Co-reporter:Xi Lu, Yan Wang, Ben Zhang, Jing-Jing Pi, Xiao-Xu Wang, Tian-Jun Gong, Bin Xiao, and Yao Fu
Journal of the American Chemical Society September 13, 2017 Volume 139(Issue 36) pp:12632-12632
Publication Date(Web):August 29, 2017
DOI:10.1021/jacs.7b06469
Herein, we described a nickel-catalyzed monofluoroalkenylation through defluorinative reductive cross-coupling of gem-difluoroalkenes with alkyl halides. Key to the success of this strategy is the combination of C–F cleavage with alkyl halides activation. This reaction enables the convenient synthesis of a large variety of functionalized monofluoroalkenes under mild reaction conditions with broad functional group compatibility and excellent Z-selectivity. The combination of Ni catalysis with (Bpin)2/K3PO4 as terminal reductant promoted the efficient C(sp2)–C(sp3) formation especially the generation of all-carbon quaternary centers with high chemoselectivity.
Co-reporter:Rui Zhu, Ju-Long Jiang, Xing-Long Li, Jin Deng, and Yao Fu
ACS Catalysis November 3, 2017 Volume 7(Issue 11) pp:7520-7520
Publication Date(Web):September 19, 2017
DOI:10.1021/acscatal.7b01569
Direct hydrogenolysis of lactone to carboxylic acid (i.e., hydrogenolysis of the Calkoxy–O bond with the carbonyl group untouched) is generally difficult, as the current strategies employing Brønsted acids as the catalyst usually require harsh conditions such as a high temperature and a high H2 pressure. Herein, we report a developed solvent-free catalytic transformation, in which W(OTf)6 is believed to promote the hydrogenolysis process. This strategy could efficiently hydrogenate lactones to carboxylic acids under extra mild conditions (e.g., a reaction temperature of <150 °C and 1 atm of H2) and showed a broad substrate scope. In addition, the catalytic protocol can be further applied to the hydrogenolysis of polyhydroxyalkanoate, as a renewable polymer, to the corresponding straight-chain carboxylic acids. An extensive mechanistic study was subsequently performed, and the density functional theory calculations revealed a reaction pattern, including the complete cleavage of the C═O bond with the assistance of the W(OTf)6 catalyst. Moreover, the key intermediate created in the mechanism, as an oxonium with an OTf moiety, was successfully detected by electrospray ionization mass spectra. Through a comparison with the Brønsted acid-catalyzed system, the study confirmed that the existence of the OTf moiety can significantly lower the barriers associated with the rearrangement and elimination processes. Meanwhile, emphasis was placed on the critical role that the anion plays, as well as the fact that the anion effect is directly related to the chemoselectivity.Keywords: hydrogenolysis; lactones; Lewis acids; metal triflate; tungsten;
Co-reporter:Wan-Min Cheng, Rui Shang, Bin Zhao, Wei-Long Xing, and Yao Fu
Organic Letters August 18, 2017 Volume 19(Issue 16) pp:
Publication Date(Web):July 28, 2017
DOI:10.1021/acs.orglett.7b01950
Decarboxylative borylation of aryl and alkenyl carboxylic acids with bis(pinacolato)diboron was achieved through N-hydroxyphthalimide esters using tert-butyl isonicotinate as a catalyst under base-free conditions. A variety of aryl carboxylic acids possessing different functional groups and electronic properties can be smoothly converted to aryl boronate esters, including those that are difficult to decarboxylate under transition-metal catalysis, offering a new method enabling use of carboxylic acid as building blocks in organic synthesis. Mechanistic analysis suggests the reaction proceeds through coupling of a transient aryl radical generated by radical decarboxylation with a pyridine-stabilized persistent boryl radical. Activation of redox active esters may proceed via an intramolecular single-electron-transfer (SET) process through a pyridine–diboron–phthalimide adduct and accounts for the base-free reaction conditions.
Co-reporter:Mian Cui, Jing-Hui Liu, Xiao-Yu Lu, Xi Lu, Zhen-Qi Zhang, Bin Xiao, Yao Fu
Tetrahedron Letters 2017 Volume 58, Issue 20(Issue 20) pp:
Publication Date(Web):17 May 2017
DOI:10.1016/j.tetlet.2017.02.090
•An efficient and convenient method for C5-benzylation of quinoline frameworks.•Affords an easy access to the synthesis of various functionalized N-based amidoquinoline derivatives.•Using inexpensive FeCl3 catalyst.An efficient and convenient method for C5-benzylation of quinoline frameworks is developed with the using of inexpensive FeCl3 catalyst. A range of N-bisbenzylic and N-monobenzylic sulfonamides smoothly react with aliphatic amides and aromatic amides, giving the corresponding products in moderate to excellent yields.Download high-res image (33KB)Download full-size image
Co-reporter:Long Yan;Xinxin Liu;Jin Deng
Green Chemistry (1999-Present) 2017 vol. 19(Issue 19) pp:4600-4609
Publication Date(Web):2017/10/02
DOI:10.1039/C7GC01720K
Deoxygenation is the central challenge in converting bio-derived fatty esters into diesel-range hydrocarbons. A new molybdenum oxide doping nickel phyllosilicate (Mo–Ni@PSi) bifunctional catalyst was synthesized and used for the deoxygenation of methyl palmitate. Compared to impregnated Ni catalysts, Mo–Ni@PSi catalysts showed obviously enhanced catalytic activity for the deoxygenation process. Several structural variations induced by the introduction of Mo species were confirmed by H2-TPR, XRD, FT-IR, TEM, XPS, and NH3-TPD characterization. With these variations, more dispersive Ni nanoparticles (Ni NPs) and acidic sites were explored over the surface of modified Mo–Ni@PSi, which provided high catalytic activity for the deoxygenation of fatty esters. Furthermore, the content of the Mo element and the influences of activation temperature on the catalyst were also investigated. Remarkably, the highest catalytic activity was observed over the 3% Mo–Ni@PSi(B) catalyst with 98.3% yield of hydrocarbons at 220 °C and 1.0 MPa H2, and the catalytic activity decreased with further increase in the Mo content. Then, hydrodeoxygenation and decarbonylation processes were both confirmed in the reaction mechanism study. Additionally, the catalyst also presented good catalytic activity after four recycle reactions. The oxidization of reduced Ni and Mo species was probably the main reason for the deactivation of the catalyst.
Co-reporter:Feng Li;Zhen-Qi Zhang;Xi Lu;Bin Xiao
Chemical Communications 2017 vol. 53(Issue 25) pp:3551-3554
Publication Date(Web):2017/03/23
DOI:10.1039/C7CC00129K
A Cu/PPh3-catalyzed propargylic substitution reaction of diborylmethane is reported. Different substituted propargyl electrophiles can be employed in this reaction, and various synthetic valuable functional groups can be tolerated. Di-deuterated diborylmethane can also be used under these conditions and generates α-deuterated alkylboronic esters in good yield.
Co-reporter:Zheng-Yang Xu;Hai-Zhu Yu
Science China Chemistry 2017 Volume 60( Issue 2) pp:165-166
Publication Date(Web):2017 February
DOI:10.1007/s11426-016-0432-3
Co-reporter:Bing Wang, Ju-long JiangHai-zhu Yu, Yao Fu
Organometallics 2017 Volume 36(Issue 3) pp:
Publication Date(Web):January 26, 2017
DOI:10.1021/acs.organomet.6b00717
Vinyl chlorides can be effectively prepared via alkyne hydrochlorination catalyzed by the [Cp*RuCl(cod)]/PPh3 system. The reaction mechanism has been elucidated by density functional theory calculations. Different from the previously proposed pathway (oxidative addition, alkyne coordination, H-/Cl-transfer via inner addition, and reductive elimination), the calculation results indicate that HCl favorably attacks the alkyne stepwise from the outer coordination sphere of the Ru center. The reaction is essentially the electrophilic addition of HCl to alkyne. Therefore, the proton tends to attack the more negatively charged carbon of the C–C triple bond, leading to the Markovnikov product.
Co-reporter:Wan-Min Cheng;Dr. Rui Shang;Ming-Chen Fu; Yao Fu
Chemistry - A European Journal 2017 Volume 23(Issue 11) pp:2537-2541
Publication Date(Web):2017/02/21
DOI:10.1002/chem.201605640
AbstractAn iridium photoredox catalyst in combination with either a stoichiometric amount of Brønsted acid or a catalytic amount of Lewis acid is capable of catalyzing regioselective alkylation of N-heteroarenes with N-(acyloxy)phthalimides at room temperature under irradiation. A broad range of N-heteroarenes can be alkylated using a variety of secondary, tertiary, and quaternary carboxylates. Mechanistic studies suggest that an IrII/IrIII redox catalytic cycle is responsible for the observed reactivity.
Co-reporter:Yuan-Ye JiangChen Wang, Yujie Liang, Xiaoping Man, Siwei Bi, Yao Fu
The Journal of Organic Chemistry 2017 Volume 82(Issue 2) pp:
Publication Date(Web):December 21, 2016
DOI:10.1021/acs.joc.6b02642
Acylborons, as a growing class of boron reagents, were successfully applied to amide ligation and showed potential in chemoselective bioconjugation reactions in recent years. In this manuscript, a density functional theory (DFT) study was performed to investigate the mechanism of the amide formation between monofluoroacylboronates and hydroxylamines. An updated pathway was clarified herein, including water-assisted hemiaminal formation, pyridine ligand dissociation, elimination via a six-membered-ring transition state, and water-assisted tautomerization. The proposed mechanism was further examined by applying it to investigate the activation barriers of other monofluoroacylboronates, and the related calculations well reproduced the experimentally reported relative reactivities. On the basis of these results, we found that the ortho substitution of the pyridine ligand destabilizes the acylboron substrates and the hemiaminal intermediates by steric effects and thus lowers the energy demand of the ligand dissociation and elimination steps. By contrast, the para substitution of the pyridine ligand with an electron-donating group enhances the coordination of the ligand by electronic effects, which is a disadvantage to the ligand dissociation and elimination steps. The ligand bearing a rigid linkage blocks the rotation of the pyridine ligand and makes ligand dissociation difficult.
Co-reporter:Ebrahim-Alkhalil M. A. Ahmed;Xi Lu;Tian-Jun Gong;Zhen-Qi Zhang;Bin Xiao
Chemical Communications 2017 vol. 53(Issue 5) pp:909-912
Publication Date(Web):2017/01/10
DOI:10.1039/C6CC07924E
We report the first copper-catalyzed/mediated borylative ring opening reaction of epoxides. This process represents a direct borylative C(sp3)–O bond cleavage of terminal epoxide substrates with commercially available diboron reagents. A wide range of epoxides with different functional groups are involved, and were subsequently converted to the corresponding β-hydroxyl boronic esters smoothly. Moreover, the ring opening product β-pinacol boronate alcohol provided a more beneficial approach for the formation of C–C and C–N bonds.
Co-reporter:Long Yan;Qian Yao
Green Chemistry (1999-Present) 2017 vol. 19(Issue 23) pp:5527-5547
Publication Date(Web):2017/11/27
DOI:10.1039/C7GC02503C
Levulinic acid (LA) is one of the most important biomass-derived platform molecules and can be produced from both C5 and C6 carbohydrates via tandem dehydration and hydrolysis reactions. Since LA has different functional groups, it would be converted into various compounds by catalyzed reactions. During the past few decades, it has been proved that the conversion of biomass materials into biofuels and chemicals with LA as intermediate is feasible. Alkyl levulinates derived from LA have similar chemical properties to LA and are also used for the synthesis of LA derived molecules. Herein, this review focuses on the transformation of levulinic acid and alkyl levulinate into biofuels and high-valued chemicals, such as γ-valerolactone, 2-methyltetrahydrofurnan, valeric acid/alkyl valerates, 1,4-pentanediol and N-substituted pyrrolidinones. Different homogeneous and heterogeneous catalysts are reviewed and compared. The ligands and additives exhibit a remarkable impact on the distribution of products in homogeneous catalytic systems. Moreover, the catalytic performances of heterogeneous catalytic systems are influenced by numerous factors, such as the size of the metal particles, surface morphology and acid density. In addition, in order to make this review more complete, the production of LA and alkyl levulinates is also included in the manuscript.
Co-reporter:Qian Yao, Lujiang Xu, Chaofang Guo, Ziguo Yuan, Ying Zhang, Yao Fu
Journal of Analytical and Applied Pyrolysis 2017 Volume 124(Volume 124) pp:
Publication Date(Web):1 March 2017
DOI:10.1016/j.jaap.2017.03.004
•Cellulose was selectively converted into pyrroles via CFP process in NH3.•Different factors on the product distribution were investigated systematically.•9.7% yield of N-containing chemicals was achieved over γ-Al2O3 catalyst at 400 °C.•The selectivity of pyrroles in N-containing chemicals reached at 89.5%.•The possible conversion pathway from cellulose to pyrroles was proposed.In this study, cellulose was selectively converted into pyrroles via catalytic fast pyrolysis under ammonia atmosphere over the γ-Al2O3 catalyst. Both in situ and ex situ lab-scale fast pyrolysis sets were designed and used for investigation, and more pyrroles were produced via in situ CFP process. In addition, the effects of catalyst, reaction temperature and catalyst-to-cellulose ratio on the product distribution were investigated systematically. All these factors played important roles in the production of pyrroles. Under the optimized in situ CFP condition, at 400 °C and catalyst-to-cellulose ratio at 2, the carbon yield of N-containing chemicals from cellulose under ammonia atmosphere reached 9.7%. The selectivity of pyrroles in N-containing chemicals was 89.5%. The possible conversion pathway from cellulose to pyrroles was also proposed, that is, cellulose was firstly converted into anhydrosugars through thermal decomposition, then anhydrosugars underwent dehydration and rearrangement reactions to form furans. Thereafter, the furans were transformed into pyrroles by reacting with ammonia.Download high-res image (121KB)Download full-size image
Co-reporter:Bakht Zada;Mengyuan Chen;Chubai Chen;Long Yan;Qing Xu
Science China Chemistry 2017 Volume 60( Issue 7) pp:853-869
Publication Date(Web):05 June 2017
DOI:10.1007/s11426-017-9067-1
Conversion of non-edible biomass into fuels and value-added chemicals has achieved great attention to cope the world’s energy requirements. Lignocellulose based sugar alcohols such as sorbitol, mannitol, xylitol, and erythritol can be potentially used as emerging fuels and chemicals. These sugar alcohols can be converted into widely used products (e.g. polymer synthesis, food and pharmaceuticals industry). The heterogeneous catalytic production of sugar alcohols from renewable biomass provides a safe and sustainable approach. Hydrolysis, coupled with hydrogenation and hydrogenolysis has been proved to be more effective strategy for sugar alcohols production from biomass. This review summarizes the recent advances in biomass upgrading reactions for the production of sugar alcohols and their comprehensive applications.
Co-reporter:Yong-Jian Xu, Jing Shi, Wei-Peng Wu, Rui Zhu, Xing-Long Li, Jin Deng, Yao Fu
Applied Catalysis A: General 2017 Volume 543(Volume 543) pp:
Publication Date(Web):5 August 2017
DOI:10.1016/j.apcata.2017.07.004
•A highly efficient Cp*Ir (III) and acid co-catalyst system to conversion of furfural compounds to cyclopentanones or straight chain ketones.•Mechanism of conversion of 5-HMF to HCPN was revised and supplemented. We found that HHD condensed via Aldol reaction to produce MCP instead of HCPN.•Brønsted acid and Lewis acid can promote conversion of furfural compounds to straight chain ketones and cyclopentanones respectively.In this paper, Cp*Ir (III) Complex and acid co-catalyst system was developed. By using Cp*Ir and γ-Al2O3 (Lewis acid), 5-hydroxymethylfurfural (5-HMF) can be converted efficiently to 3-hydroxymethyl cyclopentanone (HCPN). Meanwhile, Cp*Ir and Brønsted acid can promote conversion of 5-HMF to 1-Hydroxy-2,5-hexanedione (HHD). The effect of Lewis acid and Brønsted acid on the hydrogenation of furan derivatives was studied. Mechanism of conversion of 5-HMF to HCPN was discussed in detail and mechanism proposed by our predecessors was revised. Instead of being an intermediate for the formation of HCPN, it is believed that, HHD is a product of another reaction pathway. HHD condensed via Aldol reaction to produce 3-methylcyclopenten-2-ol-1-one (MCP) instead of HCPN. Under the promotion of Lewis acid, 5-HMF firstly convert to the precursor of HHD. After that, the reaction is through 4 π-electrocyclic ring closure process and HCPN was formed ultimately. Furthermore, we found that our Cp*Ir and acid co-catalyst system is suitable for a variety of furfural compounds. By using Cp*Ir, Brønsted acid can promote conversion of furfural compounds to straight chain ketones and Lewis acid can promote the rearrangement of furfural compounds to cyclopentanone derivatives.By using Cp*Ir, Brønsted acid and Lewis acid can promote conversion of furfural compounds to straight chain ketones and cyclopentanone derivatives, respectively.Download high-res image (208KB)Download full-size image
Co-reporter:Zhao-Jing Liu; Xi Lu; Guan Wang; Lei Li; Wei-Tao Jiang; Yu-Dong Wang; Bin Xiao
Journal of the American Chemical Society 2016 Volume 138(Issue 30) pp:9714-9719
Publication Date(Web):July 20, 2016
DOI:10.1021/jacs.6b05788
Copper-catalyzed directed decarboxylative amination of nonactivated aliphatic carboxylic acids is described. This intramolecular C–N bond formation reaction provides efficient access to the synthesis of pyrrolidine and piperidine derivatives as well as the modification of complex natural products. Moreover, this reaction presents excellent site-selectivity in the C–N bond formation step through the use of directing group. Our work can be considered as a big step toward controllable radical decarboxylative carbon–heteroatom cross-coupling.
Co-reporter:Chi Fang, Chunlei Lu, Muhua Liu, Yiling Zhu, Yao Fu, and Bo-Lin Lin
ACS Catalysis 2016 Volume 6(Issue 11) pp:7876
Publication Date(Web):October 13, 2016
DOI:10.1021/acscatal.6b01856
The formylation and methylation of amines with carbon dioxide and hydrosilanes are emerging yet important types of transformations for CO2. Catalytic methods effective for both reactions with wide substrate scopes are rare because of the difficulty in controlling the selectivity. Herein, we report that simple and readily available inorganic bases—alkali-metal carbonates, especially cesium carbonate—catalyze both the formylation and methylation reactions efficiently under mild conditions. The selectivity can be conveniently controlled by varying the reaction temperature and silane. A “cesium effect” on both reactions was observed by comparing the catalytic activity of various alkali-metal carbonates. Combined experimental and computational studies suggested the following reaction mechanism: (i) activation of Si–H by Cs2CO3, (ii) insertion of CO2 into Si–H, (iii) formylation of amines by silyl formate, and (iv) reduction of formamides to methylamines.Keywords: amines; CO2; Cs2CO3; formylation; hydrosilanes; methylation
Co-reporter:Yuan-Ye Jiang, Long Yan, Hai-Zhu Yu, Qi Zhang, and Yao Fu
ACS Catalysis 2016 Volume 6(Issue 7) pp:4399
Publication Date(Web):May 24, 2016
DOI:10.1021/acscatal.6b00239
Efficient depolymerization methods are critical to the sustainable production of fuels and chemicals from biomass. Ligand-controlled selective C(sp3)–O and Ar–C(sp3) cleavages of β-O-4 lignin model compounds were realized with vanadium catalysts under redox-neutral conditions or air atmosphere. To clarify the mechanism and the origin of selectivity, a joint theoretical and experimental study was performed herein. First, with the aid of density functional theory (DFT) calculations, an updated mechanism involving VV, VIV, and VIII complexes was discovered for the C(sp3)–O cleavage process catalyzed by the Schiff base vanadium complexes with an overall free energy barrier of 34.9 kcal/mol. Meanwhile, a detailed catalytic cycle involving novel stepwise O–O/Ar–C(sp3) cleavage was clarified for the Ar–C(sp3) cleavage process catalyzed by the bis(8-oxyquinolate) coordinated vanadium complexes, having an overall free energy barrier of 28.8 kcal/mol. Further analysis based on the energetic span model revealed that the switchable selectivity results from the different T1 (ground triplet state)–HOMO separation/charge dispersion effects of ligands and the different formal oxidation states of the TOF-determining transition state (TDTS) in the C(sp3)–O and Ar–C(sp3) cleavage processes. Finally, control experiments of base and oxygen pressure were conducted to validate the conclusions from DFT studies regarding the role of bases and the TDTS step in the Ar–C(sp3) cleavage process.Keywords: aerobic oxidation; C−C bond cleavage; C−O bond cleavage; density functional theory; lignin model compounds; spin crossover; two-state reactivity; vanadium
Co-reporter:Ming-Chen Fu, Rui Shang, Wan-Min Cheng, and Yao Fu
ACS Catalysis 2016 Volume 6(Issue 4) pp:2501
Publication Date(Web):March 10, 2016
DOI:10.1021/acscatal.6b00276
By the combination of a Ni(II) salt, a bisphosphine ligand, and a catalytic amount of carboxylic acid anhydride, atom-economic hydrocarboxylation of various alkynes with formic acid can be achieved with high selectivity and remarkable functional group compatibility, affording α,β-unsaturated carboxylic acids regio- and stereoselectively. Both terminal and internal alkynes are amenable substrates. A mechanism proceeding through carbon monoxide recycling in a catalytic amount is demonstrated to be crucial for the success of this transformation.Keywords: alkynes; carbon monoxide; formic acid; hydrocarboxylation; nickel
Co-reporter:Meng-Yuan Chen, Chu-Bai Chen, Bakht Zada and Yao Fu
Green Chemistry 2016 vol. 18(Issue 13) pp:3858-3866
Publication Date(Web):15 Mar 2016
DOI:10.1039/C6GC00432F
The hydrogenolysis of C–O and CO in 5-hydroxymethylfurfural for the production of furan biofuel 2,5-dimethylfuran (DMF) is of great importance for biomass refining. However, development of non-noble metal-based catalysts which perform stably for this process is still challenging. Here, perovskite-supported Ni catalysts were used for the hydrogenolysis of 5-hydroxymethylfurfural at 230 °C, with 98.3% yield of DMF being obtained. The effects of reaction conditions such as temperature and pressure were investigated and discussed, and the catalyst could maintain good activity after being used at least 5 times. In order to further explore the reaction mechanism, dynamic experiments at different times were carried out and a possible reaction pathway was proposed. The development of efficient perovskite-supported Ni catalysts verified their great potential in biomass conversion.
Co-reporter:Rui Zhu, Bing Wang, Minshu Cui, Jin Deng, Xinglong Li, Yingbo Ma and Yao Fu
Green Chemistry 2016 vol. 18(Issue 7) pp:2029-2036
Publication Date(Web):06 Nov 2015
DOI:10.1039/C5GC02347E
A remarkably effective method of chemoselective dehydrogenation of alcohols in lignin has been developed with an iridium catalyst. An additional operation of Zn/NH4Cl via a two-step one pot process could further promote the cleavage of the C–O bond in β-O-4 units in lignin. And this reaction system was also applicable to native lignin as the molecular weight of native lignin decreased obviously as detected by gel permeation chromatography (GPC). Additionally, this is the first to date generation of the by-product H2 from native lignin and the by-product was straightforwardly captured by 1-decene. A probable mechanistic pathway was also proposed with the help of density functional theory (DFT) calculations.
Co-reporter:Qing Xu, Xinglong Li, Tao Pan, Chuguo Yu, Jin Deng, Qingxiang Guo and Yao Fu
Green Chemistry 2016 vol. 18(Issue 5) pp:1287-1294
Publication Date(Web):02 Oct 2015
DOI:10.1039/C5GC01454A
Levulinic acid (LA) is one of the most significant cellulose-derived compounds. γ-Valerolactone (GVL) and 1,4-pentanediol (1,4-PDO) are considered to be important chemical intermediates. Direct conversion of LA to GVL and GVL to 1,4-PDO was achieved via chemoselective hydrogenation by supported copper catalysts. We studied the transformation of LA to GVL in water and alcohol, and the pathway of the reaction was also studied. LA was converted to GVL catalyzed by the Cu (30%)-WO3 (10%)/ZrO2-CP-300 catalyst at 413 K in ethanol with 81% yield, while 84% GVL was obtained with the Cu (30%)/ZrO2-OG-300 catalyst in water at 393 K. Furthermore, 1,4-PDO was produced from GVL in excellent selectivities (>90%) using the Cu-TiO2/ZrO2-CP-600 catalyst.
Co-reporter:Wei Su, Tian-Jun Gong, Qi Zhang, Qing Zhang, Bin Xiao, and Yao Fu
ACS Catalysis 2016 Volume 6(Issue 10) pp:6417
Publication Date(Web):August 30, 2016
DOI:10.1021/acscatal.6b02039
A ligand-controlled regiodivergent and stereospecific copper-catalyzed alkylboration of unactivated terminal alkynes has been developed. Anti-Markovnikov alkylboration products were obtained with dppbz as ligand; conversely, Markovnikov alkylboration products were obtained with DMAP as ligand. This reaction not only presents a direct route to construct a vinyl C(sp2)–C(sp3) bond but also provides an efficient way to synthesize both internal and external vinylboronic esters regiodivergently from a single unactivated terminal alkyne.Keywords: alkynes; carboboration; copper; regiodivergent; vinylboronic esters
Co-reporter:Zhen-Qi Zhang, Ben Zhang, Xi Lu, Jing-Hui Liu, Xiao-Yu Lu, Bin Xiao, and Yao Fu
Organic Letters 2016 Volume 18(Issue 5) pp:952-955
Publication Date(Web):February 12, 2016
DOI:10.1021/acs.orglett.5b03692
A Cu/(NHC)-catalyzed SN2′-selective substitution reaction of allylic electrophiles with gem-diborylalkanes is reported. Different substituted gem-diborylalkanes and allylic electrophiles can be employed in this reaction, and various synthetic valuable functional groups can be tolerated. The asymmetric version of this reaction was initially researched with chiral N-heterocyclic carbene (NHC) ligands.
Co-reporter:Zhi-Wei Yang, Qi Zhang, Yuan-Ye Jiang, Lei Li, Bin Xiao and Yao Fu
Chemical Communications 2016 vol. 52(Issue 40) pp:6709-6711
Publication Date(Web):27 Apr 2016
DOI:10.1039/C6CC01732K
The transition-metal-catalyzed direct triflation of naphthyl amides and naphthyl ketones has been accomplished for the first time. Benzophenone (BP) was found to be a suitable ligand for the cross-coupling reactions. Density functional theory (DFT) calculations revealed that excessive amounts of HOTf inhibit the reductive elimination of the C–F bond to realize the unusual reductive elimination of the C–OTf bond.
Co-reporter:Xiao-Yu Lu, Jing-Hui Liu, Xi Lu, Zheng-Qi Zhang, Tian-Jun Gong, Bin Xiao and Yao Fu
Chemical Communications 2016 vol. 52(Issue 30) pp:5324-5327
Publication Date(Web):22 Mar 2016
DOI:10.1039/C6CC00176A
A Ni-catalyzed Markovnikov hydroalkylation of alkynes with alkyl halides is described. The reaction proceeds smoothly without the use of sensitive organometallic reagents and shows good functional-group compatibility, enabling the efficient synthesis of a variety of 1,1-disubstituted olefins. It also provides a straightforward approach for the modification of complex organic molecules.
Co-reporter:Ahmed Ebrahim-Alkhalil, Zhen-Qi Zhang, Tian-Jun Gong, Wei Su, Xiao-Yu Lu, Bin Xiao and Yao Fu
Chemical Communications 2016 vol. 52(Issue 27) pp:4891-4893
Publication Date(Web):14 Mar 2016
DOI:10.1039/C5CC09817C
Herein, we describe a novel copper-catalyzed epoxide opening reaction with gem-diborylmethane. Aliphatic, aromatic epoxides as well as aziridines are converted to the corresponding γ-pinacolboronate alcohols or amines in moderate to excellent yields. This new reaction provides beneficial applications for classic epoxide substrates as well as interesting gem-diborylalkane reagents.
Co-reporter:Jing-Hui Liu, Mian Cui, Xiao-Yu Lu, Zhen-Qi Zhang, Bin Xiao and Yao Fu
Chemical Communications 2016 vol. 52(Issue 6) pp:1242-1245
Publication Date(Web):27 Nov 2015
DOI:10.1039/C5CC08393A
A copper-mediated directed demethylation of propionamides has been developed. This reaction proceeds predominantly at the α-methyl groups of aliphatic amides with high efficiency and provides a unique tool for the direct cleavage of unactivated C(sp3)–C(sp3) bonds. The directing groups can be smoothly removed to afford the corresponding alkyl carboxylic acids.
Co-reporter:Lujiang Xu, Qian Yao, Zheng Han, Ying Zhang, and Yao Fu
ACS Sustainable Chemistry & Engineering 2016 Volume 4(Issue 3) pp:1115
Publication Date(Web):December 24, 2015
DOI:10.1021/acssuschemeng.5b01178
In this study, polylactic acid served as raw material to produce fine chemicals (pyridines) via a thermocatalytic conversion and ammonization (TCC-A) process. Ammonia was employed as not only carrier gas but also a reactant in this process. The thermal decomposition behavior of PLA under N2 or NH3 atmosphere was investigated. Different catalysts, including MCM-41, β-zeolite, ZSM-5 (Si/Al = 50) and HZSM-5 with different Si/Al ratios (Si/Al = 25, 50, 80) were also screened. Reaction temperature and residence time, which may affect the pyridines production, were investigated systematically. It was verified that all the investigated factors, including catalyst structure, catalyst acid amounts, reaction temperature, and residence time, influenced the PLA conversion and the pyridines production. The highest pyridines yield, 24.8%, was achieved by using HZSM-5 (Si/Al = 25) at around 500 °C. The catalyst regeneration tests were carried out. It demonstrated that the catalyst was stable after five regenerations and the catalytic activity did not change significantly. A possible reaction pathway from PLA to pyridines was also proposed. PLA initially thermally decomposed to form lactic acid and some byproducts such as acetaldehyde, acetone, etc., and then lactic acid, the mixture of acetaldehyde and acetone, or other byproducts reacted with ammonia to form imines and finally underwent complicated reactions to form pyridines.Keywords: Ammonia; Polylactic acid; Pyridines; Thermocatalytic conversion and ammonization; Zeolites
