Co-reporter:Wenjun Yao;Xin Feng;Hanbing He;Changsong Wang
Industrial & Engineering Chemistry Research October 30, 2013 Volume 52(Issue 43) pp:15034-15040
Publication Date(Web):2017-2-22
DOI:10.1021/ie402630h
Na2Ti6O13 nanowhiskers with controllable morphologies were prepared via a simple molten salt evaporation method using a small quantity of NaCl–KCl as molten salt. The synthesized products were characterized by X-ray diffraction, field emission scanning electron microscope, and transmission electron microscope. The optimal growth dynamic conditions for synthesis of Na2Ti6O13 nanowhiskers were also studied and discussed. According to thermogravimetry-differential scanning calorimetry analysis, the calcination process was designed to include two stages, lower temperature for reaction and higher temperature for evaporation of molten salt. Nanowhiskers and nanorods with different diameters can be obtained under different evaporation conditions. By comparing residual amounts of NaCl–KCl on product surfaces calculated by determined kinetic equation and experimental results only using NaCl as molten salt, it was revealed that the molten salt evaporation rates could play an important role on the morphologies of Na2Ti6O13. A formation mechanism was provided based on nucleation and growth model and an oriented aggregation process to understand different morphologies of Na2Ti6O13. This simple molten salt evaporation method would be suitable for large scale synthesis.
Co-reporter:Gang Wang, Nanhua Wu, Jionghua Chen, Jinjian Wang, Jingling Shao, Xiaolei Zhu, Xiaohua Lu, Lucun Guo
Journal of Physics and Chemistry of Solids 2016 Volume 98() pp:183-189
Publication Date(Web):November 2016
DOI:10.1016/j.jpcs.2016.07.011
•The confined gold nanoparticles have face center cubic structure.•The confined gold nanoparticles freeze start from the outmost layers.•The freezing transition mechanism of a confined gold cluster is revealed.•The two-layer-GNS support with smaller hole diameter can stabilize smaller gold nanoparticles.•Some important kinetic parameters of confined gold nanoparticles are estimated.The thermodynamic and kinetic behaviors of gold nanoparticles confined between two-layer graphene nanosheets (two-layer-GNSs) are examined and investigated during heating and cooling processes via molecular dynamics (MD) simulation technique. An EAM potential is applied to represent the gold–gold interactions while a Lennard–Jones (L–J) potential is used to describe the gold–GNS interactions. The MD melting temperature of 1345 K for bulk gold is close to the experimental value (1337 K), confirming that the EAM potential used to describe gold–gold interactions is reliable. On the other hand, the melting temperatures of gold clusters supported on graphite bilayer are corrected to the corresponding experimental values by adjusting the εAu–C value. Therefore, the subsequent results from current work are reliable. The gold nanoparticles confined within two-layer GNSs exhibit face center cubic structures, which is similar to those of free gold clusters and bulk gold. The melting points, heats of fusion, and heat capacities of the confined gold nanoparticles are predicted based on the plots of total energies against temperature. The density distribution perpendicular to GNS suggests that the freezing of confined gold nanoparticles starts from outermost layers. The confined gold clusters exhibit layering phenomenon even in liquid state. The transition of order–disorder in each layer is an essential characteristic in structure for the freezing phase transition of the confined gold clusters. Additionally, some vital kinetic data are obtained in terms of classical nucleation theory.
Co-reporter:Yudan Zhu, Luzheng Zhang, Xiaohua Lu, Linghong Lu, Ximing Wu
Fluid Phase Equilibria 2014 Volume 362() pp:235-241
Publication Date(Web):25 January 2014
DOI:10.1016/j.fluid.2013.10.014
The anomalous flow behavior of nanoconfined water is attracting considerable attention. This study aimed to investigate the effect of pore wall interfacial properties on the flow behavior of water confined in a slit pore. Non-equilibrium molecular dynamics simulations were performed on water molecules confined in slit pores. By moving the two pore walls to opposite directions, the confined water molecules were made in a directed flowing. The flow resistance of the water molecules was then analyzed at the nanoscale. Two Si(1 1 1) surfaces were used to construct the slit pore model. The interaction strength, ɛSi-w, between the pore wall atom and water's oxygen atom was adjusted to represent different pore wall interfacial properties. A higher ɛSi-w indicates a more hydrophilic pore wall interface. Simulation results show that for the studied cases, more hydrophilic pore walls leads to larger flow resistance of the confined water. At the molecular level, the friction between the pore wall and water molecules increases with increased hydrophilicity of pore walls and further hampers the flow of the confined water in the slit pore. Moreover, simulation results demonstrate that water molecules confined in the slits are layered. The increase in the hydrophilicity increases hydrogen bonds between water layers, thereby enhancing flow resistance arising from the water molecules themselves.
Co-reporter:Wei Li ; Yang Bai ; Wei Zhuang ; Kwong-Yu Chan ; Chang Liu ; Zhuhong Yang ; Xin Feng
The Journal of Physical Chemistry C 2014 Volume 118(Issue 6) pp:3049-3055
Publication Date(Web):January 13, 2014
DOI:10.1021/jp408112z
Efforts are being made to improve crystalline properties of the skeletons of mesoporous TiO2. On the basis of the unique crystal structures and versatile phase evolution of K2Ti2O5, we obtain mesoporous TiO2(B) nanofibers (M-NFs) with highly crystalline and high-energy facets exposed skeletons via a cheap and scalable route. Verified by X-ray diffraction, scanning electron microscopy, high-resolution transmission electron microscopy, and N2 adsorption, these special structures come from layered structure of K2Ti2O5 and subsequent interlayer splitting and exfoliating and intralayer topotactic transformation. Because of their well-organized frameworks and high surface area, M-NFs exhibit efficient photogenerated charge transportation and hydrogen production, showing better charge mobility along one-dimensional and highly crystalline skeletons than irregularly shaped polycrystalline counterpart and more accessible reactive sites due to larger surface area than single crystal nonporous counterpart.
Co-reporter:Rong An, Yudan Zhu, Nanhua Wu, Wenlong Xie, Jiawei Lu, Xin Feng, and Xiaohua Lu
ACS Applied Materials & Interfaces 2013 Volume 5(Issue 7) pp:2692
Publication Date(Web):March 6, 2013
DOI:10.1021/am400175z
Ionic liquids based on 1-butyl-3-methylimidazolium hexafluoro-phosphate (ILs [Bmim][PF6]) has been employed to wet the mesoporous and dense titanium dioxide (TiO2) films. It has been found from atomic force microscopy (AFM) analysis that ILs [Bmim][PF6] can form a wetting phase on mesoporous TiO2 films, but nonwetting and sphere shaped droplets on dense films. AFM topography, phase images, and adhesion measurements suggest a remarkable dependence of wetting ILs [Bmim][PF6] films on the TiO2 porous geometry. On mesoporous TiO2 films, the adhesive force of ILs [Bmim][PF6] reaches at 40 nN, but only 4 nN on dense TiO2 films. The weak interacting ILs [Bmim][PF6] on dense TiO2 films forms rounded liquid spheres (contact angle as 40°), which helps to reduce friction locally but not on the whole surface. The stronger adhesive force on mesoporous TiO2 films makes ILs [Bmim][PF6] adhere to the surface tightly (contact angle as 5°), and this feature remains after five months. The stable spreading ILs [Bmim][PF6] films provide low friction coefficient (0.0025), large wetting areas, and short CO2 diffusion distance on the whole mesoporous TiO2 surface, avoiding the significant decelerating effect through equilibrium limitations to enable CO2 capture rate up to 1.6 and 10 times faster than that on dense TiO2 and pure ILs, respectively. And importantly, ILs wetted on mesoporous TiO2 shorten the time reaching to the maximum adsorption rate (2.8 min), faster than that on mesoporous TiO2 (6.1 min), and dense TiO2 (11.2 min). This work provides an important guidance for the improvement of the efficiency of CO2 capture, gas separation, and the lubrication of micro/nanoelectromechanical systems (M/NEMs).Keywords: adhesive force; AFM; CO2 capture; friction; ionic liquid; mesoporous TiO2; wetting;
Co-reporter:Wei Zhuang, Linghong Lu, Xinbing Wu, Wei Jin, Meng Meng, Yudan Zhu, Xiaohua Lu
Electrochemistry Communications 2013 Volume 27() pp:124-127
Publication Date(Web):February 2013
DOI:10.1016/j.elecom.2012.11.012
TiO2-B nanofibers with large surface area, high thermal stability and high crystallinity were synthesized by steam thermal method from K2Ti2O5. Compared to the bulk TiO2-B (TB-bulk) prepared from K2Ti4O9, these nanofibers exhibited much higher reversible capacity, cycling stability and rate capability. Such excellent electrochemical performances were derived from the facile charge transport due to the specific framework of a large specific surface area and one dimensional structure.Highlights► We prepared TiO2-B nanofibers utilizing a very simple methodology. ► The nanofibers exhibit a large specific surface area of 112 m2 g− 1. ► The nanofibers possess high crystallinity and thermal stability until 600 °C. ► The TiO2-B electrodes show excellent rate capability and cycling stability. ► This material can be considered as an alternative anode material.
Co-reporter:Cheng Wang, Wenwen Cui, Jingling Shao, Xiaolei Zhu, Xiaohua Lu
Computational and Theoretical Chemistry 2013 Volume 1006() pp:19-30
Publication Date(Web):15 February 2013
DOI:10.1016/j.comptc.2012.12.001
We perform a comprehensive investigation on the geometry, stability, aromaticity, bonding nature, and potential energy surface of low-lying isomers of planar BnC2 (n = 3–8) at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d) level. The geometries of lowest-energy isomers of CnB2 (n = 3–8) clusters are similar to the results of Wang and Zeng, respectively. CnB2 (n = 3–6) clusters are analogous to pure boron clusters in structure. Interestingly, the lowest-energy isomers of BnC2 (n = 3–8) clusters undergo polycyclic to wheel-type structure transition as the number of boron atom increases. Energy analysis reveals that BnC2 clusters with even n have relatively higher stability. The valence molecular orbital (VMO), electron localization function (ELF), and Mayer bond order (MBO) are used to reveal the bonding feature of BnC2 (n = 3–8) isomers. The aromaticity of B6C2 and B8C2 is discussed in terms of VMO, ELF, adaptive natural density partitioning (AdNDP), and nucleus-independent chemical shift (NICS) analyses. Some B3C2 and B5C2 isomers with large thermodynamic and kinetic stability are predicted at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d) level and observable in laboratory.Graphical abstractHighlights► The lowest-energy isomers of even-n BnC2 clusters have high stability. ► BnC2 (n = 3–8) clusters exhibit polycyclic to wheel-like structure transition. ► The growth pattern and bonding nature of BnC2 (n = 3–8) are revealed. ► The thermodynamic and kinetic stability of BnC2 (n = 3–8) is examined. ► The aromaticity of some BnC2 isomers (n = 3–8) is analyzed and discussed.
