Co-reporter:Dong Wang, Hairong Feng, Linna Li, Zhenlin Liu, Zhongli Yan, and Peng Yu
The Journal of Organic Chemistry October 20, 2017 Volume 82(Issue 20) pp:11275-11275
Publication Date(Web):September 27, 2017
DOI:10.1021/acs.joc.7b02063
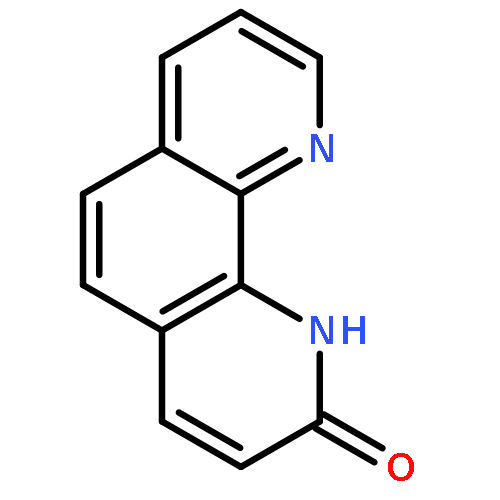
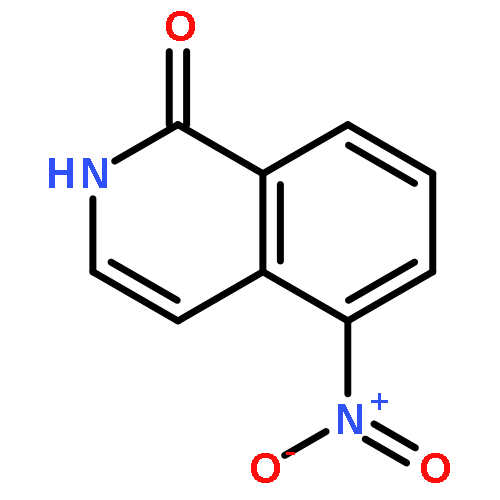
A concise and practical synthetic method has been developed for 8-azachromones, including 8-azaflavones, which have emerged as a promising class of compounds. Using commercially available nicotinates as the starting material, 8-azachromones were obtained in only three steps. The key intramolecular O-arylation reaction was achieved by nucleophilic attack of enolates to C2 of N-oxides under PyBrop or Ac2O activation conditions. These studies provide the basis for the access to 8-azachromones, enabling future work including the discovery and development of novel chromonoid drugs or other functional materials.
Co-reporter:Zhen Liu, Qi Zheng, Wenzhu Chen, Meng Wu, Guojun Pan, Ke Yang, Xuzhe Li, Shuli Man, Yuou Teng, Peng Yu, Wenyuan Gao
European Journal of Medicinal Chemistry 2017 Volume 125() pp:760-769
Publication Date(Web):5 January 2017
DOI:10.1016/j.ejmech.2016.09.066
•A combination of Paris Saponin I (PSI) and Camptothecin (CPT) was studied.•A combination of PSI and 10-hydroxycamptothecin (HCPT) was studied.•PSI enhanced the sensitivities of cancer cells to CPT/HCPT by inducing apoptosis.•PSI sensitized lung cancer cells to CPT and HCPT via MAPKs and Akt pathways.Paris Saponin I (PSI), a steroidal sponins isolated from plant, has been exhibited antitumor and many other biological activities. In this study, we investigated the role and underlying mechanisms of PSI in the synergistic regulation of antitumor activity of Camptothecin (CPT) and 10-hydroxycamptothecin (HCPT) in four types of lung cancer cells. The inhibitory evaluation showed that PSI could significantly reduce the CPT/HCPT-mediated cell proliferation and enhance the sensitivities of H1299, H460 and H446 lung cancer cells to CPT/HCPT. Mechanism study indicated that PSI improved the CPT/HCPT induced apoptosis in lung cancer cells through mitochondria pathway including cytochrome C release and activation of caspase-9 and -3 cascades. Furthermore, PSI plus CPT/HCPT also increased the up-regulation of Bax and down-regulation of Bcl-2 and Bcl-XL in H460 and H446 cells. Moreover, PSI enhanced CPT/HCPT-mediated inhibition of p38 MAPK and activation of phosphorylation of p38 MAPK in H1299 cells, and suppression of Akt and ERK pathways activation in H460 cells as well as in H446 cells. Collectively, our results demonstrated that PSI functions as a chemosensitizer by enhancing apoptosis through influencing p38 MAPK, ERK, and Akt pathways in lung cancer cells, and the combination with CPT/HCPT might be a promising strategy for the development of new therapeutic agents.
Co-reporter:Yuou Teng, Xuzhe Li, Ke Yang, Xuehui Li, Zijun Zhang, Luyao Wang, Zhijie Deng, Binbin Song, Zhihong Yan, Yongmin Zhang, Kui Lu, Peng Yu
European Journal of Medicinal Chemistry 2017 Volume 125() pp:335-345
Publication Date(Web):5 January 2017
DOI:10.1016/j.ejmech.2016.09.024
•Four ring-closing analogs of Desmethylxanthohumol were synthesized.•The catechol motif is critical for the antioxidant activity.•The dimers show better antioxidant activity than the corresponding monomers.•Compound 14d was identified as the most potent antioxidant activity.•Compound 14d decreased apoptosis in H2O2-treated PC12 cells.Four ring-closed analogs of natural prenylated chalcone desmethylxanthohumol (1) and their dimers were synthesized from the commercially available 1-(2,4,6-trihydroxyphenyl)ethan-1-one in five and six linear steps, respectively. The structures of the eight new derivatives were confirmed using1H NMR, 13C NMR and HRMS. The antioxidant activity of the new chalcone derivatives were evaluated in a PC12 cell model of H2O2-induced oxidative damage. The SAR studies suggested that the catechol motif was essential for the antioxidant activity. Moreover, the dimers showed better antioxidant activity than their corresponding monomers did. Among them, compound 14d was the most potent and increased PC12 cell viability from 25% to 85%. Flow cytometric analysis showed that compound 14d, the most potent compound, decreased the apoptotic PC12 cell percentage and significantly reduced the LDH release and 8-OHdG generation but increased the GSH levels in H2O2-treated PC12 cells. Furthermore, compound 14d had a higher FRAP value than that of gallic acid. It also reduced the stable ABTS+ free radical with a lower EC50 than that of gallic acid.Four ring-closing analogs of natural prenylated chalcone Desmethylxanthohumol (1) and their dimers were synthesized. The antioxidant activities of these new chalcone derivatives were evaluated in the PC12 cell model of hydrogen peroxide (H2O2)-induced oxidative damage.
Co-reporter:Xin Meng, Chuanming Ji, Chao Su, Di Shen, Yaxin Li, Peijie Dong, Ding Yuan, Mengya Yang, Song Bai, Demei Meng, Zhenchuan Fan, Yang Yang, Peng Yu, Tao Zhu
European Journal of Medicinal Chemistry 2017 Volume 134(Volume 134) pp:
Publication Date(Web):7 July 2017
DOI:10.1016/j.ejmech.2017.03.058
•An original neoglycoconjugate of PG-CRM197 has been synthesized.•The application of squaric acid chemistry afforded conjugates in good yield.•The conjugate without any adjuvant induced high antigen-specific IgG levels in mice.A PG-tb1 hapten from the West Beijing strains of Mycobacterium tuberculosis cell wall has been efficiently synthesized and conjugated to CRM197 in a simple way as linker-equipped carbohydrate by applying squaric acid chemistry for an original neoglycoprotein, creating a potent T-dependent conjugate vaccine. The intermediate monoester can be easily purified and the degree of incorporation can be monitored by MALDI-TOF mass spectrometry. After administered systemically in mice without any adjuvant, the conjugate induced high antigen-specific IgG levels in serum. Furthermore, following the third immunization, significant antibody titers frequently exceeding 0.8 million were observed in the sera of mice vaccinated with PG–CRM197 conjugate which showed the potential for preparation of TB vaccine.Download high-res image (218KB)Download full-size image
Co-reporter:Na Guo;Tiantian Hao;Xiuzhuan Shang;Tianle Zhang
Journal of Nanoparticle Research 2017 Volume 19( Issue 6) pp:205
Publication Date(Web):06 June 2017
DOI:10.1007/s11051-017-3897-4
A series of novel hydroxycamptothecin (HCPT) conjugates (13a–14d), which contained a polyethylene glycol moiety and disulfide bond, were designed and synthesized in five to six steps, with overall yields of 20–39%. The anticancer activities and toxicities of these new conjugates were evaluated using an in vitro MTT assay in K562, HepG2, and HT-29 cell lines and HUVECs. The conjugates displayed enhanced antitumor activity and reduced toxicity in comparison with their parent molecule, HCPT. Among these conjugates, compound 13a exhibited 100-fold better selectivity to the tumor cells than to HUVECs. TEM and DLS experiments demonstrated that 13a formed nanosized micelles with a diameter of approximately 200 nm in aqueous solution and that the conjugate could undergo glutathione-responsive degradation to release HCPT at the tumor site. The improved potency and reduced toxicity of these conjugates may be caused by the enhanced permeation and retention (EPR) effect of nanoparticles.
Co-reporter:Dong WangYuxi Wang, Junjie Zhao, Meng Shen, Jianyong Hu, Zhenlin Liu, Linna Li, Furen Xue, Peng Yu
Organic Letters 2017 Volume 19(Issue 5) pp:
Publication Date(Web):February 10, 2017
DOI:10.1021/acs.orglett.6b03771
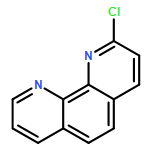
8-Azacoumarins have emerged as a promising class of compounds but are rarely explored due to challenging access. A novel, general, and practical method is provided for this class of compounds. The key lactonization step employs trans-acrylic acid attached pyridine N-oxides as the starting material, with acetic anhydride as both the activation agent and the solvent. Multiple transformations were involved in this reaction, including conjugate addition, nucleophilic aromatic substitution, and elimination. These studies provide the basis for access to 8-azacoumarins, enabling future work including the discovery and development of novel coumarin-type drugs, fluorescent probes, photolabile protecting groups, and other active molecules.
