Co-reporter:Zachary L. Niemeyer, Suresh Pindi, Dimitri A. Khrakovsky, Christian N. Kuzniewski, Cynthia M. Hong, Leo A. Joyce, Matthew S. Sigman, and F. Dean Toste
Journal of the American Chemical Society September 20, 2017 Volume 139(Issue 37) pp:12943-12943
Publication Date(Web):September 8, 2017
DOI:10.1021/jacs.7b08791
Computed descriptors for acyclic diaminocarbene ligands are developed in the context of a gold catalyzed enantioselective tandem [3,3]-sigmatropic rearrangement-[2+2]-cyclization. Surrogate structures enable the rapid identification of parameters that reveal mechanistic characteristics. The observed selectivity trends are validated in a robust multivariate analysis facilitating the development of a highly enantioselective process.
Co-reporter:Manuel Orlandi, Margaret J. Hilton, Eiji Yamamoto, F. Dean Toste, and Matthew S. Sigman
Journal of the American Chemical Society September 13, 2017 Volume 139(Issue 36) pp:12688-12688
Publication Date(Web):August 11, 2017
DOI:10.1021/jacs.7b06917
A mechanistic study of the Pd-catalyzed enantioselective 1,1-diarylation of benzyl acrylates that is facilitated by a chiral anion phase transfer (CAPT) process is presented. Kinetic analysis, labeling, competition, and nonlinear effect experiments confirm the hypothesized general mechanism and reveal the role of the phosphate counterion in the CAPT catalysis. The phosphate was found to be involved in the phase transfer step and in the stereoinduction process, as expected, but also in the unproductive reaction that provides the traditional Heck byproduct. Multivariate correlations revealed the CAPT catalyst’s structural features, affecting the production of this undesired byproduct, as well as weak interactions responsible for enantioselectivity. Such putative interactions include π-stacking and a CH···O electrostatic attraction between the substrate benzyl moiety and the phosphate. Analysis of the computed density functional theory transition structures for the stereodetermining step of the reaction supports the multivariate model obtained. The presented work provides the first comprehensive study of the combined use of CAPT and transition metal catalysis, setting the foundation for future applications.
Co-reporter:Manuel Orlandi, Jaime A. S. Coelho, Margaret J. Hilton, F. Dean Toste, and Matthew S. Sigman
Journal of the American Chemical Society May 24, 2017 Volume 139(Issue 20) pp:6803-6803
Publication Date(Web):May 5, 2017
DOI:10.1021/jacs.7b02311
The use of computed interaction energies and distances as parameters in multivariate correlations is introduced for postulating non-covalent interactions. This new class of descriptors affords multivariate correlations for two diverse catalytic systems with unique non-covalent interactions at the heart of each process. The presented methodology is validated by directly connecting the non-covalent interactions defined through empirical data set analyses to the computationally derived transition states.
Co-reporter:Jing-Yao Guo, Yury Minko, Celine B. Santiago, and Matthew S. Sigman
ACS Catalysis June 2, 2017 Volume 7(Issue 6) pp:4144-4144
Publication Date(Web):May 5, 2017
DOI:10.1021/acscatal.7b00739
The applicability of computational descriptors extracted from metal pyridine-oxazoline complexes to relate both site and enantioselectivity to structural diversity was investigated. A group of computationally derived features (e.g., metal NBO charges, steric descriptors, torsion angles) were acquired for a library of pyridine-oxazoline ligands. Correlation studies were employed to examine steric/electronic features described by each descriptor, followed by application of the said descriptors in modeling the results of two reaction types, the site-selective redox-relay Heck reaction and the enantioselective Carroll rearrangement, affording simple, well-validated models. Through experimental validation and extrapolation, parameters derived from ground state metal complexes were found to be advantageous over those from the free ligand.Keywords: ground state metal complex; parameterization; rational ligand design; selective oxidation;
Co-reporter:Katrina H. Jensen, Jonathan D. Webb, and Matthew S. Sigman
Journal of the American Chemical Society December 15, 2010 Volume 132(Issue 49) pp:17471-17482
Publication Date(Web):November 17, 2010
DOI:10.1021/ja108106h
The mechanism of an enantioselective palladium-catalyzed alkene difunctionalization reaction has been investigated. Kinetic analysis provides evidence of turnover-limiting attack of a proposed quinone methide intermediate with MeOH and suggests that copper is involved in productive product formation, not just catalyst turnover. Through examination of substrate electronic effects, a Jaffé relationship was observed correlating rate to electronic perturbation at two positions of the substrate. Ligand effects were evaluated to provide evidence of rapid ligand exchange between palladium and copper as well as a correlation between ligand electronic nature and enantioselectivity.
Co-reporter:Mitchell H. Keylor, Zachary L. Niemeyer, Matthew S. Sigman, and Kian L. Tan
Journal of the American Chemical Society August 9, 2017 Volume 139(Issue 31) pp:10613-10613
Publication Date(Web):July 17, 2017
DOI:10.1021/jacs.7b05409
A new catalyst system capable of selective chloride functionalization in the Pd-catalyzed amination of 3,2- and 5,2- Br/Cl-pyridines is reported. A reaction optimization strategy employing ligand parametrization led to the identification of 1,1′-bis[bis(dimethylamino)phosphino]ferrocene “DMAPF”, a readily available yet previously unutilized diphosphine, as a uniquely effective ligand for this transformation.
Co-reporter:Brian P. Woods, Manuel Orlandi, Chung-Yang Huang, Matthew S. Sigman, and Abigail G. Doyle
Journal of the American Chemical Society April 26, 2017 Volume 139(Issue 16) pp:5688-5688
Publication Date(Web):April 13, 2017
DOI:10.1021/jacs.7b03448
A Ni-catalyzed reductive cross-coupling of styrenyl aziridines with aryl iodides is reported. This reaction proceeds by a stereoconvergent mechanism and is thus amenable to asymmetric catalysis using a chiral bioxazoline ligand for Ni. The process allows facile access to highly enantioenriched 2-arylphenethylamines from racemic aziridines. Multivariate analysis revealed that ligand polarizability, among other features, influences the observed enantioselectivity, shedding light on the success of this emerging ligand class for enantioselective Ni catalysis.
Co-reporter:Dr. Manuel Orli; F. Dean Toste; Matthew S. Sigman
Angewandte Chemie International Edition 2017 Volume 56(Issue 45) pp:14080-14084
Publication Date(Web):2017/11/06
DOI:10.1002/anie.201707644
AbstractThe study of the oxidative amination of tetrahydroisoquinolines under chiral-anion phase-transfer (CAPT) catalysis by multidimensional correlation analysis (MCA) is revisited. The parameterization of the transition states (TSs) for the uncatalyzed reaction, the introduction of conformational descriptors, and the use of computed interaction energies and distances as parameters allowed access to a considerably simplified mathematical correlation of substrate and catalyst structure to enantioselectivity. The equation obtained is suggestive of key interactions occurring at the TS. Specifically, the CAPT catalyst is proposed to coordinate the intermediate iminium cation by P=O⋅⋅⋅H−O hydrogen-bonding and N⋅⋅⋅H−C electrostatic interactions. The conformational freedom of the benzyl substituent of the substrate was also found to be important in providing an efficient mode of molecular recognition.
Co-reporter:Dr. Zhi-Min Chen;Christine S. Nervig;Dr. Ryan J. DeLuca; Matthew S. Sigman
Angewandte Chemie International Edition 2017 Volume 56(Issue 23) pp:6651-6654
Publication Date(Web):2017/06/01
DOI:10.1002/anie.201703089
AbstractAn enantioselective redox-relay Heck alkynylation of di- and trisubstituted alkenols to construct propargylic stereocenters is disclosed using a new pyridine oxazoline ligand. This strategy allows direct access to chiral β-alkynyl carbonyl compounds employing allylic alcohol substrates in contrast to more traditional conjugate addition methods.
Co-reporter:Dr. Zhi-Min Chen;Christine S. Nervig;Dr. Ryan J. DeLuca; Matthew S. Sigman
Angewandte Chemie 2017 Volume 129(Issue 23) pp:6751-6754
Publication Date(Web):2017/06/01
DOI:10.1002/ange.201703089
AbstractAn enantioselective redox-relay Heck alkynylation of di- and trisubstituted alkenols to construct propargylic stereocenters is disclosed using a new pyridine oxazoline ligand. This strategy allows direct access to chiral β-alkynyl carbonyl compounds employing allylic alcohol substrates in contrast to more traditional conjugate addition methods.
Co-reporter:Matthew S. Sigman, Kaid C. Harper, Elizabeth N. Bess, and Anat Milo
Accounts of Chemical Research 2016 Volume 49(Issue 6) pp:1292
Publication Date(Web):May 24, 2016
DOI:10.1021/acs.accounts.6b00194
In most modern organic chemistry reports, including many of ours, reaction optimization schemes are typically presented to showcase how reaction conditions have been tailored to augment the reaction’s yield and selectivity. In asymmetric catalysis, this often involves evaluation of catalyst, solvent, reagent, and, sometimes, substrate features. Such an article will then detail the process’s scope, which mainly focuses on its successes and briefly outlines the “limitations”. These limitations or poorer-performing substrates are occasionally the result of obvious, significant changes to structure (e.g., a Lewis basic group binds to a catalyst), but frequently, a satisfying explanation for inferior performance is not clear. This is one of several reasons such results are not often reported. These apparent outliers are also commonplace in the evaluation of catalyst structure, although most of this information is placed in the Supporting Information.These practices are unfortunate because results that appear at first glance to be peculiar or poor are considerably more interesting than ones that follow obvious or intuitive trends. In other words, all of the data from an optimization campaign contain relevant information about the reaction under study, and the “outliers” may be the most revealing.Realizing the power of outliers as an entry point to entirely new reaction development is not unusual. Nevertheless, the concept that no data should be wasted when considering the underlying phenomena controlling the observations of a given reaction is at the heart of the strategy we describe in this Account. The idea that one can concurrently optimize a reaction to expose the structural features that control its outcomes would represent a transformative addition to the arsenal of catalyst development and, ultimately, de novo design.Herein we outline the development of a recently initiated program in our lab that unites optimization with mechanistic interrogation by correlating reaction outputs (e.g., electrochemical potential or enantio-, site, or chemoselectivity) with structural descriptors of the molecules involved. The ever-evolving inspiration for this program is rooted in outliers of classical linear free energy relationships. These outliers encouraged us to ask questions about the parameters themselves, suggest potential interactions at the source of the observed effects, and, of particular applicability, identify more sophisticated physical organic descriptors. Throughout this program, we have integrated techniques from disparate fields, including synthetic methodology development, mechanistic investigations, statistics, computational chemistry, and data science. The implementation of many of these strategies is described, and the resulting tools are illustrated in a wide range of case studies, which include data sets with simultaneous and multifaceted changes to the reagent, substrate, and catalyst structures. This tactic constitutes a modern approach to physical organic chemistry wherein no data are wasted and mechanistic hypotheses regarding sophisticated processes can be developed and probed.
Co-reporter:Zhi-Min Chen, Margaret J. Hilton, and Matthew S. Sigman
Journal of the American Chemical Society 2016 Volume 138(Issue 36) pp:11461-11464
Publication Date(Web):August 29, 2016
DOI:10.1021/jacs.6b06994
An enantioselective redox-relay oxidative Heck arylation of 1,1-disubstituted alkenes to construct β-stereocenters was developed using a new pyridyl-oxazoline ligand. Various 1,2-diaryl carbonyl compounds were readily obtained in moderate yield and good to excellent enantioselectivity. Additionally, analysis of the reaction outcomes using multidimensional correlations revealed that enantioselectivity is tied to specific electronic features of the 1,1-disubstituted alkenol and the extent of polarizability of the ligand.
Co-reporter:Celine B. Santiago, Anat Milo, and Matthew S. Sigman
Journal of the American Chemical Society 2016 Volume 138(Issue 40) pp:13424-13430
Publication Date(Web):September 21, 2016
DOI:10.1021/jacs.6b08799
The effects of aryl ring ortho-, meta-, and para-substitution on site selectivity and enantioselectivity were investigated in the following reactions: (1) enantioselective Pd-catalyzed redox-relay Heck reaction of arylboronic acids, (2) Pd-catalyzed β-aryl elimination of triarylmethanols, and (3) benzoylformate decarboxylase-catalyzed enantioselective benzoin condensation of benzaldehydes. Through these studies, it is demonstrated that the electronic and steric effects of various substituents on selectivities obtained in these reactions can be described by NBO charges, the IR carbonyl stretching frequency, and Sterimol values of various substituted benzoic acids. An extended compilation of NBO charges and IR carbonyl stretching frequencies of various substituted benzoic acids was used as an alternative to Hammett values. These parameters provide a correlative tool that allows for the analysis of a much greater range of substituent effects because they can also account for proximal and remote steric effects.
Co-reporter:Harshkumar H. Patel and Matthew S. Sigman
Journal of the American Chemical Society 2016 Volume 138(Issue 43) pp:14226-14229
Publication Date(Web):October 21, 2016
DOI:10.1021/jacs.6b09649
In this report, we describe the generation of remote allylic quaternary stereocenters β, γ, and δ relative to a carbonyl in high enantioselectivity. We utilize a redox-relay Heck reaction between alkenyl triflates and acyclic trisubstituted alkenols of varying chain-lengths. A wide array of terminal (E)-alkenyl triflates are suitable for this process. The utility of this functionalization is validated further by conversion of the products, via simple organic processes to access remotely functionalized chiral tertiary acid, amine, and alcohol products.
Co-reporter:Nicholas J. Race, Cristiane S. Schwalm, Takayuki Nakamuro, and Matthew S. Sigman
Journal of the American Chemical Society 2016 Volume 138(Issue 49) pp:15881-15884
Publication Date(Web):November 23, 2016
DOI:10.1021/jacs.6b11486
An enantioselective intermolecular coupling of oxygen nucleophiles and allylic alcohols to give β-aryloxycarbonyl compounds is disclosed using a chiral pyridine oxazoline-ligated palladium catalyst under mild conditions. As opposed to the formation of traditional Wacker-type products, enantioselective migratory insertion is followed by β-hydride elimination toward the adjacent alcohol. Deuterium labeling experiments suggest a syn-migratory insertion of the alkene into the Pd–O bond. A broad scope of phenols, various allylic alcohols, and an alkyl hydroperoxide are viable coupling partners in this process.
Co-reporter:Eiji Yamamoto, Margaret J. Hilton, Manuel Orlandi, Vaneet Saini, F. Dean Toste, and Matthew S. Sigman
Journal of the American Chemical Society 2016 Volume 138(Issue 49) pp:15877-15880
Publication Date(Web):November 22, 2016
DOI:10.1021/jacs.6b11367
Enantioselective 1,1-diarylation of terminal alkenes enabled by the combination of Pd catalysis with a chiral anion phase transfer (CAPT) strategy is reported herein. The reaction of substituted benzyl acrylates with aryldiazonium salts and arylboronic acids gave the corresponding 3,3-diarylpropanoates in moderate to good yields with high enantioselectivies (up to 98:2 er). Substituents on the benzyl acrylate and CAPT catalyst significantly affect the enantioselectivity, and multidimensional parametrization identified correlations suggesting structural origins for the high stereocontrol.
Co-reporter:Andrew J. Neel; Anat Milo; Matthew S. Sigman;F. Dean Toste
Journal of the American Chemical Society 2016 Volume 138(Issue 11) pp:3863-3875
Publication Date(Web):March 11, 2016
DOI:10.1021/jacs.6b00356
Enantioselectivity values represent relative rate measurements that are sensitive to the structural features of the substrates and catalysts interacting to produce them. Therefore, well-designed enantioselectivity data sets are information rich and can provide key insights regarding specific molecular interactions. However, if the mechanism for enantioselection varies throughout a data set, these values cannot be easily compared. This premise, which is the crux of free energy relationships, exposes a challenging issue of identifying mechanistic breaks within multivariate correlations. Herein, we describe an approach to addressing this problem in the context of a chiral phosphoric acid catalyzed fluorination of allylic alcohols using aryl boronic acids as transient directing groups. By designing a data set in which both the phosphoric and boronic acid structures were systematically varied, key enantioselectivity outliers were identified and analyzed. A mechanistic study was executed to reveal the structural origins of these outliers, which was consistent with the presence of several mechanistic regimes within the data set. While 2- and 4-substituted aryl boronic acids favored the (R)-enantiomer with most of the studied catalysts, meta-alkoxy substituted aryl boronic acids resulted in the (S)-enantiomer when used in combination with certain (R)-phosphoric acids. We propose that this selectivity reversal is the result of a lone pair-π interaction between the substrate ligated boronic acid and the phosphate. On the basis of this proposal, a catalyst system was identified, capable of producing either enantiomer in high enantioselectivity (77% (R)-2 to 92% (S)-2) using the same chiral catalyst by subtly changing the structure of the achiral boronic acid.
