorine substituent is placed in the backbone. X-ray structure analysis provides evidence that hyperconjugative effects are responsible for a change in conformation in the azolium precatalyst. This new N-heterocyclic carbene precursor bearing fluorine substitution in the backbone results in significantly improved enantioselectivities across a range of substrates.
Co-reporter:Rebecca Keller Friedman
Journal of the American Chemical Society 2009 Volume 131(Issue 30) pp:10775-10782
Publication Date(Web):July 1, 2009
DOI:10.1021/ja903899c
A regioselective, rhodium-catalyzed cycloaddition between a variety of internal, unsymmetrical alkynes is described. We document the impact of both steric and electronic properties of the alkyne on reaction course, efficiency, and enantioselectivity. The substituent that better stabilizes a positive charge or the larger group, all else being equal, inserts distal to the carbonyl moiety in a predictable and controllable fashion. The reaction scope is broad and the enantioselectivities are high, providing an “instruction manual” for substrate choice when utilizing this reaction as a synthetic tool.
Co-reporter:Robert T. Yu ; Rebecca Keller Friedman
Journal of the American Chemical Society 2009 Volume 131(Issue 37) pp:13250-13251
Publication Date(Web):August 27, 2009
DOI:10.1021/ja906641d
A highly enantioselective rhodium-catalyzed [4+2+2] cycloaddition of terminal alkynes and dienyl isocyanates has been developed. The cycloaddition provides a rapid entry to highly functionalized and enantioenriched bicyclic azocines. This reaction represents the first [4+2+2] cycloaddition strategy to construct nitrogen-containing eight-membered rings.
Co-reporter:Stephen P. Lathrop
Journal of the American Chemical Society 2009 Volume 131(Issue 38) pp:13628-13630
Publication Date(Web):September 4, 2009
DOI:10.1021/ja905342e
A one-pot, asymmetric multicatalytic formal [3+2] reaction between 1,3-dicarbonyls and α,β-unsaturated aldehydes is described. The multicatalytic process involves a secondary amine catalyzed Michael addition followed by a N-heterocyclic carbene catalyzed intramolecular crossed benzoin reaction to afford densely functionalized cyclopentanones with high enantioselectivities. The reaction proceeds with a variety of alkyl and aryl enals as well as a range of 1,3-dicarbonyls (diketones and β-ketoesters). The functionalized products are obtained from cheap, readily available starting materials in a rapid and efficient manner in a one-pot, one-step operation.
Co-reporter:Derek M. Dalton ; Kevin M. Oberg ; Robert T. Yu ; Ernest E. Lee ; Stéphane Perreault ; Mark Emil Oinen ; Melissa L. Pease ; Guillaume Malik
Journal of the American Chemical Society 2009 Volume 131(Issue 43) pp:15717-15728
Publication Date(Web):October 9, 2009
DOI:10.1021/ja905065j
This manuscript describes the development and scope of the asymmetric rhodium-catalyzed [2 + 2 + 2] cycloaddition of terminal alkynes and alkenyl isocyanates leading to the formation of indolizidine and quinolizidine scaffolds. The use of phosphoramidite ligands proved crucial for avoiding competitive terminal alkyne dimerization. Both aliphatic and aromatic terminal alkynes participate well, with product selectivity a function of both the steric and electronic character of the alkyne. Manipulation of the phosphoramidite ligand leads to tuning of enantio- and product selectivity, with a complete turnover in product selectivity seen with aliphatic alkynes when moving from Taddol-based to biphenol-based phosphoramidites. Terminal and 1,1-disubstituted olefins are tolerated with nearly equal efficacy. Examination of a series of competition experiments in combination with analysis of reaction outcome shed considerable light on the operative catalytic cycle. Through a detailed study of a series of X-ray structures of rhodium(cod)chloride/phosphoramidite complexes, we have formulated a mechanistic hypothesis that rationalizes the observed product selectivity.
Co-reporter:Mark Emil Oinen, Robert T. Yu and Tomislav Rovis
Organic Letters 2009 Volume 11(Issue 21) pp:4934-4937
Publication Date(Web):October 5, 2009
DOI:10.1021/ol9020805
Excess substrate has been identified as an unintended spectator ligand affecting enantioselectivity in the [2 + 2 + 2] cycloaddition of alkenyl isocyanates with tolanes. Replacement of excess substrate with an exogenous additive affords products with consistent and higher ee’s. The increase in enantioselectivity is the result of a change in composition of a proposed rhodium(III) intermediate on the catalytic cycle. The net result is a rational probe of a short-lived rhodium(III) intermediate and gives insight that may have applications in many rhodium-catalyzed reactions.
Co-reporter:Qin Liu and Tomislav Rovis
Organic Letters 2009 Volume 11(Issue 13) pp:2856-2859
Publication Date(Web):June 9, 2009
DOI:10.1021/ol901081a
A triazolinylidene carbene catalyzed intermolecular Stetter reaction of glyoxamide and alkylidene ketoamides has been developed. 1,4-Dicarbonyl products are afforded in good to excellent yields, enantioselectivities, and diastereoselectivities. Further derivatization of the products affords useful intermediates for organic synthesis.
Co-reporter:Jeffrey B. Johnson, Matthew J. Cook, Tomislav Rovis
Tetrahedron 2009 65(16) pp: 3202-3210
Publication Date(Web):
DOI:10.1016/j.tet.2008.10.075
Co-reporter:RobertT. Yu;ErnestE. Lee Dr.;Guillaume Malik
Angewandte Chemie 2009 Volume 121( Issue 13) pp:2415-2418
Publication Date(Web):
DOI:10.1002/ange.200805455
Co-reporter:BronR. Galan Dr.
Angewandte Chemie 2009 Volume 121( Issue 16) pp:2870-2874
Publication Date(Web):
DOI:10.1002/ange.200804651
Co-reporter:RobertT. Yu;ErnestE. Lee Dr.;Guillaume Malik
Angewandte Chemie International Edition 2009 Volume 48( Issue 13) pp:2379-2382
Publication Date(Web):
DOI:10.1002/anie.200805455
Co-reporter:BronR. Galan Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 16) pp:2830-2834
Publication Date(Web):
DOI:10.1002/anie.200804651
Co-reporter:Kevin M. Oberg, Ernest E. Lee, Tomislav Rovis
Tetrahedron 2009 65(26) pp: 5056-5061
Publication Date(Web):
DOI:10.1016/j.tet.2009.02.021
Co-reporter:Jeffrey B. Johnson and Tomislav Rovis
Accounts of Chemical Research 2008 Volume 41(Issue 2) pp:327
Publication Date(Web):January 31, 2008
DOI:10.1021/ar700176t
The construction of carbon−carbon bonds, particularly with concomitant control of newly formed asymmetric centers, is of paramount importance for the development of synthetic routes to complex organic molecules. While cross-coupling reactions for the generation of sp2 carbon centers are well established, similar methodology for the formation and control of sp3-hydridized carbon stereocenters is extremely limited. We suggest that the nucleophilic interception of metalacycles provides the means to achieve such a transformation, wherein the metal complex serves to activate electrophiles, facilitate nucleophile addition, and ultimately control stereochemistry. One means of accessing these intermediates is through the use of simple meso-carboxylic anhydrides, which upon reaction with transition metals readily generate the desired metalacycles. Interception of the metalacycle with an appropriate carbon-based nucleophile generates an enantioenriched ketoacid, the product of the asymmetric desymmetrization of achiral starting materials. Early successes with achiral nickel catalysts and organozinc reagents provided the foundations for our approach. Alkylation of both succinic and glutaric anhydrides proceeds with a wide range of organozinc nucleophiles, forming 1,4- and 1,5-ketoacids in excellent yields. This reaction manifold has been extensively examined with a detailed kinetic study and mechanistic investigations utilizing mixed zinc reagents and alkene directing groups. This work has highlighted a number of unusual phenomena, including rate-limiting reductive elimination to form an sp3−sp2 carbon−carbon bond. Despite excellent results with the achiral system, to date, all efforts to render the nickel-catalyzed reaction asymmetric have been limited to modest success. Palladium and rhodium complexes, with the use of chiral P−P and P−N ligands, respectively, have been identified as competent catalysts for the enantioselective addition of organozinc reagents to anhydrides. The arylation of a series of succinic anhydrides with Ph2Zn can be achieved in greater than 95% enantioselectivity using a Pd/Josiphos catalyst. Rhodium catalysts have proven amenable for the incorporation of in situ formed organozinc reagents, nucleophiles traditionally troublesome in transition metal catalysis due to the deleterious effects of residual halide ions. Highly functionalized organozinc nucleophiles, including those containing indole and furan, participate in this chemistry to provide the corresponding 1,4- and 1,5-ketoacids in excellent yield with greater than 85% enantioselectivity. This metalacycle interception methodology is currently being expanded to the use of other systems, most notably the asymmetric [2 + 2 + 2] cycloaddition of alkenes, alkynes, and isocyanates. Ongoing studies promise the extension of existing methodology toward the development of modular, fully intermolecular three-component couplings in which both metalacycle formation and nucleophilic interception can be controlled. Ultimately, we envision the use of heterocumulenes in such methodology, providing a route to complex products utilizing CO2 as an inexpensive C1 feedstock.