Co-reporter:Hua SunDong Wang, Xiaotong Song, Yazhou Zhang, Weina Ding, Xiaolin Peng, Xiaoting Zhang, Yashan Li, Ying Ma, Runling Wang, Peng Yu
Journal of Agricultural and Food Chemistry 2017 Volume 65(Issue 8) pp:
Publication Date(Web):January 29, 2017
DOI:10.1021/acs.jafc.6b05445
Inhibition of α-glucosidase and α-amylase decreases postprandial blood glucose levels and delays glucose absorption, making it a treatment strategy for type 2 diabetes. This study examined in vivo and in vitro antidiabetic activities of natural prenylchalconaringenins 1 and 2 and prenylnaringenins 3 and 4, found in hops and beer. 3′-Geranylchalconaringenin (2) competitively and irreversibly inhibited α-glucosidase (IC50 = 1.08 μM) with activity 50-fold higher than that of acarbose (IC50 = 51.30 μM) and showed moderate inhibitory activity against α-amylase (IC50 = 20.46 μM). Docking analysis substantiated these findings. In addition, compound 2 suppressed the increase in postprandial blood glucose levels and serum levels of total cholesterol and triglycerides in streptozotocin-induced diabetic mice. Taken together, these results suggest that 2 has dual inhibitory activity against α-glucosidase and α-amylase and alleviates diabetic hyperglycemia and hyperlipidemia, making it a potential functional food ingredient and drug candidate for management of type 2 diabetes.Keywords: diabetes; docking; prenylchalconaringenin; prenylnaringenin; α-amylase; α-glucosidase;
Co-reporter:Hua Sun, Weina Ding, Xiaotong Song, Dong Wang, Mingzhu Chen, Kaili Wang, Yazhou Zhang, Peng Yuan, Ying Ma, Runling Wang, Robert H. Dodd, Yongmin Zhang, Kui Lu, Peng Yu
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 15(Issue 15) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.bmcl.2017.06.040
A series of 6-hydroxyaurones and their analogues have been synthesized and evaluated for their in vitro α-glucosidase inhibitory and glucose consumption-promoting activity. These compounds exhibited varying degrees of α-glucosidase inhibitory activity, 11 of them showing higher potency than that of the control standard acarbose (IC50 = 50.30 μM). Surprisingly, analogues devoid of a substituent at C-2 but having an aryl group at C-5 were found to be highly active (e.g., 7f, IC50 = 9.88 μM). Docking analysis substantiated these findings. The kinetic analysis of compound 7f, the most potent α-glucosidase inhibitor of this study, revealed that it inhibited α-glucosidase in an irreversible and mixed competitive mode. In addition, compounds 7f and 10c exhibited significant glucose consumption promoting activity at 1 μM.Download high-res image (221KB)Download full-size image
Co-reporter:Kui Lu;Ke Yang;Xiaoliang Jia;Xing Gao;Xia Zhao;Guojun Pan;Yantao Ma;Qiyao Huang
Organic Chemistry Frontiers 2017 vol. 4(Issue 4) pp:578-586
Publication Date(Web):2017/03/28
DOI:10.1039/C6QO00726K
The first total synthesis of I3,II8-biapigenin and ridiculuflavone A via the key steps of regioselective iodination, Sonogashira reaction, and rhodium-catalyzed oxidative coupling with the overall yields of 31% and 22%, respectively, was reported. The structures of the key intermediates for the two natural products were confirmed by single-crystal X-ray analysis.
Co-reporter:Kui Lu, Yantao Ma, Meile Gao, Yan Liu, Ming Li, Chuanming Xu, Xia Zhao, and Peng Yu
Organic Letters 2016 Volume 18(Issue 19) pp:5038-5041
Publication Date(Web):September 23, 2016
DOI:10.1021/acs.orglett.6b02493
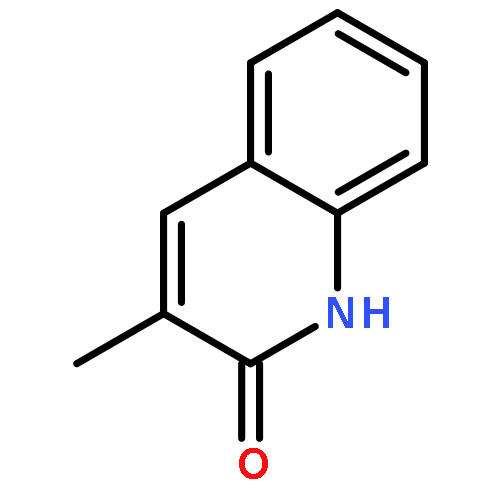
A four-component, 1,4-addition Ugi reaction using cyclic α,β-unsaturated ketones, carboxylic acids, amines, and isocyanides was developed for the first time. By combining this reaction with Michael addition, nucleophilic substitution, and C–N bond formation reactions, bicyclic and tricyclic scaffolds with pyridinone and quinolinone moieties, two basic units among a variety of natural products and pharmaceuticals, were constructed.
Co-reporter:Guojun Pan, Lianbo Zhao, Na Xiao, Ke Yang, Yantao Ma, Xia Zhao, Zhenchuan Fan, Yongmin Zhang, Qingwei Yao, Kui Lu, Peng Yu
European Journal of Medicinal Chemistry 2016 Volume 122() pp:674-683
Publication Date(Web):21 October 2016
DOI:10.1016/j.ejmech.2016.07.015
•A novel flavone 8-(6″-umbelliferyl)apigenin were synthesized for the first time.•Regio-selective iodination and Suzuki coupling reactions were used as key steps.•Analogues with different flavonoids cores were synthesized.•Effects of these compounds on glucose disposal were investigated in adipocytes.•The most potent could promote glucose consumption by 57% in 10 μΜ.The naturally occurring flavone 8-(6″-umbelliferyl)apigenin, a hybrid structure of apigenin and coumarin, as well as seven of its analogues were synthesized for the first time by using iodination and Suzuki coupling reactions as key steps. The synthesis of 8-(6″-umbelliferyl)-apigenin was achieved in seven linear steps from the commercially available 1-(2,4,6-trihydroxyphenyl)ethan-1-one and 7-hydroxyl coumarine with 31% overall yield. Effects of these compounds on glucose disposal were investigated in adipocytes. All of the flavonoid and coumarin hydrids were found to have better bioactivities than their corresponding flavonoid cores. The most potent compound 15 (10 μΜ) could promote glucose consumption by 57% which exhibited similar effect as the positive control metformin at 1 mM. Moreover, fluorescence microscopy showed that four 8-(6″-umbelliferyl)apigenin analogues 2, 15, 30 and 31 could promote the 2-NBDG uptake into 3T3-L1 cells, which consist with those observed in the regulation of glucose.A novel flavone 8-(6″-umbelliferyl)apigenin and its analogues were synthesized for the first time. The anti-diabetic activity evaluation results revealed these compounds could promote glucose consumption in adipocytes.
Co-reporter:Wen-Qiang Zhang, Yun He, Qun Yu, Hai-Peng Liu, De-Min Wang, Xiao-Bin Li, Jian Luo, Xin Meng, Hai-Juan Qin, Naomi W. Lucchi, Venkatachalam Udhayakumar, Suri S. Iyer, Yang Yang, Peng Yu
European Journal of Medicinal Chemistry 2016 Volume 121() pp:640-648
Publication Date(Web):4 October 2016
DOI:10.1016/j.ejmech.2016.05.069
•Natural product Matayoside D with antiplasmodial activity was fully synthesized.•Di-, tri-, tetra-, penta-, octa- and polyvalent glycoclusters of the natural product with modified linker was further prepared.•Polyvalent glycocluster proved to be more potent with IC50 values of less than 1.5 and 1.4 μM against 3D7 and W2 strains, respectively.An efficient and facile total synthesis of diglycoside Matayoside D isolated from the root bark of Matayba guianensis with antiplasmodial activity have been accomplished in 11 steps with 5% overall yields starting from commercially available glucose and rhamnose. Furthermore, a class of the diglycosidic derivatives with different lengths of the linker and valences were also prepared and evaluated for their antiplasmodial activities against chloroquine-susceptible (3D7) and chloroquine-resistant (W2) strains of Plasmodium falciparum. Low valent and short linker attached diglycoside show no enhancement of the antiplasmodial activity while polyvalent conjugates showed enhanced antiplasmodial activity with IC50 value at least 20 fold better than that of the corresponding diglycosidic monomer. The polyvalent diglycoside were non-cytotoxic against normal mammalian cells under 50,000 μg/L.
Co-reporter:Hua Sun, Yazhou Zhang, Weina Ding, Xue Zhao, Xiaotong Song, Dong Wang, Yashan Li, Kailin Han, Yang Yang, Ying Ma, Runling Wang, Dong Wang, Peng Yu
European Journal of Medicinal Chemistry 2016 Volume 123() pp:365-378
Publication Date(Web):10 November 2016
DOI:10.1016/j.ejmech.2016.07.044