Co-reporter:Matthew S. McCammant, Takashi Shigeta, and Matthew S. Sigman
Organic Letters 2016 Volume 18(Issue 8) pp:1792-1795
Publication Date(Web):March 28, 2016
DOI:10.1021/acs.orglett.6b00517
A Pd-catalyzed 1,3-difunctionalization of terminal alkenes using 1,1-disubstituted alkenyl nonaflates and arylboronic acid coupling partners is reported. This transformation affords allylic arene products that are difficult to selectively access using traditional Heck cross-coupling methodologies. The evaluation of seldom employed 1,1-disubstituted alkenyl nonaflate coupling partners led to the elucidation of subtle mechanistic features of π-allyl stabilized Pd-intermediates. Good stereo- and regioselectivity for the formation of 1,3-addition products can be accessed through a minimization of steric interactions that emanate from alkenyl nonaflate substitution.
Co-reporter:Harshkumar H. Patel
Journal of the American Chemical Society 2015 Volume 137(Issue 10) pp:3462-3465
Publication Date(Web):March 4, 2015
DOI:10.1021/ja5130836
We report a highly enantioselective intermolecular Heck reaction of alkenyl triflates and acyclic primary or racemic secondary alkenols. The mild reaction conditions permit installation of a wide range of alkenyl groups at positions β, γ, or δ to a carbonyl group in high enantioselectivity. The success of this reaction is attributed to the use of electron-withdrawing alkenyl triflates, which offer selective β-hydride elimination followed by migration of the catalyst through the alkyl chain to give the alkenylated carbonyl products. The synthetic utility of the process is demonstrated by a two-step modification of a reaction product to yield a tricyclic core structure, present in various natural products.
Co-reporter:Victor Mougel; Celine B. Santiago; Pavel A. Zhizhko; Elizabeth N. Bess; Jeno Varga; Georg Frater; Matthew S. Sigman;Christophe Copéret
Journal of the American Chemical Society 2015 Volume 137(Issue 20) pp:6699-6704
Publication Date(Web):May 4, 2015
DOI:10.1021/jacs.5b03344
A broad series of fully characterized, well-defined silica-supported W metathesis catalysts with the general formula [(≡SiO)W(═NAr)(═CHCMe2R)(X)] (Ar = 2,6-iPr2C6H3 (AriPr), 2,6-Cl2C6H3 (ArCl), 2-CF3C6H4 (ArCF3), and C6F5 (ArF5); X = OC(CF3)3 (OtBuF9), OCMe(CF3)2 (OtBuF6), OtBu, OSi(OtBu)3, 2,5-dimethylpyrrolyl (Me2Pyr) and R = Me or Ph) was prepared by grafting bis-X substituted complexes [W(NAr)(═CHCMe2R)(X)2] on silica partially dehydroxylated at 700 °C (SiO2-(700)), and their activity was evaluated with the goal to obtain detailed structure–activity relationships. Quantitative influence of the ligand set on the activity (turnover frequency, TOF) in self-metathesis of cis-4-nonene was investigated using multivariate linear regression analysis tools. The TOF of these catalysts (activity) can be well predicted from simple steric and electronic parameters of the parent protonated ligands; it is described by the mutual contribution of the NBO charge of the nitrogen or the IR intensity of the symmetric N–H stretch of the ArNH2, corresponding to the imido ligand, together with the Sterimol B5 and pKa of HX, representing the X ligand. This quantitative and predictive structure–activity relationship analysis of well-defined heterogeneous catalysts shows that high activity is associated with the combination of X and NAr ligands of opposite electronic character and paves the way toward rational development of metathesis catalysts.
Co-reporter:Chun Zhang; Celine B. Santiago; Lei Kou
Journal of the American Chemical Society 2015 Volume 137(Issue 23) pp:7290-7293
Publication Date(Web):June 1, 2015
DOI:10.1021/jacs.5b04289
A highly enantioselective and site-selective Pd-catalyzed arylation of alkenes linked to carbonyl derivatives to yield α,β-unsaturated systems is reported. The high site selectivity is attributed to both a solvent effect and the polarized nature of the carbonyl group, both of which have been analyzed through multidimensional analysis tools. The reaction can be performed in an iterative fashion allowing for a diastereoselective installation of two aryl groups along an alkyl chain.
Co-reporter:Chun Zhang; Celine B. Santiago; Jennifer M. Crawford
Journal of the American Chemical Society 2015 Volume 137(Issue 50) pp:15668-15671
Publication Date(Web):December 1, 2015
DOI:10.1021/jacs.5b11335
An enantioselective, intermolecular dehydrogenative Heck arylation of trisubstituted alkenes to construct remote quaternary stereocenters has been developed. Using a new chiral pyridine oxazoline ligand, good to high enantioselectivity is achieved for various combinations of indole derivatives and trisubstituted alkenes. However, some combinations of substrates led to lower enantioselectivity, which provided the impetus to use structure enantioselectivity correlations to design a better performing ligand.
Co-reporter:David P. Hickey; David A. Schiedler; Ivana Matanovic; Phuong Vy Doan; Plamen Atanassov; Shelley D. Minteer
Journal of the American Chemical Society 2015 Volume 137(Issue 51) pp:16179-16186
Publication Date(Web):December 3, 2015
DOI:10.1021/jacs.5b11252
Stable nitroxyl radical-containing compounds, such as 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) and its derivatives, are capable of electrocatalytically oxidizing a wide range of alcohols under mild and environmentally friendly conditions. Herein, we examine the structure–function relationships that determine the catalytic activity of a diverse range of water-soluble nitroxyl radical compounds. A strong correlation is described between the difference in the electrochemical oxidation potentials of a compound and its electrocatalytic activity. Additionally, we construct a simple computational model that is able to accurately predict the electrochemical potential and catalytic activity of a wide range of nitroxyl radical derivatives.
Co-reporter:Vaneet Saini; Mark O’Dair
Journal of the American Chemical Society 2015 Volume 137(Issue 2) pp:608-611
Publication Date(Web):January 2, 2015
DOI:10.1021/ja511640g
An efficient method for the construction of Csp2–Csp3 bond in a regio- and stereoselective fashion involving 1,3-terminal dienes, enol triflates/nonaflates, and sodium formate under Pd(0)-catalysis is described. The three component assembly allows trapping of a π-allyl intermediate, after the initial migratory insertion of the diene, by a hydride source that leads to structurally complex and synthetically challenging tri- and tetrasubstituted alkene building blocks.
Co-reporter:David P. Hickey, Ross D. Milton, Dayi Chen, Matthew S. Sigman, and Shelley D. Minteer
ACS Catalysis 2015 Volume 5(Issue 9) pp:5519
Publication Date(Web):August 14, 2015
DOI:10.1021/acscatal.5b01668
We demonstrate a method to simultaneously immobilize the oxidation catalyst, TEMPO, while dramatically enhancing its electrocatalytic activity toward several biologically available alcohols. TEMPO is covalently immobilized onto linear poly(ethylenimine), which is then cross-linked onto the surface of a glassy carbon electrode to form a hydrogel through which substrates can readily diffuse. The TEMPO-LPEI electrode is used as an anode capable of generating currents from 0.41 ± 0.06 mA cm–2 in the presence of 250 mM sucrose to 8.20 ± 0.04 mA cm–2 in the presence of 2 M methanol and 33.4 ± 9.4 mA cm–2 in the presence of 500 mM formate under neutral pH and at 25 °C. The newly described anode is combined with an enzymatic biocathode to construct a hybrid biofuel cell to produce 0.38 ± 0.04 mA cm–2 while using 2 M methanol as a fuel source.Keywords: alcohols; electrocatalysis; hybrid fuel cell; oxidation; TEMPO
Co-reporter:Matthew S. McCammant and Matthew S. Sigman
Chemical Science 2015 vol. 6(Issue 2) pp:1355-1361
Publication Date(Web):28 Nov 2014
DOI:10.1039/C4SC03074E
Palladium-catalyzed 1,4-difunctionalizations of isoprene that produce skipped polyenes are reported. Complex isomeric product mixtures are possible as a result of the difficult-to-control migratory insertion of isoprene into a Pd–alkenyl bond, but good site selectivity has been achieved using easily accessible pyrox ligands. Mechanistic studies suggest that the control of insertion is the result of the unique electronic asymmetry and steric properties of the ligand.
Co-reporter:Elizabeth N. Bess, David M. Guptill, Huw M. L. Davies and Matthew S. Sigman
Chemical Science 2015 vol. 6(Issue 5) pp:3057-3062
Publication Date(Web):18 Mar 2015
DOI:10.1039/C5SC00357A
Achieving selective C–H functionalization is a significant challenge that requires discrimination between many similar C–H bonds. Yet, reaction systems employing Rh2(DOSP)4 and Rh2(BPCP)4 were recently demonstrated to afford high levels of selectivity in the C–H insertion of carbenes into toluene-derived substrates. Herein, we explore the origin of this selectivity through a systematic analysis of substrate and reagent features that alter levels of selectivity from 20:1 to 1:610 for secondary (or tertiary)-to-primary benzylic C–H functionalization of toluene derivatives. Describing this variation using infrared vibrations and point charges, we have developed a mathematical model from which are identified features of the systems that determine levels of site-selectivity and are applied as predictive factors to describe the selectivity behavior of new substrate/reagent combinations.
Co-reporter:Margaret J. Hilton, Bin Cheng, Benjamin R. Buckley, Liping Xu, Olaf Wiest, Matthew S. Sigman
Tetrahedron 2015 Volume 71(Issue 37) pp:6513-6518
Publication Date(Web):16 September 2015
DOI:10.1016/j.tet.2015.05.020
The relative rates of alkenyl alcohols in the Pd-catalyzed redox-relay Heck reaction were measured in order to examine the effect of their steric and electronic properties on the rate-determining step. Competition experiments between an allylic alkenyl alcohol and two substrates with differing chain lengths revealed that the allylic alcohol reacts 3–4 times faster in either case. Competition between di- and trisubstituted alkenyl alcohols provided an interesting scenario, in which the disubstituted alkene was consumed first followed by reaction of the trisubstituted alkene. Consistent with this observation, the transition structures for the migratory insertion of the aryl group into the di- and trisubstituted alkenes were calculated with a lower barrier for the former. An internal competition between a substrate containing two alcohols with differing chain lengths demonstrated the catalyst's preference for migrating toward the closest alcohol. Additionally, it was observed that increasing the electron-density in the arene boronic acid promotes a faster reaction, which correlates with Hammett σp values to give a ρ of −0.87.
Co-reporter:Anat Milo;Andrew J. Neel;F. Dean Toste
Science 2015 Vol 347(6223) pp:737-743
Publication Date(Web):13 Feb 2015
DOI:10.1126/science.1261043
Optimizing a catalyst many ways at once
Optimization strategies are often likened to hikes in a hilly landscape. If your goal is to get to the top of the highest hill, and you only take steps toward higher ground, you might never find a peak on a route that requires a preliminary descent. So it is in chemistry, where optimizing each structural feature of a catalyst consecutively might gloss over subtle tradeoffs that in combination offer the best performance. Milo et al. use multidimensional analysis techniques to generate a predictive model of how selectivity depends on multiple characteristics of the catalyst and substrate in a C-N bond-forming reaction (see the Perspective by Lu). They then apply this model to improve the catalyst globally.
Science, this issue p. 737; see also p. 719
Co-reporter:Elizabeth N. Bess ; Ryan J. DeLuca ; Daniel J. Tindall ; Martins S. Oderinde ; Jennifer L. Roizen ; J. Du Bois
Journal of the American Chemical Society 2014 Volume 136(Issue 15) pp:5783-5789
Publication Date(Web):March 27, 2014
DOI:10.1021/ja5015508
Predicting site selectivity in C–H bond oxidation reactions involving heteroatom transfer is challenged by the small energetic differences between disparate bond types and the subtle interplay of steric and electronic effects that influence reactivity. Herein, the factors governing selective Rh2(esp)2-catalyzed C–H amination of isoamylbenzene derivatives are investigated, where modification to both the nitrogen source, a sulfamate ester, and substrate are shown to impact isomeric product ratios. Linear regression mathematical modeling is used to define a relationship that equates both IR stretching parameters and Hammett σ+ values to the differential free energy of benzylic versus tertiary C–H amination. This model has informed the development of a novel sulfamate ester, which affords the highest benzylic-to-tertiary site selectivity (9.5:1) observed for this system.
Co-reporter:David P. Hickey ; Matthew S. McCammant ; Fabien Giroud ; Matthew S. Sigman ;Shelley D. Minteer
Journal of the American Chemical Society 2014 Volume 136(Issue 45) pp:15917-15920
Publication Date(Web):October 28, 2014
DOI:10.1021/ja5098379
We demonstrate the complete electrochemical oxidation of the biofuel glycerol to CO2 using a hybrid enzymatic and small-molecule catalytic system. Combining an enzyme, oxalate oxidase, and an organic oxidation catalyst, 4-amino-TEMPO, we are able to electrochemically oxidize glycerol at a carbon electrode, while collecting up to as many as 16 electrons per molecule of fuel. Additionally, we investigate the anomalous electrocatalytic properties that allow 4-amino-TEMPO to be active under the acidic conditions that are required for oxalate oxidase to function.
Co-reporter:Liping Xu ; Margaret J. Hilton ; Xinhao Zhang ; Per-Ola Norrby ; Yun-Dong Wu ; Matthew S. Sigman ;Olaf Wiest
Journal of the American Chemical Society 2014 Volume 136(Issue 5) pp:1960-1967
Publication Date(Web):January 11, 2014
DOI:10.1021/ja4109616
The enantioselective Pd-catalyzed redox-relay Heck arylation of acyclic alkenyl alcohols allows access to various useful chiral building blocks from simple olefinic substrates. Mechanistically, after the initial migratory insertion, a succession of β-hydride elimination and migratory insertion steps yields a saturated carbonyl product instead of the more general Heck product, an unsaturated alcohol. Here, we investigate the reaction mechanism, including the relay function, yielding the final carbonyl group transformation. M06 calculations predict a ΔΔG⧧ of 1 kcal/mol for the site selectivity and 2.5 kcal/mol for the enantioselectivity, in quantitative agreement with experimental results. The site selectivity is controlled by a remote electronic effect, where the developing polarization of the alkene in the migratory insertion transition state is stabilized by the C–O dipole of the alcohol moiety. The enantioselectivity is controlled by steric repulsion between the oxazoline substituent and the alcohol-bearing alkene substituent. The relay efficiency is due to an unusually smooth potential energy surface without high barriers, where the hydroxyalkyl-palladium species acts as a thermodynamic sink, driving the reaction toward the carbonyl product. Computational predictions of the relative reactivity and selectivity of the double bond isomers are validated experimentally.
Co-reporter:Benjamin J. Stokes, Amanda J. Bischoff and Matthew S. Sigman
Chemical Science 2014 vol. 5(Issue 6) pp:2336-2339
Publication Date(Web):17 Mar 2014
DOI:10.1039/C4SC00602J
Pd-catalyzed allylic relay Suzuki cross-coupling reactions of secondary alkyl tosylates, featuring a sterically-hindered oxidative addition and precise control of β-hydride elimination, are reported. The identification of a linear free energy relationship between the relative rates of substrate consumption and the electronic nature of the substrate alkene suggests that the oxidative addition requires direct alkene involvement. A study of the effect of alkyl chain length on the reaction outcome supports a chelation-controlled oxidative addition.
Co-reporter:Benjamin J. Stokes, Longyan Liao, Aline Mendes de Andrade, Qiaofeng Wang, and Matthew S. Sigman
Organic Letters 2014 Volume 16(Issue 17) pp:4666-4669
Publication Date(Web):August 27, 2014
DOI:10.1021/ol502279u
A palladium-catalyzed intermolecular vicinal diarylation of terminal 1,3-dienes using aryldiazonium tetrafluoroborates and arylboronic acids is reported. Using this technology, two different arenes are regioselectively introduced in a vicinal fashion across the terminal alkene of a variety of terminal 1,3-dienes at ambient temperature. Through the action of a chiral bicyclo[2.2.2]octadienyl ligand at −20 °C, good enantioselectivity has also been achieved.
Co-reporter:Margaret J. Hilton, Li-Ping Xu, Per-Ola Norrby, Yun-Dong Wu, Olaf Wiest, and Matthew S. Sigman
The Journal of Organic Chemistry 2014 Volume 79(Issue 24) pp:11841-11850
Publication Date(Web):September 4, 2014
DOI:10.1021/jo501813d
The mechanism of the redox-relay Heck reaction was investigated using deuterium-labeled substrates. Results support a pathway through a low energy palladium–alkyl intermediate that immediately precedes product formation, ruling out a tautomerization mechanism. DFT calculations of the relevant transition structures at the M06/LAN2DZ+f/6-31+G* level of theory show that the former pathway is favored by 5.8 kcal/mol. Palladium chain-walking toward the alcohol, following successive β-hydride eliminations and migratory insertions, is also supported in this study. The stereochemistry of deuterium labels is determined, lending support that the catalyst remains bound to the substrate during the relay process and that both cis- and trans-alkenes form from β-hydride elimination.
Co-reporter:Elizabeth N. Bess;Amanda J. Bischoff
PNAS 2014 Volume 111 (Issue 41 ) pp:14698-14703
Publication Date(Web):2014-10-14
DOI:10.1073/pnas.1409522111
Assessment of reaction substrate scope is often a qualitative endeavor that provides general indications of substrate sensitivity
to a measured reaction outcome. Unfortunately, this field standard typically falls short of enabling the quantitative prediction
of new substrates’ performance. The disconnection between a reaction’s development and the quantitative prediction of new