Co-reporter:Arturo Orellana and Tomislav Rovis
Chemical Communications 2008 (Issue 6) pp:730-732
Publication Date(Web):04 Dec 2007
DOI:10.1039/B716445A
A rapid assembly of the tetracyclic core of FD-838, featuring a catalytic asymmetric Stetter reaction, is described.
Co-reporter:Christopher G. Nasveschuk and Tomislav Rovis
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 2) pp:240-254
Publication Date(Web):04 Dec 2007
DOI:10.1039/B714881J
The relay of stereochemistry of a breaking C–O bond into a forming C–C bond is well-known in the context of [3,3] sigmatropic shifts; however, this useful strategy is less well-known in other types of molecular rearrangements. Though the first successful example of a [1,3] O-to-C rearrangement was reported more than 100 years ago, this class of reactions has received less attention than its [3,3] counterpart. This perspective analyzes the various methods used for the activation and [1,3] rearrangement of vinyl ethers with an emphasis on mechanism and applications to stereoselective synthesis. We also highlight our own contributions to this area.
Co-reporter:JeffreyB. Johnson Dr.
Angewandte Chemie 2008 Volume 120( Issue 5) pp:852-884
Publication Date(Web):
DOI:10.1002/ange.200700278
Abstract
Olefine und Alkine sind in der Übergangsmetallkatalyse in vielfältiger Art vertreten, ob als Teil des Substrats oder des Katalysators oder als Additiv. Während der Einfluss von Metallen und Liganden relativ gut verstanden ist, wird der von Olefinen allgemein unterschätzt, obwohl zahlreiche Beispiele bekannt sind, in denen Olefine durch Steigerung der Aktivität, Stabilität oder Selektivität den Ausgang einer Reaktion beeinflussen. Dieser Aufsatz gibt eine Übersicht über die Wechselwirkung von Olefinen mit Übergangsmetallen und liefert Beispiele dafür, wie Olefine das Ergebnis von katalytischen Reaktionen, insbesondere Kreuzkupplungsreaktionen, beeinflussen. Er sollte damit die Grundlage für ein besseres Verständnis des Einflusses von Olefinen und Alkinen in übergangsmetallkatalysierten Reaktionen und für dessen Nutzung bieten.
Co-reporter:JeffreyB. Johnson Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 5) pp:840-871
Publication Date(Web):
DOI:10.1002/anie.200700278
Abstract
Olefins and alkynes are ubiquitous in transition-metal catalysis, whether introduced by the substrate, the catalyst, or as an additive. Whereas the impact of metals and ligands is relatively well understood, the effects of olefins in these reactions are generally underappreciated, even though numerous examples of olefins influencing the outcome of a reaction, through increased activity, stability, or selectivity, have been reported. This Review provides an overview of the interaction of olefins with transition metals and documents examples of olefins influencing the outcome of catalytic reactions, in particular cross-coupling reactions. It should thus provide a basis for the improved understanding and further utilization of olefin and alkyne effects in transition-metal-catalyzed reactions.
Co-reporter:Rebecca L. Rogers;Jennifer L. Moore
Angewandte Chemie International Edition 2007 Volume 46(Issue 48) pp:
Publication Date(Web):5 NOV 2007
DOI:10.1002/anie.200703124
The -enes have it! A resident alkene directs a nickel-catalyzed cross-coupling of cyclic anhydrides with diorganozinc reagents. Relative directing effects parallel the stability of nickel–alkene complexes, with less-hindered terminal olefins dominating over internal olefins.
Co-reporter:Jeffrey B. Johnson Dr.;Eric A. Bercot;Catherine M. Williams
Angewandte Chemie International Edition 2007 Volume 46(Issue 24) pp:
Publication Date(Web):11 MAY 2007
DOI:10.1002/anie.200700816
The wages ofsyn: The development of a phosphoramidite-ligated rhodium catalyst allows the enantioselective desymmetrization of cyclic anhydrides with organozinc nucleophiles formed in situ. This methodology has been utilized for a concise synthesis of eupomatilones 4 and 7 and of the putative structure of eupomatilone 6, each of which is completed in four steps in greater than 50 % overall yield.
Co-reporter:Jeffrey B. Johnson Dr.;Eric A. Bercot;Catherine M. Williams
Angewandte Chemie 2007 Volume 119(Issue 24) pp:
Publication Date(Web):11 MAY 2007
DOI:10.1002/ange.200700816
Ein Phosphoramidit-Rhodium-Katalysator ermöglicht die enantioselektive Desymmetrisierung cyclischer Anhydride mit in situ erzeugten Organozink-Nucleophilen. Dieser Ansatz wurde für eine effektive Synthese der Eupomatilone 4 und 7 sowie der vermuteten Struktur von Eupomatilon 6 genutzt, die jeweils in vier Stufen mit über 50 % Gesamtausbeute erhalten wurden.
Co-reporter:Rebecca L. Rogers;Jennifer L. Moore
Angewandte Chemie 2007 Volume 119(Issue 48) pp:
Publication Date(Web):5 NOV 2007
DOI:10.1002/ange.200703124
Periphere Alkengruppen steuern die nickelkatalysierte Kreuzkupplung von cyclischen Anhydriden mit Diorganozinkreagentien. Die dirigierende Wirkung korreliert mit der Stabilität der Nickel-Alken-Komplexe: Weniger gehinderte, terminale Doppelbindungen sind gegenüber internen Doppelbindungen bevorzugt.
Co-reporter:Christopher G. Nasveschuk
Angewandte Chemie International Edition 2005 Volume 44(Issue 21) pp:
Publication Date(Web):21 APR 2005
DOI:10.1002/anie.200500088
A room-temperature diastereoselective [1,3] rearrangement results from treatment of 2,5-dihydrooxepins with EtAlCl2 (see scheme). A modular synthesis of dihydrooxepins allows substituents to be incorporated at any position on the ring, which means that various polysubstituted cyclopentenes can be prepared through this Lewis acid mediated [1,3] ring contraction.
Co-reporter:Christopher G. Nasveschuk
Angewandte Chemie 2005 Volume 117(Issue 21) pp:
Publication Date(Web):21 APR 2005
DOI:10.1002/ange.200500088
Bei Raumtemperatur gelingt mit EtAlCl2 die diastereoselektive [1,3]-Umlagerung von 2,5-Dihydrooxepinen (siehe Schema). Eine modulare Synthese von Dihydrooxepinen ermöglicht den Einbau von Substituenten an jeder beliebigen Ringposition, sodass mit dieser Lewis-Säure-vermittelten [1,3]-Ringverengung eine Vielzahl polysubstituierter Cyclopentene zugänglich ist.
Co-reporter:Christopher G. Nasveschuk and Tomislav Rovis
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 2) pp:NaN254-254
Publication Date(Web):2007/12/04
DOI:10.1039/B714881J
The relay of stereochemistry of a breaking C–O bond into a forming C–C bond is well-known in the context of [3,3] sigmatropic shifts; however, this useful strategy is less well-known in other types of molecular rearrangements. Though the first successful example of a [1,3] O-to-C rearrangement was reported more than 100 years ago, this class of reactions has received less attention than its [3,3] counterpart. This perspective analyzes the various methods used for the activation and [1,3] rearrangement of vinyl ethers with an emphasis on mechanism and applications to stereoselective synthesis. We also highlight our own contributions to this area.
Co-reporter:Jamie M. Neely
Inorganic Chemistry Frontiers 2014 - vol. 1(Issue 8) pp:
Publication Date(Web):2014/09/08
DOI:10.1039/C4QO00187G
