Co-reporter:Haifeng Wang, Linjie Yan, Yan Wu, Fener Chen
Tetrahedron 2017 Volume 73, Issue 19(Issue 19) pp:
Publication Date(Web):11 May 2017
DOI:10.1016/j.tet.2017.03.076
We have developed the chloramphenicol base urea-catalyzed intramolecular Michael addition of γ-hydroxy-α, β-unsaturated enones. The oxidation of the resulting products provided facile access to the corresponding α-hydroxy chiral alcohols with good efficiency and enantioselectivity, with the reaction displaying broad substrate scope. The utility of this methodology was further demonstrated by the synthesis of (R)-2-hydroxy-4-phenylbutanoate, which is a key building block for the construction of the ACE inhibitor benazepril hydrochloride.Download high-res image (91KB)Download full-size image
Co-reporter:Xinlong Wang, Lingjun Xu, Fangjun Xiong, Yan Wu, Fener Chen
Tetrahedron: Asymmetry 2017 Volume 28, Issue 6(Issue 6) pp:
Publication Date(Web):15 June 2017
DOI:10.1016/j.tetasy.2017.04.013
An efficient and stereocontrolled synthesis of (20S)-camptothecin and an analogue has been developed. The key feature of this synthesis is the organocatalyzed asymmetric α-hydroxylation of the lactone precursor 4 to construct its stereocenter, providing tricyclic hydroxylactone 2 in 90% yield and with 88% enantioselectivity. The precursor 4 was efficiently synthesized from the known pyridine 5 in three steps.Download high-res image (69KB)Download full-size image
Co-reporter:Linjie Yan, Haifeng Wang, Fangjun Xiong, Yuan Tao, Yan Wu, Fener Chen
Tetrahedron: Asymmetry 2017 Volume 28, Issue 7(Issue 7) pp:
Publication Date(Web):15 July 2017
DOI:10.1016/j.tetasy.2017.05.015
The first chloramphenicol base-derived thiourea-catalyzed enantioselective Michael addition of malononitrile to α,β-unsaturated ketones is reported. The Michael adducts were obtained in good to excellent yields (up to 98% yield) and enantioselectivities (up to 94% ee). This reaction has a broad substrate scope to various α,β-unsaturated ketones. With this in mind, this methodology was successfully applied to the synthesis of a chiral piperidone, an advanced building block for dihydropyridinone P2X7 receptor antagonists.Download high-res image (161KB)Download full-size image
Co-reporter:Haihui Peng
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 30) pp:6281-6301
Publication Date(Web):2017/08/02
DOI:10.1039/C7OB01341H
Prostaglandins (PGs) are a series of hormone-like chemical messengers and play a critical role in regulating physiological activity. The diversified therapeutic activities and complex molecular architectures of PGs have attracted special attention, and huge progress has been made in asymmetric total synthesis and discovery of pharmaceutically useful drug candidates. In the last 10 years, several powerful syntheses have emerged as new solutions to the problem of building PGs and represent major breakthroughs in this area. This review highlights the advances in methodologies for the asymmetric total synthesis of prostaglandins. The application of these methodologies in the syntheses of medicinally useful prostaglandins is also described. The study has been carefully categorized according to the key procedures involved in the syntheses of various prostaglandins, aiming to give readers an easy understanding of this chemistry and provide insights for further improvements.
Co-reporter:Yafeng Wang, Guanxin Huang, Sha Hu, Kaijun Jin, Yan Wu, Fener Chen
Tetrahedron 2017 Volume 73, Issue 34(Issue 34) pp:
Publication Date(Web):24 August 2017
DOI:10.1016/j.tet.2017.05.066
A highly enantioselective synthesis of chiral β-hydroxy thioesters that uses a decarboxylative aldol reaction of malonic acid half thioesters and aldehydes catalyzed by a chloramphenicol base-derived bifunctional organocatalyst is reported. The resulting chiral β-hydroxy thioesters were obtained in high yields (up to 82%) with good to excellent enantioselectivities (up to 94% ee). The synthetic application of the methodology is illustrated by the asymmetric synthesis of the selective serotonin reuptake inhibitor dapoxetine.Download high-res image (110KB)Download full-size image
Co-reporter:Fangjun Xiong, Haifeng Wang, Lingjie Yan, Sheng Han, Yuan Tao, Yan Wu and Fener Chen
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 4) pp:1363-1369
Publication Date(Web):11 Dec 2015
DOI:10.1039/C5OB02245B
A novel, stereoselective approach towards rosuvastatin calcium from the known (S)-homoallylic alcohol has been developed. The synthesis is highlighted by a regio- and stereocontrolled ICl-induced intramolecular cyclization of chiral homoallylic carbonate to deliver the C6-formyl statin side chain with a syn-1,3-diol moiety. An improved synthesis of the rosuvastatin pyrimidine core moiety is also included. Moreover, this methodology is useful in the asymmetric synthesis of structural variants of statins such as pitavastatin calcium and atorvastatin calcium and their related analogs.
Co-reporter:Haifeng Wang, Linjie Yan, Fangjun Xiong, Yan Wu and Fener Chen
RSC Advances 2016 vol. 6(Issue 79) pp:75470-75477
Publication Date(Web):03 Aug 2016
DOI:10.1039/C6RA15304F
Several novel chiral Schiff base ligands were prepared from a commercially available chloramphenicol base and applied to the titanium-mediated asymmetric aldol reaction of diketene with various α,β-unsaturated aldehydes. This reaction proceeded in good yield with high enantioselectivity. The synthetic utility of this methodology was demonstrated in the short synthesis of atorvastatin calcium.
Co-reporter:Xinlong Wang, Lingjun Xu, Fangjun Xiong, Yan Wu and Fener Chen
RSC Advances 2016 vol. 6(Issue 44) pp:37701-37709
Publication Date(Web):11 Apr 2016
DOI:10.1039/C6RA05109J
Herein we describe the application of a series of newly developed Ru-chloramphenicol base derivative complexes as catalysts for the highly diastereo- and enantioselective transfer hydrogenation of N-Boc α-amino-β-ketoesters for the asymmetric synthesis of anti-N-Boc-β-hydroxy-α-amino esters. This report highlights the utility of this catalytic methodology for the preparation of pharmaceutical compounds bearing a N-Boc α-amino-β-hydroxy substructure with two stereocenters.
Co-reporter:Xinlong Wang, Lingjun Xu, Lingjie Yan, Haifeng Wang, Sheng Han, Yan Wu, Fener Chen
Tetrahedron 2016 Volume 72(Issue 14) pp:1787-1793
Publication Date(Web):7 April 2016
DOI:10.1016/j.tet.2016.02.045
A robust and practical method has been developed for the synthesis of florfenicol (1) starting from commercial available 4-(methylsulfonyl) benzoic acid. The key step in this synthesis was the Ru-chloramphenicol base catalyzed asymmetric transfer hydrogenation of N-Boc α-amino-β-ketoester 5 through a dynamic kinetic resolution, which afforded the key chiral building block, anti-(2S,3S)-α-Boc-amino-β-hydroxyl ester 4, with high diastereoselectivity (92% de) and enantioselectivity (78% ee). The synthesis of a series of novel chloramphenicol base ligands L1–L10 is also included. This protocol could also be used for the asymmetric synthesis of fully synthetic analogs of florfenicol.
Co-reporter:Lin-Jie Yan;Hai-Feng Wang;Dr. Wen-Xue Chen;Yuan Tao;Kai-Jun Jin ; Fen-Er Chen
ChemCatChem 2016 Volume 8( Issue 13) pp:2249-2253
Publication Date(Web):
DOI:10.1002/cctc.201600228
Abstract
The synthesis of new chloramphenicol-base-derived thiourea organocatalysts, (1S,2R)-12 a–f and (1R,2R)-15 a–c, and their use in the enantioselective alcoholysis of meso-anhydrides are described. In particular, hemiesters afforded excellent enantioselectivities if low loadings of (1S,2R)-12 a–f were used. Almost no enantioselectivities were achieved with the use of (1R,2R)-15 a–c. This technique was used to synthesize (R)-(−)-baclofen.
Co-reporter:Fangjun Xiong, Haifeng Wang, Lingjie Yan, Lingjun Xu, Yuan Tao, Yan Wu and Fener Chen
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 38) pp:9813-9819
Publication Date(Web):10 Aug 2015
DOI:10.1039/C5OB01148E
An efficient and concise asymmetric synthesis of pitavastatin calcium (1) starting from commercially available (S)-epichlorohydrin is described. A convergent C1 + C6 route allowed for the assembly of the pitavastatin C7 side chain via a Wittig reaction between phosphonium salt 2 and the enantiomerically pure C6-formyl side chain 3. The 1,3-syn-diol acetal motif in 3 was established with excellent stereo control by a diastereoselective bismuth-promoted two-component hemiacetal/oxa-Michael addition reaction of (S)-α,β-unsaturated ketone 4 with acetaldehyde.
Co-reporter:Zheng-Yong Wan, Jin Yao, Yuan Tao, Tian-Qi Mao, Xin-Long Wang, Yi-Pei Lu, Hai-Feng Wang, Hong Yin, Yan Wu, Fen-Er Chen, Erik De Clercq, Dirk Daelemans, Christophe Pannecouque
European Journal of Medicinal Chemistry 2015 Volume 97() pp:1-9
Publication Date(Web):5 June 2015
DOI:10.1016/j.ejmech.2015.04.050
•The etravirine–VRX-480773 hybrids previously disclosed were further optimized.•Novel piperidin-4-yl-aminopyrimidines were designed using molecular hybridization.•26 new compounds were designed and synthesized as the anti-drug resistant HIV NNRTIs.•Most compounds have EC50 at single-digit nanomolar concentrations against WT HIV-1.•SARs were discussed in detail.A novel series of piperidin-4-yl-aminopyrimidine derivatives were designed fusing the pharmacophore templates of etravirine–VRX-480773 hybrids our group previously described and piperidine-linked aminopyrimidines. Most compounds displayed significantly improved activity against wild-type HIV-1 with EC50 values in single-digit nanomolar concentrations compared to etravirine–VRX-480773 hybrids. Selected compounds were also evaluated for activity against reverse transcriptase, and had lower IC50 values than that of nevirapine. The improved potency observed in this in vitro model of HIV RNA replication partly validates the mechanism by which this class of allosteric pyrimidine derivatives inhibits reverse transcriptase, and represents a remarkable step forward in the development of AIDS therapeutics.
Co-reporter:Zheng-Yong Wan, Jin Yao, Tian-Qi Mao, Xin-Long Wang, Hai-Feng Wang, Wen-Xue Chen, Hong Yin, Fen-Er Chen, Erik De Clercq, Dirk Daelemans, Christophe Pannecouque
European Journal of Medicinal Chemistry 2015 Volume 102() pp:215-222
Publication Date(Web):18 September 2015
DOI:10.1016/j.ejmech.2015.08.007
•The etravirine–VRX-480773 hybrids previously disclosed were further optimized.•Novel pyrimidine sulfonylacetanilides were designed using molecular simulation.•The newly designed compounds exhibited broad spectrum activity against mutant HIV-1.•Targeting of highly conserved residue is important for overcoming HIV mutation.•SARs were discussed in detail.Based on molecular simulation, the etravirine–VRX-480773 hybrids previously disclosed by our group were optimized to yield novel pyrimidine sulfonylacetanilides 8 with improved activity against a panel of seven clinically relevant single and double mutant strains of HIV-1. The improvement in potency in this in vitro model of HIV RNA replication partly validates the mechanism by which this class of allosteric pyrimidine derivatives inhibits the reverse transcriptase (RT), and represents a remarkable step forward in the development of anti-HIV drugs.