substrates’ behavior limits the applicative usefulness of many methodologies. Herein, we present a method by which substrate
libraries can be systematically developed to enable quantitative modeling of reaction systems and the prediction of new reaction
outcomes. Presented in the context of rhodium-catalyzed asymmetric transfer hydrogenation, these models quantify the molecular
features that influence enantioselection and, in so doing, lend mechanistic insight to the modes of asymmetric induction.
Co-reporter:Kaid C. Harper ; Sarah C. Vilardi
Journal of the American Chemical Society 2013 Volume 135(Issue 7) pp:2482-2485
Publication Date(Web):February 6, 2013
DOI:10.1021/ja4001807
The effectiveness of a new asymmetric catalytic methodology is often weighed by the number of diverse substrates that undergo reaction with high enantioselectivity. Here we report a study that correlates substrate and ligand steric effects to enantioselectivity for the propargylation of aliphatic ketones. The mathematical model is shown to be highly predictive when applied to substrate/catalyst combinations outside the training set.
Co-reporter:Tian-Sheng Mei ; Erik W. Werner ; Alexander J. Burckle
Journal of the American Chemical Society 2013 Volume 135(Issue 18) pp:6830-6833
Publication Date(Web):April 22, 2013
DOI:10.1021/ja402916z
A general, highly selective asymmetric redox-relay oxidative Heck reaction using achiral or racemic acyclic alkenols and boronic acid derivatives is reported. This reaction delivers remotely functionalized arylated carbonyl products from acyclic alkenol substrates, with excellent enantioselectivity under mild conditions, bearing a range of useful functionality. A preliminary mechanistic investigation suggests that the regioselectivity of the initial migratory insertion is highly dependent on the electronic nature of the boronic acid and more subtle electronic effects of the alkenyl alcohol.
Co-reporter:Matthew S. McCammant ; Longyan Liao
Journal of the American Chemical Society 2013 Volume 135(Issue 11) pp:4167-4170
Publication Date(Web):March 8, 2013
DOI:10.1021/ja3110544
A palladium-catalyzed 1,4-addition across the commodity chemical 1,3-butadiene to afford skipped polyene products is reported. Through a palladium σ → π → σ allyl isomerization, two new carbon–carbon bonds are formed with high regioselectivity and trans stereoselectivity of the newly formed alkene. The utility of this method is highlighted by the successful synthesis of the ripostatin A skipped triene core.
Co-reporter:Elizabeth N. Bess and Matthew S. Sigman
Organic Letters 2013 Volume 15(Issue 3) pp:646-649
Publication Date(Web):January 11, 2013
DOI:10.1021/ol303465c
An iridium-catalyzed asymmetric hydrogenation of 1,1-diarylkenes is described. Employing a novel, modular phosphoramidite ligand, PhosPrOx, in this transformation affords biologically relevant 1,1-diarylmethine products in good enantiomeric ratios (96.5:3.5 to 71:29). We propose that a meta-directing group, 3,5-dimethoxyphenyl, is responsible for the observed enantioselection, the highest reported, to date, for iridium-catalyzed hydrogenation of 1,1-diarylalkenes lacking ortho-directing groups.
Co-reporter:Ryan J. DeLuca and Matthew S. Sigman
Organic Letters 2013 Volume 15(Issue 1) pp:92-95
Publication Date(Web):December 19, 2012
DOI:10.1021/ol303129p
A palladium-catalyzed hydroalkylation reaction of protected allylic alcohols using alkylzinc bromide reagents is reported. This account includes numerous allylic, homoallylic, and bishomoallylic alcohol derivatives, all with a uniform selectivity of >20:1 for the anti-Markovnikov product. The reaction features the ability to deliver enantiomerically enriched alcohols in unfunctionalized regions, which results from the catalyst avoiding β-hydride elimination at the allylic position.
Co-reporter:Vaneet Saini, Longyan Liao, Qiaofeng Wang, Ranjan Jana, and Matthew S. Sigman
Organic Letters 2013 Volume 15(Issue 19) pp:5008-5011
Publication Date(Web):September 18, 2013
DOI:10.1021/ol4023358
An efficient protocol for the one-step synthesis of biologically relevant 1,1-diarylalkanes has been described. This reaction introduces two different aryl groups across the terminal end of simple feedstock alkenes such as ethylene and allylic carbonates. The propensity to generate π-benzylpalladium intermediates dictates the exclusive 1,1-regioselectivity observed in the product.
Co-reporter:Vaneet Saini;Dr. Benjamin J. Stokes ; Matthew S. Sigman
Angewandte Chemie 2013 Volume 125( Issue 43) pp:11414-11429
Publication Date(Web):
DOI:10.1002/ange.201303916
Abstract
Ethylen ist das (volumenbezogen) in größter Menge synthetisierte organische Molekül. Es lässt sich leicht in übergangsmetallkatalysierte C-C-Kupplungsreaktionen einbinden, und zwar durch migratorische Insertion in Alkylmetall-Intermediate. Wegen der D2h-Symmetrie des Ethylens kann die Insertion nur einen einzigen Verlauf nehmen, was die Nebenproduktbildung limitiert und die Produktanalyse außerordentlich vereinfacht. Wie in diesem Kurzaufsatz beschrieben wird, wird bei vielen C-C-Kupplungen das Ethylenmolekül (oder mehrere) bei Umgebungsdruck und -temperatur eingefügt. Auch sind viele Reaktionen bekannt, bei denen ein in geeigneter Weise substituiertes Alken in das Produkt eingeführt wird.
Co-reporter:Vaneet Saini;Dr. Benjamin J. Stokes ; Matthew S. Sigman
Angewandte Chemie International Edition 2013 Volume 52( Issue 43) pp:11206-11220
Publication Date(Web):
DOI:10.1002/anie.201303916
Abstract
Ethylene, the simplest alkene, is the most abundantly synthesized organic molecule by volume. It is readily incorporated into transition-metal-catalyzed carbon–carbon bond-forming reactions through migratory insertions into alkylmetal intermediates. Because of its D2h symmetry, only one insertion outcome is possible. This limits byproduct formation and greatly simplifies analysis. As described within this Minireview, many carbon–carbon bond-forming reactions incorporate a molecule (or more) of ethylene at ambient pressure and temperature. In many cases, a useful substituted alkene is incorporated into the product.
Co-reporter:Ryan J. DeLuca, Jennifer L. Edwards, Laura D. Steffens, Brian W. Michel, Xiaoxiao Qiao, Chunyin Zhu, Silas P. Cook, and Matthew S. Sigman
The Journal of Organic Chemistry 2013 Volume 78(Issue 4) pp:1682-1686
Publication Date(Web):January 30, 2013
DOI:10.1021/jo302638v
The Pd-catalyzed TBHP-mediated Wacker-type oxidation of internal alkenes is reported. The reaction uses 2-(4,5-dihydro-2-oxazolyl)quinoline (Quinox) as ligand and TBHP(aq) as oxidant to deliver single ketone constitutional isomer products in a predictable fashion from electronically biased olefins. This methodology is showcased through its application on an advanced intermediate in the total synthesis of the antimalarial drug artemisinin.
Co-reporter:Kaid C. Harper and Matthew S. Sigman
The Journal of Organic Chemistry 2013 Volume 78(Issue 7) pp:2813-2818
Publication Date(Web):March 19, 2013
DOI:10.1021/jo4002239
A classic strategy of physical organic chemists is to probe reaction mechanisms using linear free energy relationships. Identifying such relationships in asymmetric catalytic reactions provides substantial insight into the key factors controlling enantioselectivity, which in turn increases the predictability and applicability of these reactions. The focus of this JOCSynopsis is to highlight several recent examples in which various parameters were identified and applied to the elucidation of LFERs.
Co-reporter:Matthew S. Sigman and Erik W. Werner
Accounts of Chemical Research 2012 Volume 45(Issue 6) pp:874
Publication Date(Web):November 23, 2011
DOI:10.1021/ar200236v
The functional group transformations carried out by the palladium-catalyzed Wacker and Heck reactions are radically different, but they are both alkenyl C–H bond functionalization reactions that have found extensive use in organic synthesis. The synthetic community depends heavily on these important reactions, but selectivity issues arising from control by the substrate, rather than control by the catalyst, have prevented the realization of their full potential. Because of important similarities in the respective selectivity-determining nucleopalladation and β-hydride elimination steps of these processes, we posit that the mechanistic insight garnered through the development of one of these catalytic reactions may be applied to the other. In this Account, we detail our efforts to develop catalyst-controlled variants of both the Wacker oxidation and the Heck reaction to address synthetic limitations and provide mechanistic insight into the underlying organometallic processes of these reactions.In contrast to previous reports, we discovered that electrophilic palladium catalysts with noncoordinating counterions allowed for the use of a Lewis basic ligand to efficiently promote tert-butylhydroperoxide (TBHP)-mediated Wacker oxidation reactions of styrenes. This discovery led to the mechanistically guided development of a Wacker reaction catalyzed by a palladium complex with a bidentate ligand. This ligation may prohibit coordination of allylic heteroatoms, thereby allowing for the application of the Wacker oxidation to substrates that were poorly behaved under classical conditions.Likewise, we unexpectedly discovered that electrophilic Pd-σ-alkyl intermediates are capable of distinguishing between electronically inequivalent C–H bonds during β-hydride elimination. As a result, we have developed E-styrenyl selective oxidative Heck reactions of previously unsuccessful electronically nonbiased alkene substrates using arylboronic acid derivatives. The mechanistic insight gained from the development of this chemistry allowed for the rational design of a similarly E-styrenyl selective classical Heck reaction using aryldiazonium salts and a broad range of alkene substrates. The key mechanistic findings from the development of these reactions provide new insight into how to predictably impart catalyst control in organometallic processes that would otherwise afford complex product mixtures. Given our new understanding, we are optimistic that reactions that introduce increased complexity relative to simple classical processes may now be developed based on our ability to predict the selectivity-determining nucleopalladation and β-hydride elimination steps through catalyst design.
Co-reporter:Vaneet Saini
Journal of the American Chemical Society 2012 Volume 134(Issue 28) pp:11372-11375
Publication Date(Web):July 5, 2012
DOI:10.1021/ja304344h
The 1,1-difunctionalization of ethylene, with aryl/vinyl/heteroaryl transmetalating agents and vinyl electrophiles, is reported. The reaction is high-yielding under a low pressure of ethylene, and regioselectivity is generally high for the 1,1-disubstituted product. The process is highlighted by the use of heteroaromatic cross-coupling reagents, which have not been competent reaction partners in previously reported efforts.
Co-reporter:Benjamin J. Stokes ; Susanne M. Opra
Journal of the American Chemical Society 2012 Volume 134(Issue 28) pp:11408-11411
Publication Date(Web):June 28, 2012
DOI:10.1021/ja305403s
The Pd(0)-catalyzed allylic cross-coupling of homoallylic tosylate substrates using boronic acids and pinacol esters is reported. The reaction uses 2-(4,5-dihydro-2-oxazolyl)quinoline (quinox) as a ligand and is performed at ambient temperature. The scope of the reaction is broad in terms of both the boronate transmetalating reagent and the substrate and includes secondary tosylates. Mechanistic studies support an alkene-mediated SN2-type stereoinvertive oxidative addition of unactivated primary and secondary alkyl tosylates.
Co-reporter:Ranjan Jana, Tejas P. Pathak, Katrina H. Jensen, and Matthew S. Sigman
Organic Letters 2012 Volume 14(Issue 16) pp:4074-4077
Publication Date(Web):August 8, 2012
DOI:10.1021/ol3016989
A palladium-catalyzed enantio- and diastereoselective synthesis of pyrrolidine derivatives is described. Initial intramolecular nucleopalladation of the tethered protected amine forms the pyrrolidine moiety and a quinone methide intermediate. A second nucleophile adds intermolecularly to afford diverse products in high enantio- and diastereoselectivity.
Co-reporter:Tejas P. Pathak, Jaroslaw G. Osiak, Rachel M. Vaden, Bryan E. Welm, Matthew S. Sigman
Tetrahedron 2012 68(26) pp: 5203-5208
Publication Date(Web):
DOI:10.1016/j.tet.2012.03.075
Co-reporter:Matthew S. Sigman;Tian-Sheng Mei;Alexander J. Burckle;Erik W. Werner
Science 2012 Volume 338(Issue 6113) pp:1455-1458
Publication Date(Web):14 Dec 2012
DOI:10.1126/science.1229208
Co-reporter:Ranjan Jana, Tejas P. Pathak, and Matthew S. Sigman
Chemical Reviews 2011 Volume 111(Issue 3) pp:1417
Publication Date(Web):February 14, 2011
DOI:10.1021/cr100327p
Co-reporter:Kaid C. Harper
Science 2011 Vol 333(6051) pp:1875-1878
Publication Date(Web):30 Sep 2011
DOI:10.1126/science.1206997
An unexpected synergy between a ligand's
steric bulk and its electronic structure improves a stereoselective catalyst.
Co-reporter:Erik W. Werner
Journal of the American Chemical Society 2011 Volume 133(Issue 25) pp:9692-9695
Publication Date(Web):May 31, 2011
DOI:10.1021/ja203164p
Simple, mild, and efficient conditions are reported for a Pd0-catalyzed Heck reaction that delivers high yields and selectivity for (E)-styrenyl products using electronically nonbiased olefin substrates bearing a range of useful functionality. Preliminary mechanistic studies demonstrate that the σ-donating DMA solvent is crucial for high selectivity. Further studies suggest that the catalyst distinguishes between β-hydrogens on the basis of their relative hydridic character, in contrast to previously reported PdII-catalyzed oxidative reaction conditions.
Co-reporter:Longyan Liao ; Ranjan Jana ; Kaveri Balan Urkalan
Journal of the American Chemical Society 2011 Volume 133(Issue 15) pp:5784-5787
Publication Date(Web):March 30, 2011
DOI:10.1021/ja201358b
A three-component coupling of vinyl triflates and boronic acids to alkenes catalyzed by palladium is reported. Using 1,3-dienes, selective 1,2-alkene difunction-alization is observed, whereas the use of terminal alkenes results in 1,1-alkene difunctionalization. The reaction outcome is attributed to the formation of stabilized, cationic Pd-π-allyl intermediates to regulate β-hydride elimination.
Co-reporter:Brian W. Michel ; Laura D. Steffens
Journal of the American Chemical Society 2011 Volume 133(Issue 21) pp:8317-8325
Publication Date(Web):May 9, 2011
DOI:10.1021/ja2017043
The mechanism of the tert-butylhydroperoxide-mediated, Pd(Quinox)-catalyzed Wacker-type oxidation was investigated to evaluate the hypothesis that a selective catalyst-controlled oxidation could be achieved by rendering the palladium coordinatively saturated using a bidentate amine ligand. The unique role of the Quinox ligand framework was probed via systematic ligand modifications. The modified ligands were evaluated through quantitative Hammett analysis, which supports a “push–pull” relationship between the electronically asymmetric quinoline and oxazoline ligand modules.
Co-reporter:Ryan J. DeLuca
Journal of the American Chemical Society 2011 Volume 133(Issue 30) pp:11454-11457
Publication Date(Web):July 5, 2011
DOI:10.1021/ja204080s
Palladium-catalyzed hydroalkylation of allylic amine derivatives by alkylzinc reagents is reported. This reductive cross-coupling reaction yields anti-Markovnikov products using a variety of allylic amine protecting groups. Preliminary mechanistic studies suggest that a reversible β-hydride elimination/hydride insertion process furnishes the primary Pd-alkyl intermediate, which then undergoes transmetalation followed by reductive elimination to form a new sp3–sp3 carbon–carbon bond.
Co-reporter:Tejas P. Pathak and Matthew S. Sigman
The Journal of Organic Chemistry 2011 Volume 76(Issue 22) pp:9210-9215
Publication Date(Web):October 16, 2011
DOI:10.1021/jo201789k
Ortho-quinone methides are important synthetic intermediates and widely implicated in biological processes. In this Synopsis, recent advances concerning the synthesis and utility of these intermediates are discussed with a particular emphasis on metal-catalyzed formation of quinone methide intermediates. Additionally, applications of these intermediates as partners in asymmetric synthesis will be discussed including methods we have developed that involve the enantioselective Pd-catalyzed formation of ortho-quinone methides and the trapping of aforementioned intermediates with diverse nucleophiles.
Co-reporter:Jessica R. McCombs, Brian W. Michel, and Matthew S. Sigman
The Journal of Organic Chemistry 2011 Volume 76(Issue 9) pp:3609-3613
Publication Date(Web):March 29, 2011
DOI:10.1021/jo200462a
Homoallylic alcohols are oxidized to β-hydroxy ketones using a TBHP-mediated Pd-catalyzed Wacker-type oxidation. The use of a bidentate ligand, quinoline-2-oxazoline (Quinox), and TBHP(aq) as the terminal oxidant provides good yields of the desired products with reaction times significantly reduced as compared to the Tsuji−Wacker oxidation. Additionally, bis- and tris-homoallylic alcohols are oxidized to provide cyclic peroxyketals, presumably via nucleophilic attack of the methyl ketone product.
Co-reporter:Susanne M. Podhajsky, Yasumasa Iwai, Amanda Cook-Sneathen, Matthew S. Sigman
Tetrahedron 2011 67(24) pp: 4435-4441
Publication Date(Web):
DOI:10.1016/j.tet.2011.02.027
Co-reporter:Kaid C. Harper
PNAS 2011 Volume 108 (Issue 6 ) pp:2179-2183
Publication Date(Web):2011-02-08
DOI:10.1073/pnas.1013331108
Using a modular amino acid based chiral ligand motif, a library of ligands was synthesized systematically varying the substituents
at two positions. The effects of these changes on ligand structure were probed in the enantioselective allylation of benzaldehyde,
acetophenone, and methylethyl ketone under Nozaki-Hiyama-Kishi conditions. The resulting three-dimensional datasets allowed
for the construction of mathematical surface models which describe the interplay of substituent effects on enantioselectivity
for a given reaction. The surface models were both extrapolated and manipulated to predict the enantioselective outcomes of
several previously untested ligands. Analyses were also used to predict optimal ligand structure of a minimal dataset. Within
the dataset, a linear free energy relationship was also discovered and a direct comparison of both the linear prediction as
