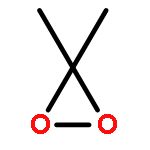
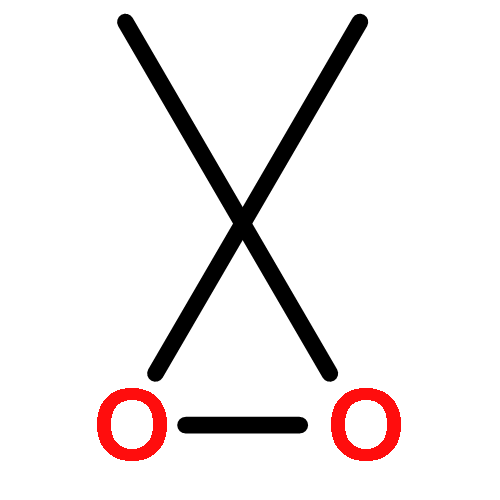
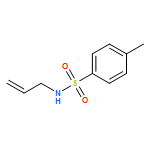
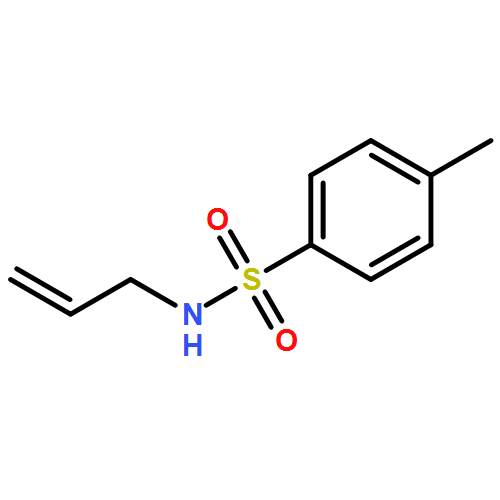
Co-reporter:Ashay Patel, Joseph R. Vella, Zhi-Xiong Ma, R. P. Hsung, and K. N. Houk
The Journal of Organic Chemistry 2015 Volume 80(Issue 23) pp:11888-11894
Publication Date(Web):October 4, 2015
DOI:10.1021/acs.joc.5b02085
Hsung et al. have reported a series of torquoselective electrocyclizations of chiral 1-azahexa-1E,3Z,5E-trienes that yield functionalized dihydropyridines. To understand the origins of the torquoselectivities of these azaelectrocyclizations, we modeled these electrocyclic ring closures using the M06-2X density functional. A new stereochemical model that rationalizes the observed 1,2 stereoinduction emerges from these computations. This model is an improvement and generalization of the “inside-alkoxy” model used to rationalize stereoselectivities of the 1,3-dipolar cycloaddition of chiral allyl ethers and emphasizes a stabilizing hyperconjugative effect, which we have termed a transition state gauche effect. This stereoelectronic effect controls the conformational preferences at the electrocyclization transition states, and only in one of the allowed disrotatory electrocyclization transition states is the ideal stereoelectronic arrangement achieved without the introduction of a steric clash. Computational experiments confirm the role of this effect as a stereodeterminant since substrates with electropositive groups and electronegative groups have different conformational preferences at the transition state and undergo ring closure with divergent stereochemical outcomes. This predicted reversal of stereoselectivity for the ring closures of several silyl substituted azatrienes have been demonstrated experimentally.
Co-reporter:Grant S. Buchanan, Kevin P. Cole, Gang Li, Yu Tang, Ling-Feng You, Richard P. Hsung
Tetrahedron 2011 67(52) pp: 10105-10118
Publication Date(Web):
DOI:10.1016/j.tet.2011.09.111
Co-reporter:Ruth Figueroa;John B. Feltenberger;Christle C. Guevarra
Science China Chemistry 2011 Volume 54( Issue 1) pp:31-42
Publication Date(Web):2011 January
DOI:10.1007/s11426-010-4176-8
Two remarkable epimerization processes were uncovered during our pursuit of an enantioselective synthesis of (+)-aigialospirol featuring a cyclic acetal tethered ring-closing metathesis. Through modeling, we were able to turn these two unexpected epimerizations to our advantage via modeling to ensure a successful and concise total synthesis, thereby firmly establishing cyclic acetal tethered RCM as a powerful strategy in natural product synthesis. Most importantly, calculations allowed us to fully understand the nature and the mechanistic course of these two epimerizations that were imperative to the total synthesis efforts.
Co-reporter:Elizabeth H. Krenske, K. N. Houk, Andrew G. Lohse, Jennifer E. Antoline and Richard P. Hsung
Chemical Science 2010 vol. 1(Issue 3) pp:387-392
Publication Date(Web):25 Jun 2010
DOI:10.1039/C0SC00280A
Chiral oxazolidinones were previously thought to control cycloaddition stereoselectivity by steric crowding of one face of the substrate. We have discovered that in (4 + 3) cycloaddition reactions of oxyallyls, the stereoinduction is caused instead by stabilising CH–π interactions that lead to reaction at the more crowded face of the oxyallyl. Density functional theory calculations on the (4 + 3) cycloadditions of oxazolidinone-substituted oxyallyls with furans establish unexpected transition state conformations and a new explanation of selectivity.
Co-reporter:Elizabeth H. Krenske, K. N. Houk, Andrew G. Lohse, Jennifer E. Antoline and Richard P. Hsung
Chemical Science (2010-Present) 2010 - vol. 1(Issue 3) pp:NaN392-392
Publication Date(Web):2010/06/25
DOI:10.1039/C0SC00280A
Chiral oxazolidinones were previously thought to control cycloaddition stereoselectivity by steric crowding of one face of the substrate. We have discovered that in (4 + 3) cycloaddition reactions of oxyallyls, the stereoinduction is caused instead by stabilising CH–π interactions that lead to reaction at the more crowded face of the oxyallyl. Density functional theory calculations on the (4 + 3) cycloadditions of oxazolidinone-substituted oxyallyls with furans establish unexpected transition state conformations and a new explanation of selectivity.

