Co-reporter:Tianwen Bai and Jun Ling
The Journal of Physical Chemistry A June 15, 2017 Volume 121(Issue 23) pp:4588-4588
Publication Date(Web):May 19, 2017
DOI:10.1021/acs.jpca.7b04278
The normal amine mechanism via the proton-transfer route (NAM-H) is widely accepted for the synthesis of polypeptides with nonionic initiators. Besides proton transfer, the trimethylsilyl (TMS) group transfer process has been found in living/controlled polymerization initiated by N-TMS amine in experiments, but the corresponding mechanism has never been proposed. In this work, we employed density functional theory (DFT) with the solvation model to investigate the details of the TMS-transfer mechanism, defined as NAM-TMS, for the ring-opening polymerization of α-amino acid N-carboxyanhydride. The TMS transfer process of NAM-TMS is thermodynamically more favored than the NAM-H mechanism according to the lower addition energy barrier observed. The rate-determining step (RDS) in NAM-TMS is the decarboxylation step, i.e., the release of CO2, rather than carbonyl addition in NAM-H because of the low dipole stable precursor enlarged energy gap of decarboxylation. It is the first calculation evidence supporting decarboxylation as RDS in the NAM mechanism.
Co-reporter:Xinfeng Tao, Botuo Zheng, Tianwen Bai, Baoku Zhu, and Jun Ling
Macromolecules April 25, 2017 Volume 50(Issue 8) pp:3066-3066
Publication Date(Web):April 4, 2017
DOI:10.1021/acs.macromol.7b00309
N-Carboxyanhydride (NCA) polymerization cannot tolerate nucleophilic groups that have the ability of initiation, e.g., hydroxyl group. In contrast, N-thiocarboxyanhydride (NTA) is a much more stable monomer to tolerate them. In this contribution, we investigate aminoalcohols including 2-amino-1-ethanol (AE), 3-amino-1-propanol (AP), 4-aminomethylbenzyl alcohol (AMB), 6-amino-1-hexanol (AH), and 12-amino-1-dodecanol (AD) as initiators for ring-opening polymerization of N-substituted glycine N-thiocarboxyanhydride (NNTA) to prepare α-hydroxyl-ω-aminotelechelic water-soluble polypeptoids. Hydroxyl groups of AE, AP, and AMB are activated by hydrogen bonding with amino groups, which results in a mixture of α,ω-diaminotelechelic and α-hydroxyl-ω-aminotelechelic polypeptoids confirmed by 1H NMR, MALDI-ToF, and SEC measurements. Pure α-hydroxyl-ω-aminotelechelic polypeptoids are synthesized for the first time initiated by AH and AD with controlled molecular weights (1.3–12.4 kg/mol) and low polydispersity indices (<1.30). Hydroxyl groups in AH and AD remain inactive to generate hydrogen bonding due to the long distance from amino groups. Water-soluble polypeptoids with special functional end groups are attractive alternatives of PEG for their nontoxicity and biocompatibility having great potential in biomedical applications.
Co-reporter:Benzhao He, Huifang Su, Tianwen Bai, Yongwei Wu, Shiwu Li, Meng Gao, Rongrong Hu, Zujin Zhao, Anjun Qin, Jun Ling, and Ben Zhong Tang
Journal of the American Chemical Society April 19, 2017 Volume 139(Issue 15) pp:5437-5437
Publication Date(Web):March 29, 2017
DOI:10.1021/jacs.7b00929
Efficient synthesis of poly(enamine)s has been a great challenge because of their poor stability, poor solubility, and low molecular weights. In this work, a spontaneous amino-yne click polymerization for the efficient preparation of poly(enamine)s was established, which could proceed with 100% atom efficiency under very mild conditions without any external catalyst. Through systematic optimization of the reaction conditions, several soluble and thermally stable poly(β-aminoacrylate)s with high molecular weights (Mw up to 64400) and well-defined structures were obtained in excellent yields (up to 99%). Moreover, the polymerization can perform in a regio- and stereospecific fashion. Nuclear magnetic resonance spectra analysis revealed that solely anti-Markovnikov additive products with 100% E-isomer were obtained. The reaction mechanism was well demonstrated with the assistance of density functional theory calculations. In addition, by introducing the tetraphenylethene moiety, the resulting polymers exhibit unique aggregation-induced emission characteristics and could be applied in explosives detection and bioimaging. This polyhydroamination is a new type of click polymerization and opens up enormous opportunities for preparing functional polymeric materials.
Co-reporter:Benzhao He, Huifang Su, Tianwen Bai, Yongwei Wu, Shiwu Li, Meng Gao, Rongrong Hu, Zujin Zhao, Anjun Qin, Jun Ling, and Ben Zhong Tang
Journal of the American Chemical Society April 19, 2017 Volume 139(Issue 15) pp:5437-5437
Publication Date(Web):March 29, 2017
DOI:10.1021/jacs.7b00929
Efficient synthesis of poly(enamine)s has been a great challenge because of their poor stability, poor solubility, and low molecular weights. In this work, a spontaneous amino-yne click polymerization for the efficient preparation of poly(enamine)s was established, which could proceed with 100% atom efficiency under very mild conditions without any external catalyst. Through systematic optimization of the reaction conditions, several soluble and thermally stable poly(β-aminoacrylate)s with high molecular weights (Mw up to 64400) and well-defined structures were obtained in excellent yields (up to 99%). Moreover, the polymerization can perform in a regio- and stereospecific fashion. Nuclear magnetic resonance spectra analysis revealed that solely anti-Markovnikov additive products with 100% E-isomer were obtained. The reaction mechanism was well demonstrated with the assistance of density functional theory calculations. In addition, by introducing the tetraphenylethene moiety, the resulting polymers exhibit unique aggregation-induced emission characteristics and could be applied in explosives detection and bioimaging. This polyhydroamination is a new type of click polymerization and opens up enormous opportunities for preparing functional polymeric materials.
Co-reporter:Xia Zhou;Weilin Sun;Zhiquan Shen
Journal of Materials Chemistry A 2017 vol. 5(Issue 20) pp:9717-9722
Publication Date(Web):2017/05/23
DOI:10.1039/C7TA00924K
Nanosheets of coordination polymers (CPs) were synthesized via a facile and one-step complexing-coprecipitation (CC) method. Upon calcination, pure CeO2 and homogeneously La3+- or Cu2+-doped CeO2 products were fabricated by using CPs as precursors. The final products not only retained a stable nano-scale sheet-shaped morphology of their CP precursors, but also possessed high catalytic activity towards CO oxidation. Superior to commercial ceria with negligible catalytic activity, the synthesized CeO2 nanosheets exhibited high activity achieving a CO conversion of 60% at a temperature of 360 °C. When doped with Cu2+, remarkable improvement of catalysis is observed owing to the homogeneous incorporation of Cu2+ into the CeO2 lattice. As a catalytically active center, uniformly dispersed CuO also produces more oxygen vacancies and improves oxygen mobility, which results in an enhancement in catalytic activity. The detected temperatures with 50% CO conversion (T50) for Cu0.1Ce0.9O2−δ and Cu0.04Ce0.96O2−δ are 83 and 95 °C, respectively. In contrast, the catalytic test for La3+ doped ceria reveals a decreased activity compared with un-doped ceria. We believe that the Cu2+-doped CeO2 with superior catalytic performance can also be applied to other catalytic systems.
Co-reporter:
Journal of Polymer Science Part A: Polymer Chemistry 2017 Volume 55(Issue 3) pp:404-410
Publication Date(Web):2017/02/01
DOI:10.1002/pola.28402
ABSTRACTN-Substituted glycine N-thiocarboxyanhydrides (NNTAs) are promising cyclic monomers to synthesize polypeptoids with the advantages of easier preparation and higher stability during purification and storage than N-substituted glycine N-carboxyanhydrides (NNCAs). NNTAs were commonly considered too stable to polymerize for their low reactivity. In this contribution, we report controlled polymerizations of N-ethylglycine NTA (NEG-NTA) and sarcosine NTA (Sar-NTA) using primary amines as initiator under proper polymerization conditions. The controllability has been fully supported by 1H NMR end group analyses, MALDI-ToF mass spectra, kinetic data, block copolymerizations by sequential monomer addition, and low polydispersities (1.14–1.17) of polypeptoids. Variation of the [NNTA]/[initiator] ratio allows well control of the molar mass, and degrees of polymerization (DPs) up to 287 can be reached for poly(N-ethylglycine) or DPs up to 262 for polysarcosine. NNTAs exhibit excellent activity and they are potential to synthesize polypeptoids with controllable polymerization. © 2016 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2017, 55, 404–410
Co-reporter:Huan Qiu, Zhening Yang, Muhammad Ijaz Shah, Zhengwei Mao, Jun Ling
Polymer 2017 Volume 128(Volume 128) pp:
Publication Date(Web):16 October 2017
DOI:10.1016/j.polymer.2017.08.040
•High molecular weight multiblock copolymers (MBCPs) are synthesized by a one-step methodology named Janus polymerization.•The well-defined MBCPs exhibit excellent thermoplastic elastic property in tensile tests.•Different microphase separation behaviors are observed by AFM responding to composition and heating histories.Multiblock copolymers (MBCPs) composing of poly(ε-caprolactone) (PCL) and poly(tetrahydrofuran-co-ε-caprolactone) (P(THF-co-CL)) segments are synthesized in a one-step process by “Janus polymerization”, a newly raised synthetic protocol. Janus polymerization includes an anionic and a cationic ring-opening polymerization on both ends of a single living polymer chain, and finally couples into MBCPs. The well-defined MBCPs have relatively high molecular weights up to 131 kg/mol and exhibit thermoplastic elastic property with excellent elongation of 2610% in tensile tests. The repeating blocks form phase separation structures observed by AFM responsible for the elasticity. The biodegradable elastomers have negligible cytotoxicity. According to the physiochemical properties observed in this work, such multiblock elastic poly(ether-ester)s are promising materials in many fields.Download high-res image (339KB)Download full-size image
Co-reporter:Yifei Wang;Zhicheng Zheng;Zhengdong Huang
Polymer Chemistry (2010-Present) 2017 vol. 8(Issue 17) pp:2659-2665
Publication Date(Web):2017/04/27
DOI:10.1039/C7PY00167C
We report a new synthetic route of a polymer backbone (polyCTA) containing chain transfer agent (CTA) pendent groups with a theoretical degree of polymerization (DP) up to 1200 and narrow polydispersity through reversible addition–fragmentation chain transfer (RAFT) polymerization of 4-vinylbenzyl acetate (VBA) followed by three modification steps. Well-defined cylindrical polymer brushes (CPBs) with polystyrene side chains are synthesized from the polyCTA via “CTA-shuttled” RAFT polymerization to prove the feasibility. Furthermore, various post-functionalizations on CPBs are investigated to demonstrate the potential of building complex architectures. CPBs with polystyrene-b-poly(N-isopropylacrylamide) side chains, amine-terminated polystyrene side chains and polystyrene-b-poly(ethylene glycol) side chains are synthesized successfully by the direct chain extension of N-isopropylacrylamide, one-pot aminolysis and Michael addition using acrylates containing a tert-butoxy carbonyl (Boc) protected amine group or mono-methyl poly(ethylene glycol), respectively. The strategy of “CTA-shuttled” R-group approach RAFT polymerization along with post-functionalization exhibits versatility in preparing tailor-made CPBs.
Co-reporter:Xia Zhou;Weilin Sun;Zhiquan Shen
Journal of Materials Chemistry A 2017 vol. 5(Issue 20) pp:9717-9722
Publication Date(Web):2017/05/23
DOI:10.1039/C7TA00924K
Nanosheets of coordination polymers (CPs) were synthesized via a facile and one-step complexing-coprecipitation (CC) method. Upon calcination, pure CeO2 and homogeneously La3+- or Cu2+-doped CeO2 products were fabricated by using CPs as precursors. The final products not only retained a stable nano-scale sheet-shaped morphology of their CP precursors, but also possessed high catalytic activity towards CO oxidation. Superior to commercial ceria with negligible catalytic activity, the synthesized CeO2 nanosheets exhibited high activity achieving a CO conversion of 60% at a temperature of 360 °C. When doped with Cu2+, remarkable improvement of catalysis is observed owing to the homogeneous incorporation of Cu2+ into the CeO2 lattice. As a catalytically active center, uniformly dispersed CuO also produces more oxygen vacancies and improves oxygen mobility, which results in an enhancement in catalytic activity. The detected temperatures with 50% CO conversion (T50) for Cu0.1Ce0.9O2−δ and Cu0.04Ce0.96O2−δ are 83 and 95 °C, respectively. In contrast, the catalytic test for La3+ doped ceria reveals a decreased activity compared with un-doped ceria. We believe that the Cu2+-doped CeO2 with superior catalytic performance can also be applied to other catalytic systems.