well as the three-dimensional prediction illustrates the potential predictive power of using a three-dimensional model approach
to asymmetric catalyst development.
Co-reporter:Longyan Liao
Journal of the American Chemical Society 2010 Volume 132(Issue 30) pp:10209-10211
Publication Date(Web):July 8, 2010
DOI:10.1021/ja105010t
A palladium-catalyzed reductive cross-coupling of 1,3-dienes with boronic esters in which a π-allyl Pd species is generated directly from a 1,3-diene via a Pd-catalyzed aerobic alcohol oxidation is reported. Both the scope of the process and the origin of a highly selective 1,2-addition are discussed.
Co-reporter:Brian J. Anderson ; John A. Keith
Journal of the American Chemical Society 2010 Volume 132(Issue 34) pp:11872-11874
Publication Date(Web):August 5, 2010
DOI:10.1021/ja1057218
The kinetics of the Pd[(−)-sparteine]Cl2 catalyzed oxidation of decene using oxygen as the sole oxidant have been studied in the absence of copper salts and high [Cl−]. Saturation kinetics are observed for [decene] as well as a third order dependence on [water]. A mechanism is proposed involving the dissociation of two chlorides and rate-limiting formation of a three-water hydrogen bridged network and subsequent oxypalladation as supported by computational studies.
Co-reporter:Tejas P. Pathak ; Keith M. Gligorich ; Bryan E. Welm
Journal of the American Chemical Society 2010 Volume 132(Issue 23) pp:7870-7871
Publication Date(Web):May 20, 2010
DOI:10.1021/ja103472a
A unique alkene difunctionalization reaction that allows rapid construction of molecular complexity around the biologically relevant indole framework has been developed. The reaction proceeds with up to 87% yield, 99:1 er, and >20:1 dr. Evaluation of several of the compounds revealed promising anticancer activity against MCF-7 cells.
Co-reporter:Jeffrey L. Gustafson, Matthew S. Sigman and Scott J. Miller
Organic Letters 2010 Volume 12(Issue 12) pp:2794-2797
Publication Date(Web):May 20, 2010
DOI:10.1021/ol100927m
Linear free-energy relationships have been found for enantioselectivity and various steric parameters in an enantioselective desymmetrization of symmetrical bis(phenol) substrates. The potential origin of this observation and the role of different steric parameters are discussed.
Co-reporter:Erik W. Werner, Kaveri B. Urkalan and Matthew S. Sigman
Organic Letters 2010 Volume 12(Issue 12) pp:2848-2851
Publication Date(Web):May 19, 2010
DOI:10.1021/ol1009575
Evaluation of the scope of a PdII-catalyzed oxidative 1,1-diarylation reaction of terminal olefins using aryl stannanes is reported. The reaction is shown to be tolerant of functionality commonly encountered in organic synthesis; however, the reaction outcome was found to be dependent on the nature of the aryl stannane used. A mechanistic rationale for the observation of this influence is provided.
Co-reporter:Brian W. Michel;Jessica R. McCombs;Andrea Winkler ;Dr. Matthew S. Sigman
Angewandte Chemie International Edition 2010 Volume 49( Issue 40) pp:7312-7315
Publication Date(Web):
DOI:10.1002/anie.201004156
Co-reporter:Brian W. Michel;Jessica R. McCombs;Andrea Winkler ;Dr. Matthew S. Sigman
Angewandte Chemie 2010 Volume 122( Issue 40) pp:7470-7473
Publication Date(Web):
DOI:10.1002/ange.201004156
Co-reporter:Katrina H. Jensen and Matthew S. Sigman
The Journal of Organic Chemistry 2010 Volume 75(Issue 21) pp:7194-7201
Publication Date(Web):October 4, 2010
DOI:10.1021/jo1013806
A modular catalyst structure was applied to evaluate the effects of catalyst acidity in a hydrogen bond-catalyzed hetero Diels−Alder reaction. Linear free energy relationships between catalyst acidity and both rate and enantioselectivity were observed, where greater catalyst acidity leads to increased activity and enantioselectivity. A relationship between reactant electronic nature and rate was also observed, although there is no such correlation to enantioselectivity, indicating the system is under catalyst control.
Co-reporter:Katrina H. Jensen ; Tejas P. Pathak ; Yang Zhang
Journal of the American Chemical Society 2009 Volume 131(Issue 47) pp:17074-17075
Publication Date(Web):November 10, 2009
DOI:10.1021/ja909030c
A sequential intramolecular−intermolecular enantioselective alkene difunctionalization reaction has been developed which is thought to proceed through Pd-catalyzed quinone methide formation. The synthesis of new chiral heterocyclic compounds with adjacent chiral centers is achieved in enantiomeric ratios up to 99:1 and diastereomeric ratios up to 10:1.
Co-reporter:Brian W. Michel ; Andrew M. Camelio ; Candace N. Cornell
Journal of the American Chemical Society 2009 Volume 131(Issue 17) pp:6076-6077
Publication Date(Web):April 13, 2009
DOI:10.1021/ja901212h
Utilizing the rapidly synthesized Quinox ligand and commercially available aqueous TBHP, a Wacker-type oxidation has been developed, which efficiently converts the traditionally challenging substrate class of protected allylic alcohols to the corresponding acyloin products. Additionally, the catalytic system is general for several other substrate classes, converting terminal olefins to methyl ketones, with short reaction times. The system is scalable (20 mmol) and can be performed with a reduced catalyst loading of 1 mol%. Enantioenriched substrates undergo oxidation with complete retention of enantiomeric excess.
Co-reporter:Kaveri Balan Urkalan and Matthew S. Sigman
Journal of the American Chemical Society 2009 Volume 131(Issue 50) pp:18042-18043
Publication Date(Web):November 24, 2009
DOI:10.1021/ja908545b
An unconventional route for the formation of sp3−sp3 C−C bonds from various styrenes and an organozinc reagent in a formal alkene hydroalkylation process is detailed. Mechanistically, this process is proposed to proceed by initial transmetalation followed by formation of a Pd−H species, which is subsequently trapped by the styrene.
Co-reporter:Keith M. Gligorich and Matthew S. Sigman
Chemical Communications 2009 (Issue 26) pp:3854-3867
Publication Date(Web):14 May 2009
DOI:10.1039/B902868D
During the past 10 years there have been significant advances in PdII-catalyzed oxidation reactions where the use of ligands has led to the development of catalytic systems capable of achieving high turnover numbers, which employ molecular oxygen as the sole stoichiometric oxidant. This Feature article will highlight some of the recent developments in direct molecular oxygen-coupled PdII-catalyzed oxidation reactions with an emphasis on enhanced catalytic systems and new reactions. Additionally, limitations of current catalytic systems, such as ligand oxidation, are presented and their implications for the development of new reactions are discussed.
Co-reporter:KaveriBalan Urkalan ;MatthewS. Sigman
Angewandte Chemie International Edition 2009 Volume 48( Issue 17) pp:3146-3149
Publication Date(Web):
DOI:10.1002/anie.200900218
Co-reporter:KaveriBalan Urkalan ;MatthewS. Sigman
Angewandte Chemie 2009 Volume 121( Issue 17) pp:3192-3195
Publication Date(Web):
DOI:10.1002/ange.200900218
Co-reporter:Keith M. Gligorich, Yasumasa Iwai, Sarah A. Cummings, Matthew S. Sigman
Tetrahedron 2009 65(26) pp: 5074-5083
Publication Date(Web):
DOI:10.1016/j.tet.2009.03.096
Co-reporter:Jeremie J. Miller, Sridhar Rajaram, Cornelia Pfaffenroth, Matthew S. Sigman
Tetrahedron 2009 65(16) pp: 3110-3119
Publication Date(Web):
DOI:10.1016/j.tet.2008.11.046
Co-reporter:Katrina H. Jensen and Matthew S. Sigman
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 22) pp:4083-4088
Publication Date(Web):09 Oct 2008
DOI:10.1039/B813246A
Alkene difunctionalization, the addition of two functional groups across a double bond, exemplifies a class of reactions with significant synthetic potential. This emerging area examines recent developments of palladium-catalyzed difunctionalization reactions, with a focus on mechanistic strategies that allow for functionalization of a common palladium alkyl intermediate.
Co-reporter:JeremieJ. Miller ;MatthewS. Sigman
Angewandte Chemie 2008 Volume 120( Issue 4) pp:783-786
Publication Date(Web):
DOI:10.1002/ange.200704257
Co-reporter:Yasumasa Iwai;KeithM. Gligorich ;MatthewS. Sigman
Angewandte Chemie 2008 Volume 120( Issue 17) pp:3263-3266
Publication Date(Web):
DOI:10.1002/ange.200705317
Co-reporter:JeremieJ. Miller ;MatthewS. Sigman
Angewandte Chemie International Edition 2008 Volume 47( Issue 4) pp:771-774
Publication Date(Web):
DOI:10.1002/anie.200704257
Co-reporter:Yasumasa Iwai;KeithM. Gligorich ;MatthewS. Sigman
Angewandte Chemie International Edition 2008 Volume 47( Issue 17) pp:3219-3222
Publication Date(Web):
DOI:10.1002/anie.200705317
Co-reporter:Katrina H. Jensen;Matthew S. Sigman
Angewandte Chemie 2007 Volume 119(Issue 25) pp:
Publication Date(Web):14 MAY 2007
DOI:10.1002/ange.200700298
Beziehungsmodell: Der Einfluss der Acidität wurde an einem modularen Oxazolinkatalysator für eine Hetero-Diels-Alder-Reaktion, die durch Wasserstoffbrückenbildung katalysiert wird, systematisch untersucht. Lineare-Freie-Energie-Beziehungen ergaben sich zwischen der Acidität des Katalysators und der Reaktionsgeschwindigkeit sowie der Enantioselektivität (siehe Bild).
Co-reporter:Katrina H. Jensen;Matthew S. Sigman
Angewandte Chemie International Edition 2007 Volume 46(Issue 25) pp:
Publication Date(Web):14 MAY 2007
DOI:10.1002/anie.200700298
Defining the relationship: The effect of catalyst acidity has been systematically probed by using a modular oxazoline catalyst in a hetero-Diels–Alder reaction catalyzed by hydrogen bonding. Linear free energy relationships were observed between the catalyst acidity and both the reaction rate and enantioselectivity (see picture).
Co-reporter:Keith M. Gligorich
Angewandte Chemie International Edition 2006 Volume 45(Issue 40) pp:
Publication Date(Web):20 SEP 2006
DOI:10.1002/anie.200602138
Which pathway? Aerobic Pd-catalyzed oxidation reactions couple organic-substrate oxidation to the reduction of O2. Two pathways are prevalent for the regeneration of the active PdII catalyst (see scheme; SubH2= substrate; SubOx=oxidized substrate), and recent studies have provided a deeper fundamental understanding of the interactions of Pd with O2.
Co-reporter:Matthew S. Sigman and Mitchell J. Schultz
Organic & Biomolecular Chemistry 2004 vol. 2(Issue 18) pp:2551-2554
Publication Date(Web):23 Aug 2004
DOI:10.1039/B409127M
Palladium(II)-catalyzed oxidations constitute a paramount reaction class but have remained immature over the past few decades. Recently, this field has reappeared at the forefront of organometallic catalysis. This emerging area article outlines recent developments in palladium(II)-catalyzed oxidation chemistry with discussion of potential future growth.
Co-reporter:David R. Jensen;Mitchell J. Schultz;Jaime A. Mueller Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 32) pp:
Publication Date(Web):28 JUL 2003
DOI:10.1002/anie.200351997
A breath of fresh air: A variety of alcohols are oxidized using 0.5–0.1 mol % of the catalyst, and in some cases the oxidation can simply be carried out open to the air (see scheme). Mechanistic insight into the mechanism is provided by a crystal structure that shows remarkable hydrogen bonds between the coordinated water and acetate ligands and an unprecedented large kinetic isotope effect.
Co-reporter:David R. Jensen;Mitchell J. Schultz;Jaime A. Mueller Dr.
Angewandte Chemie 2003 Volume 115(Issue 32) pp:
Publication Date(Web):28 JUL 2003
DOI:10.1002/ange.200351997
Ein Hauch frischer Luft: In Gegenwart von 0.5–0.1 Mol-% Katalysator konnte eine Vielfalt von Alkoholen oxidiert werden – in einigen Fällen sogar an der Luft (siehe Schema). Mechanistische Schlüsse können anhand einer Kristallstruktur, die auf ungewöhnliche Wasserstoffbrücken zwischen dem koordinierten Wasser und Acetatliganden hinweist, und eines starken kinetischen Isotopeneffekts gezogen werden.
Co-reporter:Mitchell J. Schultz, Candice C. Park and Matthew S. Sigman
Chemical Communications 2002 (Issue 24) pp:3034-3035
Publication Date(Web):14 Nov 2002
DOI:10.1039/B209344H
A simple Pd-catalyzed aerobic oxidation of benzylic and aliphatic alcohols to the corresponding aldehydes and ketones at room temperature is described.
Co-reporter:Erik W. Werner
Journal of the American Chemical Society () pp:
Publication Date(Web):
DOI:10.1021/ja1060998
A general, highly selective oxidative Heck reaction is reported. The reaction is high-yielding under mild conditions without the need for base or high temperatures, and the selectivity is excellent, without the requirement for electronically biased olefins or other specific directing groups. A preliminary mechanistic investigation suggests that the unusually high selectivity may be due to the catalyst’s sensitivity to C−H bond strength in the selectivity-determining β-hydride elimination step.
Co-reporter:Katrina H. Jensen ; Jonathan D. Webb
Journal of the American Chemical Society () pp:
Publication Date(Web):November 17, 2010
DOI:10.1021/ja108106h
The mechanism of an enantioselective palladium-catalyzed alkene difunctionalization reaction has been investigated. Kinetic analysis provides evidence of turnover-limiting attack of a proposed quinone methide intermediate with MeOH and suggests that copper is involved in productive product formation, not just catalyst turnover. Through examination of substrate electronic effects, a Jaffé relationship was observed correlating rate to electronic perturbation at two positions of the substrate. Ligand effects were evaluated to provide evidence of rapid ligand exchange between palladium and copper as well as a correlation between ligand electronic nature and enantioselectivity.
Co-reporter:Keith M. Gligorich and Matthew S. Sigman
Chemical Communications 2009(Issue 26) pp:
Publication Date(Web):
DOI:10.1039/B902868D
Co-reporter:Benjamin J. Stokes, Amanda J. Bischoff and Matthew S. Sigman
Chemical Science (2010-Present) 2014 - vol. 5(Issue 6) pp:NaN2339-2339
Publication Date(Web):2014/03/17
DOI:10.1039/C4SC00602J
Pd-catalyzed allylic relay Suzuki cross-coupling reactions of secondary alkyl tosylates, featuring a sterically-hindered oxidative addition and precise control of β-hydride elimination, are reported. The identification of a linear free energy relationship between the relative rates of substrate consumption and the electronic nature of the substrate alkene suggests that the oxidative addition requires direct alkene involvement. A study of the effect of alkyl chain length on the reaction outcome supports a chelation-controlled oxidative addition.
Co-reporter:Elizabeth N. Bess, David M. Guptill, Huw M. L. Davies and Matthew S. Sigman
Chemical Science (2010-Present) 2015 - vol. 6(Issue 5) pp:NaN3062-3062
Publication Date(Web):2015/03/18
DOI:10.1039/C5SC00357A
Achieving selective C–H functionalization is a significant challenge that requires discrimination between many similar C–H bonds. Yet, reaction systems employing Rh2(DOSP)4 and Rh2(BPCP)4 were recently demonstrated to afford high levels of selectivity in the C–H insertion of carbenes into toluene-derived substrates. Herein, we explore the origin of this selectivity through a systematic analysis of substrate and reagent features that alter levels of selectivity from 20:1 to 1:610 for secondary (or tertiary)-to-primary benzylic C–H functionalization of toluene derivatives. Describing this variation using infrared vibrations and point charges, we have developed a mathematical model from which are identified features of the systems that determine levels of site-selectivity and are applied as predictive factors to describe the selectivity behavior of new substrate/reagent combinations.
Co-reporter:Katrina H. Jensen and Matthew S. Sigman
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 22) pp:NaN4088-4088
Publication Date(Web):2008/10/09
DOI:10.1039/B813246A
Alkene difunctionalization, the addition of two functional groups across a double bond, exemplifies a class of reactions with significant synthetic potential. This emerging area examines recent developments of palladium-catalyzed difunctionalization reactions, with a focus on mechanistic strategies that allow for functionalization of a common palladium alkyl intermediate.
Co-reporter:Matthew S. McCammant and Matthew S. Sigman
Chemical Science (2010-Present) 2015 - vol. 6(Issue 2) pp:NaN1361-1361
Publication Date(Web):2014/11/28
DOI:10.1039/C4SC03074E
Palladium-catalyzed 1,4-difunctionalizations of isoprene that produce skipped polyenes are reported. Complex isomeric product mixtures are possible as a result of the difficult-to-control migratory insertion of isoprene into a Pd–alkenyl bond, but good site selectivity has been achieved using easily accessible pyrox ligands. Mechanistic studies suggest that the control of insertion is the result of the unique electronic asymmetry and steric properties of the ligand.
.jpg)
![3-Pyridinecarboxylic acid, 6-[(4S)-4-(1,1-dimethylethyl)-4,5-dihydro-2-oxazolyl]-, methyl ester](/data/chemimg/3721700/1117792-25-2.png)
![3-Pyridinecarboxylic acid, 6-[(4S)-4-(1,1-dimethylethyl)-4,5-dihydro-2-oxazolyl]-, methyl ester](/data/chemimg/3721700/1117792-25-2_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2_b.png)
![2-Azatricyclo[3.3.1.13,7]dec-2-yloxy, 1-methyl-](http://img.cochemist.com/ccimg/872600/872598-44-2.png)
![2-Azatricyclo[3.3.1.13,7]dec-2-yloxy, 1-methyl-](http://img.cochemist.com/ccimg/872600/872598-44-2_b.png)
![2-Propenoic acid, [3,5-bis(trifluoromethyl)phenyl]methyl ester](http://img.cochemist.com/ccimg/862500/862498-03-1.png)
![2-Propenoic acid, [3,5-bis(trifluoromethyl)phenyl]methyl ester](http://img.cochemist.com/ccimg/862500/862498-03-1_b.png)
![Boronic acid, [2-[4-(trifluoromethyl)phenyl]ethenyl]-](http://img.cochemist.com/ccimg/851700/851660-03-2.png)
![Boronic acid, [2-[4-(trifluoromethyl)phenyl]ethenyl]-](http://img.cochemist.com/ccimg/851700/851660-03-2_b.png)
![[1-(tert-Butoxycarbonyl)-1,2,3,6-tetrahydropyridine-4-yl]boronic acid](http://img.cochemist.com/ccimg/844600/844501-00-4.png)
![[1-(tert-Butoxycarbonyl)-1,2,3,6-tetrahydropyridine-4-yl]boronic acid](http://img.cochemist.com/ccimg/844600/844501-00-4_b.png)
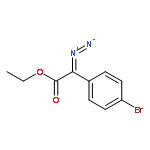
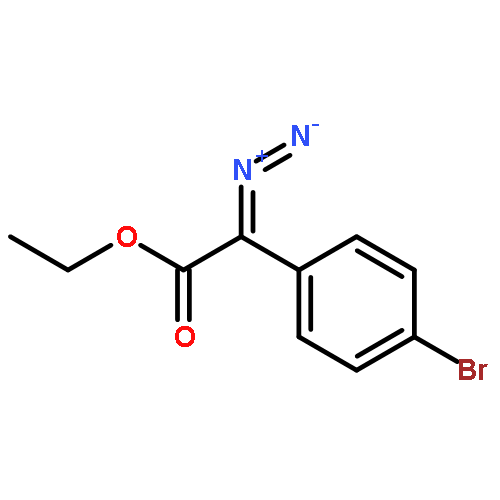
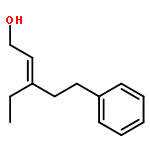
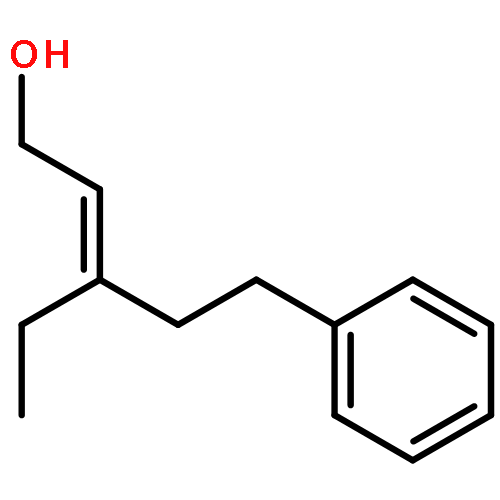
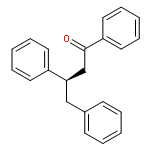
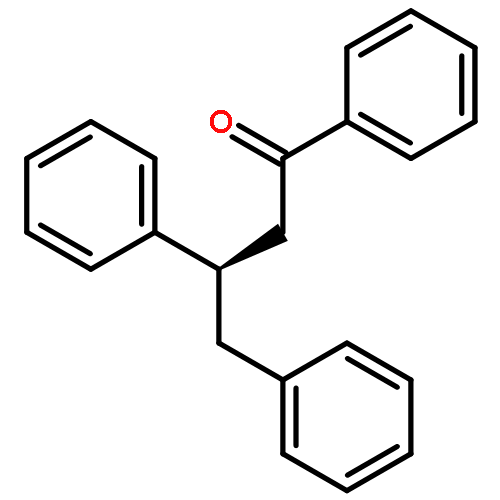