Co-reporter:Min-Shu Cui, Jin Deng, Xing-Long Li, and Yao Fu
ACS Sustainable Chemistry & Engineering 2016 Volume 4(Issue 3) pp:1707
Publication Date(Web):February 1, 2016
DOI:10.1021/acssuschemeng.5b01657
Replacing petroleum feedstocks by sustainable resources requires new and efficient methods to convert simple biomass-derived compounds to functionally useful and versatile chemicals. We discovered a new path for potential industrial-scale conversion of glycerol, a voluminously available byproduct from worldwide biodiesel production, to 5- and 4-hydroxymethylfurfurals through a cost-effective process. Staring from glycerol derivatives, glyceraldehyde (GLYD) or dihydroxyacetone (DHA), 5-HMF or 4-HMF could be obtained through base-catalyzed condensation and acid-catalyzed dehydration steps in the batch process. Effects of reaction temperature, base, and acid effectively demonstrate the product distribution. To promote this process into commercial-scale application, we also performed the two-step conversion in continuous process. Under optimal conditions, 4-HMF could be obtained in 80% isolated yield. To interpret the potential utilization of 4-HMF, we also explored the application of 4-HMF and synthesized several functional molecules for both pharmaceutical and material sciences.Keywords: Biomass; Dihydroxyacetone; Glyceraldehyde; Glycerol; Hydroxymethylfurfural; Sustainable chemistry;
Co-reporter:Qian Yao, Lujiang Xu, Ying Zhang, Yao Fu
Journal of Analytical and Applied Pyrolysis 2016 Volume 121() pp:258-266
Publication Date(Web):September 2016
DOI:10.1016/j.jaap.2016.08.005
•The effect of diluted NH3 on TCC-A of furfural was investigated.•The carbon yield of indoles increased by 203.96% compared with that in pure NH3.•The production of N-containing chemicals was enhanced by diluted NH3 from furans.•The side reaction pathway to form coke was inhibited by the dilution of NH3.•The catalyst in diluted NH3 was more stable in pure NH3.Thermo-catalytic conversion and ammonization (TCC-A) is a novel and promising thermochemical conversion process for the direct production of N-heterocycles under NH3 atmosphere, which is similar to the pyrolysis technology through introducing exogenous nitrogen. Since NH3 served as the carrier gas and reactant, it played an important part in the process. The introduction of N2 into NH3 significantly enhanced the indoles production and catalyst stability in the TCC-A process of bio-derived furans. Under the optimized TCC-A conditions of furfural, NH3 being diluted by 25% N2 at 600 °C with WHSV as 1.5 h−1 and gas flow rate at 40 ml/min, the carbon yields of N-containing chemicals and indoles reached 46.51% and 33.04%, respectively, which increased by 90.54% and 203.96% compared with those in pure NH3. Using furan derivatives as the feedstock, diluted NH3 also showed positive effect on the production of N-containing chemicals. Functional groups in the furan derivatives strongly affect the product distribution. It was found that the increase of indoles production from furfural was because the generation of 2-furonitrile via the side reaction pathway to form coke was inhibited by the dilution of NH3. The catalysts were tested via five reaction/regeneration cycles in pure and diluted NH3 atmosphere and characterized by N2 adsorption/desorption, XRD, XRF, NH3-TPD analyses and SEM. Compared in pure NH3, the catalysts in N2 diluted NH3 was more stable, which could be due to the lower degree of dealumination, structure damage, and acid site loss.The N2 diluted NH3 showed a positive effect on the production of indole from furfural through TCC-A process.
Co-reporter:Long-Can Cui, Zhen-Qi Zhang, Xi Lu, Bin Xiao and Yao Fu
RSC Advances 2016 vol. 6(Issue 57) pp:51932-51935
Publication Date(Web):27 May 2016
DOI:10.1039/C6RA09959A
The Pd-catalyzed synthesis of benzylboronic esters through coupling of aryl triflates with 1,1-diborylalkane under ambient conditions is described. Varieties of primary and secondary arylboronic esters could be successfully synthesized by this strategy. Competitive reaction demonstrated that aryl C–OTf bond was more inert than aryl bromide.
Co-reporter:Qi Zhang, Hai-Zhu Yu, and Yao Fu
Organometallics 2016 Volume 35(Issue 15) pp:2473-2479
Publication Date(Web):July 22, 2016
DOI:10.1021/acs.organomet.6b00347
Density functional theory (DFT) calculations have been performed to study the mechanism of Ir(III) pincer complex (POCOP)Ir(H)(acetone)+ (POCOP = 2,6-bis(dibutylphosphinito)phenyl) catalyzed chemoselective C1–O hydrosilylative reduction of glucose. The mechanisms for reduction of the external and internal C1–O (i.e., C1–Oext and C1–Oint) on the C1-MeO-substituted glucose (i.e., 1Me) and C1–Me2EtSiO-substituted glucose (i.e., 1Si) have been investigated. The calculation results show that both mechanisms proceed with the first concerted silyl transfer and the subsequent C1–Oext or C1–Oint bond cleavage and hydride transfer steps. In the hydride transfer step, the Ir-H moiety acts as the hydride source. The C1–O cleavage is the rate-determining step of the overall mechanism. The C1–Oext reduction is more favorable than C1–Oint reduction for the substrate 1Me, while the C1–Oint reduction is more favorable for 1Si. These results are consistent with the recent experimental outcomes. Analyzing the origin of chemoselectivity for the C1–Oext or C1–Oint cleavage, we found that the more stable precursor of C1–Oext cleavage and retention of the six-membered-ring structure result in the selective C1–Oext reduction of 1Me. Meanwhile, the higher basicity of the alkyl ether Oint atom (in comparison to the silyl ether Oext atom) and greater steric hindrance in the precursor favor the C1–Oint bond weakening. Therefore, the C1–Oint reduction occurs selectively for 1Si.
Co-reporter:Yuan-Ye Jiang;Zheng-Yang Xu;Hai-Zhu Yu
Science China Chemistry 2016 Volume 59( Issue 6) pp:724-729
Publication Date(Web):2016 June
DOI:10.1007/s11426-015-5525-4
Extracting hydrogen from methanol is a safe and cost-efficient strategy for fuel supply. This process was realized recently at a mild condition with excellent efficiency by ruthenium pincer catalysts. Despite the experimental success, the associated mechanism remains under debate. With the aid of density functional theory (DFT) calculations, an updated and self-consistent mechanism which involves MeOH-catalysed dehydrogenation of ruthenium hydride intermediate and pre-protonation of the pincer ligand was present herein. This mechanism is kinetically favoured over the previously-proposed water- or formicacid-participated ones and more consistent with the optimal experimental condition where strong base and neat methanol solvent are used.
Co-reporter:Qi Zhang, Ming-Chen Fu, Hai-Zhu Yu, and Yao Fu
The Journal of Organic Chemistry 2016 Volume 81(Issue 15) pp:6235-6243
Publication Date(Web):July 21, 2016
DOI:10.1021/acs.joc.6b00778
Mechanistic study has been carried out on the B(C6F5)3-catalyzed amine alkylation with carboxylic acid. The reaction includes acid-amine condensation and amide reduction steps. In condensation step, the catalyst-free mechanism is found to be more favorable than the B(C6F5)3-catalyzed mechanism, because the automatic formation of the stable B(C6F5)3-amine complex deactivates the catalyst in the latter case. Meanwhile, the catalyst-free condensation is constituted by nucleophilic attack and the indirect H2O-elimination (with acid acting as proton shuttle) steps. After that, the amide reduction undergoes a Lewis acid (B(C6F5)3)-catalyzed mechanism rather than a Brønsted acid (B(C6F5)3-coordinated HCOOH)-catalyzed one. The B(C6F5)3)-catalyzed reduction includes twice silyl-hydride transfer steps, while the first silyl transfer is the rate-determining step of the overall alkylation catalytic cycle. The above condensation–reduction mechanism is supported by control experiments (on both temperature and substrates). Meanwhile, the predicted chemoselectivity is consistent with the predominant formation of the alkylation product (over disilyl acetal product).
Co-reporter:Dr. Xi Lu;Dr. Bin Xiao;Dr. Lei Liu;Dr. Yao Fu
Chemistry - A European Journal 2016 Volume 22( Issue 32) pp:11161-11164
Publication Date(Web):
DOI:10.1002/chem.201602486
Abstract
Olefins and carboxylic acids are among the most important feedstock compounds. They are commonly found in natural products and drug molecules. We report a new reaction of nickel-catalyzed decarboxylative olefin hydroalkylation, which provides a novel practical strategy for the construction of C(sp3)−C(sp3) bonds. This reaction can tolerate a variety of synthetically relevant functional groups and shows good chemo- and regioselectivity. It enables cross-coupling of complex organic molecules containing olefin groups and carboxylic acid groups in a convergent fashion.
Co-reporter:Yuan-Ye Jiang, Ju-Long Jiang, and Yao Fu
Organometallics 2016 Volume 35(Issue 19) pp:3388-3396
Publication Date(Web):September 20, 2016
DOI:10.1021/acs.organomet.6b00602
Vanadium-catalyzed deoxydehydration (DODH) reactions provide a cost-effective approach for the conversion of vicinal diols to olefin and polycyclic aromatic hydrocarbons. In this paper, density functional theory (DFT) calculations employing M06 and M06-L methods were conducted to clarify the mechanism of V-catalyzed DODH. Three types of mechanisms generally proposed for transition-metal-catalyzed DODH, associated with the previously omitted spin crossover processes, were considered herein. As a result, a different catalytic cycle including a new olefin-formation mechanism was located, which is in contrast to the findings of a recent study. We found that the favorable mechanism involves the condensation of diols to form vanadium(V) diolate, reduction of the vanadium(V) diolate by PPh3, and spin-crossover steps to form a triplet vanadium(III) diolate. Thereafter, single C–O bond cleavage occurs followed by another spin crossover to form a singlet alkylvanadium(V) intermediate. The final concerted V–O/C–O bond cleavage generates an olefin and finishes the catalytic cycle. The reduction of vanadium(V) diolate by PPh3 and the extrusion of olefin have close overall free energy barriers of 34.3 and 33.7 kcal/mol, respectively. These results suggest that both steps influence the reaction rate. On the other hand, the two mechanisms starting by the reduction of the oxovanadium(V) catalyst with either PPh3 or a secondary alcohol were excluded due to their higher energy demands in the reduction and the olefin-formation stages. The good consistency between the experimental observations and the calculation results verified the proposed mechanism and also enabled us to clarify the reason for the efficiency of different reductants.
Co-reporter:Yuan-Ye Jiang, Qi Zhang, Hai-Zhu Yu, and Yao Fu
ACS Catalysis 2015 Volume 5(Issue 3) pp:1414
Publication Date(Web):January 16, 2015
DOI:10.1021/cs5018776
Traditional Wacker-type oxidations of unbiased alkenes produce ketones as major products. Recently, Grubbs’ group reported a Wacker-type oxidation system in which aldehydes (rather than ketones) have been generated predominantly in the presence of a nitrite co-catalyst. To elucidate the mechanistic origin of the aldehyde selectivity, density functional theory (DFT) studies have been conducted in this study. Two oxymetalation pathways, i.e., syn addition and anti addition pathways, were considered for various possible active species including monomeric Pd, bimetallic Pd–Pd, heterobimetallic Pd–Cu, and heterobimetallic Pd–Ag complexes. It is found that syn addition is kinetically more favored than anti addition in general. Meanwhile, the most feasible oxymetalation processes occur on the heterobimetallic Pd–Cu complexes. Investigations on the subsequent aldehyde formation process show that 1,2-H shift mechanism on tBuOH-ligated Pd–Cu complexes is superior to the betaH-elimination mechanism. Besides, the 1,2-H shift is the rate- and regioselectivity-determining step of the whole catalytic cycle. The analysis on spin density population indicates that the tBuOH-ligated Pd–Cu complex promotes a radical 1,2-H shift on the oxygenated alkene. The longer Pd–C(alkene) distance facilitates the aldehyde-selective pathway (relative to the ketone-selective pathway) due to the stronger stability of the secondary carbon radical and the smaller distortion energy therein.Keywords: aldehyde; copper; density functional theory; hydrogen transfer; palladium; radical; Wacker oxidation
Co-reporter:Qianqian Lu, Bing Wang, Haizhu Yu, and Yao Fu
ACS Catalysis 2015 Volume 5(Issue 8) pp:4881
Publication Date(Web):July 6, 2015
DOI:10.1021/acscatal.5b00891
Recently, Dong’s group [Angew. Chem., Int. Ed. 2012, 51, 7567–7571; Angew. Chem., Int. Ed. 2014, 53, 1891–1895] reported the ligand-controlled selectivity of Rh-catalyzed intramolecular coupling reaction of alkene-benzocyclobutenone: the direct coupling product (i.e., fused-rings) was formed in the DPPB-assisted system (DPPB = PPh2(CH2)4PPh2), while the decarbonylative coupling product (i.e., spirocycles) was generated in the P(C6F5)3-assited system. To explain this interesting selectivity, density functional theory (DFT) calculations have been carried out in the present study. It was found that the direct and decarbonylative couplings experience the same C(acyl)–C(sp2) activation and alkene insertion steps. The following C–C reductive elimination or β-H elimination–decarbonylation–reductive elimination leads to the direct or decarbonylative coupling reaction, respectively. The coordination features of different ligands were found to significantly influence C–C reductive elimination and decarbonylation step. The requisite phosphine dissociation of DPPB ligand from Rh center for the decarbonylation step is disfavored, and thus, the reductive elimination and direct coupling reaction are favored therein. By contrast, a free coordination site is available on the Rh center in the P(C6F5)3-assisted system, facilitating the decarbonylation process together with the generation of related decarbonylative coupling product.Keywords: benzocyclobutenone; C−C activation; DFT; ligand-controlled selectivity; Rh(I) catalysis
Co-reporter:Lujiang Xu, Yuanye Jiang, Qian Yao, Zheng Han, Ying Zhang, Yao Fu, Qingxiang Guo and George W. Huber
Green Chemistry 2015 vol. 17(Issue 2) pp:1281-1290
Publication Date(Web):24 Nov 2014
DOI:10.1039/C4GC02250E
In this study we demonstrate that indoles can be directly produced by thermo-catalytic conversion of bio-derived furans with ammonia over zeolite catalysts. MCM-41, β-zeolite, ZSM-5 (Si/Al = 50) and HZSM-5 catalysts with different Si/Al ratios (Si/Al = 25, 50, 63, 80) were screened and HZSM-5 with an Si/Al ratio of 25 showed the best reactivity for indole production due to the desired pore structure and acidity. Temperature displayed a significant effect on the product distribution. The maximum yield of indoles was obtained at moderate temperatures around 500 °C. The weight hourly space velocity (WHSV) of furan to catalyst investigation indicated that a lower WHSV could cause the overreaction of furan over the catalyst to produce more aniline and pyridines, while a higher WHSV would cause the incomplete reaction of furan. Because ammonia served as both a reactant and a carrier gas, to supply sufficient reactants and keep the desired reaction time, an appropriate ammonia to furan molar ratio was important for furan conversion to indoles. Under optimized conditions, the highest total carbon yield of indoles and their selectivity in the N-containing chemicals were 32% and 75%, respectively. 2-Methylfuran and the mixture of furan and 2-methylfuran were also studied, which demonstrated that more alkyl indoles could be selectively obtained via the coupling reaction of different bio-derived furans. Ring opening of the furan is a more favorable mechanism compared to the Diels–Alder mechanism, and the pyrrole reacting with furan is the more favorable pathway compared to pyrrole reacting with pyrrole based on our experimental and theoretical calculations.
Co-reporter:Lujiang Xu, Zheng Han, Qian Yao, Jin Deng, Ying Zhang, Yao Fu and Qingxiang Guo
Green Chemistry 2015 vol. 17(Issue 4) pp:2426-2435
Publication Date(Web):29 Jan 2015
DOI:10.1039/C4GC02235A
In this study, renewable pyridines could be directly produced from glycerol and ammonia via a thermo-catalytic conversion process with zeolites. The major factors, including catalyst, temperature, weight hourly space velocity (WHSV) of glycerol to catalyst, and the molar ratio of ammonia to glycerol, which may affect the pyridine production, were investigated systematically. The optimal conditions for producing pyridines from glycerol were achieved with HZSM-5 (Si/Al = 25) at 550 °C with a WHSV of glycerol to catalyst of 1 h−1 and an ammonia to glycerol molar ratio of 12:1. The carbon yield of pyridines was up to 35.6%. The addition of water to the feed decreased the pyridine yield, because water competed with glycerol on the acid sites of the catalyst and therefore impacted the acidity of the catalyst. After five reaction/regeneration cycles, a slight deactivation of the catalyst was observed. The catalysts were investigated by N2 adsorption/desorption, XRD, XRF and NH3-TPD and the results indicated that the deactivation could be due to the structure changes and the acid site loss of the catalyst. The reaction pathway from glycerol to pyridines was studied and the main pathway should be that glycerol was initially dehydrated to form acrolein and some by-products such as acetaldehyde, acetol, acetone, etc., and then acrolein, a mixture of acrolein and acetaldehyde, or other by-products reacted with ammonia to form imines and finally pyridines.
Co-reporter:Yao-Bing Huang, Long Yan, Meng-Yuan Chen, Qing-Xiang Guo and Yao Fu
Green Chemistry 2015 vol. 17(Issue 5) pp:3010-3017
Publication Date(Web):10 Mar 2015
DOI:10.1039/C5GC00326A
Direct hydrogenolysis of the aromatic Csp2–O bonds in both phenols and phenyl ethers to form arenes selectively is a core enabling technology that can expand greatly the scope of chemical manufacture from biomass. However, conventional hydrogenolysis of phenols typically led to aromatic ring saturation instead of the cleavage of the Csp2–O bonds. Herein, we report a recyclable Ru–WOx bifunctional catalyst that showed high catalytic activities for the hydrogenolysis of a wide range of phenols and phenyl ethers, including dimeric lignin model compounds and the primitive phenols separated from pyrolysis lignin, to form arenes selectively in water. Preliminary mechanistic studies supported that the reactions occurred via a direct cleavage of the Csp2–O bonds and the concerted effects of the hydrogenating Ru sites and the Lewis acidic W sites are the key to such an unusual reactivity.
Co-reporter:Xing-Yu Li, Rui Shang, Ming-Chen Fu and Yao Fu
Green Chemistry 2015 vol. 17(Issue 5) pp:2790-2793
Publication Date(Web):30 Mar 2015
DOI:10.1039/C5GC00556F
A metal-free hydrodeoxygenation process was developed for the production of hydrocarbons from biomass-derived fatty acids and derivatives. Biomass-derived fatty acids and derivatives were converted to alkanes and alkenes under mild reaction conditions. Furthermore, this catalytic system can also be applied to convert real biomass with satisfactory results.
Co-reporter:Meng-Yuan Chen, Yao-Bing Huang, Huan Pang, Xin-Xin Liu and Yao Fu
Green Chemistry 2015 vol. 17(Issue 3) pp:1710-1717
Publication Date(Web):24 Dec 2014
DOI:10.1039/C4GC01992J
Phenolic compounds derived from lignin are important feedstocks for the sustainable production of alkane fuels with C6–C9 carbons. Hydrodeoxygenation (HDO) is the main chemical process to remove oxygen-containing functionalities. Here, we have reported the HDO of phenols in a biphasic H2O/n-dodecane system. A series of supported Ru catalysts were prepared, characterized and explored for the reaction among which Ru/CNT showed the highest catalytic activity towards the production of alkanes. The model reaction with eugenol achieved a high conversion (>99%) and a high alkane selectivity (98%), which was much higher than the results from the monophasic system (56.5% yield of alkanes in H2O). The reaction conditions including reaction temperature, hydrogen pressure and the ratio of H2O/n-C12H26 were optimized. The kinetic experiments revealed that eugenol was first hydrogenated to 4-propyl-guaiacol, and then deoxygenated into 4-propyl-cyclohexanol which was the main detected intermediate of the reaction. After that, 4-propyl-cyclohexanol was dehydrated and hydrogenated into propylcyclohexane. Moreover, various phenols and dimeric lignin model compounds were also successfully converted into alkanes in the biphasic systems. The construction of the biphasic solvent-Ru/CNT catalyst system highlights an efficient route for the conversion of lignin-derived phenolic compounds to biofuels.
Co-reporter:Xing-Long Li, Jin Deng, Jing Shi, Tao Pan, Chu-Guo Yu, Hua-Jian Xu and Yao Fu
Green Chemistry 2015 vol. 17(Issue 2) pp:1038-1046
Publication Date(Web):23 Oct 2014
DOI:10.1039/C4GC01601G
Cu–Co catalysts, prepared by a co-precipitation method (CP) and an oxalate sol–gel method (OG), can selectively convert furfural (FFA) to cyclopentanone (CPO) or cyclopentanol (CPL), respectively. The conversion of FFA to CPO or CPL by Cu–Co catalysts were studied in aqueous solutions. We found that the product distribution was influenced by the catalyst support, Cu loading, calcination temperature, hydrogen pressure, the number of times the catalyst was reused and the preparation method of the catalyst. The surface morphology, surface area and composition of the catalysts were studied by XRD, XPS, BET, ICP-AES and TEM characterization. We found that there was a strong interaction between Cu and Co. Cu0, Cu2O and Co0 were the main active catalyst phases on the surfaces of the catalysts, but the amounts were different in the different catalysts. Cu0, Co0 and Cu2O were the active hydrogenation species, and Cu2O also played the role of an electrophile or Lewis acid to polarize the CO bond via lone pair electrons on the oxygen atom. According to XRD and XPS, the main phases on the surface of the CP catalysts were Cu0 and Cu2O. The hydrogenation activity of the CP catalyst was relatively weak and the main product was CPO. In contrast, the hydrogenation activity of the OG catalyst was high and the main product was the fully hydrogenated product CPL due to the main active phases of Co0 and Cu2O on the surface of the OG catalyst. At lower hydrogen pressure (2 MPa) and lower Cu loadings (2% for OG, 5% for CP), we obtained the highest yield of 67% CPO and 68% CPL, respectively.
Co-reporter:Guang-Zu Wang, Jian Jiang, Xiao-Song Bu, Jian-Jun Dai, Jun Xu, Yao Fu, and Hua-Jian Xu
Organic Letters 2015 Volume 17(Issue 15) pp:3682-3685
Publication Date(Web):July 16, 2015
DOI:10.1021/acs.orglett.5b01612
The cross-coupling reaction of allyl boron ester with 1°/2°/3°-halogenated alkanes in the presence of copper has been developed for the first time, which provides a mild and efficient method for the construction of saturated C(sp3)–C(sp3) bonds. This protocol shows excellent compatibility with the nonactivated primary, secondary, and even tertiary halogenated alkanes under mild conditions.
Co-reporter:Guang-Zu Wang, Rui Shang, Wan-Min Cheng, and Yao Fu
Organic Letters 2015 Volume 17(Issue 19) pp:4830-4833
Publication Date(Web):September 14, 2015
DOI:10.1021/acs.orglett.5b02392
Enabled by iridium photoredox catalysis, 2-oxo-2-(hetero)arylacetic acids were decarboxylatively added to various Michael acceptors including α,β-unsaturated ester, ketone, amide, aldehyde, nitrile, and sulfone at room temperature. The reaction presents a new type of acyl Michael addition using stable and easily accessible carboxylic acid to formally generate acyl anion through photoredox-catalyzed radical decarboxylation.
Co-reporter:Wei Su, Tian-Jun Gong, Bin Xiao and Yao Fu
Chemical Communications 2015 vol. 51(Issue 59) pp:11848-11851
Publication Date(Web):12 Jun 2015
DOI:10.1039/C4CC09790D
Rh(III)-catalyzed direct vinylic C–H cyanation reaction has been developed as a practical method for the synthesis of alkenyl nitriles. N-Cyano-N-phenyl-p-methylbenzenesulfonamide (NCTS), a user-friendly cyanation reagent, was used in the transformation. Both acrylamides and ketoximes can be employed in the new C–H cyanation process.
Co-reporter:Xiao-Yu Lu, Chu-Ting Yang, Jing-Hui Liu, Zheng-Qi Zhang, Xi Lu, Xin Lou, Bin Xiao and Yao Fu
Chemical Communications 2015 vol. 51(Issue 12) pp:2388-2391
Publication Date(Web):19 Dec 2014
DOI:10.1039/C4CC09321F
A copper-catalyzed cross-coupling reaction of epoxides with arylboronates is described. This reaction is not limited to aromatic epoxides, because aliphatic epoxides are also suitable substrates. In addition, N-sulfonyl aziridines can be successfully converted into the products. This reaction provides convenient access to β-phenethyl alcohols, which are valuable synthetic intermediates.
Co-reporter:Lujiang Xu, Qian Yao, Jin Deng, Zheng Han, Ying Zhang, Yao Fu, George W. Huber, and Qingxiang Guo
ACS Sustainable Chemistry & Engineering 2015 Volume 3(Issue 11) pp:2890
Publication Date(Web):October 7, 2015
DOI:10.1021/acssuschemeng.5b00841
Chemical conversion of biomass to value-added products provides a sustainable alternative to the current chemical industry that is predominantly dependent on fossil fuels. N-Heterocycles, including pyrroles, pyridines, and indoles, etc., are the most abundant and important classes of heterocycles in nature and widely applied as pharmaceuticals, agrochemicals, dyes, and other functional materials. However, all starting materials for the synthesis of N-heterocycles currently are derived from crude oil through complex multistep-processes and sometimes result in environmental problems. In this study, we show that N-heterocycles can be directly produced from biomass (including cellulose, lignocelluloses, sugars, starch, and chitosan) over commercial zeolites via a thermocatalytic conversion and ammonization process (TCC-A). All desired reactions occur in one single-step reactor within seconds. The production of pyrroles, pyridines, or indoles can be simply tuned by changing the reaction conditions. Meanwhile, N-containing biochar can be obtained as a valuable coproduct. We also outline the chemistry for the conversion of biomass into heterocycle molecules by the addition of ammonia into pyrolysis reactors demonstrating how industrial chemicals could be produced from renewable biomass resources. Only minimal biomass pretreatment is required for the TCC-A approach.Keywords: Biomass; N-Heterocycles; Reaction pathway; Thermocatalytic conversion and ammonization; Zeolites;
Co-reporter:Chi Fang, Jian-Jun Dai, Hua-Jian Xu, Qing-Xiang Guo, Yao Fu
Chinese Chemical Letters 2015 Volume 26(Issue 10) pp:1265-1268
Publication Date(Web):October 2015
DOI:10.1016/j.cclet.2015.07.001
An iron(III)-catalyzed selective oxidation of 5-HMF to 2,5-DFF in air at room temperature was developed. This approach gives 2,5-DFF with good selectivity and yields. Additionally, a two-step process was developed for the oxidation of 2,5-DFF to 2,5-FDCA at remarkably high substrate concentrations. This work demonstrates unequivocally the great potential of iron as a cheap and earth-abundant catalyst for the development of new protocols for the conversion of biomass to value-added chemicals.An iron(III)-catalyzed selective oxidation of 5-HMF to 2,5-DFF in air at room temperature was developed. This work demonstrates unequivocally the great potential of iron as a cheap and earth-abundant catalyst for the development of new protocols for the conversion of biomass to value-added chemicals.
Co-reporter:Nan Dai;Rui Shang;Mingchen Fu
Chinese Journal of Chemistry 2015 Volume 33( Issue 4) pp:405-408
Publication Date(Web):
DOI:10.1002/cjoc.201500035
Abstract
Conversion of biomass-derived ethyl levulinate to γ-valerolactone is realized by using homogeneous iron-catalyzed transfer hydrogenation (CTH). By utilizing Casey's catalyst and cheap isopropanol as hydrogen source, γ-valerolactone can be generated in 95% yield. Addition of catalytic amount of base is important to achieve good yield.
Co-reporter:Nan Dai;Rui Shang;Mingchen Fu
Chinese Journal of Chemistry 2015 Volume 33( Issue 4) pp:
Publication Date(Web):
DOI:10.1002/cjoc.201590009
Co-reporter:Yuanye Jiang;Haizhu Yu;Lei Liu
Science China Chemistry 2015 Volume 58( Issue 4) pp:673-683
Publication Date(Web):2015 April
DOI:10.1007/s11426-014-5178-8
Trifluoromethylation reactions are important transformations in the research and development of drugs, agrochemicals and functional materials. An oxidation/reduction process of trifluoromethyl-containing compounds is thought to be involved in many recently tested catalytic trifluoromethylation reactions. To provide helpful physical chemical data for mechanistic studies on trifluoromethylation reactions, the redox potentials of a variety of trifluoromethyl-containing compounds and trifluoromethylated radicals were studied by quantum-chemical methods. First, wB97X-D was found to be a reliable method in predicting the ionization potentials, electron affinities, bond dissociation enthalpies and redox potentials of trifluoromethyl-containing compounds. One-electron absolute redox potentials of 79 trifluoromethyl substrates and 107 trifluoromethylated radicals in acetonitrile were then calculated with this method. The theoretical results were found to be helpful for interpreting experimental observations such as the relative reaction efficiency of different trifluoromethylation reagents. Finally, the bond dissociation free energies (BDFE) of various compounds were found to have a good linear relationship with the related bond dissociation enthalpies (BDE). Based on this observation, a convenient method was proposed to predict one-electron redox potentials of neutral molecules.
Co-reporter:Jun Xu;Ebrahim-Alkhalil Ahmed;Bin Xiao;Qian-Qian Lu;Yun-Long Wang;Chu-Guo Yu; Yao Fu
Angewandte Chemie 2015 Volume 127( Issue 28) pp:8349-8353
Publication Date(Web):
DOI:10.1002/ange.201502308
Abstract