Co-reporter:Ming-jian Jiang;Xin-yuan Li;Lei-tao Sun
Chinese Journal of Polymer Science 2017 Volume 35( Issue 9) pp:1097-1109
Publication Date(Web):19 July 2017
DOI:10.1007/s10118-017-1961-2
A diquinoxalinopyrene (DQP) derivative and the corresponding oligomers (PDQPs) with Mn of ca. 3300, 4200, 5600, and 7300 have been synthesized. It is found that the band gaps and highest occupied molecular orbital (HOMO)/lowest unoccupied molecular orbital (LUMO) levels between DQP and its homopolymers only have a slight difference, implying a limited conjugation extension achieved from unimer to heptamer. The DFT calculation is in accordance with the experimental results. It reveals that the nodal planes are across the 2,11-positions in the LUMO and/or HOMO of DQP, dimer and trimer, which can be used to explain this special phenomenon.
Co-reporter:Zhening Yang, Zhengwei Mao and Jun Ling
Polymer Chemistry 2016 vol. 7(Issue 3) pp:519-522
Publication Date(Web):23 Nov 2015
DOI:10.1039/C5PY01681A
Non-ionic water-soluble poly-DL-serine (PSer) was synthesized from an activated urethane-type derivative of serine. SEC, NMR and MALDI-ToF analyses of the polypeptides revealed a controllable polymerization initiated by primary or secondary amines. PSer exhibited excellent solubility in water when the ratios of L-serine/D-serine units ranged from 5/5 to 8/2.
Co-reporter:Chao Deng
Macromolecular Rapid Communications 2016 Volume 37( Issue 16) pp:1352-1356
Publication Date(Web):
DOI:10.1002/marc.201600188
Co-reporter:Ying Chen, Zhengqing Xu, Difeng Zhu, Xinfeng Tao, Yuqian Gao, Hong Zhu, Zhengwei Mao, Jun Ling
Journal of Colloid and Interface Science 2016 Volume 483() pp:201-210
Publication Date(Web):1 December 2016
DOI:10.1016/j.jcis.2016.08.038
Polysarcosine (PS), a non-ionic hydrophilic polypeptoid whose structure is similar to polypeptides, bearing repeating units of natural α-amino acid, has been used to stabilize gold nanoparticles (AuNPs) due to its excellent hydrophilicity and biocompatibility. Disulfide functionalized polysarcosines with different molecular weight were synthesized and used to cap AuNPs by traditional ligand exchange. The grafting of PS on AuNPs was evidenced by Fourier transform infrared (FTIR) spectroscopy and the alternation of surface zeta potential. The polysarcosine coated AuNPs (Au@PS) showed good stabilities in wide pH range and saline condition. They had strong resistance to ligand competition of dithiothreitol (DTT). They showed good stability in serum, with a molecular weight dependent interaction pattern with proteins. The Au@PS had very low cytotoxicity and cell uptake in vitro. Based on the results in vitro, polysarcosine with molecular weight of 5 kD with the best ability to stabilize AuNPs was used for in vivo test. The Au@PS had a longer circulation time in blood after intravenous injection than that of Au@PEG, indicating a better stealth-like property of polysarcosine. The Au@PS did not cause obvious toxicity in vivo, suggesting potential applications in disease diagnosis and therapy.Polysarcosine stabilized gold nanoparticles have high colloidal stability, low cell recognition and uptake in vitro, long circulation time and low toxicity in vivo, suggesting potential applications of polysarcosine as a nature inspired coating of nanomaterials in biomedical field.
Co-reporter:Xia Zhou, Jun Ling, Weilin Sun and Zhiquan Shen
Dalton Transactions 2016 vol. 45(Issue 23) pp:9398-9401
Publication Date(Web):11 May 2016
DOI:10.1039/C6DT01271J
Novel nanoparticles of coordination polymers (CPs) with various morphologies are successfully prepared. The obtained products can be well-dispersed to make films on glass substrates by the colloidal deposition method and introduced into methyl cellulose to produce transparent and luminescent films.
Co-reporter:Jun Ling;Xiaoqing Wang;Lixin You ;Zhiquan Shen
Journal of Polymer Science Part A: Polymer Chemistry 2016 Volume 54( Issue 18) pp:
Publication Date(Web):
DOI:10.1002/pola.28187
ABSTRACT
Novel thermoplastic elastomers based on multi-block copolymers of poly(l-lysine) (PLL), poly(N-ε-carbobenzyloxyl-l-lysine) (PZLL), poly(ε-caprolactone) (PCL), and poly(ethylene glycol) (PEG) were synthesized by combination of ring-opening polymerization (ROP) and chain extension via l-lysine diisocyanate (LDI). SEC and 1H NMR were used to characterize the multi-block copolymers, with number-average molecular weights between 38,900 and 73,400 g/mol. Multi-block copolymers were proved to be good thermoplastic elastomers with Young's modulus between 5 and 60 MPa and tensile strain up to 1300%. The PLL-containing multi-block copolymers were electrospun into non-woven mats that exhibited high surface hydrophilicity and wettability. The polypeptide–polyester materials were biocompatible, bio-based and environment-friendly for promising wide applications. © 2016 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2016, 54, 3012–3018
Co-reporter:Chao Deng, Zhening Yang, Zhicheng Zheng, Na Liu and Jun Ling
Journal of Materials Chemistry A 2015 vol. 3(Issue 15) pp:3666-3675
Publication Date(Web):24 Feb 2015
DOI:10.1039/C5TC00318K
We report a novel fluorescent polyfluorene-containing methacrylate macromonomer (PFMA) and its well-defined copolymers with 2-(dimethylamino)ethyl methacrylate (DMAEMA) [P(PFMA-r-DMAEMA)s] via reversible addition–fragmentation chain transfer (RAFT) polymerization. These copolymers self-assemble into photoluminescent nanoparticles in aqueous solutions and act as an excellent scaffold for the incorporation of various pigments to emit tunable colors, including white, through Förster resonance energy transfer with high quantum yield up to 0.80. The directly cast films of these NPs after evaporation of water exhibit strong colorful emission on both smooth quartz and rough plaster surfaces. The NP aqueous solutions are ready for ink-jet printing to produce exquisite fluorescent pictures.
Co-reporter:Xinfeng Tao, Jianwei Du, Youxiang Wang and Jun Ling
Polymer Chemistry 2015 vol. 6(Issue 16) pp:3164-3174
Publication Date(Web):05 Mar 2015
DOI:10.1039/C5PY00191A
N-substituted glycine N-thiocarboxyanhydrides (NTAs) are alternative monomers for preparing polypeptoids. With an easy synthetic approach and stability during purification and storage, they have much more potential for mass production than the corresponding N-carboxyanhydrides (NCAs). Thermoresponsive copolypeptoids are synthesized by copolymerization of sarcosine NTA (Sar-NTA) with N-butylglycine NTA (NBG-NTA) initiated by benzylamine in THF at 60 °C. Polypeptoids with a degree of polymerization over 150 are obtained for the first time through primary amine-initiated NTA polymerizations. The molecular weights (MWs) and compositions of poly(sarcosine-r-N-butylglycine)s [P(Sar-r-NBG)s] are controlled by the feed molar ratios of [Sar]/[NBG]/[benzylamine]. The thermal behaviors of the copolypeptoids are investigated. Reactivity ratios of Sar-NTA and NBG-NTA are determined as 1.70(7) and 0.63(7), indicating a random distribution of the two monomers in the polypeptoid products. The structure and precise amino chain end of P(Sar-r-NBG) are confirmed by MALDI-ToF mass analysis. P(Sar-r-NBG)s have lower critical solution temperatures (LCST) and exhibit reversible phase transitions in aqueous solution. Their cloud point temperatures (Tcps) are tunable between 27 and 71 °C by adjusting the sarcosine molar fraction in copolymers. In addition, Tcp transitions depend on the MWs and the concentrations of the polypeptoids, as well as salt additives, to a certain degree. A biocompatibility study on P(Sar-r-NBG) reveals a controlled cytotoxicity related to the composition of polypeptoids. Easily accessible from NTA polymerizations, polypeptoids are therefore novel degradable materials with LCST for biomedical applications.
Co-reporter:Yifei Wang and Jun Ling
RSC Advances 2015 vol. 5(Issue 24) pp:18546-18553
Publication Date(Web):06 Feb 2015
DOI:10.1039/C4RA17094F
We report a new protocol to synthesize conjugates of polypeptides with vinyl polymers prepared by reversible addition–fragmentation chain transfer (RAFT) polymerization. Well-defined polystyrene-b-poly(γ-benzyl L-glutamate) (PS-b-PBLG) diblock copolymers are synthesized as a typical example of such conjugates. Polystyrene is prepared by RAFT polymerization using conventional and commonly used 2-cyano-2-propyl dodecyl trithiocarbonate as a RAFT chain transfer agent bearing an inert R group. Amine functionalization is carried out by a one-pot process combining aminolysis with the in situ thiol capping reaction by acrylate or methanethiosulfonate containing a tert-butoxy carbonyl (Boc) protected amine group. After deprotection of the Boc group, the released amino group initiates ring-opening polymerization of γ-benzyl L-glutamate N-carboxyanhydride (BLG NCA) to generate the desired block copolymers. Hydrolysis of the benzyl group affords the corresponding polystyrene-b-poly(L-glutamic acid) (PS-b-PGlu) amphiphilic block copolymers which self-assemble into spherical micelles in aqueous media. Circular dichroism (CD) spectra show secondary structural changes of PGlu in different pH conditions. When methanethiosulfonate is used as the capping agent, polystyrene and PBLG segments are conjugated by a reduction-responsive disulfide linkage, which is confirmed by size-exclusion chromatography and destruction of the micelle after treating with dithiothreitol (DTT). This protocol is a simple way to synthesize diblock copolymers of amino acids with vinyl monomers without specific design of RAFT CTA, which is practical for various polymer conjugates with tunable properties.
Co-reporter:Fangyi Cao, Zheng Yuan, Junhua Liu and Jun Ling
RSC Advances 2015 vol. 5(Issue 124) pp:102535-102541
Publication Date(Web):23 Nov 2015
DOI:10.1039/C5RA19710D
A novel Eu3+ complex coordinated by polymers with β-diketone pendant groups (Eu3+–PDKMA) has been synthesized for Cu2+ cation and acid–base vapor detections. Its sensitive and selective response to Cu2+ is confirmed by fluorescence quenching of characteristic emission of Eu3+ at 612 nm wavelength. The fluorescence intensity of Eu3+–PDKMA obeys linear relationship at the presence of Cu2+ with the concentration in the order of 10−7 mol L−1. The detection limit of Cu2+ is as low as 2.0 × 10−8 mol L−1 at natural pH, which makes Eu3+–PDKMA an excellent luminescent sensor. Portable strips containing Eu3+–PDKMA can be easily prepared to detect Cu2+ in aqueous solution. Moreover, the same strips can also be used to recognize and sense acidic and basic environments by naked eye. The red emission can be switched “off” and “on” when the strips contact acid and base vapors, respectively. The produced strips exhibit relatively fast response time less than 10 s, and good reusability over 10 times. The Eu3+–PDKMA materials are promising in biological and environmental applications.
Co-reporter:Ao-kai Zhang 凌君;Yu-wei Sun
Chinese Journal of Polymer Science 2015 Volume 33( Issue 5) pp:721-731
Publication Date(Web):2015 May
DOI:10.1007/s10118-015-1620-4
Three-dimensional (3-D) coarse-grained Monte Carlo algorithms were used to simulate the conformations of swollen hydrogels formed by copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC). The simulation consists of three successive steps including diffusion, cross-linking and relaxation. The cross-linking of multifunctional reaction sites is simulated instantly followed by fast crosslinking. In order to explore the validity of this approach pristine poly(ethylene glycol) (PEG) hydrogels with tri- and tetra-functional reaction sites (G3 and G4 respectively) were prepared and characterized. The data from the simulations were found to be in good agreement with experimental results such as PEG lengths between crosslinks, pore volume and pore radius distribution, indicating the validity of the modeling algorithm. The calculated PEG lengths in G3 and G4 networks are close (≈ 4.6 nm). The 3-D visual topological structure of the hydrogel network suggests that the “ideal” hydrogel is far from cubic, diamond or any well defined structures of regular repeating cells.
Co-reporter:Junhua Liu and Jun Ling
The Journal of Physical Chemistry A 2015 Volume 119(Issue 27) pp:7070-7074
Publication Date(Web):June 18, 2015