Co-reporter:Tian-Qi Mao, Qiu-Qin He, Zheng-Yong Wan, Wen-Xue Chen, Fen-Er Chen, Gang-Feng Tang, Erik De Clercq, Dirk Daelemans, Christophe Pannecouque
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 13) pp:3860-3868
Publication Date(Web):1 July 2015
DOI:10.1016/j.bmc.2015.03.037
A molecular hybridization approach is a powerful tool in the design of new molecules with improved affinity and efficacy. In this context, a series of diarylpyrimidine–quinolone hybrids were synthesized and evaluated against both wt HIV-1 and mutant viral strains. The most active hybrid 5a displayed an EC50 value of 0.28 ± 0.07 μM against HIV-1 IIIB. A couple of enzyme-based assays clearly pinpoint a RT-targeted mechanism of action. Docking studies revealed that these hybrids could be well located in the NNIBP of HIV-1 RT despite the bulky and polar properties of a quinolone 3-carboxylic acid moiety in the molecules.
Co-reporter:Chen Zheng;Hai-Feng Wang;Wei-Qi Chen;Wen-Xue Chen
Asian Journal of Organic Chemistry 2015 Volume 4( Issue 7) pp:619-621
Publication Date(Web):
DOI:10.1002/ajoc.201500111
Abstract
The highly efficient asymmetric vinylogous Michael addition of 3-(2-oxoindolin-3-ylidene)butanoates to nitroalkenes, catalyzed by a cinchona-based squaramide, is described. The enantioselectivity (up to 99 % ee) and diastereoselectivity (>20:1 d.r.) of the reaction is excellent.
Co-reporter:Chen Zheng;Wen-Xue Chen
Asian Journal of Organic Chemistry 2015 Volume 4( Issue 10) pp:1044-1046
Publication Date(Web):
DOI:10.1002/ajoc.201500299
Abstract
Here we report the asymmetric amination of 3-(2-oxoindolin-3-ylidene)butanoates catalyzed by a cinchona-derived alkaloid, yielding products with an N-containing stereocenter in high yields and moderate enantioselectivity. The developed process could be an attractive approach for the synthesis of chiral amino acids.
Co-reporter:Weiqi Chen, Fangjun Xiong, Qian Liu, Lingjun Xu, Yan Wu, Fener Chen
Tetrahedron 2015 Volume 71(Issue 29) pp:4730-4737
Publication Date(Web):22 July 2015
DOI:10.1016/j.tet.2015.05.053
A novel approach to synthesize pitavastatin calcium (1), an effective HMG-CoA reductase inhibitor, based on readily available and attractively functionalized (R)-3-chloro-1,2-propanediol is reported. This work highlights an intermolecular diastereoselective bromine-induced cyclization of homoallylic carbonate to meet stereochemical challenges in the synthesis of statins. An efficient route to a new triphenylphosphonium tetrafluoroborate salt of a quinoline core is also presented.
Co-reporter:Shuang-Xi Gu;Yuan-Yuan Zhu;Erik De Clercq
Medicinal Chemistry Research 2015 Volume 24( Issue 1) pp:220-225
Publication Date(Web):2015 January
DOI:10.1007/s00044-014-1119-5
As a continuing and exploratory work on diarylpyrimidines (DAPYs) as human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) inhibitors, bulky and electron-rich naphthyl was introduced into the structure of DAPYs to replace the phenyl left wing of DAPYs, which is aimed to improve the π–π stacking interactions between inhibitors and some aromatic amino acid residues within the binding pocket of RT. The title compound 1a, with a 1-naphthyl left wing, displayed good inhibitory activity against wild-type HIV-1 (EC50 = 0.071 μM), along with moderate inhibitory activity against HIV-2 (EC50 = 6.5 μM).
Co-reporter:Hai-Qiu Wu, Jin Yao, Qiu-Qin He, Wen-Xue Chen, Fen-Er Chen, Christophe Pannecouque, Erik De Clercq, Dirk Daelemans
Bioorganic & Medicinal Chemistry 2015 23(3) pp: 624-631
Publication Date(Web):
DOI:10.1016/j.bmc.2014.11.032
Co-reporter:Zheng-Yong Wan, Yuan Tao, Ya-Feng Wang, Tian-Qi Mao, Hong Yin, Fen-Er Chen, Hu-Ri Piao, Erik De Clercq, Dirk Daelemans, Christophe Pannecouque
Bioorganic & Medicinal Chemistry 2015 23(15) pp: 4248-4255
Publication Date(Web):
DOI:10.1016/j.bmc.2015.06.048
Co-reporter:Ge Meng, Yang Liu, Aqun Zheng, Fener Chen, Wenxue Chen, Erik De Clercq, Christophe Pannecouque, Jan Balzarini
European Journal of Medicinal Chemistry 2014 Volume 82() pp:600-611
Publication Date(Web):23 July 2014
DOI:10.1016/j.ejmech.2014.05.059
•18 new compounds were designed, synthesized as the anti-drug resistant HIV NNRTIs.•All the new molecules were evaluated for their biological activities against HIV.•Compound 1d displayed anti HIV-1 activities 47 fold than that of AZT.•Compound 1d also showed activities against double mutated HIV-1 and HIV-2 strain.•SAR study including the docking analysis was also investigated deeply.This article reports the design, synthesis and antiviral evaluation of a new series of non-nucleoside reverse transcriptase inhibitors (NNRTIs). The basic skeleton of these target 18 molecules is diarylpyrimidine featuring a substituted amino group between the pyrimidine scaffold and the aryl wing. All of the new compounds have been characterized by spectra analysis. The entire target molecules were evaluated for their in vitro anti-HIV activity with controlling group of FDA approved drugs. Most of them showed good to potent activities against wild-type (WT) HIV-1 with IC50 values in the range of 0.0175–69.21 μM. 2-(4-Cyanophenylamino)-4-(2-cyanovinylphenylhydrazonomethyl)pyrimidine (1d) displayed potent anti-HIV-1 activity against WT HIV-1 with a selectivity index (SI) of 106367 and an IC50 value of 1.75 nM, which was 47 fold lower than that of AZT. Compound 1d also showed a broad-spectrum inhibitory activity, with an IC50 value of 5.33 μM and 5.05 μM against both HIV-1 double-mutated (K103N/Y181C) strain and HIV-2 strain, respectively. The preliminary structure–activity relationship (SAR) was also investigated. The binding modes with HIV-1 RT for both the wild type and mutant type have also been discussed.A series of new diarylpyrimidine analogues featuring a functional substituted amino group between the pyrimidine scaffold and the aryl wing have been designed and synthesized, which were also evaluated as anti-drug-resistant HIV reverse transcriptase inhibitors.
Co-reporter:Zi-Hong Yan, Xia-Yun Huang, Hai-Qiu Wu, Wen-Xue Chen, Qiu-Qin He, Fen-Er Chen, Erik De Clercq, Christophe Pannecouque
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 8) pp:2535-2541
Publication Date(Web):15 April 2014
DOI:10.1016/j.bmc.2014.02.030
Co-reporter:Chen Zheng, Fen-Er Chen
Chinese Chemical Letters 2014 Volume 25(Issue 1) pp:1-8
Publication Date(Web):January 2014
DOI:10.1016/j.cclet.2013.11.025
The asymmetric desymmetrization of cyclic anhydrides via the addition of carbon-based nucleophiles has been the focus of considerable levels of interest because it leads to optically active products. Over the past 20 years, a variety of different catalytic asymmetric alkylation reactions have been developed for the desymmetrization of cyclic anhydrides using different metal reagents as nucleophiles and using chiral ligands. The purpose of this review is to provide an overview of significant developments in this field.The asymmetric opening of anhydrides with carbon-based nucleophiles has been extensively investigated due to its great potential in the asymmetric synthesis of important chiral intermediates and natural products. This review summarizes the desymmetrizations of anhydrides via carbon-based nucleophiles.
Co-reporter:Hai-Qiu Wu, Christophe Pannecouque, Zi-Hong Yan, Wen-Xue Chen, Qiu-Qin He, Fen-Er Chen, Jan Balzarini, Dirk Daelemans and Erik De Clercq
MedChemComm 2014 vol. 5(Issue 4) pp:468-473
Publication Date(Web):09 Jan 2014
DOI:10.1039/C3MD00247K
A series of conformationally restricted dihydro-alkylthio-benzyl-oxopyrimidine (S-DABO) hybrids, which combined the structural features of C6-α-methylbenzyl-thio-DABOs (α-methyl-S-DABOs) and C6-α-cyanobenzyl-thio-DABOs (CN-S-DABOs), has been synthesized and biologically evaluated for their anti-HIV activity against wild-type HIV-1 strain IIIB, double RT mutant (K103N + Y181C) strain RES056 and HIV-2 strain ROD in MT-4 cell cultures. Most of these compounds exhibited inhibitory activity (wild-type) within the range of EC50 values from micromolar to nanomolar. Among them, compound 1s displayed the highest anti-HIV-1 activity with an EC50 value of 91 nM and a selectivity index (SI) of 548, which was more potent than zalcitabine and comparable to nevirapine and delavirdine in the same assay. The HIV-1 reverse transcriptase inhibitory (RT) assay confirmed that these conformationally restricted S-DABO hybrids targeted HIV-1 RT. The preliminary structure–activity relationship (SAR) and molecular docking analysis of this new series of conformationally constrained CN-S-DABO hybrids were also investigated.