Cycloaddition reactions are powerful tools in synthetic chemistry. Incorporation of a nitrogen-containing component via cycloaddition represents an attractive strategy for assembling aza-heterocycles. One approach takes advantage of [4 + 2] cycloaddition reactions of 1-azadiene derivatives and 2-carbon π-components to access pyridines, a particularly significant subset of aza-heterocycles. This highlight discusses two distinct modes of reactivity within this class: the thermal pericyclic or hetero-Diels Alder (hDA) reaction and the transition metal-catalysed formal [4 + 2] cycloaddition reaction.
Co-reporter:Arturo Orellana and Tomislav Rovis
Chemical Communications 2008(Issue 6) pp:NaN732-732
Publication Date(Web):2007/12/04
DOI:10.1039/B716445A
A rapid assembly of the tetracyclic core of FD-838, featuring a catalytic asymmetric Stetter reaction, is described.
Co-reporter:Stéphane Perreault and Tomislav Rovis
Chemical Society Reviews 2009 - vol. 38(Issue 11) pp:NaN3159-3159
Publication Date(Web):2009/09/15
DOI:10.1039/B816702H
Cycloaddition reactions are attractive strategies for the rapid formation of molecular complexity in organic synthesis, as multiple bonds are formed in a single process. To this end, several research groups have been actively involved in the development of catalytic methods to activate readily accessible π-components to achieve cycloadditions. However, the use of C–N π-components for the formation of heterocycles by these processes is less well developed. It has been previously demonstrated that the combination of different isocyanates with two alkynes yields pyridones of several types by metal-catalyzed [2 + 2 + 2] cycloadditions. The potential of this chemistry has been extended to alkenes as C–C π-components, allowing the formation of sp3-stereocenters. In this tutorial review directed towards [n + 2 + 2] cycloadditions of heterocumulenes, alkynes and alkenes, the recent advances in the catalytic asymmetric synthesis of indolizidine, quinolizidine and azocine skeletons are discussed.
Co-reporter:Todd K. Hyster and Tomislav Rovis
Chemical Communications 2011 - vol. 47(Issue 43) pp:NaN11848-11848
Publication Date(Web):2011/10/10
DOI:10.1039/C1CC15248C
The synthesis of pyridines from readily available α,β-unsaturated oximes and alkynes under mild conditions and low temperatures using Rh(III) catalysis has been developed. It was found that the use of sterically different ligands allows for complementary selectivities to be achieved.
Co-reporter:Derek M. Dalton, Anthony K. Rappé and Tomislav Rovis
Chemical Science (2010-Present) 2013 - vol. 4(Issue 5) pp:NaN2070-2070
Publication Date(Web):2013/03/05
DOI:10.1039/C3SC50271F
Perfluorinated Taddol-based phosphoramidite, CKphos, is a highly selective ligand for formation of vinylogous amide cycloadducts in the Rh(I) catalyzed [2 + 2 + 2] cycloaddition of alkenyl isocyanates and alkynes. CKphos overrides substrate bias of product selectivity in the cycloaddition, providing indolizidinones in excellent yields and enantioselectivities. The excellent selectivities are attributed to a shortened Rh–P bond and coordination of one C6F5 to rhodium via a Z-type interaction, making the phosphoramidite a bidentate L,Z-ligand on rhodium. Evidence for the shortened Rh–P and C6F5 coordination is provided by X-ray, NMR and DFT computational analyses. Additionally, an anionic cobalt complex with CKphos was synthesized and two Co–C6F5 interactions are seen. The Rh(C2H4)Cl·CKphos catalyst in the [2 + 2 + 2] cycloaddition of alkenyl isocyanates and alkynes represents a rare example of metal–C6F5 Z-type interaction affecting selectivity in transition metal catalysis.
Co-reporter:Benjamin L. Kohn and Tomislav Rovis
Chemical Science (2010-Present) 2014 - vol. 5(Issue 7) pp:NaN2892-2892
Publication Date(Web):2014/05/22
DOI:10.1039/C4SC00743C
Perfluoroketones are useful products and intermediates in medicinal chemistry. Herein, cobalt-mediated fluoroacylation of 1,3-dienes is described using perfluorinated anhydrides such as TFAA. The reaction is thought to proceed through a fluoroacylcobalt reagent formed in situ. Perfluoroacylation of 1,3 dienes can also be performed to attain longer chain perfluorinated ketones.
Co-reporter:Todd K. Hyster, Derek M. Dalton and Tomislav Rovis
Chemical Science (2010-Present) 2015 - vol. 6(Issue 1) pp:NaN258-258
Publication Date(Web):2014/10/01
DOI:10.1039/C4SC02590C
We report the regioselective synthesis of dihydroisoquinolones from aliphatic alkenes and O-pivaloyl benzhydroxamic acids mediated by a Rh(III) precatalyst bearing sterically bulky substituents. While the prototypical Cp* ligand provides product with low selectivity, sterically bulky Cpt affords product with excellent regioselectivity for a range of benzhydroxamic acids and alkenes. Crystallographic evidence offers insight as to the source of the increased regioselectivity.
Co-reporter:Philip Wheeler, Harit U. Vora and Tomislav Rovis
Chemical Science (2010-Present) 2013 - vol. 4(Issue 4) pp:NaN1679-1679
Publication Date(Web):2013/02/13
DOI:10.1039/C3SC00089C
The scope of the NHC-redox amidation has been expanded to include a variety of α,β-unsaturated aldehydes, including α-fluoro α,β-unsaturated aldehydes which give rise to enantioenriched α-fluoroamides in good to excellent yield and enantioselectivity (up to 97% ee). Enantioenriched amines may be elaborated to either diastereomer of the product in high diastereoselectivity (up to 99:1). Functionalization of the amide products to amines and fluorohydrins is also demonstrated with retention of enantioenrichment at the fluorine stereocenter.
Co-reporter:Ya Du, Todd K. Hyster and Tomislav Rovis
Chemical Communications 2011 - vol. 47(Issue 44) pp:NaN12076-12076
Publication Date(Web):2011/10/17
DOI:10.1039/C1CC15843K
An efficient strategy for the oxidative carbonylation of aromatic amidesvia C–H/N–H activation to form phthalimides using an Rh(III) catalyst has been developed. The reaction shows a preference for C–H bonds of electron-rich aromatic amides and tolerates a variety of functional groups.
Co-reporter:Kerem E. Ozboya and Tomislav Rovis
Chemical Science (2010-Present) 2011 - vol. 2(Issue 9) pp:NaN1838-1838
Publication Date(Web):2011/06/16
DOI:10.1039/C1SC00175B
Herein we report an enantioselective synthesis of complex cyclopentanones using aliphatic aldehydes and activated enones. With the combination of a chiral secondary amine and a chiral triazolium catalyst, high diastereoselectivity and excellent enantioselectivity can be achieved. We present evidence of a clear cooperative effect when these two catalysts are present simultaneously in the system.
Co-reporter:Todd K. Hyster and Tomislav Rovis
Chemical Communications 2015 - vol. 51(Issue 26) pp:NaN5778-5778
Publication Date(Web):2015/03/09
DOI:10.1039/C5CC90120K
Correction for ‘Pyridine synthesis from oximes and alkynes via rhodium(III) catalysis: Cp* and Cpt provide complementary selectivity’ by Todd K. Hyster et al., Chem. Commun., 2011, 47, 11846–11848.
Co-reporter:Todd K. Hyster and Tomislav Rovis
Chemical Science (2010-Present) 2011 - vol. 2(Issue 8) pp:NaN1610-1610
Publication Date(Web):2011/05/16
DOI:10.1039/C1SC00235J
We have developed a method for preparing pyridones from the coupling reaction of acrylamides and alkynes with either stoichometric Cu(OAc)2 or catalyticCu(OAc)2 and air as oxidants. In the course of these studies, it was found that a larger ligand, 1,3-di-tert-butylcyclopentadienyl (termed Cpt) results in higher degrees of regioselectivity in the alkyneinsertion event. The transformation tolerates a broad variety of alkynes and acrylamides. Furthermore, Cpt and Cp* demonstrate similar catalytic activity. This similarity allows for mechanistic studies to be undertaken which suggest a difference in mechanism between this reaction and the previously studied benzamide system.
.jpg)
![(R)-5-isopropyl-2-(perfluorophenyl)-6,7-dihydro-5H-pyrrolo[2,1-c][1,2,4]triazol-2-ium tetrafluoroborate](http://img.cochemist.com/ccimg/1175100/1175052-07-9.png)
![(R)-5-isopropyl-2-(perfluorophenyl)-6,7-dihydro-5H-pyrrolo[2,1-c][1,2,4]triazol-2-ium tetrafluoroborate](http://img.cochemist.com/ccimg/1175100/1175052-07-9_b.png)
![1,3-DIOXEPIN, 4,7-DIHYDRO-2-[2-(PHENYLTHIO)ETHYL]-](http://img.cochemist.com/ccimg/909400/909399-18-4.png)
![1,3-DIOXEPIN, 4,7-DIHYDRO-2-[2-(PHENYLTHIO)ETHYL]-](http://img.cochemist.com/ccimg/909400/909399-18-4_b.png)