An unprecedented Pd-catalyzed regioselective activation of gem-difluorinated cyclopropanes induced by CC bond cleavage is reported. It provides a general and efficient access to a variety of 2-fluoroallylic amines, ethers, esters, and alkylation products in high Z-selectivity, which are important skeletons in many biologically active molecules. In addition, the transformation represents the first general application of gem-difluorinated cyclopropanes as reaction partners in transition-metal-catalyzed cross-coupling reaction.
Co-reporter:Jun Xu;Ebrahim-Alkhalil Ahmed;Bin Xiao;Qian-Qian Lu;Yun-Long Wang;Chu-Guo Yu; Yao Fu
Angewandte Chemie International Edition 2015 Volume 54( Issue 28) pp:8231-8235
Publication Date(Web):
DOI:10.1002/anie.201502308
Abstract
An unprecedented Pd-catalyzed regioselective activation of gem-difluorinated cyclopropanes induced by CC bond cleavage is reported. It provides a general and efficient access to a variety of 2-fluoroallylic amines, ethers, esters, and alkylation products in high Z-selectivity, which are important skeletons in many biologically active molecules. In addition, the transformation represents the first general application of gem-difluorinated cyclopropanes as reaction partners in transition-metal-catalyzed cross-coupling reaction.
Co-reporter:Mazloom Shah;Jian-Jun Dai;Qing-Xiang Guo
Science China Chemistry 2015 Volume 58( Issue 7) pp:1110-1121
Publication Date(Web):2015 July
DOI:10.1007/s11426-015-5397-7
With the depletion of fossil resources, there is a need to find alternative resources of fuels and chemicals. The use of renewable feedstock such as those from seed oil processing is one of the best available resources that have come to the fore-front recently. This paper critically analyzes and highlights major factors in the biodiesel industry, such as seeds oil composition, production methods, properties of biodiesel, problems and potential solutions of using vegetable seed oil, the composition, quality and effective utilization of crude glycerol, the catalytic conversion of glycerol into possible fuels and chemicals.
Co-reporter:Qi Zhang;Haizhu Yu
Science China Chemistry 2015 Volume 58( Issue 8) pp:1316-1322
Publication Date(Web):2015 August
DOI:10.1007/s11426-015-5360-7
A theoretical study is carried out on Gaunt’s palladium-catalyzed selective C(sp3)-H activation of unprotected aliphatic amines. In this reaction, the methyl group is proposed to be activated through a four-membered cyclometallation pathway even though an ethyl group is present in the substrate. Our calculation shows that the methyl and ethyl activation processes proceed in nitrogen-atom-directed pathway rather than carbonyl-directed one. More important, methyl activation is more favorable than ethyl activation with nitrogen atom as the directing group. Further studies on the structural parameters show that the lactone structure in cyclic substrate is the origin of the selective methyl activation. When the lactone moiety is changed to ketone, ether or alkyl, the selectivity could be reversed so that the ethyl activation becomes more favorable. This result validates the proposal that lactone structure is key to selective methyl activation.
Co-reporter:Qianqian Lu ; Haizhu Yu
Journal of the American Chemical Society 2014 Volume 136(Issue 23) pp:8252-8260
Publication Date(Web):May 13, 2014
DOI:10.1021/ja4127455
Itami et al. recently reported the C–O electrophile-controlled chemoselectivity of Ni-catalyzed coupling reactions between azoles and esters: the decarbonylative C–H coupling product was generated with the aryl ester substrates, while C–H/C–O coupling product was generated with the phenol derivative substrates (such as phenyl pivalate). With the aid of DFT calculations (M06L/6-311+G(2d,p)-SDD//B3LYP/6-31G(d)-LANL2DZ), the present study systematically investigated the mechanism of the aforementioned chemoselective reactions. The decarbonylative C–H coupling mechanism involves oxidative addition of C(acyl)–O bond, base-promoted C–H activation of azole, CO migration, and reductive elimination steps (C–H/Decar mechanism). This mechanism is partially different from Itami’s previous proposal (Decar/C–H mechanism) because the C–H activation step is unlikely to occur after the CO migration step. Meanwhile, C–H/C–O coupling reaction proceeds through oxidative addition of C(phenyl)–O bond, base-promoted C–H activation, and reductive elimination steps. It was found that the C–O electrophile significantly influences the overall energy demand of the decarbonylative C–H coupling mechanism, because the rate-determining step (i.e., CO migration) is sensitive to the steric effect of the acyl substituent. In contrast, in the C–H/C–O coupling mechanism, the release of the carboxylates occurs before the rate-determining step (i.e., base-promoted C–H activation), and thus the overall energy demand is almost independent of the acyl substituent. Accordingly, the decarbonylative C–H coupling product is favored for less-bulky group substituted C–O electrophiles (such as aryl ester), while C–H/C–O coupling product is predominant for bulky group substituted C–O electrophiles (such as phenyl pivalate).
Co-reporter:Tian-Jun Gong, Wei Su, Zhao-Jing Liu, Wan-Min Cheng, Bin Xiao, and Yao Fu
Organic Letters 2014 Volume 16(Issue 2) pp:330-333
Publication Date(Web):January 3, 2014
DOI:10.1021/ol403407q
RhIII-catalyzed C–H activation with allenes produces highly unsaturated conjugated olefins. The reaction is applicable to both olefin and arene C(sp2)–H and is compatible with a variety of functional groups. The products can be further transformed into other important skeletons through Diels–Alder reaction and intramolecular transesterification.
Co-reporter:Zhen-Qi Zhang, Chu-Ting Yang, Lu-Jun Liang, Bin Xiao, Xi Lu, Jing-Hui Liu, Yan-Yan Sun, Todd B. Marder, and Yao Fu
Organic Letters 2014 Volume 16(Issue 24) pp:6342-6345
Publication Date(Web):December 1, 2014
DOI:10.1021/ol503111h
The first copper-catalyzed/promoted sp3-C Suzuki–Miyaura coupling reaction of gem-diborylalkanes with nonactivated electrophilic reagents is reported. Not only 1, 1-diborylalkanes but also some other gem-diborylalkanes can be coupled with nonactivated primary alkyl halides, offering a new method for sp3C–sp3C bond formation and, simultaneously, providing a new strategy for the synthesis of alkylboronic esters.
Co-reporter:Yan-Yan Sun, Jun Yi, Xi Lu, Zhen-Qi Zhang, Bin Xiao and Yao Fu
Chemical Communications 2014 vol. 50(Issue 75) pp:11060-11062
Publication Date(Web):31 Jul 2014
DOI:10.1039/C4CC05376A
A copper-catalyzed Suzuki–Miyaura coupling of benzyl halides with arylboronates is described. Varieties of primary benzyl halides as well as more challenging secondary benzyl halides with β hydrogens or steric hindrance could be successfully converted into the corresponding products. Thus it provides access to diarylmethanes, diarylethanes and triarylmethanes.
Co-reporter:Jun Xu, Yun-Long Wang, Tian-Jun Gong, Bin Xiao and Yao Fu
Chemical Communications 2014 vol. 50(Issue 85) pp:12915-12918
Publication Date(Web):01 Sep 2014
DOI:10.1039/C4CC05692B
A new copper-catalyzed trifluoromethylarylation reaction of alkynes has been developed. The transformation represents the first example of endo-type carbotrifluoromethylation of unsaturated carbon–carbon bonds and provides efficient access to a variety of CF3-substituted dihydronaphthalenes and chromenes.
Co-reporter:Jin Deng, Qiu-Ge Zhang, Tao Pan, Qing Xu, Qing-Xiang Guo and Yao Fu
RSC Advances 2014 vol. 4(Issue 52) pp:27541-27544
Publication Date(Web):16 Jun 2014
DOI:10.1039/C4RA04567J
Glutamic acid was transformed into succinimide in a two step procedure involving a dehydration in water to pyroglutamic acid followed by an oxidative decarboxylation using a silver catalyst.
Co-reporter:Jiang Li, Dao-jun Ding, Lu-jiang Xu, Qing-xiang Guo and Yao Fu
RSC Advances 2014 vol. 4(Issue 29) pp:14985-14992
Publication Date(Web):11 Feb 2014
DOI:10.1039/C3RA47923D
A biphasic system consisting of THF and water was studied to achieve the integrated conversion of cellulose and hemicellulose in lignocellulosic biomass to levulinic acid. As compared to previous studies using GVL as solvent, the utilization of a lower boiling point solvent, THF, also achieves the simultaneous hydrolysis of C6 and C5 carbohydrates in lignocellulosic biomass, and the results of simultaneous hydrolysis are comparable. Furthermore, it offers an alternative operation procedure after the hydrolysis. A distillation process is not only used to achieve the effective separation of the solid residue from the desired products, but it also helps in the complete isolation of furfural and formic acid from levulinic acid. Consequently, the utilization of by-product formic acid in the hydrogenation of furfural to furfuryl alcohol is explored, and the process is achieved with both model substrates and the feed from the lignocellulosic biomass feedstock. The hydrolysis of furfuryl alcohol gave C5 carbohydrate-derived levulinic acid. We finally explored the integrated conversion with five biomass raw materials, and the total yield of levulinic acid was quite obviously promoted by the additional conversion of pentose.
Co-reporter:Tian-Jun Gong, Yuan-Ye Jiang, Yao Fu
Chinese Chemical Letters 2014 Volume 25(Issue 3) pp:397-400
Publication Date(Web):March 2014
DOI:10.1016/j.cclet.2014.01.006
Rhodium-catalyzed cross-coupling reactions of unactivated primary alkyl chlorides with diboron reagents have been developed as practical methods for the synthesis of alkylboronic esters. These reactions expand the concept and utility of Rh(I)-catalyzed cross-coupling of aliphatic electrophiles.Rhodium-catalyzed cross-coupling reactions of unactivated primary alkyl chlorides with diboron reagents have been developed as practical methods for the synthesis of alkylboronic esters. These reactions expands the concept and utility of Rh(I)-catalyzed cross-coupling of aliphatic electrophiles.
Co-reporter:Tian-Jun Gong, Wan-Min Cheng, Wei Su, Bin Xiao, Yao Fu
Tetrahedron Letters 2014 Volume 55(Issue 11) pp:1859-1862
Publication Date(Web):12 March 2014
DOI:10.1016/j.tetlet.2013.11.065
A practical Rh-catalyzed reaction was developed to achieve 2-alkyl-substituted indole synthesis. The reaction can tolerate a variety of synthetically important functional groups. The indole products can also be transformed into other important skeletons. Two bioactive compounds, that is indomethacin and pravadoline were prepared using the new method.A practical Rh-catalyzed reaction was developed to achieve 2-alkyl-substituted indole synthesis. The reaction can tolerate a variety of synthetically important functional groups (aryl-Br, heterocycle, amino acid). The indole products can also be transformed into other important skeletons. Two bioactive compounds, that is, indomethacin and pravadoline were prepared using the new method.
Co-reporter:Jun Xu, Xiaoyang Liu, Yao Fu
Tetrahedron Letters 2014 Volume 55(Issue 3) pp:585-594
Publication Date(Web):15 January 2014
DOI:10.1016/j.tetlet.2013.11.108
•We focus on the cases of transition-metal-mediated C(sp3)–CF3 bond formation.•Trifluoromethylation of sp3-hybridized C–X bonds.•Trifluoromethylation of alkyl organometallic reagents.•Trifluoromethylation of sp3-hybridized C–H Bonds.•Trifluoromethylation of alkene to form C(sp3)–CF3 bonds.In the past 5 years, transition-metal-mediated trifluoromethylation for the construction of various CF3-containing building blocks has been the focus of recent research in both industrial and academic communities. Progresses in the construction of C(sp2)–CF3 bonds and C(sp)–CF3 have been well reviewed. This Letter will focus on the cases of transition-metal-mediated C(sp3)–CF3 bond formation, which involves the trifluoromethylation of sp3-hybridized C–X bonds, alkyl organometallic reagents, sp3-hybridized C–H bonds, and alkene derivatives.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Jing-Hui Liu;Chu-Ting Yang;Xiao-Yu Lu;Zhen-Qi Zhang;Ling Xu;Mian Cui;Xi Lu;Bin Xiao;Dr. Yao Fu;Dr. Lei Liu
Chemistry - A European Journal 2014 Volume 20( Issue 47) pp:15334-15338
Publication Date(Web):
DOI:10.1002/chem.201405223
Abstract
A copper-catalyzed reductive cross-coupling reaction of nonactivated alkyl tosylates and mesylates with alkyl and aryl bromides was developed. It provides a practical method for efficient and cost-effective construction of aryl–alkyl and alkyl–alkyl CC bonds with stereocontrol from readily available substrates. When used in an intramolecular fashion, the reaction enables convenient access to various substituted carbo- or heterocycles, such as 2,3-dihydrobenzofuran and benzochromene derivatives.
Co-reporter:Yao-Bing Huang;Meng-Yuan Chen;Long Yan; Qing-Xiang Guo ; Yao Fu
ChemSusChem 2014 Volume 7( Issue 4) pp:1068-1072
Publication Date(Web):
DOI:10.1002/cssc.201301356
Abstract
The development of new catalytic systems for the conversion of biomass-derived molecules into liquid fuels has attracted much attention. We propose a non-noble bimetallic catalyst based on nickel–tungsten carbide for the conversion of the platform molecules 5-(hydroxymethyl)furfural into the liquid-fuel molecule 2,5-dimethylfuran (DMF). Different catalysts, metal ratios and reaction conditions have been tested and give rise to a 96% yield of DMF. The catalysts have been characterized and are discussed. The reaction mechanism is also explored through capture of reaction intermediates. The analysis of the reaction mixture over different catalysts is presented and helps to understand the role of nickel and tungsten carbide during the reaction.
Co-reporter:Yuan-Ye Jiang, Hai-Zhu Yu, and Yao Fu
Organometallics 2014 Volume 33(Issue 22) pp:6577-6584
Publication Date(Web):November 12, 2014
DOI:10.1021/om500921d
Recently, the Li group reported the first Ag-catalyzed hydrogenation of aldehydes in water, demonstrating the utility of Ag complexes in homogeneous catalytic transformations through hydrogen activation. In the present study, density functional theory methods have been used to study the mechanism of Ag-catalyzed hydrogen activation. Three possible pathways, including base-assisted hydrogen activation, ligand-assisted hydrogen activation, and oxidative addition were investigated. The ligand-assisted hydrogen activation is disfavored because the neutral biaryl phosphine ligand XPhos is not a competent proton acceptor and results in the destruction of the aromaticity of an aryl group. Oxidative addition of H2 on AgI complexes was also found to be unlikely. The resulting AgIII hydride complexes are highly unstable and can undergo spontaneous reduction due to the weakly electron-donating ligand and the relatively low electronegativity of hydrogen. By contrast, the base-assisted hydrogen activation mechanism is more favored. This mechanism mainly includes three steps: base-assisted heterolytic H–H bond cleavage, hydride transfer, and protonation. Hydride transfer is the rate-determining step of the whole catalytic cycle. In addition, the ligand XPhos was found to coordinate with the Ag center by both the phosphine and the isopropyl-substituted phenyl groups, and this coordination mode is able to facilitate hydrogen activation.
Co-reporter:Tian-Jun Gong ; Bin Xiao ; Wan-Min Cheng ; Wei Su ; Jun Xu ; Zhao-Jing Liu ; Lei Liu
Journal of the American Chemical Society 2013 Volume 135(Issue 29) pp:10630-10633
Publication Date(Web):July 3, 2013
DOI:10.1021/ja405742y
A Rh-catalyzed directed C–H cyanation reaction was developed for the first time as a practical method for the synthesis of aromatic nitriles. N-Cyano-N-phenyl-p-toluenesulfonamide, a user-friendly cyanation reagent, was used in the transformation. Many different directing groups can be used in this C–H cyanation process, and the reaction tolerates a variety of synthetically important functional groups.
Co-reporter:Jian-Jun Dai ; Chi Fang ; Bin Xiao ; Jun Yi ; Jun Xu ; Zhao-Jing Liu ; Xi Lu ; Lei Liu
Journal of the American Chemical Society 2013 Volume 135(Issue 23) pp:8436-8439
Publication Date(Web):May 29, 2013
DOI:10.1021/ja404217t
A copper-promoted trifluoromethylation reaction of aromatic amines is described. This transformation proceeds smoothly under mild conditions and exhibits good tolerance of many synthetically relevant functional groups. It provides an alternative approach for the synthesis of trifluoromethylated arenes and heteroarenes. It also constitutes a new example of the Sandmeyer reaction.
Co-reporter:Bin Xiao ; Zhao-Jing Liu ; Lei Liu
Journal of the American Chemical Society 2013 Volume 135(Issue 2) pp:616-619
Publication Date(Web):January 2, 2013
DOI:10.1021/ja3113752
An unexpected C–H activation/C–C cross-coupling reaction has been found to occur between pyridine N-oxides and general nonactivated secondary and even tertiary alkyl bromides. It provides a practically useful approach for the synthesis of alkylated pyridine derivatives. Experimental observations indicated that the C–Br cleavage step involves a radical-type process. Thus, the title reaction provides a rather extraordinary example of Pd-catalyzed cross-coupling of secondary and tertiary aliphatic electrophiles.
Co-reporter:Tao Pan, Jin Deng, Qing Xu, Yang Xu, Qing-Xiang Guo and Yao Fu
Green Chemistry 2013 vol. 15(Issue 10) pp:2967-2974
Publication Date(Web):13 Aug 2013
DOI:10.1039/C3GC40927A
The development of the catalytic conversion of biomass-based platform molecules into oxygenated fuel molecules is of great significance in order to reduce the dependence on fossil resources and to solve environmental problems. Alkyl valerate esters were proven to have the potential to be renewable additives of gasoline and diesel. In this work, we studied the hydrogenation of levulinic acid (LA) to valerate esters over supported Ru catalysts, and found that the acidity was an important factor for the catalyst performance. A bifunctional catalyst Ru/SBA-SO3H was developed as an active catalyst, and a highest yield of 94% to ethyl valerate (EV) was achieved. The catalyst was characterized by nitrogen adsorption/desorption methods, X-ray power diffraction (XRD), transmission electron spectroscopy (TEM) and X-ray photoelectron spectroscopy (XPS). The effects of reaction conditions were comprehensively investigated and probable reaction pathways were proposed and verified. The conversion of LA to various alkyl valerate esters can also be catalyzed by the bifunctional catalyst. In addition, supported Cu and Ni catalysts were also screened under similar reaction conditions as Ru-based catalysts, and the combination of Ni/SBA-15 and SBA-SO3H exhibited activity for the conversion of LA to EV.
Co-reporter:Yao-Bing Huang and Yao Fu
Green Chemistry 2013 vol. 15(Issue 5) pp:1095-1111
Publication Date(Web):28 Feb 2013
DOI:10.1039/C3GC40136G
As the main component of lignocelluloses, cellulose is a biopolymer consisting of many glucose units connected through β-1,4-glycosidic bonds. Breakage of the β-1,4-glycosidic bonds by acids leads to the hydrolysis of cellulose polymers, resulting in the sugar molecule glucose or oligosaccharides. Mineral acids, such as HCl and H2SO4, have been used in the hydrolysis of cellulose. However, they suffer from problems of product separation, reactor corrosion, poor catalyst recyclability and the need for treatment of waste effluent. The use of heterogeneous solid acids can solve some of these problems through the ease of product separation and good catalyst recyclability. This review summarizes recent advances in the hydrolysis of cellulose by different types of solid acids, such as sulfonated carbonaceous based acids, polymer based acids and magnetic solid acids. The acid strength, acid site density, adsorption of the substance and micropores of the solid material are all key factors for effective hydrolysis processes. Methods used to promote reaction efficiency such as the pretreatment of cellulose to reduce its crystallinity and the use of ionic liquids or microwave irradiation to improve the reaction rate are also discussed.
Co-reporter:Qianqian Lu, Haizhu Yu and Yao Fu
Chemical Communications 2013 vol. 49(Issue 92) pp:10847-10849
Publication Date(Web):17 Sep 2013
DOI:10.1039/C3CC46069J
DFT calculations indicate a linear correlation between the C–H activation barrier and the C–Cu/C–H bond dissociation energy gap in Cu-promoted C–H activation of heteroarenes.
Co-reporter:Zhen Yang, Yao-Bing Huang, Qing-Xiang Guo and Yao Fu
Chemical Communications 2013 vol. 49(Issue 46) pp:5328-5330
Publication Date(Web):24 Apr 2013
DOI:10.1039/C3CC40980E
A catalytic transfer hydrogenation process was developed for the production of γ-valerolactone (GVL) from ethyl levulinate (EL) and a H-donor at room temperature. Ethyl levulinate was almost quantitatively converted to γ-valerolactone. Further, a two step process for producing GVL from biomass derived platform molecules was also reported.
Co-reporter:Qi Zhang, Hai-Zhu Yu, Yi-Tong Li, Lei Liu, Yong Huang and Yao Fu
Dalton Transactions 2013 vol. 42(Issue 12) pp:4175-4184
Publication Date(Web):06 Feb 2013
DOI:10.1039/C3DT31898B
A systematic theoretical study on the Rh-catalyzed oxidative Heck-coupling of phenol carbamates with alkenes is carried out. Two possible mechanisms (i.e. arene activation-first and alkene activation-first mechanisms) are examined. As to the C–H activation step, four mechanisms including oxidative addition, electrophilic substitution, concerted metallation-deprotonation (CMD), and σ-bond metathesis are evaluated. The calculation results indicate that the arene activation-first mechanism is more favorable for the overall catalytic cycle. This mechanism involves three steps: arene C–H activation at the position ortho to the carbamate directing group affording a six-membered rhodiacycle intermediate, insertion of the alkene double bond into the Rh(III)–aryl bond, and a final β-H elimination step to release the product and re-generate the catalyst. The rate determining step of the overall catalytic cycle is the arene C–H activation step, which is found to proceed through the acetate-assisted CMD mechanism.
Co-reporter:Chen Wang, Hai-Zhu Yu, Yao Fu and Qing-Xiang Guo
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 13) pp:2140-2146
Publication Date(Web):16 Jan 2013
DOI:10.1039/C3OB27367A
Arylboronic acids were found to be efficient catalysts for the amidation reactions between carboxylic acids and amines. Theoretical calculations have been carried out to investigate the mechanism of this catalytic process. It is found that the formation of the acyloxyboronic acid intermediates from the carboxylic acid and the arylboronic acid is kinetically facile but thermodynamically unfavorable. Removal of water (as experimentally accomplished by using molecular sieves) is therefore essential for overall transformation. Subsequently C–N bond formation between the acyloxyboronic acid intermediates and the amine occurs readily to generate the desired amide product. The cleavage of the C–O bond of the tetracoordinate acyl boronate intermediates is the rate-determining step in this process. Our analysis indicates that the mono(acyloxy)boronic acid is the key intermediate. The high catalytic activity of ortho-iodophenylboronic acid is attributed to the steric effect as well as the orbital interaction between the iodine atom and the boron atom.
Co-reporter:Haizhu Yu;Dingjia Liu;Zhimin Dang;Dongrui Wang
Chinese Journal of Chemistry 2013 Volume 31( Issue 2) pp:200-208
Publication Date(Web):
DOI:10.1002/cjoc.201200753
Abstract
The recent development of Au-catalyzed reactions has greatly enriched the methods of organic synthesis. The phosphine or phosphate-coordinated Au complexes have shown high efficiency in catalyzing various CC and CH activations. In many of these reactions, the AuP bond strength was found to play an important role in determining the catalytic efficiency. In the present study, the accuracy of different theoretical methods for prediction of AuP strengths has been examined on basis of the experimental enthalpy changes between different ClAu(PR3) and ClAu(THT) (THT=tetrahydrothiophene). By comparing the different DFT functionals (e.g. B3LYP, TPSS, M06), different basis sets (including the different effective core potentials on Au and the total electron basis sets on all other atoms), and different solution phase single point calculations, we found that the TPSS/(CPE-121G+f:6-311+G(d,p)(SMD)//TPSS/(CPE-121G:6-31G(d) (M1) method gives the best correlations with the available experimental results. Meanwhile, the calculations with B3LYP//TPSS and M05//TPSS also give comparable calculation results. Finally, the specified method (M1) is used to calculate the AuP bond dissociation enthalpies and energies of different ClAu(PR3)/ClAu(P(OR)3) complexes. By accurately reproducing the available experimental results, we believe that the method (M1) is reliable for the theoretical analysis of Au-P systems.
Co-reporter:Tao Pan;Jin Deng;Qing Xu;Yong Zuo; Qing-Xiang Guo ;Dr. Yao Fu
ChemSusChem 2013 Volume 6( Issue 1) pp:47-50
Publication Date(Web):
DOI:10.1002/cssc.201200652
Co-reporter:Zhiwei Yang;Dr. Haizhu Yu; Yao Fu
Chemistry - A European Journal 2013 Volume 19( Issue 36) pp:12093-12103
Publication Date(Web):
DOI:10.1002/chem.201203666
Abstract
Density functional theory (DFT) calculations have been performed to study the mechanism of the recently reported Co-catalyzed ligand-controlled hydroarylation of styrenes as a means of preparing 1,1- or 1,2-diarylalkanes. The present study corroborates the previously proposed three-step mechanism, comprising CH activation (CH oxidative addition), styrene insertion, and reductive elimination. In the CH activation and reductive elimination steps, our calculations suggest that styrene does not coordinate to the Co center. In the insertion step, styrene is inserted into the CoH bond rather than the CoC bond. Furthermore, the rate- and regiodetermining step is found to be CC reductive elimination. It is significant that the regioselectivity observed experimentally has been successfully reproduced by our calculations. More importantly, in analyzing the origin of the ligand-controlled regioselectivity, we have found that the steric effects of different ligands mainly determine the observed regioselectivity. Both the shape (i.e., “umbrella-up” or “umbrella-down”) and bulkiness of the ligand contribute to the steric effect.
Co-reporter:Qi Zhang, Hai-Zhu Yu, and Yao Fu
Organometallics 2013 Volume 32(Issue 15) pp:4165-4173
Publication Date(Web):July 29, 2013
DOI:10.1021/om400370v
A theoretical study has been carried out on the palladium-catalyzed C(sp3)–H activation/amidation reaction of carbamoyl chloride precursors (Takemoto, Y. Angew. Chem. Int. Ed. 2012, 51, 2763 ). In Takemoto’s reaction, although the C(sp2)–H bond of naphthalene was present in the substrate, the benzylic C(sp3)–H bond was activated exclusively. Mechanistic calculations have been performed on the two possible pathways: the C(sp3)–H activation/amidation pathway (Path-sp3) and the C(sp2)–H activation/amidation pathway (Path-sp2). Calculation results show that both paths include three steps: oxidative addition (via the mono-phosphine mechanism), C–H activation involving the PivNHO– anion (via the CMD mechanism), and final reductive elimination. The calculations indicate that the Path-sp3 mechanism is kinetically favored, and the C(sp3)–H amidated product is predicted to be the main product. This conclusion is consistent with Takemoto’s experimental observations. The rate-determining step of Path-sp3 is the oxidative addition step, and the C(sp3)–H bond activation step determines the selectivity. Further examination on the origin of the selective C(sp3)–H activation shows that the higher acidity of the benzylic C(sp3)–H (in comparison to the naphthalene C(sp2)–H in this system) is the main reason for the chemoselectivity. The additive might promote the reaction by forming a more soluble organic base (PivNHOCs) via reaction with Cs2CO3.
Co-reporter:Yuan-Ye Jiang, Hai-Zhu Yu, and Yao Fu
Organometallics 2013 Volume 32(Issue 3) pp:926-936
Publication Date(Web):January 29, 2013
DOI:10.1021/om301263s
Recently the Chatani group reported the Rh(I)-catalyzed borylation of nitriles, which provided an efficient protocol for transformation of the C–CN bond to the C–B bond (J. Am. Chem. Soc.2012, 134, 115). Although an unconventional β-carbon elimination mechanism was proposed in their study, the other previously proposed mechanisms, i.e., oxidative addition, deinsertion, and initial C–H bond activation, cannot be excluded. To clarify the dominant mechanism of this reaction, a density functional theory study on borylation of PhCN and BnCN catalyzed by [Rh(XantPhos)(B(nep))] (nep = neopentylglycolate, XantPhos = 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene) was conducted. The computational results indicated that the deinsertion mechanism (2,1-insertion of the Rh–B bond into the C≡N bond occurs first, followed by the insertion of the metal center into C–CN bond) is favored over oxidative addition, β-carbon elimination, and the initial C–H bond activation mechanism within all the investigated reactions. The activation of the C–CN bond is a facile step in the deinsertion mechanism, and the oxidative addition of the diboron reagent is the rate-determining step. On this basis, the mechanism of borylation of PhCN catalyzed by a similar Ir–B complex ([Ir(XantPhos)(B(nep))]) was also examined. The deinsertion mechanism was found to be the most favorable. The overall energy barrier of the Ir–B complex-catalyzed borylation of benzonitriles was slightly higher than that of the same Rh–B complex-catalyzed reaction (by 1.1 kcal/mol), indicating that [Ir(XantPhos)(B(nep))] could act as an alternative catalyst for borylation of nitriles.
Co-reporter:ShiYa Tang;TianJun Gong
Science China Chemistry 2013 Volume 56( Issue 5) pp:619-632
Publication Date(Web):2013 May
DOI:10.1007/s11426-012-4795-3