•Three series of tetracyclic oxindole derivatives were synthesized and evaluated for α-glucosidase inhibitory activity.•Compound 6t displayed the most potent inhibitory activity against α-glucosidase (IC50 = 0.7 μM).•Compound 6t inhibited α-glucosidase in an irreversible and mixed manner.•Docking studies of compound 6t into α-glucosidase.α-Glucosidase inhibitors are known to prevent the digestion of carbohydrates and reduce the impact of carbohydrates on blood glucose. Three series of tetracyclic oxindole derivatives were designed, synthesized and evaluated for α-glucosidase inhibitory activity in vitro. Compound 6t exhibited the most potent inhibitory activity with IC50 0.7 μM and was about 170 times as active as acarbose (IC50 = 115.8 μM). The kinetic analysis of compound 6t revealed it inhibited α-glucosidase in an irreversible and mixed manner. Fluorescence spectra indicated that 6t directly bound to α-glucosidase. Docking simulation showed the existence of potential H-bonding, van der Waals, Pi and Sigma-Pi interactions between 6t and α-glucosidase.
Co-reporter:Yu-Ou Teng, Hong-Ye Zhao, Jing Wang, Huan Liu, Mei-Le Gao, Yao Zhou, Kai-Lin Han, Zhen-Chuan Fan, Yong-Min Zhang, Hua Sun, Peng Yu
European Journal of Medicinal Chemistry 2016 Volume 112() pp:145-156
Publication Date(Web):13 April 2016
DOI:10.1016/j.ejmech.2015.12.050
•The antitumor SAR studies of fourty-three novel isatin derivatives were performed.•The combination of C-5 and N-substitution may enhance their cytotoxicy.•Compound 2h displayed the most potent cytotoxic activity (IC50 = 0.03 μM).•2h inhibited Jurkat’ proliferation by inducing mitochondrial-mediated apoptosis.A series of novel di- or trisubstituted isatin derivatives were designed and synthesized in 5–6 steps in 25–45% overall yields. Their structures were confirmed by 1H NMR and 13C NMR as well as LC-MS. The anticancer activity of the fourty-three new isatin derivatives against human T lymphocyte cells Jurkat was evaluated by MTT assay in vitro. SAR study suggested that the combination of 1-benzyl and 5-[trans-2-(methoxycarbonyl)ethen-1-yl] substitution greatly enhanced their cytotoxic activity. Among them, compound 2h was shown to have a significant cytotoxic activity with an IC50 value of 0.03 μM, more than 330-fold higher than that of it's mother molecule isatin. Investigation of the cell morphology changes and annexin-V/PI staining study demonstrated that compound 2h inhibited the proliferation of Jurkat cells by inducing apoptosis. Since compound 2h induced the dissipation of mitochondrial membrane potential and the activation of caspase-3, it was obvious that compound 2h inhibited the proliferation of Jurkat cells through the mitochondrial apoptotic pathway. Other than this, compound 2h exerted inhibition effect to many other tumor cells and only showed weak cytotoxic to human normal cells suggesting that compound 2h possessed a broad range of anticancer spectrum and high safety to normal cells.A series of di-or trisubstituted novel isatin derivatives were synthesized. The anti-cancer activity evaluation results revealed these isatin derivatives inhibit Jurkat proliferation by inducing mitochondrial-mediated apoptosis.
Co-reporter:Zhao-Liang Yang, Xiong-Fei Zeng, Hai-Peng Liu, Qun Yu, Xin Meng, Zhong-Li Yan, Zhen-Chuan Fan, Hai-Xia Xiao, Suri S. Iyer, Yang Yang, Peng Yu
Tetrahedron Letters 2016 Volume 57(Issue 24) pp:2579-2582
Publication Date(Web):15 June 2016
DOI:10.1016/j.tetlet.2016.04.079
•Mono-, di-, tetra-, and octa-valent difluorinated zanamivir analogs as potent inhibitors of influenza virus were synthesized.•Neuraminidase inhibition assay was used to evaluate their anti-influenza virus activity against H7N9 virus like particle.•Octa-valent difluorinated zanamivir (OFax) displayed the highest anti-influenza activity with an IC50 at 30.59 nM.An efficient synthesis of mono-, di-, tetra-, and octa-valent difluorinated zanamivir analogs as potent inhibitors of influenza virus is reported. The mono difluorinated zanamivir with an azide linker attached at the C-7 position was synthesized in good yield from sialic acid using Selectfluor® and (diethylaminosulfur trifluoride) DAST as the fluorination reagent. This key intermediate was attached on various alkynlated scaffolds via “Click Chemistry” to afford multivalent glycoclusters. A neuraminidase inhibition assay was used to evaluate the compounds for their inhibitory activity using H7N9 virus like particle. These multivalent sialosides show enhanced inhibition with IC50 values ∼100-fold better than the monomer indicative of the strength of using a multivalent approach.
Co-reporter:Yang Yang, Hai-Peng Liu, Qun Yu, Mei-Bing Yang, De-Min Wang, Tian-Wei Jia, Hao-Jie He, Yun He, Hai-Xia Xiao, Suri S. Iyer, Zhen-Chuan Fan, Xin Meng, Peng Yu
Carbohydrate Research 2016 Volume 435() pp:68-75
Publication Date(Web):29 November 2016
DOI:10.1016/j.carres.2016.09.017
•Di-, tri- and tetra-valent S-sialosides as moderate NA inhibitors were synthesized.•Different density of S-sialoside coated protein as potent HA inhibitor were prepared.•Sialylation of proteins does not change their secondary structures.•The conjugates showed no cytotoxicity with the concentrations up to 100 μM.A new class of S-sialoside Human Serum Albumin (HSA) and Bovine Serum Albumin (BSA) conjugates were prepared to enhance the binding affinity to hemagglutinin (HA) and neuraminidase (NA). The valency of glycoconjugates was controlled by the reaction ratio of the S-sialoside monomer and protein. Hemagglutination inhibition assay showed that these synthetic glycoproteins have higher affinity to HA than the small clusters of sialosides with lower valency, due to multivalent effect and optimized three dimensional presentation of sialosides on the protein platform. The results of fluorescent NA inhibition assay showed that some of the conjugates have moderate NA inhibitory activity, in comparison to the monomer and low valent conjugates with weak or none inhibitory activity. These synthetic sialylated proteins were not cytotoxic with concentrations up to 100 μM, since the sialylation did not change the secondary structure of protein. This new kind of conjugates can be used as lead compounds for antiviral drug design and the construction of pseudo sialoside-protein conjugates library to investigate the carbohydrate-HA/NA recognition process and a platform for the influenza virus capturing.
Co-reporter:Dong Wang, Yuxi Wang, Junjie Zhao, Linna Li, Longfei Miao, Dong Wang, Hua Sun, Peng Yu
Tetrahedron 2016 Volume 72(Issue 38) pp:5762-5768
Publication Date(Web):22 September 2016
DOI:10.1016/j.tet.2016.07.083
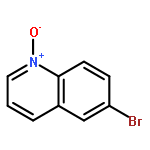
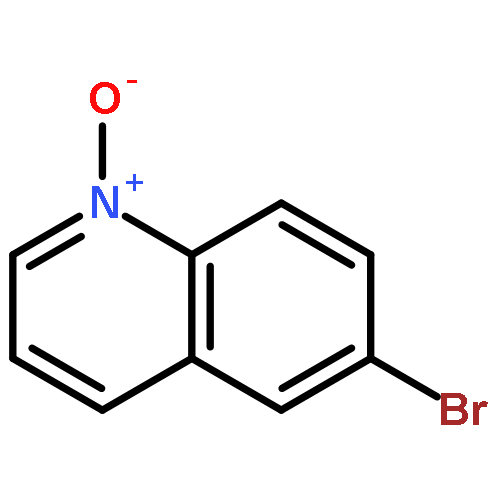
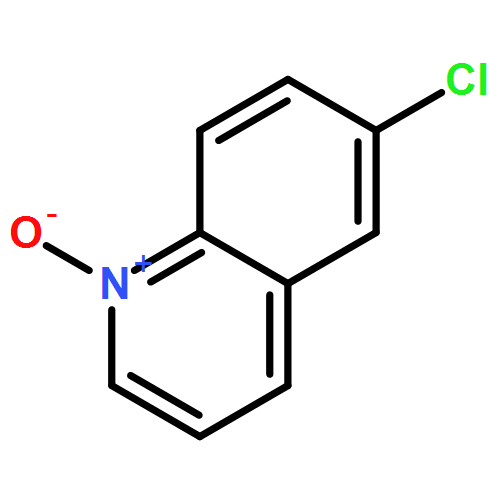
A novel, simple and practical method for the regioselective halogenation of fused heterocyclic N-oxides has been developed. It employs Vilsmeier reagent, generated in situ by POX3 and DMF, as both the activating agent and the nucleophilic halide source. The method is amenable across a broad range of substrates, including quinolines, isoquinolines and the diazine N-oxides, possessing a variety of substitution patterns. Furthermore, all of the reagents associated are cheap and easy to obtain. The potential extension of this method to a one-pot oxidation/halogenation sequence that obviates the need for isolation of the N-oxide intermediates is also presented.A novel, mild and practical method for the regioselective halogenation of fused azine N-oxides and diazine N-oxides has been developed. The procedure is operationally simple, compatible with a broad range of functional groups, and all of the reagents associated are cheap and easy to obtain. The potential extension of this method to a one-pot oxidation/bromination sequence that obviates the need for isolation of the N-oxide intermediates is also presented.
Co-reporter:Hao-Meng Wang, Li Zhang, Jiang Liu, Zhao-Liang Yang, Hong-Ye Zhao, Yao Yang, Di Shen, Kui Lu, Zhen-Chuan Fan, Qing-Wei Yao, Yong-Min Zhang, Yu-Ou Teng, Yu Peng
European Journal of Medicinal Chemistry 2015 Volume 92() pp:439-448
Publication Date(Web):6 March 2015
DOI:10.1016/j.ejmech.2015.01.007