![ACETALDEHYDE, 2-[(1,2,6-TRIMETHYL-4-OXO-2,5-CYCLOHEXADIEN-1-YL)OXY]-](http://img.cochemist.com/ccimg/881200/881181-69-7.png)
![ACETALDEHYDE, 2-[(1,2,6-TRIMETHYL-4-OXO-2,5-CYCLOHEXADIEN-1-YL)OXY]-](http://img.cochemist.com/ccimg/881200/881181-69-7_b.png)
![ACETALDEHYDE, 2-[[1,3,5-TRIS(1,1-DIMETHYLETHYL)-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-67-5.png)
![ACETALDEHYDE, 2-[[1,3,5-TRIS(1,1-DIMETHYLETHYL)-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-67-5_b.png)
![ACETALDEHYDE, 2-[[3,5-BIS(1,1-DIMETHYLETHYL)-1-METHYL-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-65-3.png)
![ACETALDEHYDE, 2-[[3,5-BIS(1,1-DIMETHYLETHYL)-1-METHYL-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-65-3_b.png)
![ACETALDEHYDE, 2-[[3,5-BIS(METHOXYMETHYL)-1-METHYL-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-63-1.png)
![ACETALDEHYDE, 2-[[3,5-BIS(METHOXYMETHYL)-1-METHYL-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-63-1_b.png)
![ACETALDEHYDE, 2-[(1,3,5-TRIMETHYL-4-OXO-2,5-CYCLOHEXADIEN-1-YL)OXY]-](http://img.cochemist.com/ccimg/881200/881181-61-9.png)
![ACETALDEHYDE, 2-[(1,3,5-TRIMETHYL-4-OXO-2,5-CYCLOHEXADIEN-1-YL)OXY]-](http://img.cochemist.com/ccimg/881200/881181-61-9_b.png)
![ACETALDEHYDE, 2-[[1-(2-METHOXYETHYL)-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-57-3.png)
![ACETALDEHYDE, 2-[[1-(2-METHOXYETHYL)-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-57-3_b.png)
![ACETALDEHYDE, 2-[[1-[(ACETYLOXY)METHYL]-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-55-1.png)
![ACETALDEHYDE, 2-[[1-[(ACETYLOXY)METHYL]-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-55-1_b.png)
![ACETALDEHYDE, 2-[[1-(4-BROMOPHENYL)-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-53-9.png)
![ACETALDEHYDE, 2-[[1-(4-BROMOPHENYL)-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-53-9_b.png)



