Co-reporter:Lu Wang, Yuji Eguchi, and Eugene Y.-X. Chen
Industrial & Engineering Chemistry Research October 11, 2017 Volume 56(Issue 40) pp:11380-11380
Publication Date(Web):September 25, 2017
DOI:10.1021/acs.iecr.7b02920
A bisfuran dibromide has been established as the versatile and common intermediate for the high-yield synthesis of the three important classes of bisfuran monomers for furan-based renewable materials, bisfuran diacids, diols, and diamines. The general synthetic route involves a coupling reaction of 2-methylfuran with a ketone (acetone or cyclohexanone) under acidic conditions and a bromination reaction of the resulting bisfuran dimethyl compound to produce the bisfuran dibromide intermediate. This dibromide intermediate is subsequently converted to the corresponding bisfuran diacid (via oxidation reaction with KMnO4 under basic conditions), bisfuran diol (by hydrolysis reaction under mild basic conditions), and bisfuran diamine (through the Gabriel reaction). The versatility of the bisfuran dibromide intermediate and the effective transformation into the monomers with high to quantitative yield typically without the need for further purification highlight the two attractive features and potential for large-scale production.
Co-reporter:Miao Hong, Xiaoyan Tang, Brian S. Newell, and Eugene Y.-X. Chen
Macromolecules November 14, 2017 Volume 50(Issue 21) pp:8469-8469
Publication Date(Web):October 25, 2017
DOI:10.1021/acs.macromol.7b02174
Despite several anticipated advantages of the bioderived γ-butyrolactone (γ-BL) as an effective comonomer to modulate materials properties of its copolyesters, the currently unmet challenge hinders access to such copolyesters with high γ-BL incorporations due to unfavorable thermodynamics toward the ring-opening polymerization of the highly stable, typically referred to as “nonstrained”, γ-BL. Here we report the effective copolymerization of γ-BL with two common cyclic esters with very different monomer thermodynamic polymerizability, ε-caprolactone (ε-CL) and δ-valerolactone (δ-VL), leading to a series of relatively high molecular weight (Mn up to 135 kg/mol) random copolyesters with unprecedented levels of γ-BL incorporations (up to 84.0 mol %) and thus providing access to γ-BL-based copolyesters in the entire composition range needed for comprehensive investigations into the composition-dependent physical properties and degradation behavior of the resulting copolyesters. This copolymerization was enabled by the judiciously chosen metal and organic catalysts that exhibit different kinetic behavior or monomer selectivity, designed to more effectively compete the “nonstrained” γ-BL against the relatively high-strained lactones toward ring-opening. The successful synthesis of the copolyesters with high γ-BL incorporations of >50 mol % led to the discovery of the eutectic phase of the γ-BL/ε-CL copolymer with a eutectic temperature Teu of 11.0 °C and a eutectic composition Xeu of 66.0% γ-BL; thus, at this composition, the copolymer becomes a viscous liquid at room temperature, although the two constituent homopolymers are semicrystalline solids. Other important composition-dependent properties of γ-BL-based copolyesters, including thermal transitions, cocrystallization, as well as thermal and hydrolytic degradation behaviors, have also been examined.
Co-reporter:Jian-Bo Zhu, Xiaoyan Tang, Laura Falivene, Lucia Caporaso, Luigi Cavallo, and Eugene Y.-X. Chen
ACS Catalysis June 2, 2017 Volume 7(Issue 6) pp:3929-3929
Publication Date(Web):May 3, 2017
DOI:10.1021/acscatal.7b00794
In the presence of a N-heterocyclic carbene (NHC) in THF, Br-substituted l-lactide (Br-LA) unexpectedly undergoes exclusive coupling with THF to form a chiral ω-bromo-α-keto-diester. This coupling reaction is completely selective (in a precise 1:1 fashion), readily scalable (>20 g scale), and extremely efficient (with only 50 ppm of NHC loading). Other cyclic ethers and carbonates can also undergo similar coupling with Br-LA, thus offering a class of Br-functionalized chiral diesters with various functions and chain lengths. Combined experimental and computational studies led to a coupling mechanism that proceeds through an anion (bromide)-mediated catalytic cycle, rather than an apparent NHC-catalyzed cycle.Keywords: bromo-lactide; chiral diesters; coupling; N-heterocyclic carbene; organocatalysis;
Co-reporter:Fernando Vidal and Eugene Y.-X. Chen
Organometallics August 14, 2017 Volume 36(Issue 15) pp:2922-2922
Publication Date(Web):July 12, 2017
DOI:10.1021/acs.organomet.7b00358
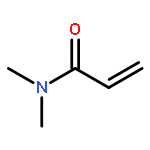
This work investigates the reactivity of neutral and cationic complexes of both bridged ansa-zirconocenes, rac-[C2H4(Ind)2]ZrMe[OC(OiPr)═CMe2] (1) and rac-[C2H4(Ind)2]Zr+(THF)[OC(OiPr)═CMe2][MeB(C6F5)3]− (1+), and nonbridged zirconocenes, Cp*(nPrCp)ZrMe[OC(OiPr)═CMe2] (13) and Cp*(nPrCp)Zr(THF)[OC(OiPr)═CMe2]+[MeB(C6F5)3]− (13+), toward biorenewable itaconic dialkyl esters (itaconates) and anhydride (IA). Behaving similarly, both cationic complexes 1+ and 13+ react readily with itaconates to form cleanly single monomer addition products, eight-membered-ring metallacycles 2 and 15, respectively, and neutral enolate complexes 1 and 13 insert 1 equiv of IA to afford single-IA-addition products 5 and 17. Behaving differently, eight-membered-ring chelates 2 derived from the bridged metallocene framework undergo slow isomerization at room temperature via ligand exchange between the coordinated and uncoordinated ester groups to form thermodynamically favored seven-membered-ring chelates 4, while eight-membered chelates 15 derived from the sterically more crowded unbridged metallocene framework are stable at room temperature and do not undergo such isomerization. The above cationic complexes exhibit no reactivity toward further additions of itaconates. Replacing itaconates with more basic monomers such as N,N-dimethylacrylamide that can ring open the chelating resting intermediate, however, brings about effective and controlled polymerization by eight-membered Zr-itaconate metallacycles 2 and 15, but not seven-membered 4, producing either highly isotactic polymers (>99% mm, by 2) or polymers with narrow molecular weight distributions (Đ < 1.19, by 15). These results further highlight the ansa effects in the metallocene polymerization chemistry and the importance of the formation and ring opening of the eight-membered chelating intermediates involved in the metallocene-mediated conjugate-addition polymerization.
Co-reporter:Jing Tang, Eugene Y.-X. Chen
European Polymer Journal 2017 Volume 95(Volume 95) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.eurpolymj.2017.05.043
•First study on the homopolymerization of furanones by organic catalysts.•Uncovering the complexity of the polymerization of trifunctional gamma-butyrolactones.•Discovery of unique trimerization of 3-methylfuran-2(5H)-one.•Mechanistic understanding of branched polymer formation by the trifunctional monomer.This contribution investigates organopolymerization of five multifunctional γ-butyrolactone-based monomers, including bifunctional (endocyclic double bond, lactone ring) dihydrofuran-2(3H)-one (FO), 3-methylfuran-2(5H)-one (3-MFO), and 5-methylfuran-2(5H)-one (5-MFO), as well as trifunctional (endocyclic or exocyclic double bond, lactone ring, hydroxyl group) 3-(hydroxymethyl) furan-2(5H)-one (3-HMFO) and β-hydroxy-α-methylene-γ-butyrolactone (βHMBL). The complexity of the reaction under nucleophilic and basic conditions using N-heterocyclic carbene (NHC) and superbase organic catalysts increases dramatically on going from the bifunctional monomers to the trifunctional ones. Thus, the polymerization of the parent FO leads to a vinyl-addition polymer, while the reaction of the base catalysts with the two methyl-substituted derivatives, 3-MFO and 5-MFO, affords predominately a trimer and dimer, respectively. The polymerization of trifunctional 3-HMFO gives a poly(vinyl–ether lactone) copolymer structure, via two different types of base activation mechanisms and a combination of Michael and ox-Michael additions and proton transfer processes. The polymerization of βHMBL has the highest degree of the complexity in this monomer series, due to its presence of both the reactive exocyclic double bond and hydroxyl group, producing a branched vinyl–ether lactone copolymer structure having six different types of substructural units. The results reveal multiple types of reaction pathways and their mechanistic crossovers involved in the βHMBL polymerization, including conjugate Michael and oxa-Michael additions and proton transfer processes, as well as ene-type dehydration reactions, enabled by proton transfer.Download high-res image (124KB)Download full-size image
Co-reporter:Miao Hong
Green Chemistry (1999-Present) 2017 vol. 19(Issue 16) pp:3692-3706
Publication Date(Web):2017/08/14
DOI:10.1039/C7GC01496A
The current practices in the generation and disposal of synthetic polymers are largely unsustainable. As part of the solution, the development of biodegradable polymers, which constitute a class of “green polymers” according to green chemistry principles, has been intensively pursued in the past two decades. However, the degradation of such polymers in Earth's landfills typically leads to no recovery of the materials’ value, and their degradation in the Oceans could create new or unintended environmental consequences. Industrial mechanical recycling always suffers from a significant quality loss. The proposed more sustainable solution is to develop chemically recyclable polymers that not only solve the end-of-life issue of polymers, but also provide a direct approach to establish a circular materials economy. Accordingly, this critical review article captures some selected highlights of the emerging area of recyclable “green polymers” by focusing on the major progress made and the technical and environmental benefits obtained in the development of repurposing and depolymerization processes for chemical recycling of polymers at the end of their useful life.
Co-reporter:Xiaoyan Tang, Miao Hong, Laura Falivene, Lucia Caporaso, Luigi Cavallo, and Eugene Y.-X. Chen
Journal of the American Chemical Society 2016 Volume 138(Issue 43) pp:14326-14337
Publication Date(Web):October 4, 2016
DOI:10.1021/jacs.6b07974
α-Methylene-γ-butyrolactone (MBL), a naturally occurring and biomass-sourced bifunctional monomer, contains both a highly reactive exocyclic C═C bond and a highly stable five-membered γ-butyrolactone ring. Thus, all previous work led to exclusive vinyl-addition polymerization (VAP) product P(MBL)VAP. Now, this work reverses this conventional chemoselectivity to enable the first ring-opening polymerization (ROP) of MBL, thereby producing exclusively unsaturated polyester P(MBL)ROP with Mn up to 21.0 kg/mol. This elusive goal was achieved through uncovering the thermodynamic, catalytic, and processing conditions. A third reaction pathway has also been discovered, which is a crossover propagation between VAP and ROP processes, thus affording cross-linked polymer P(MBL)CLP. The formation of the three types of polymers, P(MBL)VAP, P(MBL)CLP, and P(MBL)ROP, can be readily controlled by adjusting the catalyst (La)/initiator (ROH) ratio, which is determined by the unique chemoselectivity of the La–X (X = OR, NR2, R) group. The resulting P(MBL)ROP is degradable and can be readily postfunctionalized into cross-linked or thiolated materials but, more remarkably, can also be fully recycled back to its monomer thermochemically. Computational studies provided the theoretical basis for, and a mechanistic understanding of, the three different polymerization processes and the origin of the chemoselectivity.
Co-reporter:Miao Hong; Xiaoyan Tang; Laura Falivene; Lucia Caporaso; Luigi Cavallo
Journal of the American Chemical Society 2016 Volume 138(Issue 6) pp:2021-2035
Publication Date(Web):January 18, 2016
DOI:10.1021/jacs.5b13019
This contribution presents a full account of experimental and theoretical/computational investigations into the N-heterocyclic carbene (NHC)-catalyzed proton-transfer polymerization (HTP) that converts common dimethacrylates (DMAs) containing no protic groups into unsaturated polyesters. This new HTP proceeds through the step-growth propagation cycles via enamine intermediates, consisting of the proposed conjugate addition–proton transfer–NHC release fundamental steps. This study examines the monomer and catalyst scopes as well as the fundamental steps involved in the overall HTP mechanism. DMAs having six different types of linkages connecting the two methacrylates have been polymerized into the corresponding unsaturated polyesters. The most intriguing unsaturated polyester of the series is that based on the biomass-derived furfuryl dimethacrylate, which showed a unique self-curing ability. Four MeO- and Cl-substituted TPT (1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene) derivatives as methanol insertion products, RxTPT(MeO/H) (R = MeO, Cl; x = 2, 3), and two free carbenes (catalysts), OMe2TPT and OMe3TPT, have been synthesized, while OMe2TPT(MeO/H) and OMe2TPT have also been structurally characterized. The structure/reactivity relationship study revealed that OMe2TPT, being both a strong nucleophile and a good leaving group, exhibits the highest HTP activity and also produced the polyester with the highest Mn, while the Cl-substituted TPT derivatives are least active and efficient. Computational studies have provided mechanistic insights into the tail-to-tail dimerization coupling step as a suitable model for the propagation cycle of the HTP. The extensive energy profile was mapped out, and the experimentally observed unicity of the TPT-based catalysts was satisfactorily explained with the thermodynamic formation of key spirocyclic species.
Co-reporter:Jiawei Chen; Laura Falivene; Lucia Caporaso; Luigi Cavallo
Journal of the American Chemical Society 2016 Volume 138(Issue 16) pp:5321-5333
Publication Date(Web):April 4, 2016
DOI:10.1021/jacs.6b01497
This contribution reports the first example of highly selective reduction of CO2 into CH4 via tandem hydrosilylation with mixed main-group organo-Lewis acid (LA) catalysts [Al(C6F5)3 + B(C6F5)3] {[Al] + [B]}. As shown by this comprehensive experimental and computational study, in this unique tandem catalytic process, [Al] effectively mediates the first step of the overall reduction cycle, namely the fixation of CO2 into HCOOSiEt3 (1) via the LA-mediated C═O activation, while [B] is incapable of promoting the same transformation. On the other hand, [B] is shown to be an excellent catalyst for the subsequent reduction steps 2–4, namely the hydrosilylation of the more basic intermediates [1 to H2C(OSiEt3)2 (2) to H3COSiEt3 (3) and finally to CH4] through the frustrated Lewis pair (FLP)-type Si–H activation. Hence, with the required combination of [Al] and [B], a highly selective hydrosilylative reduction of CO2 system has been developed, achieving high CH4 production yield up to 94%. The remarkably different catalytic behaviors between [Al] and [B] are attributed to the higher overall Lewis acidity of [Al] derived from two conflicting factors (electronic and steric effects), which renders the higher tendency of [Al] to form stable [Al]–substrate (intermediate) adducts with CO2 as well as subsequent intermediates 1, 2, and 3. Overall, the roles of [Al] and [B] are not only complementary but also synergistic in the total reduction of CO2, which render both [Al]-mediated first reduction step and [B]-mediated subsequent steps catalytic.
Co-reporter:Fernando Vidal; Laura Falivene; Lucia Caporaso; Luigi Cavallo
Journal of the American Chemical Society 2016 Volume 138(Issue 30) pp:9533-9547
Publication Date(Web):July 7, 2016
DOI:10.1021/jacs.6b04064
The successful synthesis of highly syndiotactic polar vinyl polymers bearing the reactive pendant vinyl group on each repeat unit, which is enabled by perfectly chemoselective and highly syndiospecific coordination polymerization of divinyl polar monomers developed through this work, has allowed the construction of robust cross-linked supramolecular stereocomplexes and C60 inclusion complexes. The metal-mediated coordination polymerization of three representative polar divinyl monomers, including vinyl methacrylate (VMA), allyl methacrylate (AMA), and N,N-diallyl acrylamide (DAA) by Cs-ligated zirconocenium ester enolate catalysts under ambient conditions exhibits complete chemoselectivity and high stereoselectivity, thus producing the corresponding vinyl-functionalized polymers with high (92% rr) to quantitative (>99% rr) syndiotacticity. A combined experimental (synthetic, kinetic, and mechanistic) and theoretical (DFT) investigation has yielded a unimetallic, enantiomorphic-site-controlled propagation mechanism. Postfunctionalization of the obtained syndiotactic vinyl-functionalized polymers via the thiol–ene click and photocuring reactions readily produced the corresponding thiolated polymers and flexible cross-linked thin-film materials, respectively. Complexation of such syndiotactic vinyl-functionalized polymers with isotactic poly(methyl methacrylate) and fullerene C60 generates supramolecular crystalline helical stereocomplexes and inclusion complexes, respectively. Cross-linking of such complexes affords robust cross-linked stereocomplexes that are solvent-resistant and also exhibit considerably enhanced thermal and mechanical properties compared with the un-cross-linked stereocomplexes.
Co-reporter:Ravikumar R. Gowda and Eugene Y.-X. Chen
ACS Macro Letters 2016 Volume 5(Issue 6) pp:772
Publication Date(Web):June 8, 2016
DOI:10.1021/acsmacrolett.6b00370
Achieving complete chemoselectivity in the polymerization of multivinyl polar monomers is an important yet challenging task, currently achievable only by metal- or metalloid-mediated polymerization processes but in a noncatalytic fashion. Now this work shows that organic N-heterocyclic carbene (NHC) catalysts effect rapid, chemoselective, and catalytic polymerization of multivinyl-functionalized γ-butyrolactones, particularly γ-vinyl-α-methylene-γ-butyrolactone (VMBL). Thus, the NHC-catalyzed polymerization of VMBL not only is quantitatively chemoselective, proceeding exclusively via polyaddition across the conjugated α-methylene double bond while leaving the γ-vinyl double bond intact, but also requires only an exceptionally low catalyst loading of 50 ppm, thus, exhibiting a remarkably high catalyst turnover frequency of 80000 h–1 and producing on average 33.6 polymer chains of Mn = 73.8 kg/mol per NHC molecule. The resulting PVMBL can be either thermally cured into cross-linked materials or postfunctionalized with the thiol–ene “click” reaction to achieve complete conversion of the pendant vinyl group on every repeat unit into the corresponding thioether.
Co-reporter:Zehuai Mou, Shuo (Kelvin) Feng and Eugene Y. X. Chen
Polymer Chemistry 2016 vol. 7(Issue 8) pp:1593-1602
Publication Date(Web):18 Jan 2016
DOI:10.1039/C5PY02032H
A triol monomer, 5,5′-dihydroxymethyl furoin (DHMF), was prepared from the biomass platform chemical 5-hydroxymethylfurfural (HMF) in 95% yield via organocatalysis. Selective oxidation and reduction of DHMF afforded a new diol monomer, 5,5′-bihydroxymethyl furil (BHMF), and a new tetraol, 5,5′-bihydroxymethyl hydrofuroin (BHMH), respectively. The catalyzed polyaddition of the diol BHMF with various diisocyanates produced linear polyurethanes (PUs), whereas catalyzed polyadditions using the triol and tetraol monomers led to cross-linked PUs. Especially interesting is the PU material derived from BHMF and aromatic diisocyanates such as diphenylmethane diisocyanate, which exhibited a Mn of 39.8 kg mol−1, an onset decomposition temperature of 234 °C, and a Tg of 140 °C. Various PU thin films have also been prepared by in situ polyaddition of these three polyols with diisocyanates in various ratios through solvent casting, affording PU materials ranging from being brittle to flexible with a high strain at break of 300%.
Co-reporter:Jedediah Wilson and Eugene Y.-X. Chen
ACS Sustainable Chemistry & Engineering 2016 Volume 4(Issue 9) pp:4927
Publication Date(Web):August 3, 2016
DOI:10.1021/acssuschemeng.6b01235
To synthesize the still missing C11 furoins as potential trifunctional difuranic building blocks for biopolymers and biofuels, the route based on the cross-coupling of two biofuraldehydes, furfural (FF) and 5-hydroxymethylfurfural (HMF), has been investigated using organic N-heterocyclic carbene (NHC) catalysts. This NHC-catalyzed coupling reaction in solution gives a statistical mixture of products including homocoupled C10 and C12 furoins 1 and 4, as well as cross-coupled C11 furoins 2 and 3, regardless of the electronics or sterics of the NHC precatalyst used, but a slight preference for cross-coupled products (∼60% 2 + 3 combined) under neat conditions has been achieved. Cross-coupled products 2 and 3 have been isolated in 48.1% yield and further separated into their pure state in 22.2% and 19.9% yield for 2 and 3, respectively. The isolated 2 and 3 have been fully characterized by NMR, HRMS, and single crystal X-ray diffraction. A simple metal-free/in-air oxidation reaction converts these two isomeric furoins into the single α-diketone furanil structure 5, another new C11 bioderived building block.Keywords: 5-Hydroxymethylfurfural; Cross-coupling reaction; Furfural; Furoin; NHC; Organocatalysis
Co-reporter:Jiawei Chen and Eugene Y.-X. Chen
Dalton Transactions 2016 vol. 45(Issue 14) pp:6105-6110
Publication Date(Web):16 Nov 2015
DOI:10.1039/C5DT03895B
Alkyl/aryl ligand exchange between AlEt3 and B(C6F5)3 in hexanes enables the formation and isolation of the unsolvated Al(C6F5)3 as a crystalline solid, the structure of which has been determined by single-crystal X-ray diffraction analysis. Instead of forming the anticipated Al⋯F contacts with the seemingly more accessible meta- and para-F's of –C6F5 groups, two Al(C6F5)3 molecules form a dimeric structure with double Al⋯F interactions between the Al center of one molecule and the ortho-F atom of the –C6F5 group on the other molecule. This mode of interactions is apparently linked to the thermal and shock sensitivity of the unsolvated Al(C6F5)3 in the solid state. To compare with the B(C6F5)3/ferrocene frustrated Lewis pair system, the complexation between Al(C6F5)3 and ferrocene has also been studied, which affords a stable adduct formed through the η1-coordination of Al to one of the CCp atoms, similar to the alane–toluene or benzene complex.
Co-reporter:Jiawei Chen, Ravikumar R. Gowda, Jianghua He, Yuetao Zhang, and Eugene Y.-X. Chen
Macromolecules 2016 Volume 49(Issue 21) pp:8075-8087
Publication Date(Web):October 24, 2016
DOI:10.1021/acs.macromol.6b01654