![2,6-Nonadienal, 9-[(1R,6S)-4-hydroxy-1,6-dimethyl-2-oxo-3-cyclohexen-1-yl]-3,7-dimethyl-, (6E)-rel- (9CI)](/data/chemimg/1353400/873782-36-6.png)
![2,6-Nonadienal, 9-[(1R,6S)-4-hydroxy-1,6-dimethyl-2-oxo-3-cyclohexen-1-yl]-3,7-dimethyl-, (6E)-rel- (9CI)](/data/chemimg/1353400/873782-36-6_b.png)
![2H-Pyran, 2-[(7-bromo-1-methyl-2-heptyn-1-yl)oxy]tetrahydro-](/data/chemimg/201800/1268717-97-0.png)
![2H-Pyran, 2-[(7-bromo-1-methyl-2-heptyn-1-yl)oxy]tetrahydro-](/data/chemimg/201800/1268717-97-0_b.png)


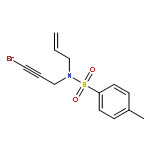
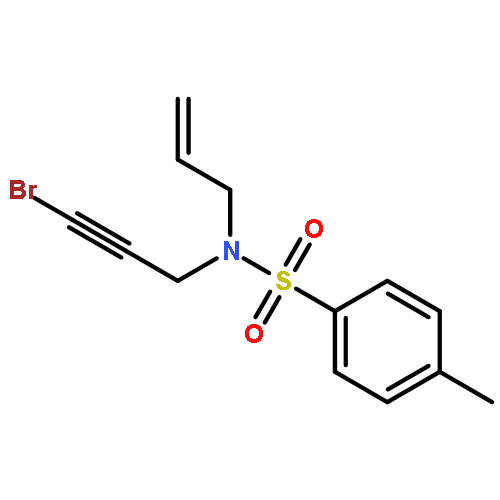
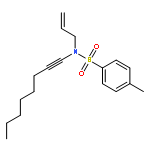

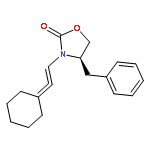
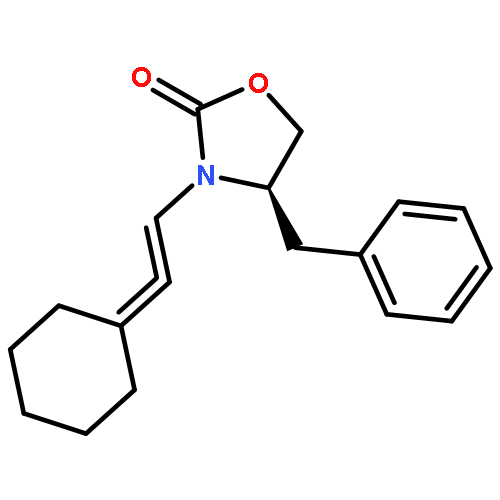
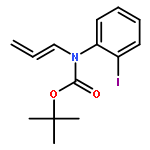
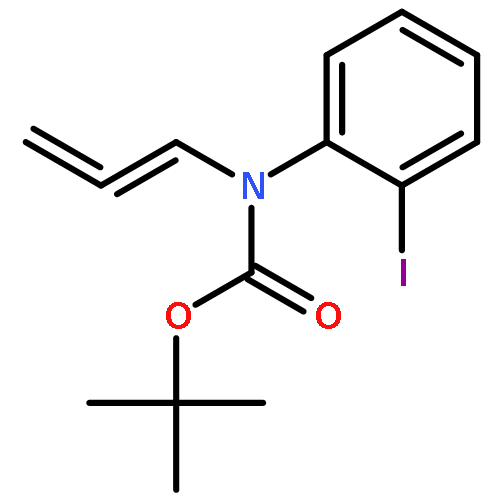
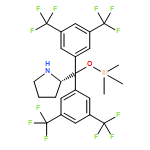

![SILANE, (1,1-DIMETHYLETHYL)[[(2E)-4-IODO-2-BUTENYL]OXY]DIPHENYL-](http://img.cochemist.com/ccimg/808800/808771-15-5.png)
![SILANE, (1,1-DIMETHYLETHYL)[[(2E)-4-IODO-2-BUTENYL]OXY]DIPHENYL-](http://img.cochemist.com/ccimg/808800/808771-15-5_b.png)
![1,2-Pentanediol, 5-(phenylmethoxy)-4-[(phenylmethoxy)methyl]-](http://img.cochemist.com/ccimg/718000/717919-06-7.png)
![1,2-Pentanediol, 5-(phenylmethoxy)-4-[(phenylmethoxy)methyl]-](http://img.cochemist.com/ccimg/718000/717919-06-7_b.png)