Intramolecular ortho-C-H activation and C-N/C-O cyclizations of phenyl amidines and amides have recently been achieved under Cu catalysis. These reactions provide important examples of Cu-catalyzed functionalization of inert C-H bonds, but their mechanisms remain poorly understood. In the present study the several possible mechanisms including electrophilic aromatic substitution, concerted metalation-deprotonation (CMD), Friedel-Crafts mechanism, radical mechanism, and proton-coupled electron transfer have been theoretically examined. Cu(II)-assisted CMD mechanism is found to be the most feasible for both C-O and C-N cyclizations. This mechanism includes three steps, i.e. CMD with Cu(II), oxidation of the Cu(II) intermediate, and reductive elimination from Cu(III). Our calculations show that Cu(II) mediates the C-H activation through an six-membered ring CMD transition state similar to that proposed for many Pd-catalyzed C-H activation reactions. It is also interesting to find that the rate-limiting steps are different for C-N and C-O cyclizations: for the former it is concerted metalation-deprotonation with Cu(II), whereas for the latter it is reductive elimination from Cu(III). The above conclusions are consistent with the experimental kinetic isotope effects (1.0 and 2.1 for C-O and C-N cyclizations, respectively), substituent effects, and the reactions under O2-free conditions.
Co-reporter:Jin Deng;Yan Wang;Tao Pan;Qing Xu; Qing-Xiang Guo ;Dr. Yao Fu
ChemSusChem 2013 Volume 6( Issue 7) pp:1163-1167
Publication Date(Web):
DOI:10.1002/cssc.201300245
Co-reporter:Yao-Bing Huang;Zhen Yang;Meng-Yuan Chen;Jian-Jun Dai; Qing-Xiang Guo ; Yao Fu
ChemSusChem 2013 Volume 6( Issue 8) pp:1348-1351
Publication Date(Web):
DOI:10.1002/cssc.201300190
Co-reporter:Zheng Huang, Rui Shang, Zi-Rong Zhang, Xiao-Dan Tan, Xiao Xiao, and Yao Fu
The Journal of Organic Chemistry 2013 Volume 78(Issue 9) pp:4551-4557
Publication Date(Web):April 11, 2013
DOI:10.1021/jo400616r
A copper-catalyzed decarboxylative coupling reaction of potassium alkynyl carboxylates with 1,1-dibromo-1-alkenes was developed for the synthesis of unsymmetrical 1,3-diyne and 1,3,5-triyne derivatives. Diverse aryl, alkenyl, alkynyl, and alkyl substituted 1,1-dibromo-1-alkenes can react smoothly with aryl and alkyl substituted propiolates to produce unsymmetrical 1,3-diynes and 1,3,5-triynes with high selectivity and good functional group compatibility.
Co-reporter:Xian-fa Li, Yong Zuo, Ying Zhang, Yao Fu, Qing-xiang Guo
Fuel 2013 Volume 113() pp:435-442
Publication Date(Web):November 2013
DOI:10.1016/j.fuel.2013.06.008
•A new method of in situ preparing carbon loading K2CO3 as solid catalyst is proposed.•Kraft lignin was used as precursor of solid catalyst for biodiesel production.•This solid catalyst is low-cost, efficient, environmentally friendly and reusable.•The solid catalysts were characterized to reveal the textures and catalytic performance.In this study, the catalysts K2CO3 supported Kraft lignin activated carbon (K2CO3/KLC) were prepared in situ by simply mixing K2CO3 with technical Kraft lignin (KL) and subsequently activating at 800 °C for 2 h under N2 flow. The precursor and catalysts were characterized by thermal gravity analysis (TGA), Fourier transform infrared (FTIR) spectroscopy, X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM) and Brunauer–Emmett–Teller (BET) surface area. The catalytic performance of prepared catalysts was evaluated by transesterification of rapeseed oil with methanol. The effects of various parameters on biodiesel yield were investigated. The biodiesel yield of 99.6% was achieved by using the catalyst prepared by 0.6 of K2CO3/KL mass ratio and activation at 800 °C, under the transesterification condition of 65 °C, 2 h, methanol to rapeseed oil molar ratio of 15:1 and 3.0 wt.% catalyst (relative to the weight of rapeseed oil). The solid catalyst can be reused for 4 times and biodiesel yield remained over 82.1% for the fourth time.
Co-reporter:Zhen-Zhen Yang, Jin Deng, Tao Pan, Qing-Xiang Guo and Yao Fu
Green Chemistry 2012 vol. 14(Issue 11) pp:2986-2989
Publication Date(Web):29 Aug 2012
DOI:10.1039/C2GC35947B
We have demonstrated an efficient, selective and environmentally benign heterogeneous catalyst (K-OMS-2) for aerobic oxidation of 5-HMF to 2,5-DFF. In addition, a combination of Fe3O4-SBA-SO3H and K-OMS-2 successfully catalyzed direct synthesis of 2,5-DFF from fructose via acid-catalyzed dehydration and successive aerobic oxidation in one-pot reaction.
Co-reporter:Rui Shang;Zheng Huang;Xiao Xiao;Xi Lu;Lei Liu
Advanced Synthesis & Catalysis 2012 Volume 354( Issue 13) pp:2465-2472
Publication Date(Web):
DOI:10.1002/adsc.201200383
Abstract
The palladium-catalyzed decarboxylative benzylation of α-cyano aliphatic carboxylate salts with benzyl electrophiles was discovered. This reaction exhibits good functional group compatibility and proceeds under relatively mild conditions. A diverse range of quaternary, tertiary and secondary β-aryl nitriles can be conveniently prepared by this method.
Co-reporter:Yao-Bing Huang, Zhen Yang, Jian-Jun Dai, Qing-Xiang Guo and Yao Fu
RSC Advances 2012 vol. 2(Issue 30) pp:11211-11214
Publication Date(Web):19 Sep 2012
DOI:10.1039/C2RA22008C
A new route of converting the whole of lignocellulose biomass derived molecules into high quality fuels was developed with C–C bond formation as the key step. The fuel precursors with 10–14 carbons could be easily obtained though direct self-coupling of furfural, 5-methyfurfural or aromatic aldehyde, the main components of lignocellulose, under mild conditions in water. After dehydration/hydrogenation, straight or branched alkanes with 8–14 carbons within diesel ranged fuels were obtained in moderate to high yield.
Co-reporter:Dong-Fen Luo, Jun Xu, Yao Fu, Qing-Xiang Guo
Tetrahedron Letters 2012 Volume 53(Issue 22) pp:2769-2772
Publication Date(Web):30 May 2012
DOI:10.1016/j.tetlet.2012.03.107
A copper-catalyzed method for trifluoromethylation of terminal alkynes with Umemoto’s reagent has been developed. The reaction is conducted at room temperature and shows good tolerance to a variety of functional groups.
Co-reporter:Dr. Zhe Li;Yuan-Ye Jiang ;Dr. Yao Fu
Chemistry - A European Journal 2012 Volume 18( Issue 14) pp:4345-4357
Publication Date(Web):
DOI:10.1002/chem.201103882
Abstract
Ni-catalyzed cross-coupling of unactivated secondary alkyl halides with alkylboranes provides an efficient way to construct alkyl–alkyl bonds. The mechanism of this reaction with the Ni/L1 (L1=trans-N,N′-dimethyl-1,2-cyclohexanediamine) system was examined for the first time by using theoretical calculations. The feasible mechanism was found to involve a NiI–NiIII catalytic cycle with three main steps: transmetalation of [NiI(L1)X] (X=Cl, Br) with 9-borabicyclo[3.3.1]nonane (9-BBN)R1 to produce [NiI(L1)(R1)], oxidative addition of R2X with [NiI(L1)(R1)] to produce [NiIII(L1)(R1)(R2)X] through a radical pathway, and CC reductive elimination to generate the product and [NiI(L1)X]. The transmetalation step is rate-determining for both primary and secondary alkyl bromides. KOiBu decreases the activation barrier of the transmetalation step by forming a potassium alkyl boronate salt with alkyl borane. Tertiary alkyl halides are not reactive because the activation barrier of reductive elimination is too high (+34.7 kcal mol−1). On the other hand, the cross-coupling of alkyl chlorides can be catalyzed by Ni/L2 (L2=trans-N,N′-dimethyl-1,2-diphenylethane-1,2-diamine) because the activation barrier of transmetalation with L2 is lower than that with L1. Importantly, the Ni0–NiII catalytic cycle is not favored in the present systems because reductive elimination from both singlet and triplet [NiII(L1)(R1)(R2)] is very difficult.
Co-reporter:Dr. Haizhu Yu; Yao Fu
Chemistry - A European Journal 2012 Volume 18( Issue 52) pp:16765-16773
Publication Date(Web):
DOI:10.1002/chem.201202623
Abstract
Mechanistic studies have been performed for the recently developed, Ni-catalysed selective cross-coupling reaction between aryl and alkyl aldehydes. A mono-carbonyl activation (MCA) mechanism (in which one of the carbonyl groups is activated by oxidative addition) was found to be the most favourable pathway, and the rate-determining step is oxidative addition. Analysing the origin of the observed cross-coupling selectivity, we found the most favourable carbonyl activation step requires both coordination of the aryl aldehyde and oxidative addition of the alkyl aldehyde. Therefore, the stronger π-accepting ability of the aryl aldehyde (relative to alkyl aldehyde) and the ease of oxidative addition of the alkyl aldehyde (relative to aryl aldehyde) are responsible for the cross-coupling selectivity.
Co-reporter:YuanYe Jiang;Lei Liu
Science China Chemistry 2012 Volume 55( Issue 10) pp:2057-2062
Publication Date(Web):2012 October
DOI:10.1007/s11426-012-4672-0
Recently we reported Pd-catalyzed decarboxylative cross-coupling of cyanoacetate salts with aryl halides and triflates. This reaction shows good functional group tolerance and is useful for the synthesis of α-aryl nitriles. To elucidate the mechanism for this reaction, we now carry out a density functional theory study on the cross-coupling of potassium cyanoacetate with bromobenzene. Our results show that the decarboxylation transition state involving the interaction of Pd with the α-carbon atom has a very high energy barrier of +34.5 kcal/mol and therefore, must be excluded. Decarboxylation of free ion (or tight-ion-pair) also causes a high energy increase and should be ruled out. Thus the most favored decarboxylation mechanism corresponds to a transition state in which Pd interacts with the cyano nitrogen. The energy profile of the whole catalytic cycle shows that decarboxylation is the rate-determining step. The total energy barrier is +27.5 kcal/mol, which is comprised of two parts, i.e. the energy barrier for decarboxylation and the energy cost for transmetallation.
Co-reporter:Jun Xu;Bin Xiao;Chuan-Qi Xie;Dong-Fen Luo; Lei Liu; Yao Fu
Angewandte Chemie International Edition 2012 Volume 51( Issue 50) pp:12551-12554
Publication Date(Web):
DOI:10.1002/anie.201206681
Co-reporter:Dr. Zhe Li;Yuan-Ye Jiang;Dr. Andrew A. Yeagley;James P. Bour; Lei Liu; Jason J. Chruma; Yao Fu
Chemistry - A European Journal 2012 Volume 18( Issue 45) pp:14527-14538
Publication Date(Web):
DOI:10.1002/chem.201201425
Abstract
The Pd-catalyzed decarboxylative allylation of α-(diphenylmethylene)imino esters (1) or allyl diphenylglycinate imines (2) is an efficient method to construct new C(sp3)C(sp3) bonds. The detailed mechanism of this reaction was studied by theoretical calculations [ONIOM(B3LYP/LANL2DZ+p:PM6)] combined with experimental observations. The overall catalytic cycle was found to consist of three steps: oxidative addition, decarboxylation, and reductive allylation. The oxidative addition of 1 to [(dba)Pd(PPh3)2] (dba=dibenzylideneacetone) produces an allylpalladium cation and a carboxylate anion with a low activation barrier of +9.1 kcal mol−1. The following rate-determining decarboxylation proceeds via a solvent-exposed α-imino carboxylate anion rather than an O-ligated allylpalladium carboxylate with an activation barrier of +22.7 kcal mol−1. The 2-azaallyl anion generated by this decarboxylation attacks the face of the allyl ligand opposite to the Pd center in an outer-sphere process to produce major product 3, with a lower activation barrier than that of the minor product 4. A positive linear Hammett correlation [ρ=1.10 for the PPh3 ligand] with the observed regioselectivity (3 versus 4) supports an outer-sphere pathway for the allylation step. When Pd combined with the bis(diphenylphosphino)butane (dppb) ligand is employed as a catalyst, the decarboxylation still proceeds via the free carboxylate anion without direct assistance of the cationic Pd center. Consistent with experimental observations, electron-withdrawing substituents on 2 were calculated to have lower activation barriers for decarboxylation and, thus, accelerate the overall reaction rates.
Co-reporter:Jian-Jun Dai;Yao-Bing Huang;Chi Fang; Qing-Xiang Guo ; Yao Fu
ChemSusChem 2012 Volume 5( Issue 4) pp:617-620
Publication Date(Web):
DOI:10.1002/cssc.201100776
Co-reporter:Jiang Li;Dao-Jun Ding;Li Deng; Qing-Xiang Guo ;Dr. Yao Fu
ChemSusChem 2012 Volume 5( Issue 7) pp:1313-1318
Publication Date(Web):
DOI:10.1002/cssc.201100466
Abstract
An efficient catalytic system for biomass oxidation to form formic acid was developed. The conversion of glucose to formic acid can reach up to 52 % yield within 3 h when catalyzed by 5 mol % of H5PV2Mo10O40 at only 373 K using air as the oxidant. Furthermore, the heteropolyacid can be used as a bifunctional catalyst in the conversion of cellulose to formic acid (yield=35 %) with air as the oxidant.
Co-reporter:Jun Xu;Bin Xiao;Chuan-Qi Xie;Dong-Fen Luo; Lei Liu; Yao Fu
Angewandte Chemie 2012 Volume 124( Issue 50) pp:12719-12722
Publication Date(Web):
DOI:10.1002/ange.201206681
Co-reporter:Da-ming Lai, Li Deng, Qing-xiang Guo and Yao Fu
Energy & Environmental Science 2011 vol. 4(Issue 9) pp:3552-3557
Publication Date(Web):25 Jul 2011
DOI:10.1039/C1EE01526E
A magnetic solid acid with mesoporous structure was synthesised for biomass saccharification and levulinic acid production. The catalyst shows improved performance for the hydrolysis of β-1,4-glucan and carbohydrate dehydration than a conventional solid acid. Moreover, the catalyst can be easily separated from the reaction residues with an external magnetic force.
Co-reporter:Jun Xu, Yao Fu, Dong-Fen Luo, Yuan-Ye Jiang, Bin Xiao, Zhao-Jing Liu, Tian-Jun Gong, and Lei Liu
Journal of the American Chemical Society 2011 Volume 133(Issue 39) pp:15300-15303
Publication Date(Web):September 13, 2011
DOI:10.1021/ja206330m
An unprecedented type of reaction for Cu-catalyzed trifluoromethylation of terminal alkenes is reported. This reaction represents a rare instance of catalytic trifluoromethylation through C(sp3)–H activation. It also provides a mechanistically unique example of Cu-catalyzed allylic C–H activation/functionalization. Both experimental and theoretical analyses indicate that the trifluoromethylation may occur via a Heck-like four-membered-ring transition state.
Co-reporter:Yan Zhao, Li Deng, Shi-Ya Tang, Da-Ming Lai, Bing Liao, Yao Fu, and Qing-Xiang Guo
Energy & Fuels 2011 Volume 25(Issue 8) pp:3693
Publication Date(Web):July 5, 2011
DOI:10.1021/ef200648s
A highly selective decomposition of formic acid in aqueous medium was achieved with immobilized catalysts. In the gas product of all of the experiments, only hydrogen and carbon dioxide were detected. The activity of different immobilized catalysts for the decomposition of formic acid was checked, and the highest turnover frequency (TOF) was observed using Pd–S–SiO2. Moreover, we have found that the addition of sulfates could significantly promote the activities of mercapto-functionalized catalysts by 30–70%, which is attractive for practical applications.
Co-reporter:Jun Yi, Yao Fu, Bin Xiao, Wei-Chen Cui, Qing-Xiang Guo
Tetrahedron Letters 2011 Volume 52(Issue 2) pp:205-208
Publication Date(Web):12 January 2011
DOI:10.1016/j.tetlet.2010.10.128
A Pd-catalyzed coupling reaction of ArBr/ArCl/ArOTf with sodium thiosulfate takes place in presence of Cs2CO3 at 80 °C. The reaction mixture is directly treated with Zn/HCl to afford aryl thiols in good to excellent yields.
Co-reporter:Chen Wang;Qing-Xiang Guo ; Yao Fu
Chemistry – An Asian Journal 2011 Volume 6( Issue 5) pp:1241-1251
Publication Date(Web):
DOI:10.1002/asia.201000760
Abstract
The method of native chemical ligation between an unprotected peptide α-thioester and an N-terminal cysteine–peptide to give a native peptide in aqueous solution is one of the most effective peptide ligation methods. In this work, a systematic theoretical study was carried out to fully understand the detailed mechanism of ligation. It was found that for the conventional native chemical ligation reaction between a peptide thioalkyl ester and a cysteine in combination with an added aryl thiol as catalyst, both the thiol-thioester exchange step and the transthioesterification step proceed by an anionic concerted SN2 displacement mechanism, whereas the intramolecular rearrangement proceeds by an addition–elimination mechanism, and the rate-limiting step is the thiol-thioester exchange step. The theoretical method was then extended to study the detailed mechanism of the auxiliary-mediated peptide ligation between a peptide thiophenyl ester and an N-2-mercaptobenzyl peptide in which both the thiol-thioester exchange step and intramolecular acyl-transfer step proceed by a concerted SN2-type displacement mechanism. The energy barrier of the thiol-thioester exchange step depends on the side-chain steric hindrance of the C-terminal amino acid, whereas that of the acyl-transfer step depends on the side-chain steric hindrance of the N-terminal amino acid.
Co-reporter:Da-ming Lai;Li Deng;Jiang Li;Bing Liao; Qing-xiang Guo;Dr. Yao Fu
ChemSusChem 2011 Volume 4( Issue 1) pp:55-58
Publication Date(Web):
DOI:10.1002/cssc.201000300
Co-reporter:Shi-Ya Tang;Dr. Qing-Xiang Guo ;Dr. Yao Fu
Chemistry - A European Journal 2011 Volume 17( Issue 49) pp:13866-13876
Publication Date(Web):
DOI:10.1002/chem.201101587
Abstract
The use of ligands to control regioselectivity in transition-metal-catalyzed CH activation/functionalization is a highly desirable but challenging task. Recently, Itami et al. reported an important finding relating to Pd-catalyzed ligand-controlled α/β-selective CH arylation of thiophenes. Specifically, the use of the 2,2′-bipyridyl ligand resulted in α-arylation, whereas the use of the bulky fluorinated phosphine ligand P[OCH(CF3)2]3 resulted in β-arylation. Understanding of this surprising ligand-controlled α/β-selectivity could provide important insights into the development of more efficient catalyst systems for selective CH arylation, and so we carried out a detailed computational study on the problem with use of density functional theory methods. Three mechanistic possibilities—SEAr and migration, metalation/deprotonation, and Heck-type arylation mechanisms—were examined. The results showed that the SEAr and migration mechanism might not be plausible, because the key Wheland intermediates could not be obtained. On the other hand, our study indicated that the metalation/deprotonation and Heck-type arylation mechanisms were both involved in Itami’s reactions. In the metalation/deprotonation pathway the α-selective product (C5-product) was preferred, whereas in the Heck-type arylation mechanism the β-selective product (C4-product) was favored. The ligands played crucial roles in tuning the relative barriers of the two different pathways. In the 2,2′-bipyridyl-assisted system, the metalation/deprotonation pathway was energetically advantageous, leading to α-selectivity. In the P[OCH(CF3)2]3-assisted system, on the other hand, the Heck-type arylation mechanism was kinetically favored, leading to β-selectivity. An interesting finding was that P[OCH(CF3)2]3 could produce a CH⋅⋅⋅O hydrogen bond in the catalyst system, which was crucial for stabilization of the Heck-type transition state. In comparison, this CH⋅⋅⋅O hydrogen bond was absent with the other phosphine ligands [i.e., P(OMe)3, PPh3, PCy3] and these phosphine ligands therefore favored the metalation/deprotonation pathway leading to α-selectivity. Furthermore, in this study we have provided theoretical evidence showing that the Heck-type arylation reaction could proceed through an anti-β-hydride elimination process.
Co-reporter:Yao-Bing Huang;Jian-Jun Dai;Xiao-Jian Deng;Yan-Chao Qu; Qing-Xiang Guo ; Yao Fu
ChemSusChem 2011 Volume 4( Issue 11) pp:1578-1581
Publication Date(Web):
DOI:10.1002/cssc.201100344
Co-reporter:Yao-Bing Huang, Chu-Ting Yang, Jun Yi, Xiao-Jian Deng, Yao Fu, and Lei Liu
The Journal of Organic Chemistry 2011 Volume 76(Issue 3) pp:800-810
Publication Date(Web):December 31, 2010
DOI:10.1021/jo101917x
Resin-bound organic ionic bases (RBOIBs) were developed in which tetraalkyl-ammonium or phosphonium cations are covalently attached to solid resins. The application tests showed that the performance of the tetraalkyl-ammonium-type RBOIBs is slightly better than that of the corresponding Cs salts in Cu-catalyzed C−N cross-couplings, while the tetraalkylphosphonium-type RBOIBs are significantly better than all the inorganic bases. With these newly developed RBOIBs, room-temperature Cu-catalyzed C−N coupling with various nonactivated aryl iodides and even aryl bromides can be readily accomplished. Moreover, RBOIBs can be easily recycled and reused for a number of times without much drop of activity. The good performances of RBOIBs are proposed to arise from the relatively weak binding forces between the cationic polymer backbone and basic anions, as opposed to the strong metal−anion interactions in the inorganic bases. Further applications of RBOIBs in Ni-catalyzed Suzuki-type couplings at room temperature, Cu-catalyzed C−N couplings at −30 °C, a Pd-catalyzed Heck reaction at 60 °C, and Cu-catalyzed C−S couplings at room temperature demonstrate that RBOIBs are generally applicable bases with improved performance for many other types of organic transformations.
Co-reporter:Hai-Zhu Yu ; Yuan-Ye Jiang ; Yao Fu ;Lei Liu
Journal of the American Chemical Society 2010 Volume 132(Issue 51) pp:18078-18091
Publication Date(Web):December 6, 2010
DOI:10.1021/ja104264v
The ligand-dependent selectivities in Ullmann-type reactions of amino alcohols with iodobenzene by β-diketone- and 1,10-phenanthroline-ligated CuI complexes were recently explained by the single-electron transfer and iodine atom transfer mechanisms (Jones, G. O., Liu, P., Houk, K. N., and Buchwald, S. L. J. Am. Chem. Soc. 2010, 132, 6205.). The present study shows that an alternative, oxidative addition/reductive elimination mechanism may also explain the selectivities. Calculations indicate that a CuI complex with a negatively charged β-diketone ligand is electronically neutral, so that oxidative addition of ArI to a β-diketone-ligated CuI prefers to occur (and occur readily) in the absence of the amino alcohol. Thus, coordination of the amino alcohol in its neutral form can only occur at the CuIII stage where N-coordination is favored over O-coordination. The coordination step is the rate-limiting step and the outcome is that N-arylation is favored with the β-diketone ligand. On the other hand, a CuI complex with a neutral 1,10-phenanthroline ligand is positively charged, so that oxidative addition of ArI to a 1,10-phenanthroline-ligated CuI has to get assistance from a deprotonated amino alcohol substrate. This causes oxidative addition to become the rate-limiting step in the 1,10-phenanthroline-mediated reaction. The immediate product of the oxidative addition step is found to undergo facile reductive elimination to provide the arylation product. Because O-coordination of a deprotonated amino alcohol is favored over N-coordination in the oxidative addition transition state, O-arylation is favored with the 1,10-phenanthroline ligand.
Co-reporter:Yan Zhao, Li Deng, Bin Liao, Yao Fu, and Qing-Xiang Guo
Energy & Fuels 2010 Volume 24(Issue 10) pp:5735-5740
Publication Date(Web):September 29, 2010
DOI:10.1021/ef100896q
Herein we reported a promising method for the production of aromatics from pyrolytic lignin (PL). Compared to the lignins derived from pulping process, the PLs obtained from bio-oil give more aromatics (40% in carbon yield) and do not generate effluvial gas containing sulfur. More importantly, phenols are the main products with selectivity over 90% at 600 °C without catalyst. In the presence of ZSM-5, the coke deposition was not obvious and the selectivity for aromatic hydrocarbons is more than 87%, indicating robustness of ZSM-5 for deoxygenating and high tolerance to PLs. Therefore we have demonstrated the catalytic pyrolysis of PLs is an alternative way to produce fuel additives and useful chemicals.
Co-reporter:Jun Xu, Yao Fu, Bin Xiao, Tianjun Gong, Qingxiang Guo
Tetrahedron Letters 2010 Volume 51(Issue 41) pp:5476-5479
Publication Date(Web):13 October 2010
DOI:10.1016/j.tetlet.2010.08.029
Room-temperature N-vinylation of various substituted sulfonamides and acylamides with vinyl acetate was achieved for the first time with a palladium/carbene catalyst system. This reaction provides a useful method for synthesis of enamides under mild conditions.
Co-reporter:Huajing Wang;Xiongyi Huang;Rong Shen;Lei Rui
Chinese Journal of Chemistry 2010 Volume 28( Issue 1) pp:72-80
Publication Date(Web):
DOI:10.1002/cjoc.201090038
Abstract
Several 1-benzyl-1,4-dihydronicotinamide derivatives, which are important NADH model compounds were studied theoretically in acetonitrile. The performances of various DFT methods including B3LYP, B1B95, B3PW91, MPW1B95, MPWKCIS, and M06 were tested to calculate the redox potentials. The first theoretical protocol to predict the redox potentials of these derivatives is B1B95, whose reliability has been tested against almost all the available experimental data. Strikingly, the mean absolute derivations (MAD) and root mean square (RMS) error of the current theoretical model equal 0.015 and 0.017, respectively. By using this method, the important thermodynamic properties of BNAHs were investigated and the mechanisms of hydride transfer progress were explained. Besides, para-substituents that have a big effect on these redox potentials of BNAH were systematically studied and carefully demonstrated.
Co-reporter:Li Deng;Yan Zhao;Jiang Li; Yao Fu; Bing Liao; Qing-Xiang Guo
ChemSusChem 2010 Volume 3( Issue 10) pp:1172-1175
Publication Date(Web):
DOI:10.1002/cssc.201000163
Co-reporter:Chen Wang ;Qing-Xiang Guo ;Lei Liu
Chemistry - A European Journal 2010 Volume 16( Issue 8) pp:2586-2598
Publication Date(Web):
DOI:10.1002/chem.200902484
Abstract
Quantitative nucleophilicity scales are fundamental to organic chemistry and are usually constructed on the basis of Mayr’s equation [log k=s(N+E)] by using benzhydrylium ions as reference electrophiles. Here an ab initio protocol was developed for the first time to predict the nucleophilicity parameters N of various π nucleophiles in CH2Cl2 through transition-state calculations. The optimized theoretical model (BH&HLYP/6-311++G(3df,2p)//B3LYP/6-311+G(d,p)/PCM/UAHF) could predict the N values of structurally unrelated π nucleophiles within a precision of ca. 1.14 units and therefore may find applications for the prediction of nucleophilicity of compounds that are not readily amenable to experimental characterization. The success in predicting N parameters from first principles also allowed us to analyze in depth the electrostatic, steric, and solvation energies involved in electrophile–nucleophile reactions. We found that solvation does not play an important role in the validity of Mayr’s equation. On the other hand, the correlations of the E, N, and log k values with the energies of the frontier molecular orbitals indicated that electrostatic/charge-transfer interactions play vital roles in Mayr’s equation. Surprising correlations observed between the electrophile–nucleophile CC distances in the transition state, the activation energy barriers, and the E and N parameters indicate the importance of steric interactions in Mayr’s equation. A method is then proposed to separate the attraction and repulsion energies in the nucleophile–electrophile interaction. It was found that the attraction energy correlated with N+E, whereas the repulsion energy correlated to the s parameter.
Co-reporter:Song-Lin Zhang ; Yao Fu ; Rui Shang ; Qing-Xiang Guo ;Lei Liu
Journal of the American Chemical Society 2009 Volume 132(Issue 2) pp:638-646
Publication Date(Web):December 28, 2009
DOI:10.1021/ja907448t
Transition-metal-catalyzed decarboxylative coupling presents a new and important direction in synthetic chemistry. Mechanistic studies on decarboxylative coupling not only improve the understanding of the newly discovered transformations, but also may have valuable implications for the development of more effective catalyst systems. In this work, a comprehensive theoretical study was conducted on the mechanism of Myers’ Pd-catalyzed decarboxylative Heck reaction. The catalytic cycle was found to comprise four steps: decarboxylation, olefin insertion, β-hydride elimination, and catalyst regeneration. Decarboxylation was the rate-limiting step, and it proceeded through a dissociative pathway in which Pd(II) mediated the extrusion of CO2 from an aromatic carboxylic acid to form a Pd(II)−aryl intermediate. Further analysis was conducted on the factors that might control the efficiency of Myers’ decarboxylative Heck reaction. These factors included Pd salts, ligands, acid substrates, and metals. (1) Regarding Pd salts, PdCl2 and PdBr2 were worse catalysts than Pd(TFA)2, because the exchange of Cl or Br by a carboxylate from Pd was thermodynamically unfavorable. (2) Regarding ligands, DMSO provided the best compromise between carboxyl exchange and decarboxylation. Phosphines and N-heterocarbenes disfavored decarboxylation because of their electron richness, whereas pyridine ligands disfavored carboxyl exchange. (3) Regarding acid substrates, a good correlation was observed between the energy barrier of R−COOH decarboxylation and the R−H acidity. Substituted benzoic acids showed deviation from the correlation because of the involvement of π(substituent)−σ(Cipso−Pd) interaction. (4) Regarding metals, Ni and Pt were worse catalysts than Pd because of the less favorable carboxyl exchange and/or DMSO removal steps in Ni and Pt catalysis.
Co-reporter:Rui Shang ; Yao Fu ; Jia-Bin Li ; Song-Lin Zhang ; Qing-Xiang Guo ;Lei Liu
Journal of the American Chemical Society 2009 Volume 131(Issue 16) pp:5738-5739
Publication Date(Web):April 1, 2009
DOI:10.1021/ja900984x
Pd-catalyzed decarboxylative cross-coupling of aryl iodides, bromides, and chlorides with potassium oxalate monoesters has been discovered. This reaction is potentially useful for laboratory-scale synthesis of aryl and alkenyl esters. Bulky, electron-rich bidentate phosphine ligands are preferred in the reaction, whereas Cu is not needed for decarboxylation. Theoretical calculations suggest a five-coordinate Pd(II) transition state for decarboxylation with an energy barrier of ∼30 kcal/mol.
Co-reporter:Wan-Ming Xiong, Man-Zhou Zhu, Li Deng, Yao Fu and Qing-Xiang Guo
Energy & Fuels 2009 Volume 23(Issue 4) pp:2278-2283
Publication Date(Web):March 16, 2009
DOI:10.1021/ef801021j