DOI:10.1021/acs.jpca.5b04654
Is decarboxylation the rate-determining step of amine-mediated ring-opening polymerizations (ROPs) of α-amino acid N-carboxyanhydride (NCA) and N-substituted glycine N-carboxyanhydride (NNCA)? Can a secondary amine initiate living a ROP of NCA to produce primary amine as an active chain end? We investigate the normal amine mechanism (NAM) of polymerizations of l-alanine-NCA (Ala-NCA) and sarcosine-NCA (Sar-NCA) principally by means of density functional theory (DFT) computation. Detailed Gibbs free energy profiles of elemental reactions in NAM of Ala-NCA and Sar-NCA ROPs have been successfully obtained. In addition, the reaction pathways are checked by DFT, Møller–Plesset perturbation theory, and coupled cluster theory. The rate-determining steps in all ROPs of NCA and NNCA initiated by either secondary or primary amine are amine addition on a 5-carbonyl group rather than decarboxylation. The energy barrier in the case of a secondary amine is lower than that of a primary amine with quite a small difference, which confirms that the secondary amine can initiate the ROPs of NCA and NNCA as fast as, if not faster than, a primary amine does, which supports the experimental results. All the results obtained from various calculation methods show that the reactivity ratios (r) of Ala-NCA and Sar-NCA as well as the product of r1 × r2 are close to 1, indicating a random copolymerization of the two different kinds of NCAs.
Co-reporter:Ce Tian 凌君;You-qing Shen
Chinese Journal of Polymer Science 2015 Volume 33( Issue 8) pp:1186-1195
Publication Date(Web):2015 August
DOI:10.1007/s10118-015-1669-0
Polypeptides and polypeptoids were widely used as biomedical materials because of their good biocompatibility. In this work we reported a series of pH-responsive copolypeptides and polypeptide-polypeptoid block copolymers, i.e. random copolymers of L-glutamic acid (Glu) with L-leucine (Leu) [poly(Glu-r-Leu)s], as well as their block copolymers with polysarcosine (polySar). Well-defined poly(Glu-r-Leu)s with predictable compositions and molecular weights were synthesized by ring opening polymerization of corresponding N-carboxyanhydride monomers. We investigated the relationship between hydrophilicity-hydrophobicity transition and copolymer composition. With increasing Leu fraction, both the pH value of cloud point and the micellar size increased. Poly(Glu-r-Leu) with 60% Leu exhibited a cloud point at the pH of 5.0 to 6.0 the same as that in endosome and lysosome. Poly(Glu-r-Leu)-b-polySars assembled in phosphate buffer and performed pH-responsive morphology change from orbicular micelles at high pH to worm-like micelles at low pH. They were potential pH-responsive carriers for drug and gene delivery to enhance cargo release in cellules.
Co-reporter:Yangwei Deng, Tao Zou, Xinfeng Tao, Vincent Semetey, Sylvain Trepout, Sergio Marco, Jun Ling, and Min-Hui Li
Biomacromolecules 2015 Volume 16(Issue 10) pp:
Publication Date(Web):September 21, 2015
DOI:10.1021/acs.biomac.5b00930
Biocompatible amphiphilic block copolymers composed of polysarcosine (PSar) and poly(ε-caprolactone) (PCL) were synthesized using ring-opening polymerization of sarcosine N-thiocarboxyanhydride initiated by oxyamine-ended PCL and characterized by NMR, SEC, and DSC. Self-assembling of two triblock copolymers PSar8-b-PCL28-b-PSar8 (CS7) and PSar16-b-PCL40-b-PSar16 (CS10) in dilute solution was studied in detail toward polymersome formation using thin-film hydration and nanoprecipitation techniques. A few giant vesicles were obtained by thin-film hydration from both copolymers and visualized by confocal laser scanning microscope. Unilamellar sheets and nanofibers (with 8–10 nm thickness or diameter) were obtained by nanoprecipitation at room temperature and observed by Cryo-TEM. These lamellae and fibrous structures were transformed into worm-like cylinders and spheres (D ∼ 30–100 nm) after heating to 65 °C (>Tm,PCL). Heating CS10 suspensions to 90 °C led eventually to multilamellar polymersomes (D ∼ 100–500 nm). Mechanism II, where micelles expand to vesicles through water diffusion and hydrophilic core forming, was proposed for polymersome formation. A cell viability test confirmed the self-assemblies were not cytotoxic.
Co-reporter:Xinfeng Tao;Chao Deng
Macromolecular Rapid Communications 2014 Volume 35( Issue 9) pp:875-881
Publication Date(Web):
DOI:10.1002/marc.201400066
Co-reporter:Jun Ling;Zhicheng Zheng;Anna Köhler;Axel H. E. Müller
Macromolecular Rapid Communications 2014 Volume 35( Issue 1) pp:52-55
Publication Date(Web):
DOI:10.1002/marc.201300785
Co-reporter:Zhicheng Zheng;Axel H. E. Müller
Macromolecular Rapid Communications 2014 Volume 35( Issue 2) pp:234-241
Publication Date(Web):
DOI:10.1002/marc.201300578
Co-reporter:Lixin You, Zhiquan Shen, Jie Kong, Jun Ling
Polymer 2014 Volume 55(Issue 10) pp:2404-2410
Publication Date(Web):13 May 2014
DOI:10.1016/j.polymer.2014.03.032
A series of rare earth triflates (RE(OTf)3, RE = Sc, Y and Lu) were used for the first time as moisture-stable precursors to generate rare earth alkoxide complexes through an in situ reaction with sodium alkoxides (NaOR) in tetrahydrofuran. 1H NMR and 13C NMR results confirmed the fast ligands exchange process and the formation of rare earth–oxygen (RE–OR) bond. The in situ formed catalysts displayed high reactivity toward living ring-opening polymerization (ROP) of ε-caprolactone (CL). For instance, Lu(OTf)3/sodium isopropoxide (NaOiPr)-catalyzed ROP of CL with the [CL]0/[NaOiPr]0/[Lu(OTf)3]0 feeding ratio of 300/3/1 produced poly(ε-caprolactone) (PCL) with controlled molecular weight (Mn,exp = 11.9 kDa vs Mn,theo = 11.8 kDa) and narrow polydispersity (PDI) of 1.08 within 3 min at 25 °C. The kinetic studies and chain extension confirmed the controlled/living nature for the Lu(OTf)3/NaOiPr-catalyzed ROP of CL. In addition, end-functionalized PCLs bearing vinyl or alkynyl group with narrow PDIs were obtained by using functional sodium alkoxides in the presence of Lu(OTf)3. 1H NMR and MALDI-ToF MS analyses of the obtained PCLs clearly indicated the presence of the residue of OR groups at the chain ends. A coordination–insertion polymerization mechanism was proposed including a fast ligand exchange between Lu(OTf)3 and NaOR giving the respective lutetium alkoxide complexes, and a CL insertion into RE–OR bond via acyl-oxygen cleavage.
Co-reporter:Lixin You and Jun Ling
Macromolecules 2014 Volume 47(Issue 7) pp:2219-2225
Publication Date(Web):March 17, 2014
DOI:10.1021/ma500173c
Is it possible to combine cationic and (coordinated) anionic ring-opening polymerizations (CROP and AROP) into a unimolecular chain? In this work, we design polymerizations of ε-caprolactone (CL) and tetrahydrofuran (THF) using a single catalytic system of lutetium triflate/propylene oxide (Lu(OTf)3/PO) in which a cationic copolymerization of THF with CL and an AROP of CL perform at two chain ends of a growing chain. Kinetic and mechanistic studies demonstrate that the two chain-growth ROPs which have no mutual interference up to 90% of CL conversion keep a living manner producing diblock copolymers of PCL-b-P(THF-co-CL) with narrow polydispersities below 1.2. Afterward, the depletion of CL triggers a final step-growth polymerization via intermolecular coupling reaction of cationic and anionic chain ends, developing multiblock polyester–polyether elastomers of [PCL-b-P(THF-co-CL)]m. A concept of “Janus polymerization” is defined as a combination of cationic and anionic polymerizations on the two ends of a single growing chain, involving an initially living chain-growth polymerization and a subsequently self-triggered step-growth polycondensation.
Co-reporter:Xinfeng Tao, Yangwei Deng, Zhiquan Shen, and Jun Ling
Macromolecules 2014 Volume 47(Issue 18) pp:6173-6180
Publication Date(Web):September 10, 2014
DOI:10.1021/ma501131t
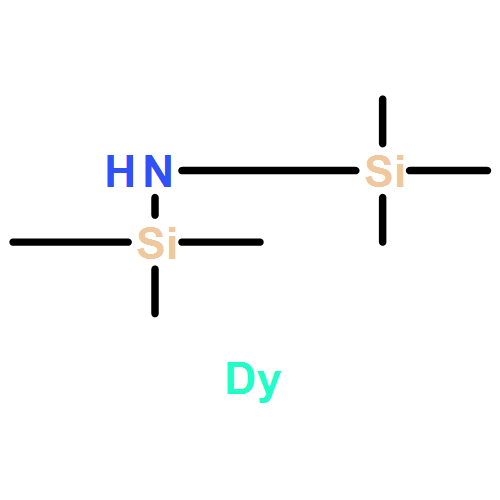
N-substituted glycine N-thiocarboxyanhydrides (NTAs) are alternative monomers to prepare polypeptoids with large-scale producing potential compared to the corresponding N-carboxyanhydrides (NCAs) due to their easily synthetic approach and stability during purification and storage. Novel monomer N-butylglycine NTA (NBG-NTA) has been synthesized and well characterized for the first time. Rare earth borohydrides [RE(BH4)3(THF)3, RE = Sc, Y, La, Nd, Dy, and Lu] have been first applied in the polymerization of sarcosine NTA (Sar-NTA) and NBG-NTA to achieve high molecular weight (MW) hydrophilic and hydrophobic polypeptoids. Polysarcosines (PSars), poly(N-butylglycine)s (PNBGs), and their copolymers with high yields, high MWs, and moderate MW distributions are synthesized at 60 °C by using RE(BH4)3(THF)3 initiators. MWs of polypeptoids are controlled by feed molar ratios. For instance, PSar with an absolute Mn of 27.7 kDa (DP = 390) and PDI of 1.14 is produced successfully from Sar-NTA. Thermoresponsive random copolypeptoids poly(sarcosine-r-N-butylglycine)s [P(Sar-r-NBG)s] have reversible phase transitions (cloud point temperature) in aqueous solution and minimal cytotoxicity comparable to PEG and PSar, which is promising in various biomedical and biotechnological applications. Thermal properties of homo- and co-polypeptoids are investigated by TGA and DSC measurements.
Co-reporter:Hui Peng;Wan-li Chen;Jie Kong;Zhi-quan Shen
Chinese Journal of Polymer Science 2014 Volume 32( Issue 6) pp:743-750
Publication Date(Web):2014 June
DOI:10.1007/s10118-014-1445-6
It is reported that alkali-metal borohydrides (MBH4, M = Li, Na and K) are efficient catalysts for ring opening polymerization (ROP) of α-amino acid N-carboxyanhydrides (NCAs). Polypeptides are prepared in quantitative yields with relatively narrow molecular weight distributions (MWDs = 1.1∼1.5) which depend on the reaction temperature. End groups of the produced polypeptide are studied in detail by MALDI-ToF MS, 1H-NMR, 13C-NMR, 1H-1H COSY and 1H-13C HMQC analyses. The results indicate that α-hydroxy-ω-aminotelechelic polypeptides are formed which are suitable for postpolymerization functionalization.
Co-reporter:Wei Lu, Ganhong Du, Keyuan Liu, Liming Jiang, Jun Ling, and Zhiquan Shen
The Journal of Physical Chemistry A 2014 Volume 118(Issue 1) pp:283-292
Publication Date(Web):December 16, 2013
DOI:10.1021/jp410370q
We propose a new strategy to construct chiral molecular switches with highly reversible and sensitive chiroptical responses to variations in the external environment. Its fundamental concept involves a stimuli-triggered exchange of two conformations presenting significantly different chiroptical properties through the rotation of a carbon–carbon single bond, as demonstrated by chiral Schiff bases s-1, s-2, and a salicylamide analogue s-3. Upon addition of base in solution, the circular dichroism (CD) spectra of these molecular switches displayed unique changes featuring an inversion of the Cotton effect’s signs, and the original CD profiles can be recoverd by acidification. Various spectroscopic studies as well as the conformational analysis combining with TDDFT computations allowed clear elucidation of the chiroptical inversion mechanism. It is expected that this kind of chiroptical switches is of great interest for molecular recognition, chemosensing, and the construction of molecular-scale devices. Furthermore, the present study indicates that the use of the conformational transition about a single bond may serve as the basis for designing chiroptical inversion systems.
Co-reporter:Zhicheng Zheng, Alexander Daniel, Wei Yu, Birgit Weber, Jun Ling, and Axel H. E. Müller
Chemistry of Materials 2013 Volume 25(Issue 22) pp:4585
Publication Date(Web):October 21, 2013
DOI:10.1021/cm402776d
A novel template-directed approach based on core–shell cylindrical polymer brushes (CPBs) has been developed to prepare rare-earth metal cations (Ln3+) incorporated silica hybrid nanoparticles (NPs) with predictable dimensions. Tight chelation of Ln3+ ions in the core of the CPB template and a cross-linked silica layer deposited on the shell provide a very stable encapsulation of Ln3+ ions within the hybrid NPs and thus a high biocompatibility. As expected, the silica hybrid NPs obtain unique and diverse properties from the incorporated Ln3+ ions. That is, the hybrid NPs with Tb3+ or Eu3+ incorporation exhibit characteristic photoluminescence in visible light range, while the Gd3+- and Tb3+-containing hybrid NPs show paramagnetic behavior. Especially, the Gd3+-containing silica hybrid NPs show a remarkable longitudinal relaxation time (T1) shortening effect as well as minimal cytotoxicity, suggesting the application potential of these NPs as effective magnetic resonance imaging (MRI) contrast agents. This novel template-directed approach succeeds in combining different functional centers via loading in situ mixed Ln3+ ions (e.g. Tb3+ and Gd3+) into individual CPBs resulting in multicomponent hybrid NPs, which possess both visible photoluminescence and T1 contrast enhancement and can thus be applied as multimodal bioimaging probes.Keywords: cylindrical polymer brushes; cytotoxicity; MRI contrast agent; paramagnetic; photoluminescence; rare-earth cations; template-directed;