![[1,1'-Biphenyl]-3-carbonitrile, 4-fluoro-](http://img.cochemist.com/ccimg/544700/544673-44-1.png)
![[1,1'-Biphenyl]-3-carbonitrile, 4-fluoro-](http://img.cochemist.com/ccimg/544700/544673-44-1_b.png)
![2-Propenoic acid, 2-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/500200/500131-06-6.png)
![2-Propenoic acid, 2-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/500200/500131-06-6_b.png)


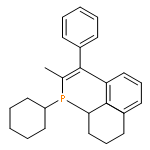
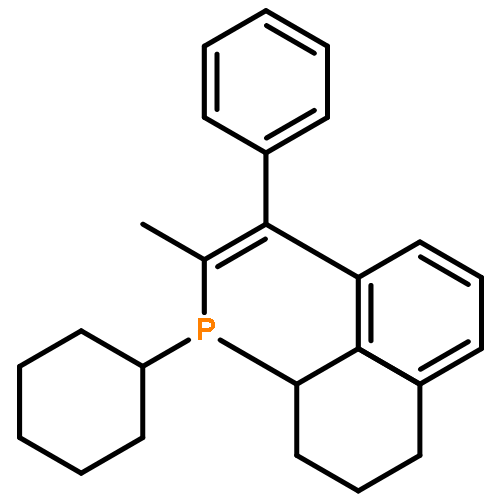
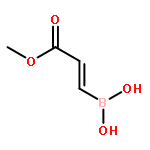
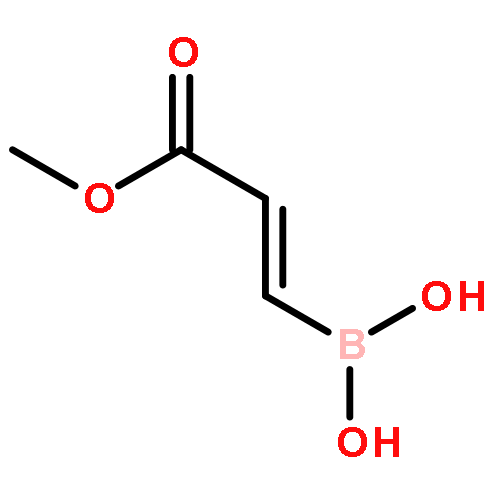
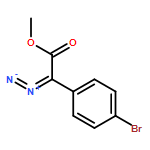