![(4aR,9aS)-4,4a,9,9a-Tetrahydroindeno[2,1-b][1,4]oxazin-3(2H)-one](http://img.cochemist.com/ccimg/862100/862095-79-2.png)
![(4aR,9aS)-4,4a,9,9a-Tetrahydroindeno[2,1-b][1,4]oxazin-3(2H)-one](http://img.cochemist.com/ccimg/862100/862095-79-2_b.png)




![(2S)-2-METHYL-2-[2-(4-NITROPHENYL)-2-OXOETHYL]CYCLOPENTAN-1-ONE](http://img.cochemist.com/ccimg/740900/740816-34-6.png)
![(2S)-2-METHYL-2-[2-(4-NITROPHENYL)-2-OXOETHYL]CYCLOPENTAN-1-ONE](http://img.cochemist.com/ccimg/740900/740816-34-6_b.png)



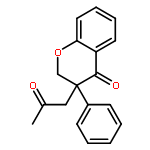
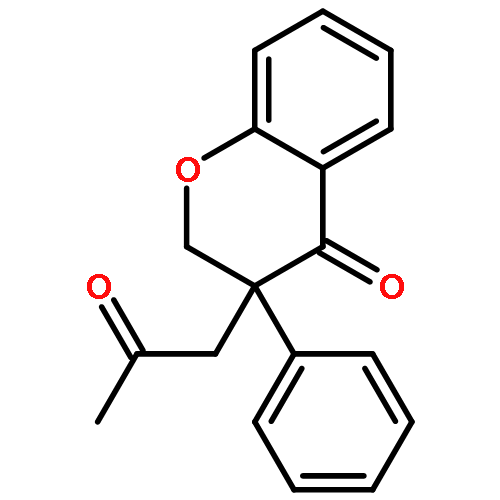
![Benzaldehyde, 2-[[(2E)-4-oxo-2-phenyl-2-pentenyl]oxy]-](http://img.cochemist.com/ccimg/740900/740816-19-7.png)
![Benzaldehyde, 2-[[(2E)-4-oxo-2-phenyl-2-pentenyl]oxy]-](http://img.cochemist.com/ccimg/740900/740816-19-7_b.png)