Co-reporter:Zi-Hong Yan, Hai-Qiu Wu, Wen-Xue Chen, Yan Wu, Hu-Ri Piao, Qiu-Qin He, Fen-Er Chen, Erik De Clercq, Christophe Pannecouque
Bioorganic & Medicinal Chemistry 2014 22(12) pp: 3220-3226
Publication Date(Web):
DOI:10.1016/j.bmc.2014.03.020
Co-reporter:Fang-Jun Xiong, Jie Li, Xiao-Fei Chen, Wen-Xue Chen, Fen-Er Chen
Tetrahedron: Asymmetry 2014 Volume 25(16–17) pp:1205-1208
Publication Date(Web):15 September 2014
DOI:10.1016/j.tetasy.2014.07.002
An improved and practical synthesis of tert-butyl ((4R,6R)-6-aminoethyl-2,2-dimethyl-1,3-dioxan-4-yl)acetate 3 has been developed for supplying this key chiral side-chain of atorvastatin by using a Blaise reaction of (S)-4-chloro-3-((trimethylsilyl)oxy)butanenitrile 7 and the Raney Ni catalyzed hydrogenation of tert-butyl 2-((4R,6R)-6-(-2-oximeethyl)-2,2-dimethyl-1,3-dioxan-4-yl)acetate 12 as the key steps. This nine-step route from (R)-epichlorohydrin afforded the target compound in 55% overall yield of high chemical and enantiomeric purity.(R)-3-((Trimethylsilyl)oxy)hex-5-enenitrileC9H17NOSi[α]D21 = −2.2 (c 1.0, MeOH).Source of chirality: (R)-epichlorohydrinAbsolute configuration: (3R)(R)-tert-Butyl 5-hydroxy-3-oxooct-7-enoateC12H20O4[α]D21 = −10.3 (c 1.0, MeOH)Source of chirality: (R)-epichlorohydrinAbsolute configuration: (5R)(3R,5R)-tert-Butyl 3,5-dihydroxyoct-7-enoateC12H22O4[α]D21 = −10.5 (c 1.0, MeOH)Source of chirality: (R)-epichlorohydrin; Stereoselective reductionAbsolute configuration: (3R,5R)tert-Butyl 2-((4R,6R)-6-allyl-2,2-dimethyl-1,3-dioxan-4-yl)acetateC15H26O4dr = 99:1[α]D21 = +0.7 (c 1.0, MeOH)Source of chirality: (R)-epichlorohydrin; Stereoselective reductionAbsolute configuration: (4R,6R)tert-Butyl 2-((4R,6R)-6-(-2-oximeethyl)-2,2-dimethyl-1,3-dioxan-4-yl)acetateC14H25NO5[α]D24 = −0.6 (c 1.0, MeOH)Source of chirality: (R)-epichlorohydrin; Stereoselective reductionAbsolute configuration: (4R,6R)tert-Butyl ((4R,6R)-6-aminoethyl-2,2-dimethyl-1,3-dioxan-4-yl)acetateC14H27NO4[α]D14 = +16.9 (c 0.34, CHCl3)Source of chirality: (R)-epichlorohydrin; Stereoselective reductionAbsolute configuration: (4R,6R)
Co-reporter:Chen Zheng, Fen-er Chen
Tetrahedron: Asymmetry 2014 Volume 25(10–11) pp:792-795
Publication Date(Web):31 May 2014
DOI:10.1016/j.tetasy.2014.04.016
An efficient and convenient process has been developed for the resolution of VANOL using commercially available (1S,2S)-(+)-diaminocyclohexane as the resolving reagent, with (R)- and (S)-VANOL being isolated in 86% and 74% yields, respectively. The resolving reagent was recovered in 91% yield and could be reused without any decrease in its efficiency. The X-ray structural analysis of a molecular crystal consisting of (R)-VANOL and (1S,2S)-(+)-diaminocyclohexane revealed that hydrogen bonding interactions between the functional groups of the host and guest molecules dominated the stereochemistry of the guest in the molecular complex.
Co-reporter:Xiaofei Chen, Fangjun Xiong, Wenxue Chen, Qiuqin He, and Fener Chen
The Journal of Organic Chemistry 2014 Volume 79(Issue 6) pp:2723-2728
Publication Date(Web):February 27, 2014
DOI:10.1021/jo402829b
An efficient asymmetric synthesis of atorvastatin calcium has been achieved from commercially available diethyl 3-hydroxyglutarate through a novel approach that involves an organocatalytic enantioselective cyclic anhydride desymmetrization to establish C(3) stereogenicity and cyanide-free assembly of C7 amino type side chain via C5+C2 strategy as the key transformations.
Co-reporter:Shiqiong Yang, Christophe Pannecouque, Dirk Daelemans, Xiao-Dong Ma, Yang Liu, Fen-Er Chen, Erik De Clercq
European Journal of Medicinal Chemistry 2013 Volume 65() pp:134-143
Publication Date(Web):July 2013
DOI:10.1016/j.ejmech.2013.04.052
•Hybrid the special structural features of diarylpyrimidines and benzophenones.•Synthesize a series of BP-O-DAPY hybrids and O-DAPY derivatives as NNRTIs.•Evaluate of the synthesized compounds against HIV-1 and HIV-2.•Study the potential binding mode by molecular docking.This paper reports the synthesis and antiviral evaluation of a series of non-nucleoside reverse transcriptase inhibitors (NNRTIs) that combine the peculiar structural features of diarylpyrimidine derivatives (DAPYs) and benzophenone derivatives (BPs). The DAPY derivatives bearing benzoyl or alkoxyl substitutes on the A-ring showed the inhibitory activity against wild-type HIV-1 at the cellular level within the range of EC50 values from micromolar to nanomolar. Among these compounds, 1u exhibited the most potent anti-HIV-1 activity (EC50 = 0.06 ± 0.01 μM, SI > 6260), which were about 1.8-fold more active than nevirapine (NVP) and delavirdine (DLV). In addition, the binding modes with HIV-1 RT and the preliminary SAR studies of these derivatives were also considered for further investigation.
Co-reporter:Qian Liu, Fang-Jun Xiong, Qiu-Qin He, and Fen-Er Chen
Organic Process Research & Development 2013 Volume 17(Issue 12) pp:1540-1542
Publication Date(Web):November 8, 2013
DOI:10.1021/op400271k
An improved multikilogram-scale process for the production of (S)-1,2,4-butanetriol has been developed. This process involves the efficient removal of residual boric acid and the decomposition of the borate complexes formed during the reduction of (−)-dimethyl malate with sodium borohydride by methanolysis using a circular distillation-coupled hydrolysis apparatus.
Co-reporter:Hong-Jun Yang;Fang-Jun Xiong;Xiao-Fei Chen
European Journal of Organic Chemistry 2013 Volume 2013( Issue 21) pp:4495-4498
Publication Date(Web):
DOI:10.1002/ejoc.201300464
Abstract
Asymmetric thiolysis of prochiral cyclic anhydrides was achieved with our developed chiral sulfonamide and squaramide bifunctional organocatalysts based on amino alcohol scaffolds. The corresponding thioesters were obtained in high yields with excellent enantioselectivities. The usefulness of this methodology was demonstrated in the enantioselective synthesis of pregabalin.
Co-reporter:Hai-Qiu Wu, Zi-Hong Yan, Wen-Xue Chen, Qiu-Qin He, Fen-Er Chen, Erik De Clercq, Jan Balzarini, Dirk Daelemans, Christophe Pannecouque
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 21) pp:6477-6483
Publication Date(Web):1 November 2013
DOI:10.1016/j.bmc.2013.08.040
A series of C6-rigid S-DABO analogs characterized by a substituted benzoyl group at C6 position of the pyrimidine ring has been synthesized and biological evaluation as NNRTIs against wild-type HIV-1 strain IIIB, double RT mutant (K103N + Y181C) strain RES056 as well as HIV-2 strain ROD in MT-4 cell cultures. Most of the compounds exhibited moderate antiviral activities. Among them, compound 7q displayed the highest anti-HIV-1 activity with an EC50 value of 0.26 μM and a selectivity index (SI) of 541. The preliminary structure–activity relationship (SAR) of these new S-DABOs was investigated, the target RT was confirmed and docking study was performed.
Co-reporter:Hong-Jun Yang, Fang-Jun Xiong, Jie Li, Fen-Er Chen
Chinese Chemical Letters 2013 Volume 24(Issue 7) pp:553-558
Publication Date(Web):July 2013
DOI:10.1016/j.cclet.2013.04.009
A family of novel squaramides/sulfamides based on 1,2-alkamine was developed as chiral bifunctional catalysts to promote the asymmetric alcoholysis of meso cyclic anhydrides. The hemiesters were obtained in high yield with up to 93% ee. The usefulness of this methodology was demonstrated in the asymmetric synthesis of the key intermediate of P2X7 receptor antagonists.A family of novel squaramides/sulfamides based on 1,2-alkamine was developed as chiral bifunctional catalysts to promote the asymmetric alcoholysis of meso cyclic anhydrides. The hemiesters were obtained in high yield with up to 93% ee.
Co-reporter:Fei Xiong;Fang-Jun Xiong;Wen-Xue Chen;Hui-Qing Jia
Journal of Heterocyclic Chemistry 2013 Volume 50( Issue 5) pp:1078-1082
Publication Date(Web):
DOI:10.1002/jhet.1512
An enantioselective asymmetric total synthesis of (+)−biotin (1) via the Hoffmann–Roche lactone–thiolactone strategy has been accomplished from commercially available cis-1,3-dibenzyl-2-imidazolidone-4,5-dicarboxylic acid (2). Strategic transformations include a cinchona alkaloid-based bifunctional thiourea mediated methanolytic desymmetrization of prochiral cyclic anhydride 3 to produce the enantiomerically enriched precursor of Roche lactone 5 and an improved introduction of the 4-carboxybutyl side chain at C-4 position of Roche thiolactone 6 via Grignard reaction.
Co-reporter:Lemeng Hu, Fangjun Xiong, Xiaofei Chen, Wenxue Chen, Qiuqin He, Fener Chen
Tetrahedron: Asymmetry 2013 Volume 24(Issue 4) pp:207-211
Publication Date(Web):28 February 2013
DOI:10.1016/j.tetasy.2012.12.009
A short and cyanide-free enantioselective synthesis of atorvastatin calcium has been achieved starting from a commercially available highly substituted 1,4-diketone in an overall yield of 40%. The key step in this approach is the asymmetric aldol reaction of an aldehyde with diketene in the presence of Ti(O-i-Pr)4–Schiff base complex to create the (5R)-stereochemistry of atorvastatin calcium.(R)-Isopropyl 7-[2-(4-fluorophenyl)-5-(1-methylethyl)-3-pheny1-4-[(phenylamino)carbonyl]-1H-pyrrol-1-yl]-5-hydroxy-3-oxoheptanoateC36H39FN2O5Ee = 82%[α]D20=+9.1 (c 1.0, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration (R)(3R,5R)-Isopropyl 7-[2-(4-fluorophenyl)-5-(1-methylethyl)-3-phenyl-4-[(phenylamino)carbonyl]-1H-pyrrol-1-yl]-3,5-dihydroxyheptanoateC36H41FN2O5Ee = 99%[α]D20=+14.3 (c 1.0, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration (3R,5R)Isopropyl 2-((4R,6R)-6-(2-(2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(phenylcarbamoyl)-1H-pyrrol-1-yl)ethyl)-2,2-dimethyl-1,3-dioxan-4-yl)acetateC39H45FN2O5[α]D20=+5.8 (c 1.0, CHCl3)Source of chirality: the precursorAbsolute configuration (4R,6R)Atorvastatin calciumC33H40CaFN2O8[α]D20=-7.5 (c 1.0, DMSO)Source of chirality: the precursorAbsolute configuration (3R,5R)
Co-reporter:Shuang-Xi Gu, Zhi-Ming Li, Xiao-Dong Ma, Shi-Qiong Yang, Qiu-Qin He, Fen-Er Chen, Erik De Clercq, Jan Balzarini, Christophe Pannecouque
European Journal of Medicinal Chemistry 2012 Volume 53() pp:229-234
Publication Date(Web):July 2012
DOI:10.1016/j.ejmech.2012.04.004
(+)-3a and (−)-3a were successfully separated from racemate (±)-3a by the chiral technique of supercritical fluid chromatography (SCF) with enantiomeric excess (ee%) >99% and purity >99%, and assigned for their absolute configuration as R and S, respectively, by the experimental electronic circular dichroism (ECD) spectrum and simulated ECD spectra calculated by time-dependent density functional theory (TDDFT) calculations. (+)-(R)-3a displayed excellent activity with an EC50 of 5.3 nM against wild-type HIV-1, which was 12-fold more potent than (−)-(S)-3a. However, (−)-(S)-3a showed higher potency than (+)-(R)-3a against the double HIV-1 RT mutant (K103N + Y181C) as well as HIV-2 strain ROD. The possible reason for the difference of (R)- and (S)-3a in anti-HIV-1 activity was interpreted by molecular docking.Graphical AbstractDiarylpyrimidines (+)-3a and (−)-3a were separated from racemate (±)-3a, assigned for their absolute configurations, and evaluated for their anti-HIV activity.Highlights► Diarylpyrimidines (+)- and (−)-3a were separated from (±)-3a. ► The absolute configurations of (+)- and (−)-3a were confirmed by ECD spectroscopy. ► (+)-(R)-3a and (−)-(S)-3a were evaluated for their anti-HIV activity.
