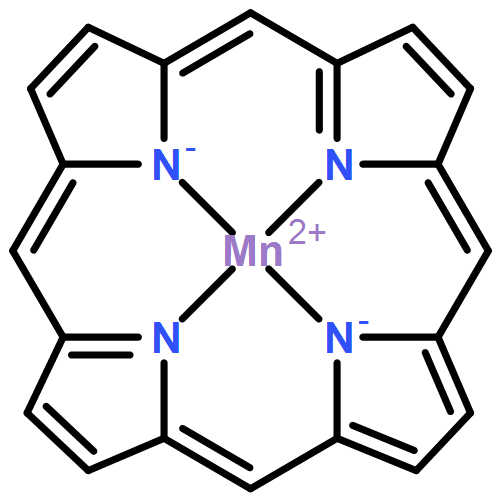
B3LYP density functional theory calculations were performed to quantify the binding affinities of six divalent first-row transition metals (Cr2+, Mn2+, Fe2+, Co2+, Ni2+, and Cu2+) for three well-known macrocyclic ligands (porphine, corrin, and 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane [TMC]). Our calculations show that, as expected from the neutral, monoanionic, and dianionic characters of the TMC, corrin, and porphine ligands, respectively, the binding energy increases in the order TMC < corrin < porphine. This is because a more anionic ligand gives rise to greater electrostatic stabilization upon interaction with the metal cations. For all ligands, the binding energy increases in the order Mn2+ < Cr2+ ∼ Fe2+ < Co2+ < Ni2+ < Cu2+. Single occupation of all five d orbitals in the high-spin Mn2+ complexes does not afford large stabilization due to either ligand-to-metal or metal-to-ligand charge transfer, thereby resulting in the minimum binding energies observed for Mn2+ among the six different metal ions considered.Graphical abstractB3LYP density functional theory calculations were performed to quantify the binding affinities of six divalent first-row transition metals (Cr2+, Mn2+, Fe2+, Co2+, Ni2+, and Cu2+) for three well-known macrocyclic ligands (porphine, corrin, and 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane [TMC]).
![Image for unlabelled figure]()
Highlights► Metal–ligand binding energy was calculated. ► Interactions between three ligands and six divalent metal ions were investigated. ► Different binding energies and spin-state preferences were observed.