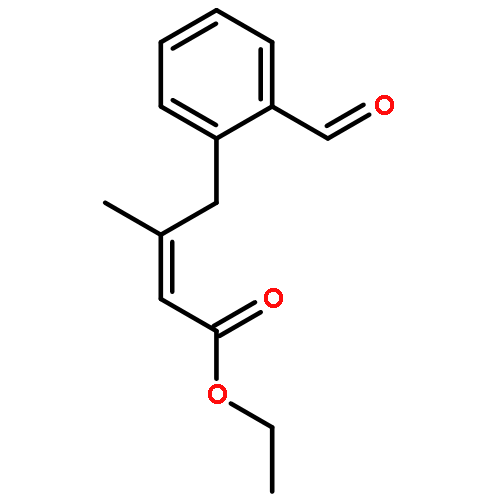
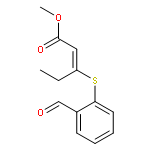

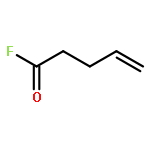
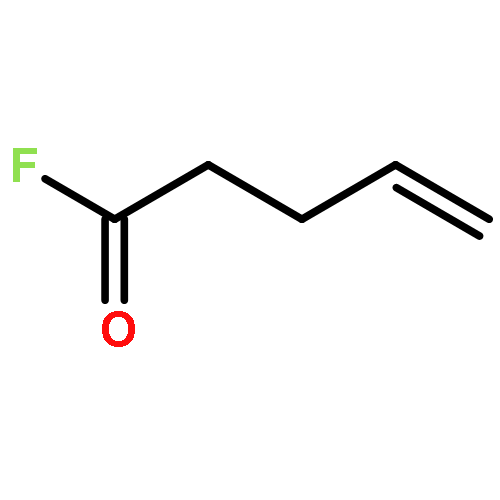

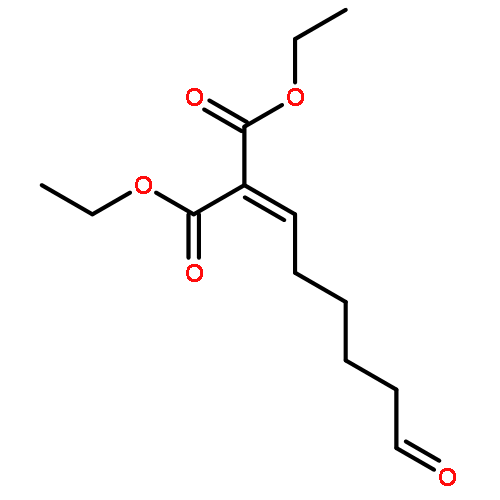
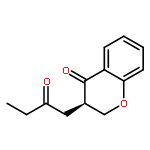
![BENZALDEHYDE, 2-[[(2E)-4-OXO-2-HEXENYL]OXY]-](http://img.cochemist.com/ccimg/640800/640737-17-3.png)
![BENZALDEHYDE, 2-[[(2E)-4-OXO-2-HEXENYL]OXY]-](http://img.cochemist.com/ccimg/640800/640737-17-3_b.png)
















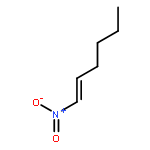
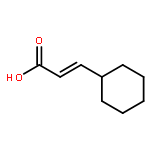
![Cyclohexane, [(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/132900/132839-80-6.png)
![Cyclohexane, [(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/132900/132839-80-6_b.png)









































![(3aR,9aS)-3a,4,9,9a-tetrahydronaphtho[2,3-c]furan-1,3-dione](http://img.cochemist.com/ccimg/1500/1459-16-1.png)
![(3aR,9aS)-3a,4,9,9a-tetrahydronaphtho[2,3-c]furan-1,3-dione](http://img.cochemist.com/ccimg/1500/1459-16-1_b.png)





![5-{[3'-(6-FLUORO-2,3-DIHYDRO-1H-INDOL-1-YL)-4-BIPHENYLYL]METHYL}-1,2,5-THIADIAZOLIDIN-3-ONE 1,1-DIOXIDE](http://img.cochemist.com/ccimg/1013500/1013400-92-4.png)
![5-{[3'-(6-FLUORO-2,3-DIHYDRO-1H-INDOL-1-YL)-4-BIPHENYLYL]METHYL}-1,2,5-THIADIAZOLIDIN-3-ONE 1,1-DIOXIDE](http://img.cochemist.com/ccimg/1013500/1013400-92-4_b.png)





![1,3-Dioxepin, 4,7-dihydro-2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/909400/909399-19-5.png)
![1,3-Dioxepin, 4,7-dihydro-2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/909400/909399-19-5_b.png)


![Oxazole,2-[2-(diphenylphosphino)phenyl]-4,5-dihydro-4-(1-methylethyl)-, (4S)-](/data/chemimg/130100/148461-14-7.png)
![Oxazole,2-[2-(diphenylphosphino)phenyl]-4,5-dihydro-4-(1-methylethyl)-, (4S)-](/data/chemimg/130100/148461-14-7_b.png)