Group transfer polymerization (GTP) is an important ambient-temperature living polymerization method using silyl ketene acetal (SKA) or related initiators. Although several different GTP systems have been developed for polymerizing acrylic monomers, they all require the use of a catalyst to activate the SKA initiator, commonly believed to be ineffective on its own. Now, this work shows that, in fact, the neutral SKA alone mediates either controlled or extremely rapid polymerization of acrylic monomers such as methyl methacrylate (MMA) in polar donor solvents such as DMF, depending on the nuclearity of the SKA and the chelating pendant group on Si. In the case of a mono-SKA such as Me2C═C(OMe)OSiMe3, the GTP of MMA in DMF is relatively slow (several hours to completion) but is controlled and remarkably efficient, producing PMMA with Mn values close to those predicted on the basis of the [M]/[I] ratio, low Đ values (≤1.2), and high initiation efficiencies (≥80%). In sharp contrast, the di-SKAs linked by an oxo, ferrocenyl, or binaphthyl bridge, as well as the mono-SKA with a donor chelating methoxy pendant group on Si, mediate extremely rapid polymerization (a few seconds to completion), affording an extremely high turnover frequency up to 1.92 × 105 h–1, but the polymerization is uncontrolled. Several lines of evidence obtained through mechanistic studies indicate that the polymerization by the mono-SKA and di-SKA in DMF proceeds through a dissociative pathway with the released enolate anion being the highly active species and the polymerization characteristics are highly dependent on the amount of free enolate anions in solution. In this mechanism, the donor ability of the solvent plays a critical role in promoting the activity through activation of the Si site of the neutral SKA by forming the pentacoordinate Si intermediate.
Co-reporter:Zehuai Mou and Eugene Y.-X. Chen
ACS Sustainable Chemistry & Engineering 2016 Volume 4(Issue 12) pp:
Publication Date(Web):October 12, 2016
DOI:10.1021/acssuschemeng.6b02007
This contribution investigates the impact of rigid and flexible difuranic polyols on the resulting polyester (PE) and poly(ester-urethane) (PEU) properties. Three biobased difuranic polyol monomers, 5,5′-bihydroxymethyl furil (BHMF), 5,5′-dihydroxymethyl furoin (DHMF), and bis[5-(hydroxymethyl)furan-2-yl)methyl]adipate (BHFA), all derived from the biomass platform chemical 5-hydroxymethylfurfural (HMF), were employed for the synthesis of a series of new linear and cross-linked PEs as well as amorphous and semicrystalline PEUs. The polycondensations of diols (rigid BHMF and flexible BHFA) with various diacyl chlorides afford linear PEs, whereas the rigid triol (DHMF) reacts with diacyl chlorides to form cross-linked PEs. Among these difuranic PEs, the most intriguing PE is the one containing C═C double bonds, derived from BHFA and fumaryl chloride, which exhibits the unique self-curing ability via the Diels–Alder reaction. Furthermore, the catalyzed polyaddition of BHFA with various diiscyanates produces novel PEUs, the most interesting of which is the one derived from BHFA and hexamethylene diisocyanate, a semicrystalline material displaying a high melting-transition temperature of 135.8 °C.Keywords: 5-Hydroxymethylfurfural; Poly(ester-urethane); Polyaddition; Polycondensation; Polyester; Polyurethane;
Co-reporter:Dr. Ravikumar R. Gowda ;Dr. Eugene Y.-X. Chen
ChemSusChem 2016 Volume 9( Issue 2) pp:181-185
Publication Date(Web):
DOI:10.1002/cssc.201501402
Abstract
Nanoparticles (NPs) derived from earth-abundant metal(0) carbonyls catalyze conversion of bio-derived levulinic acid into γ-valerolactone in up to 93 % isolated yield. This sustainable and green route uses non-precious metal catalysts and can be performed in aqueous or ethanol solution without using hydrogen gas as the hydrogen source. Generation of metal NPs using microwave irradiation greatly enhances the rate of the conversion, enables the use of ethanol as both solvent and hydrogen source without forming the undesired ethyl levulinate, and affords recyclable polymer-stabilized NPs.
Co-reporter:Dr. Miao Hong ;Dr. Eugene Y.-X. Chen
Angewandte Chemie International Edition 2016 Volume 55( Issue 13) pp:4188-4193
Publication Date(Web):
DOI:10.1002/anie.201601092
Abstract
The first effective organopolymerization of the biorenewable “non-polymerizable” γ-butyrolactone (γ-BL) to a high-molecular-weight metal-free recyclable polyester is reported. The superbase tert-Bu-P4 is found to directly initiate this polymerization through deprotonation of γ-BL to generate reactive enolate species. When combined with a suitable alcohol, the tert-Bu-P4-based system rapidly converts γ-BL into polyesters with high monomer conversions (up to 90 %), high molecular weights (Mn up to 26.7 kg mol−1), and complete recyclability (quantitative γ-BL recovery).
Co-reporter:Dr. Miao Hong ;Dr. Eugene Y.-X. Chen
Angewandte Chemie International Edition 2016 Volume 55( Issue 13) pp:
Publication Date(Web):
DOI:10.1002/anie.201681361
Co-reporter:Dr. Miao Hong ;Dr. Eugene Y.-X. Chen
Angewandte Chemie 2016 Volume 128( Issue 13) pp:4260-4265
Publication Date(Web):
DOI:10.1002/ange.201601092
Abstract
The first effective organopolymerization of the biorenewable “non-polymerizable” γ-butyrolactone (γ-BL) to a high-molecular-weight metal-free recyclable polyester is reported. The superbase tert-Bu-P4 is found to directly initiate this polymerization through deprotonation of γ-BL to generate reactive enolate species. When combined with a suitable alcohol, the tert-Bu-P4-based system rapidly converts γ-BL into polyesters with high monomer conversions (up to 90 %), high molecular weights (Mn up to 26.7 kg mol−1), and complete recyclability (quantitative γ-BL recovery).
Co-reporter:Dr. Miao Hong ;Dr. Eugene Y.-X. Chen
Angewandte Chemie 2016 Volume 128( Issue 13) pp:
Publication Date(Web):
DOI:10.1002/ange.201681361
Co-reporter:Fernando Vidal; Ravikumar R. Gowda
Journal of the American Chemical Society 2015 Volume 137(Issue 29) pp:9469-9480
Publication Date(Web):July 8, 2015
DOI:10.1021/jacs.5b05811
This contribution reports the first chemoselective, stereospecific, and living polymerization of polar divinyl monomers, enabled by chiral ansa-zirconocenium catalysts through an enantiomorphic-site controlled coordination–addition polymerization mechanism. Silyl-bridged-ansa-zirconocenium ester enolate 2 has been synthesized and structurally characterized, but it exhibits low to negligible activity and stereospecificity in the polymerization of polar divinyl monomers including vinyl methacrylate (VMA), allyl methacrylate (AMA), 4-vinylbenzyl methacrylate (VBMA), and N,N-diallyl acrylamide (DAA). In contrast, ethylene-bridged-ansa-zirconocenium ester enolate 1 is highly active and stereospecific in the polymerization of such monomers including AMA, VBMA, and DAA. The polymerization by 1 is perfectly chemoselective for all four polar divinyl monomers, proceeding exclusively through conjugate addition across the methacrylic C═C bond, while leaving the pendant C═C bonds intact. The polymerization of DAA is most stereospecific and controlled, producing essentially stereoperfect isotactic PDAA with [mmmm] > 99%, Mn matching the theoretical value (thus a quantitative initiation efficiency), and a narrow molecular weight distribution (Đ = 1.06–1.16). The stereospecificity is slightly lower for the AMA polymerization but still leading to highly isotactic poly(allyl methacrylate) (PAMA) with 95–97% [mm]. The polymerization of VBMA is further less stereospecific, affording PVBMA with 90–94% [mm], while the polymerization VMA is least stereospecific. Several lines of evidence from both homo- and block copolymerization results have demonstrated living characteristics of the AMA polymerization by 1. Mechanistic studies of this polymerization have yielded a monometallic coordination–addition polymerization mechanism involving the eight-membered chelating intermediate. Post-functionalization of isotactic polymers bearing the pendant vinyl group on every repeating unit via the thiol–ene “click” reaction achieves a full conversion of all the pendant double bonds to the corresponding thioether bonds. Photocuring of such isotactic polymers is also successful, producing an elastic material readily characterizable by dynamic mechanical analysis.
Co-reporter:Jian-Bo Zhu
Journal of the American Chemical Society 2015 Volume 137(Issue 39) pp:12506-12509
Publication Date(Web):September 21, 2015
DOI:10.1021/jacs.5b08658
B/N Lewis pairs have been discovered to catalyze rapid epimerization of meso-lactide (LA) or LA diastereomers quantitatively into rac-LA. The obtained rac-LA is kinetically polymerized into poly(l-lactide) and optically resolved d-LA, with a high stereoselectivity kL/kD of 53 and an ee of 91% at 50.6% monomer conversion, by newly designed bifunctional chiral catalyst 4 that incorporates three key elements (β-isocupreidine core, thiourea functionality, and chiral BINAM) into a single organic molecule. The epimerization and enantioselective polymerization can be coupled into a one-pot process for transforming meso-LA directly into poly(l-lactide) and d-LA.
Co-reporter:Lu Wang and Eugene Y.-X. Chen
ACS Catalysis 2015 Volume 5(Issue 11) pp:6907
Publication Date(Web):October 21, 2015
DOI:10.1021/acscatal.5b01410
Two highly efficient and recyclable heterogeneous azolium catalyst systems, one grafted (g) onto the inorganic oxide (Silica) and the other onto the organic polymer [Merrifield’s peptide or chloromethylated polystyrene (PS) resin], have been developed and employed to catalyze quantitative self-coupling (umpolung condensation) reactions of furfural and 5-hydroxymethylfurfural (HMF) into C10 and C12 furoins, respectively. Supported benzimidazolium ([BI]) salts bearing a long-chain alkyl substituent (i.e., C12 dodecyl) on the azolium nitrogen atom, upon activation with a suitable base to generate the corresponding N-heterocyclic carbene (NHC) catalyst, are found to be far more effective catalysts for furaldehyde self-coupling reactions than the analogous catalysts carrying a short-chain alkyl substituent (i.e., C1 methyl). Thus, supported NHC catalysts generated in situ from Silica-g-[BI]-C12 or PS-g-[BI]-C12-benzyl/base afford the C10 and C12 furoins in about 97% and 94% yield, respectively. By adopting a catalyst recycling procedure that involves activation of the precatalyst with a base to generate the NHC catalyst, catalysis in conversion of furaldehydes into furoins, and recycle of the catalyst by quenching the reaction with HCl to convert the catalyst back to the precatalyst, excellent recyclability has been achieved without loss of the catalytic activity after 10 cycles by maintaining essentially a constant furoin yield of 96–97% for all 10 cycles performed with both supported catalyst systems.Keywords: furfural; HMF; N-heterocyclic carbene; self-coupling (condensation) reaction; supported (heterogeneous) catalyst
Co-reporter:Lu Wang and Eugene Y.-X. Chen
Green Chemistry 2015 vol. 17(Issue 12) pp:5149-5153
Publication Date(Web):18 Aug 2015
DOI:10.1039/C5GC01648G
Intercalation of benzimidazolium cations [BI]+ into the nanogalleries of Na+/montmorillonite (MMT) clay leads to generation of recyclable supported precatalysts [BI]+/MMT, which, upon treatment with a base, catalyze furfural self-condensation coupling reaction into furoin in almost constant yields of >96% over the three cycles investigated. This catalyst system combines the best features of both homogeneous and heterogeneous catalyst systems, as it performs the homogeneous molecular catalysis by the discharged N-heterocyclic carbene catalyst in solution and then recovers the catalyst through in situ heterogenization after the reaction via re-intercalation of the charged precatalyst. The [12,12BI]+/MMT catalyst system carrying two long-chain C12 dodecyl substituents on the [BI] nitrogen atoms is particularly effective for achieving both high product yield and catalyst recyclability.
Co-reporter:Miao Hong and Eugene Y.-X. Chen
Polymer Chemistry 2015 vol. 6(Issue 10) pp:1741-1750
Publication Date(Web):02 Jan 2015
DOI:10.1039/C4PY01488J
Rapid formation of thermally robust polymeric Lewis adducts between the poly(N-heterocyclic carbene) [poly(NHC)] Lewis base and the carbon Lewis acid C60 is utilized to develop a powerful synthetic strategy for the convenient and efficient synthesis of C60-containing polymers with an unprecedented level of C60 incorporation (up to 69.3 wt%). The resulting polymer Lewis adducts, poly(NHC–C60), exhibit several intriguing properties pertaining to their molecular weights, thermal stability, cross-linking, and morphologies of donor(D)/acceptor(A) blends. Most significantly, the morphology of the D/A bulk heterojunction (BHJ) using poly(NHC–C60) appears to be more controlled and the C60 domain size is apparently much smaller and more uniform, as compared to the D/A BHJ directly using C60 with the same D/A ratio.
Co-reporter:Jiawei Chen
Israel Journal of Chemistry 2015 Volume 55( Issue 2) pp:216-225
Publication Date(Web):
DOI:10.1002/ijch.201400136
Abstract
This work has uncovered the first highly active and efficient Lewis pair polymerization (LPP) system based on N-heterocyclic carbene (NHC)/B(C6F5)3 pairs for converting acrylic monomers into medium- to high-molecular weight polymers. The study has systematically examined steric and electronic effects of three 1,3-dialkyl(Me, iPr, tBu)imidazol-2-ylidene NHCs on the LPP of three classes of acrylic monomers, including linear methyl methacrylate (MMA), cyclic biorenewable γ-methyl-α-methylene-γ-butyrolactone (γMMBL), and difunctional allyl methacrylate (AMA). For MMA polymerization, IiPr is not only the most active (∼3× and ∼120× more active than IMe and ItBu, respectively), but also the most effective NHC, especially under low NHC loading conditions. Kinetic results are consistent with a bimolecular, activated monomer propagation mechanism. In the case of the more reactive γMMBL, the polymerization by NHC/B(C6F5)3 in CH2Cl2 is extremely rapid, with all three NHCs achieving quantitative monomer conversion in 1 min and thus reaching a high turnover frequency of≥48,000 h−1. The molecular weight (MW) of PγMMBL can be tuned by adjusting the [γMMBL]/[NHC] ratio, and thus high MW polymers with relatively narrow MW distributions can be readily synthesized (e.g., from Mn=1.41×105 g mol−1, Đ=1.08 to Mn=4.89×105 g mol−1, Đ=1.20). The LPP by NHC/B(C6F5)3 is completely chemoselective, as demonstrated by the polymerization of AMA, which selectively polymerizes the conjugated vinyl group while leaving the non-conjugated vinyl group in the allyl moiety intact, thanks to its activated monomer propagation mechanism. The resulting PAMA is syndiotactic (rr=83 %), uncross-linked, and soluble in common solvents, thus suitable for further functionalization. This quantitatively chemoselective polymerization by NHC/B(C6F5)3 should provide a facile, yet powerful, approach to functional acrylic polymers.
Co-reporter:Shuo (Kelvin) Feng;Meghan Schmitt
Macromolecular Chemistry and Physics 2015 Volume 216( Issue 13) pp:1421-1430
Publication Date(Web):
DOI:10.1002/macp.201500079
Co-reporter:Dr. Jiawei Chen ;Dr. Eugene Y.-X. Chen
Angewandte Chemie International Edition 2015 Volume 54( Issue 23) pp:6842-6846
Publication Date(Web):
DOI:10.1002/anie.201502400
Abstract
The super acidity of the unsolvated Al(C6F5)3 enabled isolation of the elusive silane–alane complex [SiH⋅⋅⋅Al], which was structurally characterized by spectroscopic and X-ray diffraction methods. The Janus-like nature of this adduct, coupled with strong silane activation, effects multifaceted frustrated-Lewis-pair-type catalysis. When compared with the silane–borane system, the silane–alane system offers unique features or clear advantages in the four types of catalytic transformations examined in this study, including: ligand redistribution of tertiary silanes into secondary and quaternary silanes, polymerization of conjugated polar alkenes, hydrosilylation of unactivated alkenes, and hydrodefluorination of fluoroalkanes.
Co-reporter:Garret M. Miyake;Yuetao Zhang
Journal of Polymer Science Part A: Polymer Chemistry 2015 Volume 53( Issue 12) pp:1523-1532
Publication Date(Web):
DOI:10.1002/pola.27629
ABSTRACT
This work examines the stereochemical control and polymerizability of exo-methylene-lactide (MLA) or (6S)-3-methylene-6-methyl-1,4-dioxane-2,5-dione, a chiral monomer derived from l-lactide, toward vinyl-addition and ring-opening polymerization (ROP) pathways, respectively. Currently, no information on the stereochemistry of the vinyl-addition polymerization of MLA is known, and the possible ROP pathway is unexplored. Accordingly, this work first investigated the stereochemical control and other characteristics of the radical polymerization of MLA and its copolymerization with an analogous exo-methylene-lactone, γ-methyl-α-methylene-γ-butyrolactone (MMBL), and di-methylene-lactide (DMLA) or 3,6-dimethylene-1,4-dioxane-2,5-dione. The MLA homopolymerization produced optically active, but atactic, vinyl-type polymers having a specific rotation of [α]23D = −42 ± 4°, a high Tg from 229 to 254 °C, and a medium (Mw = 76.3 kg/mol, Đ = 1.16) to high (Mw = 358 kg/mol, Đ = 2.83) molecular weight, depending on the solvent. The copolymerization of MLA with MMBL afforded copolymers exhibiting enhanced thermal stability, while its copolymerization with DMLA led to cross-linked polymers. The results obtained from the model reactions designed to probe the possible ROP indicate that the nonpolymerizability of MLA by initiators or catalysts comprising acidic, protic, and/or nucleophilic reagents is due to the high sensitivity of MLA toward such common ROP reagents that trigger decomposition or other types of transformations of MLA forming nonpolymerizable derivatives. © 2015 Wiley Periodicals, Inc. J. Polym. Sci. Part A: Polym. Chem. 2015, 53, 1523–1532
Co-reporter:Tieqi Xu
Journal of Polymer Science Part A: Polymer Chemistry 2015 Volume 53( Issue 16) pp:1895-1903
Publication Date(Web):
DOI:10.1002/pola.27641
ABSTRACT
Silylium ions (“R3Si+”) are found to catalyze both 1,4-hydrosilylation of methyl methacrylate (MMA) with R3SiH to generate the silyl ketene acetal initiator in situ and subsequent living polymerization of MMA. The living characteristics of the MMA polymerization initiated by R3SiH (Et3SiH or Me2PhSiH) and catalyzed by [Et3Si(L)]+[B(C6F5)4]– (L = toluene), which have been revealed by four sets of experiments, enabled the synthesis of the polymers with well-controlled Mn values (identical or nearly identical to the calculated ones), narrow molecular weight distributions (Đ = 1.05–1.09), and well defined chain structures {H[MMA]nH}. The polymerization is highly efficient too, with quantitative or near quantitative initiation efficiencies (I* = 96–100%). Monitoring of the reaction of MMA + Me2PhSiH + [Et3Si(L)]+[B(C6F5)4]– (0.5 mol%) by 1H NMR provided clear evidence for in situ generation of the corresponding SKA, Me2CC(OMe)OSiMe2Ph, via the proposed “Et3Si+”-catalyzed 1,4-hydrosilylation of monomer through “frustrated Lewis pair” type activation of the hydrosilane in the form of the isolable silylium-silane complex, [Et3SiHSiR3]+[B(C6F5)4]–. © 2015 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2015, 53, 1895–1903
Co-reporter:Garret M. Miyake;Yuetao Zhang
Journal of Polymer Science Part A: Polymer Chemistry 2015 Volume 53( Issue 17) pp:
Publication Date(Web):
DOI:10.1002/pola.27704
Co-reporter:Dr. Jiawei Chen ;Dr. Eugene Y.-X. Chen
Angewandte Chemie 2015 Volume 127( Issue 23) pp:6946-6950
Publication Date(Web):
DOI:10.1002/ange.201502400
Abstract
The super acidity of the unsolvated Al(C6F5)3 enabled isolation of the elusive silane–alane complex [SiH⋅⋅⋅Al], which was structurally characterized by spectroscopic and X-ray diffraction methods. The Janus-like nature of this adduct, coupled with strong silane activation, effects multifaceted frustrated-Lewis-pair-type catalysis. When compared with the silane–borane system, the silane–alane system offers unique features or clear advantages in the four types of catalytic transformations examined in this study, including: ligand redistribution of tertiary silanes into secondary and quaternary silanes, polymerization of conjugated polar alkenes, hydrosilylation of unactivated alkenes, and hydrodefluorination of fluoroalkanes.
Co-reporter:Jianghua He;Yuetao Zhang
Macromolecular Symposia 2015 Volume 349( Issue 1) pp:104-114
Publication Date(Web):
DOI:10.1002/masy.201400018
Summary
This study investigates the characteristics of the recently developed catalytic H-shuttling polymerization (HSP) of polar vinyl monomers mediated by cationic zirconocenium catalysts, through examining the scopes of chain transfer agent, catalyst, and monomer. Amongst the three classes of chain transfer agents (metal hydrides, metal aryl/alkyls, organics) investigated for the MMA polymerization catalyzed by Cs-ligated catalyst 1, the Lewis acid B(C6F5)3, metal hydrides MH (M = K, Na, Li) and the organic PhNMe2 promote efficient catalytic polymerization, while metal alkyls such as Et3Al and Et2Zn serve only as the scavenger and/or monomer activator and thus do not promote chain transfer. In the presence of 5 equivalents of KH, both Cs-ligated catalyst 1 and achiral C2v-ligated catalyst 3 promote catalytic MMA polymerization through effective chain transfer, whereas C2-ligated catalyst 2 shows no chain transfer. Of several other conjugated polar vinyl monomers investigated, including acrylamides and α-methylene-γ-butyrolactones, only N,N-diphenylacrylamide (DPAA) exhibits catalytic polymerization behavior. Thus, in the presence of enantiopure pre-catalyst (S,S)-4 and excess B(C6F5)3, DPAA is polymerized catalytically and asymmetrically to optically active poly(DPAA) with a narrow MWD of 1.02.
Co-reporter:Tieqi Xu
Journal of the American Chemical Society 2014 Volume 136(Issue 5) pp:1774-1777
Publication Date(Web):January 13, 2014
DOI:10.1021/ja412445n
The first highly active phosphine (P)/borane (B) Lewis pair polymerization is promoted unexpectedly by P–B adducts. The P and B site cooperativity is essential for achieving effective polymerization, as shown by this study examining the reactivity of a library of P/B Lewis pairs toward polymerization of a renewable acrylic monomer.
Co-reporter:Dajiang (D. J.) Liu and Eugene Y.-X. Chen
Green Chemistry 2014 vol. 16(Issue 3) pp:964-981
Publication Date(Web):12 Nov 2013
DOI:10.1039/C3GC41934G
Organocatalysis using small-molecule organic compounds as catalysts has risen to prominence in organic synthesis and polymer synthesis. However, its application in biorefining for catalytic biomass conversion and upgrading into sustainable chemicals, materials, and biofuels has come to light only recently. This emergence of applying organocatalysis for biorefining has not only broadened the scope of organocatalysis and offered metal-free “greener” alternatives for biomass conversion and upgrading, it has also showed some unique activity and selectivity in such transformations as compared to metal-mediated processes. This review captures highlights of this emerging area by focusing on utilization of organocatalytic means for catalytic conversions of cellulose, glucose and fructose, upgrading of furaldehydes, and organocatalytic polymerization of biomass feedstocks.