Co-reporter:Yudan Zhu;Jian Zhou;Xiaojing Guo
Microfluidics and Nanofluidics 2013 Volume 15( Issue 2) pp:191-205
Publication Date(Web):2013 August
DOI:10.1007/s10404-013-1143-7
Nanoporous materials applications have been increasingly applied in energy and environmental fields. Nanoconfined water behaviors play important roles in the application of nanoporous materials and molecular simulation is an effective approach to investigate these roles. We reviewed the selected related works and the recent research progress of our group to understand what indeed we can learn from molecular simulations of nanoconfined water molecule behaviors and how these understandings could promote the nanoporous material applications. This review is organized into two parts. First, the understanding of nanoconfined water molecule behaviors in biology using molecular simulation sets up the performance benchmarks for designing nanoporous materials. Second, molecular simulation studies in carbon nanotubes reveal useful structure–property relationships of confined water molecules for the preparation and applications of carbon nanotube membranes in flux and selectivity. This review shows that roles of molecular simulation studies are to discover the key factor at the nanoscale which is usually ignored, and to provide an understanding that will break the conventional view of nanoporous material design and application. The difficulties in the present study are also discussed.
Co-reporter:Jinjian Wang, Yin Wang, Xiaolei Zhu, and Xiaohua Lu
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 10) pp:1718-1722
Publication Date(Web):April 30, 2013
DOI:10.1021/jz4008855
Poly(ethylene glycol) dimethyl ether (PEGDME) polymers are widely used as drug solid dispersion reagents. They can cause the amorphization of drugs and improve their solubility, stability, and bioavailability. However, the mechanism about why amorphous PEGDME 2000 polymer is highly stable is unclear so far. Molecular dynamics (MD) simulation is a unique key technique to solve it. In the current work, we systematically investigate structure, aggregate state, and thermodynamic and kinetic behaviors during the phase-transition processes of the PEGDME polymers with different polymerization degree in terms of MD simulations. The melting and glass-transition temperatures of the polymers are in good agreement with experimental values. The amorphous PEGDME2000 exhibits high stability, which is consistent with the recent experiment results and can be ascribed to a combination of two factors, that is, a high thermodynamic driving force for amorphization and a relatively low molecular mobility.Keywords: configurational entropy; glass transition; melting; molecular mobility; phase transition; polymerization degree; stability;
Co-reporter:YuanHui Ji;XiaoYan Ji;YongMing Tu
Science China Chemistry 2013 Volume 56( Issue 6) pp:821-830
Publication Date(Web):2013 June
DOI:10.1007/s11426-013-4834-8
To investigate long-term CO2 behavior in geological formations and quantification of possible CO2 leaks, it is crucial to investigate the potential mobility of CO2 dissolved in brines over a wide range of spatial and temporal scales and density distributions in geological media. In this work, the mass transfer of aqueous CO2 in brines has been investigated by means of a chemical potential gradient model based on non-equilibrium thermodynamics in which the statistical associating fluid theory equation of state was used to calculate the fugacity coefficient of CO2 in brine. The investigation shows that the interfacial concentration of aqueous CO2 and the corresponding density both increase with increasing pressure and decreasing temperature; the effective diffusion coefficients decrease initially and then increase with increasing pressure; and the density of the CO2-disolved brines increases with decreasing CO2 pressure in the CO2 dissolution process. The aqueous CO2 concentration profiles obtained by the chemical potential gradient model are considerably different from those obtained by the concentration gradient model, which shows the importance of considering non-ideality, especially when the pressure is high.
Co-reporter:Wei Li, Yang Bai, Fujun Li, Chang Liu, Kwong-Yu Chan, Xin Feng and Xiaohua Lu
Journal of Materials Chemistry A 2012 vol. 22(Issue 9) pp:4025-4031
Publication Date(Web):25 Jan 2012
DOI:10.1039/C2JM14847A
Carbon-coated TiO2 fibers were synthesized as core–shell structured supports for highly dispersed Pt nanoparticles. The catalyst samples were characterized by XRD, Raman, TGA, SEM, TEM and EDX. Performance of methanol oxidation was evaluated in aqueous H2SO4 solutions with methanol by cyclic voltammetry and chronoamperometry. The TiO2 nanofibers were coated with carbon shells mostly between 5 and 10 nm in thickness. Platinum nanoparticles around 2 nm were evenly deposited onto the as-synthesized carbon-coated TiO2 fibers, denoted as Pt–TiO2/C. Electrochemical experiments showed that the peak current density of methanol oxidation in the forward scan was significantly increased by 7.3 and 2.5 times on Pt–TiO2/C compared with those of Pt–TiO2 and Pt–C (Vulcan XC-72), respectively. Furthermore, the Pt–TiO2/C electro-catalyst exhibited a lower onset potential and slower current decay than Pt–C, suggesting higher catalytic activity and better stability. In photo-electrochemical experiments, the electro-catalytic and photo-catalytic properties of Pt–TiO2/C have been synergistically coupled to boost the performance of methanol oxidation. Under UV irradiation, the total peak current density of methanol oxidation on Pt–TiO2/C is enhanced 2.5 times as that in the dark. In brief, the cooperation between Pt, carbon shell and TiO2 support promotes methanol oxidation on Pt–TiO2/C with and without UV illumination.
Co-reporter:Licheng Li, Yudan Zhu, Xiaohua Lu, Mingjie Wei, Wei Zhuang, Zhuhong Yang and Xin Feng
Chemical Communications 2012 vol. 48(Issue 94) pp:11525-11527
Publication Date(Web):09 Oct 2012
DOI:10.1039/C2CC36157D
In this work, we report a novel surface modification which can improve the desorption of a hydrodesulfurization product (H2S) from mesoporous TiO2. The corresponding catalyst exhibits a significantly enhanced hydrodesulfurization performance compared with an unmodified catalyst, and the dibenzothiophene conversion increases from 65% to 98%.
Co-reporter:Ming-Jie Wei, Luzheng Zhang, Linghong Lu, Yudan Zhu, Keith E. Gubbins and Xiaohua Lu
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 48) pp:16536-16543
Publication Date(Web):28 May 2012
DOI:10.1039/C2CP40687J
It is well known that titanium dioxide (TiO2) is biocompatible and environmentally friendly. Consequently, TiO2 is widely applied in many fields, such as implant materials, photocatalysis, pigments, cosmetic additives, etc. Mesoporous TiO2 finds many industrial applications, because of its high surface area and stable structure. However, the strong interaction between TiO2 and water molecules sometimes limits its application to solution environments. Our previous computational work showed that changes to the surface chemistry of TiO2 can affect the hydrogen bond network of water molecules on the TiO2 surface, and so influence the diffusion of water in the slits. Thus, a carbon-modified TiO2 surface could be an alternative way to avoid this limitation. In this work, a slit pore model with a modified TiO2 surface (pore widths 1.2 nm, 1.6 nm and 2.0 nm) with varying carbon coverages (0%, 7%, 47%, 53%, 93% and 100%) was presented. Molecular dynamics (MD) simulations were then performed to investigate the sorption and diffusion of water in these slits. Simulation results showed that the interfacial water molecules on bare TiO2 regions were little affected by the neighboring carbon, and they have the same properties as those on bare TiO2 surfaces. However, the diffusion of water molecules in the center of the slit was enhanced on increase of carbon coverage, because the carbon layer broke the hydrogen bond network between the interfacial water molecules and those on the bare TiO2 surface. It was found that in the slits (>1.2 nm) fully covered by carbon the diffusion coefficients of water are larger than that of bulk water. Moreover, large pore sizes caused an increase in the mobility of water molecules in carbon-modified TiO2, in agreement with previous experimental work.
Co-reporter:Rong An, Qiuming Yu, Luzheng Zhang, Yudan Zhu, Xiaojing Guo, Shuangqin Fu, Licheng Li, Changsong Wang, Ximing Wu, Chang Liu, and Xiaohua Lu
Langmuir 2012 Volume 28(Issue 43) pp:15270-15277
Publication Date(Web):October 9, 2012
DOI:10.1021/la3029325
A simple physical strategy to reduce the frictional and adhesive forces on TiO2 films was proposed by constructing mesoporous TiO2 films with heterogeneously distributed nanopores on the film surfaces. In comparison, TiO2 films with densely packed nanoparticles were also prepared. The crystal structure and morphology of the films were characterized with Raman spectroscopy, field emission scanning electron microscopy (FESEM), and atomic force microscopy (AFM). It was found that the TiO2(B) phase exists in the mesoporuos TiO2 films but not in the densely packed films. The existence of TiO2(B) plays a significant role in creating and maintaining the nanopores in the mesoporous TiO2 films. The frictional and adhesive forces were measured on both films using AFM. The mesoporous films exhibit two typical adhesion forces of around 3 and 12 nN in the force distribution profile whereas the densely packed films show only one around 12 nN. The frictional coefficients were 2.6 × 10–3 and 6.7 × 10–2 for the mesoporous and densely packed TiO2 films, respectively. A model based on the atomic structures of a thin film of water molecules adsorbed on TiO2 surfaces leading to hydrophobic effects was proposed to understand the lower frictional and adhesive forces observed on the mesoporous TiO2 films. This simple physical approach to reducing the frictional and adhesive forces on TiO2 films could have broad applications to a variety of surface coatings.
Co-reporter:Wei Li, Yang Bai, Weijia Liu, Chang Liu, Zhuhong Yang, Xin Feng, Xiaohua Lu and Kwong-Yu Chan
Journal of Materials Chemistry A 2011 vol. 21(Issue 18) pp:6718-6724
Publication Date(Web):31 Mar 2011
DOI:10.1039/C1JM10115C
Single-crystalline anatase TiO2 nanofibers with highly reactive {001} facets were synthesized from layered potassium titanate K2Ti2O5via topotactic transformation in ion-exchange and dehydration. The cuboid fibers showed one-dimensional (1-D) orientation in the [010] direction and {001} as well as {100} facets enclosing along the longitudinal dimension. The structural evolution was deduced from XRD, SEM and TEM characterizations, which revealed that the highly reactive {001} facets were derived from the interlayer splitting and exfoliation of the layered precursors in a surfactant-free way. Photoluminescence (PL) measurements witnessed the efficient separation and transfer of photoinduced charge carriers in the single-crystalline and reactive facets enclosed TiO2 nanofibers. Sequently, highly efficient photocatalytic property of the nanofibers was demonstrated by phenol degradation and H2 evolution. Phenol degradation rate of nanofibers is 2.7 times of the irregular-shaped nanoparticle counterparts with the same crystal phase and similar specific surface area. In photocatalytic evolution of H2, nanofibers presented much higher and more stable activity than both nanoparticle counterparts and P25 benchmark. Charge carrier highways provided by single-crystalline 1-D structures and efficient surface reactivity offered by exposed facets are the two key factors for the enhanced properties.