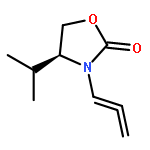
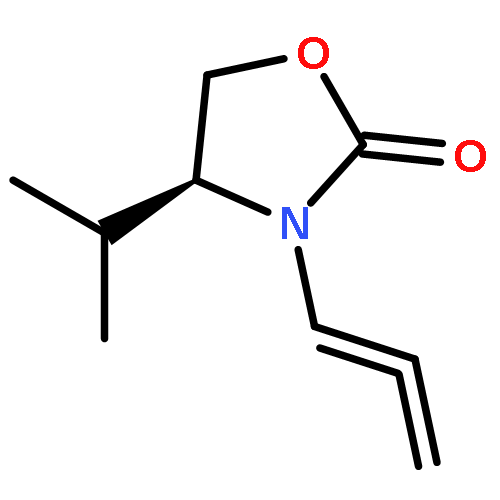
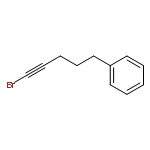
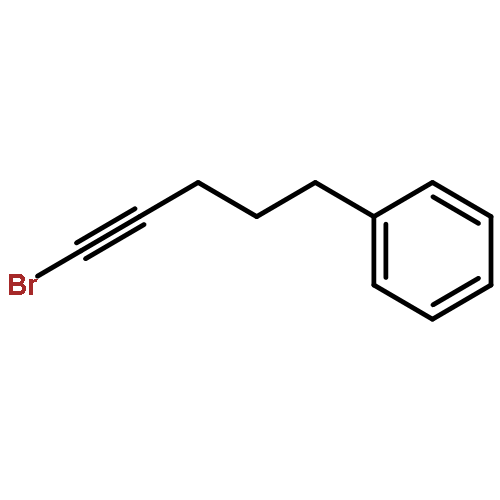
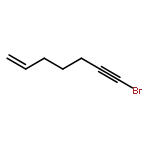
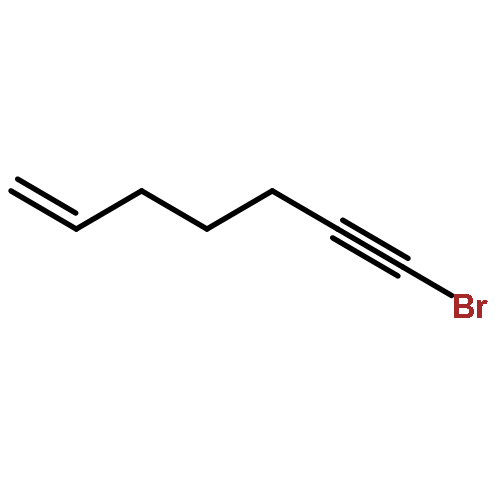

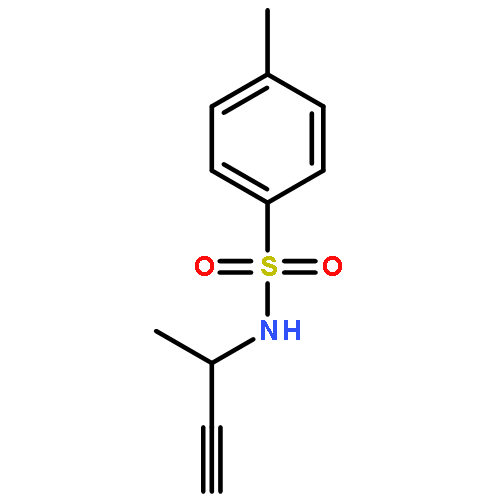

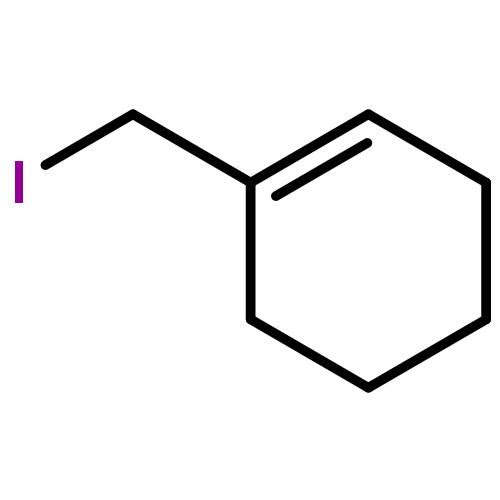
![2-Penten-1-ol, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-, (2E)-](http://img.cochemist.com/ccimg/468800/468745-31-5.png)
![2-Penten-1-ol, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-, (2E)-](http://img.cochemist.com/ccimg/468800/468745-31-5_b.png)
![b-D-Glucopyranosiduronic acid,4-[(1E)-2-(3,5-dihydroxyphenyl)ethenyl]phenyl](http://img.cochemist.com/ccimg/387400/387372-20-5.png)
![b-D-Glucopyranosiduronic acid,4-[(1E)-2-(3,5-dihydroxyphenyl)ethenyl]phenyl](http://img.cochemist.com/ccimg/387400/387372-20-5_b.png)
![b-D-Glucopyranosiduronic acid,3-hydroxy-5-[(1E)-2-(4-hydroxyphenyl)ethenyl]phenyl](http://img.cochemist.com/ccimg/387400/387372-17-0.png)
![b-D-Glucopyranosiduronic acid,3-hydroxy-5-[(1E)-2-(4-hydroxyphenyl)ethenyl]phenyl](http://img.cochemist.com/ccimg/387400/387372-17-0_b.png)