Co-reporter:Dajiang (D. J.) Liu and Eugene Y.-X. Chen
ACS Catalysis 2014 Volume 4(Issue 5) pp:1302
Publication Date(Web):March 18, 2014
DOI:10.1021/cs500058p
Report herein is an integrated catalytic process for conversion and upgrading of biomass feedstocks into 5,5′-dihydroxymethyl furoin (DHMF), through self-coupling of 5-hydroxymethyl furfural (HMF) via organocatalysis, and subsequently into n-C12H26 alkane fuel via metal–acid tandem catalysis. The first step of the process involves semicontinuous organocatalytic conversion of biomass (fructose, in particular) to the high-purity HMF. N-Heterocyclic carbenes (NHCs) are found to catalyze glucose-to-fructose isomerization, and the relatively inexpensive thiazolium chloride [TM]Cl, a Vitamin B1 analog, catalyzes fructose dehydration to HMF of good purity (>99% by HPLC), achieving a constant HMF yield of 72% over 10 semicontinuous extraction batch runs. Crystallization of the crude HMF from toluene yields the spectroscopically and analytically pure HMF as needle crystals. The second step of the process is the NHC-catalyzed coupling of C6 HMF produced by the semicontinuous process to C12 DHMF; the most effective organic NHC catalyst produces DHMF in 93% or 91% isolated yield with an NHC loading of 0.70 mol % or 0.10 mol % at 60 °C for 3 h under solvent-free conditions. The third step of the process converts C12 DHMF to linear alkanes via hydrodeoxygenation. With a bifunctional catalyst system consisting of Pd/C + acetic acid + La(OTf)3 at 250 °C and 300 psi H2 for 16 h, DHMF has been transformed to liquid hydrocarbon fuel (78% alkanes), with a 64% selectivity to n-C12H26 and an overall C/H/O % ratio of 84/11/5.0.Keywords: biofuel; biomass; biomass upgrading; cellulose; fructose; glucose; organocatalysis
Co-reporter:Meghan Schmitt, Laura Falivene, Lucia Caporaso, Luigi Cavallo and Eugene Y.-X. Chen
Polymer Chemistry 2014 vol. 5(Issue 9) pp:3261-3270
Publication Date(Web):28 Jan 2014
DOI:10.1039/C3PY01579C
The organic phosphazene superbase, 1-tert-butyl-4,4,4-tris(dimethylamino)-2,2-bis[tris(dimethylamino)phosphoranylid-enamino]-2λ5,4λ5-catenadi(phosphazene) (t-Bu-P4), is found to directly initiate high-speed polymerization of the biomass-derived renewable γ-methyl-α-methylene-γ-butyrolactone (MMBL), in contrast to other polymerization systems using t-Bu-P4 which typically require addition of an organic acid or a nucleophile as a co-initiating component. This MMBL polymerization by t-Bu-P4 alone is extremely rapid; even with a low t-Bu-P4 loading of 0.1 mol% or 0.02 mol%, quantitative monomer conversion is achieved in 20 s or 1 min, respectively, affording medium to high molecular weight PMMBL bioplastics in a catalytic fashion. The combined experimental and theoretical/computational studies have yielded mechanisms of chain initiation through abstraction of a proton from a monomer by t-Bu-P4, essentially barrier-less chain propagation through rapid conjugate addition of the enolate anion stabilized by the nano-size cation [t-Bu-P4H]+ to the monomer, and chain termination through chain transfer to the monomer which generates a saturated termination chain end and the [t-Bu-P4H]+-stabilized anionic active species that starts a new chain.
Co-reporter:Jianghua He, Yuetao Zhang, Laura Falivene, Lucia Caporaso, Luigi Cavallo, and Eugene Y.-X. Chen
Macromolecules 2014 Volume 47(Issue 22) pp:7765-7774
Publication Date(Web):November 7, 2014
DOI:10.1021/ma5019389
A combined experimental and theoretical study on mechanistic aspects of polymerization of conjugated polar alkenes by frustrated Lewis pairs (FLPs) based on N-heterocyclic carbene (NHC) and Al(C6F5)3 pairs is reported. This study consists of three key parts: structural characterization of active propagating intermediates, propagation kinetics, and chain-termination pathways. Zwitterionic intermediates that simulate the active propagating species in such polymerization have been generated or isolated from the FLP activation of monomers such as 2-vinylpyridine and 2-isopropenyl-2-oxazoline—one of which, IMes+-CH2C(Me)═(C3H2NO)Al(C6F5)3– (2), has been structurally characterized. Kinetics performed on the polymerization of 2-vinylpyridine by ItBu/Al(C6F5)3 revealed that the polymerization follows a zero-order dependence on monomer concentration and a first-order dependence on initiator (ItBu) and activator [Al(C6F5)3] concentrations, indicating a bimolecular, activated monomer propagation mechanism. The Lewis pair polymerization of conjugate polar alkenes such as methacrylates is accompanied by competing chain-termination side reactions; between the two possible chain-termination pathways, the one that proceeds via intramolecular backbiting cyclization involving nucleophilic attack of the activated ester group of the growing polymer chain by the O-ester enolate active chain end to generate a six-membered lactone (δ-valerolactone)-terminated polymer chain is kinetically favored, but thermodynamically disfavored, over the pathway leading to the β-ketoester-terminated chain, as revealed by computational studies.
Co-reporter:Ravikumar R. Gowda, Lucia Caporaso, Luigi Cavallo, and Eugene Y.-X. Chen
Organometallics 2014 Volume 33(Issue 15) pp:4118-4130
Publication Date(Web):July 31, 2014
DOI:10.1021/om500661y
Group 4 tetrabenzyl compounds MBn4 (M = Zr, Ti), upon protonolysis with an equimolar amount of the tetradentate amine-tris(phenol) ligand N[(2,4-tBu2C6H2(CH2)OH]3 in toluene from −30 to 25 °C, unexpectedly lead to amine-bis(phenoxy) dibenzyl complexes, BnCH2N[(2,4-tBu2C6H2(CH2)O]2MBn2 (M = Zr (1), Ti (2)) in 80% (1) and 75% (2) yields. This reaction involves an apparent cleavage of the >NCH2–ArOH bond (loss of the phenol in the ligand) and formation of the >NCH2–CH2Bn bond (gain of the benzyl group in the ligand). Structural characterization of 1 by X-ray diffraction analysis confirms that the complex formed is a bis(benzyl) complex of Zr coordinated by a newly derived tridentate amine-bis(phenoxy) ligand arranged in a mer configuration in the solid state. The abstractive activation of 1 and 2 with B(C6F5)3·THF in CD2Cl2 at room temperature generates the corresponding benzyl cations {BnCH2N[(2,4-tBu2C6H2(CH2)O]2MBn(THF)}+[BnB(C6F5)3]− (M = Zr (3), Ti, (4)). These cationic complexes, along with their analogues derived from (imino)phenoxy tri- and dibenzyl complexes, [(2,6-iPr2C6H3)N═C(3,5-tBu2C6H2)O]ZrBn3 (5) and [2,4-Br2C6H2(O)(6-CH2(NC5H9))CH2N═CH(2-adamantyl-4-MeC6H2O)]ZrBn2 (6), have been found to effectively polymerize the biomass-derived renewable β-methyl-α-methylene-γ-butyrolactone (βMMBL) at room temperature into the highly stereoregular polymer PβMMBL with an isotacticity up to 99% mm. A combined experimental and DFT study has yielded a mechanistic pathway for the observed unusual C–C bond cleavage in the present protonolysis reaction between ZrBn4 and N[(2,4-tBu2C6H2(CH2)OH]3 for the formation of complex 1, which involves the benzyl radical and the Zr(III) species, resulting from thermal and photochemical decomposition of ZrBn4, followed by a series of reaction sequences consisting of protonolysis, tautomerization, H-transfer, oxidation, elimination, and radical coupling.
Co-reporter:Dr. Miao Hong ;Dr. Eugene Y.-X. Chen
Angewandte Chemie International Edition 2014 Volume 53( Issue 44) pp:11900-11906
Publication Date(Web):
DOI:10.1002/anie.201406630
Abstract
A new polymerization termed proton (H)-transfer polymerization (HTP) has been developed to convert dimethacrylates to unsaturated polyesters. HTP is catalyzed by a selective N-heterocyclic carbene capable of promoting intermolecular Umpolung condensation through proton transfer and proceeds through the step-growth propagation cycles via enamine intermediates. The role of the added suitable phenol, which is critical for achieving an effective HTP, is twofold: shutting down the radically induced chain-growth addition polymerization under HTP conditions (typically at 80–120 °C) and facilitating proton transfer after each monomer enchainment. The resulting unsaturated polyesters have a high thermal stability and can be readily cross-linked to robust polyester materials.
Co-reporter:Dr. Miao Hong ;Dr. Eugene Y.-X. Chen
Angewandte Chemie 2014 Volume 126( Issue 44) pp:12094-12100
Publication Date(Web):
DOI:10.1002/ange.201406630
Abstract
A new polymerization termed proton (H)-transfer polymerization (HTP) has been developed to convert dimethacrylates to unsaturated polyesters. HTP is catalyzed by a selective N-heterocyclic carbene capable of promoting intermolecular Umpolung condensation through proton transfer and proceeds through the step-growth propagation cycles via enamine intermediates. The role of the added suitable phenol, which is critical for achieving an effective HTP, is twofold: shutting down the radically induced chain-growth addition polymerization under HTP conditions (typically at 80–120 °C) and facilitating proton transfer after each monomer enchainment. The resulting unsaturated polyesters have a high thermal stability and can be readily cross-linked to robust polyester materials.
Co-reporter:Miao Hong and Eugene Y.-X. Chen
Macromolecules 2014 Volume 47(Issue 11) pp:3614-3624
Publication Date(Web):May 29, 2014
DOI:10.1021/ma5007717
Reported herein is the first coordination–insertion ring-opening copolymerization of α-methylene-γ-butyrolactone (MBL) and ε-caprolactone (ε-CL) catalyzed by f-block lanthanide (Ln) catalysts, Ln[N(SiMe3)2]3, that produce exclusively an unsaturated copolyester PMBL-co-PCL without coproducing any homopolymer PMBL. Accomplishing such synthesis requires effective strategies to meet two key challenges: ring-opening of the γ-butyrolactone (γ-BL) ring in MBL—the five-membered lactone well recognized for its nonpolymerizability—and shutting down the vinyl-addition pathway via conjugate addition across the double bond—the exocyclic C═C moiety in MBL known for its high reactivity toward vinyl addition. Remarkably, the current Ln coordination catalyst system, coupled with judiciously chosen reaction conditions (relatively nonpolar solvent and low temperature, 0 or −20 °C), effectively copolymerizes MBL and ε-CL to produce the ring-opening copolyester PMBL-co-PCL, with the MBL incorporation up to 40 mol % and without any detectable PMBL formation, even when employing a large excess of MBL in feed. Successful ring-opening homopolymerization of γ-BL by the Ln catalyst has also been realized at −78 °C under ambient pressure, producing the polyester PBL with a Tm of 63.0 °C and a Tg of −53.7 °C. Investigation into the thermal property of the resulting copolyester reveals an overall depression of Tm of the copolyester as increasing the MBL incorporation, indicating that the ring-opened MBL (unsaturated) polyester incorporated in the random copolymer is noncrystallizable and disrupts the crystallization process of the crystallizable, ring-opened ε-CL (saturated) polyester segment. Mechanistic studies provide key evidence for a coordination–insertion ring-opening copolymerization mechanism.
Co-reporter:Yuetao Zhang, Meghan Schmitt, Laura Falivene, Lucia Caporaso, Luigi Cavallo, and Eugene Y.-X. Chen
Journal of the American Chemical Society 2013 Volume 135(Issue 47) pp:17925-17942
Publication Date(Web):November 18, 2013
DOI:10.1021/ja4088677
This contribution presents a full account of experimental and theoretical/computational investigations into the mechanisms of chain initiation, propagation, and termination of the recently discovered N-heterocyclic carbene (NHC)-mediated organocatalytic conjugate-addition polymerization of acrylic monomers. The current study specifically focuses on three commonly used NHCs of vastly different nucleophilicity, 1,3-di-tert-butylimidazolin-2-ylidene (ItBu), 1,3-dimesitylimidazolin-2-ylidene (IMes), and 1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene (TPT), and two representative acrylic monomers, the linear methyl methacrylate (MMA) and its cyclic analog, biomass-derived renewable γ-methyl-α-methylene-γ-butyrolactone (MMBL). For MMA, there exhibits an exquisite selectivity of the NHC structure for the three types of reactions it promotes: enamine formation (single-monomer addition) by IMes, dimerization (tail-to-tail) by TPT, and polymerization by ItBu. For MMBL, all three NHCs promote no dimerization but polymerization, with the polymerization activity being highly sensitive to the NHC structure and the solvent polarity. Thus, ItBu is the most active catalyst of the series and converts quantitatively 1000–3000 equiv of MMBL in 1 min or 10 000 equiv in 5 min at room temperature to MMBL-based bioplastics with a narrow range of molecular weights of Mn = 70–85 kg/mol, regardless of the [MMBL]/[ItBu] ratio employed. The ItBu-catalyzed MMBL polymerization reaches an exceptionally high turnover frequency up to 122 s–1 and a high initiator efficiency value up to 1600%. Unique chain-termination mechanisms have been revealed, accounting for the production of relative high-molecular-weight linear polymers and the catalytic nature of this NHC-mediated conjugate-addition polymerization. Computational studies have provided mechanistic insights into reactivity and selectivity between two competing pathways for each NHC-monomer zwitterionic adduct, namely enamine formation/dimerization through proton transfer vs polymerization through conjugate addition, and mapped out extensive energy profiles for chain initiation, propagation, and termination steps, thereby satisfactorily explaining the experimental observations.
Co-reporter:Ravikumar R. Gowda and Eugene Y.-X. Chen
Dalton Transactions 2013 vol. 42(Issue 25) pp:9263-9273
Publication Date(Web):27 Mar 2013
DOI:10.1039/C3DT50430A
Protonolysis of M(Bn)4 (M = Zr, Ti; Bn = benzyl) with equimolar 2,4-di-tert-butyl-6-[(2,6-diisopropylphenylimino)methyl]phenol [(2,6-iPr2C6H3)NC(3,5-tBu2C6H2)OH] in toluene at −30 °C to 25 °C cleanly affords the corresponding achiral (imino)phenoxy-tribenzyl complexes, [(2,6-iPr2C6H3)NC(3,5-tBu2C6H2)O]Zr(Bn)3 (1) and [(2,6-iPr2C6H3)NC(3,5-tBu2C6H2)O]Ti(Bn)3 (2). A chiral dibenzyl complex 3 incorporating the unsymmetric, tetradentate amino(imino)bis(phenoxy) ligand, [2,4-Br2C6H2(O)(6-CH2(NC5H9))CH2NCH(2-adamantyl-4-MeC6H2O)]Zr(Bn)2 (3), has also been prepared using the same protonolysis protocol. Abstractive activation of 1 with B(C6F5)3·THF in CD2Cl2 at room temperature (RT) affords clean, quantitative formation of the corresponding zirconium cation [((2,6-iPr2C6H3)NC(3,5-tBu2C6H2)O)Zr(Bn)2(THF)]+[BnB(C6F5)3]− (4). Likewise, benzyl abstraction of 2 with B(C6F5)3·THF in CD2Cl2 at RT generates the cationic titanium complex [((2,6-iPr2C6H3)NC(3,5-tBu2C6H2)O)Ti(Bn)2(THF)]+[BnB(C6F5)3]− (5), accompanied by a small amount of decomposed species as a result of C6F5 transfer. The dibenzyl cations 4 and 5 have been characterized spectroscopically, and their structures have been confirmed by single crystal X-ray diffraction analysis. Characteristics of the coordination polymerization of renewable α-methylene-γ-butyrolactone monomers by the cationic catalysts derived from achiral complexes 1 and 2 as well as chiral complex 3 have been investigated, representing the first study of such polymerization by non-metallocene catalysts.
Co-reporter:Dr. Jianghua He;Dr. Yuetao Zhang ;Dr. Eugene Y.-X. Chen
ChemSusChem 2013 Volume 6( Issue 1) pp:61-64
Publication Date(Web):
DOI:10.1002/cssc.201200795
Co-reporter:Dajiang Liu;Dr. Eugene Y.-X. Chen
ChemSusChem 2013 Volume 6( Issue 12) pp:2236-2239
Publication Date(Web):
DOI:10.1002/cssc.201300476
Co-reporter:Jianghua He;Yuetao Zhang ;Eugene Y. X. Chen
Journal of Polymer Science Part A: Polymer Chemistry 2013 Volume 51( Issue 13) pp:2793-2803
Publication Date(Web):
DOI:10.1002/pola.26679
ABSTRACT
Biomass-derived furfuryl methacrylate (FMA) has been successfully polymerized for the first time by anionic polymerization to produce atactic (at-), isotactic (it-), or syndiotactic (st-) poly(furfuryl methacrylate) (PFMA), depending on initiator structure and reaction conditions. Thermal properties of the PFMA materials are strongly affected by the polymer tacticity. Most notably, while both isotactic and syndiotactic polymers can undergo inter- or intrachain crosslinking reactions when heated to 290 °C, there is no evidence for the atactic polymer to perform the same reaction. Furthermore, the PFMA tacticity also greatly affects the amount of stable carbonaceous materials it produces when heated to 650 °C, with st-PFMA forming the largest amount of such materials (26.9%), as compared to only 5.6% by at-PFMA. Using the Diels–Alder (DA) “click reaction” between the reactive furfuryl group within the PFMA polymers as the diene equivalent and a bismaleimide as the dienophile, thermoreversible smart polymers have been successfully prepared. Thermoreversibility of the preformed crosslinked polymers has been demonstrated, thanks to the facile retro-DA reaction upon heating and the DA reaction upon cooling of such self-healing materials. © 2013 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 2793–2803
Co-reporter:Xia Chen ; Lucia Caporaso ; Luigi Cavallo
Journal of the American Chemical Society 2012 Volume 134(Issue 17) pp:7278-7281
Publication Date(Web):April 23, 2012
DOI:10.1021/ja301811s
Coordination polymerization of renewable α-methylene-γ-(methyl)butyrolactones by chiral C2-symmetric zirconocene catalysts produces stereo-random, highly stereo-regular, or perfectly stereo-regular polymers, depending on the monomer and catalyst structures. Computational studies yield a fundamental understanding of the stereocontrol mechanism governing these new polymerization reactions mediated by chiral metallocenium catalysts.
Co-reporter:Dajiang (D. J.) Liu, Yuetao Zhang and Eugene Y.-X. Chen
Green Chemistry 2012 vol. 14(Issue 10) pp:2738-2746
Publication Date(Web):12 Sep 2012
DOI:10.1039/C2GC36265A
The present study of rapid degradation of the key biorefining building block 5-hydroxymethylfurfural (HMF) in an ionic liquid (IL), 1-ethyl-3-methylimidazolium acetate ([EMIM]OAc), has led to highly selective and efficient upgrading of HMF to 5,5′-di(hydroxymethyl)furoin (DHMF), a promising C12 kerosene/jet fuel intermediate. This HMF upgrading reaction is carried out under industrially favourable conditions (i.e., ambient atmosphere and 60–80 °C), catalyzed by N-heterocyclic carbenes (NHCs), and complete within 1 h; this process selectively produces DHMF with yields up to 98% (by HPLC or NMR) or 87% (unoptimized, isolated yield). Mechanistic studies have yielded four lines of evidence that support the proposed carbene catalytic cycle for this upgrading transformation catalyzed by the acetate IL and NHCs.
Co-reporter:Wei Zhao, Yang Wang, Xinli Liu, Xuesi Chen, Dongmei Cui and Eugene Y.-X. Chen
Chemical Communications 2012 vol. 48(Issue 51) pp:6375-6377
Publication Date(Web):09 May 2012
DOI:10.1039/C2CC32680A
A highly efficient strategy for one-pot synthesis of programmable, crystalline–amorphous stereomultiblock PLA from rac-lactide.
Co-reporter:Nicole C. Escudé, Yalan Ning and Eugene Y.-X. Chen
Polymer Chemistry 2012 vol. 3(Issue 12) pp:3247-3255
Publication Date(Web):10 Aug 2012
DOI:10.1039/C2PY20525D
This contribution reports the first in situ stereocomplexing polymerization of methyl methacrylate (MMA) using a pair of diastereospecific coordination polymerization catalysts for rapid, high-yield, ambient-temperature production of crystalline stereocomplex poly(methyl methacrylate), sc-PMMA, with high Tm up to 217 °C. The diastereospecific catalyst pair is conveniently generated by in situ activation of a mixture of C2- and Cs-ligated metallocene bis(ester enolate)s with [Ph3C][B(C6F5)4], which is highly active, stereospecific, and controlled for coordination–addition polymerization of MMA to afford crystalline sc-PMMA in typically high to quantitative yields within only minutes of reaction. The isotactic/syndiotactic (it/st) composition of the sc-PMMA materials can be modulated by simply adjusting the relative ratio of the diastereospecific catalysts derived from pre-catalysts such as C2-ligated rac-(EBI)ZrMe[OC(OiPr)CMe2] and Cs-ligated [Ph2C(Cp)(2,7-tBu2-Flu)]Zr[OC(OiPr)CMe2]2; such catalysts have also been used separately to produce highly stereoregular it- and st-PMMAs and their corresponding sc-PMMA transparent film that exhibits nearly idealized characteristics: Mn = 44.2 kDa (vs. the calculated 40.1 kDa), PDI = 1.14, Tg = 96 °C, Tm = 213 °C, mm = 29.2%, rr = 62.3% (it/st ≈ 1/2), and visible transmittance = 92–95%. The in situ PMMA stereocomplexation can also occur in an it/st ratio deviating from the commonly recognized ratio of 1/2. Analysis of two such sc-PMMA materials with it/st ≈ 1/1 and ≈ 1/3 by wide-angle X-ray scattering (WAXS) has revealed essentially the same WAXS profile with nearly identical crystalline diffraction peaks attributed to sc-PMMA. The dynamic light scattering (DLS) results of the in situ stereocomplexing polymerization by a diastereospecific catalyst pair, obtained by monitoring the reaction in real time with DLS, indicate that stereocomplexation occurs as the diastereomeric PMMA chains are continuously growing. The presence of nanocages such as POSS and C60, which can be encapsulated by st-PMMA, in the stereocomplexing MMA polymerization system can completely disrupt or have no effect on the stereocomplexation, or enable both stereocomplexation and inclusion complexation processes to occur, depending on the type of nanocage employed.
Co-reporter:Yuetao Zhang, Garret M. Miyake, Mallory G. John, Laura Falivene, Lucia Caporaso, Luigi Cavallo and Eugene Y.-X. Chen
Dalton Transactions 2012 vol. 41(Issue 30) pp:9119-9134
Publication Date(Web):22 May 2012
DOI:10.1039/C2DT30427A