•Four natural chalcones bearing prenyl or geranyl groups were synthesized.•Eleven 3′ or/and 5′-prenylated/geranylated chalcones analogs were prepared.•5′-prenylation/geranylation of the chalcones enhanced the cytotoxicy 7–10 folds.•These chalcone derivatives inhibited K562' proliferation by inducing apoptosis.Four natural chalcones bearing prenyl or geranyl groups, i.e., bavachalcone (1a), xanthoangelol (1b), isobavachalcone (1c), and isoxanthoangelol (1d) were synthesized by using a regio-selective iodination and the Suzuki coupling reaction as key steps. The first total synthesis of isoxanthoangelol (1d) was achieved in 36% overall yield. A series of diprenylated and digeranylated chalcone analogs were also synthesized by alkylation, regio-selective iodination, aldol condensation, Suzuki coupling and [1,3]-sigmatropic rearrangement. The structures of the 11 new derivatives were confirmed by 1H NMR, 13C NMR and HRMS. The anticancer activity of these new chalcone derivatives against human tumor cell line K562 were evaluated by MTT assay in vitro. SAR studies suggested that the 5′-prenylation/geranylation of the chalcones significantly enhance their cytotoxic activity. Among them, Bavachalcone (1a) displayed the most potent cytotoxic activity against K562 with IC50 value of 2.7 μM. The morphology changes and annexin-V/PI staining studies suggested that those chalcone derivatives inhibited the proliferation of K562 cells by inducing apoptosis.A series of prenyl and geranyl substituted chalcone natural products and their derivatives were synthesized. The anti-cancer activity evaluation results revealed these chalcone derivatives inhibit K562 proliferation by inducing apoptosis.
Co-reporter:Xia Zhao, Fang Ding, Jingyu Li, Kui Lu, Xiaoyu Lu, Bin Wang, Peng Yu
Tetrahedron Letters 2015 Volume 56(Issue 3) pp:511-513
Publication Date(Web):14 January 2015
DOI:10.1016/j.tetlet.2014.12.029
A mild method was developed for the direct C–H iodination of 1,3-azoles catalysed by CuBr2. Compared with the traditional metalation/iodination sequences carried out with nBuLi or TMPLi (TMP = 2,2,6,6-tetramethylpiperidino), a relatively weaker base, LiOtBu, was used in the presence of 1,10-phenanthroline. Five series of 1,3-azoles, including benzoxazole, benzothiozole, N-methyl-benzoimidazole, 5-phenyloxazole and 2-phenyl-1,3,4-oxadiazole were tested and afforded the corresponding iodination products.
Co-reporter:Kailin Han, Yashan Li, Yazhou Zhang, Yuou Teng, Ying Ma, Meiyan Wang, Runling Wang, Weiren Xu, Qingwei Yao, Yongmin Zhang, Haijuan Qin, Hua Sun, Peng Yu
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 7) pp:1471-1475
Publication Date(Web):1 April 2015
DOI:10.1016/j.bmcl.2015.02.031
A series of novel tetracyclic oxindole derivatives were synthesized via tandem Suzuki coupling–Michael addition reaction catalyzed by palladium. Twenty derivatives were designed and synthesized in 6–8 steps in 8–20% overall yields. Their structures were confirmed by 1H, 13C NMR and LC/MS. These compounds were evaluated for α-glucosidase inhibitory activity in vitro. Compounds 7c, 7d, 7e, 7g, 7h, and 7i exhibited IC50 values of 32.3, 12.1, 15.7, 29.0, 16.0, and 4.8 μM, respectively, with potency all higher than that of the control standard acarbose (IC50 = 115.8 μM). Molecular docking studies revealed the existence of potential hydrogen bonding and hydrophobic interaction between the enzyme and the active compound 7i.
Co-reporter:Guojun Pan, Yantao Ma, Ke Yang, Xia Zhao, Hui Yang, Qingwei Yao, Kui Lu, Tao Zhu, Peng Yu
Tetrahedron Letters 2015 Volume 56(Issue 30) pp:4472-4475
Publication Date(Web):22 July 2015
DOI:10.1016/j.tetlet.2015.05.096
Two novel flavones (±)-Anastatins A and B, each containing a benzofuran moiety, were synthesized for the first time by using bromination, Suzuki coupling reaction, and an oxidation/Oxa-Michael reaction cascade as the key steps. The syntheses of (±)-Anastatins A and B were achieved in eight steps from the commercially available phloroglucinol with 9% and 10% overall yield, respectively.
Co-reporter:Hua Sun, Yashan Li, Xiaoting Zhang, Yanan Lei, Weina Ding, Xue Zhao, Haomeng Wang, Xiaotong Song, Qingwei Yao, Yongmin Zhang, Ying Ma, Runling Wang, Tao Zhu, Peng Yu
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 20) pp:4567-4571
Publication Date(Web):15 October 2015
DOI:10.1016/j.bmcl.2015.08.059
Three series of prenylated and/or geranylated flavonoids were synthesized and evaluated for their α-glucosidase inhibitory activity. The 3′,5′-digeranylated chalcone (16) was identified as a new α-glucosidase inhibitor whose activity (IC50 = 0.90 μM) was 50-fold more than that of acarbose (IC50 = 51.32 μM). Molecular docking studies revealed the existence of strong hydrophobic interaction and H-bonding between compound 16 and α-glucosidase’s active site. The inhibitory mode analysis showed that 16 exhibited a competitive inhibitory mode.
Co-reporter:Kailin Han, Yao Zhou, Fengxi Liu, Qiannan Guo, Pengfei Wang, Yao Yang, Binbin Song, Wei Liu, Qingwei Yao, Yuou Teng, Peng Yu
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 2) pp:591-594
Publication Date(Web):15 January 2014
DOI:10.1016/j.bmcl.2013.12.001
Forty four di- or trisubstituted novel isatin derivatives were designed and synthesized in 5–6 steps in 25–45% overall yields. Their structures were confirmed by 1H NMR and 13C NMR as well as LC–MS. The anticancer activity of these new isatin derivatives against three human tumor cell lines, K562, HepG2 and HT-29, were evaluated by MTT assay in vitro. SAR studies suggested that the combination of 1-benzyl and 5-[trans-2-(methoxycarbonyl)ethen-1-yl] substitution greatly enhance their cytotoxic activity, whereas an intact carbonyl functionality on C-3 as present in the parent ring is required to such a potency. This study leads to the identification of two highly active molecules, compounds 2h (IC50 = 3 nM) and 2k (IC50 = 6 nM), against human leukemia K562 cells.
Co-reporter:Haomeng Wang, Zhihong Yan, Yanan Lei, Kai Sheng, Qingwei Yao, Kui Lu, Peng Yu
Tetrahedron Letters 2014 Volume 55(Issue 4) pp:897-899
Publication Date(Web):22 January 2014
DOI:10.1016/j.tetlet.2013.12.044
Highlights•First total synthesis of isoxanthoangelol was reported in 36% overall yield.•The improved syntheses of three natural C-alkyl chalcones were reported.•Regio-selective iodination and Suzuki coupling reaction were used as key steps.Four natural chalcones bearing prenyl or geranyl groups, i.e., isobavachalcone (1), bavachalcone (2), xanthoangelol (3), and 2′,4′,4-trihydroxy-5′-geranylchalcone (isoxanthoangelol, 4) were synthesized by using a regio-selective iodination and the Suzuki coupling reaction as key steps. Among them, the first total synthesis of 2′,4′,4-trihydroxy-5′-geranylchalcone was achieved in 36% overall yield. Comparing with the reported methods based on C-alkylation or O-alkylation followed by Claisen rearrangement to introduce the side chain, this new strategy capitalizes on a precious regiochemical control during iodination. The overall yields for the synthesis of the first three chalcones were improved from 17% to 53%, 12% to 35%, and 28% to 50%, respectively.
Co-reporter:Kui Lu, Jie Chu, Haomeng Wang, Xiaoli Fu, Dewu Quan, Hongxia Ding, Qingwei Yao, Peng Yu
Tetrahedron Letters 2013 Volume 54(Issue 47) pp:6345-6348
Publication Date(Web):20 November 2013
DOI:10.1016/j.tetlet.2013.09.051
Regioselective synthesis of C-6 and C-8 monoiodo flavonoids, which are important intermediates for the synthesis of flavonoid natural products and drug molecules, was achieved by iodination of suitably alkylated flavonoids with N-iodosuccinimide (NIS) in DMF. The iodination gives either a C-6 or C-8 iodo flavonoid in high yield, depending on the protection pattern of the C-5 and C-7 OH groups. The mild and neutral conditions render this novel protocol particularly useful for the regioselective iodination of acid-sensitive substrates.
Co-reporter:Jinling Cai, Minglang Chen, Guangce Wang, Guanghua Pan, Peng Yu
Algal Research (November 2015) Volume 12() pp:295-299
Publication Date(Web):November 2015
DOI:10.1016/j.algal.2015.09.014
Co-reporter:Kui Lu, Ke Yang, Xiaoliang Jia, Xing Gao, Xia Zhao, Guojun Pan, Yantao Ma, Qiyao Huang and Peng Yu
Inorganic Chemistry Frontiers 2017 - vol. 4(Issue 4) pp:
Publication Date(Web):
DOI:10.1039/C6QO00726K