Co-reporter:Xiao-Dong Ma, Qiu-Qin He, Xuan Zhang, Shi-Qiong Yang, Liu-Meng Yang, Shuang-Xi Gu, Yong-Tang Zheng, Fen-Er Chen, Hui-Fang Dai
European Journal of Medicinal Chemistry 2012 Volume 58() pp:504-512
Publication Date(Web):December 2012
DOI:10.1016/j.ejmech.2012.03.032
A series of N-phenylarylformamide derivatives (PAFAs) with anti-wild-type HIV-1 activity (EC50 values) ranging from 0.3 nM to 5.1 nM and therapeutic index (TI) ranging from 10 616 to 271 000 were identified as novel non-nucleoside reverse transcriptase inhibitors. Among them, compound 13g (EC50 = 0.30 nM, TI = 184 578), 13l (EC50 = 0.37 nM, TI = 212 819), 13m (EC50 = 0.32 nM, TI = 260 617) and 13r (EC50 = 0.27 nM, TI = 271 000) displayed the highest activity against this type virus nearly as potent as lead compound GW678248. Moreover, all of them were also active to inhibit the double mutant strain A17 (K103N + Y181C) with EC50 values of 0.29 μM, 0.14 μM, 0.10 μM and 0.27 μM, respectively. In particular, compound 13m, which showed broad-spectrum anti-HIV activity, was also effective to inhibit the HIV-2 ROD replication within 4.37 μM concentration.Led by novel benzophenone derivative GW678248, a series of N-phenylarylformamide derivatives (PAFAs) were identified as potent non-nucleoside HIV reverse transcriptase inhibitors.Highlights► N-Phenylarylformamide derivatives (PAFAs) as novel NNRTIs. ► The anti-wild-type HIV-1 activity (EC50 values) ranging from 0.3 nM to 5.1 nM. ► The preliminary SARs and the docking modes were investigated.
Co-reporter:Lei Zhao, Xiao-Dong Ma, and Fen-Er Chen
Organic Process Research & Development 2012 Volume 16(Issue 1) pp:57-60
Publication Date(Web):November 1, 2011
DOI:10.1021/op2002003
Two scalable processes for the synthesis of 4-amino-5-aminomethyl-2-methylpyrimidine (2) are described. In the first approach, the less expensive 2-cyanoacetamide was reacted with Vilsmeier reagent to afford enamine 18, followed by the condensation with acetamidine to produce the 4-amino-2-methylpyrimidine-5-carbonitrile (6); subsequent hydrogenation gave 2 in 65% overall yield. In the second approach, malononitrile was treated with the ionic salt 21, prepared in situ from DMF and dimethyl sulfate, to give 18, which, without isolation was reacted with acetamidine hydrochloride to afford the common intermediate 6. Overall yield of this approach was 70%. Both methods are performed in a convenient manner suitable for industrial use.
Co-reporter:Wenxue Chen;Yukun Zu;Qi Huang;Fener Chen;Guifang Wang;Wenxian Lan;Chunxue Bai;Shaohua Lu;Yong Yue;Feng Deng
Magnetic Resonance in Medicine 2011 Volume 66( Issue 6) pp:1531-1540
Publication Date(Web):
DOI:10.1002/mrm.22957
Abstract
Lung cancer causes serious health problems. Clinical diagnosis of lung cancer relies on histopathological evalution of tissue specimen. However, extensive knowledge of the metabolic biochemistry of tumors can potentially provide important information for accurate diagnosis of lung cancer. High resolution magic-angle spinning NMR spectroscopy has emerged and be widely acknowledged as an excellent tool in investigating tissue metabolism. Moreover, the combination of high resolution magic-angle spinning NMR technique and multivariate data analysis has become an important metabonomics platform for studying the intact biological tissues. This study reported the metabonomic characteristics of 51 lung tissues from 17 patients with lung cancer using the high resolution magic-angle spinning 1H NMR spectroscopy and the multivariate data analysis methods including principal component analysis and orthogonal partial least squares-discriminant analysis. Clear differences among the metabonomic characteristics of lung cancer tissues at various sites were disclosed. Compared with the adjacent noninvolved tissues, the lung cancer tissues had significantly high levels of aspartate, phosphocholine, glycerophosphocholine and lactate but significantly low levels of glucose and valine. Furthermore, significantly positive (or negative) correlations were observed between the levels of some metabolites such as lactate, fatty acids, valine, phosphocholine, and glycerophosphocholine. Magn Reson Med, 2011. © 2011 Wiley Periodicals, Inc.
Co-reporter:Le Dai;Hongjun Yang ;Fener Chen
European Journal of Organic Chemistry 2011 Volume 2011( Issue 26) pp:5071-5076
Publication Date(Web):
DOI:10.1002/ejoc.201100403
Abstract
A highly enantioselective sulfa-Michael addition (SMA) of various thiols to α,β-unsaturated N-acylated oxazolidinones has been achieved with a chiral squaramide catalyst under very mild reaction conditions. In addition, the conversion of the β-thio-substituted adduct to the corresponding methyl β-tosylbutanoate was demonstrated in high yield through a methanolysis/oxidation sequence.
Co-reporter:Shuang-Xi Gu, Qiu-Qin He, Shi-Qiong Yang, Xiao-Dong Ma, Fen-Er Chen, Erik De Clercq, Jan Balzarini, Christophe Pannecouque
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 17) pp:5117-5124
Publication Date(Web):1 September 2011
DOI:10.1016/j.bmc.2011.07.023
A series of 26 diarylpyrimidines, characterized by the hydroxymethyl linker between the left wing benzene ring and the central pyrimidine, were synthesized and evaluated for in vitro anti-HIV activity. Most of the compounds exhibited moderate to excellent activities against wild-type HIV-1. Among them, compound 10i, bearing a chlorine atom at the C-2 position of left benzene ring, was the best congener and showed potent activity against wild-type HIV-1 with an EC50 value of 0.009 μM, along with moderate activities against the double RT mutant (K103N + Y181C) HIV-1(IIIB) and HIV-2(ROD) with an EC50 value of 6.2 and 6.0 μM, respectively. The preliminary structure–activity relationship (SAR) of this new series of compounds was also investigated.
Co-reporter:Shuang-Xi Gu, Xuan Zhang, Qiu-Qin He, Liu-Meng Yang, Xiao-Dong Ma, Yong-Tang Zheng, Shi-Qiong Yang, Fen-Er Chen
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 14) pp:4220-4226
Publication Date(Web):15 July 2011
DOI:10.1016/j.bmc.2011.05.060
A novel series of naphthyl phenyl ether analogues (NPEs) have been synthesized and evaluated for their in vitro activities against HIV in C8166 cells. Most of the compounds exhibited moderate to excellent anti-HIV activities. Among them the most active compound 12o showed excellent activities against wild-type HIV-1 with an EC50 value of 4.60 nM, along with moderate activities against the double mutant strain HIV-1IIIB A17 (K103N+Y181C) and HIV-2 strain ROD with an EC50 value of 0.82 and 4.40 μM, respectively. Preliminary structure–activity relationship (SAR) among the newly synthesized NPEs was also investigated.
Co-reporter:Shuang-Xi Gu, Shi-Qiong Yang, Qiu-Qin He, Xiao-Dong Ma, Fen-Er Chen, Hui-Fang Dai, Erik De Clercq, Jan Balzarini, Christophe Pannecouque
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 23) pp:7093-7099
Publication Date(Web):1 December 2011
DOI:10.1016/j.bmc.2011.10.002
A series of 18 cycloalkyl arylpyrimidines (CAPYs) were designed from lead compounds diarylpyrimidines (DAPYs), synthesized and evaluated for in vitro anti-HIV activity. Among them, the compound 1p displayed potent anti-HIV-1 activity against WT HIV-1 with an EC50 value of 0.055 μM and a selectivity index (SI) >7290. The preliminary structure–activity relationship (SAR) of this new series of compounds was also investigated, which enriched the SAR of diarylpyrimidines (DAPYs).
Co-reporter:Qiu-Qin He, Xuan Zhang, Hai-Qiu Wu, Shuang-Xi Gu, Xiao-Dong Ma, Liu-Meng Yang, Yong-Tang Zheng, Fen-Er Chen
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 18) pp:5553-5558
Publication Date(Web):15 September 2011
DOI:10.1016/j.bmc.2011.07.037
A series of new 5-hydroxylquinolone-3-carboxylic acids (HQCAs) with various aryl or benzyl substituents on N-1 position were synthesized and evaluated for their anti-HIV activity in C8166 cell culture. Most of the target compounds displayed activity against wide-type HIV-1 in the low micromolar range in infected C8166 cells. The most active compound 5g exhibited activity against wild-type HIV-1 and HIV-1 mutant virus A17 with an EC50 value of 3.17 and 17.88 μM, respectively. The biological results and the docking study revealed that the substitution pattern on N-1 position of the quinolone core might contribute to physicochemical properties of HQCAs and resulted in great influence on their antiviral potency.A series of new 5-hydroxylquinolone-3-carboxylic acids (HQCAs) with various aryl or benzyl substituents on N-1 position were synthesized with the aim of contributing to a better understanding of the structure–activity relationships of HQCAs.
Co-reporter:Qiu-Qin He, Shuang-Xi Gu, Jia Liu, Hai-Qiu Wu, Xuan Zhang, Liu-Meng Yang, Yong-Tang Zheng, Fen-Er Chen
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 16) pp:5039-5045
Publication Date(Web):15 August 2011
DOI:10.1016/j.bmc.2011.06.020
A series of new quinolone-3-carboxylic acids featuring a hydroxyl group at C-5 position were synthesized and evaluated for their in vitro activity against HIV in C8166 cell culture. All the compounds showed anti-HIV-1 activity with low micromolar to submicromolar EC50 values. The most active compound 2k exhibited activity against wild-type HIV-1 with an EC50 value of 0.13 μΜ. Preliminary structure–activity relationship of the newly synthesized quinolone analogues was also investigated. Further docking study revealed that the anti-HIV activity of these compounds might involve a two-metal chelating mechanism.A series of novel 5-hydroxylquinolone-3-carboxylic acids were synthesized in an attempt to investigate whether the introduction of OH at C-5 position would provide an additional metal binding site and result in an improved activity against HIV-1.