Co-reporter:Shasha Qian, Changsong Wang, Weijia Liu, Yinhua Zhu, Wenjun Yao and Xiaohua Lu
Journal of Materials Chemistry A 2011 vol. 21(Issue 13) pp:4945-4952
Publication Date(Web):11 Feb 2011
DOI:10.1039/C0JM03508D
To accomplish the more effective coupling of cadmium sulfide quantum dots (CdS QDs), the mesoporous TiO2 substrate and bifunctional linker, mercaptopropionic acid (MPA), were used to disperse and stabilize the CdS QDs. Due to the porous nano-architecture on the TiO2 substrate with large surface area and high crystallinity, the efficiency of degradation of organic compounds in aqueous solution under visible light irradiation is greatly enhanced, compared to CdS loaded anatase TiO2 without porous structure and common commercial P25. Furthermore, the bifunctional linking molecule, MPA, could effectively disperse and stabilize CdS nanoparticles. CdS/TiO2 with the linking molecule CdS-MPA-TiO2(m) exhibits much more stability and activity than CdS-TiO2(m) which is prepared by direct deposition. After 3 cycling tests of degradation of MB (methylene blue), the loss ratio of CdS on CdS-TiO2(m) is 70.6%, much larger than that of 17.8% on CdS-MPA-TiO2(m). This work may give ideas for the synthesis of other stable and active supported catalysts in many fields.
Co-reporter:Ming-Jie Wei, Jian Zhou, Xiaohua Lu, Yudan Zhu, Weijia Liu, Linghong Lu, Luzheng Zhang
Fluid Phase Equilibria 2011 Volume 302(1–2) pp:316-320
Publication Date(Web):15 March 2011
DOI:10.1016/j.fluid.2010.09.044
Titanium dioxide (TiO2) is well applied in implant materials because of its biocompatibility while carbon is usually regarded as toxicant to cells. Actually, water-surface interaction has significant influence on the biocompatibility of implant materials. To understand the difference between TiO2 and graphite on the biocompatibility, molecular dynamics simulations were performed to study the structure and diffusion of water in nano-slits formed by rutile TiO2(1 1 0) and graphite(0 0 0 1) with the separation distance ranging from 0.8 to 2.0 nm. Simulation results show that the residence time of water at TiO2 surface is considerably longer than that at graphite surface, indicating the stronger interaction between water and TiO2. Moreover, the microstructure of water molecules absorbed at the TiO2 and graphite surfaces were analyzed; the bound water molecules at the TiO2 surface and the hydrogen bond network reduce the diffusivity of water through the TiO2 slits. Simulation results show that the surface chemistry is crucial to the diffusion of water in nanoscale pores, while geometric size effect may just enhance the chemical effect.
Co-reporter:Rongwei Shi ; Jingling Shao ; Xiaolei Zhu
The Journal of Physical Chemistry C 2011 Volume 115(Issue 7) pp:2961-2968
Publication Date(Web):February 3, 2011
DOI:10.1021/jp109689m
A MD simulation method is used to simulate the melting and freezing of Au−Pt nanoparticles confined in armchair single-walled carbon tubes ((n,n)-SWNTs), applying the second-moment approximation of the tight-binding potentials for metal−metal interactions and Lennard-Jones potential for the metal−carbon interactions. The carbon atoms on the SWNTs are taken to be fixed. The structures, total energies, Lindemman indices, and radial and axial density distributions are used to examine the behaviors of melting and freezing for Au−Pt nanoparticles confined in (n,n)-SWNTs. The nucleation analysis is carried out in terms of classical nucleation theory. The simulation results demonstrate that the solid Au−Pt clusters confined within (n,n)-SWNTs have multishell structures, even in a melted cluster. In addition, for the confined Au−Pt nanoparticles, Pt atoms tend to stay at the position close to the SWNT wall, which is different from free Au−Pt nanoparticles. Simulation results reveal that SWNTs and compositions of nanoparticles have significant effects on the structures and physical properties of the confined Au−Pt nanoparticles. On the other hand, some important thermodynamic and dynamic parameters are estimated based on MD simulations and compared with available theoretical and experimental results.
Co-reporter:Wei Fang ; Weijia Liu ; Xiaojing Guo ; Xiaohua Lu ;Linghong Lu
The Journal of Physical Chemistry C 2011 Volume 115(Issue 17) pp:8622-8629
Publication Date(Web):April 11, 2011
DOI:10.1021/jp110825y
CO adsorption on both clean and hydroxylated TiO2-B (100) surfaces with terminal and bridging hydroxyl groups is investigated via first-principles density functional theory calculations. The adsorption mechanisms of CO molecules on both clean and hydroxylated surfaces are discussed. CO molecules preferentially adsorb at five coordinated Ti sites of TiO2 through C atoms. The calculated adsorption energies range from 13.11 to 43.03 kJ/mol. Moreover, lower concentrations of CO gas can strongly bind to the surface. From structure point of view, CO molecules interact with the surface mainly via its 2π* state. The adsorption is accompanied by electron transfers (0.02−0.08 e) between the CO molecule and the surface. Both the terminal and bridging hydroxyl groups can slightly facilitate CO adsorption, however, in different levels. When CO molecules adsorb near the bridging hydroxyl groups (Eads = 43.56 kJ/mol), it can increase more CO adsorption than the terminal hydroxyl groups (Eads = 33.87 kJ/mol). Furthermore, our calculations indicates that the surface donates electrons to the CO molecule when the latter is adsorbed near the bridging hydroxyl groups, which is different from observations made for adsorption onto the clean surface and near the terminal hydroxyl groups. This unique mechanism provides a possible explanation for the larger increment in CO adsorption near the bridging hydroxyl groups.
Co-reporter:Rongwei Shi;Jingling Shao;Cheng Wang;Xiaolei Zhu
Journal of Molecular Modeling 2011 Volume 17( Issue 5) pp:1007-1016
Publication Date(Web):2011 May
DOI:10.1007/s00894-010-0801-x
We have systematically explored and investigated the geometrical structures, stability, growth pattern, bonding character, and potential energy surface (PES) of the possible isomers of each cluster for planar BnP (n = 1 ∼ 7) at the CCSD(T)/6-311+;G(d)//B3LYP/6-311+G(d) level. A large number of planar structures for the possible isomers of BnP (n = 1 ∼ 7) and transition states are located. Isomers 1a ∼ 7a of BnP are the lowest-energy structures and 2a, 4a, as well as 6a are more stable than their neighbors. For the lowest-energy structures (1a ∼ 7a) of BnP, P atom lies at the apex and tends to form two B-P bonds with boron atoms. They exhibit planar zigzag growth feature or approximately spherical-like growth pattern. Results from molecular orbital analysis demonstrate that the formation of the delocalized π MOs and the σ-radial and σ-tangential MOs plays a critical role in stabilizing the structures of lowest-energy isomers (2a ∼ 7a) of BnP. Importantly, isomers 3a, 3c, 3d, 4a, 4b, 5b, and 5c of BnP are stable both thermodynamically and kinetically at the CCSD(T)/6-311+G(d)// B3LYP/6-311+G(d) level and detectable in laboratory, which is valuable for further experimental studies of BnP.
Co-reporter:YuanHui Ji;WenJuan Huang;ZhuHong Yang;Xin Feng
Science China Chemistry 2011 Volume 54( Issue 3) pp:559-564
Publication Date(Web):2011 March
DOI:10.1007/s11426-010-4209-3
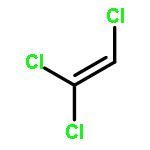
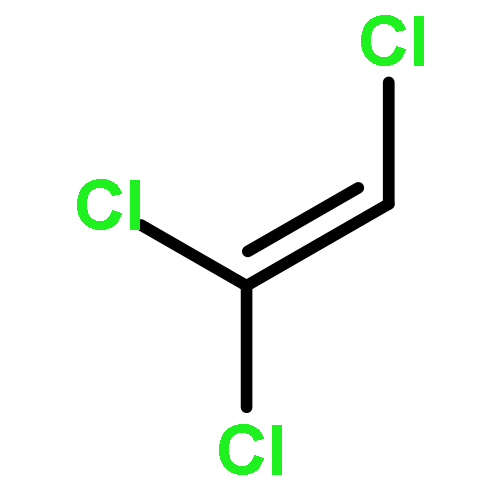
It is widely stated that most organic contaminants could be completely mineralized by Advanced Oxidation Processes (AOPs). This statement means that the concentration of the organic contaminant at equilibrium (limiting concentration, LC) is low enough to be neglected. However, for environmental safety, especially drinking water safety, this statement needs to be verified from chemical engineering thermodynamic analysis. In this paper, trichloromethane (CHCl3) and dichloromethane (CH2Cl2) are selected as the model systems, and the equilibrium concentration (theoretical limiting concentration, TLC) for the mineralization of chlorinated methanes in aqueous solutions at the different initial concentrations of chlorinated methanes, pH values and ·OH concentrations by AOPs are investigated by thermodynamic analysis. The results in this paper show that the TLC for the mineralization of CHCl3 and CH2Cl2 with ·OH increases with increasing initial concentrations of CHCl3 and CH2Cl2, decreases with increasing concentration of ·OH, and the TLC for the mineralization of CHCl3 decreases with increasing pH values except that the pH value changes from 3.0 to 3.5. For the mineralization of CH2Cl2 with ·OH, at the concentrations of ·OH obtained from the literature, there is no obvious change of the TLC with pH values, while as the concentrations of ·OH increase by 10 and 100 times, the TLC decreases with the increasing pH values from 2.0 to 3.0 and from 3.5 to 4.5, and increases with the increasing pH values from 3.0 to 3.5 and from 4.5 to 5.0. The investigations in this paper imply that high concentration of ·OH, a bit higher pH values (4.0–5.0) in acid environment and low initial concentrations of the organic contaminants are beneficial for the complete mineralization of chlorinated methanes by AOPs.
Co-reporter:XiaoHua Lu;YuanHui Ji;HongLai Liu
Science China Chemistry 2011 Volume 54( Issue 10) pp:
Publication Date(Web):2011 October
DOI:10.1007/s11426-011-4308-9
Interfacial transfer plays an important role in multi-phase chemical processes. However, it is difficult to describe the complex interfacial transport behavior by the traditional mass transfer model. In this paper, we describe an interfacial mass transfer model based on linear non-equilibrium thermodynamics for the analysis of the rate of interfacial transport. The interfacial transfer process rate J depends on the interface mass transfer coefficient K, interfacial area A and chemical potential gradient Δµ at the interface. Potassium compounds were selected as model systems. A model based on linear non-equilibrium thermodynamics was established in order to describe and predict the transport rate at the solid-solution interface. Together with accurate experimental kinetic data for potassium ions obtained using ion-selective electrodes, a general model which can be used to describe the dissolution rate was established and used to analyze ways of improving the process rate.
Co-reporter:Rongwei Shi;Jinyu Li;Xiaoning Cao;Xiaolei Zhu
Journal of Molecular Modeling 2011 Volume 17( Issue 8) pp:1941-1951
Publication Date(Web):2011 August
DOI:10.1007/s00894-010-0903-5
Human P450 protein CYP2C9 is one of the major drug-metabolizing isomers, contributing to the oxidation of 16% of the drugs currently in clinical use. To examine the interaction mechanisms between CYP2C9 and proton pump inhibitions (PPIs), we used molecular docking and molecular dynamics (MD) simulation methods to investigate the conformations and interactions around the binding sites of PPIs/CYPP2C9. Results from molecular docking and MD simulations demonstrate that nine PPIs adopt two different conformations (extended and U-bend structures) at the binding sites and position themselves far above the heme of 2C9. The presence of PPIs changes the secondary structures and residue flexibilities of 2C9. Interestingly, at the binding sites of all PPI–CYP2C9 complexes except for Lan/CYP2C9, there are hydrogen-bonding networks made of PPIs, water molecules, and some residues of 2C9. Moreover, there are strong hydrophobic interactions at all binding sites for PPIs/2C9, which indicate that electrostatic interactions and hydrophobic interactions appear to be important for stabilizing the binding sites of most PPIs/2C9. However, in the case of Lan/2C9, the hydrophobic interactions are more important than the electrostatic interactions for stabilizing the binding site. In addition, an interesting conformational conversion from extended to U-bend structures was observed for pantoprazole, which is attributed to an H-bond interaction in the binding pocket, an internal π–π stacking interaction, and an internal electrostatic interaction of pantoprazole.