Classical and frustrated Lewis pairs (LPs) of the strong Lewis acid (LA) Al(C6F5)3 with several Lewis base (LB) classes have been found to exhibit exceptional activity in the Lewis pair polymerization (LPP) of conjugated polar alkenes such as methyl methacrylate (MMA) as well as renewable α-methylene-γ-butyrolactone (MBL) and γ-methyl-α-methylene-γ-butyrolactone (γ-MMBL), leading to high molecular weight polymers, often with narrow molecular weight distributions. This study has investigated a large number of LPs, consisting of 11 LAs as well as 10 achiral and 4 chiral LBs, for LPP of 12 monomers of several different types. Although some more common LAs can also be utilized for LPP, Al(C6F5)3-based LPs are far more active and effective than other LA-based LPs. On the other hand, several classes of LBs, when paired with Al(C6F5)3, can render highly active and effective LPP of MMA and γ-MMBL; such LBs include phosphines (e.g., PtBu3), chiral chelating diphosphines, N-heterocyclic carbenes (NHCs), and phosphazene superbases (e.g., P4-tBu). The P4-tBu/Al(C6F5)3 pair exhibits the highest activity of the LP series, with a remarkably high turn-over frequency of 9.6 × 104 h−1 (0.125 mol% catalyst, 100% MMA conversion in 30 s, Mn = 2.12 × 105 g mol−1, PDI = 1.34). The polymers produced by LPs at RT are typically atactic (PγMMBL with ∼47% mr) or syndio-rich (PMMA with ∼70–75% rr), but highly syndiotactic PMMA with rr ∼91% can be produced by chiral or achiral LPs at −78 °C. Mechanistic studies have identified and structurally characterized zwitterionic phosphonium and imidazolium enolaluminates as the active species of the current LPP system, which are formed by the reaction of the monomer·Al(C6F5)3 adduct with PtBu3 and NHC bases, respectively. Kinetic studies have revealed that the MMA polymerization by the tBu3P/Al(C6F5)3 pair is zero-order in monomer concentration after an initial induction period, and the polymerization is significantly catalyzed by the LA, thus pointing to a bimetallic, activated monomer propagation mechanism. Computational study on the active species formation as well as the chain initiation and propagation events involved in the LPP of MMA with some of the most representative LPs has added our understanding of fundamental steps of LPP. The main difference between NHC and PR3 bases is in the energetics of zwitterion formation, with the NHC-based zwitterions being remarkably more stable than the PR3-based zwitterions. Comparison of the monometallic and bimetallic mechanisms for MMA addition shows a clear preference for the bimetallic mechanism.
Co-reporter:Yang Wang, Wei Zhao, Xinli Liu, Dongmei Cui, and Eugene Y.-X. Chen
Macromolecules 2012 Volume 45(Issue 17) pp:6957-6965
Publication Date(Web):August 23, 2012
DOI:10.1021/ma3007625
A simple, inexpensive, and convenient catalyst system consisting of supporting ligand-free MgnBu2 in combination with an alcohol, isopropanol (iPrOH), benzyl methanol (PhCH2OH), diphenylmethanol (Ph2CHOH), or triphenylmethanol (Ph3COH), generates a convenient catalyst system to promote the polymerization of l-LA. In particular, the binary system MgnBu2/Ph2CHOH demonstrates an unprecedentedly high activity in the presence of a large excess amount of Ph2CHOH with the [OH]0/[Mg]0 ratio varying from 2 to 500, producing up to 500 polylactide (PLA) chains per Mg center and thus showing a typical nature of immortal polymerization. The molecular weights of the obtained PLAs with a broad range of monomer-to-metal ratios ([l-LA]0/[Mg]0 = 200–5000) are rather accurately controlled by the Ph2CHOH loading, relative to [Mg]0, while the molecular weight distributions remain nearly constant with polydispersity index (PDI) = 1.08–1.18. Moreover, the active polymerization intermediate has been isolated from the stoichiometric reaction between MgnBu2 and Ph2CHOH and structurally characterized as a tetranuclear complex, Mg4(Ph2CHO)8(THF)2 (1). Complex 1 remains the tetranuclear structure in solution or in the presence of excess Ph2CHOH as determined by 2D DOSY. On the basis of structural information about the active intermediates and polymerization kinetics, a coordination–insertion polymerization mechanism is proposed.
Co-reporter:Dr. Yangjian Hu;Garret M. Miyake;Baoli Wang;Dr. Dongmei Cui;Dr. Eugene Y.-X. Chen
Chemistry - A European Journal 2012 Volume 18( Issue 11) pp:3345-3354
Publication Date(Web):
DOI:10.1002/chem.201102677
Abstract
Two ansa-half-sandwich rare-earth-metal (REM) dialkyl complexes supported by an ethylene-bridged fluorenyl (Flu)-N-heterocyclic carbene (NHC) ligand, [M{C2H4(η5-Flu-κ1-NHC)}(CH2SiMe3)2] (M=Y, 1; Lu, 2), and a chiral ansa-sandwich samarocene incorporating a C2 ligand, [Sm(η5-C12H8)2(thf)2] (3), have been investigated for the coordination–addition polymerization of renewable methylene butyrolactones, α-methylene-γ-butyrolactone (MBL) and γ-methyl-α-methylene-γ-butyrolactone (γMMBL). Both ansa-half-sandwich complexes 1 and 2 exhibit exceptional activity for the polymerization of γMMBL at room temperature in dimethylformamide (DMF); with a 0.25 mol % catalyst loading, quantitative monomer conversion can be achieved under 1 min, giving a high turn-over frequency (TOF) of 24 000 h−1. This TOF value represents a rate enhancement, by a factor of 8, 22, or 2400, over the polymerizations by unbridged samarocene [Sm(Cp*)2(thf)2] (Cp*=η5-pentamethylcyclopentadienyl), by bridged ansa-samarocene 3 with C2 ligation, or by the corresponding REM trialkyls without the ansa-Flu-NHC ligation, respectively. Complexes 1 and 2 are also highly active for the polymerization of β-methyl-α-methylene-γ-butyrolactone (βMMBL), realizing the first example of the metal-mediated coordination polymerization of this monomer and its copolymerization with γMMBL. More remarkably, the resulting PβMMBL homopolymer is highly stereoregular (91 % mm) and exhibits a high Tg of 290 °C. In sharp contrast, catalysts 1 and 2 have poor activity and efficiency in the polymerization of the parent MBL or the acyclic analog methyl methacrylate. Polymerization and kinetic studies using the most active catalyst (1) of the series have uncovered characteristics of its γMMBL polymerization and yielded a unimolecular propagation mechanism. A surprising chain-initiation pathway for the polymerization in DMF by 1 has been revealed, and catalytic polymerization in the presence of an organoacid has also been examined.
Co-reporter:Dr. Yuetao Zhang ;Dr. Eugene Y.-X. Chen
Angewandte Chemie 2012 Volume 124( Issue 10) pp:2515-2519
Publication Date(Web):
DOI:10.1002/ange.201108019
Co-reporter:Dr. Yuetao Zhang ;Dr. Eugene Y.-X. Chen
Angewandte Chemie International Edition 2012 Volume 51( Issue 10) pp:2465-2469
Publication Date(Web):
DOI:10.1002/anie.201108019
Co-reporter:Yuetao Zhang ; Lucia Caporaso ; Luigi Cavallo
Journal of the American Chemical Society 2011 Volume 133(Issue 5) pp:1572-1588
Publication Date(Web):January 6, 2011
DOI:10.1021/ja109775v
Activation of 12 group IV metallocene bis(ester enolate) complexes with B(C6F5)3 at room temperature (RT) affords quantitatively the corresponding isolable cationic eight-membered ester enolate metallacycles. This rapid two-step reaction consists of vinylogous hydride abstraction to form the anion [HB(C6F5)3]−, and nucleophilic addition of the second enolate ligand to the methacrylate resulted from loss of a hydride in the first enolate ligand to form the chelating cation. This activation methodology for generating the active species (structural models for resting intermediates involved in methacrylate polymerization) is rather general, as demonstrated by a broad substrate scope examined in this study, including group IV metallocene bis(ester enolate) complexes that varied metals (Ti, Zr, Hf), bridging atoms (Ph2C<, Ph2Si<, Me2C<, −CH2CH2−), substituents (tBu, Et3Si), substitution patterns (on 3-Cp and 2,7-Flu ring positions), and ligand symmetries (C2, C2v, C1, and Cs), all of which lead to the clean formation of their corresponding cationic metallacycles. Comparative methyl methacrylate (MMA) polymerization studies have identified metallacycle 4, {[Ph2C(Cp)(2,7-tBu2−Flu)]Zr[OC(OiPr)═CMeCH2C(Me2)C(OiPr)═O]}+[HB(C6F5)3]−, as being the most active, efficient, and syndiospecific catalyst within the Cs-ligated catalysts. Kinetic experiments at room temperature show that the MMA polymerization by 4 follows first-order kinetics in both [MMA] and [Zr], consistent with a monometallic, intramolecular coordination−addition mechanism that involves the eight-membered ester enolate chelate resting state. Thermodynamic experiments at varied temperatures yield activation parameters of ΔH⧧ = 6.23 kcal/mol, ΔS⧧ = −41.7 eu, and ΔG⧧ = 17.6 kcal/mol (273 K). As compared to ansa-Flu-Cp ligated chelating cations paired with more commonly used weakly coordinating anions such as [MeB(C6F5)3]− and [B(C6F5)4]−, the same cations paired with the anion [HB(C6F5)3]− behave differently in MMA polymerization in terms of activity, stereospecificity, and sensitivity to solvent polarity. Most uniquely, [HB(C6F5)3]−-based catalysts effect substantial internal chain-transfer reactions, especially for polymerizations carried out in toluene and in the presence of excess B(C6F5)3, thus releasing polymer chains with a terminal double bond and achieving a catalytic polymerization. Computational results show the thermodynamics feasibility of the activation steps and the reversibility of the hydride abstraction step during activation, thus indicating that [HB(C6F5)3]− can uniquely act as a weak hydride donor. The picture emerging from the combined experimental and theoretical study has led to a new hydride-shuttling chain-transfer mechanism promoted by the hydridoborate anion, involving a hydride addition and abstraction sequence through the borane center.
Co-reporter:Yuetao Zhang ; Laura O. Gustafson
Journal of the American Chemical Society 2011 Volume 133(Issue 34) pp:13674-13684
Publication Date(Web):August 5, 2011
DOI:10.1021/ja2053573
Novel dinuclear silylium-enolate active species, consisting of an electrophilic silylium catalyst site and a nucleophilic silicon enolate initiating site that are covalently linked as single molecules, and their unique polymerization characteristics and kinetics are reported. Such unimolecular, bifunctional propagating species are conveniently generated from activation of ethyl- and oxo-bridged disilicon enolate (i.e., disilyl ketene acetal, di-SKA) compounds with [Ph3C][B(C6F5)4]. Both the ethyl- and oxo-bridged dinuclear species are much more active for the polymerization of methyl methacrylate (MMA) than the mononuclear SKA-based active species, exhibiting an approximate rate enhancement by a factor of 12 and 44, respectively. The oxo-bridged silylium-enolate species is considerably more active and controlled than the ethyl-bridged one, with their differences being even more pronounced in polymerizing a renewable monomer, γ-methyl-α-methylene-γ-butyrolactone. The polymerization by the oxo-bridged silylium-enolate active species follows first-order kinetics in both monomer and silylium catalyst concentrations, indicating a unimolecular propagation mechanism which involves an intramolecular delivery of the polymeric enolate nucleophile to the monomer activated by the silylium ion electrophile being placed in proximity in the same catalyst molecule. Highly stereoregular poly(methyl methacrylate) (PMMA), with a syndiotacticity up to 92% rr, can be produced in quantitative yield using the oxo-bridged propagator at low temperature.
Co-reporter:Garret M. Miyake and Eugene Y.-X. Chen
Polymer Chemistry 2011 vol. 2(Issue 11) pp:2462-2480
Publication Date(Web):03 Aug 2011
DOI:10.1039/C1PY00245G
Progress in the synthesis of highly stereoregular polymers with syndiotacticity ≥90% from an array of monomer substrates is surveyed, with a focus being placed on the use of syndiospecific discrete catalyst or initiator systems also exhibiting high activity and efficiency in the polymerization reactions. The monomer scope encompasses nonpolar α-olefins (propylene, styrene, and higher α-olefins), conjugated diolefins, bicyclic olefins, polar conjugated olefins (acrylic monomers such as methacrylates and (meth)acrylamides), and cyclic esters (lactides and β-lactones), while the polymerization catalysts or initiators enabling such syndiospecific polymerizations cover those discrete molecular complexes of main-group, early transition, and lanthanide metals. Several free-radical polymerization systems capable of producing highly syndiotactic polymers are also highlighted.
Co-reporter:Yangjian Hu;Laura O. Gustafson;Hongping Zhu
Journal of Polymer Science Part A: Polymer Chemistry 2011 Volume 49( Issue 9) pp:2008-2017
Publication Date(Web):
DOI:10.1002/pola.24628
Abstract
Three resorbable potassium salts of hydride (K[H]), enolate Me2CC(OiPr)OK (K[E]), and allyl K[1,3-(SiMe3)2C3H3] (K[A]) have been investigated for controlled anionic polymerization of methyl methacrylate (MMA) and its cyclic analogs, naturally renewable methylene butyrolactones including α-methylene-γ-butyrolactone (MBL) and γ-methyl-α-methylene-γ-butyrolactone (MMBL). When used alone at ambient temperature in toluene, these salts exhibit no (K[H]) to low (K[A]) to modest (K[E]) polymerization activity. Mixing of K[H] and Al(C6F5)3 leads to the formation of an “ate” complex, K+[HAl(C6F5)3]−, which has been structurally characterized by X-ray diffraction; this complex has a high polymerization activity producing atactic PMMA, but addition of another equiv of Al(C6F5)3 further enhances both the rate and the efficiency of the polymerization, now producing syndiotactic PMMA with a narrow molecular weight (MW) distribution of 1.04. The K[H]/2Al(C6F5)3 system also exhibits high activity for polymerization of (M)MBL. In sharp contrast, addition of Al(C6F5)3 to K[A] shuts down the polymerization at various temperatures. The most active, controlled, and syndioselective polymerization system in this series is K[E]/2Al(C6F5)3. Accordingly, the polymerization control and kinetics of this most effective system have been examined in more detail. © 2011 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2011
Co-reporter:Yuetao Zhang, Yalan Ning, Lucia Caporaso, Luigi Cavallo and Eugene Y.-X. Chen
Journal of the American Chemical Society 2010 Volume 132(Issue 8) pp:2695-2709
Publication Date(Web):February 3, 2010
DOI:10.1021/ja908818y
This contribution reports a combined synthetic, kinetic, mechanistic, and theoretical/computational study of the recently discovered catalyst-site-controlled coordination polymerization of polar vinyl monomers [such as methyl methacrylate (MMA) and N,N-dimethylacrylamide (DMAA)] into highly syndiotactic polymers. Among the 12 Cs-ligated ansa-cyclopentadienyl (Cp)-R2E(C,Si)-fluorenyl (Flu) group 4 metallocene catalyst systems examined—which varied in metal center, anion structure, bridging atom and substituents, and ligand substitution pattern—cationic ansa-metallocene ester enolate catalyst 6+[B(C6F5)4]−, derived from the activation of the precatalyst [Ph2C(Cp)(2,7-tBu2-Flu)]Zr[OC(OiPr)═CMe2]2 with [Ph3C][B(C6F5)4], stood out as the best catalyst in all aspects of the MMA polymerization at room temperature, including the highest activity (1554 h−1 TOF), efficiency (98% I*), syndiotacticity (94% rr), and control (predicted number-average molecular weight and 1.14 molecular weight distribution). Kinetic and mechanistic results are consistent with a catalyst-site-controlled, monometallic coordination−addition mechanism, involving fast intramolecular addition within the catalyst−monomer complex leading to the resting eight-membered ester enolate chelate, followed by the rate-limiting ring-opening of the chelate to regenerate the active species. This work has also uncovered several unique features of this polymerization system that are in marked contrast to the propylene polymerization by analogous Cs-ligated cationic alkyl catalysts: a constant syndiotacticity of PMMA produced over a wide polymerization temperature range (i.e., from 0 °C, 94% rr to 25 °C, 94% rr to 50 °C, 93% rr); insensitivity of its high activity, degree of control, and stereoselectivity to solvent polarity and structure of weakly coordinating anions; and deviation from a pure site-control mechanism at high [MMA]/[catalyst] ratios. Computational results provide theoretical support for the proposed monomer-assisted, catalyst-site epimerization, after an enantiofacial mistake, to a thermodynamically more stable resting state, which accounts for the observed higher than expected [mr] contents based on a pure site-controlled mechanism. DFT calculations rationalize why the Ph2C< bridged catalyst 6 exhibits higher stereoselectivity than other catalysts with the Me2C< or Me2Si< bridge: the bridge rigidity pushes the η3-bound Flu ligand closer to the growing chain and the monomer, thereby increasing ΔEstereo between the competing transition states for the addition of a monomer molecule to the opposite (correct and wrong) enantiofaces of the enolate growing chain. The relative polymerization activity of this catalyst series is shown to correlate with the relative energetics of the back-biting of the penultimate unit and ion-pair formation.
Co-reporter:Garret M. Miyake, Stacie E. Newton, Wesley R. Mariott and Eugene Y.-X. Chen
Dalton Transactions 2010 vol. 39(Issue 29) pp:6710-6718
Publication Date(Web):25 Mar 2010
DOI:10.1039/C001909G
This contribution reports the first study of coordination-addition polymerization of renewable butyrolactone-based vinyl monomers, MBL (α-methylene-γ-butyrolactone) and MMBL (γ-methyl-α-methylene-γ-butyrolactone), using neutral lanthanocene(II), non-lanthanocene(III), and cationic group 4 metallocene catalysts. The samarocene(II) catalyst, Cp*2Sm(THF)2, promotes a rapid, efficient, and controlled polymerization of MBL and MMBL in DMF at ambient temperature, exhibiting a high TOF of 3000 h−1, typically near quantitative initiator efficiency, and the ability to control the polymer MW. The resulting atactic PMBL and PMMBL have high Tg's of 194 °C and 227 °C, respectively; when compared to atactic PMMA having comparable MW, the Tg and onset decomposition temperatures of the PMMBL produced are substantially higher (by ∼120 °C and 40 °C, respectively). Owing to the living/controlled characteristics of this polymerization, well-defined random and block copolymers of MBL with MMA and MMBL can be readily synthesized. Results of the kinetic and polymerization studies indicate that the true active species is the trivalent samarocene centers attached to the single growing polymer chain, derived presumably from a redox-then-radical-coupling process. In comparison, the polymerizations by non-lanthanocene(III) silylamides, Ln[N(SiMe3)2]3 (Ln = La, Nd, Sm, Er), and by cationic group 4 metallocene and half-metallocene catalysts incorporating C2 and Cs symmetric ligands are much slower and less effective. Catalytic polymerization of MBL by Cp*2Sm(THF)2 has also been realized in the presence of an enolizable organo acid as a suitable chain transfer agent.
Co-reporter:Yuetao Zhang, Hongbo Du, Xianghong Qian and Eugene Y.-X. Chen
Energy & Fuels 2010 Volume 24(Issue 4) pp:2410-2417
Publication Date(Web):March 10, 2010
DOI:10.1021/ef1000198
Under relatively mild conditions (≤140 °C, 1 atm) and in the absence of added acid catalysts typically employed in biomass conversion, cellulose dissolved in certain ionic liquids (ILs) has been converted into water-soluble reducing sugars in high total reducing sugar yield (up to 97%), or directly into the biomass platform chemical 5-hydroxymethyl furfural (HMF) in high conversion (up to 89%) when CrCl2 is added. The combined study of experimental methods and ab initio calculations demonstrates that the significantly increased Kw by ILs in the IL−water mixture is responsible for the catalysis seen in the current efficient biomass conversion system without added acid catalysts. The finding that the water in ILs under mild conditions can exhibit high Kw values (up to 3 orders of magnitude higher than the pure water under ambient conditions) is significant because such high Kw values are typically achievable by the water under harsh high-temperature or subcritical water conditions.
Co-reporter:Yuetao Zhang, Eugene Y.-X. Chen
Journal of Organometallic Chemistry 2010 695(10–11) pp: 1464-1471
Publication Date(Web):
DOI:10.1016/j.jorganchem.2010.02.030
Co-reporter:Dr. Yuetao Zhang;Frank Lay;Dr. Pilar García-García;Dr. Benjamin List;Dr. EugeneY.-X. Chen
Chemistry - A European Journal 2010 Volume 16( Issue 34) pp:10462-10473
Publication Date(Web):
DOI:10.1002/chem.201000961
Abstract
This contribution describes the development and demonstration of the ambient-temperature, high-speed living polymerization of polar vinyl monomers (M) with a low silylium catalyst loading (≤ 0.05 mol % relative to M). The catalyst is generated in situ by protonation of a trialkylsilyl ketene acetal (RSKA) initiator (I) with a strong Brønsted acid. The living character of the polymerization system has been demonstrated by several key lines of evidence, including the observed linear growth of the chain length as a function of monomer conversion at a given [M]/[I] ratio, near-precise polymer number-average molecular weight (Mn, controlled by the [M]/[I] ratio) with narrow molecular weight distributions (MWD), absence of an induction period and chain-termination reactions (as revealed by kinetics), readily achievable chain extension, and the successful synthesis of well-defined block copolymers. Fundamental steps of activation, initiation, propagation, and catalyst “self-repair” involved in this living polymerization system have been elucidated, chiefly featuring a propagation “catalysis” cycle consisting of a rate-limiting CC bond formation step and fast release of the silylium catalyst to the incoming monomer. Effects of acid activator, catalyst and monomer structure, and reaction temperature on polymerization characteristics have also been examined. Among the three strong acids incorporating a weakly coordinating borate or a chiral disulfonimide anion, the oxonium acid [H(Et2O)2]+[B(C6F5)4]− is the most effective activator, which spontaneously delivers the most active R3Si+, reaching a high catalyst turn-over frequency (TOF) of 6.0×103 h−1 for methyl methacrylate polymerization by Me3Si+ or an exceptionally high TOF of 2.4×105 h−1 for n-butyl acrylate polymerization by iBu3Si+, in addition to its high (>90 %) to quantitative efficiencies and a high degree of control over Mn and MWD (1.07–1.12). An intriguing catalyst “self-repair” feature has also been demonstrated for the current living polymerization system.
Co-reporter:Garret M. Miyake, Daniel A. DiRocco, Qin Liu, Kevin M. Oberg, Ercan Bayram, Richard G. Finke, Tomislav Rovis, and Eugene Y.-X. Chen
Macromolecules 2010 Volume 43(Issue 18) pp:7504-7514
Publication Date(Web):September 7, 2010
DOI:10.1021/ma101310n