Co-reporter:Xiao-Dong Ma, Xuan Zhang, Hui-Fang Dai, Shi-Qiong Yang, Liu-Meng Yang, Shuang-Xi Gu, Yong-Tang Zheng, Qiu-Qin He, Fen-Er Chen
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 15) pp:4601-4607
Publication Date(Web):1 August 2011
DOI:10.1016/j.bmc.2011.06.007
A novel series of benzophenone derivatives with B-ring substituted by naphthyl ring has been synthesized and evaluated as non-nucleoside HIV-1 reverse transcriptase inhibitors. Most of these compounds showed good to moderate activity against wild-type HIV-1 and mutated viruses. In particular, the analogue 10i demonstrated the most potent activity against wild-type HIV-1 with an EC50 value of 4.8 nM, and with a high selectivity index up to 10347.9, it also proved to be active against the HIV-1 double mutant strain A17 (K103N+Y181C) with an EC50 value of 2.1 μM. In addition, the molecular modeling study was used to explore the major interactions between the potent inhibitors with the HIV-1 RT. The investigation of the structure–activity relationships may serve as an important lead for the further optimization.
Co-reporter:Wenxue Chen;Xiaoyan Zhou;Dan Huang;Fener Chen;Xiang Du
Chinese Journal of Chemistry 2011 Volume 29( Issue 11) pp:2511-2519
Publication Date(Web):
DOI:10.1002/cjoc.201180423
Abstract
Colorectal cancer (CRC) is the third commonest malignancy cancer worldwide. Clear understandings of global metabolic profiling of the normal mucosa and cancer tissues are vitally important to aid optimizing the clinical management strategy and understanding CRC biology. We studied metabolic characteristics of 20 CRC and 20 distant normal mucosa tissues extracts from 20 patients using high resolution 1H NMR spectroscopy in conjunction with multivariate analyses, such as principal component analysis (PCA). Compared with distant normal mucosa tissues, lactate, taurine, ornithine and polyamine were present at significantly higher levels in CRC tissue extracts whereas myo-inositol was present at significantly lower level. Two metabolites ratios such as myo-inositol/taurine and myo-inositol/(ornithine+polyamine) appear to be the most valuable biomarkers for the differentiation CRC from normal mucosa tissues. Our data suggested that HR 1H NMR spectroscopy combined with multivariate analyses is a potentially useful technology for detecting malignant changes in the normal mucosa tissues, the technique may be further exploited for future CRC biomarker research or identification of targets for therapeutic manipulations.
Co-reporter:Xiao-Dong Ma, Xuan Zhang, Shi-Qiong Yang, Hui-Fang Dai, Liu-Meng Yang, Shuang-Xi Gu, Yong-Tang Zheng, Qiu-Qin He, Fen-Er Chen
Bioorganic & Medicinal Chemistry 2011 19(16) pp: 4704-4709
Publication Date(Web):
DOI:10.1016/j.bmc.2011.07.003
Co-reporter:Feng Li, Zhong-Hua Wang, Lei Zhao, Fang-Jun Xiong, Qiu-Qin He, Fen-Er Chen
Tetrahedron: Asymmetry 2011 Volume 22(Issue 12) pp:1337-1341
Publication Date(Web):30 June 2011
DOI:10.1016/j.tetasy.2011.07.011
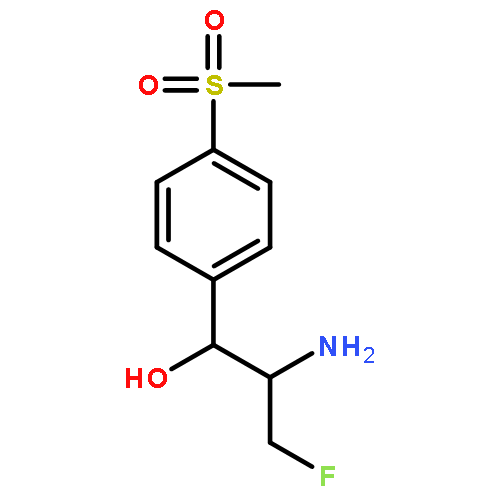
An efficient and highly enantioselective synthesis of florfenicol 1 was achieved with 37% overall yield starting from commercially available 4-methylthiobenzaldehyde. A key feature of the synthesis is the vanadium-catalyzed asymmetric epoxidation of allylic alcohol 5 with aq tert-butyl hydroperoxide to form (2S,3S)-epoxide 6.(S,S)-3-[4-(Methylsulfonyl)phenyl]-2,3-epoxypropyl alcoholC10H12O4SEe = 91%[α]D20=-33.1 (c 1.0, CHCl3)Source of chirality: asymmetric epoxidationAbsolute configuration: (2S,3S)(R)-3-Benzyl-4-[(S)-hydroxy(4-methylsulfonylphenyl)methyl]oxazolidin-2-oneC18H19NO5SDr >20:1[α]D20=-1.1 (c 1.0, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (3R,4S)(R)-((R)-3-Benzyl-2-oxooxazolidin-4-yl)[4-(methylsulfonyl)phenyl]methyl acetateC20H21NO6SDr >20:1[α]D20=-3.5 (c 1.0, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (3R,4R)(2R,3R)-2-Benzylamino-3-(4-methylsulfonylphenyl)-propane-1,3-diolC17H21NO4S[α]D20=-46.6 (c 1.0, ethanol)Source of chirality: asymmetric synthesisAbsolute configuration: (2R,3R)(1R,2R)-2-Amino-1-[4-(methylsulfonyl)phenyl]-1,3-propanediolC10H15NO4S[α]D25=-20.1 (c 1.0, ethanol)Source of chirality: asymmetric synthesisAbsolute configuration: (1R,2R)(4R,5R)-2-(Dichloromethyl)-5-[4-(methylsulfonyl)phenyl]oxazole-4-methanolC12H13Cl2NO4S[α]D20=+10.9 (c 1.0, ethanol)Source of chirality: asymmetric synthesisAbsolute configuration: (4R,5R)FlorfenicolC12H13Cl2NO4S[α]D20=-18.1 (c 1.0, DMF)Source of chirality: asymmetric synthesisAbsolute configuration: (1R,2R)
Co-reporter:Zhonghua Wang, Feng Li, Lei Zhao, Qiuqin He, Fener Chen, Chen Zheng
Tetrahedron 2011 67(47) pp: 9199-9203
Publication Date(Web):
DOI:10.1016/j.tet.2011.09.052
Co-reporter:Dr. Xiao-Dong Ma;Shi-Qiong Yang;Shuang-Xi Gu;Qiu-Qin He; Fen-Er Chen; Erik DeClercq; Jan Balzarini; Christophe Pannecouque
ChemMedChem 2011 Volume 6( Issue 12) pp:2225-2232
Publication Date(Web):
DOI:10.1002/cmdc.201100334
Abstract
A series of novel diarylpyrimidines (DAPYs) with a ketone hydrazone substituent on the methylene linker between the pyrimidine nucleus and the aryl moiety at the C-4 position were synthesized, and their antiviral activity against human immunodeficiency virus (HIV)-1 in MT-4 cells was evaluated. Most compounds of this class exhibited excellent activity against wild-type HIV-1, with EC50 values in the range of 1.7–13.2 nM. Of these compounds, 2-bromophenyl-2-[(4-cyanophenyl)amino]-4-pyrimidinone hydrazone (9 k) displayed the most potent anti-HIV-1 activity (EC50=1.7±0.6 nM), with excellent selectivity for infected over uninfected cells (SI=5762). In addition, the 4-methyl phenyl analogue 9 d (EC50=2.4±0.2 nM, SI=18461) showed broad spectrum HIV inhibitory activity, with EC50 values of 2.4±0.2 nM against wild-type HIV-1, 5.3±0.4 μM against HIV-1 double-mutated strain RES056 (K103N+Y181C), and 5.5 μM against HIV-2 ROD strain. Furthermore, structure–activity relationship (SAR) data and molecular modeling results for these compounds are also discussed.
Co-reporter:Le Dai;Su-Xi Wang
Advanced Synthesis & Catalysis 2010 Volume 352( Issue 13) pp:2137-2141
Publication Date(Web):
DOI:10.1002/adsc.201000334
Abstract
A chiral squaramide catalysts-promoted asymmetric sulfa-Michael conjugated addition of thiols to trans-chalcones is presented. Moderate to excellent yields and high enantioselectivities (up to 99% ee) were achieved under mild conditions.
Co-reporter:Jian-Ping Huang, Xu-Xiang Chen, Shuang-Xi Gu, Lei Zhao, Wen-Xue Chen and Fen-Er Chen
Organic Process Research & Development 2010 Volume 14(Issue 4) pp:939-941
Publication Date(Web):June 10, 2010
DOI:10.1021/op100094p
An improved palladium(II)-catalyzed cleavage of the allyl group in allyl faropenem 2 into faropenem sodium 1 is described. The development of an efficient method for the removal of palladium impurities from the crude product 1 upon treatment with polystyrene-bound 2,4,6-trimercapto-s-triazine (polystyrene-bound TMT) led to a drastic decrease of residual palladium level from 1500−1600 ppm to less than 10 ppm in the final isolated product. The palladium(II)-catalyzed cleavage and palladium removal process demonstrated on 10 kg scale are highly convenient and efficient.
Co-reporter:Zhao-Sen Zeng, Qiu-Qin He, Yong-Hong Liang, Xiao-Qing Feng, Fen-Er Chen, Erik De Clercq, Jan Balzarini, Christophe Pannecouque
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 14) pp:5039-5047
Publication Date(Web):15 July 2010
DOI:10.1016/j.bmc.2010.05.081
Molecular hybridization of the known anti-HIV-1 template DPC083 and etravirine based on docking model overlay has been generated a novel series of diarylbenzopyrimidine analogues (DABPs) (5a–z). These new hybrids were assessed for their activity against HIV in MT-4 cell cultures. Most of these compounds showed good activity against wild-type HIV-1 and mutant viruses. In particular, compound 5r showed the most potent activity against wild-type HIV-1 with an EC50 value of 1.8 nM, and with a selectivity index up to 111,954. It also proved more active against mutant L100I, K103N, Y188L, and K103N + Y181C RT HIV-1 strains than efavirenz.Molecular hybridization of the known anti-HIV-1 template DPC083 and etravirine based on docking modeling overlay has been set up to generate a novel series of diarylbenzopyrimidine analogues (DABPs).
Co-reporter:Yong-Hong Liang, Qiu-Qin He, Zhao-Sen Zeng, Zhi-Qian Liu, Xiao-Qing Feng, Fen-Er Chen, Jan Balzarini, Christophe Pannecouque, Erik De Clercq
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 13) pp:4601-4605
Publication Date(Web):1 July 2010
DOI:10.1016/j.bmc.2010.05.036
Nine newly 6-cynao-2-naphthyl substituted diarylpyrimidines (DAPY) were synthesized as non-nucleoside reverse transcriptase inhibitors on the basis of our previous work. The antiviral and cytotoxicity evaluation indicated that these compounds displayed strong activity against wild-type HIV-1 at nanomolar concentrations with selectivity index SI greater than 23 779. The most active compounds 3c and 3e exhibited activity against the double mutant (103N+181C) strains at an EC50 of 0.16 and 0.15 μM, and were more activity than that of efavirenz.Nine newly 6-cyano-2-naphthyl substituted DAPY analogues were synthesized and evaluated as inhibitors of the HIV-1 wild-type and double mutant (K103N+Y181C) strains in this paper.