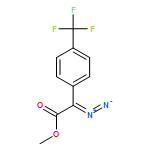

![Boronic acid, [2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/215000/214907-25-2.png)
![Boronic acid, [2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/215000/214907-25-2_b.png)
![Rhodium, tetrakis[m-[1-[(4-dodecylphenyl)sulfonyl]-L-prolinato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/179200/179162-34-6.png)
![Rhodium, tetrakis[m-[1-[(4-dodecylphenyl)sulfonyl]-L-prolinato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/179200/179162-34-6_b.png)
![1-Piperidinyloxy, 2,2,6,6-tetramethyl-4-[(phenylmethyl)amino]-](http://img.cochemist.com/ccimg/178500/178445-77-7.png)
![1-Piperidinyloxy, 2,2,6,6-tetramethyl-4-[(phenylmethyl)amino]-](http://img.cochemist.com/ccimg/178500/178445-77-7_b.png)
![1H-Isoindole-1,3(2H)-dione, 2-[(5E)-7-hydroxy-5-heptenyl]-](http://img.cochemist.com/ccimg/174200/174151-99-6.png)
![1H-Isoindole-1,3(2H)-dione, 2-[(5E)-7-hydroxy-5-heptenyl]-](http://img.cochemist.com/ccimg/174200/174151-99-6_b.png)