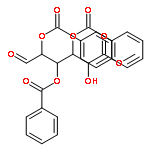
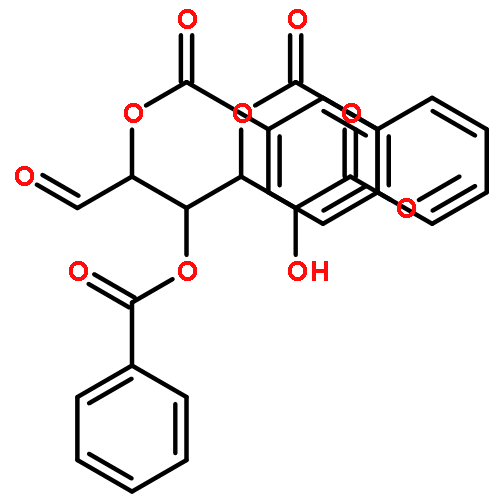
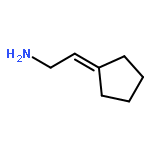
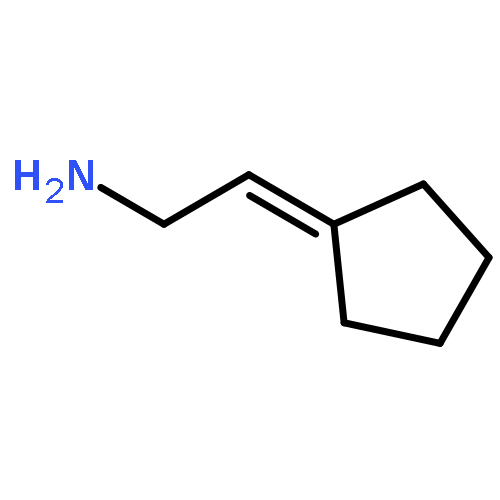



![1H-Isoindole-1,3(2H)-dione, 2-[(3-methyl-2-butenyl)oxy]-](http://img.cochemist.com/ccimg/157200/157128-91-1.png)
![1H-Isoindole-1,3(2H)-dione, 2-[(3-methyl-2-butenyl)oxy]-](http://img.cochemist.com/ccimg/157200/157128-91-1_b.png)
![2-Cyclohexen-1-one, 3-[(diphenylmethyl)amino]-](http://img.cochemist.com/ccimg/148300/148278-39-1.png)
![2-Cyclohexen-1-one, 3-[(diphenylmethyl)amino]-](http://img.cochemist.com/ccimg/148300/148278-39-1_b.png)
![Acetic acid, [(diethoxyphosphino)oxy]-, ethyl ester](http://img.cochemist.com/ccimg/144200/144152-65-8.png)
![Acetic acid, [(diethoxyphosphino)oxy]-, ethyl ester](http://img.cochemist.com/ccimg/144200/144152-65-8_b.png)
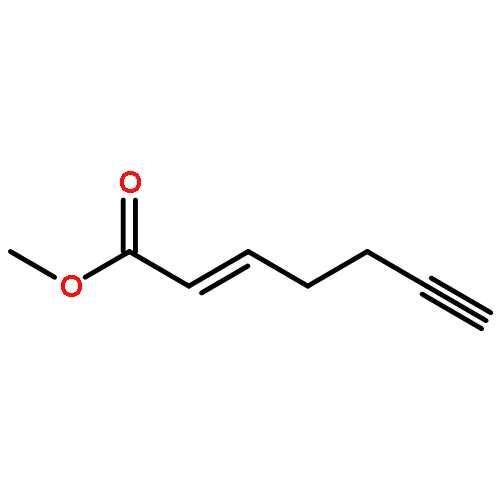
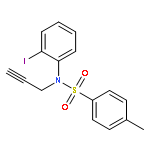
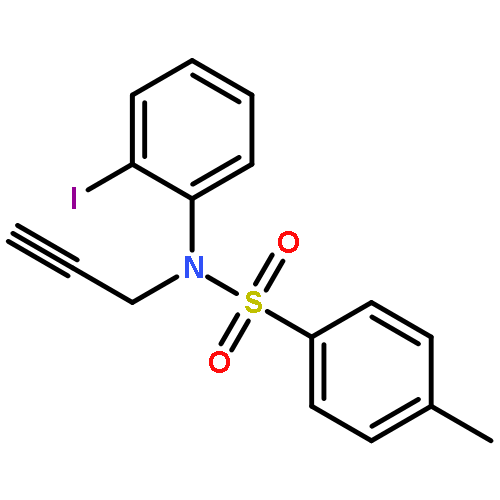
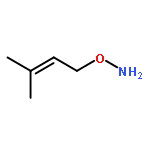
![1-Hexen-3-one, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/132700/132621-86-4.png)
![1-Hexen-3-one, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/132700/132621-86-4_b.png)
![2-Butenal, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/131400/131380-48-8.png)
![2-Butenal, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/131400/131380-48-8_b.png)
![Octanimidamide, N,N-diethyl-N'-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/126800/126746-56-3.png)
![Octanimidamide, N,N-diethyl-N'-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/126800/126746-56-3_b.png)
dimethyl-](http://img.cochemist.com/ccimg/124700/124696-14-6.png)
dimethyl-](http://img.cochemist.com/ccimg/124700/124696-14-6_b.png)
![Hexanal, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/118800/118794-69-7.png)
![Hexanal, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/118800/118794-69-7_b.png)
![Stannane,tributyl[(methoxymethoxy)methyl]-](http://img.cochemist.com/ccimg/100100/100045-83-8.png)
![Stannane,tributyl[(methoxymethoxy)methyl]-](http://img.cochemist.com/ccimg/100100/100045-83-8_b.png)
![2H-Pyran, 2-[[(2Z)-4-bromo-2-butenyl]oxy]tetrahydro-](http://img.cochemist.com/ccimg/98300/98234-53-8.png)
![2H-Pyran, 2-[[(2Z)-4-bromo-2-butenyl]oxy]tetrahydro-](http://img.cochemist.com/ccimg/98300/98234-53-8_b.png)
![2H-Pyran, tetrahydro-2-[[(2Z)-4-iodo-2-butenyl]oxy]-](http://img.cochemist.com/ccimg/94600/94518-15-7.png)
![2H-Pyran, tetrahydro-2-[[(2Z)-4-iodo-2-butenyl]oxy]-](http://img.cochemist.com/ccimg/94600/94518-15-7_b.png)
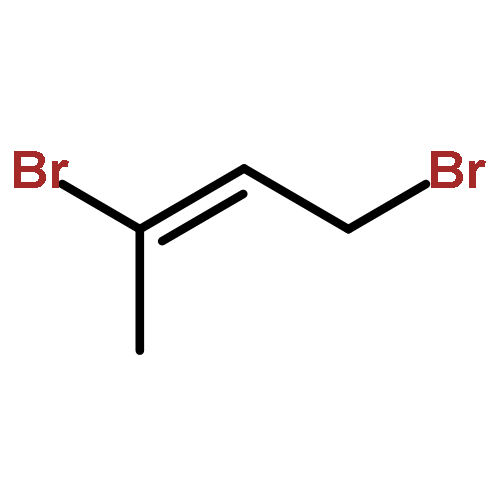
![Benzene, [(1E)-3-iodo-1-propenyl]-](http://img.cochemist.com/ccimg/82900/82859-39-0.png)
![Benzene, [(1E)-3-iodo-1-propenyl]-](http://img.cochemist.com/ccimg/82900/82859-39-0_b.png)
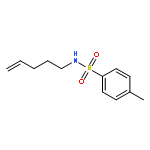
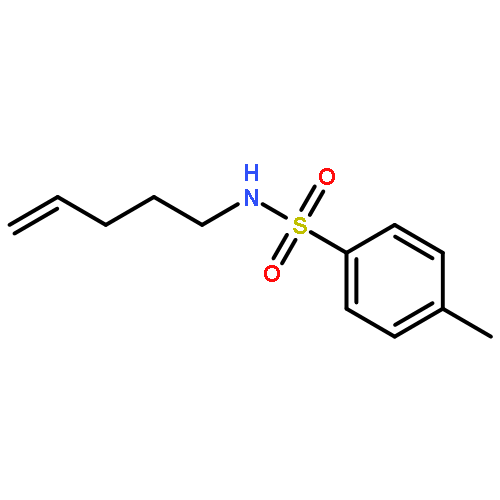
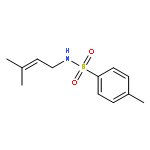