![Benzene, [(1E)-2-iodo-1-methylethenyl]-](http://img.cochemist.com/ccimg/66700/66644-32-4.png)
![Benzene, [(1E)-2-iodo-1-methylethenyl]-](http://img.cochemist.com/ccimg/66700/66644-32-4_b.png)
![Benzene, [(1E)-2-iodoethenyl]-](http://img.cochemist.com/ccimg/42600/42599-24-6.png)
![Benzene, [(1E)-2-iodoethenyl]-](http://img.cochemist.com/ccimg/42600/42599-24-6_b.png)


![Bicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic acid,dimethyl ester,(endo,endo)](http://img.cochemist.com/ccimg/17900/17812-27-0.png)
![Bicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic acid,dimethyl ester,(endo,endo)](http://img.cochemist.com/ccimg/17900/17812-27-0_b.png)
![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0.png)
![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0_b.png)




![5H-Pyrrolo[2,1-c]-1,2,4-triazolium,6,7-dihydro-2-phenyl-, chloride (1:1)](http://img.cochemist.com/ccimg/829000/828914-68-7.png)
![5H-Pyrrolo[2,1-c]-1,2,4-triazolium,6,7-dihydro-2-phenyl-, chloride (1:1)](http://img.cochemist.com/ccimg/829000/828914-68-7_b.png)

















![3-Oxabicyclo[3.2.0]heptane-2,4-dione](http://img.cochemist.com/ccimg/4500/4462-96-8.png)
![3-Oxabicyclo[3.2.0]heptane-2,4-dione](http://img.cochemist.com/ccimg/4500/4462-96-8_b.png)
![2(3H)-Furanone, dihydro-3-[(1R)-2-oxocyclopentyl]-, (3R)-](http://img.cochemist.com/ccimg/852300/852248-23-8.png)
![2(3H)-Furanone, dihydro-3-[(1R)-2-oxocyclopentyl]-, (3R)-](http://img.cochemist.com/ccimg/852300/852248-23-8_b.png)




![ACETALDEHYDE, 2-[(4-OXO-1-PHENYL-2,5-CYCLOHEXADIEN-1-YL)OXY]-](http://img.cochemist.com/ccimg/881200/881181-51-7.png)
![ACETALDEHYDE, 2-[(4-OXO-1-PHENYL-2,5-CYCLOHEXADIEN-1-YL)OXY]-](http://img.cochemist.com/ccimg/881200/881181-51-7_b.png)
![ACETALDEHYDE, 2-[[1-(1,1-DIMETHYLETHYL)-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-49-3.png)
![ACETALDEHYDE, 2-[[1-(1,1-DIMETHYLETHYL)-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-49-3_b.png)
![ACETALDEHYDE, 2-[[1-(1-METHYLETHYL)-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-47-1.png)
![ACETALDEHYDE, 2-[[1-(1-METHYLETHYL)-4-OXO-2,5-CYCLOHEXADIEN-1-YL]OXY]-](http://img.cochemist.com/ccimg/881200/881181-47-1_b.png)
![ACETALDEHYDE, 2-[(1-ETHYL-4-OXO-2,5-CYCLOHEXADIEN-1-YL)OXY]-](http://img.cochemist.com/ccimg/881200/881181-45-9.png)
![ACETALDEHYDE, 2-[(1-ETHYL-4-OXO-2,5-CYCLOHEXADIEN-1-YL)OXY]-](http://img.cochemist.com/ccimg/881200/881181-45-9_b.png)
![ACETALDEHYDE, 2-[(1-METHYL-4-OXO-2,5-CYCLOHEXADIEN-1-YL)OXY]-](http://img.cochemist.com/ccimg/881200/881181-43-7.png)
![ACETALDEHYDE, 2-[(1-METHYL-4-OXO-2,5-CYCLOHEXADIEN-1-YL)OXY]-](http://img.cochemist.com/ccimg/881200/881181-43-7_b.png)
![(5AR,10BS)-(+)-5A,10B-DIHYDRO-2-(PENTAFLUOROPHENYL)-4H,6H-INDENO[2,1-B][1,2,4]TRIZOLO[4,3-D][1,4]OXAZINIUM TETRAFLUOROBORATE](http://img.cochemist.com/ccimg/872200/872143-57-2.png)
![(5AR,10BS)-(+)-5A,10B-DIHYDRO-2-(PENTAFLUOROPHENYL)-4H,6H-INDENO[2,1-B][1,2,4]TRIZOLO[4,3-D][1,4]OXAZINIUM TETRAFLUOROBORATE](http://img.cochemist.com/ccimg/872200/872143-57-2_b.png)




![1-Hexen-3-one, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-](/data/chemimg/3771000/1416725-15-9.png)
![1-Hexen-3-one, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-](/data/chemimg/3771000/1416725-15-9_b.png)









![1H-Indole, 3-[(1S)-2-nitro-1-phenylethyl]-](http://img.cochemist.com/ccimg/863400/863301-32-0.png)
![1H-Indole, 3-[(1S)-2-nitro-1-phenylethyl]-](http://img.cochemist.com/ccimg/863400/863301-32-0_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2_b.png)
![(5AS, 10BR)-(-)-5A,10B-DIHYDRO-2-(PENTAFLUOROPHENYL)-4H,6H-INDENO[2,1-B][1,2,4]TRIZOLO[4,3-D][1,4]OXAZINIUM TETRAFLUOROBORATE](http://img.cochemist.com/ccimg/740900/740816-14-2.png)
![(5AS, 10BR)-(-)-5A,10B-DIHYDRO-2-(PENTAFLUOROPHENYL)-4H,6H-INDENO[2,1-B][1,2,4]TRIZOLO[4,3-D][1,4]OXAZINIUM TETRAFLUOROBORATE](http://img.cochemist.com/ccimg/740900/740816-14-2_b.png)





