![ETHANONE, 1-[2,6-DIHYDROXY-4-(METHOXYMETHOXY)PHENYL]-](http://img.cochemist.com/ccimg/257700/257621-57-1.png)
![ETHANONE, 1-[2,6-DIHYDROXY-4-(METHOXYMETHOXY)PHENYL]-](http://img.cochemist.com/ccimg/257700/257621-57-1_b.png)













![2-Propen-1-one,3-(4-hydroxyphenyl)-1-[2,4,6-trihydroxy-3-(3-methyl-2-buten-1-yl)phenyl]-,(2E)-](http://img.cochemist.com/ccimg/115100/115063-39-3.png)
![2-Propen-1-one,3-(4-hydroxyphenyl)-1-[2,4,6-trihydroxy-3-(3-methyl-2-buten-1-yl)phenyl]-,(2E)-](http://img.cochemist.com/ccimg/115100/115063-39-3_b.png)
![2H-Pyrano[2,3-b]quinolin-2-one](http://img.cochemist.com/ccimg/101400/101382-63-2.png)
![2H-Pyrano[2,3-b]quinolin-2-one](http://img.cochemist.com/ccimg/101400/101382-63-2_b.png)



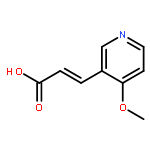
![3-(1-oxy-[3]pyridyl)-propionic acid](http://img.cochemist.com/ccimg/98500/98491-89-5.png)
![3-(1-oxy-[3]pyridyl)-propionic acid](http://img.cochemist.com/ccimg/98500/98491-89-5_b.png)