Co-reporter:Linghong Lu, Yudan Zhu, Fujun Li, Wei Zhuang, Kwong Yu Chan and Xiaohua Lu
Journal of Materials Chemistry A 2010 vol. 20(Issue 36) pp:7645-7651
Publication Date(Web):06 Aug 2010
DOI:10.1039/C0JM00054J
Titania carbon composites were prepared via in situ carbonization on mesoporous titania whiskers. Their microstructures were characterized by scanning electron microscopy (SEM) and X-ray diffraction (XRD), showing that the composites, after carbonization, still retain the original morphology of the whiskers and the crystalline structure of titania. Based on N2 sorption isotherms, the average pore sizes of the as-prepared composites were found to depend on the amount of filled carbon. The electrochemical capacitance performance of the as-prepared composites was investigated by cyclic voltammetry, electrochemical impedance spectroscopy (EIS) and galvanostatic charge–discharge cycles. Although the specific surface area of the composite TiO2/0.252C is moderate at 156 m2 g−1, its specific volumetric capacitance of 25 F cm−3 was much higher than the value of 10 F cm−3 for Vulcan XC-72, which has a specific surface area of 236 m2 g−1. This enhanced capacitance may come from the composite mesopores derived from porous titania whiskers. They provide readily accessible diffusion pathways for electrolyte ions. There is better conductivity with carbon in the composite. After 2000 cycles, we observed a change of −2.8%, −2.6% and −1.9% decrease in the specific volumetric capacitance compared to the values at the 100th cycle of the composites TiO2/0.252C, TiO2/0.143C and TiO2/0.08C, respectively. This decrease is small and significantly less than the 10% decrease of capacitance in Vulcan XC-72 in the same period. The more consistent capacitance in the composite suggests a more stable interface between titania, carbon filling and electrolyte compared to that of Vulcan XC-72 without titania.
Co-reporter:Weijia Liu, Jian-guo Wang, Wei Li, Xiaojing Guo, Linghong Lu, Xiaohua Lu, Xin Feng, Chang Liu and Zhuhong Yang
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 31) pp:8721-8727
Publication Date(Web):16 Jun 2010
DOI:10.1039/B920128A
By means of density functional theory (DFT) calculations, we study the water adsorption behavior on two common surfaces, (001) and (100) TiO2-B, which maintains the monoclinic structure as high as ∼550 °C or higher in ambient conditions. The two surfaces show totally different activity for water dissociation. The dissociative chemisorption of water on TiO2-B (100) is identified at both submonolayer and monolayer coverages, which indicates considerable reactivity. In contrast, the non-dissociative molecular adsorption of water is the most stable state on TiO2-B (001) which suggests no special activity. Furthermore, we compare the structural features of different surfaces with diverse crystal structures, such as rutile, anatase, brookite, TiO2-B etc. Keeping a close eye on the exposed atoms on the surface, we conclude a more general criterion for a quick evaluation of reactivities of different TiO2 surfaces merely based on local surface structure features.
Co-reporter:Dong Li, Kui Xiong, Wei Li, Zhuhong Yang, Chang Liu, Xin Feng and Xiaohua Lu
Industrial & Engineering Chemistry Research 2010 Volume 49(Issue 18) pp:8397-8405
Publication Date(Web):July 28, 2010
DOI:10.1021/ie100277g
A scale-up model for photoreactors based on a comparative study of the photocatalytic efficiency of suspended and immobilized systems was developed. The model is independent of reactor size and configurations, and it assumes that photocatalytic efficiency is the same when normalized per unit of illuminated catalyst area in both systems. In all cases, phenol/TiO2 (Degussa P25) was selected as the photodegradation system. First, a kinetic model was built in an immobilized system based on the corresponding experimental data, and then predicted rates of phenol degradation in the suspended system were calculated using the above kinetic model combined with a simplified radiation model, which was expressed as an apparent form of the Lambert law. Second, to obtain experimental rates, experiments conducted in the suspended system were carried out under the same conditions used in the immobilized system. Ratios between experimental rates and predicted rates were obtained, revealing the differences in efficiency between the suspended and immobilized systems. The typical value of the ratio was 2.5−9.2, suggesting that the efficiency of the suspended system was 2.5−9.2 times higher than that of the immobilized system. The ratio decreased with increasing concentrations of both phenol and catalyst. When the catalyst concentration and initial concentration of phenol were set, the ratio became constant within the range of the light intensity of 1.71−3.60 mW cm−2. Finally, for photoreactor scale-up, the proposed model was validated in a larger photoreactor operated in the suspended system, and good agreements were obtained with errors less than 5%. This methodology provides an alternative to the scale-up of photoreactors, which allows for easier engineering applications.
Co-reporter:Wenjuan Huang, Yuanhui Ji, Zhuhong Yang, Xin Feng, Chang Liu, Yinhua Zhu and Xiaohua Lu
Industrial & Engineering Chemistry Research 2010 Volume 49(Issue 13) pp:6243-6249
Publication Date(Web):May 27, 2010
DOI:10.1021/ie100116c
It has great significance to investigate the degradation of aromatic contaminants as they are highly carcinogenic and nondegradable pollutants in drinking water. In this paper, the mineralization orders of the representative nitro/chloro/methyl/amino-aromatic contaminants with oxidants (·OH, H2O2, ·O−, O2,O3) in advanced oxidation processes (AOPs) are investigated based on the calculated standard molar Gibbs free energy changes of reaction (ΔrGm0) and the results are consistent with those from previous experimental results, electrophilic substitution orientation rules of the Hammett equation, and predicted results with a quantitative structure−activity relationship (QSAR). In addition, the quantitative function relationship between the degradation rate (r) of the aromatic contaminants and the thermodynamic driving force (ΔrGm0) is analyzed in order to investigate the degradation kinetics more rigorously.
Co-reporter:Bin Cao, Wenjun Yao, Changsong Wang, Xuanxuan Ma, Xin Feng, Xiaohua Lu
Materials Letters 2010 Volume 64(Issue 16) pp:1819-1821
Publication Date(Web):31 August 2010
DOI:10.1016/j.matlet.2010.05.014
Rutile TiO2 whiskers with a length of 30 μm and a diameter of 1.5 μm were prepared via a facile sintering technique with H2Ti4O9.0.25H2O as a precursor under strong acidic condition. Scanning electron microscopy images showed that the whiskers have a uniform shape and a straight rod morphology with a square cross section. X-ray diffraction and transmission electron microscopy combined with a selected-area electron diffraction pattern showed that the TiO2 whiskers are single crystalline rutile structures grown along the [002] direction and the circumferential faces have four equivalent (110) planes. Compared with rutile microparticles, rutile whiskers exhibit higher activity for the photocatalytic degradation of methylene blue.
Co-reporter:Jingling Shao, Chunyan He, Rongwei Shi, Cheng Wang, Xiaolei Zhu, Xiaohua Lu
Journal of Molecular Structure: THEOCHEM 2010 Volume 961(1–3) pp:17-28
Publication Date(Web):15 December 2010
DOI:10.1016/j.theochem.2010.08.036
The structures and relative stabilities of CnB3 (n = 1–8) clusters are explored and analyzed at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d) level. For low-lying isomers of CnB3 (n = 1–8), cyclic structures are more favorable in energy than branch as well as linear structures, and rings with exocyclic chains. For most isomers of CnB3 (n = 1–8), the structures with doublet states are lower in energy than the ones with quartet states. The lowest-energy isomers of CnB3 are all cyclic structures. The lowest-energy structures of CnB3 (n = 1–3) appear to be similar to those of pure boron clusters which have polycylic structures, while the lowest-energy structures of CnB3 (n = 4–8) tend to be analogous to the corresponding geometries of pure carbon, CnB, and CnB2 clusters which possess single-ring structures. The most stable structures of planar CnB3 (n = 3–8) clusters can be derived from combining a B–C–B–C–C–B building unit and a carbon chain. Some physical properties, such as the binding energy per atom, incremental binding energy, and second order energy difference suggest that for CnB3 clusters, odd-n clusters are more stable than even-n clusters. Results from NBO and molecular orbital analysis illustrate that the formation of the delocalized π MOs, the σ-radial and σ-tangential MOs are favorable to the stabilization of structures for lowest-energy isomers. It is interesting to find from the valence molecular orbital and NBO analyses that for the most stable isomers of CnB3 (n = 1–8), the numbers of π electrons for CnB3 (n = 1–8) is (n + 1) with an exception of C4B3 (6 π electrons), suggesting that CB3, C4B3 and C5B3 may have aromaticity, which is consistent with the results of the nucleus independent chemical shifts (NICS).
Co-reporter:Yuanhui Ji;Hongliang Qian;Chang Liu
Frontiers of Chemical Science and Engineering 2010 Volume 4( Issue 1) pp:52-56
Publication Date(Web):2010 March
DOI:10.1007/s11705-009-0301-7
In this paper, the research framework for specific structure crystallization modeling has been proposed in which four steps are required in order to investigate the rigorous crystallization modeling by thermodynamics. The first is the activity coefficient model of the solution, the second is Solid-Liquid equilibrium, the third and fourth are the dissolution and crystallization kinetics modeling, respectively. Our investigations show that the mechanisms of complex structure formation and microphase transition can be analyzed by combining the dissolution and crystallization kinetics modeling. Moreover, the formation mechanism of the porous KCl has been analyzed, which may provide a reference for the porous structure formation in the advanced material synthesis.
Co-reporter:Jingling Shao, Cao Yang, Xiaolei Zhu and Xiaohua Lu
The Journal of Physical Chemistry C 2010 Volume 114(Issue 7) pp:2896-2902
Publication Date(Web):February 4, 2010
DOI:10.1021/jp910289c
The structure, phase transition, and nucleation of Au nanoparticles confined within armchair single-walled carbon nanotubes ((n,n)-SWNTs) are investigated using molecular dynamics (MD) simulation technique. The Au−Au interactions are described by the TB-SMA potentials and the Au-SWNT interactions are represented by Lennard-Jones potential. SWNTs are approximately considered to be rigid. The total energies, structures, Lindemman indices, and radial density distributions are used to reveal the feature of phase transition for Au nanoparticles confined in (n,n)-SWNTs. The classical nucleation theory is applied to perform nucleation analysis. Results demonstrate that confined AuN exhibit multishell structures. The order−disorder transformation of atoms in each layer is an important structure feature of phase transition. Interestingly, the melting starts from the innermost layer and freezing starts from outermost layer for confined Au nanoparticles. SWNTs have a significant effect on the structures and stabilities of the confined Au nanoparticles. On the other hand, some important thermodynamic and dynamic parameters are estimated and compared with available experimental and calculated results. This work provides the primary physical insights into the phase transition and nucleation process of confined Au nanoparticles.