Co-reporter:Xiao-Qing Feng, Zhao-Sen Zeng, Yong-Hong Liang, Fen-Er Chen, Christophe Pannecouque, Jan Balzarini, Erik De Clercq
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 7) pp:2370-2374
Publication Date(Web):1 April 2010
DOI:10.1016/j.bmc.2010.03.007
A series of novel diarylpyrimidine analogues featuring a hydroxyiminomethyl group between the pyrimidine scaffold and the aryl wing I have been synthesized and tested in MT-4 cells culture as non-nucleoside reverse transcriptase inhibitors against human immunodeficiency virus (HIV). Most of these new congeners exhibited moderate to excellent activity against wild-type virus with an EC50 value ranging from 0.569 μM to 0.005 μM. 4-(4-((Hydroxyimino) (3-methoxyphenyl)methyl)pyrimidin-2-ylamino)benzonitrile (12n) was identified as the most active compound of this new series (EC50 = 0.025 μM, SI >1223) associated with moderate activity against HIV-1 double mutant strains (K103N + Y181C) (EC50 = 8.72 μM) in addition to its anti-HIV-2 activity with an EC50 value of 8.31 μM. Preliminary structure–activity relationship (SAR) among the newly synthesized DAPYs was also investigated.
Co-reporter:Fei Xiong, Xu-Xiang Chen, Zhi-Qian Liu, Fen-Er Chen
Tetrahedron Letters 2010 Volume 51(Issue 28) pp:3670-3672
Publication Date(Web):14 July 2010
DOI:10.1016/j.tetlet.2010.05.035
A concise and efficient TEA-mediated desymmetrization of meso-thioanhydride 6 with 5-ethoxy-5-oxopentylzinc bromine has been developed, which affords a convenient strategy for the stereospecific total synthesis of (±)-biotin 1.A concise and efficient TEA-mediated desymmetrization of meso-thioanhydride with 5-ethoxy-5-oxopentylzinc bromine has been developed, which affords a convenient strategy for the stereospecific total synthesis of (±)-biotin.
Co-reporter:Yun-Yan Kuang;Jing-Ze Niu
Helvetica Chimica Acta 2010 Volume 93( Issue 10) pp:2094-2099
Publication Date(Web):
DOI:10.1002/hlca.201000049
Abstract
A concise and efficient asymmetric process for the total synthesis of (20S)-7-ethyl-10-hydroxycamptothecin (=SN-38; 1f), an active metabolic form of the prodrug irinotecan, is described. This approach features the enantioselective cyanosilylation of indolizinone 4 into the corresponding cyanohydrin 5, mediated by a bifunctional thiourea-based cinchona alkaloid under mild conditions, and I2-catalyzed Friedländer condensation of the tricyclic lactone 6 and 2-amino-5-hydroxy propiophenone (=1-(2-amino-5-hydroxyphenyl)propan-1-one).
Co-reporter:Zhao-Sen Zeng Dr.;Yong-Hong Liang Dr.;Xiao-Qing Feng Dr. ;Christophe Pannecouque ;Jan Balzarini ;Erik DeClercq
ChemMedChem 2010 Volume 5( Issue 6) pp:837-840
Publication Date(Web):
DOI:10.1002/cmdc.201000045
Co-reporter:Fei Xiong, Xu-Xiang Chen, Fen-Er Chen
Tetrahedron: Asymmetry 2010 Volume 21(Issue 6) pp:665-669
Publication Date(Web):8 April 2010
DOI:10.1016/j.tetasy.2010.03.041
The highly enantioselective total synthesis of (+)-biotin 1 via the Hoffmann–Roche lactone–thiolactone strategy has been achieved starting from cis-1,3-dibenzyl-2-imidazolidone-4,5-dicarboxylic acid 2 with an overall yield of 35%. Two contiguous stereogenic centers at C-3a and C-6a were established through a rapid cinchona alkaloid-based sulfonamide-mediated enantioselective alcoholysis of meso-cyclic anhydride 3 to afford (4S,5R)-cinnamyl hemiester 4h, the direct precursor to (3aS,6aR)-lactone 5 with high enantioselectivity. A one-pot installation of the 4-carboxybutyl side chain was accomplished by a Fukuyama coupling reaction of (3aS,6aR)-thiolactone 6 with the organozinc reagent prepared from ethyl 5-bromopentanoate.(4S,5R)-1,3-Dibenzyl-2-oxo-5-[(3-phenylallyloxy)-carbonyl]imidazolidine-4-carboxylic acidC28H26N2O5Ee = 98%[α]D22.1=+7.6 (c 1.0, CH3OH)Source of chirality: asymmetric synthesisAbsolute configuration: (4S,5R)Ethyl(3aS,4S,6aR)-5-(1,3-Dibenzyl-2,3,3a,4,6,6a-hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-5-yl)pentanoateC26H32N2O3S[α]D22.1=-24.1 (c 1.0, CH3OH)Source of chirality: asymmetric synthesisAbsolute configuration: (3aS,4S,6aR)(+)-BiotinC10H16N2O3S[α]D22.3=+91.3 (c 1.0, 0.1 N NaOH)Source of chirality: asymmetric synthesis
Co-reporter:Su-Xi Wang
Advanced Synthesis & Catalysis 2009 Volume 351( Issue 4) pp:547-552
Publication Date(Web):
DOI:10.1002/adsc.200800761
Co-reporter:Yue-Ping Wang, Fen-Er Chen, Erik De Clercq, Jan Balzarini, Christophe Pannecouque
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 3) pp:1016-1023
Publication Date(Web):March 2009
DOI:10.1016/j.ejmech.2008.06.028
A series of new 5-alkyl-2-benzylsulfanylpyrimidin-4(3H)-ones (5a–y) bearing different substituted arylmethyl moieties at the C-6 position of the pyrimidine core have been synthesized and evaluated for their in vitro activities against HIV-1 and HIV-2 in MT-4 cell cultures. The majority of the title compounds showed moderate to good activities against HIV-1 with an IC50 range from 6.67 μM to 0.12 μM. Among them, 6-(3,5-dimethylbenzyl) analogue 5q exhibited the most potent anti-HIV-1 activity (IC50 = 0.12 μM, SI > 2642), which was about 40-fold more active than the reference compounds 1-[(2-hydroxyethoxy)methyl]-6-(phenylsulfanyl)thymine (HEPT) and 2′,3′-dideoxyinosine (DDI). The structure–activity relationships (SARs) of these new congeners were further discussed.
Co-reporter:Yong-Hong Liang, Fen-Er Chen
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 2) pp:625-631
Publication Date(Web):February 2009
DOI:10.1016/j.ejmech.2008.03.021
For the activity values against HIV-1 RT wild-type and mutant strains (L100I, Y181C and Y188N) of 34 diarylpyrimidines (DAPYs) taken from literature, multiple linear regression analysis and the crossvalidation of Leave-One-Out method are carried out against the activity values with the hydrophobicity index log P, the modified steric parameter LY, the Muliken charge of nitrogen atom on the right wing NC and the indicator index I for the substituents R3′ and R1 on the left wing, and four good QSAR models are established: R = 0.8211(N = 34), R = 0.8599 (N = 33), R = 0.8711(N = 30) and R = 0.9079 (N = 29) for the cases of wild type, and mutant strain forms L100I, Y181C and Y188N, respectively. Additionally, the results indicate that log P plays a vital role in the prediction of the activity of DAPYs, and the cyano group on the left wing is important for inhibiting the mutant forms L100I and Y188N.
Co-reporter:Yun-Yan Kuang
Helvetica Chimica Acta 2009 Volume 92( Issue 5) pp:897-902
Publication Date(Web):
DOI:10.1002/hlca.200800385
Co-reporter:Jian Huang;Fei Xiong;Zhong-Hua Wang
Helvetica Chimica Acta 2009 Volume 92( Issue 7) pp:1445-1449
Publication Date(Web):
DOI:10.1002/hlca.200800435
Abstract
The reaction of vinylmagnesium bromide with the (3aS,6aR)-γ-thiolactone 2 in THF afforded, after unexpected ring expansion of the γ-thiolactone moiety, the seven-membered-ring ketone 5 in excellent yield, instead of the expected tertiary alcohol 3.
Co-reporter:Yuan-Zhen Xiong;Jan Balzarini;Erik DeClercq;Christophe Pannecouque
Chemistry & Biodiversity 2009 Volume 6( Issue 4) pp:561-568
Publication Date(Web):
DOI:10.1002/cbdv.200800163
Co-reporter:Yong-Hong Liang;Xiao-Qing Feng;Zhao-Sen Zeng Dr.;Jan Balzarini Dr.;Christophe Pannecouque Dr.;Erik DeClercq Dr.
ChemMedChem 2009 Volume 4( Issue 9) pp:1537-1545
Publication Date(Web):
DOI:10.1002/cmdc.200900212
Abstract
A series of 38 2-naphthyl-substituted diarylpyrimidine (DAPY) analogues, characterized by various substitution patterns on the pyrimidine and naphthalene rings, was synthesized in a straightforward fashion by means of parallel synthesis and evaluated as inhibitors of the HIV-1 wild-type and double mutant (K103N+Y181C) strains. Most of the compounds displayed strong activity against wild-type HIV-1. The most active compound, with a cyano group at position C6 on the naphthalene ring, exhibited activity against wild-type HIV-1 with an EC50 value of 0.002 μM and against the double mutant strain with an EC50 value of 0.24 μM; the selectivity index (SI) against wild-type is >180 000, the highest SI value among DAPY analogues. The structure–activity relationship (SAR) of the newly synthesized DAPYs is presented herein.
Co-reporter:Xiao-Qing Feng;Yong-Hong Liang;Zhao-Sen Zeng Dr.;Jan Balzarini Dr.;Christophe Pannecouque Dr.;Erik DeClercq Dr.