Co-reporter:Zhicheng Zheng, Markus Müllner, Jun Ling, and Axel H. E. Müller
ACS Nano 2013 Volume 7(Issue 3) pp:2284
Publication Date(Web):February 13, 2013
DOI:10.1021/nn3054347
A tapping-mode AFM investigation of core–shell cylindrical polymer brushes (CPBs) on mica shows that they can be ruptured upon spin-coating. Three different CPBs were synthesized, having a methacrylate backbone, carrying branches of poly[oligo(ethylene glycol)methacrylate] (POEGMA), POEGMA-block-poly[2-(dimethylamino)ethyl methacrylate] (POEGMA-b-PDMAEMA), and POEGMA-block-poly[2-(methacryloyloxy)ethyl trimethylammoniumiodide] (POEGMA-b-PMETAI). The polymer backbone of core–shell CPB with POEGMA-b-PDMAEMA or POEGMA-b-PMETAI branches is ruptured upon drying on a mica surface, while they are stable in aqueous solution. We propose that the scission behavior is induced by Coulomb interactions between PDMAEMA or PMETAI corona and the solid surface and that this interaction is stronger than one or more carbon–carbon single bonds. We control this scission behavior by tuning the surface interactions through switching the surface nature, varying pH, or adding multivalent counterions. Our study demonstrates that core–shell CPB serves as a template to directly compare the weak intermolecular forces with the strong carbon–carbon covalent bonds.Keywords: carbon−carbon bond energy; cylindrical polymer brushes; intermolecular attractions; polyelectrolytes; scission behavior; surface charges; surface interaction energy
Co-reporter:Jin-Ou Lin, Wanli Chen, Zhiquan Shen, and Jun Ling
Macromolecules 2013 Volume 46(Issue 19) pp:7769-7776
Publication Date(Web):September 17, 2013
DOI:10.1021/ma401218p
Biobased and environmental-friendly polylactones provide a solution to the increasing crisis of fossil-sourced products. Ring-opening polymerization (ROP) of ε-decalactone (DL) catalyzed by lanthanum tris(2,6-di-tert-butyl-4-methylphenolate) [La(OAr)3] and lanthanum tris(borohydride) [La(BH4)3(THF)3] is reported for the first time in this paper. Both of them exhibit good activities producing poly(ε-decalactone) (PDL) with molecular weight (MW) up to 26.4 kg/mol and polydispersity index (PDI) as low as 1.10. PLLA-b-PDL-b-PEG-b-PDL-b-PLLA pentablock copolymers with predictable MWs and relatively narrow PDIs (1.19–1.28) are synthesized by sequential ROP of DL and l-lactide (LLA) catalyzed by La(OAr)3 in the presence of poly(ethylene glycol) (PEG). Chain extension reactions of the obtained pentablock copolymers are carried out using l-lysine diisocyanate (LDI) to produce multiblock copolymers with relatively high MW. The thermal behaviors studied by DSC and DMA measurements indicate that PDL is completely amorphous under ambient temperature and the copolymers with two Tgs suggest microphase separation of hard and soft domains. We employ tensile tests to assess mechanical properties and find excellent elongation up to 723% of the chain-extended samples. Considering the biorenewable resource of DL and LLA, a novel, biobased, biodegradable, and biocompatible elastomer is successfully synthesized.
Co-reporter:Xin Li;Yinghong Zhu;Zhiquan Shen
Macromolecular Rapid Communications 2012 Volume 33( Issue 11) pp:1008-1013
Publication Date(Web):
DOI:10.1002/marc.201100848
Abstract
Unmodified β-cyclodextrin has been directly used to initiate ring-opening polymerization of ϵ-caprolactone in the presence of yttrium trisphenolate. Well-defined cyclodextrin (CD)-centered star-shaped poly(ϵ-caprolactone)s have been successfully synthesized containing definite average numbers of arms (Narm = 4–6) and narrow polydispersity indexes (below 1.10). The number-average molecular weight () and average molecular weight per arm () are controlled by the feeding molar ratio of monomer to initiator. The prepared star-PCL with of 2.7 × 103 is in fully amorphous and that with of 13.3 × 103 is crystallized. In addition, the obtained poly(e-caprolactone) (PCL) stars with various molecular weights have different solubilities in methanol and tetrahydrofuran, which can be applied for further modifications.
Co-reporter:Jun Ling;Lixin You;Yifei Wang ;Zhiquan Shen
Journal of Applied Polymer Science 2012 Volume 124( Issue 3) pp:2537-2540
Publication Date(Web):
DOI:10.1002/app.34226
Abstract
In this article, we provide a concept of a two-phase polymerization system consisting of immiscible monomer and room temperature ionic liquid (IL). The catalyst is immobilized in the IL phase where polymerization takes place. The produced polymer is extracted by the monomer, and the remaining IL phase is catalytically active for more polymerizations. Thus, common volatile organic solvents are no longer needed. Ring-opening polymerization of cyclohexene oxide (CHO) in 1-n-butyl-3-methylimidazolium tetrafluoroborate IL ([bmim][BF4]) using scandium triflate [Sc(OTf)3] catalyst serves as a realistic example of such concept. The yield of polyCHO in [bmim][BF4] is higher than that in bulk. IL containing Sc(OTf)3 can be used for at least three times. A circulatory polymerization process is carried out with added catalyst to keep a relatively high yield in following circulation processes. The assignments of proton signals of polyCHO in 1H NMR are discussed in detail. © 2011 Wiley Periodicals, Inc. J Appl Polym Sci, 2012
Co-reporter:Lixin You, Thieo E. Hogen-Esch, Yinghong Zhu, Jun Ling, Zhiquan Shen
Polymer 2012 Volume 53(Issue 19) pp:4112-4118
Publication Date(Web):31 August 2012
DOI:10.1016/j.polymer.2012.07.047
We report a series of novel, highly efficient, hydrolytically stable and recyclable Lewis acid rare earth triflate catalysts for the living/controlled bulk ring-opening polymerization (ROP) of tetrahydrofuran (THF) in the presence of epoxides. The rare earth triflates [RE(OTf)3] include that of Sc, Y, La, Nd, Dy and Lu. Epoxides include propylene oxide (PO), cyclohexene oxide (CHO) or styrene oxide (SO). Especially RE(OTf)3/PO shows high activities, producing PTHF with up to 62 percent conversion and relatively narrow molecular weight distributions (MWDs) (as low as 1.14). Molecular weights (MWs) can be readily controlled from 1.7 to 56.1 kDa by changing monomer to catalyst molar ratios. The Sc(OTf)3/PO catalyst gives an α,ω-dihydroxyl telechelic PTHF with MWs close to calculated values and MWDs below 1.2. The rare earth catalysts are easily recovered by simple water extractions. For instance, Sc(OTf)3 can be used at least six times without an obvious decrease of catalytic activity. A polymerization mechanism involving the first two propagation steps of the alkyltetrahydrofuranium cation is proposed to account for the induction periods observed.Graphical abstract
Co-reporter:Dan Chen;Wei Lu;Ganhong Du;Liming Jiang;Zhiquan Shen
Journal of Polymer Science Part A: Polymer Chemistry 2012 Volume 50( Issue 20) pp:4191-4197
Publication Date(Web):
DOI:10.1002/pola.26230
Abstract
A new kind of polymeric chemosensor containing chiral naphthaldimine moiety in the side chain was synthesized by the reversible addition-fragmentation chain transfer polymerization of N-{[2-(4-vinylbenzyloxy)-1-naphthyl]-methylene}-(S)-2-phenylglycinol (VNP). The resulting polymers (PVNP) showed high selectivity for hydrogen sulfate relative to other anions including F−, Cl−, Br−, H2PO, CH3CO, and NO in tetrahydrofuran (THF) solution as judged from UV−vis, fluorescence, and circular dichroism spectrophotometric titrations. Compared with its monomer, the polymer has proven to be more attractive for detection of HSO in terms of sensitivity and reproducibility. Upon addition of the anion it gives remarkable spectral responses concomitant with detectable color change from colorless to pale yellow. Furthermore, the HSO-induced CD or fluorescence signal can be totally reversed with addition of base and eventually recovered the initial state, leading to a reproducible molecular switch with two distinguished “on” and “off” states. © 2012 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2012
Co-reporter:Hui Peng;Zhiquan Shen
Journal of Polymer Science Part A: Polymer Chemistry 2012 Volume 50( Issue 6) pp:1076-1085
Publication Date(Web):
DOI:10.1002/pola.25848
Abstract
Five rare earth complexes are first introduced to catalyze ring opening polymerizations (ROPs) of γ-benzyl-L-glutamate N-carboxyanhydride (BLG NCA) and L-alanine NCA (ALA NCA) including rare earth isopropoxide (RE(OiPr)3), rare earth tris(2,6-di-tert-butyl-4-methylphenolate) (RE(OAr)3), rare earth tris(borohydride) (RE(BH4)3(THF)3), rare earth tris[bis(trimethylsilyl)amide] (RE(NTMS)3), and rare earth trifluoromethanesulfonate. The first four catalysts exhibit high activities in ROPs producing polypeptides with quantitative yields (>90%) and moderate molecular weight (MW) distributions ranging from 1.2 to 1.6. In RE(BH4)3(THF)3 and RE(NTMS)3 catalytic systems, MWs of the produced polypeptides can be controlled by feeding ratios of monomer to catalyst, which is in contrast to the systems of RE(OiPr)3 and RE(OAr)3 with little controllability over the MWs. End groups of the polypeptides are analyzed by MALDI-TOF MS and polymerization mechanisms are proposed accordingly. With ligands of significant steric hindrance in RE(OiPr)3 and RE(OAr)3, deprotonation of 3-NH of NCA is the only initiation mode producing a N-rare earth metallated NCA (i) responsible for further chain growth, resulting in α-carboxylic-ω-aminotelechelic polypeptides after termination. In the case of RE(BH4)3(THF)3 with small ligands, another initiation mode at 5-CO position of NCA takes place simultaneously, resulting in α-hydroxyl-ω-aminotelechelic polypeptides. In RE(NTMS)3 system, the protonated ligand hexamethyldisilazane (HMDS) initiates the polymerization and produces α-amide-ω-aminotelechelic polypeptides. © 2011 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2012
Co-reporter:Hui Peng;Yinghong Zhu;Lixin You;Zhiquan Shen
Journal of Polymer Science Part A: Polymer Chemistry 2012 Volume 50( Issue 15) pp:3016-3029
Publication Date(Web):
DOI:10.1002/pola.26077
Abstract
In this work, rare earth tris(borohydride) complexes, Ln(BH4)3(THF)3 (Ln = Sc, Y, La, and Dy), have been used to catalyze the ring-opening polymerization of γ-benzyl-L-glutamate N-carboxyanhydride (BLG NCA). All the catalysts show high activities and the resulting poly(γ-benzyl-L-glutamate)s (PBLGs) are recovered with high yields (≥90%). The molecular weights (MWs) of PBLG can be controlled by the molar ratios of monomer to catalyst, and the MW distributions (MWDs) are relatively narrow (as low as 1.16) depending on the rare earth metals and reaction temperatures. Block copolypeptides can be easily synthesized by the sequential addition of two monomers. The obtained P(γ-benzyl-L-glutamate-b-ε-carbobenzoxy-L-lysine) [P(BLG-b-BLL)] and P(γ-benzyl-L-glutamate-b-alanine) [P(BLG-b-ALA)] have been well characterized by NMR, gel permeation chromatography, and differential scanning calorimetry measurements. A random copolymer P(BLG-co-BLL) with a narrow MWD of 1.07 has also been synthesized. The polymerization mechanisms have been investigated in detail. The results show that both nucleophilic attack at the 5-CO of NCA and deprotonation of 3-NH of NCA in the initiation process take place simultaneously, resulting in two active centers, that is, an yttrium ALA carbamate derivative [H2BOCH2(CH)NHC(O)OLn] and a N-yttriumlated ALA NCA. Propagation then proceeds on these centers via both normal monomer insertion and polycondensation. After termination, two kinds of telechelic polypeptide chains, that is, α-hydroxyl-ω-aminotelechelic chains and α-carboxylic-ω-aminotelechelic ones, are formed as characterized by MALDI-TOF MS, 1H NMR, 13C NMR, 1H–1H COSY, and 1H–13C HMQC measurements. By decreasing the reaction temperature, the normal monomer insertion pathway can be exclusively selected, forming an unprecedented α-hydroxyl-ω-aminotelechelic polypeptide. © 2012 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2012
Co-reporter:Jun Ling;Hui Peng ;Zhiquan Shen
Journal of Polymer Science Part A: Polymer Chemistry 2012 Volume 50( Issue 18) pp:3743-3749