A dicationic ionic liquid C6(mim)2−HSO4 was synthesized and used as the catalyst for bio-oil upgrading through the esterificartion reaction of organic acids and ethanol at room temperature. The reaction system turned into two layers when the reaction was complete. No coke and deactivation on the catalyst were observed. The yield of upgraded oil was about 49%, and its properties were significantly improved. The higher heating value approached to 24.6 MJ/kg, the pH value increased from 2.9 to 5.1, and the moisture decreased from 29.8 to 8.2 wt %. The chemical composition of the upgraded bio-oil was analyzed with GC-MS. The results showed that organic acids were successfully converted into esters. Thus our data indicated that the dicationic ionic liquid was a promising catalyst for esterification to upgrade bio-oil.
Co-reporter:Li Deng, Zhao Yan, Yao Fu and Qing-Xiang Guo
Energy & Fuels 2009 Volume 23(Issue 6) pp:3337
Publication Date(Web):May 28, 2009
DOI:10.1021/ef9002268
Co-reporter:Li Deng, Yao Fu and Qing-Xiang Guo
Energy & Fuels 2009 Volume 23(Issue 1) pp:564
Publication Date(Web):December 16, 2008
DOI:10.1021/ef800692a
In this paper, we proposed a novel method to upgrade the acid-rich phase of bio-oil via ketonic condensation over weak base catalysts. Most acetic acid could be transformed to acetone in model reactions, and CeO2 was proved to be suitable as an active phase for catalyzing this reaction. The effects of water and three model components (phenol, p-methoxyphenol, and frufural) on acid transformation over four CeO2-based catalysts were investigated. As a result, with feeds containing water and phenols with liquid hourly space velocity (LHSV) of 4 cm3 g−1 h−1, this reaction could proceed efficiently and adequately. However, furfural deactivated catalysts significantly, and therefore, furans must be treated before this transformation.
Co-reporter:Chu-Ting Yang ;Yao-Bing Huang;Jun Yi;Qing-Xiang Guo ;Lei Liu
Angewandte Chemie 2009 Volume 121( Issue 40) pp:7534-7537
Publication Date(Web):
DOI:10.1002/ange.200903158
Co-reporter:Li Deng;Jiang Li;Da-Ming Lai, ;Qing-Xiang Guo
Angewandte Chemie International Edition 2009 Volume 48( Issue 35) pp:6529-6532
Publication Date(Web):
DOI:10.1002/anie.200902281
Co-reporter:Hua-Jian Xu, Xiao-Yang Zhao, Jin Deng, Yao Fu, Yi-Si Feng
Tetrahedron Letters 2009 50(4) pp: 434-437
Publication Date(Web):
DOI:10.1016/j.tetlet.2008.11.029
Co-reporter:Chu-Ting Yang ;Yao-Bing Huang;Jun Yi;Qing-Xiang Guo ;Lei Liu
Angewandte Chemie International Edition 2009 Volume 48( Issue 40) pp:7398-7401
Publication Date(Web):
DOI:10.1002/anie.200903158
Co-reporter:Li Deng;Jiang Li;Da-Ming Lai, ;Qing-Xiang Guo
Angewandte Chemie 2009 Volume 121( Issue 35) pp:6651-6654
Publication Date(Web):
DOI:10.1002/ange.200902281
Co-reporter:Haizhu Yu, Yao Fu, Qingxiang Guo and Zhenyang Lin
Organometallics 2009 Volume 28(Issue 15) pp:4507-4512
Publication Date(Web):June 1, 2009
DOI:10.1021/om900329f
DFT calculations have been carried out to study the mechanisms of Pd(OAc)2-catalyzed intramolecular diamination reactions in the presence of bases and oxidants. On the basis of the calculation results, a mechanism involving an anti-aminopalladation/syn-Csp3−N bond formation was proposed.
Co-reporter:Haizhu Yu, Yao Fu, Qingxiang Guo and Zhenyang Lin
Organometallics 2009 Volume 28(Issue 15) pp:4443-4451
Publication Date(Web):July 16, 2009
DOI:10.1021/om9002957
DFT calculations have been carried out to study the mechanisms for reactions of O2 with a series of metal complexes, including d6 CpRuL2, d6 ML5, and d8 ML4 complexes. The calculation results indicate that the reaction is initiated by an end-on coordination of O2 to the metal center, which gives an (η1-O2)[M] intermediate. The uncoordinated oxygen atom of the η1-O2 ligand then approaches the metal center to give a new η1-O2 intermediate in which the η1-O2 ligand is oriented approximately the same as the one defined in the product. An intersystem conversion from the triplet to singlet energy surface (MECP) then occurs to enable the metal peroxide product to be formed.
Co-reporter:Hua-Jing Wang, Yao Fu
Journal of Molecular Structure: THEOCHEM 2009 Volume 893(1–3) pp:67-72
Publication Date(Web):15 January 2009
DOI:10.1016/j.theochem.2008.09.017
Bond dissociation energies of a series of substituted silanes were studied with the density functional theory methods. The performances of six different density functional methods including B3LYP, B3P86, BH&HLYP, B1LYP, PBE1KCIS, and TPSSLYP1W were examined for the prediction of Si–H bond dissociation energies. The results showed that B3P86 was the most accurate theoretical procedure among these six DFT methods. Using the B3P86 method, we then carried out a systematic study about the substituent effects on Si–H bond dissociation energies, with a focus to identify the possible approaches to weaken the Si–H bond strength. On the basis of the knowledge learned from the systematic study on model systems, we proposed some new silicon-based radical reducing reagents which may be used to replace toxic tin hydride reagents.
Co-reporter:Wen-Rui Zheng, Yao Fu and Qing-Xiang Guo
Journal of Chemical Theory and Computation 2008 Volume 4(Issue 8) pp:1324-1331
Publication Date(Web):July 3, 2008
DOI:10.1021/ct800070y
On the basis of systematic examinations it was found that the BMK functional significantly outperformed the other popular density functional theory methods including B3LYP, B3P86, KMLYP, MPW1P86, O3LYP, and X3LYP for the calculation of bond dissociation enthalpies (BDEs). However, it was also found that even the BMK functional might dramatically fail in predicting the BDEs of some chemical bonds. To solve this problem, a new composite ab initio method named G3//BMK was developed by combining the strengths of both the G3 theory and BMK. G3//BMK was found to outperform the G3 and G3//B3LYP methods. It could accurately predict the BDEs of diverse types of chemical bonds in various organic molecules within a precision of ca. 1.2 kcal/mol.
Co-reporter:Chen WANG;Zhe LI ;Qing-Xiang GUO
Chinese Journal of Chemistry 2008 Volume 26( Issue 2) pp:358-362
Publication Date(Web):
DOI:10.1002/cjoc.200890069
Abstract
A computational study with the B3PW91 density functional theory was carried out on the activation process of palladacycles as catalysts in the Heck reaction. Two possible pathways (i.e. anion reductive cleavage of the Pd–C bond, and olefin insertion into the Pd–C bond followed by β-H elimination) were taken into consideration. Computational results indicate that the palladacycles are activated via olefin insertion into the Pd–C bond followed by β-H elimination in the reaction conditions.
Co-reporter:Xiang-Ming MENG;Lu-Feng ZOU;Miao XIE
Chinese Journal of Chemistry 2008 Volume 26( Issue 4) pp:787-793
Publication Date(Web):
DOI:10.1002/cjoc.200890147
Abstract
The available experimental αC-H BDEs of a variety of amine-containing molecules were examined by using the G3B3 and CBS-Q methods. The verified values were employed to benchmark and calibrate the density functional theory methods. It was found that the (U)BHandH/6-311++G(2df, 2p)//(U)B3LYP/6-31G(d) method was a fast and accurate method for calculating C–H BDEs at nitrogen α-positions. By using the newly benchmarked BHandH method, the αC–H BDEs in a number of nitrogen-containing drug molecules were calculated, where a dramatic variation of the αC–H BDEs was discovered. To understand this variation, the effects of mono- and double-substitution at both carbon and nitrogen atoms on the αC-H BDEs were systematically studied. The origin of the substitution effects was thoroughly discussed in terms of four categories of substituents.
Co-reporter:Ming FANG, Ming;Zhe LI
Chinese Journal of Chemistry 2008 Volume 26( Issue 6) pp:1122-1128
Publication Date(Web):
DOI:10.1002/cjoc.200890200
Abstract
Six density function theory methods (B3LYP, B3P86, MPWB1K1, MPWPW91, PBEPBE, TPSS1KCIS3) were used to calculate bond dissociation enthalpies of nitro compounds, where the B3P86 method was found to give the most accurate predictions. Using the B3P86 method meta- and para-substituted nitroaromatics were systematically studied for the first time. The remote substituent effects, Hammett relationships, and the origin of the substituent effects were discussed on the basis of the calculated results. Both meta- and para-substituted nitromethyl-benzenes showed significant substituent effects and a fair correlation against substituent constants σp+ The ground state effects were found to play the major role in determining the overall substituent effects. Meanwhile, nitroamino- benzenes showed irregular substituent effects and a poorer Hammett correlation, where both ground and radical state effects contributed to the overall substituent effects.
Co-reporter:Chen ZHU;Lei RUI
Chinese Journal of Chemistry 2008 Volume 26( Issue 8) pp:1493-1500
Publication Date(Web):
DOI:10.1002/cjoc.200890270
Abstract
The homolytic C–C and C–H bond dissociation enthalpyies (BDE) of highly crowded alkanes were calculated by using an ONIOM-G3B3 method. Geometric parameters such as bond length, bond angle and molecular volume were carefully investigated, as most of the acyclic alkanes in this study were not yet synthesized. These parameters reflect the influence of steric effect on BDE. Good correlations were found between the rapid decrease of BDE and the increase of molecular volumes. The correlations can be applied to the prediction of the possible existence of many highly strained compounds.
Co-reporter:Xiao-Lu Li, Yao Fu
Journal of Molecular Structure: THEOCHEM 2008 Volume 856(1–3) pp:112-118
Publication Date(Web):15 May 2008
DOI:10.1016/j.theochem.2008.01.029
The reduction potentials of 28 flavin derivatives in aqueous solution have been calculated using quantum chemistry methods. This is the first time that the method is applied on the task to systematically study the reduction potentials of flavins, and the average deviation between the calculated values and the experimental ones is less than 0.06 eV. It is a relatively good result in consideration of the complication of the compounds. Armed with the abundant calculation results including the geometry parameters, ionization potentials and reduction potentials, some useful properties of flavin derivatives such as substituent effects are systematically studied.
Co-reporter:Miao Xie, Yao Fu, Qian Chen
Journal of Molecular Structure: THEOCHEM 2008 Volume 855(1–3) pp:111-119
Publication Date(Web):30 April 2008
DOI:10.1016/j.theochem.2008.01.018
To elucidate the mechanism of alkaline hydrolysis and aminolysis of cephalosporin (Cep) and its thioxo analogue, thioxocephalosporin (Thi), density functional theory and continuum dielectric method were used to map out the solution-phase free-energy profiles. The hydrolysis reactivity of Cep was similar to that of Thi, but Cep was much slower in aminolysis than Thi due to different expulsion between the incoming hydroxide anion and amine. Furthermore, the rate-limiting step for Cep, in both hydrolysis and aminolysis, was the approaching of the negatively charged ion toward the anionic reactant to form a tetrahedral intermediate, and the breakdown of four-membered β-lactam for Thi. The predicted rate-limiting step and relative energy barrier for the hydrolysis and aminolysis of Cep and Thi were in agreement with the experimental results. By establishing model systems, the different reactivity between Cep and Thi was rationalized through amide resonance.
Co-reporter:Zhe Li ; Yao Fu ; Qing-Xiang Guo ;Lei Liu
Organometallics 2008 Volume 27(Issue 16) pp:4043-4049
Publication Date(Web):July 22, 2008
DOI:10.1021/om701065f
The mechanism of oxidative addition of aryl halides to Pd(PR3)2 (R = Me, Et, iPr, tBu, Ph) was investigated by using density functional theory methods enhanced with a polarized continuum solvation model. Different reaction pathways were discussed on the basis of Gibbs free-energy profiles in a tetrahydrofuran solution. The calculations indicated that monophosphine PdPR3 was catalytically more active than bisphosphine Pd(PR3)2 for oxidative addition. However, among different PR3 ligands (R = Me, Et, iPr, tBu, Ph), the free-energy barriers for oxidative addition to PdPR3 did not change significantly (i.e., less than 2 kcal/mol). This gave rise to an important question: why was P(t-Bu)3 the only catalytically active ligand toward aryl chlorides among the above five ligands? It was proposed on the basis of the calculated data that the difference of the dissociation energies from PdL2 to PdL and L (L = ligand) between the various PR3 ligands dictated their dissimilar reactivity in oxidative addition.
Co-reporter:Yi-Yun Yu;Lei Liu;Qing-Xiang Guo
Chinese Journal of Chemistry 2007 Volume 25(Issue 7) pp:
Publication Date(Web):16 JUL 2007
DOI:10.1002/cjoc.200790162
In the study, the X–H (XCH2, NH, O) bond dissociation energies (BDE) of para-substituted azulene (Y-C10H8X-H) were predicted theoretically for the first time using Density Functronal Theory (DFT) methods at UB3LYP/6-311++g(2df,2p)// UB3LYP/6-31+g(d) level. It was found that the substituents exerted similar effects on the X–H BDE of azulene as those on benzene, except for 6-substituted 2-methylazulene. Owing to the substituent-dipole interaction, the reaction constants (ρ+) of 2- and 6-Y-C10H8X-H (XNH and O only) varied violently. The origin of the substituent effects on the X–H BDE of azulene was found, by both GE/RE and SIE theory, to be directly associated with variation of the radical effects, although the ground effects also played a modest role in determining the net substituent effects.
Co-reporter:Hua Jian Xu, Xiao Lan Xu, Yao Fu, Yi Si Feng
Chinese Chemical Letters 2007 Volume 18(Issue 12) pp:1471-1475
Publication Date(Web):December 2007
DOI:10.1016/j.cclet.2007.10.009
Photocatalytic oxidation of primary and secondary benzyl alcohol to corresponding benzaldehyde or acetophenone using Acr+ClO4− or PhAcr+ClO4− as photocatalysts under visible light irradiation at room temperature.
Co-reporter:Lu-Feng Zou;Kuang Shen;Qing-Xiang Guo
Journal of Physical Organic Chemistry 2007 Volume 20(Issue 10) pp:754-763
Publication Date(Web):10 AUG 2007
DOI:10.1002/poc.1236
Experimental studies showed that sulfur radicals play the vital role in petroleum formation.1 Sulfur- centered radicals also exhibit activities in antioxidant functions. Here we conduct a theoretical investigation of their precursor-disulfides. By investigation into substituent effect on sulfursulfur bond dissociation enthalpies (SS BDEs), we would like to find the most effective provider for sulfur radicals. In the present work, 50 alpha-substituted disulfides and 16 para-substituted aryl disulfides are studied systematically, with the general formula XS-SX or HS-SX. The substituent effect on SS BDEs is found to be very eminent, ranging from 33.2 to 75.0 kcal/mol for alpha-substituted disulfide, and from 43.7 to 59.7 kcal/mol for para-substituted phenyl disulfides. We also evaluate the performance of 44 density functional methods to get an accurate prediction. A further study indicates that substituents play a major role in radical energies, instead of molecule energies, which is substantiated by the good linearity between XS-SX bond dissociation enthalpy (BDE) and HS-SX BDE. Copyright © 2007 John Wiley & Sons, Ltd.
Co-reporter:Wenrui Zheng, Yao Fu, Lei Liu, Qingxiang Guo
Acta Physico-Chimica Sinica 2007 Volume 23(Issue 7) pp:1018-1024
Publication Date(Web):July 2007
DOI:10.1016/S1872-1508(07)60057-6
The hydrogen bonding interactions between ureas or thioureas and different carbonyl compounds were studied using the Moller-Plesset perturbation theory (MP2). The natural bond orbital analysis further disclosed the essence of the hydrogen bonding interaction. In addition, the substituent effects were investigated and the results indicated that both the electron-withdrawing groups on ureas (thioureas) and electron-donating groups on carbonyl compounds can facilitate the hydrogen bonding formation. Two possible hydrogen bonding complex conformations, cis and trans, were discussed and the energy gaps between them were analyzed, in combination with the catalytic reactions.
Co-reporter:Wen-Rui Zheng, Yao Fu, Kuang Shen, Lei Liu, Qing-Xiang Guo
Journal of Molecular Structure: THEOCHEM 2007 Volume 822(1–3) pp:103-110
Publication Date(Web):15 November 2007
DOI:10.1016/j.theochem.2007.07.023
The hydrogen bonding interaction between ureas and thioureas with imines was investigated using the MP2 method. In combination with the catalytic reactions, we discussed two different hydrogen bonding patterns, which have the same typical hydrogen bonding feature of red shift effect. Based on it, the substituent effect of the more stable pattern was researched. The results indicated that electron-withdrawing groups on ureas or thioureas and electron-donating groups on imines can both facilitate the hydrogen bonding formation. We also studied the cis–trans isomerization of this pattern which is associated with the catalysis mechanism. The NBO analysis gave a more profound understanding of the hydrogen bonding interaction and appeared quite significant in view of their importance for understanding the intermolecular interaction leading to hydrogen bonding.
Co-reporter:Min-Jie Li, Lei Liu, Yao Fu, Qing-Xiang Guo
Journal of Molecular Structure: THEOCHEM 2007 Volume 815(1–3) pp:1-9
Publication Date(Web):1 August 2007
DOI:10.1016/j.theochem.2007.03.012
Radical-scavenging antioxidants play vital roles in the prevention of oxidative damage caused by free radicals, which is involved in many important chemical and biological processes. Using the ONIOM-G3B3 method, the bond dissociation enthalpies (BDEs) of coenzyme Q, flavonoids, olives, curcumins, indolinonic hydroxylamines, phenothiazines, edaravones and antioxidants used as food additives are predicted in the present study. On the basis of the computed BDE values, discussions were then made about their antioxidant activities, structure–activity relationships, and radical scavenging mechanisms. This work may be useful to clarify the radical scavenging mechanism of antioxidants and to design novel antioxidants.
Co-reporter:Lu-Feng Zou, Yao Fu, Kuang Shen, Qing-Xiang Guo
Journal of Molecular Structure: THEOCHEM 2007 Volume 807(1–3) pp:87-92
Publication Date(Web):1 April 2007
DOI:10.1016/j.theochem.2006.12.014
The sulfur–sulfur bond dissociation enthalpies in 12 disulfide compounds were calculated using high-level ab initio methods including G3, G3B3, CBS-Q, CBS-4M, CCSD(T), and ROMP2. The theoretical bond dissociation enthalpies calculated by different methods generally agreed well with each other. However, when compared to the available experimental data, only seven experimental values were consistent with the theoretical predictions, whereas the theoretical values for three molecules, i.e. FSSF, ClSSCl, HSSSSH, differed dramatically from the experimental ones. The origin for the inconsistency between the theory and experiment was discussed. Furthermore, the electronic effect of the substituents on sulfur–sulfur bond dissociation enthalpies was investigated.
Co-reporter:Bin Xiao ; Yao Fu ; Jun Xu ; Tian-Jun Gong ; Jian-Jun Dai ; Jun Yi ;Lei Liu
Journal of the American Chemical Society () pp:
Publication Date(Web):December 18, 2009
DOI:10.1021/ja909818n
Although nitrogen-containing group-directed cyclopalladation reactions have been well-known, Pd(II) insertion into C−H bonds promoted by coordination of an oxygen-only group to the palladium remains rather rare. In the present study, the first cyclopalladation complex formed from a simple phenol ester was characterized by X-ray crystallography. A promising protocol for the ortho C−H activation/aryl−aryl coupling of phenol esters that was not sensitive to moisture or air was then established. The utility of the reaction was demonstrated for the synthesis of useful phenol derivatives.
Co-reporter:Ebrahim-Alkhalil M. A. Ahmed, Xi Lu, Tian-Jun Gong, Zhen-Qi Zhang, Bin Xiao and Yao Fu
Chemical Communications 2017 - vol. 53(Issue 5) pp:NaN912-912
Publication Date(Web):2016/12/23
DOI:10.1039/C6CC07924E
We report the first copper-catalyzed/mediated borylative ring opening reaction of epoxides. This process represents a direct borylative C(sp3)–O bond cleavage of terminal epoxide substrates with commercially available diboron reagents. A wide range of epoxides with different functional groups are involved, and were subsequently converted to the corresponding β-hydroxyl boronic esters smoothly. Moreover, the ring opening product β-pinacol boronate alcohol provided a more beneficial approach for the formation of C–C and C–N bonds.
Co-reporter:Feng Li, Zhen-Qi Zhang, Xi Lu, Bin Xiao and Yao Fu
Chemical Communications 2017 - vol. 53(Issue 25) pp:NaN3554-3554
Publication Date(Web):2017/03/13
DOI:10.1039/C7CC00129K
A Cu/PPh3-catalyzed propargylic substitution reaction of diborylmethane is reported. Different substituted propargyl electrophiles can be employed in this reaction, and various synthetic valuable functional groups can be tolerated. Di-deuterated diborylmethane can also be used under these conditions and generates α-deuterated alkylboronic esters in good yield.
Co-reporter:Qi Zhang, Hai-Zhu Yu and Yao Fu
Inorganic Chemistry Frontiers 2014 - vol. 1(Issue 6) pp:NaN624-624
Publication Date(Web):2014/05/09
DOI:10.1039/C4QO00036F
A systematic theoretical study has been carried out on the NHC-catalyzed homoenolate reaction of enals and nitroalkenes. The detailed mechanism of these nitroalkenes participated homoenolate reactions, the chemoselectivity of the homoenolate (vs. Stetter) pathway and the intriguing syn- (vs. anti-) stereoselectivity have been investigated. Calculations show that the homoenolate and Stetter reaction (Path-homo and Path-set) first undergo the same formation process of the Breslow intermediate, which proceeds via a HOAc mediated stepwise mechanism. The subsequent process of Path-homo consists of five steps: nucleophilic attack of the Breslow intermediate (C3 atom) on the nitroalkene, intramolecular proton transfer, HOAc involved protonation of C2, concerted nucleophilic attack of OEt on the carbonyl, and final carbene dissociation. The subsequent process of Path-set consists of three steps: nucleophilic attack of the Breslow intermediate (C1 atom) on the nitroalkene, intramolecular proton transfer and carbene dissociation. Path-homo is more feasible than Path-set. The highly exoergonic nucleophilic attack, the facile intramolecular proton transfer and attack of OEt on the carbonyl in Path-homo result in its feasibility. The stereoselectivity determining step of Path-homo is the attack of the Breslow intermediate on the nitroalkene, in which the steric hindrance and hydrogen bonding determine the syn stereoselectivity.
Co-reporter:Jing-Hui Liu, Mian Cui, Xiao-Yu Lu, Zhen-Qi Zhang, Bin Xiao and Yao Fu
Chemical Communications 2016 - vol. 52(Issue 6) pp:NaN1245-1245
Publication Date(Web):2015/11/27
DOI:10.1039/C5CC08393A
A copper-mediated directed demethylation of propionamides has been developed. This reaction proceeds predominantly at the α-methyl groups of aliphatic amides with high efficiency and provides a unique tool for the direct cleavage of unactivated C(sp3)–C(sp3) bonds. The directing groups can be smoothly removed to afford the corresponding alkyl carboxylic acids.
Co-reporter:Xiao-Yu Lu, Jing-Hui Liu, Xi Lu, Zheng-Qi Zhang, Tian-Jun Gong, Bin Xiao and Yao Fu
Chemical Communications 2016 - vol. 52(Issue 30) pp:NaN5327-5327
Publication Date(Web):2016/03/22
DOI:10.1039/C6CC00176A
A Ni-catalyzed Markovnikov hydroalkylation of alkynes with alkyl halides is described. The reaction proceeds smoothly without the use of sensitive organometallic reagents and shows good functional-group compatibility, enabling the efficient synthesis of a variety of 1,1-disubstituted olefins. It also provides a straightforward approach for the modification of complex organic molecules.
Co-reporter:Zhi-Wei Yang, Qi Zhang, Yuan-Ye Jiang, Lei Li, Bin Xiao and Yao Fu
Chemical Communications 2016 - vol. 52(Issue 40) pp:NaN6711-6711
Publication Date(Web):2016/04/27
DOI:10.1039/C6CC01732K
The transition-metal-catalyzed direct triflation of naphthyl amides and naphthyl ketones has been accomplished for the first time. Benzophenone (BP) was found to be a suitable ligand for the cross-coupling reactions. Density functional theory (DFT) calculations revealed that excessive amounts of HOTf inhibit the reductive elimination of the C–F bond to realize the unusual reductive elimination of the C–OTf bond.
Co-reporter:Xiao-Yu Lu, Chu-Ting Yang, Jing-Hui Liu, Zheng-Qi Zhang, Xi Lu, Xin Lou, Bin Xiao and Yao Fu
Chemical Communications 2015 - vol. 51(Issue 12) pp:NaN2391-2391
Publication Date(Web):2014/12/19
DOI:10.1039/C4CC09321F
A copper-catalyzed cross-coupling reaction of epoxides with arylboronates is described. This reaction is not limited to aromatic epoxides, because aliphatic epoxides are also suitable substrates. In addition, N-sulfonyl aziridines can be successfully converted into the products. This reaction provides convenient access to β-phenethyl alcohols, which are valuable synthetic intermediates.
Co-reporter:Wei Su, Tian-Jun Gong, Bin Xiao and Yao Fu
Chemical Communications 2015 - vol. 51(Issue 59) pp:NaN11851-11851
Publication Date(Web):2015/06/12
DOI:10.1039/C4CC09790D
Rh(III)-catalyzed direct vinylic C–H cyanation reaction has been developed as a practical method for the synthesis of alkenyl nitriles. N-Cyano-N-phenyl-p-methylbenzenesulfonamide (NCTS), a user-friendly cyanation reagent, was used in the transformation. Both acrylamides and ketoximes can be employed in the new C–H cyanation process.
Co-reporter:Yan-Yan Sun, Jun Yi, Xi Lu, Zhen-Qi Zhang, Bin Xiao and Yao Fu
Chemical Communications 2014 - vol. 50(Issue 75) pp:NaN11062-11062
Publication Date(Web):2014/07/31
DOI:10.1039/C4CC05376A
A copper-catalyzed Suzuki–Miyaura coupling of benzyl halides with arylboronates is described. Varieties of primary benzyl halides as well as more challenging secondary benzyl halides with β hydrogens or steric hindrance could be successfully converted into the corresponding products. Thus it provides access to diarylmethanes, diarylethanes and triarylmethanes.
Co-reporter:Zhen Yang, Yao-Bing Huang, Qing-Xiang Guo and Yao Fu
Chemical Communications 2013 - vol. 49(Issue 46) pp:NaN5330-5330
Publication Date(Web):2013/04/24
DOI:10.1039/C3CC40980E
A catalytic transfer hydrogenation process was developed for the production of γ-valerolactone (GVL) from ethyl levulinate (EL) and a H-donor at room temperature. Ethyl levulinate was almost quantitatively converted to γ-valerolactone. Further, a two step process for producing GVL from biomass derived platform molecules was also reported.
Co-reporter:Qi Zhang, Hai-Zhu Yu, Yi-Tong Li, Lei Liu, Yong Huang and Yao Fu
Dalton Transactions 2013 - vol. 42(Issue 12) pp:NaN4184-4184
Publication Date(Web):2013/02/06
DOI:10.1039/C3DT31898B
A systematic theoretical study on the Rh-catalyzed oxidative Heck-coupling of phenol carbamates with alkenes is carried out. Two possible mechanisms (i.e. arene activation-first and alkene activation-first mechanisms) are examined. As to the C–H activation step, four mechanisms including oxidative addition, electrophilic substitution, concerted metallation-deprotonation (CMD), and σ-bond metathesis are evaluated. The calculation results indicate that the arene activation-first mechanism is more favorable for the overall catalytic cycle. This mechanism involves three steps: arene C–H activation at the position ortho to the carbamate directing group affording a six-membered rhodiacycle intermediate, insertion of the alkene double bond into the Rh(III)–aryl bond, and a final β-H elimination step to release the product and re-generate the catalyst. The rate determining step of the overall catalytic cycle is the arene C–H activation step, which is found to proceed through the acetate-assisted CMD mechanism.
Co-reporter:Chen Wang, Hai-Zhu Yu, Yao Fu and Qing-Xiang Guo
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 13) pp:NaN2146-2146
Publication Date(Web):2013/01/16
DOI:10.1039/C3OB27367A
Arylboronic acids were found to be efficient catalysts for the amidation reactions between carboxylic acids and amines. Theoretical calculations have been carried out to investigate the mechanism of this catalytic process. It is found that the formation of the acyloxyboronic acid intermediates from the carboxylic acid and the arylboronic acid is kinetically facile but thermodynamically unfavorable. Removal of water (as experimentally accomplished by using molecular sieves) is therefore essential for overall transformation. Subsequently C–N bond formation between the acyloxyboronic acid intermediates and the amine occurs readily to generate the desired amide product. The cleavage of the C–O bond of the tetracoordinate acyl boronate intermediates is the rate-determining step in this process. Our analysis indicates that the mono(acyloxy)boronic acid is the key intermediate. The high catalytic activity of ortho-iodophenylboronic acid is attributed to the steric effect as well as the orbital interaction between the iodine atom and the boron atom.
Co-reporter:Qianqian Lu, Haizhu Yu and Yao Fu
Chemical Communications 2013 - vol. 49(Issue 92) pp:NaN10849-10849
Publication Date(Web):2013/09/17
DOI:10.1039/C3CC46069J
DFT calculations indicate a linear correlation between the C–H activation barrier and the C–Cu/C–H bond dissociation energy gap in Cu-promoted C–H activation of heteroarenes.
Co-reporter:Jun Xu, Yun-Long Wang, Tian-Jun Gong, Bin Xiao and Yao Fu
Chemical Communications 2014 - vol. 50(Issue 85) pp:NaN12918-12918
Publication Date(Web):2014/09/01
DOI:10.1039/C4CC05692B
A new copper-catalyzed trifluoromethylarylation reaction of alkynes has been developed. The transformation represents the first example of endo-type carbotrifluoromethylation of unsaturated carbon–carbon bonds and provides efficient access to a variety of CF3-substituted dihydronaphthalenes and chromenes.
Co-reporter:Ahmed Ebrahim-Alkhalil, Zhen-Qi Zhang, Tian-Jun Gong, Wei Su, Xiao-Yu Lu, Bin Xiao and Yao Fu
Chemical Communications 2016 - vol. 52(Issue 27) pp:NaN4893-4893
Publication Date(Web):2016/03/14
DOI:10.1039/C5CC09817C
Herein, we describe a novel copper-catalyzed epoxide opening reaction with gem-diborylmethane. Aliphatic, aromatic epoxides as well as aziridines are converted to the corresponding γ-pinacolboronate alcohols or amines in moderate to excellent yields. This new reaction provides beneficial applications for classic epoxide substrates as well as interesting gem-diborylalkane reagents.