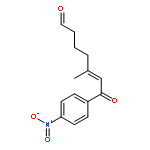
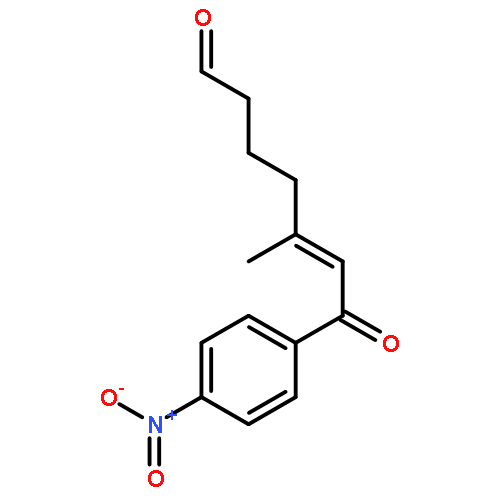
![Benzene, [(3E)-4-nitro-3-butenyl]-](http://img.cochemist.com/ccimg/169800/169775-70-6.png)
![Benzene, [(3E)-4-nitro-3-butenyl]-](http://img.cochemist.com/ccimg/169800/169775-70-6_b.png)










![5-Methoxybenzo[d]oxazole](http://img.cochemist.com/ccimg/132300/132227-03-3.png)
![5-Methoxybenzo[d]oxazole](http://img.cochemist.com/ccimg/132300/132227-03-3_b.png)
![Butanal, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/127800/127793-62-8.png)
![Butanal, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/127800/127793-62-8_b.png)

![2-Butanone, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/120600/120591-36-8.png)
![2-Butanone, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/120600/120591-36-8_b.png)


![Propanal, 3-[[tris(1-methylethyl)silyl]oxy]-](http://img.cochemist.com/ccimg/112000/111998-84-6.png)
![Propanal, 3-[[tris(1-methylethyl)silyl]oxy]-](http://img.cochemist.com/ccimg/112000/111998-84-6_b.png)

![4-Methylbenzo[d]oxazole](http://img.cochemist.com/ccimg/107200/107165-67-3.png)
![4-Methylbenzo[d]oxazole](http://img.cochemist.com/ccimg/107200/107165-67-3_b.png)

![Silane, (1,1-dimethylethyl)[[1-(2-furanyl)-2-hexynyl]oxy]dimethyl-](http://img.cochemist.com/ccimg/104400/104376-30-9.png)
![Silane, (1,1-dimethylethyl)[[1-(2-furanyl)-2-hexynyl]oxy]dimethyl-](http://img.cochemist.com/ccimg/104400/104376-30-9_b.png)



![(s)-2-amino-3-[4-(phenylmethoxy)phenyl]-1-propanol](http://img.cochemist.com/ccimg/85900/85803-44-7.png)
![(s)-2-amino-3-[4-(phenylmethoxy)phenyl]-1-propanol](http://img.cochemist.com/ccimg/85900/85803-44-7_b.png)






![1-HEXEN-3-OL, 6-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-2-METHYL-](http://img.cochemist.com/ccimg/73900/73835-58-2.png)
![1-HEXEN-3-OL, 6-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-2-METHYL-](http://img.cochemist.com/ccimg/73900/73835-58-2_b.png)















![Benzenamine, N-[(2E)-3-phenyl-2-propenylidene]-, (E)-](http://img.cochemist.com/ccimg/56200/56110-07-7.png)
![Benzenamine, N-[(2E)-3-phenyl-2-propenylidene]-, (E)-](http://img.cochemist.com/ccimg/56200/56110-07-7_b.png)











![BENZENE, 4-[(4-METHOXYPHENYL)ETHYNYL]-4-(TRIFLUOROMETHYL)-](http://img.cochemist.com/ccimg/40500/40474-01-9.png)
![BENZENE, 4-[(4-METHOXYPHENYL)ETHYNYL]-4-(TRIFLUOROMETHYL)-](http://img.cochemist.com/ccimg/40500/40474-01-9_b.png)
![Benzene, [(1E)-2-methyl-1,3-butadienyl]-](http://img.cochemist.com/ccimg/39700/39600-69-6.png)
![Benzene, [(1E)-2-methyl-1,3-butadienyl]-](http://img.cochemist.com/ccimg/39700/39600-69-6_b.png)

























![7-Methylbenzo[d]oxazole](http://img.cochemist.com/ccimg/10600/10531-82-5.png)
![7-Methylbenzo[d]oxazole](http://img.cochemist.com/ccimg/10600/10531-82-5_b.png)









![Benzenamine, 4-methoxy-N-[(4-nitrophenyl)methylene]-](/data/chemimg/94300/5455-87-8.png)
![Benzenamine, 4-methoxy-N-[(4-nitrophenyl)methylene]-](/data/chemimg/94300/5455-87-8_b.png)













































![1-Hexen-3-one, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/132700/132621-86-4.png)
![1-Hexen-3-one, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/132700/132621-86-4_b.png)


![1-[(1r,2r)-2-benzoylcyclopropyl]ethanone](http://img.cochemist.com/ccimg/64400/64390-04-1.png)
![1-[(1r,2r)-2-benzoylcyclopropyl]ethanone](http://img.cochemist.com/ccimg/64400/64390-04-1_b.png)























![(5R,7R)-7-FLUORO-2-(2,3,4,5,6-PENTAFLUOROPHENYL)-5-PROPAN-2-YL-6,7-DIHYDRO-5H-PYRROLO[2,1-C][1,2,4]TRIAZOL-4-IUM;TETRAFLUOROBORATE](http://img.cochemist.com/data/images/1311991-39-5.png)
![(5R,7R)-7-FLUORO-2-(2,3,4,5,6-PENTAFLUOROPHENYL)-5-PROPAN-2-YL-6,7-DIHYDRO-5H-PYRROLO[2,1-C][1,2,4]TRIAZOL-4-IUM;TETRAFLUOROBORATE](http://img.cochemist.com/data/images/1311991-39-5_b.png)
![1H-ISOINDOLE-1,3(2H)-DIONE, 2-[(1-PHENYLETHENYL)OXY]-](http://img.cochemist.com/ccimg/91600/91523-92-1.png)
![1H-ISOINDOLE-1,3(2H)-DIONE, 2-[(1-PHENYLETHENYL)OXY]-](http://img.cochemist.com/ccimg/91600/91523-92-1_b.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)