Publication Date(Web):
DOI:10.1002/pola.26156
Abstract
Reaction of yttrium tris[bis(trimethylsilyl)amide] [(TMSN)3Y] with equivalent L-alanine N-carboxyanhydride (ALA NCA) yields yttrium α-isocyanato carboxylate (II), yttrium ketenyl carbamate (III), and hexamethyldisilazane (V). The products indicate that 4-CH group of ALA NCA monomer is deprotonated in addition to 3-NH group, which has been neglected in NCA chemistry for decades. This result proves the acidity of 4-CH in NCA and provides the first direct evidence for racemization phenomenon of NCA in strong base in microscopic aspect. Rare earth tris[bis(trimethylsilyl)amide] (TMSN)3Ln (Ln = Sc, Y, La, Dy, and Lu) compounds are high efficient catalysts for ring-opening polymerizations of NCAs. Polypeptides can be produced in quantitative yields with narrow molecular weight distributions below 1.3, and block copolypeptides can be facilely prepared by sequential addition method. © 2012 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2012
Co-reporter:Jun Ling;Jinzhi Liu;Zhiquan Shen;Thieo E. Hogen-Esch
Journal of Polymer Science Part A: Polymer Chemistry 2011 Volume 49( Issue 9) pp:2081-2089
Publication Date(Web):
DOI:10.1002/pola.24637
Abstract
Although the ring-opening polymerization (ROP) of ε-caprolactone (CL) in toluene at 100 °C can be initiated by yttrium trisphenolate (Y(OC6H5)3), in the presence of 1,2-propanediol (PD) the ROP gives much better, that is, controlled polymerizations. In this case, the molecular weights (MWs) are controlled by the CL/PD molar ratios with primary and secondary hydroxyl groups both initiating the ROP and the MW distributions are narrow. The chain transfers between the active yttrium alkoxides and the residual hydroxyl groups on the PD and/or the chain ends appear to be much faster than chain propagation, consistent with the living character of the ROP. Computational studies support these facile reactions with estimated activation free energies in the 3.0–4.5 kcal/mol range compared with about 25–30 kcal/mol for the polymerization. Intramolecular transfer within the PD is predicted to be negligible having a calculated activation energy of 19 kcal /mol. © 2011 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2011
Co-reporter:Min-Min Shi, Yi Chen, Ya-Xiong Nan, Jun Ling, Li-Jian Zuo, Wei-Ming Qiu, Mang Wang, and Hong-Zheng Chen
The Journal of Physical Chemistry B 2011 Volume 115(Issue 4) pp:618-623
Publication Date(Web):December 13, 2010
DOI:10.1021/jp109683h
To investigate the relationship between π−π stacking and charge transport property of organic semiconductors, a highly soluble violanthrone derivative, 16,17-bis(2-ethylhexyloxy)anthra[9,1,2-cde-]benzo[rst]pentaphene-5,10-dione (3), is designed and synthesized. The π−π stacking behavior and the aggregation of compound 3 in both solution and thin film were studied in detail by 1H nuclear magnetic resonance (NMR) spectroscopy, ultraviolet−visible (UV−vis) absorption, X-ray diffraction (XRD), and atomic force microscopy (AFM). When 1H NMR spectroscopy and theoretical modeling results were combined, the arrangements of compound 3 molecules in the aggregates are demonstrated, where the dipole moments of the two adjacent molecules are nearly reversed to achieve efficient intermolecular π−π overlapping. Furthermore, it is interesting to find that the π−π stacking of compound 3, in both solution and thin films, can be enhanced by introducing a poor solvent n-hexane into the dilute chloroform solution. The resulting film exhibits more red-shifted absorption and higher crystallinity than the film made from pure chloroform solvent, suggesting that π−π interactions in the solid state are intensified by the poor solvent. Organic field-effect transistors (OFETs) with compound 3 film as the transportation layer were fabricated. It is disclosed that the compound 3 film obtained from the chloroform/n-hexane mixed solvents exhibits 1 order of magnitude higher hole mobility than that from the pure chloroform solvent because of the enhanced π−π interactions and the higher crystallinity in the former film. This work provided us valuable information in the improvement of electronic and optoelectronic performances of organic semiconductors by tuning their aggregate structures.
Co-reporter:Hui Peng, Jun Ling, Jinzhi Liu, Ning Zhu, Xufeng Ni, Zhiquan Shen
Polymer Degradation and Stability 2010 Volume 95(Issue 4) pp:643-650
Publication Date(Web):April 2010
DOI:10.1016/j.polymdegradstab.2009.12.005
Co-reporter:Jun Ling;Yanxin Huang
Macromolecular Chemistry and Physics 2010 Volume 211( Issue 15) pp:
Publication Date(Web):
DOI:10.1002/macp.201090034
Co-reporter:Jun Ling;Yanxin Huang
Macromolecular Chemistry and Physics 2010 Volume 211( Issue 15) pp:1708-1711
Publication Date(Web):
DOI:10.1002/macp.201000115
Co-reporter:Ning Zhu;Yinghong Zhu;Weilin Sun;Zhiquan Shen
Journal of Polymer Science Part A: Polymer Chemistry 2010 Volume 48( Issue 19) pp:4366-4369
Publication Date(Web):
DOI:10.1002/pola.24233
Co-reporter:Jun Ling;Yongbo Dai;Yinghong Zhu;Weilin Sun;Zhiquan Shen
Journal of Polymer Science Part A: Polymer Chemistry 2010 Volume 48( Issue 17) pp:3807-3815
Publication Date(Web):
DOI:10.1002/pola.24166
Abstract
Ring-opening polymerization of 1-methyltrimethylene carbonate (MTMC) initiated by highly active single-component rare earth tris(2,6-di-tert-butyl-4-methylphenolate)s [Ln(OAr)3, Ln = La, Dy, Y] or yttrium isopropoxide [Y(OiPr)3] is reported for the first time. PolyMTMC (Mw = 8.4 × 104, molecular weight distributions = 1.5) initiated by La(OAr)3 at [MTMC]/[initiator] = 1000 was obtained with the yield over 99% in toluene within 1 h at 30 °C. Random and block copolymers of MTMC with ε-caprolactone (CL), 2,2-dimethyltrimethylene carbonate (DTC) or polyethylene glycol (PEG) including poly(MTMC-r-CL), poly(MTMC-b-CL), poly(MTMC-r-DTC), poly(MTMC-b-DTC), and poly(MTMC-b-PEG-b-MTMC) were synthesized. The differential scanning calorimetry results show that thermal behaviors of the polymers sensitively depend on their compositions and chain structures. Furthermore, the measurements of 1H-1H COSY and density functional theory calculation are applied to investigate the mechanism. The polymerization of MTMC takes place according to a coordination-insertion mechanism, and the ring is opened via acyl-oxygen bond cleavage resulting in a LnO active center. There exist two ring-opening modes of MTMC in which mode b, breaking the CH2OCO bond, is the major pathway. © 2010 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 48: 3807–3815, 2010
Co-reporter:Jinzhi Liu, Jun Ling, Xin Li, Zhiquan Shen
Journal of Molecular Catalysis A: Chemical 2009 300(1–2) pp: 59-64
Publication Date(Web):
DOI:10.1016/j.molcata.2008.10.038
Co-reporter:Jun Ling, Jingguo Shen, Thieo E. Hogen-Esch
Polymer 2009 50(15) pp: 3575-3581
Publication Date(Web):
DOI:10.1016/j.polymer.2009.06.006
Co-reporter:Jun Ling, Nadezda Fomina, Golam Rasul and Thieo E. Hogen-Esch
The Journal of Physical Chemistry B 2008 Volume 112(Issue 33) pp:10116-10122
Publication Date(Web):July 17, 2008
DOI:10.1021/jp800440z
The dilute solution properties of poly(9,9-dihexylfluorene-2,7-diyl) (PDHF) were studied by coupled SEC/light scattering and MALDI−TOF over a large molecular weight (MW) span ranging from PDHF oligomers (1−8-mer) to high MW polymer. The results were compared with Monte Carlo simulations based on realistic PDHF models obtained from X-ray data and density functional theory (DFT) calculations and with a DFT based Kratky−Porod−Benoit−Doty (KPBD) worm-like chain. The simulations called “selective random walk” (SRW) and the corresponding “selective self-avoiding random walk” (SSAW) explicitly take into account the rotationally labile bonds between the fluorene units in that four distinct torsion angles (±37.5 and ± 143°) between the units are chosen randomly. The simulations better account than the KPBD model for the experimental data obtained by us and others for various poly(9,9-dialkylfluorene-2,7-diyl) polymers but still give somewhat larger values for the radii of gyration and hydrodynamic volumes. The torsion angle selectivity of the SRW and SSAW simulations predict long chain sections punctuated by sudden sharp loops.
Co-reporter:Chao Deng, Zhening Yang, Zhicheng Zheng, Na Liu and Jun Ling
Journal of Materials Chemistry A 2015 - vol. 3(Issue 15) pp:NaN3675-3675
Publication Date(Web):2015/02/24
DOI:10.1039/C5TC00318K
We report a novel fluorescent polyfluorene-containing methacrylate macromonomer (PFMA) and its well-defined copolymers with 2-(dimethylamino)ethyl methacrylate (DMAEMA) [P(PFMA-r-DMAEMA)s] via reversible addition–fragmentation chain transfer (RAFT) polymerization. These copolymers self-assemble into photoluminescent nanoparticles in aqueous solutions and act as an excellent scaffold for the incorporation of various pigments to emit tunable colors, including white, through Förster resonance energy transfer with high quantum yield up to 0.80. The directly cast films of these NPs after evaporation of water exhibit strong colorful emission on both smooth quartz and rough plaster surfaces. The NP aqueous solutions are ready for ink-jet printing to produce exquisite fluorescent pictures.
Co-reporter:Xia Zhou, Jun Ling, Weilin Sun and Zhiquan Shen
Journal of Materials Chemistry A 2017 - vol. 5(Issue 20) pp:NaN9722-9722
Publication Date(Web):2017/04/13
DOI:10.1039/C7TA00924K
Nanosheets of coordination polymers (CPs) were synthesized via a facile and one-step complexing-coprecipitation (CC) method. Upon calcination, pure CeO2 and homogeneously La3+- or Cu2+-doped CeO2 products were fabricated by using CPs as precursors. The final products not only retained a stable nano-scale sheet-shaped morphology of their CP precursors, but also possessed high catalytic activity towards CO oxidation. Superior to commercial ceria with negligible catalytic activity, the synthesized CeO2 nanosheets exhibited high activity achieving a CO conversion of 60% at a temperature of 360 °C. When doped with Cu2+, remarkable improvement of catalysis is observed owing to the homogeneous incorporation of Cu2+ into the CeO2 lattice. As a catalytically active center, uniformly dispersed CuO also produces more oxygen vacancies and improves oxygen mobility, which results in an enhancement in catalytic activity. The detected temperatures with 50% CO conversion (T50) for Cu0.1Ce0.9O2−δ and Cu0.04Ce0.96O2−δ are 83 and 95 °C, respectively. In contrast, the catalytic test for La3+ doped ceria reveals a decreased activity compared with un-doped ceria. We believe that the Cu2+-doped CeO2 with superior catalytic performance can also be applied to other catalytic systems.
Co-reporter:Xia Zhou, Jun Ling, Weilin Sun and Zhiquan Shen
Dalton Transactions 2016 - vol. 45(Issue 23) pp:NaN9401-9401
Publication Date(Web):2016/05/11
DOI:10.1039/C6DT01271J
Novel nanoparticles of coordination polymers (CPs) with various morphologies are successfully prepared. The obtained products can be well-dispersed to make films on glass substrates by the colloidal deposition method and introduced into methyl cellulose to produce transparent and luminescent films.
Co-reporter:Xia Zhou, Jun Ling, Weilin Sun and Zhiquan Shen
Journal of Materials Chemistry A 2017 - vol. 5(Issue 20) pp:NaN9722-9722
Publication Date(Web):2017/04/13
DOI:10.1039/C7TA00924K
Nanosheets of coordination polymers (CPs) were synthesized via a facile and one-step complexing-coprecipitation (CC) method. Upon calcination, pure CeO2 and homogeneously La3+- or Cu2+-doped CeO2 products were fabricated by using CPs as precursors. The final products not only retained a stable nano-scale sheet-shaped morphology of their CP precursors, but also possessed high catalytic activity towards CO oxidation. Superior to commercial ceria with negligible catalytic activity, the synthesized CeO2 nanosheets exhibited high activity achieving a CO conversion of 60% at a temperature of 360 °C. When doped with Cu2+, remarkable improvement of catalysis is observed owing to the homogeneous incorporation of Cu2+ into the CeO2 lattice. As a catalytically active center, uniformly dispersed CuO also produces more oxygen vacancies and improves oxygen mobility, which results in an enhancement in catalytic activity. The detected temperatures with 50% CO conversion (T50) for Cu0.1Ce0.9O2−δ and Cu0.04Ce0.96O2−δ are 83 and 95 °C, respectively. In contrast, the catalytic test for La3+ doped ceria reveals a decreased activity compared with un-doped ceria. We believe that the Cu2+-doped CeO2 with superior catalytic performance can also be applied to other catalytic systems.