Acryloyl and vinyl monomers functionalized with a chiral oxazolidinone auxiliary have been successfully polymerized in a stereospecific fashion to highly isotactic, optically active polymers, through either the previously established isospecific coordination polymerization (for acryloyl monomers) or a novel isospecific cationic polymerization (for vinyl monomers). Specifically, conjugated chiral acryloyloxazolidinones, N-acryloyl-(R or S)-4-phenyl-2-oxazolidinone [(R or S)-AOZ], are readily polymerized by chiral ansa-zirconocenium coordination catalysts, (R,R-, S,S-, or R,R/S,S)-[C2H4(η5-Ind)2]Zr+(THF)[OC(OiPr)═CMe2][MeB(C6F5)3]− (1), in an isospecific manner through a catalyst-site-controlled mechanism, producing the corresponding optically active chiral polymers, (R or S)-PAOZ. Owing to the nature of stereocontrol dictated by the chiral catalyst site, even the coordination polymerization of the parent AOZ, without the chiral side group, also affords PAOZ with nearly quantitative isotacticity. A series of experiments have shown that the chiral polymers (R or S)-PAOZ exhibit no chiral amplifications, despite having stereoregularly placed stereogenic centers in the main chain, and the optical activity of the polymers arises solely from their chiral auxiliary, a consequence of adopting a random-coil secondary structure and thus having a cryptochiral chain. In sharp contrast, the chiral isotactic polymers derived from nonconjugated chiral vinyl oxazolidinones, N-vinyl-(R)-4-phenyl-2-oxazolidinone [(R)-VOZ] and its p-hexyloxyphenyl derivative (R)-HVOZ (designed to solve the solubility issue of the resulting polymer), exhibit substantial chiral amplifications by virtue of adopting a solution-stable, one-handed helical conformation. The synthesis of such helical vinyl polymers has been accomplished by the development of a novel isospecific cationic polymerization using Lewis and Brønsted acids, such as [Ph3C][B(C6F5)4], BF3·Et2O, and [H(Et2O)2][B(C6F5)4], through a chiral auxiliary-controlled mechanism. Noteworthy is the combination of the near-quantitative isotactic placement of the stereogenic centers of the polymer main chain with the chiral side groups located near those stereocenters that renders one-handed helicity of (R)-PVOZ and (R)-PHVOZ. Significantly, this novel cationic polymerization process, operating at ambient temperature, effectively assembles two elements of polymer local chirality—side-chain chirality and main-chain chirality—into global chirality in the form of excess one-handed helicity. Furthermore, the resulting chiral helical vinyl polymers exhibit considerably higher thermal decomposition temperatures and polymer crystallinity, in comparison to the random-coil chiral acryloyl polymers, having a similarly high degree of main-chain stereoregularity.
Co-reporter:Garret M. Miyake, Yuetao Zhang and Eugene Y.-X. Chen
Macromolecules 2010 Volume 43(Issue 11) pp:4902-4908
Publication Date(Web):May 12, 2010
DOI:10.1021/ma100615t
Naturally renewable butyrolactone-based vinylidene monomers, α-methylene-γ-butyrolactone (MBL) and γ-methyl-α-methylene-γ-butyrolactone (MMBL), have been successfully polymerized in a rapid and living fashion, using ambiphilic silicon propagating species consisting of both the nucleophilic silyl ketene acetal (SKA) initiating moiety and the electrophilic silylium catalyst. Uniquely, the R3Si+ catalyst is derived directly from the SKA initiator upon in situ oxidative activation with a catalytic amount of the trityl borate activator. Investigations into effects of SKA (thus the resulting R3Si+ catalyst) and activator (thus the resulting counteranion) structures have revealed that the Me2C═C(OMe)OSiiBu3/Ph3CB(C6F5)4 combination is the most active and controlled system for (M)MBL polymerizations. Thus, under ambient conditions and with a low catalyst loading (0.05 mol % relative to monomer), this polymerization system rapidly (within 10 min) and completely converts MMBL to PMMBL with controlled low to high (Mn = 5.43 × 105 kg/mol) MW’s and narrow MW distributions (1.01−1.06). Well-defined block copolymers of MBL and MMBL with MMA as well as block and statistical copolymers of MBL with MMBL have also been readily synthesized. Atactic homopolymers, PMBL and PMMBL, produced herein exhibit high glass transition temperatures (Tg’s) of 194 and 225 °C, respectively, representing Tg enhancements of ∼90 °C (for PMBL) and ∼120 °C (for PMMBL) over the Tg of the typical atactic PMMA. The critical MW of PMMBL has been estimated to be ∼47 kg/mol.
Co-reporter:Yangjian Hu, Xin Xu, Yuetao Zhang, Yaofeng Chen, and Eugene Y.-X. Chen
Macromolecules 2010 Volume 43(Issue 22) pp:9328-9336
Publication Date(Web):October 22, 2010
DOI:10.1021/ma101901y
Four discrete half-sandwich dialkyl rare earth metal (REM) complexes incorporating a disilylated indenyl ligand, (1,3-(SiMe3)2C9H5)M(CH2SiMe3)2(THF) (M = Sc, Y, Dy, Lu), have been investigated for the coordination−addition polymerization of naturally renewable methylene butyrolactones, α-methylene-γ-butyrolactone (MBL) and γ-methyl-α-methylene-γ-butyrolactone (MMBL). Initial screening for the polymerization of methyl methacrylate highlighted several differences in catalytic behavior between these half-sandwich REM catalysts and well-studied sandwich REM catalysts in terms of reactivity trend, polymer tacticity, and solvent dependence. Most significantly, all four catalysts herein exhibit exceptional activity for polymerization of MMBL in DMF, achieving quantitative monomer conversion in <1 min with a 0.20 mol % catalyst loading and giving a high turnover frequency of >30 000 h−1. Slower polymerizations occur in CH2Cl2, allowing for establishment of the activity trend within this REM series, which follows: Dy (largest ion) ≥ Y > Lu > Sc (smallest ion). The most active and effective Dy catalyst has been examined in detail, demonstrating its ability to control the polymerization for producing PMMBL with high Tg (221 °C) and with molecular weight ranging from a medium Mn of 1.89 × 104 Da to a high Mn of 1.63 × 105 Da, programmed by the [MMBL]/[Dy] ratio. Kinetic experiments have revealed a first-order dependence on [monomer] and a second-order dependence on [REM]. These kinetic results, coupled to catalyst efficiencies, NMR studies, as well as with chain-end group analysis by MALDI-TOF mass spectrometry, have yielded a chain initiation mechanism that involves both alkyl groups on each metal center and a bimolecular chain propagation that involves two metal centers in the rate-limiting C−C bond forming step. The Dy catalyst response to enolizable organo acids, externally added as chain-transfer agents, has also been examined.
Co-reporter:Dr. Yuetao Zhang;Garret M. Miyake ;Dr. Eugene Y.-X. Chen
Angewandte Chemie 2010 Volume 122( Issue 52) pp:10356-10360
Publication Date(Web):
DOI:10.1002/ange.201005534
Co-reporter:Dr. Yuetao Zhang;Garret M. Miyake ;Dr. Eugene Y.-X. Chen
Angewandte Chemie 2010 Volume 122( Issue 52) pp:
Publication Date(Web):
DOI:10.1002/ange.201007438
Co-reporter:Dr. Yuetao Zhang;Garret M. Miyake ;Dr. Eugene Y.-X. Chen
Angewandte Chemie International Edition 2010 Volume 49( Issue 52) pp:10158-10162
Publication Date(Web):
DOI:10.1002/anie.201005534
Co-reporter:Dr. Yuetao Zhang;Garret M. Miyake ;Dr. Eugene Y.-X. Chen
Angewandte Chemie International Edition 2010 Volume 49( Issue 52) pp:
Publication Date(Web):
DOI:10.1002/anie.201007438
Co-reporter:Eugene Y.-X. Chen
Chemical Reviews 2009 Volume 109(Issue 11) pp:5157
Publication Date(Web):September 9, 2009
DOI:10.1021/cr9000258
Co-reporter:Nicole C. Escudé and Eugene Y.-X. Chen
Chemistry of Materials 2009 Volume 21(Issue 24) pp:5743
Publication Date(Web):November 12, 2009
DOI:10.1021/cm902599w
This contribution reports the first examples of stereoregular (isotactic, it-, and syndiotactic, st-) methacrylate-POSS (polyhedral oligomeric silsesquioxane) hybrid polymers and their derived nanostructured assemblies. The polymerization of methyl methacrylate (MMA) by isospecific and syndiospecific living metallocene catalysts, when end-capped with methacrylisobutyl POSS (MA-POSS) or simultaneously copolymerized with MA-POSS at ambient temperature, readily produces highly stereoregular (94% it- and st-) MA-POSS end-capped PMMA (PMMA-POSS) or statistical copolymers PMMA-co-P(MA-POSS). The MA-POSS incorporation in the it-copolymers ranges from a low 2.6 mol % (20 wt %) to a high, maximum 24 mol % (75 wt %), whereas the incorporation in the st-copolymers is relatively lower with the same comonomer feed ratio due to the formation of a crystalline inclusion complex in which the POSS nanocages are encapsulated within the helical st-PMMA cavity. The it-copolymers with high POSS contents (>20 mol %) show evidence for interchain association through POSS aggregation and display a ∼20 °C Tg enhancement over the pristine it-PMMA. MA-POSS end-capped diastereomeric PMMAs exhibit versatile nanostructured assemblies, including micelle-like core−shell nanostructures (Rh up to 186 nm derived from the 5.7 nm it-PMMA-POSS unimer) through POSS inorganic collapse in selective solvents, helical stereocomplexes (Rh = 11 nm) between two or three polymer chains through organic self-organization of the diastereomeric PMMAs, and large core−shell nanospheres (Rh up to 636 nm) through synergistic organic PMMA self-organization and inorganic POSS nanocluster aggregation. Thermal annealing of the it-/st-PMMA-POSS blend in a 1:2 ratio also generates the crystalline stereocomplex with a high Tm of 213 °C.
Co-reporter:Eugene Y.-X. Chen
Dalton Transactions 2009 (Issue 41) pp:8784-8793
Publication Date(Web):21 Aug 2009
DOI:10.1039/B912640F
Chiral metallocene cations, discrete main group anions, dually active ion pairs and nucleophile/electrophile pairs have been utilized to transform the polymerization of polar vinyl monomers from one type to another. These discrete or hybrid catalysts have enabled the development of five types of such polymerizations, including stereospecific and living coordination polymerization, asymmetric coordination polymerization, single-site anionic polymerization, bimolecular polymer-transfer polymerization, and diastereospecific ion-pairing polymerization.
Co-reporter:Garret Miyake, Lucia Caporaso, Luigi Cavallo and Eugene Y.-X. Chen
Macromolecules 2009 Volume 42(Issue 5) pp:1462-1471
Publication Date(Web):February 13, 2009
DOI:10.1021/ma802697z
This contribution reports the first successful coordination−addition polymerization of N,N-dialkylmethacrylamides and the first example of kinetic resolution of a racemic methacrylamide by chiral metallocene catalysts. The polymerization of methacryloyl-2-methylaziridine (MMAz) by rac-(EBI)Zr+(THF)[OC(OiPr)═CMe2][MeB(C6F5)3]− (1) is stereospecific and also exhibits a high degree of control over polymerization. This polymerization follows first-order kinetics in both concentrations of monomer and catalyst, consistent with a monometallic propagation mechanism involving the fast step of intramolecular conjugate addition within the catalyst−monomer coordination complex leading to the eight-membered-ring resting intermediate. Substituents on the highly strained aziridine ring stabilize the aziridine moiety against thermally induced cross-linking through its ring-opening reaction; thus, the polymer derived from methacryloyl-tetramethyleneaziridine (MTMAz) exhibits greatly enhanced resistance toward thermal cross-linking over poly(MMAz), marking 57 and 42 °C higher onset cross-linking and maximum cross-linking temperatures, respectively. Enantiomeric catalyst (S,S)-1 demonstrates experimentally and theoretically its ability to kinetically resolve the racemic MMAz monomer with a low stereoselectivity factor s of 1.8. Polymerizability of several methacrylamide monomers has been investigated via a combined experimental and theoretical (DFT) study that examines the degree of conjugation between the vinyl and carbonyl double bonds, relative polymerization reactivity, and relative energy for the formation of amide−enolate intermediates.
Co-reporter:Dajiang (D.J.) Liu, Eugene Y.-X. Chen
Biomass and Bioenergy (January 2013) Volume 48() pp:181-190
Publication Date(Web):January 2013
DOI:10.1016/j.biombioe.2012.11.020
Co-reporter:Jing Tang and Eugene Y.-X. Chen
Inorganic Chemistry Frontiers 2015 - vol. 2(Issue 12) pp:NaN1631-1631
Publication Date(Web):2015/11/06
DOI:10.1039/C5QO00262A
The first successful polymerization of the naturally occurring, OH-containing, tri-functional monomer Tulipalin B (βHMBL) is established. N-Heterocyclic carbene and phosphazene superbase catalysts effectively polymerize βHMBL into polymers with Mn up to 13.2 kg mol−1. The possible polymer structure is thought to be a branched copolymer of poly(vinyl-ether lactone)s, derived from proposed crossovers between conjugate Michael and oxa-Michael additions, enabled by proton transfer.
Co-reporter:Garret M. Miyake
Macromolecules () pp:
Publication Date(Web):May 12, 2011
DOI:10.1021/ma2007199
This work investigates, for the first time, cinchona alkaloids such as cinchonidine (CD) and β-isocupreidine (ICD) consisting of both a chiral nucleophilic amine catalyst site and an electrophilic hydroxy moiety, as bifunctional, stereoselective organocatalysts for the ring-opening polymerization (ROP) of l-lactide (l-LA) and rac-lactide (rac-LA). While the ROP of l-LA by CD is ineffective, the ROP of l-LA by ICD proceeds without noticeable epimerization, thus affording polylactide (PLA) with a quantitative isotacticity. The measured number-average molecular weight (Mn) is much higher than the expected Mn due to sluggish initiation by ICD, but when benzyl alcohol is added as an external protic initiator, the ROP is highly efficient and proceeds to high conversions without undesired transesterification reactions, thus producing PLA with a controlled Mn and a narrow molecular weight distribution of 1.12. More significantly, the ROP of rac-LA by ICD/alcohol affords crystalline isotactic-rich, stereogradient PLA that exhibits multiple melting-transition temperatures as a result of a partial kinetic resolution polymerization that preferentially polymerizes l-LA and kinetically resolves d-LA. Overall, this study uncovers the first kinetic resolution polymerization of rac-LA by an organocatalyst.
Co-reporter:Ravikumar R. Gowda and Eugene Y.-X. Chen
Inorganic Chemistry Frontiers 2014 - vol. 1(Issue 3) pp:NaN234-234
Publication Date(Web):2014/03/05
DOI:10.1039/C3QO00089C
An efficient, high-yielding route to β-methyl-α-methylene-γ-butyrolactone (βMMBL)—a monomer for the production of high-performance engineering bioplastics—from biorenewable and inexpensive itaconic acid has been developed. The monomer prepared by this new route is of high purity as isolated without purification, as evidenced by NMR as well as small and large-scale polymerization tests.
Co-reporter:Garret M. Miyake, Stacie E. Newton, Wesley R. Mariott and Eugene Y.-X. Chen
Dalton Transactions 2010 - vol. 39(Issue 29) pp:NaN6718-6718
Publication Date(Web):2010/03/25
DOI:10.1039/C001909G
This contribution reports the first study of coordination-addition polymerization of renewable butyrolactone-based vinyl monomers, MBL (α-methylene-γ-butyrolactone) and MMBL (γ-methyl-α-methylene-γ-butyrolactone), using neutral lanthanocene(II), non-lanthanocene(III), and cationic group 4 metallocene catalysts. The samarocene(II) catalyst, Cp*2Sm(THF)2, promotes a rapid, efficient, and controlled polymerization of MBL and MMBL in DMF at ambient temperature, exhibiting a high TOF of 3000 h−1, typically near quantitative initiator efficiency, and the ability to control the polymer MW. The resulting atactic PMBL and PMMBL have high Tg's of 194 °C and 227 °C, respectively; when compared to atactic PMMA having comparable MW, the Tg and onset decomposition temperatures of the PMMBL produced are substantially higher (by ∼120 °C and 40 °C, respectively). Owing to the living/controlled characteristics of this polymerization, well-defined random and block copolymers of MBL with MMA and MMBL can be readily synthesized. Results of the kinetic and polymerization studies indicate that the true active species is the trivalent samarocene centers attached to the single growing polymer chain, derived presumably from a redox-then-radical-coupling process. In comparison, the polymerizations by non-lanthanocene(III) silylamides, Ln[N(SiMe3)2]3 (Ln = La, Nd, Sm, Er), and by cationic group 4 metallocene and half-metallocene catalysts incorporating C2 and Cs symmetric ligands are much slower and less effective. Catalytic polymerization of MBL by Cp*2Sm(THF)2 has also been realized in the presence of an enolizable organo acid as a suitable chain transfer agent.
Co-reporter:Ravikumar R. Gowda and Eugene Y.-X. Chen
Dalton Transactions 2013 - vol. 42(Issue 25) pp:NaN9273-9273
Publication Date(Web):2013/03/27
DOI:10.1039/C3DT50430A
Protonolysis of M(Bn)4 (M = Zr, Ti; Bn = benzyl) with equimolar 2,4-di-tert-butyl-6-[(2,6-diisopropylphenylimino)methyl]phenol [(2,6-iPr2C6H3)NC(3,5-tBu2C6H2)OH] in toluene at −30 °C to 25 °C cleanly affords the corresponding achiral (imino)phenoxy-tribenzyl complexes, [(2,6-iPr2C6H3)NC(3,5-tBu2C6H2)O]Zr(Bn)3 (1) and [(2,6-iPr2C6H3)NC(3,5-tBu2C6H2)O]Ti(Bn)3 (2). A chiral dibenzyl complex 3 incorporating the unsymmetric, tetradentate amino(imino)bis(phenoxy) ligand, [2,4-Br2C6H2(O)(6-CH2(NC5H9))CH2NCH(2-adamantyl-4-MeC6H2O)]Zr(Bn)2 (3), has also been prepared using the same protonolysis protocol. Abstractive activation of 1 with B(C6F5)3·THF in CD2Cl2 at room temperature (RT) affords clean, quantitative formation of the corresponding zirconium cation [((2,6-iPr2C6H3)NC(3,5-tBu2C6H2)O)Zr(Bn)2(THF)]+[BnB(C6F5)3]− (4). Likewise, benzyl abstraction of 2 with B(C6F5)3·THF in CD2Cl2 at RT generates the cationic titanium complex [((2,6-iPr2C6H3)NC(3,5-tBu2C6H2)O)Ti(Bn)2(THF)]+[BnB(C6F5)3]− (5), accompanied by a small amount of decomposed species as a result of C6F5 transfer. The dibenzyl cations 4 and 5 have been characterized spectroscopically, and their structures have been confirmed by single crystal X-ray diffraction analysis. Characteristics of the coordination polymerization of renewable α-methylene-γ-butyrolactone monomers by the cationic catalysts derived from achiral complexes 1 and 2 as well as chiral complex 3 have been investigated, representing the first study of such polymerization by non-metallocene catalysts.
Co-reporter:Eugene Y.-X. Chen
Dalton Transactions 2009(Issue 41) pp:NaN8793-8793
Publication Date(Web):2009/08/21
DOI:10.1039/B912640F
Chiral metallocene cations, discrete main group anions, dually active ion pairs and nucleophile/electrophile pairs have been utilized to transform the polymerization of polar vinyl monomers from one type to another. These discrete or hybrid catalysts have enabled the development of five types of such polymerizations, including stereospecific and living coordination polymerization, asymmetric coordination polymerization, single-site anionic polymerization, bimolecular polymer-transfer polymerization, and diastereospecific ion-pairing polymerization.
Co-reporter:Yuetao Zhang, Garret M. Miyake, Mallory G. John, Laura Falivene, Lucia Caporaso, Luigi Cavallo and Eugene Y.-X. Chen
Dalton Transactions 2012 - vol. 41(Issue 30) pp:NaN9134-9134
Publication Date(Web):2012/05/22
DOI:10.1039/C2DT30427A
Classical and frustrated Lewis pairs (LPs) of the strong Lewis acid (LA) Al(C6F5)3 with several Lewis base (LB) classes have been found to exhibit exceptional activity in the Lewis pair polymerization (LPP) of conjugated polar alkenes such as methyl methacrylate (MMA) as well as renewable α-methylene-γ-butyrolactone (MBL) and γ-methyl-α-methylene-γ-butyrolactone (γ-MMBL), leading to high molecular weight polymers, often with narrow molecular weight distributions. This study has investigated a large number of LPs, consisting of 11 LAs as well as 10 achiral and 4 chiral LBs, for LPP of 12 monomers of several different types. Although some more common LAs can also be utilized for LPP, Al(C6F5)3-based LPs are far more active and effective than other LA-based LPs. On the other hand, several classes of LBs, when paired with Al(C6F5)3, can render highly active and effective LPP of MMA and γ-MMBL; such LBs include phosphines (e.g., PtBu3), chiral chelating diphosphines, N-heterocyclic carbenes (NHCs), and phosphazene superbases (e.g., P4-tBu). The P4-tBu/Al(C6F5)3 pair exhibits the highest activity of the LP series, with a remarkably high turn-over frequency of 9.6 × 104 h−1 (0.125 mol% catalyst, 100% MMA conversion in 30 s, Mn = 2.12 × 105 g mol−1, PDI = 1.34). The polymers produced by LPs at RT are typically atactic (PγMMBL with ∼47% mr) or syndio-rich (PMMA with ∼70–75% rr), but highly syndiotactic PMMA with rr ∼91% can be produced by chiral or achiral LPs at −78 °C. Mechanistic studies have identified and structurally characterized zwitterionic phosphonium and imidazolium enolaluminates as the active species of the current LPP system, which are formed by the reaction of the monomer·Al(C6F5)3 adduct with PtBu3 and NHC bases, respectively. Kinetic studies have revealed that the MMA polymerization by the tBu3P/Al(C6F5)3 pair is zero-order in monomer concentration after an initial induction period, and the polymerization is significantly catalyzed by the LA, thus pointing to a bimetallic, activated monomer propagation mechanism. Computational study on the active species formation as well as the chain initiation and propagation events involved in the LPP of MMA with some of the most representative LPs has added our understanding of fundamental steps of LPP. The main difference between NHC and PR3 bases is in the energetics of zwitterion formation, with the NHC-based zwitterions being remarkably more stable than the PR3-based zwitterions. Comparison of the monometallic and bimetallic mechanisms for MMA addition shows a clear preference for the bimetallic mechanism.
Co-reporter:Jiawei Chen and Eugene Y.-X. Chen
Dalton Transactions 2016 - vol. 45(Issue 14) pp:NaN6110-6110
Publication Date(Web):2015/11/16
DOI:10.1039/C5DT03895B
Alkyl/aryl ligand exchange between AlEt3 and B(C6F5)3 in hexanes enables the formation and isolation of the unsolvated Al(C6F5)3 as a crystalline solid, the structure of which has been determined by single-crystal X-ray diffraction analysis. Instead of forming the anticipated Al⋯F contacts with the seemingly more accessible meta- and para-F's of –C6F5 groups, two Al(C6F5)3 molecules form a dimeric structure with double Al⋯F interactions between the Al center of one molecule and the ortho-F atom of the –C6F5 group on the other molecule. This mode of interactions is apparently linked to the thermal and shock sensitivity of the unsolvated Al(C6F5)3 in the solid state. To compare with the B(C6F5)3/ferrocene frustrated Lewis pair system, the complexation between Al(C6F5)3 and ferrocene has also been studied, which affords a stable adduct formed through the η1-coordination of Al to one of the CCp atoms, similar to the alane–toluene or benzene complex.
Co-reporter:Wei Zhao, Yang Wang, Xinli Liu, Xuesi Chen, Dongmei Cui and Eugene Y.-X. Chen
Chemical Communications 2012 - vol. 48(Issue 51) pp:NaN6377-6377
Publication Date(Web):2012/05/09
DOI:10.1039/C2CC32680A
A highly efficient strategy for one-pot synthesis of programmable, crystalline–amorphous stereomultiblock PLA from rac-lactide.
.png)
![3-(2,4,6-trimethylphenyl)-5,6,7,8-tetrahydro-4h-cyclohepta[d][1,3]thiazol-3-ium;perchlorate](http://img.cochemist.com/ccimg/1062200/1062158-63-7.png)
![3-(2,4,6-trimethylphenyl)-5,6,7,8-tetrahydro-4h-cyclohepta[d][1,3]thiazol-3-ium;perchlorate](http://img.cochemist.com/ccimg/1062200/1062158-63-7_b.png)