ChemMedChem 2009 Volume 4( Issue 2) pp:219-224
Publication Date(Web):
DOI:10.1002/cmdc.200800334
Co-reporter:Hui-Fang Dai;Wen-Xue Chen;Lei Zhao;Fei Xiong;Hao Sheng
Advanced Synthesis & Catalysis 2008 Volume 350( Issue 10) pp:1635-1641
Publication Date(Web):
DOI:10.1002/adsc.200800151
Abstract
A practical and asymmetric process for the total synthesis of (+)-biotin (1) has been accomplished starting from cis-1,3-dibenzyl-2-oxoimidazolidine-4,5-dicarboxylic acid (2). This approach features a highly enantioselective alcoholysis of meso-cyclic anhydride 3 into (4S,5R)-cinnamyl hemiester 4 mediated by Cinchona alkaloids. Another attractive feature of this synthesis involves the use of recyclable palladium nanoparticles-catalyzed assembly of the 4-carboxybutyl chain at C-4 in (3aS,6aR)-thiolactone 7 employing an improved Fukuyama-type cross-coupling reaction
Co-reporter:Yuan-Zhen Xiong, Fen-Er Chen, Jan Balzarini, Erik De Clercq, Christophe Pannecouque
European Journal of Medicinal Chemistry 2008 Volume 43(Issue 6) pp:1230-1236
Publication Date(Web):June 2008
DOI:10.1016/j.ejmech.2007.08.001
A series of novel 6-naphthyloxy substituted DATA analogues bearing different substituents on the C-6 position of triazine ring were synthesized and evaluated for their in vitro anti-HIV activity in MT-4 cells. The results demonstrated that most of the compounds in this series are potent activity against HIV-1 with moderate to high selectivity. Among these analogues, two compounds exhibited excellent effect in inhibiting HIV-1 replication at nanomolar concentration (for compound 9h: IC50 = 9.3 nM, SI = 15,385; for compound 9i: IC50 = 9.4 nM, SI = 14,094), which are about 15-fold more active than nevirapine. In addition, several compounds are active against both HIV-1 and HIV-2, whose mechanism may be different from typical NNRTIs.A number of diaryltriazine derivatives (DATAs) 8a–i and 9a–t were prepared and evaluated for their anti-HIV activity. Among which 9h and 9i were the most potent compounds with noteworthy selectivity against HIV-1 RT.
Co-reporter:Yue-Ping Wang, Fen-Er Chen, Erik De Clercq, Jan Balzarini, Christophe Pannecouque
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 7) pp:3887-3894
Publication Date(Web):1 April 2008
DOI:10.1016/j.bmc.2008.01.039
A novel series of 2-arylcarbonylmethylthio-6-arylmethylpyrimidin-4(3H)-ones have been synthesized and evaluated for in vitro anti-HIV activities in MT-4 cells. Most of these new compounds showed moderate to potent activities against wild-type HIV-1 with an EC50 range from 8.97 μM to 0.010 μM. Among them, the 6-(3,5-dimethylbenzyl) analogue 5p was identified as the most promising compound (EC50 = 0.010 μM, SI > 31,800) associated with moderate activity against the HIV-1 double mutant RT strain K103N + Y181C. The structure–activity relationships of these new congeners were further discussed.
Co-reporter:Xiongjie Yu, Suxi Wang and Fener Chen
ACS Combinatorial Science 2008 Volume 10(Issue 4) pp:605
Publication Date(Web):June 18, 2008
DOI:10.1021/cc800069t
An efficient and convergent solid-phase strategy for the total synthesis of All-E solanesol is described. This method features avoidance of iterative and difficult purifications comparing with solution-phase synthesis and is suitable for the preparation of other oligoprenols.
Co-reporter:Xiong-Jie Yu;Hao Zhang;Fang-Jun Xiong;Xu-Xiang Chen
Helvetica Chimica Acta 2008 Volume 91( Issue 10) pp:1967-1974
Publication Date(Web):
DOI:10.1002/hlca.200890211
Abstract
A practical and highly regio- and stereoselective synthesis of oligoprenols starting from commercially available geraniol is described. The convergent synthetic strategy features the iterative allyl-allyl coupling of monomers easily derived from geraniol that contain one reacting terminal functional group and the repetitive reductive elimination of the p-toluenesulfonyl (Ts) groups.
Co-reporter:Li-Peng Zhang;Yong Bao;Yun-Yan Kuang
Helvetica Chimica Acta 2008 Volume 91( Issue 11) pp:2057-2061
Publication Date(Web):
DOI:10.1002/hlca.200890218
Abstract
A six-step asymmetric total synthesis of (20S)-camptothecin (1) has been accomplished in 25% overall yield starting from the known pyridone 3. The key steps in this synthesis are the chemoselective Ni-catalyzed hydrogenation of 3-cyanopyridone 6 to 3-formylpyridone 7 in AcOH/pyridine/H2O and the Davis asymmetric hydroxylation of tricyclic lactone 4 utilizing a chiral N-sulfonyloxaziridine into (4′S)-tricyclic hydroxylactone 2.
Co-reporter:Yueping Wang;Jan Balzarini;Erik DeClercq;Christophe Pannecouque
Chemistry & Biodiversity 2008 Volume 5( Issue 1) pp:168-176
Publication Date(Web):
DOI:10.1002/cbdv.200890008
Abstract
A series of structurally related 2,5-disubstituted 6-(1-naphthylmethyl)-pyrimidin-4(3H)-ones, compounds 6a–6r, were synthesized and evaluated for their in vitro anti-HIV activities in MT-4 cells. Most of the new compounds investigated showed moderate-to-good activities against wild-type HIV-1, with IC50 values in the range 5.64–0.21 μM. Compound 6d was the most potent congener (IC50=0.21 μM, SI=724) in inhibiting HIV-1 replication, which is ca. 25 times more effective than the reference compound 2′,3′-dideoxyinosine (DDI). Preliminary structure–activity relationship (SAR) studies revealed that both modulation of the amino function at C(2) and of the alkyl group at C(5) of the pyrimidine ring are crucial for high anti-HIV-1 activity.
Co-reporter:Jian Huang, Fei Xiong, Fen-Er Chen
Tetrahedron: Asymmetry 2008 Volume 19(Issue 12) pp:1436-1443
Publication Date(Web):30 June 2008
DOI:10.1016/j.tetasy.2008.05.020
A concise asymmetric total synthesis of (+)-biotin 1 has been accomplished in which the absolute stereochemistry of C3a, C6a of 1 was established by utilizing an efficient and practical quinine-mediated asymmetric alcoholysis of meso-cyclic anhydride 2 in a single step, the C4 stereochemistry was installed by a direct stereoselective ionic hydrogenation of the thiolactol 7.(4S,5R)-1,3-Dibenzyl-5-(propargyloxycarbonyl)-2-oxo-imidazolidine-4-carboxylic acidC22H20N2O5Ee = 97%[α]D25=+15.6 (c 1.0, CHCl3)Absolute configuration: (4S,5R)Benzyl(3aS,4S,6aR)-5-(1,3-dibenzyl-2,3,3a,4,6,6a-hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-5-yl)pentanoateC31H34N2O3S[α]D25=-20.7 (c 1.0, MeOH)Absolute configuration: (3aS,4S,6aR)Benzyl(3aS,4Z,6aR)-5-(1,3-dibenzyl-2,3,3a,4,6,6a-hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-5-ylidene)pentanoateC31H32N2O3S[α]D25=+154.4 (c 1.0, MeOH)Absolute configuration: (3aS,4Z,6aR)
Co-reporter:Lei Ji, Fen-Er Chen, Bin Xie, Erik De Clercq, Jan Balzarini, Christophe Pannecouque
European Journal of Medicinal Chemistry 2007 Volume 42(Issue 2) pp:198-204
Publication Date(Web):February 2007
DOI:10.1016/j.ejmech.2006.09.018
The synthesis and anti-HIV activity evaluation of a new series of 2,4-pyrimidinediones bearing a 6-(1-naphthoyl) group are described. In general, it was found that most of the title compounds showed good activities against human immunodeficiency virus type-1 (HIV-1). In particular, compound 26 displayed the most potent anti-HIV-1 activity (IC50 = 0.11 ± 0.05 μM), inhibiting HIV-1 replication in MT-4 cells more effectively than HEPT (by 45-fold) and DDI (by 50-fold).The synthesis and anti-HIV activities of a new series of 2,4-pyrimidinediones bearing a 6-(1-naphthoyl) group are reported. It was found that most of the title compounds showed good anti-HIV-1 activity.
Co-reporter:Xiong-Jie Yu;Hui-Fang Dai
Helvetica Chimica Acta 2007 Volume 90(Issue 5) pp:967-971
Publication Date(Web):18 MAY 2007
DOI:10.1002/hlca.200790099
An efficient and stereoselective approach to the synthesis of coenzyme Q10 is described (Scheme). The MeOCH2-protected p-hydroquinone 4 containing the C5 (E)-allyl (tert-butyl)dimethylsilyl ether moiety was obtained via a halogen–lithium exchange of the MeOCH2-proctected 2-bromo-5,6-dimethoxy-3-methylhydroquinone 2 and subsequent addition to (E)-(tBuMe2Si)-OCH2C(Me)=CHCH2Br (3). The reductive desulfonylation of compound 8, obtained from 4via5–7, was successfully carried out by employing Li/EtNH2.
Co-reporter:Jian Huang
Helvetica Chimica Acta 2007 Volume 90(Issue 7) pp:1366-1372
Publication Date(Web):16 JUL 2007
DOI:10.1002/hlca.200790138
A highly stereoselective route to the polysubstituted chiral octahydrobenzofuran 12, a potential synthon for the E-ring core in the (−)-reserpine synthesis, is described. The α-bromo acetal 11 was obtained from inexpensive (−)-shikimic acid (3) through a series of highly stereoselective chemical reactions (Scheme). Et3B/Bu3SnH-Mediated intramolecular radical cyclization of 11 led to compound 12 with the required configuration.
Co-reporter:Hui-Fang Dai;Xiong-Jie Yu
Helvetica Chimica Acta 2006 Volume 89(Issue 7) pp:1317-1321
Publication Date(Web):26 JUL 2006
DOI:10.1002/hlca.200690130
An improved route to coenzyme Q10 (1) starting from commercially available coenzyme Q1 is described. The key steps in this synthesis are the SeO2-mediated oxidation of the protected isoprenylhydroquinone 3 into the (E)-allyl alcohol 5 without the formation of undesired stereoisomer and the one-pot reductive elimination of the phenylsulfonyl and dibenzyl groups in 7 by using naphthalenyllithium.
Co-reporter:Fen-Er Chen;Xu-Xiang Chen;Hui-Fang Dai;Yun-Yan Kuang;Bin Xie;Jian-Feng Zhao
Advanced Synthesis & Catalysis 2005 Volume 347(Issue 4) pp:
Publication Date(Web):28 FEB 2005
DOI:10.1002/adsc.200404311
A practical chemoenzymatic method for the asymmetric total synthesis of d-biotin (1) starting from the commercially available cis-1,3-dibenzyl-2-imidazolidone-4,5-dicarboxylic acid (2) has been developed. The key step of the synthesis is the highly enantioselective hydrolysis of meso-dicarboxylic esters by a polymer-supported pig liver esterase and introduction of a formyl group at the C-4 position in 4via a Grignard reaction. The polymer-supported PLE can be recovered quantitatively from the reaction mixture by simple filtration and reused without significant loss of activity.