![Ethanone, 1-[2-hydroxy-4,6-bis(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/65500/65490-09-7.png)
![Ethanone, 1-[2-hydroxy-4,6-bis(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/65500/65490-09-7_b.png)
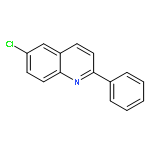
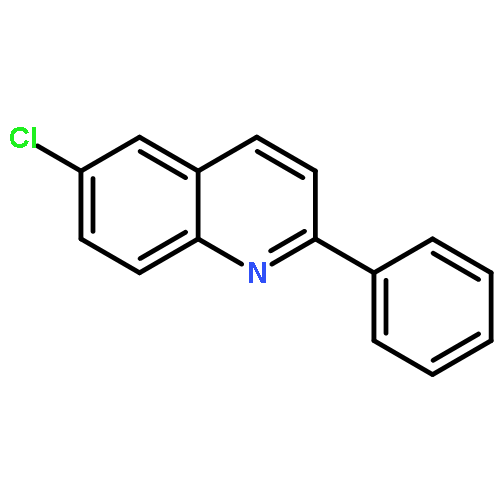


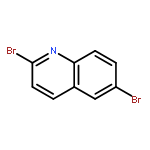
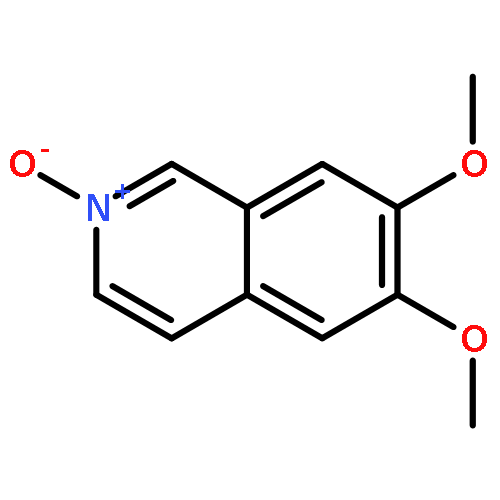
![(R)-(6-methoxy-1-oxido-quinolin-1-ium-4-yl)-[(2S,5R)-5-vinylquinuclidin-2-yl]methanol](http://img.cochemist.com/ccimg/54900/54821-44-2.png)
![(R)-(6-methoxy-1-oxido-quinolin-1-ium-4-yl)-[(2S,5R)-5-vinylquinuclidin-2-yl]methanol](http://img.cochemist.com/ccimg/54900/54821-44-2_b.png)
















































![1-[(e)-2-(4-chlorophenyl)vinyl]-3,5-dimethoxybenzene](http://img.cochemist.com/ccimg/1032600/1032508-03-4.png)
![1-[(e)-2-(4-chlorophenyl)vinyl]-3,5-dimethoxybenzene](http://img.cochemist.com/ccimg/1032600/1032508-03-4_b.png)
![Benzene, 1,3-dimethoxy-5-[(1E)-2-(3-nitrophenyl)ethenyl]-](http://img.cochemist.com/ccimg/877200/877143-36-7.png)
![Benzene, 1,3-dimethoxy-5-[(1E)-2-(3-nitrophenyl)ethenyl]-](http://img.cochemist.com/ccimg/877200/877143-36-7_b.png)







![Benzenamine, 4-[(1E)-2-(3,5-dimethoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/586500/586410-15-3.png)
![Benzenamine, 4-[(1E)-2-(3,5-dimethoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/586500/586410-15-3_b.png)