Co-reporter:Jingling Shao;Rongwei Shi;Cheng Wang;Xiaolei Zhu
Journal of Molecular Modeling 2010 Volume 16( Issue 5) pp:939-950
Publication Date(Web):2010 May
DOI:10.1007/s00894-009-0604-0
The geometrical structures, potential energy surface, stability, and bonding character of low-energy isomers of planar C3B3 were systematically explored and investigated at the B3LYP/6-311+G(d)// CCSD(T)/6-311+G(d) level for the first time. A large number of planar structures for low-energy isomers of C3B3 are located and reported. In particular, isomers 1 (Cs,2A’) and 2 (Cs,2A’), with a belt-like structure corresponding to the lowest-energy structures of planar C3B3, are revealed. Based on molecular orbital (MO) and natural bond orbital (NBO) analyses, delocalized σ MOs, multi-centered σ MOs, and delocalized π MOs play an important role in stabilizing the structures of low-energy isomers of C3B3. It is interesting to note from isomerization analysis that the interconversion of isomers 2 and 7 can be realized through two isomerization channels. The results demonstrate that isomers 1, 2, 3, 4, 7, 9, 12, 17, 19, and 20 of C3B3 are stable both thermodynamically and kinetically at the B3LYP/ 6-311+G(d)//CCSD(T)/ 6-311+G(d) level, and that they are observable in the laboratory, which is helpful for future experimental studies of C3B3.
Co-reporter:Yang Bai, Wei Li, Chang Liu, Zhuhong Yang, Xin Feng, Xiaohua Lu and Kwong-Yu Chan
Journal of Materials Chemistry A 2009 vol. 19(Issue 38) pp:7055-7061
Publication Date(Web):04 Aug 2009
DOI:10.1039/B910240J
A novel metal–semiconductor nanocomposite with stable metal nanoparticles and efficient photocatalytic performance has been prepared. It consists of highly dispersed ∼2 nm platinum (Pt) nanoparticles loaded on mesoporous and bicrystalline TiO2 fibers (Pt/mb-TiO2). Due to the well organized porous Pt/TiO2 nanoarchitecture, rate of photodegradation of CHCl3 was nearly doubled compared to both P25 and Pt/P25. And in photocatalytic production of H2 (with methanol as an electron donor), Pt/mb-TiO2 presented much more stable activity than Pt/P25. In a 20 h H2 production, Pt nanoparticles on P25 agglomerated and grew remarkably from 2.0 ± 0.5 nm to 4.5 ± 2.5 nm, while Pt nanoparticles on mb-TiO2 changed slightly from 2.0 ± 0.5 nm to 2.2 ± 0.5 nm. Moreover, the loss ratio of Pt on P25 is 36%, which is much larger than that of 7% on mb-TiO2. The efficient charge transfer in the interfaces of Pt/TiO2(B) and TiO2(B)/anatase was discussed. We concluded that the high photocatalytic performance of Pt/mb-TiO2 can be attributed to stable Pt nanoparticles supported on the mesoporous masonry frameworks of TiO2 and efficient charge transfer in the interfaces.
Co-reporter:Wei Li, Yang Bai, Chang Liu, Zhuhong Yang, Xin Feng, Xiaohua Lu, Nicole K. van der Laak and Kwong-Yu Chan
Environmental Science & Technology 2009 Volume 43(Issue 14) pp:5423-5428
Publication Date(Web):June 8, 2009
DOI:10.1021/es8037005
In the absence of any doping and modification, the anatase-to-rutile phase transformation was inhibited at high temperatures giving rise to highly thermal stable and highly crystalline anatase TiO2 fibers. The initial formation of the TiO2(B) phase is found to be key in inhibiting this transformation. The intermediate structure of the TiO2 fiber comprises an inner anatase core with an outer TiO2(B) shell, which has a specific crystallographic orientation with respect to the anatase structure. During the calcination process from 300 to 800 °C, both the TiO2(B) shell and the bulk anatase crystal structure was preserved. At temperatures of 800−900 °C the TiO2(B)-to-anatase transformation was finished and a near-pure and thermally stable anatase fiber was obtained. This final product shows the same activity as a standard commercial photocatalyst Degussa P-25 when measured against unit mass, and 5 times the activity when measured with respect to the unit surface area. The anatase TiO2 fibers presented here have considerable interest as practical photocatalysts for water purification, as they can be easily recycled without a decrease in their photocatalytic activity and can be prepared at large scale and at low cost.
Co-reporter:Yuanhui Ji, Zhuhong Yang, Xiaoyan Ji, Xin Feng, Wenjuan Huang, Chang Liu, Wei Li and Xiaohua Lu
Industrial & Engineering Chemistry Research 2009 Volume 48(Issue 23) pp:10728-10733
Publication Date(Web):September 18, 2009
DOI:10.1021/ie900620n
There is a growing demand for the efficient treatment of organic polluted wastewaters by advanced oxidation processes (AOPs) which calls for the determination of the mineralization order of ease for the organic contaminants with oxidants. The mineralization abilities of organic contaminants in AOPs are investigated in this work. Photocatalytic experiments for three representative organic contaminants are carried out, and their corresponding reaction rates are determined experimentally. Meanwhile, molar Gibbs free energy changes ΔrGm° for the reactions of 31 organic contaminants (10 chlorinated hydrocarbons, four brominated hydrocarbons, 11 aromatic hydrocarbons and their derivatives, three chloroacetic acid, and three chloroacetyl chloride) with oxidants of •OH, H2O2, •O−, O3, and O2 are calculated, and the mineralization order of ease is determined theoretically on the basis of ΔrGm°. The agreement of the theoretical and experimental mineralization abilities for most of the organic contaminants investigated implies the reliability of the determination of the mineralization ability from the magnitude of ΔrGm° for the mineralization of trace organic contaminants. Results also show that for most of the organic contaminants studied, the mineralization abilities are •OH > H2O2 > •O− > O3 > O2, and the mineralization ability of the organic contaminants depends on not only the oxidants but also the structure and properties of the organic contaminants themselves, and the degradation reaction products.
Co-reporter:Yang Bai, Yan Zhang, Wei Li, Xuefeng Zhou, Changsong Wang, Xin Feng, Luzheng Zhang, Xiaohua Lu
Applied Surface Science 2009 Volume 255(Issue 22) pp:9296-9300
Publication Date(Web):30 August 2009
DOI:10.1016/j.apsusc.2009.06.115
Abstract
We report an effective approach to fabricate nanopatterns of alkylsilane self-assembly monolayers (SAMs) with desirable coverage and feature size by gradient photocatalysis in TiO2 aqueous suspension. Growth and photocatalytic degradation of octadecyltrichlorosilane (OTS) were combined to fabricate adjustable monolayered nanopatterns on mica sheet in this work. Systematic atomic force microscopy (AFM) analysis showed that OTS-SAMs that have similar area coverage with different feature sizes and similar feature size with different area coverages can be fabricated by this approach. Contact angle measurement was applied to confirm the gradually varied nanopatterns contributed to the gradient of UV light illumination. Since this approach is feasible for various organic SAMs and substrates, a versatile method was presented to prepare tunable nanopatterns with desirable area coverage and feature size in many applications, such as molecular and biomolecular recognition, sensor and electrode modification.
Co-reporter:Yuanhui Ji, Zhuhong Yang, Xiaoyan Ji, Wenjuan Huang, Xin Feng, Chang Liu, Linghong Lu, Xiaohua Lu
Fluid Phase Equilibria 2009 Volume 277(Issue 1) pp:15-19
Publication Date(Web):15 March 2009
DOI:10.1016/j.fluid.2008.10.020
This paper is to investigate the degradation abilities of various chlorinated aliphatics, benzene and its derivatives in order to treat organic polluted wastewaters efficiently by advanced oxidation processes (AOPs). A thermodynamic method is proposed to calculate the standard molar Gibbs energy of formation for aqueous organic species. Using the method proposed, the standard molar Gibbs energies of formation for 31 aqueous organic species are obtained. Moreover, the molar Gibbs energy change of reaction ΔrGm0 for the organic species with hydroxyl radicals is calculated from the standard molar Gibbs energy of formation for aqueous organic species to determine the degradation order of ease for the organic species. New photocatalytic experiments are carried out for the model verification. The calculation results of the model agree with the available and new experimental results. This work shows that the thermodynamics of the degradation reaction for the organic pollutants in AOPs can find the corresponding relationships with the degradation reaction rate by experimental measurements. The work in this paper represents a success of thermodynamics for the application in environmental area.
Co-reporter:Yudan Zhu, Mingjie Wei, Qing Shao, Linghong Lu, Xiaohua Lu and Wenfeng Shen
The Journal of Physical Chemistry C 2009 Volume 113(Issue 3) pp:882-889
Publication Date(Web):2017-2-22
DOI:10.1021/jp8089006
The behavior of water molecules under nanoscale confinement has received considerable attention, especially for the influence caused by the modified groups of pores. To better design bionic nanodevices for future research, we anchored carboxyl acid (−COOH) groups onto the inner wall of a single-walled armchair carbon nanotube’s (CNT’s) central region to model the pore shape of aquaporin-1 and investigated the effect of modified groups on the structure of water molecules. The orientations and density distributions of water molecules in the CNTs and near the tube mouths have been studied by molecular dynamics simulation. The results indicate that water molecules confined inside the two unmodified regions have opposite and steady preferential dipole orientations pointing toward the −COOH groups on the central region of the CNT. Meanwhile the orientations of water molecules near the tube mouths which are certain distances away from the −COOH groups are also affected. This phenomenon becomes stronger as the number of −COOH groups increases and the CNT diameter decreases. In addition, the results show that the −COOH groups on the inner wall of the central region have a slight effect on the axial density distribution of the water molecules near the tube mouths, but a strong impact on that of water molecules inside the CNTs. Different distances between the −COOH groups and tube mouths can create diverse axial density distributions of water molecules.
Co-reporter:Wei Li, Chang Liu, Yaxin Zhou, Yang Bai, Xin Feng, Zhuhong Yang, Linghong Lu, Xiaohua Lu and Kwong-Yu Chan
The Journal of Physical Chemistry C 2008 Volume 112(Issue 51) pp:20539-20545
Publication Date(Web):2017-2-22
DOI:10.1021/jp808183q
A bicrystalline titanium dioxide nanofiber with enhanced photocatalytic activity was synthesized from potassium titanate K2Ti2O5 via ion exchange and calcination. The nanofiber has a core−shell crystalline structure with a thin TiO2(B) phase sheathing the anatase core, as characterized by X-ray diffraction, Raman spectroscopy, and high-resolution transmission microscopy (HRTEM). From HRTEM and local electron diffraction patterns, the two crystalline phases form a coherent interface with the 0.34-nm spacing between the (102) planes of TiO2(B) matching that between the anatase (101) lattice planes. The core−shell anatase/TiO2(B) nanofiber shows enhanced photocatalytic activity in iodine oxidation reaction with a 20−50% increase in extent of reaction compared to either single-crystal anatase or single-crystal TiO2(B) nanofibers. Anatase and TiO2(B) have the same band gap value of 3.2 eV, while theoretical calculations show the conduction band (CB) and valence band (VB) energies in anatase are both lower than the corresponding CB and VB energies in TiO2(B). The enhanced photocatalytic property may be due to enhanced and concerted charge mobility toward or away from the anatase/TiO2(B) interface. The special structure−property relationship can provide a new strategy to design and fabricate efficient photocatalytic and photovoltaic materials.