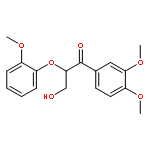
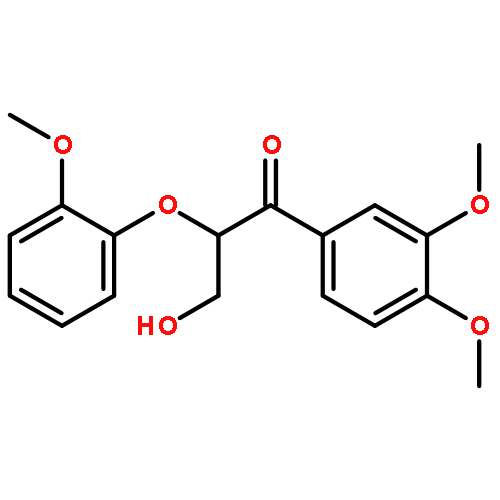



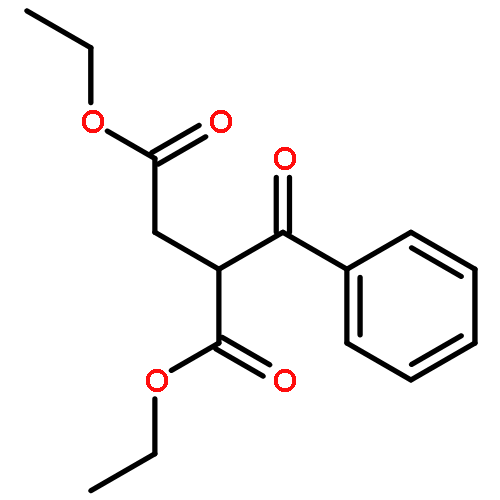
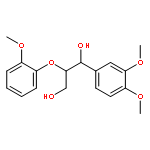