![Benzamide, N-[3-(1H-benzimidazol-2-yl)phenyl]-4-methoxy-](http://img.cochemist.com/ccimg/477500/477493-04-2.png)
![Benzamide, N-[3-(1H-benzimidazol-2-yl)phenyl]-4-methoxy-](http://img.cochemist.com/ccimg/477500/477493-04-2_b.png)
![BENZAMIDE, N-[3-(1H-BENZIMIDAZOL-2-YL)PHENYL]-2-METHYL-](http://img.cochemist.com/ccimg/404900/404831-36-3.png)
![BENZAMIDE, N-[3-(1H-BENZIMIDAZOL-2-YL)PHENYL]-2-METHYL-](http://img.cochemist.com/ccimg/404900/404831-36-3_b.png)
![1-[3-(2-PYRIDINYL)-1,2,4-OXADIAZOL-5-YL]ETHANAMINE](http://img.cochemist.com/ccimg/391700/391613-98-2.png)
![1-[3-(2-PYRIDINYL)-1,2,4-OXADIAZOL-5-YL]ETHANAMINE](http://img.cochemist.com/ccimg/391700/391613-98-2_b.png)
![Benzene, 1,3-dimethoxy-5-[(1E)-2-(2-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/354900/354803-31-9.png)
![Benzene, 1,3-dimethoxy-5-[(1E)-2-(2-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/354900/354803-31-9_b.png)


![Benzamide, N-[3-(1H-benzimidazol-2-yl)phenyl]-2,6-dimethoxy-](http://img.cochemist.com/ccimg/305400/305358-08-1.png)
![Benzamide, N-[3-(1H-benzimidazol-2-yl)phenyl]-2,6-dimethoxy-](http://img.cochemist.com/ccimg/305400/305358-08-1_b.png)




















![2,4-Imidazolidinedione, 5-[(4-fluorophenyl)methylene]-, (Z)-](http://img.cochemist.com/ccimg/140900/140894-74-2.png)
![2,4-Imidazolidinedione, 5-[(4-fluorophenyl)methylene]-, (Z)-](http://img.cochemist.com/ccimg/140900/140894-74-2_b.png)




![Phosphonic acid, [(3,4,5-trimethoxyphenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/134100/134029-74-6.png)
![Phosphonic acid, [(3,4,5-trimethoxyphenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/134100/134029-74-6_b.png)
![Benzene, 1,2,3-trimethoxy-5-[(1E)-2-(4-nitrophenyl)ethenyl]-](/data/chemimg/1681200/134029-66-6.png)
![Benzene, 1,2,3-trimethoxy-5-[(1E)-2-(4-nitrophenyl)ethenyl]-](/data/chemimg/1681200/134029-66-6_b.png)
![Benzene, 5-[2-(4-chlorophenyl)ethenyl]-1,2,3-trimethoxy-, (E)-](/data/chemimg/132100/134029-64-4.png)
![Benzene, 5-[2-(4-chlorophenyl)ethenyl]-1,2,3-trimethoxy-, (E)-](/data/chemimg/132100/134029-64-4_b.png)
![Benzene,1,2,3-trimethoxy-5-[(1E)-2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/134100/134029-62-2.png)
![Benzene,1,2,3-trimethoxy-5-[(1E)-2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/134100/134029-62-2_b.png)


![Benzene, 1-methoxy-2-[(1E)-2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/123900/123871-49-8.png)
![Benzene, 1-methoxy-2-[(1E)-2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/123900/123871-49-8_b.png)




![2H-Indol-2-one, 3-[(3,4-dichlorophenyl)methylene]-1,3-dihydro-](http://img.cochemist.com/ccimg/114800/114727-43-4.png)
![2H-Indol-2-one, 3-[(3,4-dichlorophenyl)methylene]-1,3-dihydro-](http://img.cochemist.com/ccimg/114800/114727-43-4_b.png)

















![Ethanone, 1-[2-hydroxy-4,6-bis(1-methylethoxy)phenyl]-](http://img.cochemist.com/ccimg/93400/93344-48-0.png)
![Ethanone, 1-[2-hydroxy-4,6-bis(1-methylethoxy)phenyl]-](http://img.cochemist.com/ccimg/93400/93344-48-0_b.png)








![L-Proline, N-[(1,1-dimethylethoxy)carbonyl]-L-tryptophyl-, methyl ester](http://img.cochemist.com/ccimg/85500/85416-63-3.png)
![L-Proline, N-[(1,1-dimethylethoxy)carbonyl]-L-tryptophyl-, methyl ester](http://img.cochemist.com/ccimg/85500/85416-63-3_b.png)


![1,2-Benzisothiazol-3(2H)-one, 2-[2-(4-methoxyphenyl)-2-oxoethyl]-,1,1-dioxide](http://img.cochemist.com/ccimg/75000/74921-64-5.png)
![1,2-Benzisothiazol-3(2H)-one, 2-[2-(4-methoxyphenyl)-2-oxoethyl]-,1,1-dioxide](http://img.cochemist.com/ccimg/75000/74921-64-5_b.png)
![Benzene, 5-[(1E)-2-(4-bromophenyl)ethenyl]-1,2,3-trimethoxy-](/data/chemimg/1960800/74809-50-0.png)
![Benzene, 5-[(1E)-2-(4-bromophenyl)ethenyl]-1,2,3-trimethoxy-](/data/chemimg/1960800/74809-50-0_b.png)











![Ethanone, 1-[2-hydroxy-4-(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/65500/65490-08-6.png)
![Ethanone, 1-[2-hydroxy-4-(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/65500/65490-08-6_b.png)
![(2E)-1-{3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-2,4-dihydroxyphenyl}-3-(4-hydroxyphenyl)prop-2-en-1-one](http://img.cochemist.com/ccimg/63000/62949-76-2.png)
![(2E)-1-{3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-2,4-dihydroxyphenyl}-3-(4-hydroxyphenyl)prop-2-en-1-one](http://img.cochemist.com/ccimg/63000/62949-76-2_b.png)
![(z)-7-[(1r,2r,3r)-3-hydroxy-2-[(e,3r)-3-hydroxy-4-phenoxybut-1-enyl]-5-oxocyclopentyl]-n-methylsulfonylhept-5-enamide](http://img.cochemist.com/ccimg/60400/60325-46-4.png)
![(z)-7-[(1r,2r,3r)-3-hydroxy-2-[(e,3r)-3-hydroxy-4-phenoxybut-1-enyl]-5-oxocyclopentyl]-n-methylsulfonylhept-5-enamide](http://img.cochemist.com/ccimg/60400/60325-46-4_b.png)
![Benzene, 1-bromo-4-[(1E)-2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/58400/58358-51-3.png)
![Benzene, 1-bromo-4-[(1E)-2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/58400/58358-51-3_b.png)


![3-[(4-METHOXYPHENYL)METHYLIDENE]-1H-INDOL-2-ONE](http://img.cochemist.com/ccimg/55200/55160-02-6.png)
![3-[(4-METHOXYPHENYL)METHYLIDENE]-1H-INDOL-2-ONE](http://img.cochemist.com/ccimg/55200/55160-02-6_b.png)







![(3S,8aS)-3-(1H-indol-3-ylmethyl)hexahydropyrrolo[1,2-a]pyrazine-1,4-dione](http://img.cochemist.com/ccimg/38200/38136-70-8.png)
![(3S,8aS)-3-(1H-indol-3-ylmethyl)hexahydropyrrolo[1,2-a]pyrazine-1,4-dione](http://img.cochemist.com/ccimg/38200/38136-70-8_b.png)
![Benzene,[(isocyanomethyl)sulfonyl]-](http://img.cochemist.com/ccimg/36700/36635-63-9.png)
![Benzene,[(isocyanomethyl)sulfonyl]-](http://img.cochemist.com/ccimg/36700/36635-63-9_b.png)