![Acetamide, N-[2-(2-propenyl)phenyl]-](http://img.cochemist.com/ccimg/68300/68267-69-6.png)
![Acetamide, N-[2-(2-propenyl)phenyl]-](http://img.cochemist.com/ccimg/68300/68267-69-6_b.png)
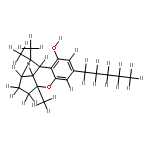
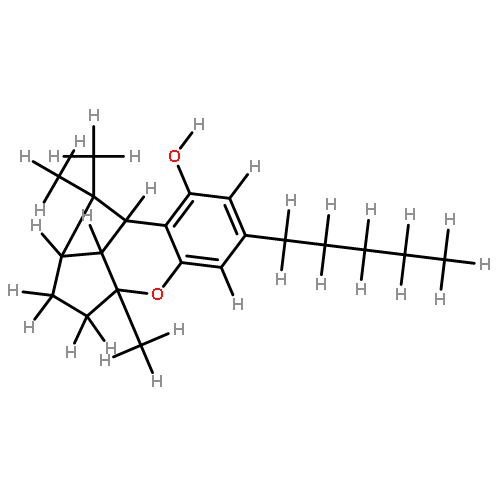






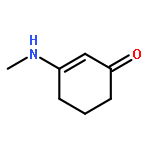
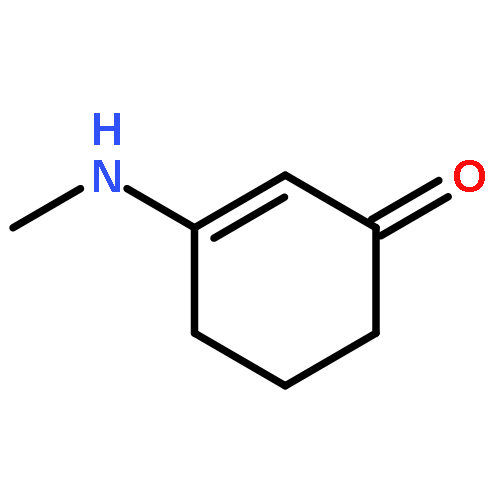


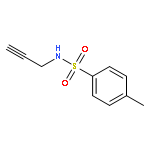
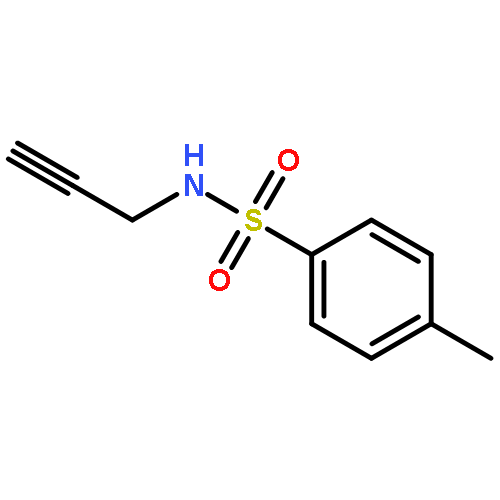