![[1,1'-Biphenyl]-2-amine, N,N-dimethyl-](http://img.cochemist.com/ccimg/6600/6590-81-4.png)
![[1,1'-Biphenyl]-2-amine, N,N-dimethyl-](http://img.cochemist.com/ccimg/6600/6590-81-4_b.png)






![L-Alanine,N-[(phenylmethoxy)carbonyl]glycyl-L-prolyl- (9CI)](http://img.cochemist.com/ccimg/5900/5891-41-8.png)
![L-Alanine,N-[(phenylmethoxy)carbonyl]glycyl-L-prolyl- (9CI)](http://img.cochemist.com/ccimg/5900/5891-41-8_b.png)





![1-methyl-4-[(2-methylprop-2-en-1-yl)oxy]benzene](http://img.cochemist.com/ccimg/5500/5471-86-3.png)
![1-methyl-4-[(2-methylprop-2-en-1-yl)oxy]benzene](http://img.cochemist.com/ccimg/5500/5471-86-3_b.png)


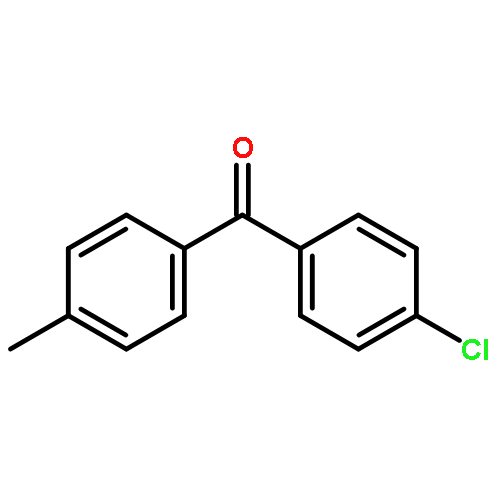






![(2R,3R,3aR,6R,6aR)-hexahydro-2,6-epoxyfuro[3,2-b]furan-3-ol](http://img.cochemist.com/ccimg/4500/4451-30-3.png)
![(2R,3R,3aR,6R,6aR)-hexahydro-2,6-epoxyfuro[3,2-b]furan-3-ol](http://img.cochemist.com/ccimg/4500/4451-30-3_b.png)








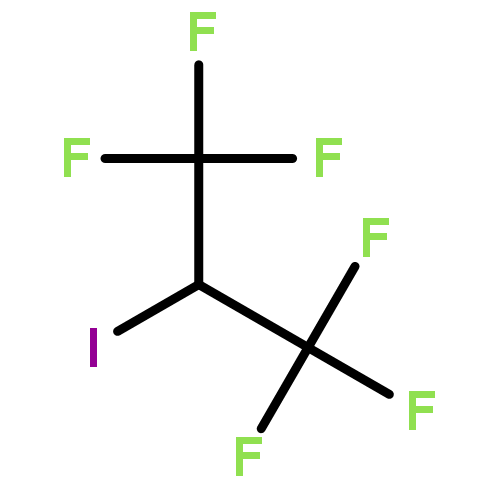









![2-(4-(Benzo[d][1,3]dioxol-5-ylmethyl)piperazin-1-yl)pyrimidine](http://img.cochemist.com/ccimg/3700/3605-01-4.png)
![2-(4-(Benzo[d][1,3]dioxol-5-ylmethyl)piperazin-1-yl)pyrimidine](http://img.cochemist.com/ccimg/3700/3605-01-4_b.png)


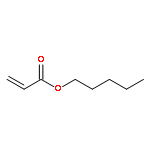
![Acetic acid, [(2,6-dimethylphenyl)amino]oxo-, ethyl ester](http://img.cochemist.com/ccimg/3000/2903-46-0.png)
![Acetic acid, [(2,6-dimethylphenyl)amino]oxo-, ethyl ester](http://img.cochemist.com/ccimg/3000/2903-46-0_b.png)


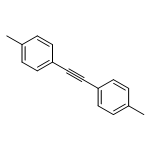












![Oxirane,2-[(4-bromophenoxy)methyl]-](http://img.cochemist.com/ccimg/2300/2212-06-8.png)
![Oxirane,2-[(4-bromophenoxy)methyl]-](http://img.cochemist.com/ccimg/2300/2212-06-8_b.png)




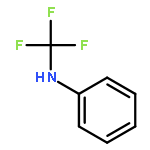
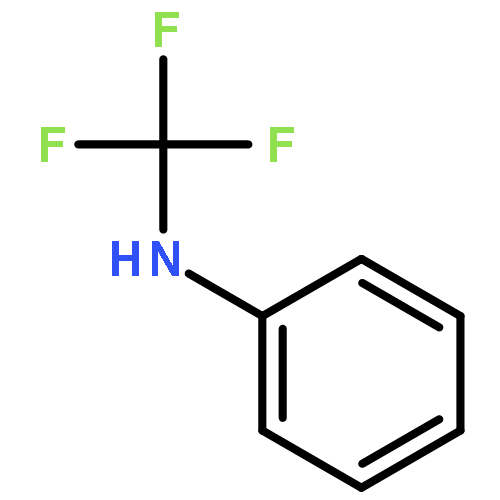





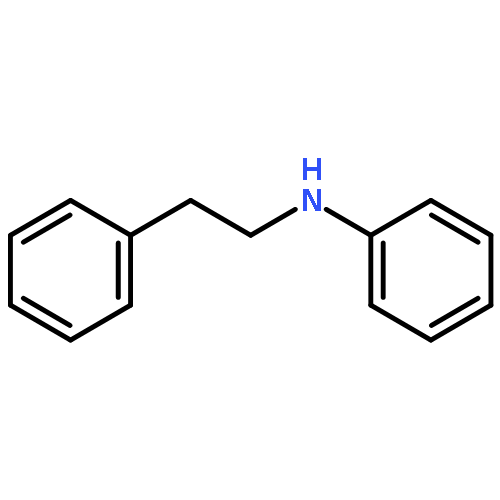






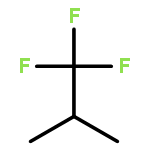
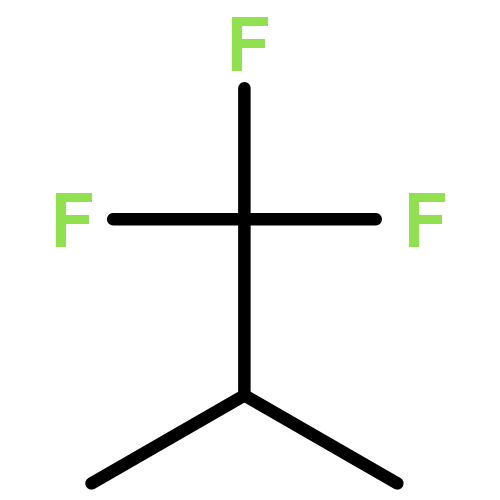




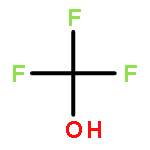
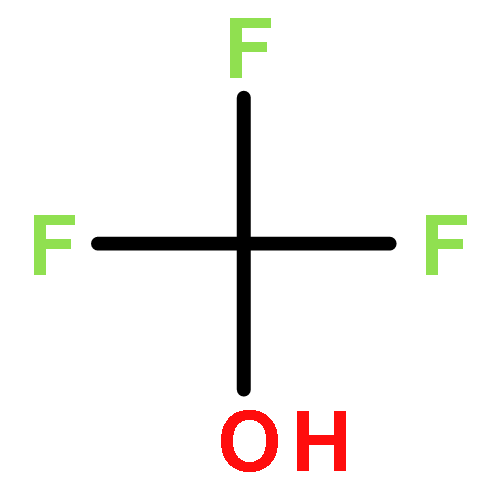


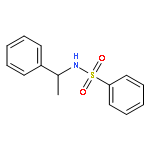
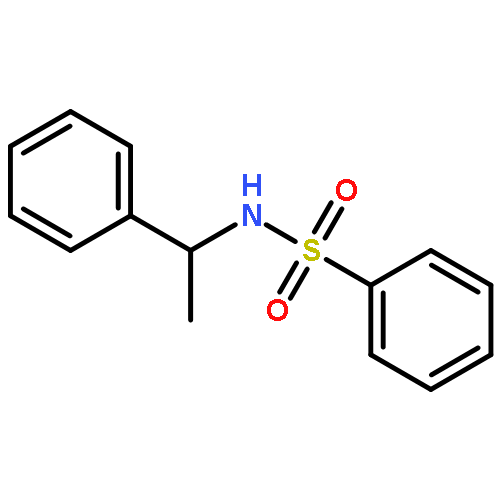


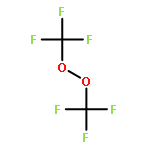


![OXIRANE, [(4-IODOPHENOXY)METHYL]-](http://img.cochemist.com/ccimg/900/828-25-1.png)
![OXIRANE, [(4-IODOPHENOXY)METHYL]-](http://img.cochemist.com/ccimg/900/828-25-1_b.png)
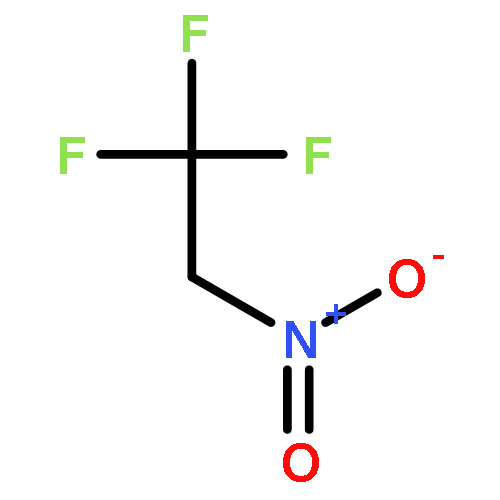











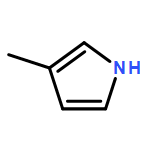





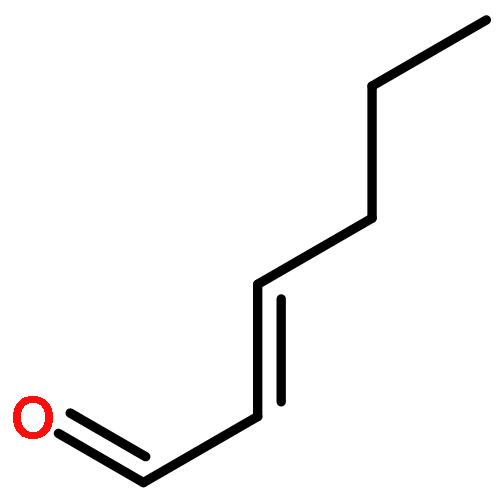


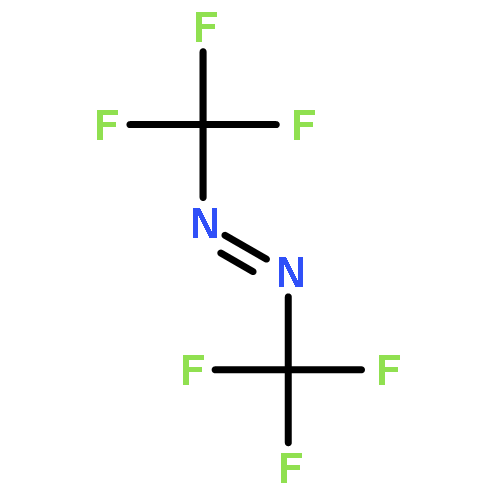
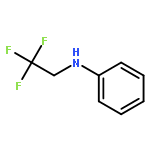
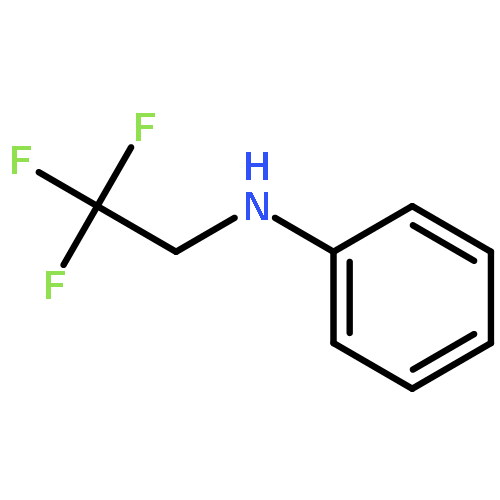
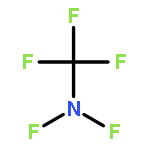
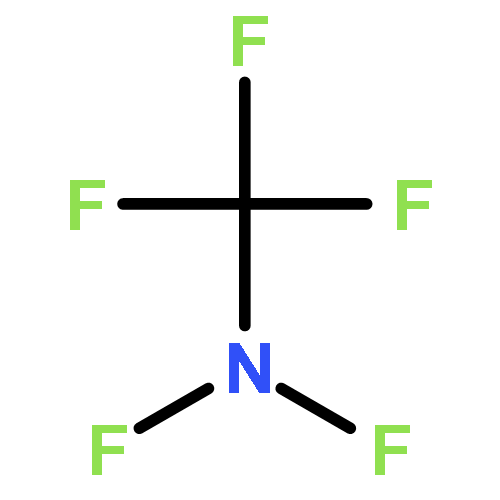
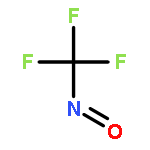
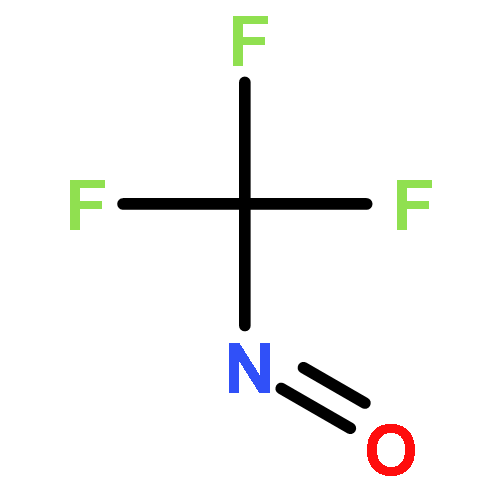
![[5-(HYDROXYMETHYL)OXOLAN-2-YL]METHANOL](http://img.cochemist.com/ccimg/200/104-80-3.png)
![[5-(HYDROXYMETHYL)OXOLAN-2-YL]METHANOL](http://img.cochemist.com/ccimg/200/104-80-3_b.png)
![Pyrrolidine, 3-iodo-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/919300/919284-59-6.png)
![Pyrrolidine, 3-iodo-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/919300/919284-59-6_b.png)