Co-reporter:Yaxin Zhou;Chang Liu;Ming He;Xin Feng
Journal of Materials Science 2008 Volume 43( Issue 1) pp:155-163
Publication Date(Web):2008 January
DOI:10.1007/s10853-007-2140-6
The splitting behavior and structural transformation process of K2Ti6O13 whiskers in various hydrothermal solutions were investigated by the X-ray diffraction technique, scanning electron microscopy, transmission electron microscopy, and atomic force microscopy. TiO2 (B) particle aggregates and rutile twinned crystals were produced respectively in diluted and concentrated HCl solutions via “dissolution-precipitation” mechanism, while no changes were observed in deionized water. In contrary to the chemical inertia of K2Ti6O13 whiskers in KOH solution, trititanate nanowires were synthesized by splitting the bulk K2Ti6O13 whiskers in NaOH solution. The driving force for the formation of nanowires originated from the intrinsic strain induced by the phase transition from K2Ti6O13 with a tunnel structure to layered trititanate.
Co-reporter:Qinghua Qian;Yuyan Hu;Gaofei Wen;Xin Feng
Frontiers of Chemical Science and Engineering 2008 Volume 2( Issue 3) pp:308-314
Publication Date(Web):2008 September
DOI:10.1007/s11705-008-0047-7
A new smooth potassium dititanate film was prepared by sol-gel method and characterized by thermogravimetry (TG) and differential scanning calorimetry (DSC), X-ray diffraction (XRD), atomic force microscopy (AFM), UV-Visible diffuse reflectance and Raman spectroscopy. The gaseous photocatalytic activity of smooth K2Ti2O5 films was studied using contact angle analysis from the photocatalytic decomposition of octadecyltrichlorosilane (OTS) based self-assembled monolayers (SAMs) formed on K2Ti2O5 films. The photocurrent response of the film was determined by an electrochemical method. It was shown that the films were smooth, compact, and transparent when formed on glass. Compared with TiO2 film, the K2Ti2O5 film showed wide absorption in the ultraviolet and visible region. It was found that the monolayers on K2Ti2O5 decomposed much faster than those on TiO2 under UV irradiation of 254 nm in air. The film also exhibited a stronger photoresponse and a more stable anodic photocurrent. The K2Ti2O5 film efficiently decomposes the alkylsiloxane monolayers under UVirradiation in air and it was found to be a good photocatalyst for gaseous organic pollutant treatment.
Co-reporter:Jingling Shao, Xiaolei Zhu, Xiaohua Lu, Rongwei Shi
Journal of Molecular Structure: THEOCHEM 2008 Volume 855(1–3) pp:82-91
Publication Date(Web):30 April 2008
DOI:10.1016/j.theochem.2008.01.013
The DFT(B3LYP)/6-311+G(d) method is used to study the geometries, growth feature, and relative stability of 106 lower-energy isomers of monocyclic CnB4 (n = 2–9). Vibrational frequency analysis is performed to ensure the optimized isomers are stable at the same level. It is interesting to find that the formation of the lowest-energy isomers of monocyclic CnB4 (n = 2–9) is through joining –Cm– (m = 0–2) and –Cn– (n = 0–5) carbon chains by two B–C–B bridges under the constraint of C2v symmetry (or D2h symmetry if m = n). The stability of global minimum isomers is discussed based on their binding energies per atom, the incremental binding energy, second order energy difference, vertical ionization energy, vertical electron affinity, and average polarizability.
Co-reporter:Chang Liu, Yuanhui Ji, Yang Bai, Fangqin Cheng, Xiaohua Lu
Fluid Phase Equilibria 2007 Volume 261(1–2) pp:300-305
Publication Date(Web):1 December 2007
DOI:10.1016/j.fluid.2007.07.052
The fractional crystallization of carnallite (KMgCl3·6H2O) in pure water, KCl aqueous solution and MgCl2 aqueous solution was studied. When carnallite was dissolved in pure water, porous crystals of KCl were formed. However, when the fractional crystallization in KCl or MgCl2 aqueous solution occured, the solid crystals of KCl with different morphologies were formed. A theoretical model was established to explore this phenomenon. The ionic product of carnallite and KCl with distance far from the surface of carnallite was analyzed in which the activity coefficient of KCl, MgCl2 and water were calculated with Lu–Maurer model. The dissolution of carnallite, the diffusion of KCl and MgCl2 in bulk solutions and the nucleation as well as crystal growth of KCl were investigated in detail. It was concluded that the coupling of the high dissolution rate of carnallite and the high nucleation rate of KCl nearby the surface of carnallite resulted in the porous crystals of KCl. TG and SEM analysis shows that high water content in KCl crystals prepared by fractional crystallization of carnallite in water results from the surface porous structure of crystals.
Co-reporter:Linghong Lu, Xiaohua Lu, Yuping Chen, Liangliang Huang, Qing Shao, Qi Wang
Fluid Phase Equilibria 2007 Volume 259(Issue 2) pp:135-145
Publication Date(Web):15 October 2007
DOI:10.1016/j.fluid.2007.06.030
Grand canonical Monte Carlo and configurational bias Monte Carlo techniques were employed to simulate the adsorption of binary mixtures of butane isomers and quaternary mixtures in nine zeolites at 300 K. For binary mixtures the results show there is a critical pore size, which is 10-membered-ring about 5.6 Å. The channel sizes of BEA, ISV, MOR and CFI are larger than this critical pore size, they prefer i-butane than n-butane, whereas TON with smaller channel size than critical pore size prefers n-butane than i-butane, but its selectivity decreases with pressure increasing. MFI, MEL and TER prefer i-butane than n-butane at low pressure, but with pressure increasing, the selectivity is reversed. BOG prefers i-butane than n-butane but the selectivity decreased with pressure increasing. It demonstrates that the adsorption and selectivity are controlled by both pore size and pore structure. The n-butane–i-butane–n-pentane–2-methylbutane quaternary mixtures adsorbed in these nine zeolites were studied, and the results show alkane chain length dependence at low pressure, but the adsorption is controlled by pore size and structure with pressure increasing in all the zeolites except for TON and BOG.
Co-reporter:Ying Chen, Hao Wang, Changsong Wang, Xin Feng, Xiaohua Lu
Acta Physico-Chimica Sinica 2007 Volume 23(Issue 8) pp:1168-1172
Publication Date(Web):August 2007
DOI:10.1016/S1872-1508(07)60062-X
The wetting behaviors of ethanol/water solutions with different surface tensions were studied on the rough superhydrophobic polytetrafluoroetylene (PTFE) coating. Air fraction was calculated using Cassie equation, and the forces were compared. The result indicated that the ethanol/water solutions gradually filled in the multilevel structures of the PTFE coating with the decrease in the surface tension. Only the stripe structure was filled when the surface tension was greater than 28 mN·m−1. When the surface tension was less than 28 mN·m−1, the papillary structure was also filled. Because it has little contribution to the air fraction, the surface could not be wetted when the stripe structure was filled. However, the surface could be wetted by the solutions when the papillary structure, which had a considerable contribution to the air fraction, was filled.
Co-reporter:Liang-Liang Huang, Qing Shao, Ling-Hong Lu, Xiao-Hua Lu, Lu-Zheng Zhang, Jun Wang and Shao-yi Jiang
Physical Chemistry Chemical Physics 2006 vol. 8(Issue 33) pp:3836-3844
Publication Date(Web):13 Jun 2006
DOI:10.1039/B604585P
Carbon nanotubes show exceptional properties that render them promising candidates as building blocks for nanostructured materials. Many ambitious applications, ranging from molecular detection to membrane separation, require the delivery of fluids, in particular aqueous solutions, through the interior of carbon nanotubes (CNT). To foster such applications, an understanding of the properties of water molecules confined in carbon nanotubes at the molecular level is needed. In this work we report a study of temperature and helicity effects on static properties of water molecules confined in modified CNT by molecular dynamics simulations. It was found that the temperature has little effect on the confined water molecules in carbon nanotubes. But on the other hand, the simulation results showed that because of the difference in helicity between (6, 6) and (10, 0) CNTs, the modification by hydrophilic carboxyl acid functional groups (–COOH) results in a different response to the CNTs, which in turn have control over the flow direction of water molecules in these CNTs.
Co-reporter:Lei Yu, Ming He, Chang Liu, Xiao-Hua Lu, Xin Feng
Materials Chemistry and Physics 2005 Volume 93(2–3) pp:342-347
Publication Date(Web):15 October 2005
DOI:10.1016/j.matchemphys.2005.03.021
The mixing mode of K2CO3 with anatase and hydrous titanium dioxide (TiO2·nH2O) was studied using thermoanalytical methods, XRD and TEM characterization. XRD results revealed a rather poor inter-contact in the anatase–K2CO3 system since there were no peaks attributed to potassic salts. On the contrary, the counterpart of the TiO2·nH2O–K2CO3 system demonstrated the well-defined peaks ascribed to KHCO3 and K2CO3·1.5H2O, at 25 and 300 °C, respectively. TEM characterization showed that TiO2 crystallites in the latter tended to disperse in a favourable way and the crystallite sizes were conspicuously smaller. These results suggested that the potassic salts could enter into the framework of TiO2·nH2O and restricted the further crystallite growth. Furthermore, the thermogravimetric analyses demonstrated the preferable nano-scale mixing status resulting from the reaction of K2CO3 with hydroxyl in the framework of TiO2·nH2O.
Co-reporter:Chang Liu, Xiaohua Lu, Gang Yu, Xin Feng, Qitu Zhang, Zhongzi Xu
Materials Chemistry and Physics 2005 Volume 94(2–3) pp:401-407
Publication Date(Web):15 December 2005
DOI:10.1016/j.matchemphys.2005.05.022
The reaction mechanism between hydrous titanium oxide and potassium carbonate at low temperatures was investigated by using XRD, TG, Raman spectra and TEM. In spite of the TiO2/K2O mole ratio in mixtures, the solid state reactions always consisted of two stages. In the first stage (500–640 °C), K2Ti3O7 nanocrystallites with layered structure were formed by the direct reaction of TiO2 and K2CO3 which was clearly confirmed by TG studies in CO2 atmosphere. In the second stage (≥640 °C), the TiO2/K2O mole ratio in mixtures affected the solid state reaction mechanism remarkably. A new transformation mechanism was found for mixtures with TiO2/K2O mole ratio of 4. The separation between the two stages could be used to control the morphologies and microstructures of potassium titanates by adjusting CO2 partial pressure in atmosphere.