Co-reporter:Guang-Fu Sun;Yun-Yan Kuang;Erik De Clercq;Jan Balzarini;Christophe Pannecouque
Archiv der Pharmazie 2005 Volume 338(Issue 10) pp:
Publication Date(Web):7 OCT 2005
DOI:10.1002/ardp.200400961
2-(Arylcarbonylmethyl)thio-6α-naphthylmethyl derivatives of dihydro-alkoxy-benzyl-oxopyrimidines (DABO) were newly found to exhibit activity against both HIV-1 and HIV-2. To further explore their structure-activity relationship, the modified S-DABO analogues (5a–g and 6e–f) with a 1-naphthylthio or phenylthio group at the C-6 position were synthesized. S-Alkylation of 5-ethyl-2-thiouracil with substituted 2-bromo-acetophenones provided crude 2-[(arylcarbonylmethyl)thio]-5-ethyl-(3H)-uracil 2a–e, which was directly subjected to toluenesulfonylation with TsCl to afford disulfonate 4a–e. Substitution of 4a–e with arylthiol afforded the desired S-DABO analogues 5a–g and 6e–f. The compounds were evaluated for their in vitro anti-HIV activity in MT-4 cells. The IC50 values for anti-HIV-1 activity fall into the range 0.37–29.50 μM, and the IC50 values for anti-HIV-2 activity fall into the range 23.11–181.07 μM. The results indicated that these compounds are moderately active against HIV-1 and HIV-2.
Co-reporter:Xiong-Jie Yu;Xu-Xiang Chen;Hui-Fang Dai;Yun-Yan Kuang;Bin Xie
Helvetica Chimica Acta 2005 Volume 88(Issue 10) pp:2575-2581
Publication Date(Web):28 OCT 2005
DOI:10.1002/hlca.200590197
A practical, highly stereoselective ten-step synthesis of coenzyme Q10 (1) has been accomplished (overall yield ca. 28%), starting from commercially available 2,3-dimethoxy-5-methylbenzoquinone (Scheme). The introduction of the first side-chain isoprenyl group with (E)-configuration (compound 6) was realized by means of a coupling reaction of the aromatic system 3 with oxirane, followed by Swern oxidation and Wittig olefination. The tosyl (Ts) group in the sulfone 9 was selectively removed with sodium naphthalenide in THF to afford 1.
Co-reporter:Yanping He, Fener Chen, Guangfu Sun, Yueping Wang, Erik De Clercq, Jan Balzarini, Christophe Pannecouque
Bioorganic & Medicinal Chemistry Letters 2004 Volume 14(Issue 12) pp:3173-3176
Publication Date(Web):21 June 2004
DOI:10.1016/j.bmcl.2004.04.008
The introduction of a β-carbonyl group to the C-2 side chain of S-DABO led to the finding of a series of novel potent anti-HIV agent. Some derivatives proved to be highly effective in inhibiting HIV-1 replication at nanomolar concentrations. Furthermore, the novel S-DABOs differ from the classical NNRTIs in that some compounds are active against both HIV-1 and HIV-2. They might interfere with another target or at least act on RT in a different way as compared to typical NNRTIs.The introduction of a β-carbonyl group to the C-2 side chain of S-DABO led to the finding of a series of novel high potent anti-HIV agent. Interestingly, some of the novel S-DABOs show activity against both HIV-1 and HIV-2.
Co-reporter:Haifeng Wang; Linjie Yan; Yan Wu; Yipei Lu;Fener Chen
Organic Letters () pp:
Publication Date(Web):October 28, 2015
DOI:10.1021/acs.orglett.5b02813
A novel asymmetric synthesis has been developed for the construction of the A-ring of a chiral precursor to calcifediol. The highlights of this synthesis include (i) the introduction of the stereochemistry at the C5-position of the A-ring through the organocatalytic enantioselective desymmetrization of a prochiral cyclic anhydride using a bifunctional urea catalyst and (ii) the introduction of the exo-cyclic (Z)-dienol side chain by a tandem Claisen rearrangement/sulfoxide thermolysis of an allylic alcohol.
Co-reporter:Haihui Peng and Fen-Er Chen
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 30) pp:NaN6301-6301
Publication Date(Web):2017/07/10
DOI:10.1039/C7OB01341H
Prostaglandins (PGs) are a series of hormone-like chemical messengers and play a critical role in regulating physiological activity. The diversified therapeutic activities and complex molecular architectures of PGs have attracted special attention, and huge progress has been made in asymmetric total synthesis and discovery of pharmaceutically useful drug candidates. In the last 10 years, several powerful syntheses have emerged as new solutions to the problem of building PGs and represent major breakthroughs in this area. This review highlights the advances in methodologies for the asymmetric total synthesis of prostaglandins. The application of these methodologies in the syntheses of medicinally useful prostaglandins is also described. The study has been carefully categorized according to the key procedures involved in the syntheses of various prostaglandins, aiming to give readers an easy understanding of this chemistry and provide insights for further improvements.
Co-reporter:Fangjun Xiong, Haifeng Wang, Lingjie Yan, Sheng Han, Yuan Tao, Yan Wu and Fener Chen
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 4) pp:NaN1369-1369
Publication Date(Web):2015/12/11
DOI:10.1039/C5OB02245B
A novel, stereoselective approach towards rosuvastatin calcium from the known (S)-homoallylic alcohol has been developed. The synthesis is highlighted by a regio- and stereocontrolled ICl-induced intramolecular cyclization of chiral homoallylic carbonate to deliver the C6-formyl statin side chain with a syn-1,3-diol moiety. An improved synthesis of the rosuvastatin pyrimidine core moiety is also included. Moreover, this methodology is useful in the asymmetric synthesis of structural variants of statins such as pitavastatin calcium and atorvastatin calcium and their related analogs.
Co-reporter:Fangjun Xiong, Haifeng Wang, Lingjie Yan, Lingjun Xu, Yuan Tao, Yan Wu and Fener Chen
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 38) pp:NaN9819-9819
Publication Date(Web):2015/08/10
DOI:10.1039/C5OB01148E
An efficient and concise asymmetric synthesis of pitavastatin calcium (1) starting from commercially available (S)-epichlorohydrin is described. A convergent C1 + C6 route allowed for the assembly of the pitavastatin C7 side chain via a Wittig reaction between phosphonium salt 2 and the enantiomerically pure C6-formyl side chain 3. The 1,3-syn-diol acetal motif in 3 was established with excellent stereo control by a diastereoselective bismuth-promoted two-component hemiacetal/oxa-Michael addition reaction of (S)-α,β-unsaturated ketone 4 with acetaldehyde.



![Benzaldehyde,3-(1,1-dimethylethyl)-5-[(4-ethenylphenyl)methoxy]-2-hydroxy-](http://img.cochemist.com/ccimg/192900/192803-38-6.png)
![Benzaldehyde,3-(1,1-dimethylethyl)-5-[(4-ethenylphenyl)methoxy]-2-hydroxy-](http://img.cochemist.com/ccimg/192900/192803-38-6_b.png)









![1,3-Propanediol,2-amino-1-[4-(methylsulfonyl)phenyl]-, (1R,2R)-](http://img.cochemist.com/ccimg/51500/51458-28-7.png)
![1,3-Propanediol,2-amino-1-[4-(methylsulfonyl)phenyl]-, (1R,2R)-](http://img.cochemist.com/ccimg/51500/51458-28-7_b.png)


![[S(R*,R*)]-2-(benzylideneamino)-1-(4-nitrophenyl)propane-1,3-diol](http://img.cochemist.com/ccimg/40900/40830-68-0.png)
![[S(R*,R*)]-2-(benzylideneamino)-1-(4-nitrophenyl)propane-1,3-diol](http://img.cochemist.com/ccimg/40900/40830-68-0_b.png)




























![1H-Pyrrole-1-heptanoicacid, 2-(4-fluorophenyl)-b,d-dihydroxy-5-(1-methylethyl)-3-phenyl-4-[(phenylamino)carbonyl]-,calcium salt, hydrate (2:1:3), (bR,dR)-](http://img.cochemist.com/ccimg/344500/344423-98-9.png)
![1H-Pyrrole-1-heptanoicacid, 2-(4-fluorophenyl)-b,d-dihydroxy-5-(1-methylethyl)-3-phenyl-4-[(phenylamino)carbonyl]-,calcium salt, hydrate (2:1:3), (bR,dR)-](http://img.cochemist.com/ccimg/344500/344423-98-9_b.png)


![Ethanol,2-[(5S)-5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methylenecyclohexylidene]-,(2Z)-](http://img.cochemist.com/ccimg/96700/96685-53-9.png)
![Ethanol,2-[(5S)-5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methylenecyclohexylidene]-,(2Z)-](http://img.cochemist.com/ccimg/96700/96685-53-9_b.png)
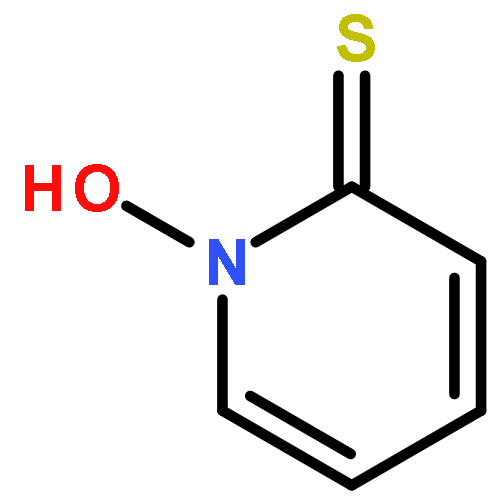


































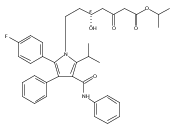
































































































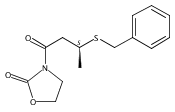


![2-Oxazolidinone, 3-[(3S)-1-oxo-3-(phenylthio)butyl]-](http://img.cochemist.com/ccimg/249700/249603-44-9.png)
![2-Oxazolidinone, 3-[(3S)-1-oxo-3-(phenylthio)butyl]-](http://img.cochemist.com/ccimg/249700/249603-44-9_b.png)




![[3,3'-Biphenanthrene]-4,4'-diol,2,2'-diphenyl-, (3R)-](http://img.cochemist.com/ccimg/147800/147702-16-7.png)
![[3,3'-Biphenanthrene]-4,4'-diol,2,2'-diphenyl-, (3R)-](http://img.cochemist.com/ccimg/147800/147702-16-7_b.png)
![[2,2'-Binaphthalene]-1,1'-diol,3,3'-diphenyl-, (2R)-](http://img.cochemist.com/ccimg/147800/147702-13-4.png)
![[2,2'-Binaphthalene]-1,1'-diol,3,3'-diphenyl-, (2R)-](http://img.cochemist.com/ccimg/147800/147702-13-4_b.png)








![2-Oxazolidinone, 3-[(2E)-1-oxo-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/109300/109299-93-6.png)
![2-Oxazolidinone, 3-[(2E)-1-oxo-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/109300/109299-93-6_b.png)
![3-[(e)-2-butenoyl]-1,3-oxazolidin-2-one](http://img.cochemist.com/ccimg/109300/109299-92-5.png)
![3-[(e)-2-butenoyl]-1,3-oxazolidin-2-one](http://img.cochemist.com/ccimg/109300/109299-92-5_b.png)