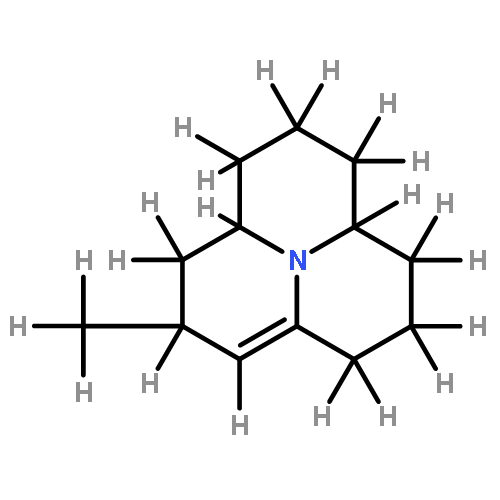
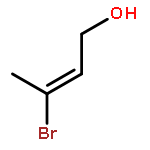
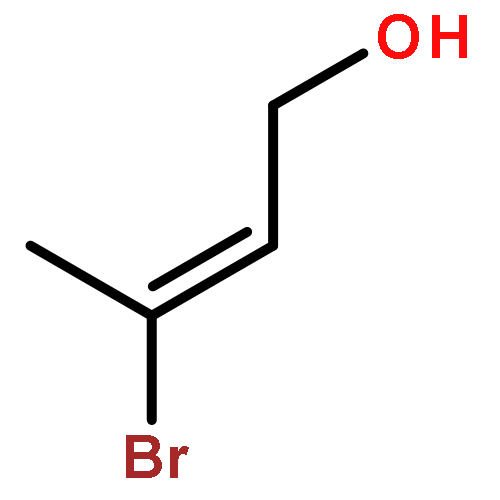
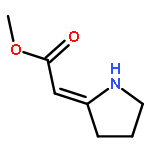




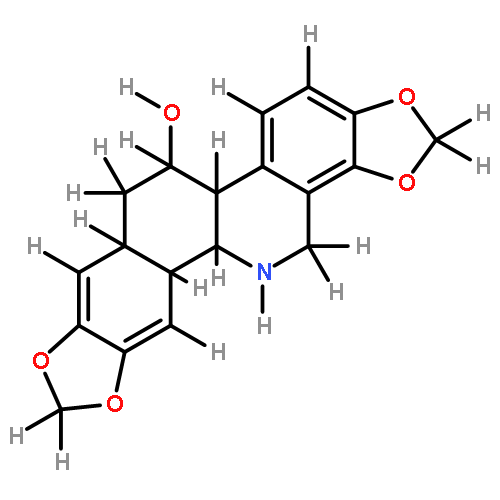
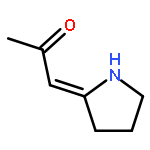
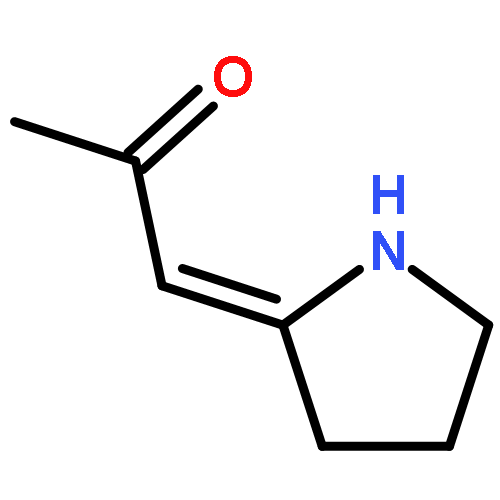
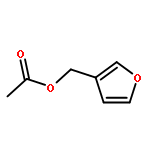












![1H-4-Oxabenzo[f]cyclobut[cd]inden-8-ol,1a,2,3,3a,8b,8c-hexahydro-1,1,3a-trimethyl-6-pentyl-, (1aS,3aR,8bR,8cR)-](http://img.cochemist.com/ccimg/21400/21366-63-2.png)
![1H-4-Oxabenzo[f]cyclobut[cd]inden-8-ol,1a,2,3,3a,8b,8c-hexahydro-1,1,3a-trimethyl-6-pentyl-, (1aS,3aR,8bR,8cR)-](http://img.cochemist.com/ccimg/21400/21366-63-2_b.png)

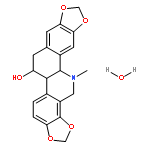
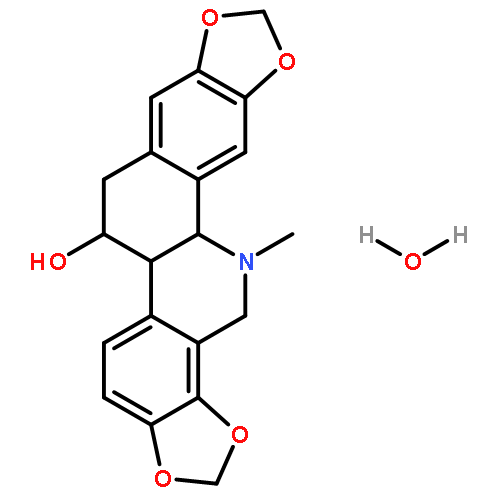

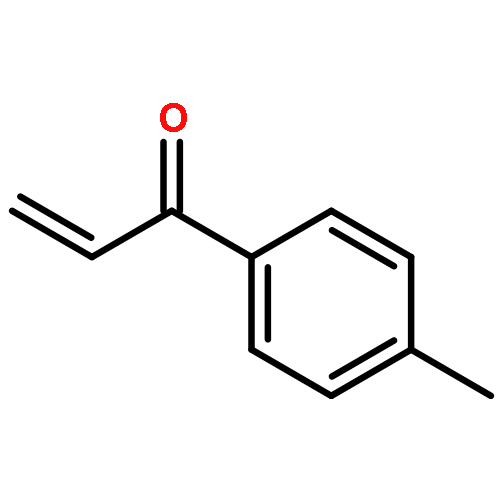
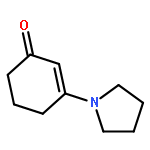