Co-reporter:Ming He, Xiao-Hua Lu, Xin Feng, Lei Yu and Zhu-Hong Yang
Chemical Communications 2004 (Issue 19) pp:2202-2203
Publication Date(Web):19 Aug 2004
DOI:10.1039/B408609K
A new approach is developed to synthesize mesoporous fibrous titania from the sintered product of K2Ti2O5, which involves a novel hydrolytic reaction for the formation of potassium-rich nanophase and the generation of an amorphous intermediate.
Co-reporter:Hanbing He, Chang Liu, Xiaohua Lu
Chinese Journal of Chemical Engineering (October 2014) Volume 22(Issue 10) pp:1105-1110
Publication Date(Web):1 October 2014
DOI:10.1016/j.cjche.2013.04.001
The formation mechanism of K2Ti2O5 was investigated with TiO2 microparticles and nanoparticles as precursors by the thermogravimetric (TG) technique. A method of direct multivariate non-linear regression was applied for simultaneous calculation of solid-state reaction kinetic parameters from TG curves. TG results show more regular decrease from initial reaction temperature with TiO2 nanoparticles as raw material compared with TiO2 microparticles, while mass losses finish at similar temperatures under the experimental conditions. From the mechanism and kinetic parameters, the reactions with the two materials are complex consecutive processes, and reaction rate constants increase with temperature and decrease with conversion. The reaction proceedings could be significantly hindered when the diffusion process of reactant species becomes rate-limiting in the later stage of reaction process. The reaction active sites on initial TiO2 particles and formation of product layers may be responsible to the changes of reaction rate constant. The calculated results are in good agreement with experimental ones.A method of direct multivariate non-linear regression was applied for comparison of K2Ti2O5 kinetic parameters using TiO2 microparticles and nanoparticles as precursors. An obvious decrease in initial reaction temperature was observed using TiO2 nanoparticles as raw material. Both reactions are complex consecutive processes. The reaction mechanism and kinetic parameters were determined. The reaction active sites on initial TiO2 particles and product layer formation may be responsible to the changes of reaction rate constant.Download full-size image
Co-reporter:Dong Li, Kui Xiong, Zhuhong Yang, Chang Liu, Xin Feng, Xiaohua Lu
Catalysis Today (25 October 2011) Volume 175(Issue 1) pp:322-327
Publication Date(Web):25 October 2011
DOI:10.1016/j.cattod.2011.04.007
The process intensification (PI) of heterogeneous photocatalysis using the Kenics static mixer was investigated and its mechanism was proposed. Three model compounds (phenol, Cr(VI), and acid orange 7 (AO7)) with different photocatalytic reaction mechanisms were selected. The use of the Kenics static mixer increased the degradation rate of phenol from 20% to 150%, but appeared to have no effect on the photodegradation of Cr(VI) and AO7. However, with the addition of formic acid and NaF to the Cr(VI) and AO7 systems, respectively, the reaction mechanism shifted from a surface-mediated reaction to a radical-mediated reaction, and the photoreduction of Cr(VI) and photo-oxidation of AO7 using the Kenics static mixer exhibited higher reaction rates. In addition, the results of experiments with the terephthalic acid (TA) fluorescence probe indicated that the Kenics static mixer increased the yield of hydroxyl radicals. Based on the reaction mechanisms, we propose that the Kenics static mixer played a role in heterogeneous photocatalysis by creating intense mixing and increasing the interfacial mass transfer, which resulted in the enhanced mobility of reactive radicals from the catalyst surface or boundary layer to the solution. This approach intensified the heterogeneous photocatalysis process by enhancing the mass transfer of the reactive species rather than the reactant substrate, provided an alternative to the PI of heterogeneous photocatalysis, and allowed for easier engineering applications.Graphical abstractDownload high-res image (144KB)Download full-size imageHighlights► Static mixer is used to intensify heterogeneous photocatalysis. ► The intensification effects of five different systems are investigated. ► This method allows for easier engineering applications. ► A deep insight into the mechanism of heterogeneous photocatalytic process. ► Therefore, we hope our manuscript could hit the general interest of readers of Catalysis Today.
Co-reporter:Weijia Liu, Jian-guo Wang, Xiaojing Guo, Wei Fang, Mingjie Wei, Xiaohua Lu, Linghong Lu
Catalysis Today (16 May 2011) Volume 165(Issue 1) pp:32-40
Publication Date(Web):16 May 2011
DOI:10.1016/j.cattod.2011.01.016
The adsorption of methanol on hydroxylated TiO2-B (1 0 0) surface with bridging and terminal hydroxyl groups has been studied by first principle calculations. On both clean and hydroxylated surfaces with bridging OH group (OHbr), the O–H bond scission is the most favorable dissociation of methanol and the C–O bond scission is also feasible. This indicates the OHbr has little influence on the adsorption of methanol. The terminal OH group (OHt) plays a major role in the C–H scission of methanol, which is important for the applications associated with the direct use of hydrogen, such as in situ hydrogenation, and hydrogen generation via the photocatalytic reaction. The dissociative adsorption of methanol via C–H scission, which is an endothermic adsorption on other TiO2 surfaces, is identified as exothermic adsorption with adsorption energy in the range of −1.54 eV to −1.91 eV around OHt on TiO2-B (1 0 0) surface. The lowest activation barrier for C–H scission is ∼0.80 eV, which is lower than the release heat of molecular adsorption. Moreover, the hydrogen atoms in methanol are easily transferred to the OHt and then move to nearby O2c sites to regenerate the hydroxyl group. This proton migration process could result in extra stable chemi-sorption of methanol with an adsorption energy as low as −2.23 eV, which is above twice that of methanol molecularly adsorbed on the surface. Thus, the proton channel feature of OHt on the surface is borne out by our calculations.
Co-reporter:Yuanhui Ji, Xiaoyan Ji, Chang Liu, Xin Feng, Xiaohua Lu
Chemical Engineering Science (1 May 2010) Volume 65(Issue 9) pp:2649-2655
Publication Date(Web):1 May 2010
DOI:10.1016/j.ces.2009.12.045
Microstructure technologies have attracted interests in chemistry, chemical engineering, and biotechnology. To investigate the mass transfer of ions and crystallization of crystals in microscale and then to explain the formation mechanism of the porous structure materials, a microscale mathematical model for mass transfer processes coupling with local reactions is proposed in which the chemical potential gradient Δμ is used as the driving force to avoid the discontinuity of the kinetics equations in the micro-channels. Meanwhile, the dissolution kinetics of KCl at 298.15 K is measured to determine the dissolution rate constant kd and the average area of crystals Ac. The investigation for the fractional crystallization process of carnallite shows that the calculated mixing time versus channel width agree with the Einstein diffusion equation, which validates that the model can be used to describe the ion diffusion very well. Meanwhile, to have an accurate Δμ of KCl, in the channel width of or narrower than 2.0×10−6 m, it is enough to consider the diffusion only, while in the channel width of or wider than 2.0×10−5 m, diffusion should be coupled with reaction. The investigation also shows the vital of the consideration of the ionic activity coefficient for the investigated systems in micron scales. Moreover, the new formation mechanism of the porous structures in the inorganic material fabrication will be proposed from the process simulation for the synthesis of porous KCl, which will provide a reference for the porous structure formation in the advanced inorganic material synthesis.
Co-reporter:Qing Shao ; Jian Zhou ; Linghong Lu ; Xiaohua Lu ; Yudan Zhu ;Shaoyi Jiang
Nano Letter () pp:
Publication Date(Web):February 10, 2009
DOI:10.1021/nl803044k
We performed molecular dynamics simulations of the hydration of Na+ and K+ in infinitely long single-walled armchair carbon nanotubes (CNTs) at 298 K. Simulation results indicate that the preferential orientation of water molecules in coordination shells of these two cations presents an anomalous change in the CNTs and causes a diameter-dependent variation for the interaction energy between the cation and water molecules in its coordination shell. In the five CNTs of this work, it is energetically favorable for confining a hydrated K+ inside the two narrow CNTs with diameters of 0.60 and 0.73 nm, whereas the situation is reverse inside the wide CNTs with diameters of 0.87, 1.0, and 1.28 nm. This finding is important for CNT applications in ionic systems that control the selectivity and the ionic flow.
Co-reporter:Shasha Qian, Changsong Wang, Weijia Liu, Yinhua Zhu, Wenjun Yao and Xiaohua Lu
Journal of Materials Chemistry A 2011 - vol. 21(Issue 13) pp:NaN4952-4952
Publication Date(Web):2011/02/11
DOI:10.1039/C0JM03508D
To accomplish the more effective coupling of cadmium sulfide quantum dots (CdS QDs), the mesoporous TiO2 substrate and bifunctional linker, mercaptopropionic acid (MPA), were used to disperse and stabilize the CdS QDs. Due to the porous nano-architecture on the TiO2 substrate with large surface area and high crystallinity, the efficiency of degradation of organic compounds in aqueous solution under visible light irradiation is greatly enhanced, compared to CdS loaded anatase TiO2 without porous structure and common commercial P25. Furthermore, the bifunctional linking molecule, MPA, could effectively disperse and stabilize CdS nanoparticles. CdS/TiO2 with the linking molecule CdS-MPA-TiO2(m) exhibits much more stability and activity than CdS-TiO2(m) which is prepared by direct deposition. After 3 cycling tests of degradation of MB (methylene blue), the loss ratio of CdS on CdS-TiO2(m) is 70.6%, much larger than that of 17.8% on CdS-MPA-TiO2(m). This work may give ideas for the synthesis of other stable and active supported catalysts in many fields.
Co-reporter:Weijia Liu, Jian-guo Wang, Wei Li, Xiaojing Guo, Linghong Lu, Xiaohua Lu, Xin Feng, Chang Liu and Zhuhong Yang
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 31) pp:NaN8727-8727
Publication Date(Web):2010/06/16
DOI:10.1039/B920128A
By means of density functional theory (DFT) calculations, we study the water adsorption behavior on two common surfaces, (001) and (100) TiO2-B, which maintains the monoclinic structure as high as ∼550 °C or higher in ambient conditions. The two surfaces show totally different activity for water dissociation. The dissociative chemisorption of water on TiO2-B (100) is identified at both submonolayer and monolayer coverages, which indicates considerable reactivity. In contrast, the non-dissociative molecular adsorption of water is the most stable state on TiO2-B (001) which suggests no special activity. Furthermore, we compare the structural features of different surfaces with diverse crystal structures, such as rutile, anatase, brookite, TiO2-B etc. Keeping a close eye on the exposed atoms on the surface, we conclude a more general criterion for a quick evaluation of reactivities of different TiO2 surfaces merely based on local surface structure features.