![Propanoic acid, 2-methyl-2-[(phenylthioxomethyl)thio]-, 2-propyn-1-yl ester](/data/chemimg/3782000/1027346-89-9.png)
![Propanoic acid, 2-methyl-2-[(phenylthioxomethyl)thio]-, 2-propyn-1-yl ester](/data/chemimg/3782000/1027346-89-9_b.png)















![2-Furancarboxylic acid, 5,5'-[[2,5-bis(2-ethylhexyl)-2,3,5,6-tetrahydro-3,6-dioxopyrrolo[3,4-c]pyrrole-1,4-diyl]di-5,2-thiophenediyl]bis-, 2,2'-diethyl ester](/data/chemimg/3777600/1597492-86-8.png)
![2-Furancarboxylic acid, 5,5'-[[2,5-bis(2-ethylhexyl)-2,3,5,6-tetrahydro-3,6-dioxopyrrolo[3,4-c]pyrrole-1,4-diyl]di-5,2-thiophenediyl]bis-, 2,2'-diethyl ester](/data/chemimg/3777600/1597492-86-8_b.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3-(5-bromo-2-thienyl)-2,5-bis(2-ethylhexyl)-2,5-dihydro-6-(5-phenyl-2-thienyl)-](/data/chemimg/3777600/1421711-97-8.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3-(5-bromo-2-thienyl)-2,5-bis(2-ethylhexyl)-2,5-dihydro-6-(5-phenyl-2-thienyl)-](/data/chemimg/3777600/1421711-97-8_b.png)
![[2,2'-Bithiophene]-5-carboxylic acid, 5',5'''-[2,5-bis(2-ethylhexyl)-2,3,5,6-tetrahydro-3,6-dioxopyrrolo[3,4-c]pyrrole-1,4-diyl]bis-, 5,5''-diethyl ester](/data/chemimg/3782000/1416418-07-9.png)
![[2,2'-Bithiophene]-5-carboxylic acid, 5',5'''-[2,5-bis(2-ethylhexyl)-2,3,5,6-tetrahydro-3,6-dioxopyrrolo[3,4-c]pyrrole-1,4-diyl]bis-, 5,5''-diethyl ester](/data/chemimg/3782000/1416418-07-9_b.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3-(5-bromo-2-thienyl)-2,5-bis(2-ethylhexyl)-2,5-dihydro-6-(2-thienyl)-](/data/chemimg/139400/1308671-90-0.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3-(5-bromo-2-thienyl)-2,5-bis(2-ethylhexyl)-2,5-dihydro-6-(2-thienyl)-](/data/chemimg/139400/1308671-90-0_b.png)