![Thiourea, N'-[3,5-bis(trifluoromethyl)phenyl]-N-cyclohexyl-](/data/chemimg/108000/454203-57-7.png)
![Thiourea, N'-[3,5-bis(trifluoromethyl)phenyl]-N-cyclohexyl-](/data/chemimg/108000/454203-57-7_b.png)













![Tris[N,N-bis(trimethylsilyl)amide]yttrium (III)](http://img.cochemist.com/ccimg/41900/41836-28-6.png)
![Tris[N,N-bis(trimethylsilyl)amide]yttrium (III)](http://img.cochemist.com/ccimg/41900/41836-28-6_b.png)















![Phenol, 4-methyl-2-[[(2-piperidinylmethyl)imino]methyl]-6-tricyclo[3.3.1.13,7]dec-1-yl-](/data/chemimg/3769300/1442078-90-1.png)
![Phenol, 4-methyl-2-[[(2-piperidinylmethyl)imino]methyl]-6-tricyclo[3.3.1.13,7]dec-1-yl-](/data/chemimg/3769300/1442078-90-1_b.png)







![1H-IMIDAZOLIUM, 1-[3-(ETHOXYDIHYDROXYSILYL)PROPYL]-3-METHYL-, CHLORIDE](http://img.cochemist.com/ccimg/887300/887276-31-5.png)
![1H-IMIDAZOLIUM, 1-[3-(ETHOXYDIHYDROXYSILYL)PROPYL]-3-METHYL-, CHLORIDE](http://img.cochemist.com/ccimg/887300/887276-31-5_b.png)
![N-[3,5-Bis(trifluoromethyl)phenyl]-N-[(8a,9S)-6-methoxy-9-cinchonanyl]thiourea](/data/chemimg/103500/852913-16-7.png)
![N-[3,5-Bis(trifluoromethyl)phenyl]-N-[(8a,9S)-6-methoxy-9-cinchonanyl]thiourea](/data/chemimg/103500/852913-16-7_b.png)