Co-reporter:Ming-Jie Wei, Luzheng Zhang, Linghong Lu, Yudan Zhu, Keith E. Gubbins and Xiaohua Lu
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 48) pp:NaN16543-16543
Publication Date(Web):2012/05/28
DOI:10.1039/C2CP40687J
It is well known that titanium dioxide (TiO2) is biocompatible and environmentally friendly. Consequently, TiO2 is widely applied in many fields, such as implant materials, photocatalysis, pigments, cosmetic additives, etc. Mesoporous TiO2 finds many industrial applications, because of its high surface area and stable structure. However, the strong interaction between TiO2 and water molecules sometimes limits its application to solution environments. Our previous computational work showed that changes to the surface chemistry of TiO2 can affect the hydrogen bond network of water molecules on the TiO2 surface, and so influence the diffusion of water in the slits. Thus, a carbon-modified TiO2 surface could be an alternative way to avoid this limitation. In this work, a slit pore model with a modified TiO2 surface (pore widths 1.2 nm, 1.6 nm and 2.0 nm) with varying carbon coverages (0%, 7%, 47%, 53%, 93% and 100%) was presented. Molecular dynamics (MD) simulations were then performed to investigate the sorption and diffusion of water in these slits. Simulation results showed that the interfacial water molecules on bare TiO2 regions were little affected by the neighboring carbon, and they have the same properties as those on bare TiO2 surfaces. However, the diffusion of water molecules in the center of the slit was enhanced on increase of carbon coverage, because the carbon layer broke the hydrogen bond network between the interfacial water molecules and those on the bare TiO2 surface. It was found that in the slits (>1.2 nm) fully covered by carbon the diffusion coefficients of water are larger than that of bulk water. Moreover, large pore sizes caused an increase in the mobility of water molecules in carbon-modified TiO2, in agreement with previous experimental work.
Co-reporter:Wei Li, Yang Bai, Weijia Liu, Chang Liu, Zhuhong Yang, Xin Feng, Xiaohua Lu and Kwong-Yu Chan
Journal of Materials Chemistry A 2011 - vol. 21(Issue 18) pp:NaN6724-6724
Publication Date(Web):2011/03/31
DOI:10.1039/C1JM10115C
Single-crystalline anatase TiO2 nanofibers with highly reactive {001} facets were synthesized from layered potassium titanate K2Ti2O5via topotactic transformation in ion-exchange and dehydration. The cuboid fibers showed one-dimensional (1-D) orientation in the [010] direction and {001} as well as {100} facets enclosing along the longitudinal dimension. The structural evolution was deduced from XRD, SEM and TEM characterizations, which revealed that the highly reactive {001} facets were derived from the interlayer splitting and exfoliation of the layered precursors in a surfactant-free way. Photoluminescence (PL) measurements witnessed the efficient separation and transfer of photoinduced charge carriers in the single-crystalline and reactive facets enclosed TiO2 nanofibers. Sequently, highly efficient photocatalytic property of the nanofibers was demonstrated by phenol degradation and H2 evolution. Phenol degradation rate of nanofibers is 2.7 times of the irregular-shaped nanoparticle counterparts with the same crystal phase and similar specific surface area. In photocatalytic evolution of H2, nanofibers presented much higher and more stable activity than both nanoparticle counterparts and P25 benchmark. Charge carrier highways provided by single-crystalline 1-D structures and efficient surface reactivity offered by exposed facets are the two key factors for the enhanced properties.
Co-reporter:Wei Li, Yang Bai, Fujun Li, Chang Liu, Kwong-Yu Chan, Xin Feng and Xiaohua Lu
Journal of Materials Chemistry A 2012 - vol. 22(Issue 9) pp:
Publication Date(Web):
DOI:10.1039/C2JM14847A
Co-reporter:Licheng Li, Yudan Zhu, Xiaohua Lu, Mingjie Wei, Wei Zhuang, Zhuhong Yang and Xin Feng
Chemical Communications 2012 - vol. 48(Issue 94) pp:NaN11527-11527
Publication Date(Web):2012/10/09
DOI:10.1039/C2CC36157D
In this work, we report a novel surface modification which can improve the desorption of a hydrodesulfurization product (H2S) from mesoporous TiO2. The corresponding catalyst exhibits a significantly enhanced hydrodesulfurization performance compared with an unmodified catalyst, and the dibenzothiophene conversion increases from 65% to 98%.
Co-reporter:Yang Bai, Wei Li, Chang Liu, Zhuhong Yang, Xin Feng, Xiaohua Lu and Kwong-Yu Chan
Journal of Materials Chemistry A 2009 - vol. 19(Issue 38) pp:NaN7061-7061
Publication Date(Web):2009/08/04
DOI:10.1039/B910240J
A novel metal–semiconductor nanocomposite with stable metal nanoparticles and efficient photocatalytic performance has been prepared. It consists of highly dispersed ∼2 nm platinum (Pt) nanoparticles loaded on mesoporous and bicrystalline TiO2 fibers (Pt/mb-TiO2). Due to the well organized porous Pt/TiO2 nanoarchitecture, rate of photodegradation of CHCl3 was nearly doubled compared to both P25 and Pt/P25. And in photocatalytic production of H2 (with methanol as an electron donor), Pt/mb-TiO2 presented much more stable activity than Pt/P25. In a 20 h H2 production, Pt nanoparticles on P25 agglomerated and grew remarkably from 2.0 ± 0.5 nm to 4.5 ± 2.5 nm, while Pt nanoparticles on mb-TiO2 changed slightly from 2.0 ± 0.5 nm to 2.2 ± 0.5 nm. Moreover, the loss ratio of Pt on P25 is 36%, which is much larger than that of 7% on mb-TiO2. The efficient charge transfer in the interfaces of Pt/TiO2(B) and TiO2(B)/anatase was discussed. We concluded that the high photocatalytic performance of Pt/mb-TiO2 can be attributed to stable Pt nanoparticles supported on the mesoporous masonry frameworks of TiO2 and efficient charge transfer in the interfaces.
Co-reporter:Linghong Lu, Yudan Zhu, Fujun Li, Wei Zhuang, Kwong Yu Chan and Xiaohua Lu
Journal of Materials Chemistry A 2010 - vol. 20(Issue 36) pp:NaN7651-7651
Publication Date(Web):2010/08/06
DOI:10.1039/C0JM00054J
Titania carbon composites were prepared via in situ carbonization on mesoporous titania whiskers. Their microstructures were characterized by scanning electron microscopy (SEM) and X-ray diffraction (XRD), showing that the composites, after carbonization, still retain the original morphology of the whiskers and the crystalline structure of titania. Based on N2 sorption isotherms, the average pore sizes of the as-prepared composites were found to depend on the amount of filled carbon. The electrochemical capacitance performance of the as-prepared composites was investigated by cyclic voltammetry, electrochemical impedance spectroscopy (EIS) and galvanostatic charge–discharge cycles. Although the specific surface area of the composite TiO2/0.252C is moderate at 156 m2 g−1, its specific volumetric capacitance of 25 F cm−3 was much higher than the value of 10 F cm−3 for Vulcan XC-72, which has a specific surface area of 236 m2 g−1. This enhanced capacitance may come from the composite mesopores derived from porous titania whiskers. They provide readily accessible diffusion pathways for electrolyte ions. There is better conductivity with carbon in the composite. After 2000 cycles, we observed a change of −2.8%, −2.6% and −1.9% decrease in the specific volumetric capacitance compared to the values at the 100th cycle of the composites TiO2/0.252C, TiO2/0.143C and TiO2/0.08C, respectively. This decrease is small and significantly less than the 10% decrease of capacitance in Vulcan XC-72 in the same period. The more consistent capacitance in the composite suggests a more stable interface between titania, carbon filling and electrolyte compared to that of Vulcan XC-72 without titania.

![1H-Benzimidazole,2-[[(4-methoxy-3-methyl-2-pyridinyl)methyl]sulfinyl]-6-(1H-pyrrol-1-yl)-](http://img.cochemist.com/ccimg/172200/172152-36-2.png)
![1H-Benzimidazole,2-[[(4-methoxy-3-methyl-2-pyridinyl)methyl]sulfinyl]-6-(1H-pyrrol-1-yl)-](http://img.cochemist.com/ccimg/172200/172152-36-2_b.png)






![Cyclohexanone, 2,6-bis[(4-hydroxyphenyl)methylene]-, (2E,6E)-](http://img.cochemist.com/ccimg/247100/247051-33-8.png)
![Cyclohexanone, 2,6-bis[(4-hydroxyphenyl)methylene]-, (2E,6E)-](http://img.cochemist.com/ccimg/247100/247051-33-8_b.png)


![Cyclopentanone, 2,5-bis[(3,5-diethyl-4-hydroxyphenyl)methylene]-](http://img.cochemist.com/ccimg/195500/195440-25-6.png)
![Cyclopentanone, 2,5-bis[(3,5-diethyl-4-hydroxyphenyl)methylene]-](http://img.cochemist.com/ccimg/195500/195440-25-6_b.png)






![Benzenamine,2-[(1H-benzimidazol-2-ylsulfinyl)methyl]-N-methyl-N-(2-methylpropyl)-](http://img.cochemist.com/ccimg/104400/104340-86-5.png)
![Benzenamine,2-[(1H-benzimidazol-2-ylsulfinyl)methyl]-N-methyl-N-(2-methylpropyl)-](http://img.cochemist.com/ccimg/104400/104340-86-5_b.png)
![2-[(4-ethylsulfanyl-3-methylpyridin-2-yl)methylsulfinyl]-1h-benzimidazole](http://img.cochemist.com/ccimg/99500/99499-40-8.png)
![2-[(4-ethylsulfanyl-3-methylpyridin-2-yl)methylsulfinyl]-1h-benzimidazole](http://img.cochemist.com/ccimg/99500/99499-40-8_b.png)



















![Benzenesulfonic acid,4-[2-[4-(dimethylamino)phenyl]diazenyl]-](http://img.cochemist.com/ccimg/600/502-02-3.png)
![Benzenesulfonic acid,4-[2-[4-(dimethylamino)phenyl]diazenyl]-](http://img.cochemist.com/ccimg/600/502-02-3_b.png)










![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)




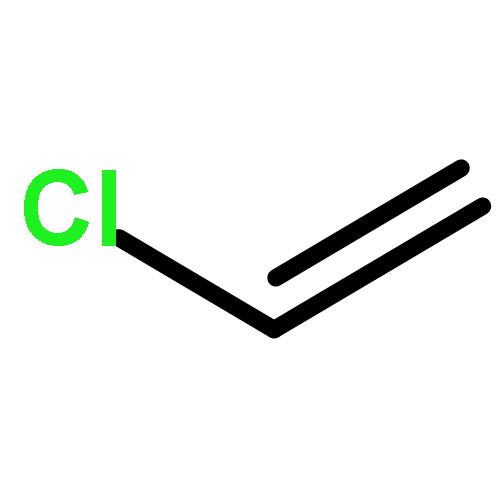


![Poly[(1,3-dihydro-1,3-dioxo-2H-isoindole-2,5-diyl)oxy(1,3-dihydro-1,3-dioxo-2H-isoindole-5,2-diyl)-1,4-phenyleneoxy-1,4-phenylene]](http://img.cochemist.com/ccimg/25800/25735-00-6.png)
![Poly[(1,3-dihydro-1,3-dioxo-2H-isoindole-2,5-diyl)oxy(1,3-dihydro-1,3-dioxo-2H-isoindole-5,2-diyl)-1,4-phenyleneoxy-1,4-phenylene]](http://img.cochemist.com/ccimg/25800/25735-00-6_b.png)