![4H-Dithieno[3,2-b:2',3'-d]pyrrole, 4-(2-ethylhexyl)-2,6-bis(trimethylstannyl)-](/data/chemimg/139500/1234499-56-9.png)
![4H-Dithieno[3,2-b:2',3'-d]pyrrole, 4-(2-ethylhexyl)-2,6-bis(trimethylstannyl)-](/data/chemimg/139500/1234499-56-9_b.png)
![2,5-bis(2-hexyldecyl)-3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione](http://img.cochemist.com/ccimg/1044600/1044598-80-2.png)
![2,5-bis(2-hexyldecyl)-3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione](http://img.cochemist.com/ccimg/1044600/1044598-80-2_b.png)
![Poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta[2,1-b;3,4-b']dithiophene)-alt-4,7(2,1,3-benzothiadiazole)]](/data/chemimg/102300/920515-34-0.png)
![Poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta[2,1-b;3,4-b']dithiophene)-alt-4,7(2,1,3-benzothiadiazole)]](/data/chemimg/102300/920515-34-0_b.png)




![SILANE, [1,2-ETHENEDIYLBIS(4,1-PHENYLENE-2,1-ETHYNEDIYL)]BIS[TRIMETHYL-](http://img.cochemist.com/ccimg/862100/862007-18-9.png)
![SILANE, [1,2-ETHENEDIYLBIS(4,1-PHENYLENE-2,1-ETHYNEDIYL)]BIS[TRIMETHYL-](http://img.cochemist.com/ccimg/862100/862007-18-9_b.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3,6-bis(4-bromophenyl)-2,5-bis(2-ethylhexyl)-2,5-dihydro-](/data/chemimg/102300/852434-82-3.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3,6-bis(4-bromophenyl)-2,5-bis(2-ethylhexyl)-2,5-dihydro-](/data/chemimg/102300/852434-82-3_b.png)
![L-Glutamic acid, 5-(phenylmethyl) ester, polymer with N6-[(phenylmethoxy)carbonyl]-L-lysine, diblock](/data/chemimg/3779200/733046-25-8.png)
![L-Glutamic acid, 5-(phenylmethyl) ester, polymer with N6-[(phenylmethoxy)carbonyl]-L-lysine, diblock](/data/chemimg/3779200/733046-25-8_b.png)