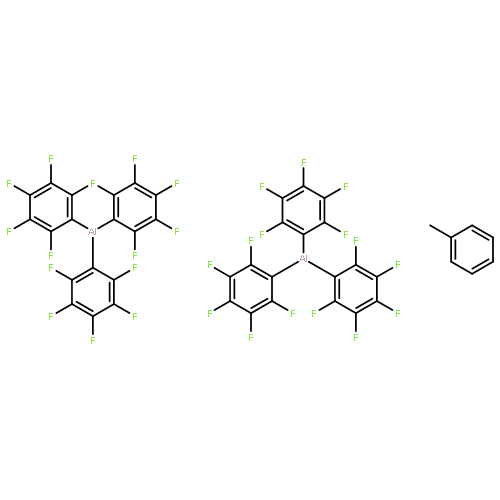
![Phenol, 2,2',2''-[nitrilotris(methylene)]tris[4,6-bis(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/350500/350484-86-5.png)
![Phenol, 2,2',2''-[nitrilotris(methylene)]tris[4,6-bis(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/350500/350484-86-5_b.png)





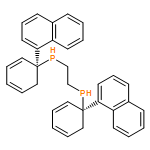
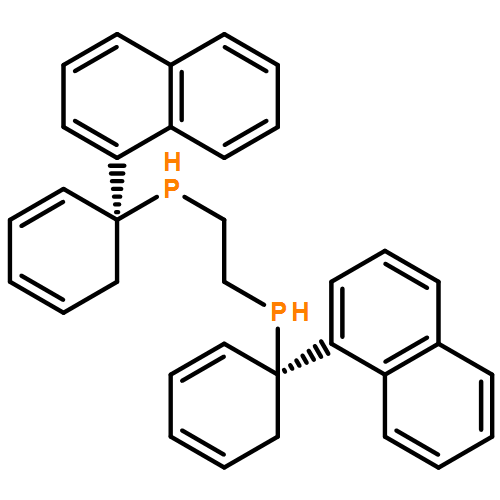
![Benzaldehyde, 2-hydroxy-5-methyl-3-tricyclo[3.3.1.13,7]dec-1-yl-](/data/chemimg/3769300/231963-91-0.png)
![Benzaldehyde, 2-hydroxy-5-methyl-3-tricyclo[3.3.1.13,7]dec-1-yl-](/data/chemimg/3769300/231963-91-0_b.png)
![Titanium,dichloro[h10-2,4-cyclopentadien-1-ylidene(diphenylmethylene)-9H-fluoren-9-ylidene]-(9CI)](http://img.cochemist.com/ccimg/225000/224951-46-6.png)
![Titanium,dichloro[h10-2,4-cyclopentadien-1-ylidene(diphenylmethylene)-9H-fluoren-9-ylidene]-(9CI)](http://img.cochemist.com/ccimg/225000/224951-46-6_b.png)