![2-(4-BROMOPHENOXY)-N-[2-(DIETHYLAMINO)ETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/79700/79676-59-8.png)
![2-(4-BROMOPHENOXY)-N-[2-(DIETHYLAMINO)ETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/79700/79676-59-8_b.png)
![9-Azabicyclo[3.3.1]nonan-3-amine, 9-(phenylmethyl)-, (3-endo)-](/data/chemimg/1968700/76272-58-7.png)
![9-Azabicyclo[3.3.1]nonan-3-amine, 9-(phenylmethyl)-, (3-endo)-](/data/chemimg/1968700/76272-58-7_b.png)
![1H-Benz[g]indole, 1-methyl-](http://img.cochemist.com/ccimg/57600/57582-30-6.png)
![1H-Benz[g]indole, 1-methyl-](http://img.cochemist.com/ccimg/57600/57582-30-6_b.png)











![Benzene, [(1E)-2-iodoethenyl]-](http://img.cochemist.com/ccimg/42600/42599-24-6.png)
![Benzene, [(1E)-2-iodoethenyl]-](http://img.cochemist.com/ccimg/42600/42599-24-6_b.png)


![BENZOIC ACID, 4-[2,2,2-TRIFLUORO-1,1-BIS(TRIFLUOROMETHYL)ETHYL]-](http://img.cochemist.com/ccimg/41200/41125-54-6.png)
![BENZOIC ACID, 4-[2,2,2-TRIFLUORO-1,1-BIS(TRIFLUOROMETHYL)ETHYL]-](http://img.cochemist.com/ccimg/41200/41125-54-6_b.png)




![8-Azabicyclo[3.2.1]octan-3-one, 1,5-dimethyl-](http://img.cochemist.com/ccimg/38400/38390-77-1.png)
![8-Azabicyclo[3.2.1]octan-3-one, 1,5-dimethyl-](http://img.cochemist.com/ccimg/38400/38390-77-1_b.png)
![8-Azabicyclo[3.2.1]oct-8-yloxy,1,5-dimethyl-3-oxo-](http://img.cochemist.com/ccimg/34100/34061-60-4.png)
![8-Azabicyclo[3.2.1]oct-8-yloxy,1,5-dimethyl-3-oxo-](http://img.cochemist.com/ccimg/34100/34061-60-4_b.png)


















![9-WEI <SUP>1</SUP>-OXIDANYL-9-AZABICYCLO[3.3.1]NONAN-3-ONE](http://img.cochemist.com/ccimg/7200/7123-92-4.png)
![9-WEI <SUP>1</SUP>-OXIDANYL-9-AZABICYCLO[3.3.1]NONAN-3-ONE](http://img.cochemist.com/ccimg/7200/7123-92-4_b.png)


![BENZOIC ACID, 4-[(TRIMETHYLSILYL)OXY]-](http://img.cochemist.com/ccimg/6700/6651-63-4.png)
![BENZOIC ACID, 4-[(TRIMETHYLSILYL)OXY]-](http://img.cochemist.com/ccimg/6700/6651-63-4_b.png)




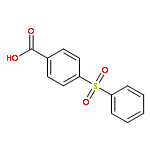
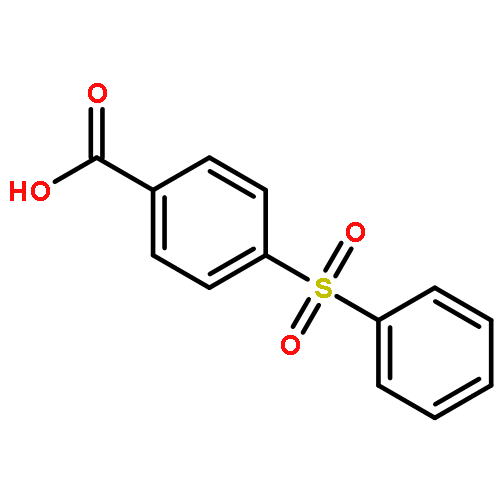
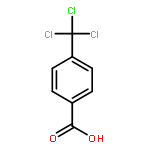
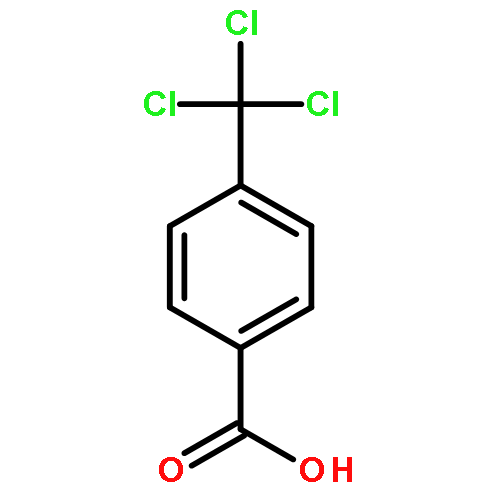






![Benzoic acid, 3-[(trifluoromethyl)sulfonyl]-](http://img.cochemist.com/ccimg/1000/952-69-2.png)
![Benzoic acid, 3-[(trifluoromethyl)sulfonyl]-](http://img.cochemist.com/ccimg/1000/952-69-2_b.png)



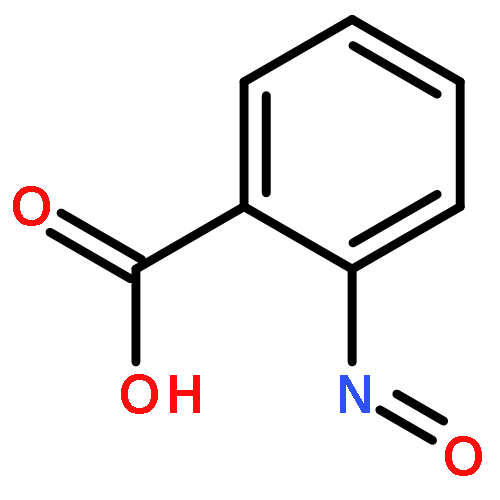
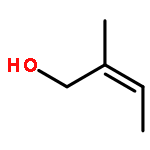
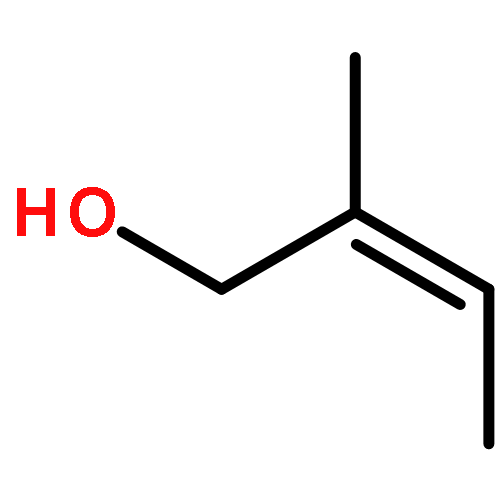
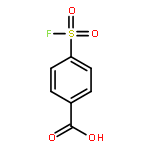

![4-[(3-CHLOROPHENOXY)METHYL]-1-PHENYL-1H-1,2,3-TRIAZOLE](http://img.cochemist.com/ccimg/400/312-22-1.png)
![4-[(3-CHLOROPHENOXY)METHYL]-1-PHENYL-1H-1,2,3-TRIAZOLE](http://img.cochemist.com/ccimg/400/312-22-1_b.png)












![1,3,2-Dioxaborolane, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/157800/157731-58-3.png)
![1,3,2-Dioxaborolane, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/157800/157731-58-3_b.png)
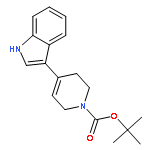
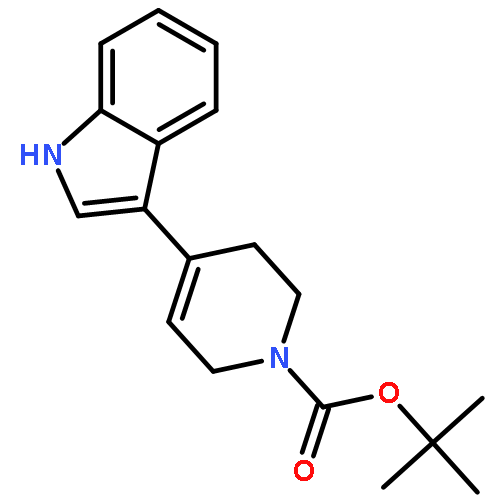




![(+)-2,2'-Isopropylidenebis[(4R)-4-phenyl-2-oxazoline]](/data/chemimg/116500/150529-93-4.png)
![(+)-2,2'-Isopropylidenebis[(4R)-4-phenyl-2-oxazoline]](/data/chemimg/116500/150529-93-4_b.png)


![1-Propanol, 3-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/147600/147509-22-6.png)
![1-Propanol, 3-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/147600/147509-22-6_b.png)
![Pyrrolo[2,1-a]isoquinoline-1-carboxylic acid, methyl ester](http://img.cochemist.com/ccimg/145500/145474-62-0.png)
![Pyrrolo[2,1-a]isoquinoline-1-carboxylic acid, methyl ester](http://img.cochemist.com/ccimg/145500/145474-62-0_b.png)


![2-Butenoic acid, 3-[[(trifluoromethyl)sulfonyl]oxy]-, ethyl ester](http://img.cochemist.com/ccimg/137300/137273-43-9.png)
![2-Butenoic acid, 3-[[(trifluoromethyl)sulfonyl]oxy]-, ethyl ester](http://img.cochemist.com/ccimg/137300/137273-43-9_b.png)
![1-[2-(DIMETHYLAMINO)-1-NAPHTHYL]-N,N-DIMETHYL-NAPHTHALEN-2-AMINE](http://img.cochemist.com/ccimg/135800/135759-57-8.png)
![1-[2-(DIMETHYLAMINO)-1-NAPHTHYL]-N,N-DIMETHYL-NAPHTHALEN-2-AMINE](http://img.cochemist.com/ccimg/135800/135759-57-8_b.png)




![INDENO[1,2-B]INDOLE, 5,10-DIHYDRO-10-METHYL-](http://img.cochemist.com/ccimg/133600/133505-09-6.png)
![INDENO[1,2-B]INDOLE, 5,10-DIHYDRO-10-METHYL-](http://img.cochemist.com/ccimg/133600/133505-09-6_b.png)
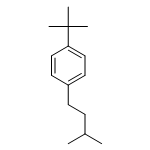
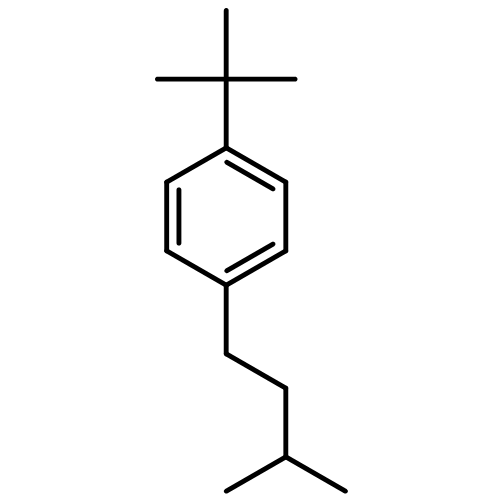










![2-Butenoic acid, 3-[[(trifluoromethyl)sulfonyl]oxy]-, ethyl ester, (2E)-](http://img.cochemist.com/ccimg/122200/122135-96-0.png)
![2-Butenoic acid, 3-[[(trifluoromethyl)sulfonyl]oxy]-, ethyl ester, (2E)-](http://img.cochemist.com/ccimg/122200/122135-96-0_b.png)
![2-Butenoic acid, 3-[[(trifluoromethyl)sulfonyl]oxy]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/122200/122135-84-6.png)
![2-Butenoic acid, 3-[[(trifluoromethyl)sulfonyl]oxy]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/122200/122135-84-6_b.png)


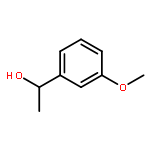
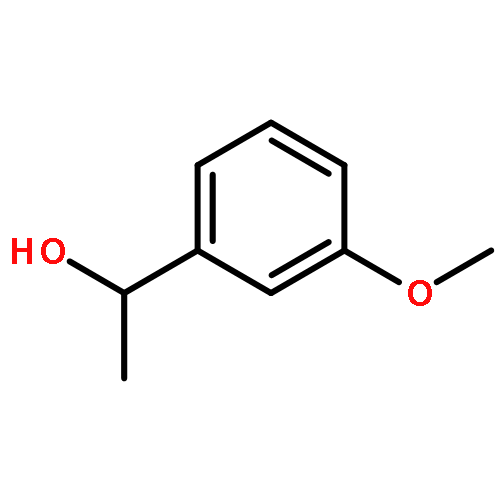
![5-Hepten-2-ol, 7-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-6-methyl-, (E)-(±)-](http://img.cochemist.com/ccimg/119800/119769-89-0.png)
![5-Hepten-2-ol, 7-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-6-methyl-, (E)-(±)-](http://img.cochemist.com/ccimg/119800/119769-89-0_b.png)