![2-BROMO-N-[6-(2-BROMOPROPANOYLAMINO)HEXYL]PROPANAMIDE](http://img.cochemist.com/ccimg/300800/300707-30-6.png)
![2-BROMO-N-[6-(2-BROMOPROPANOYLAMINO)HEXYL]PROPANAMIDE](http://img.cochemist.com/ccimg/300800/300707-30-6_b.png)








![Poly[oxycarbonyloxy(methyl-1,3-propanediyl)]](http://img.cochemist.com/ccimg/193500/193425-66-0.png)
![Poly[oxycarbonyloxy(methyl-1,3-propanediyl)]](http://img.cochemist.com/ccimg/193500/193425-66-0_b.png)
![Phenol, 2,4-bis(1,1-dimethylethyl)-6-[[[(1S)-1-phenylethyl]imino]methyl]-](http://img.cochemist.com/ccimg/191100/191087-34-0.png)
![Phenol, 2,4-bis(1,1-dimethylethyl)-6-[[[(1S)-1-phenylethyl]imino]methyl]-](http://img.cochemist.com/ccimg/191100/191087-34-0_b.png)


![Poly[oxy(1-butyl-6-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/159400/159350-72-8.png)
![Poly[oxy(1-butyl-6-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/159400/159350-72-8_b.png)
![Phenol, 2,4-bis(1,1-dimethylethyl)-6-[[(4-methylphenyl)imino]methyl]-](http://img.cochemist.com/ccimg/154300/154289-86-8.png)
![Phenol, 2,4-bis(1,1-dimethylethyl)-6-[[(4-methylphenyl)imino]methyl]-](http://img.cochemist.com/ccimg/154300/154289-86-8_b.png)





![Phenol, 2,4-bis(1,1-dimethylethyl)-6-[[(phenylmethyl)imino]methyl]-](http://img.cochemist.com/ccimg/97800/97746-38-8.png)
![Phenol, 2,4-bis(1,1-dimethylethyl)-6-[[(phenylmethyl)imino]methyl]-](http://img.cochemist.com/ccimg/97800/97746-38-8_b.png)
![3,6-bis(4-bromophenyl)-2,5-dihydro-Pyrrolo[3,4-c]pyrrole-1,4-dione](/data/chemimg/1637500/84632-54-2.png)
![3,6-bis(4-bromophenyl)-2,5-dihydro-Pyrrolo[3,4-c]pyrrole-1,4-dione](/data/chemimg/1637500/84632-54-2_b.png)
![Phenol, 2-[[[(1R)-1-phenylethyl]imino]methyl]-](http://img.cochemist.com/ccimg/78500/78419-84-8.png)
![Phenol, 2-[[[(1R)-1-phenylethyl]imino]methyl]-](http://img.cochemist.com/ccimg/78500/78419-84-8_b.png)

![Propanedinitrile, 2-[2-[2-[4-(dimethylamino)phenyl]ethenyl]-6-methyl-4H-pyran-4-ylidene]-](/data/chemimg/2138200/51325-91-8.png)
![Propanedinitrile, 2-[2-[2-[4-(dimethylamino)phenyl]ethenyl]-6-methyl-4H-pyran-4-ylidene]-](/data/chemimg/2138200/51325-91-8_b.png)





![Lanthanum Tris[bis(trimethylsilyl)amide]](http://img.cochemist.com/ccimg/35800/35788-99-9.png)
![Lanthanum Tris[bis(trimethylsilyl)amide]](http://img.cochemist.com/ccimg/35800/35788-99-9_b.png)
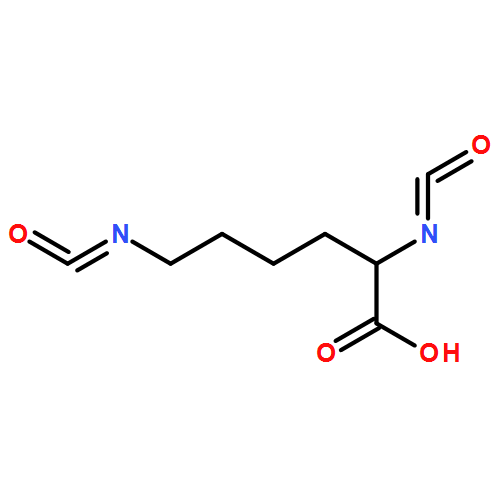

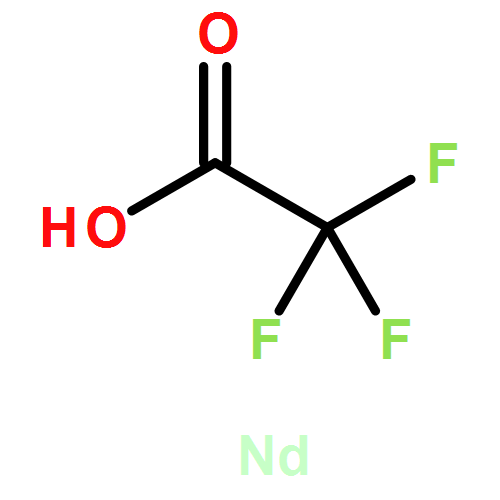
![Poly[oxy(1,6-dioxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/27000/26968-29-6.png)
![Poly[oxy(1,6-dioxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/27000/26968-29-6_b.png)


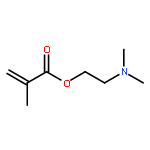
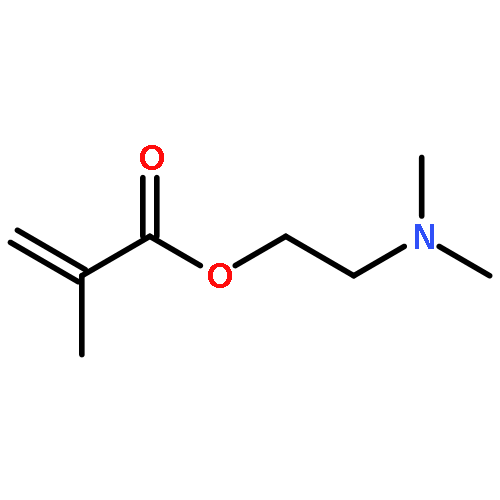
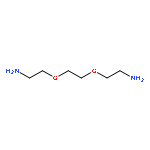
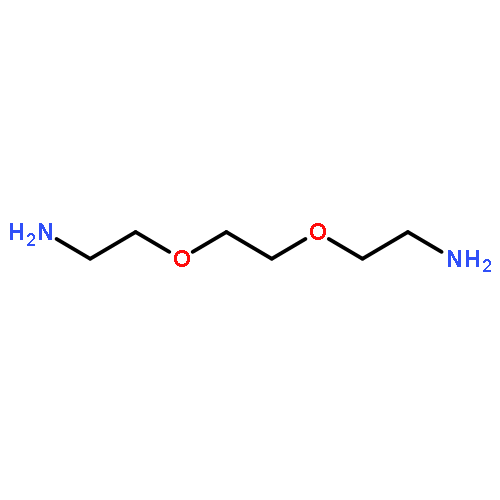
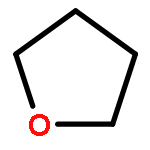
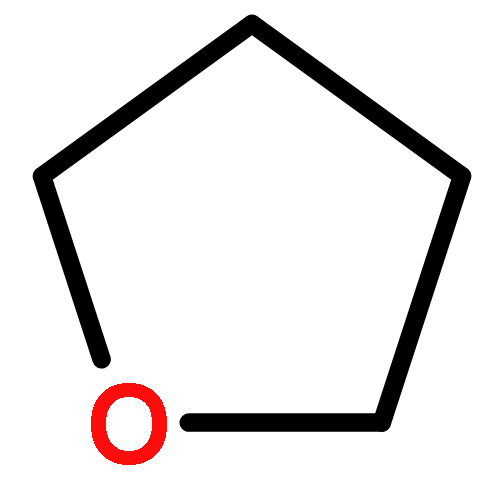



























![2,5-Bis(2-ethylhexyl)-3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione](http://img.cochemist.com/ccimg/1185900/1185885-86-2.png)
![2,5-Bis(2-ethylhexyl)-3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione](http://img.cochemist.com/ccimg/1185900/1185885-86-2_b.png)






![Tris[N,N-bis(trimethylsilyl)amide]yttrium (III)](http://img.cochemist.com/ccimg/41900/41836-28-6.png)
![Tris[N,N-bis(trimethylsilyl)amide]yttrium (III)](http://img.cochemist.com/ccimg/41900/41836-28-6_b.png)
![L-Glutamic acid, 5-(phenylmethyl) ester, polymer with N6-[(phenylmethoxy)carbonyl]-L-lysine](/data/chemimg/3779400/30900-52-8.png)
![L-Glutamic acid, 5-(phenylmethyl) ester, polymer with N6-[(phenylmethoxy)carbonyl]-L-lysine](/data/chemimg/3779400/30900-52-8_b.png)
![2-Naphthalenol, 1-[[[(1S)-1-phenylethyl]imino]methyl]-](http://img.cochemist.com/ccimg/10400/10349-23-2.png)
![2-Naphthalenol, 1-[[[(1S)-1-phenylethyl]imino]methyl]-](http://img.cochemist.com/ccimg/10400/10349-23-2_b.png)
![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3.png)
![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3_b.png)
![Phenol, 2-[[[(1S)-1-phenylethyl]imino]methyl]-](http://img.cochemist.com/ccimg/3300/3266-80-6.png)
![Phenol, 2-[[[(1S)-1-phenylethyl]imino]methyl]-](http://img.cochemist.com/ccimg/3300/3266-80-6_b.png)






![Acetic acid,2-[(ethoxythioxomethyl)thio]-](http://img.cochemist.com/ccimg/25600/25554-84-1.png)
![Acetic acid,2-[(ethoxythioxomethyl)thio]-](http://img.cochemist.com/ccimg/25600/25554-84-1_b.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4_b.png)
![Poly[imino[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/25300/25213-34-7.png)
![Poly[imino[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/25300/25213-34-7_b.png)


![Poly[imino[(2S)-1-oxo-2-[3-oxo-3-(phenylmethoxy)propyl]-1,2-ethanediyl
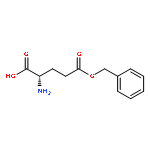
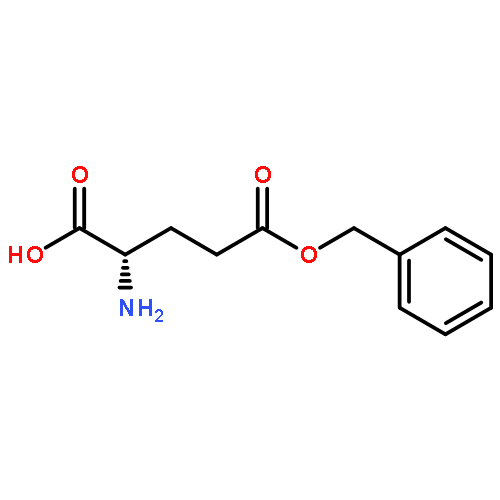
]]](http://img.cochemist.com/ccimg/25100/25038-53-3.png)
![Poly[imino[(2S)-1-oxo-2-[3-oxo-3-(phenylmethoxy)propyl]-1,2-ethanediyl
]]](http://img.cochemist.com/ccimg/25100/25038-53-3_b.png)