![3-[[4-(TRIFLUOROMETHYL)PHENYL]METHYLIDENE]-1H-INDOL-2-ONE](http://img.cochemist.com/ccimg/35400/35315-56-1.png)
![3-[[4-(TRIFLUOROMETHYL)PHENYL]METHYLIDENE]-1H-INDOL-2-ONE](http://img.cochemist.com/ccimg/35400/35315-56-1_b.png)
![2-Propen-1-one,1-[2,4-dihydroxy-5-(3-methyl-2-buten-1-yl)phenyl]-3-(4-hydroxyphenyl)-, (2E)-](http://img.cochemist.com/ccimg/28500/28448-85-3.png)
![2-Propen-1-one,1-[2,4-dihydroxy-5-(3-methyl-2-buten-1-yl)phenyl]-3-(4-hydroxyphenyl)-, (2E)-](http://img.cochemist.com/ccimg/28500/28448-85-3_b.png)









![(e)-1-[2,4-dihydroxy-3-(3-methylbut-2-enyl)phenyl]-3-(4-hydroxyphenyl)prop-2-en-1-one](http://img.cochemist.com/ccimg/20800/20784-50-3.png)
![(e)-1-[2,4-dihydroxy-3-(3-methylbut-2-enyl)phenyl]-3-(4-hydroxyphenyl)prop-2-en-1-one](http://img.cochemist.com/ccimg/20800/20784-50-3_b.png)
![Benzene, 1-chloro-4-[(1E)-2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/18900/18878-89-2.png)
![Benzene, 1-chloro-4-[(1E)-2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/18900/18878-89-2_b.png)




![2-(Hydroxymethyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide](http://img.cochemist.com/ccimg/14000/13947-20-1.png)
![2-(Hydroxymethyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide](http://img.cochemist.com/ccimg/14000/13947-20-1_b.png)
![Phenol, 4-[(1E)-2-phenylethenyl]-, acetate](http://img.cochemist.com/ccimg/13100/13041-73-1.png)
![Phenol, 4-[(1E)-2-phenylethenyl]-, acetate](http://img.cochemist.com/ccimg/13100/13041-73-1_b.png)







![ETHANONE, 1-[2,4-BIS(METHOXYMETHOXY)PHENYL]-](http://img.cochemist.com/ccimg/6600/6515-05-5.png)
![ETHANONE, 1-[2,4-BIS(METHOXYMETHOXY)PHENYL]-](http://img.cochemist.com/ccimg/6600/6515-05-5_b.png)
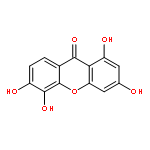

![1-[(e)-2-(4-methoxyphenyl)ethenyl]-4-nitrobenzene](http://img.cochemist.com/ccimg/4700/4648-33-3.png)
![1-[(e)-2-(4-methoxyphenyl)ethenyl]-4-nitrobenzene](http://img.cochemist.com/ccimg/4700/4648-33-3_b.png)
![Benzenamine,4-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/4400/4309-66-4.png)
![Benzenamine,4-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/4400/4309-66-4_b.png)





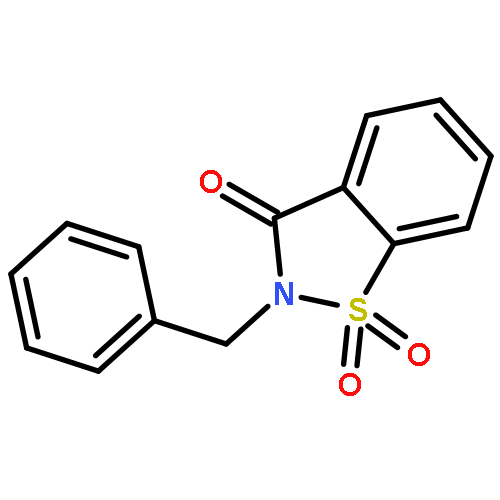


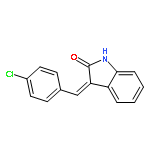

![4H-PYRAZOLO[1,5-C][1,3]THIAZOLE-2-CARBALDEHYDE](http://img.cochemist.com/ccimg/3300/3296-03-5.png)
![4H-PYRAZOLO[1,5-C][1,3]THIAZOLE-2-CARBALDEHYDE](http://img.cochemist.com/ccimg/3300/3296-03-5_b.png)
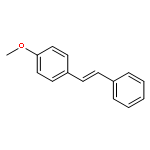






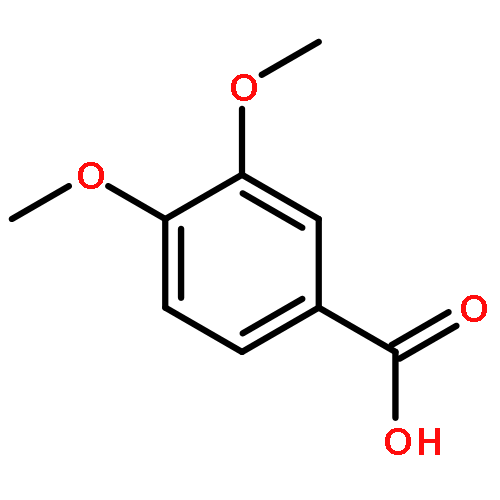
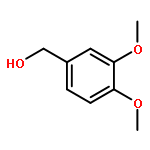
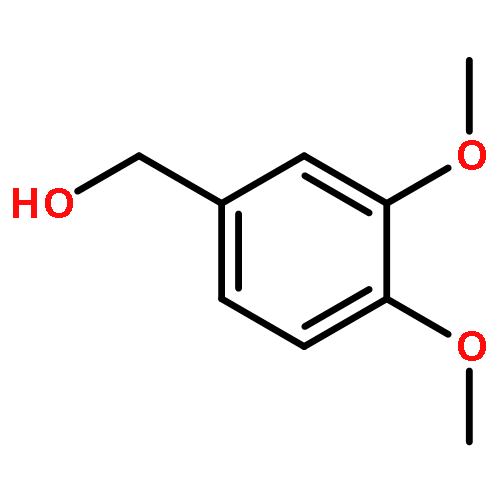






![1,3,2-Dioxaborolane, 2-[(2E)-3,7-dimethyl-2,6-octadien-1-yl]-4,4,5,5-tetramethyl-](/data/chemimg/3761300/1057662-85-7.png)
![1,3,2-Dioxaborolane, 2-[(2E)-3,7-dimethyl-2,6-octadien-1-yl]-4,4,5,5-tetramethyl-](/data/chemimg/3761300/1057662-85-7_b.png)
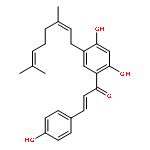













![1-Propyne, 3,3'-[[2,2-bis[(2-propyn-1-yloxy)methyl]-1,3-propanediyl]bis(oxy)]bis-](/data/chemimg/2234900/127751-08-0.png)
![1-Propyne, 3,3'-[[2,2-bis[(2-propyn-1-yloxy)methyl]-1,3-propanediyl]bis(oxy)]bis-](/data/chemimg/2234900/127751-08-0_b.png)