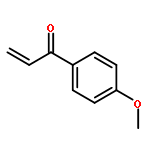
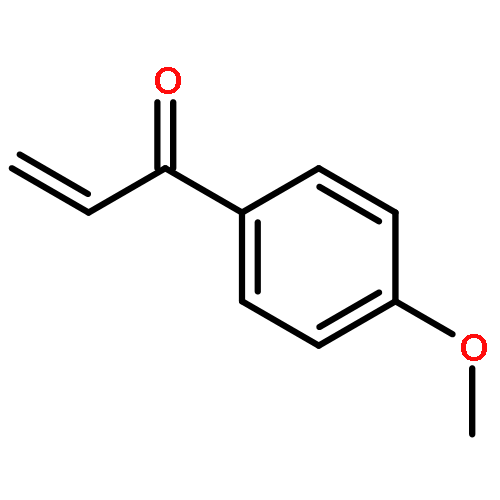






![2-Propenal, 3-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-, (2E)-](http://img.cochemist.com/ccimg/4800/4757-80-6.png)
![2-Propenal, 3-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-, (2E)-](http://img.cochemist.com/ccimg/4800/4757-80-6_b.png)
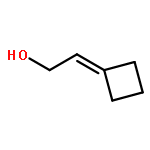
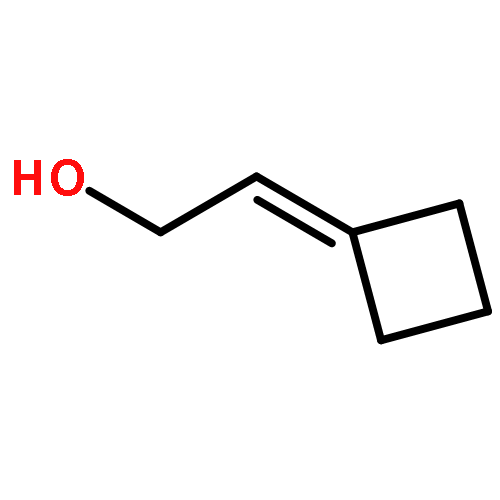
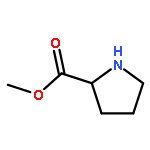
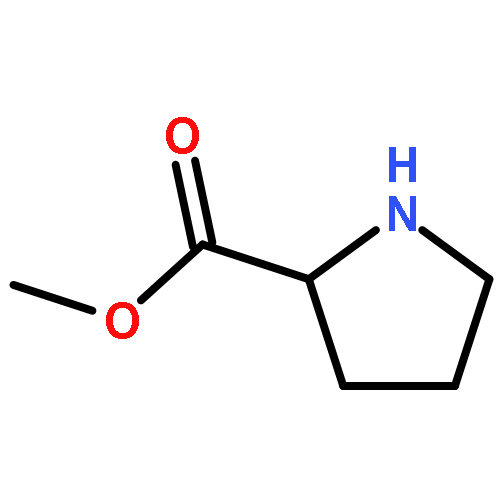
![[1,1':3',1''-Terphenyl]-2'-ol](http://img.cochemist.com/ccimg/2500/2432-11-3.png)
![[1,1':3',1''-Terphenyl]-2'-ol](http://img.cochemist.com/ccimg/2500/2432-11-3_b.png)
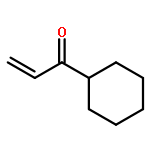
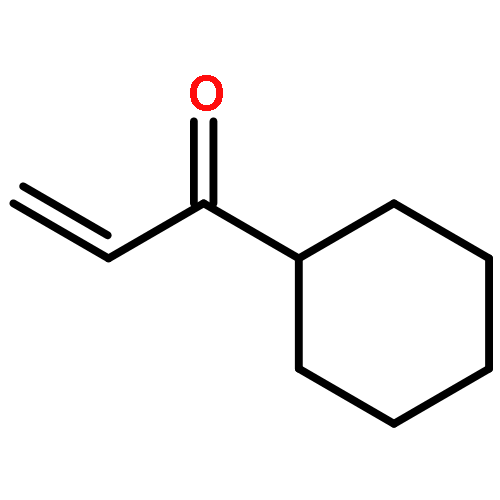

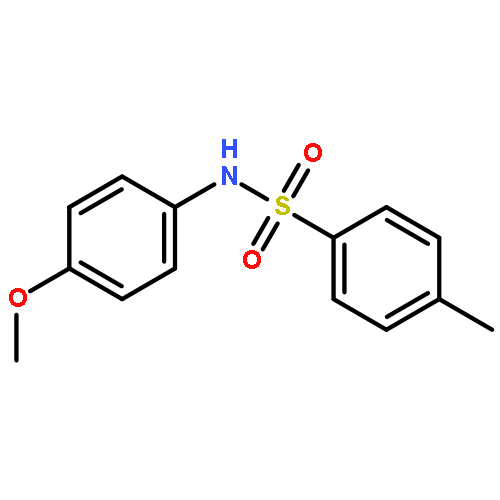
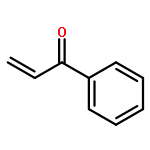
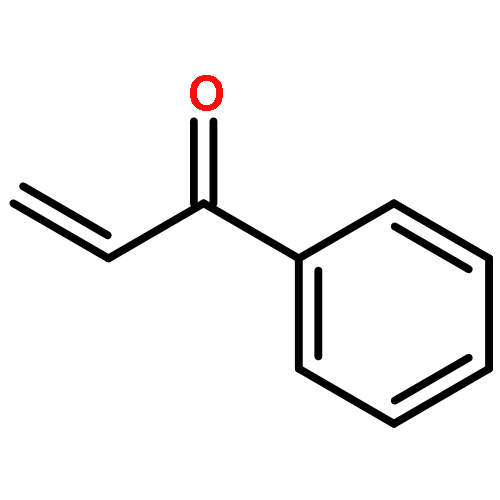
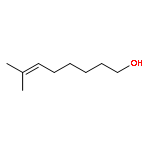
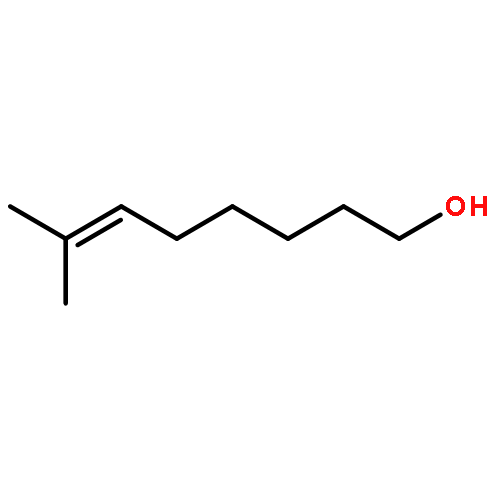