![Aziridine, 2-butyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/117000/116905-61-4.png)
![Aziridine, 2-butyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/117000/116905-61-4_b.png)
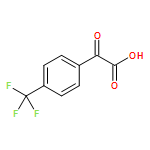
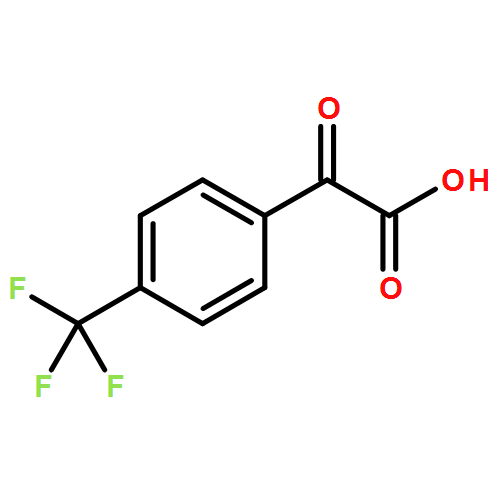
![Oxirane, [(2-bromo-4-chlorophenoxy)methyl]-](http://img.cochemist.com/ccimg/68300/68224-01-1.png)
![Oxirane, [(2-bromo-4-chlorophenoxy)methyl]-](http://img.cochemist.com/ccimg/68300/68224-01-1_b.png)

![Benzene, [(5-hexenyloxy)methyl]-](http://img.cochemist.com/ccimg/59200/59137-50-7.png)
![Benzene, [(5-hexenyloxy)methyl]-](http://img.cochemist.com/ccimg/59200/59137-50-7_b.png)
![Oxirane,2-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/51500/51410-45-8.png)
![Oxirane,2-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/51500/51410-45-8_b.png)






![Benzo[b]thiophene-2-acetic acid, α-oxo-](/data/chemimg/1762800/18021-47-1.png)
![Benzo[b]thiophene-2-acetic acid, α-oxo-](/data/chemimg/1762800/18021-47-1_b.png)
![2-[(2-fluorophenoxy)methyl]oxirane](http://img.cochemist.com/ccimg/15700/15620-80-1.png)
![2-[(2-fluorophenoxy)methyl]oxirane](http://img.cochemist.com/ccimg/15700/15620-80-1_b.png)




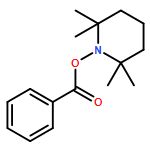
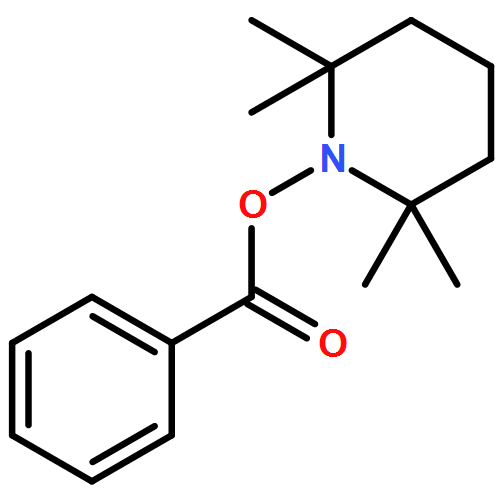
![Oxirane,2-[(2-naphthalenyloxy)methyl]-](http://img.cochemist.com/ccimg/5300/5234-06-0.png)
![Oxirane,2-[(2-naphthalenyloxy)methyl]-](http://img.cochemist.com/ccimg/5300/5234-06-0_b.png)


![Oxirane, [(3-methoxyphenoxy)methyl]-](http://img.cochemist.com/ccimg/2300/2210-75-5.png)
![Oxirane, [(3-methoxyphenoxy)methyl]-](http://img.cochemist.com/ccimg/2300/2210-75-5_b.png)
![2-{[3-(trifluoromethyl)phenoxy]methyl}oxirane](http://img.cochemist.com/ccimg/600/585-45-5.png)
![2-{[3-(trifluoromethyl)phenoxy]methyl}oxirane](http://img.cochemist.com/ccimg/600/585-45-5_b.png)


![Benzoic acid, 4-[(phenylmethyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/61500/61439-53-0.png)
![Benzoic acid, 4-[(phenylmethyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/61500/61439-53-0_b.png)
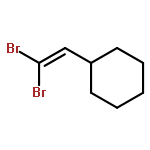









![3-Pyridinecarboxamide, 1,4-dihydro-1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/56200/56133-30-3.png)
![3-Pyridinecarboxamide, 1,4-dihydro-1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/56200/56133-30-3_b.png)
![3-Pyridinecarboxamide, 1,4-dihydro-1-[(4-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/56200/56133-29-0.png)
![3-Pyridinecarboxamide, 1,4-dihydro-1-[(4-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/56200/56133-29-0_b.png)
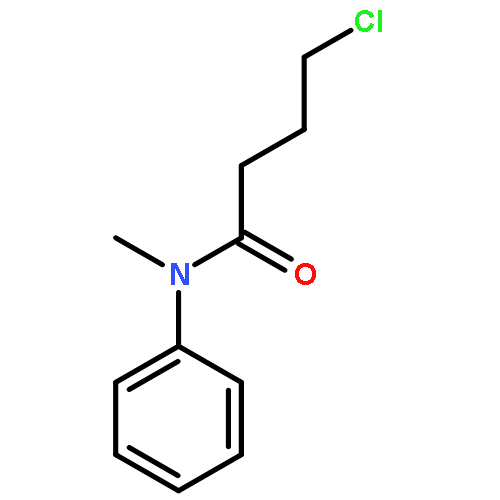
![1-[4-(2-BROMOETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/55400/55368-24-6.png)
![1-[4-(2-BROMOETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/55400/55368-24-6_b.png)


![PHOSPHINE, DIPHENYL[3-(TRIMETHOXYSILYL)PROPYL]-](http://img.cochemist.com/ccimg/52100/52090-19-4.png)
![PHOSPHINE, DIPHENYL[3-(TRIMETHOXYSILYL)PROPYL]-](http://img.cochemist.com/ccimg/52100/52090-19-4_b.png)



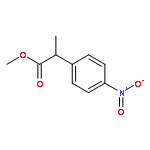
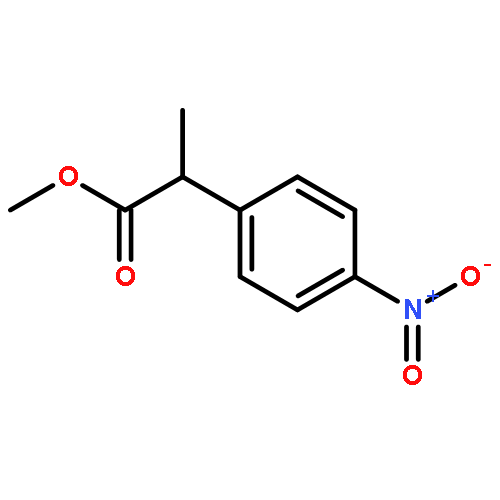


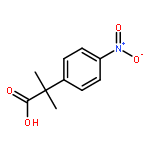
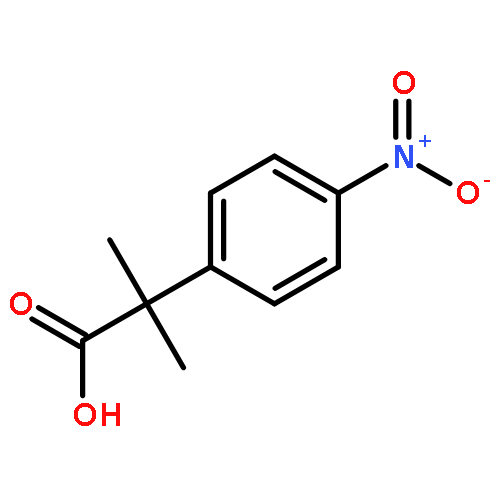
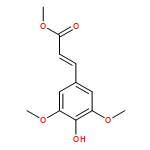
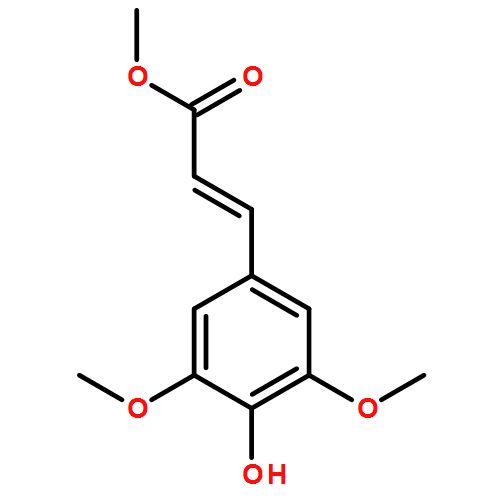
![2-[(2-METHYLPROPAN-2-YL)OXY]ETHYL METHANESULFONATE](http://img.cochemist.com/ccimg/41100/41018-73-9.png)
![2-[(2-METHYLPROPAN-2-YL)OXY]ETHYL METHANESULFONATE](http://img.cochemist.com/ccimg/41100/41018-73-9_b.png)
![N-[4-(3-BROMOPROPOXY)PHENYL]ACETAMIDE](http://img.cochemist.com/ccimg/41000/40981-73-5.png)
![N-[4-(3-BROMOPROPOXY)PHENYL]ACETAMIDE](http://img.cochemist.com/ccimg/41000/40981-73-5_b.png)




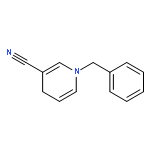
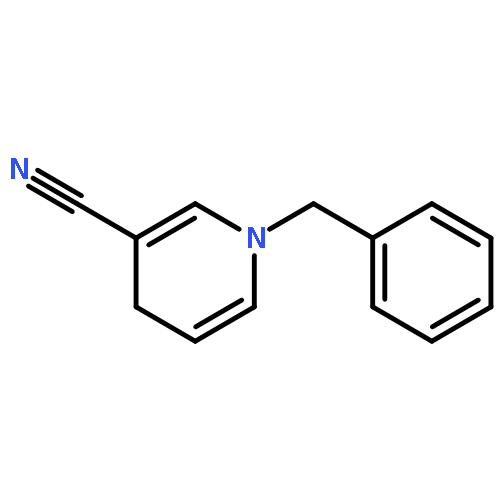
![1-[(6-bromohexyl)oxy]-4-chlorobenzene](http://img.cochemist.com/ccimg/37200/37136-99-5.png)
![1-[(6-bromohexyl)oxy]-4-chlorobenzene](http://img.cochemist.com/ccimg/37200/37136-99-5_b.png)




![Benzene, 1-chloro-4-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/30300/30203-94-2.png)
![Benzene, 1-chloro-4-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/30300/30203-94-2_b.png)
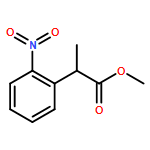
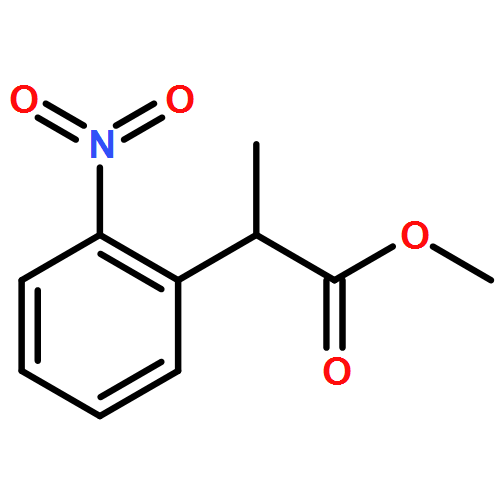

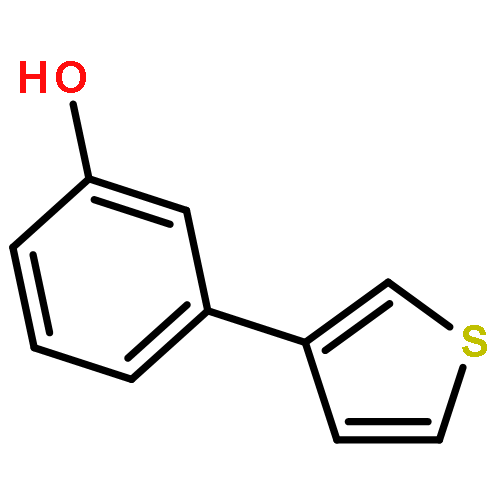
![Bicyclo[2.2.1]heptane,2-bromo-](http://img.cochemist.com/ccimg/29400/29342-65-2.png)
![Bicyclo[2.2.1]heptane,2-bromo-](http://img.cochemist.com/ccimg/29400/29342-65-2_b.png)




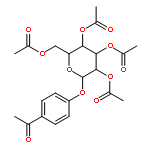









![Benzene, 1-chloro-4-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/22900/22865-55-0.png)
![Benzene, 1-chloro-4-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/22900/22865-55-0_b.png)




![Benzene,1-[(6-bromohexyl)oxy]-4-methoxy-](http://img.cochemist.com/ccimg/20800/20744-11-0.png)
![Benzene,1-[(6-bromohexyl)oxy]-4-methoxy-](http://img.cochemist.com/ccimg/20800/20744-11-0_b.png)







![3-Pyridinecarboxamide, 1-[(4-chlorophenyl)methyl]-1,4-dihydro-](http://img.cochemist.com/ccimg/19400/19350-55-1.png)
![3-Pyridinecarboxamide, 1-[(4-chlorophenyl)methyl]-1,4-dihydro-](http://img.cochemist.com/ccimg/19400/19350-55-1_b.png)
![Ethanone, 1-[4-(oxiranylmethoxy)phenyl]-](http://img.cochemist.com/ccimg/19200/19152-55-7.png)
![Ethanone, 1-[4-(oxiranylmethoxy)phenyl]-](http://img.cochemist.com/ccimg/19200/19152-55-7_b.png)
![benzo[h]quinoline 1-oxide](http://img.cochemist.com/ccimg/17200/17104-70-0.png)
![benzo[h]quinoline 1-oxide](http://img.cochemist.com/ccimg/17200/17104-70-0_b.png)
![1H-Imidazole, 4,5-dihydro-1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/16100/16054-55-0.png)
![1H-Imidazole, 4,5-dihydro-1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/16100/16054-55-0_b.png)

















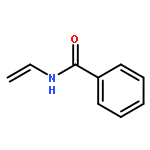

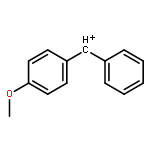
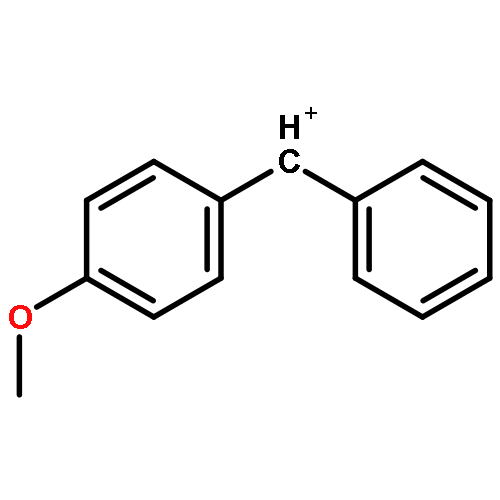
![1-(4'-Methoxy-[1,1'-biphenyl]-4-yl)ethanone](http://img.cochemist.com/ccimg/13100/13021-18-6.png)
![1-(4'-Methoxy-[1,1'-biphenyl]-4-yl)ethanone](http://img.cochemist.com/ccimg/13100/13021-18-6_b.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)




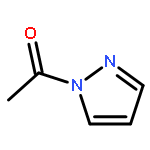
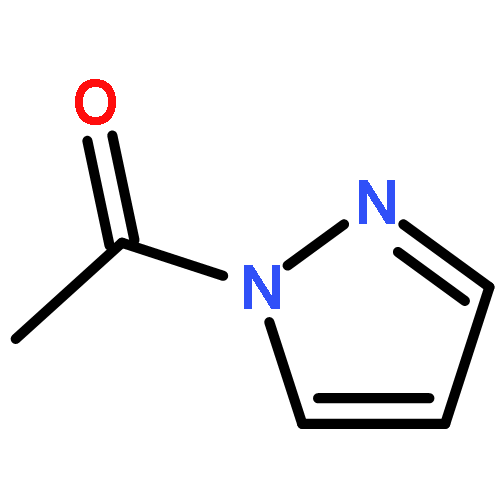


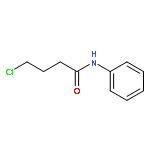

































![6-Chloro-2-methylbenzo[d]thiazole](http://img.cochemist.com/ccimg/4200/4146-24-1.png)
![6-Chloro-2-methylbenzo[d]thiazole](http://img.cochemist.com/ccimg/4200/4146-24-1_b.png)








![(NE)-N-[(2-METHYLPHENYL)METHYLIDENE]HYDROXYLAMINE](http://img.cochemist.com/ccimg/3400/3376-32-7.png)
![(NE)-N-[(2-METHYLPHENYL)METHYLIDENE]HYDROXYLAMINE](http://img.cochemist.com/ccimg/3400/3376-32-7_b.png)




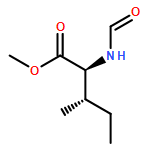
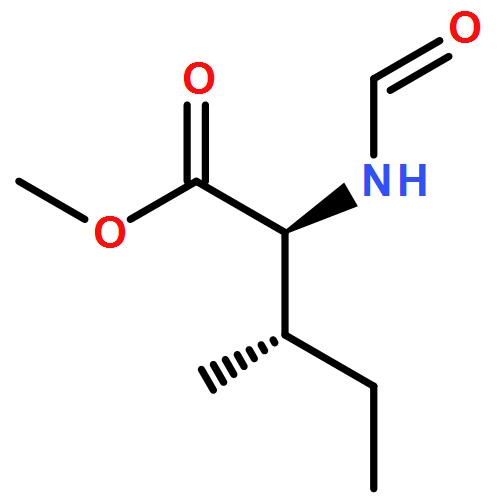
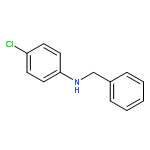







![Acetamide,N-[3-(trifluoromethoxy)phenyl]-](http://img.cochemist.com/ccimg/2000/1956-85-0.png)
![Acetamide,N-[3-(trifluoromethoxy)phenyl]-](http://img.cochemist.com/ccimg/2000/1956-85-0_b.png)














![Tungstate(3-),tetracosa-m-oxododecaoxo[m12-[phosphato(3-)-kO:kO:kO:kO':kO':kO':kO'':kO'':kO'':kO''':kO''':kO''']]dodeca-,hydrogen (1:3)](http://img.cochemist.com/ccimg/1400/1343-93-7.png)
![Tungstate(3-),tetracosa-m-oxododecaoxo[m12-[phosphato(3-)-kO:kO:kO:kO':kO':kO':kO'':kO'':kO'':kO''':kO''':kO''']]dodeca-,hydrogen (1:3)](http://img.cochemist.com/ccimg/1400/1343-93-7_b.png)
![1-[(e)-2-phenylvinyl]-4-(trifluoromethyl)benzene](http://img.cochemist.com/ccimg/1200/1149-56-0.png)
![1-[(e)-2-phenylvinyl]-4-(trifluoromethyl)benzene](http://img.cochemist.com/ccimg/1200/1149-56-0_b.png)






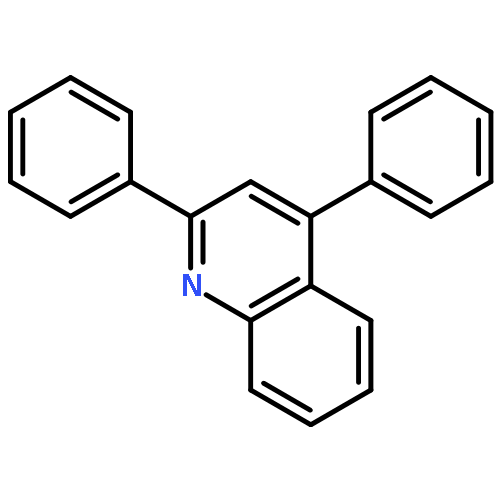
















![5-(tert-Butyl)-[1,1'-biphenyl]-2-ol](http://img.cochemist.com/ccimg/600/577-92-4.png)
![5-(tert-Butyl)-[1,1'-biphenyl]-2-ol](http://img.cochemist.com/ccimg/600/577-92-4_b.png)




![N-ETHYL-N-[(7-HYDROXY-4-OXO-2-PHENYL-4H-CHROMEN-8-YL)METHYL]ETHAN<WBR />AMINIUM CHLORIDE](http://img.cochemist.com/ccimg/400/351-70-2.png)
![N-ETHYL-N-[(7-HYDROXY-4-OXO-2-PHENYL-4H-CHROMEN-8-YL)METHYL]ETHAN<WBR />AMINIUM CHLORIDE](http://img.cochemist.com/ccimg/400/351-70-2_b.png)

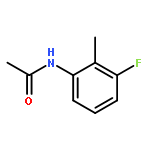
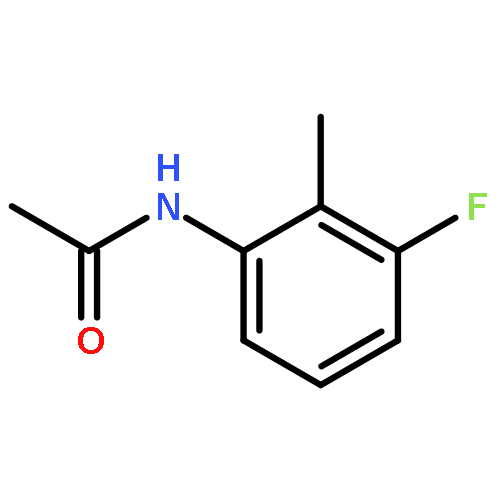
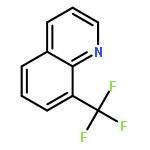
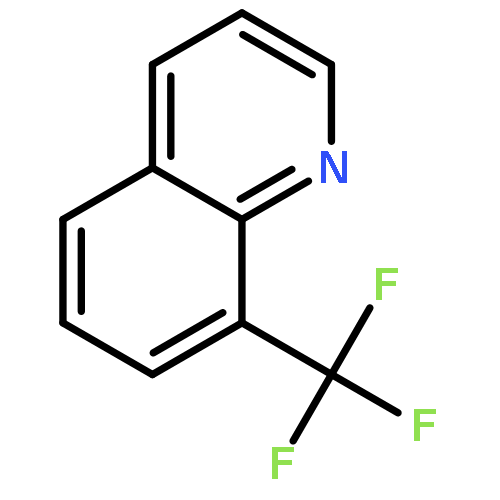











![Piperidine, 1-[(4-methylphenyl)sulfonyl]-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-](/data/chemimg/182500/1382358-37-3.png)
![Piperidine, 1-[(4-methylphenyl)sulfonyl]-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-](/data/chemimg/182500/1382358-37-3_b.png)

























{kind=link}