![Phenol, 2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]-4,6-bis(1,1-dimethylethyl)-](/data/chemimg/135600/210882-24-9.png)
![Phenol, 2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]-4,6-bis(1,1-dimethylethyl)-](/data/chemimg/135600/210882-24-9_b.png)




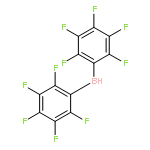
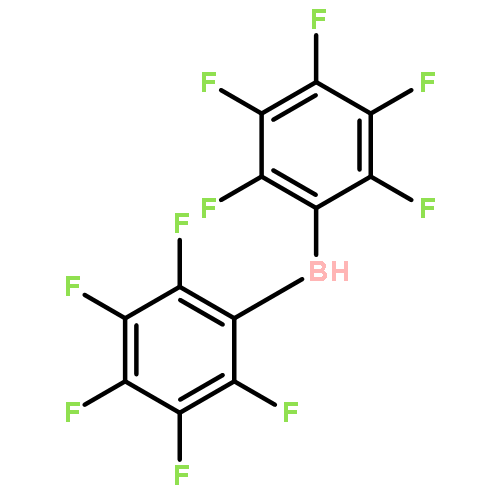

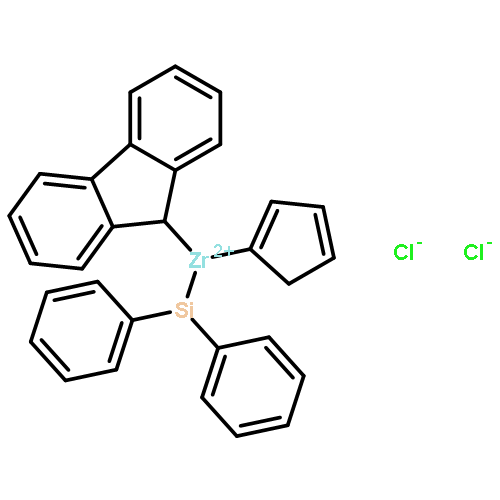









![Benzeneacetic acid, α-[[(1,1-dimethylethoxy)carbonyl]amino]-4-hydroxy-, methyl ester, (αR)-](/data/chemimg/125100/141518-55-0.png)
![Benzeneacetic acid, α-[[(1,1-dimethylethoxy)carbonyl]amino]-4-hydroxy-, methyl ester, (αR)-](/data/chemimg/125100/141518-55-0_b.png)
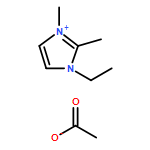
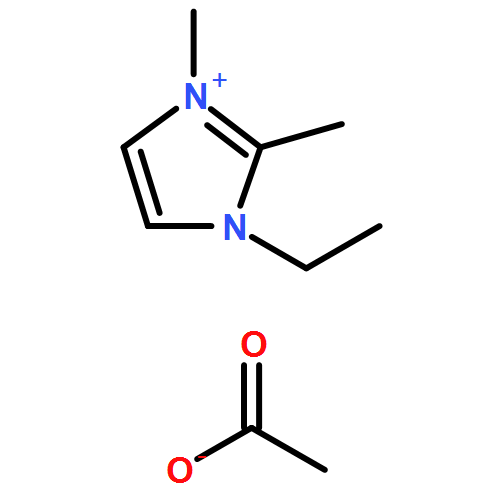






![Poly[oxy(2-methylene-1-oxo-1,4-butanediyl)]](http://img.cochemist.com/ccimg/108200/108150-16-9.png)
![Poly[oxy(2-methylene-1-oxo-1,4-butanediyl)]](http://img.cochemist.com/ccimg/108200/108150-16-9_b.png)

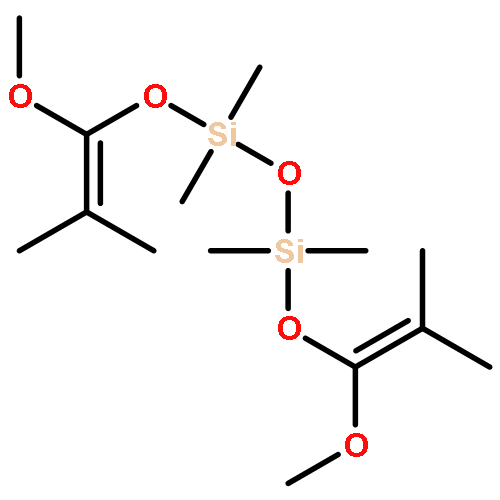
![Erbium Tris[bis(trimethylsilyl)amide]](http://img.cochemist.com/ccimg/103500/103457-72-3.png)
![Erbium Tris[bis(trimethylsilyl)amide]](http://img.cochemist.com/ccimg/103500/103457-72-3_b.png)




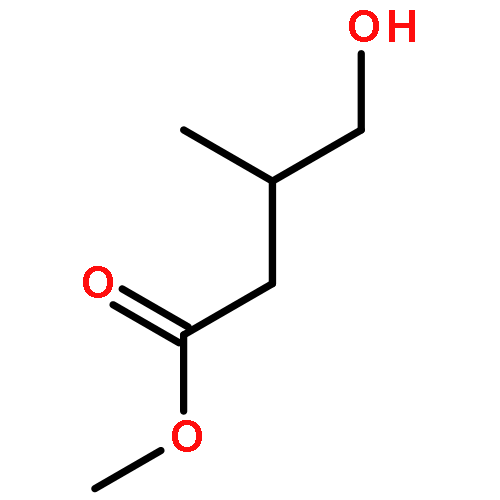
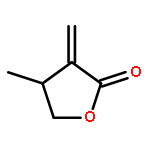
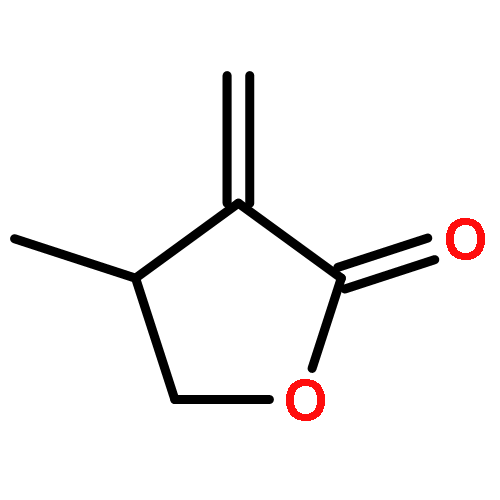

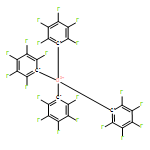
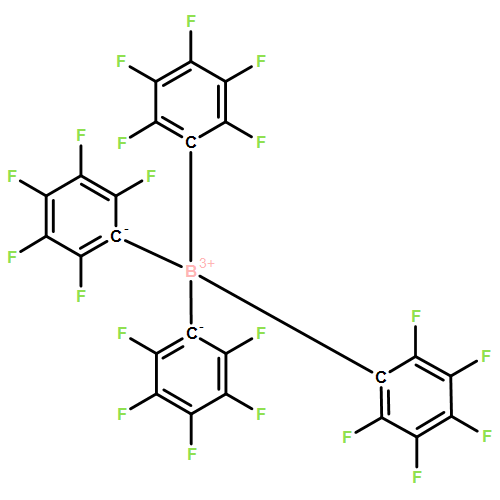


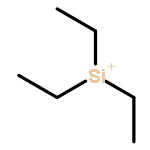
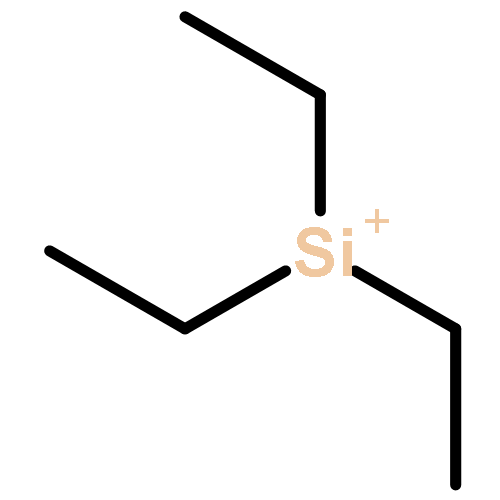


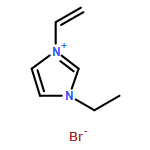
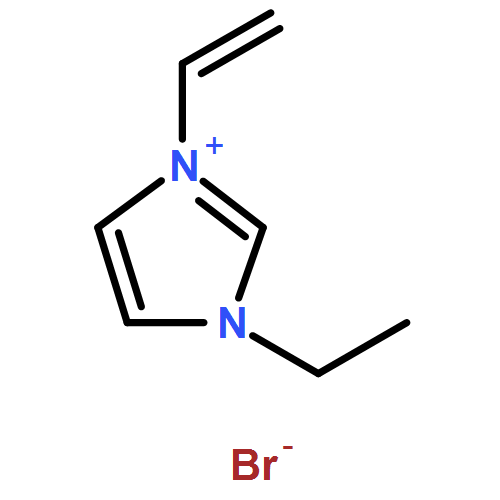




![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2.png)
![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2_b.png)
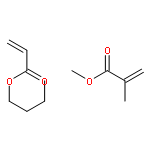
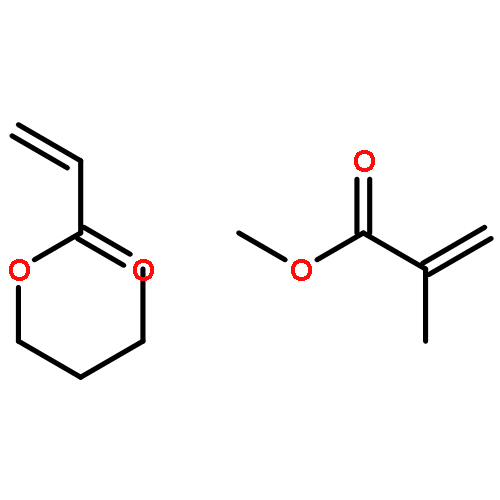



![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4_b.png)












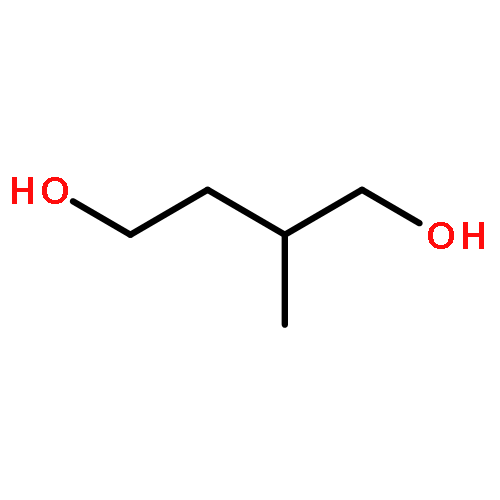







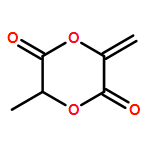

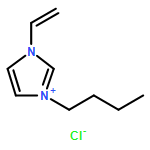
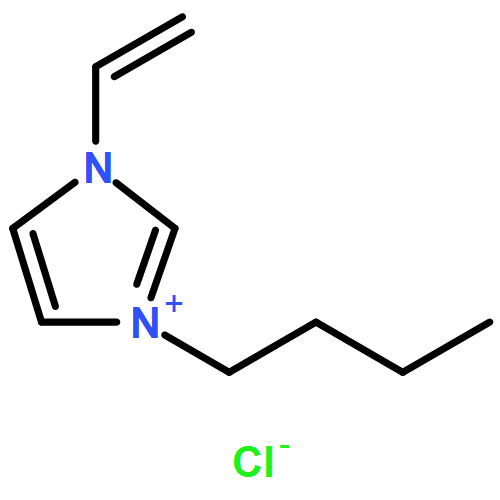

![Lanthanum Tris[bis(trimethylsilyl)amide]](http://img.cochemist.com/ccimg/35800/35788-99-9.png)
![Lanthanum Tris[bis(trimethylsilyl)amide]](http://img.cochemist.com/ccimg/35800/35788-99-9_b.png)