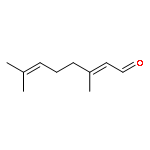
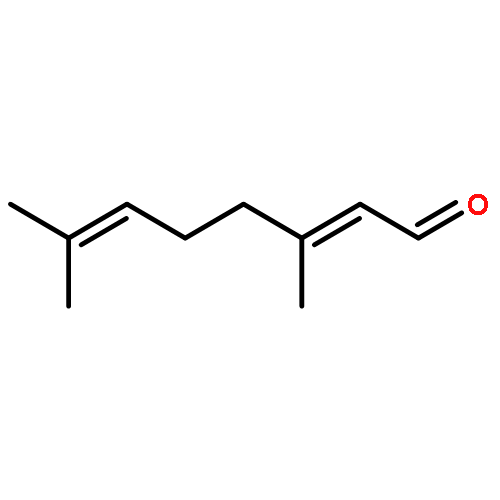
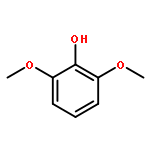
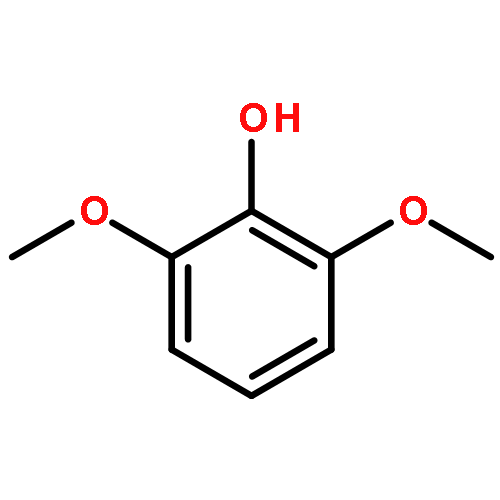


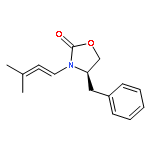


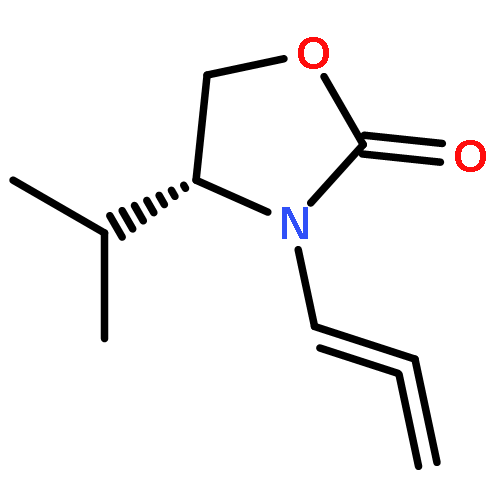




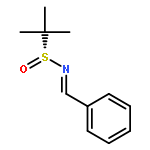
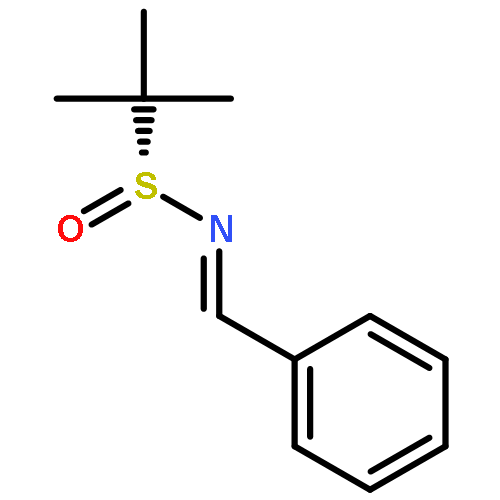




![4,13-Ethano-3,6-methano-3H-furo[3,4-c]oxacyclododecin-3,16-diol,1,4,6,7,8,11,12,13-octahydro-6,10,13,14-tetramethyl-,(3S,4R,6S,9E,13S,14R,16S)-](http://img.cochemist.com/ccimg/130600/130595-24-3.png)
![4,13-Ethano-3,6-methano-3H-furo[3,4-c]oxacyclododecin-3,16-diol,1,4,6,7,8,11,12,13-octahydro-6,10,13,14-tetramethyl-,(3S,4R,6S,9E,13S,14R,16S)-](http://img.cochemist.com/ccimg/130600/130595-24-3_b.png)
![Benzene, [(1Z)-3-bromo-1-propenyl]-](http://img.cochemist.com/ccimg/115200/115117-88-9.png)
![Benzene, [(1Z)-3-bromo-1-propenyl]-](http://img.cochemist.com/ccimg/115200/115117-88-9_b.png)