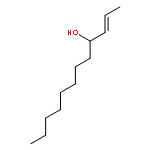
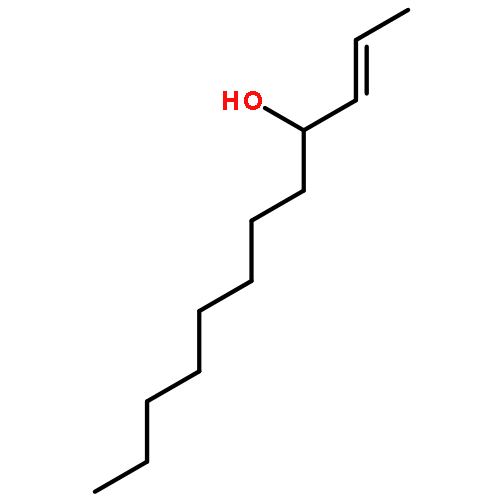


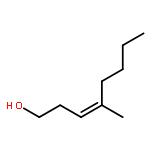
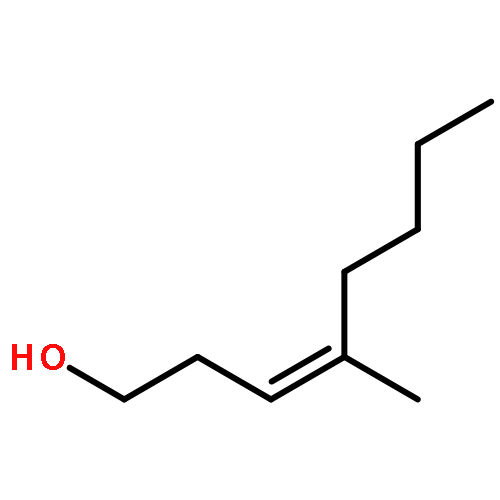
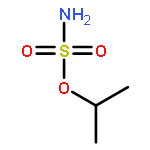
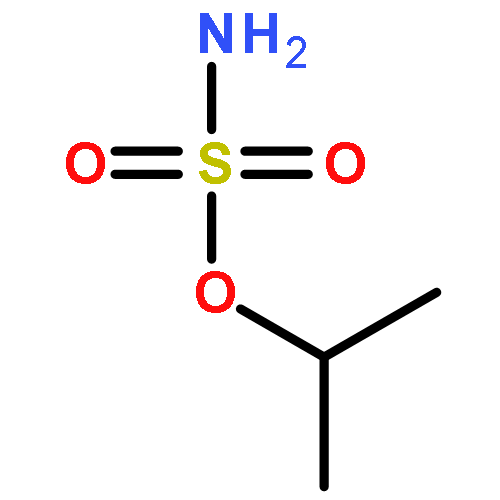
![1-Propanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/118000/117932-70-4.png)
![1-Propanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/118000/117932-70-4_b.png)

















![Isoquinoline, 2-[4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]-1,2,3,4-tetrahydro-, (S)- (9CI)](/data/chemimg/1033300/102922-30-5.png)
![Isoquinoline, 2-[4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]-1,2,3,4-tetrahydro-, (S)- (9CI)](/data/chemimg/1033300/102922-30-5_b.png)
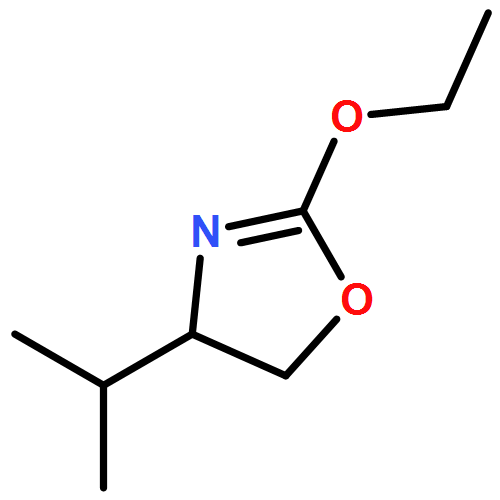











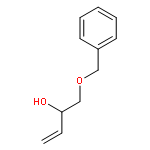
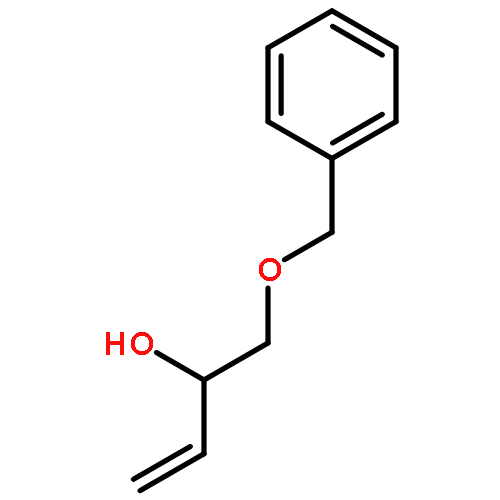
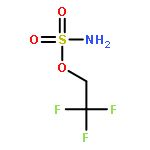
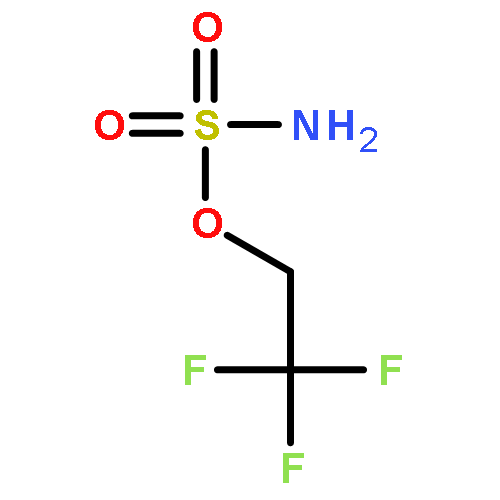
![4-[2-ethyl-1-(4-hydroxyphenyl)butyl]phenol](http://img.cochemist.com/ccimg/92600/92569-29-4.png)
![4-[2-ethyl-1-(4-hydroxyphenyl)butyl]phenol](http://img.cochemist.com/ccimg/92600/92569-29-4_b.png)
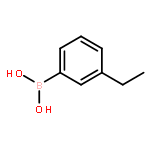



![Methanesulfonic acid, 1,1,1-trifluoro-, 1,7,7-trimethylbicyclo[2.2.1]hept-2-en-2-yl ester](/data/chemimg/2010400/86544-72-1.png)
![Methanesulfonic acid, 1,1,1-trifluoro-, 1,7,7-trimethylbicyclo[2.2.1]hept-2-en-2-yl ester](/data/chemimg/2010400/86544-72-1_b.png)




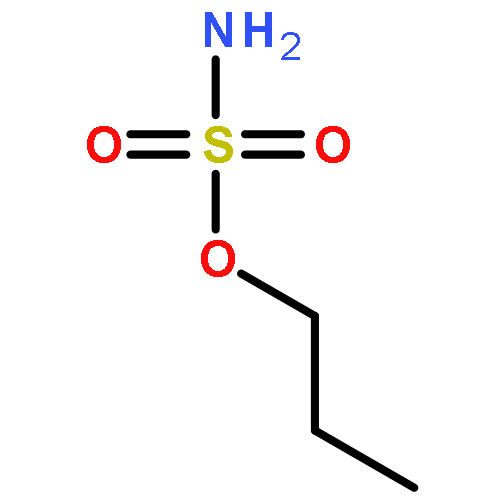

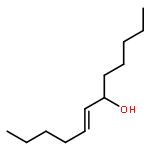
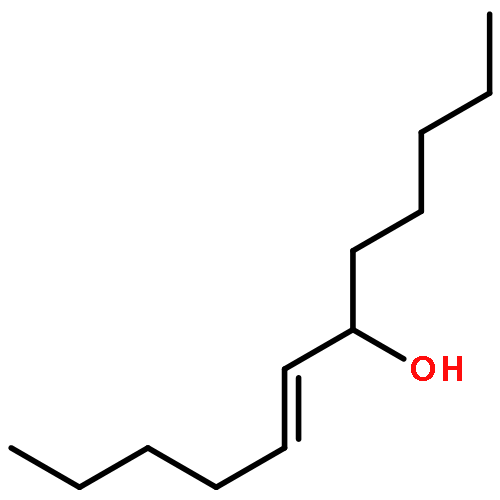




![Aluminum, dimethyl[(1E)-2-methyl-1-hexenyl]-](http://img.cochemist.com/ccimg/78000/77958-35-1.png)
![Aluminum, dimethyl[(1E)-2-methyl-1-hexenyl]-](http://img.cochemist.com/ccimg/78000/77958-35-1_b.png)


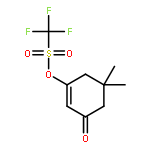
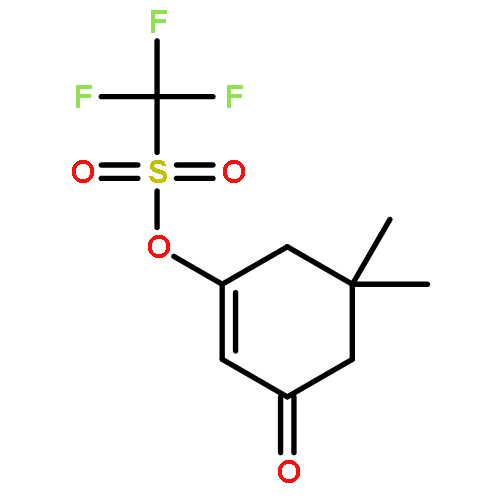








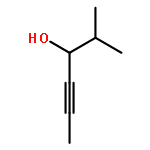
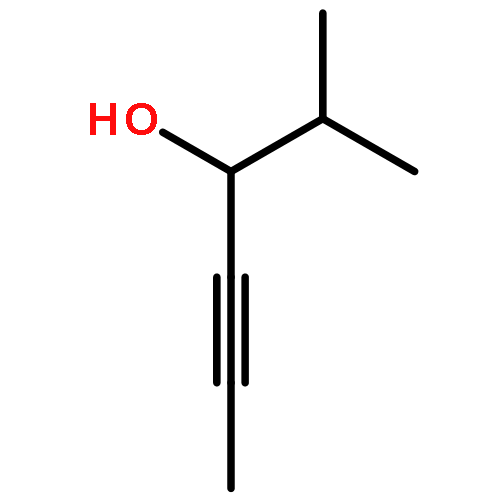
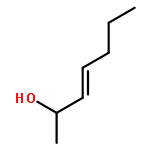
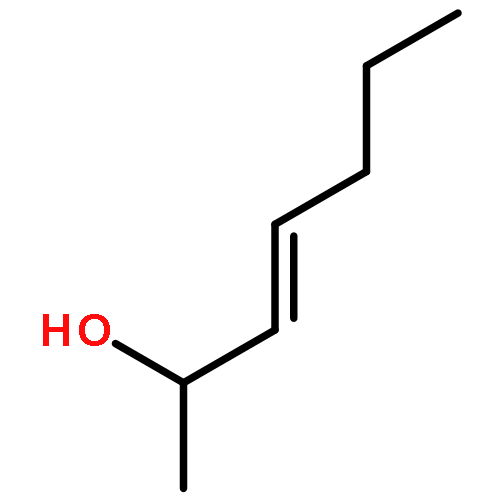
![Carbamic acid, [(4-methylphenyl)sulfonyl]-, 3-butynyl ester](http://img.cochemist.com/ccimg/64000/63924-49-2.png)
![Carbamic acid, [(4-methylphenyl)sulfonyl]-, 3-butynyl ester](http://img.cochemist.com/ccimg/64000/63924-49-2_b.png)
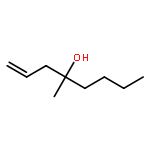
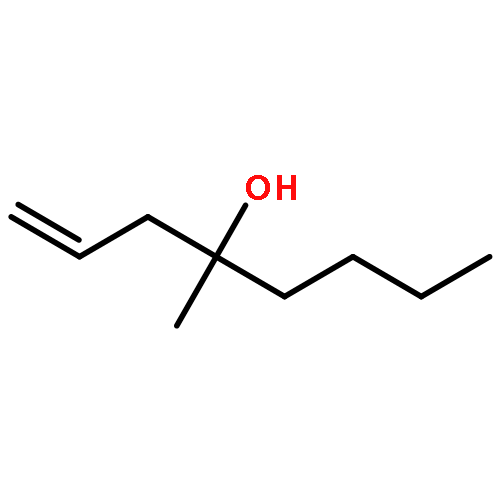


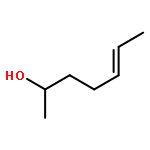
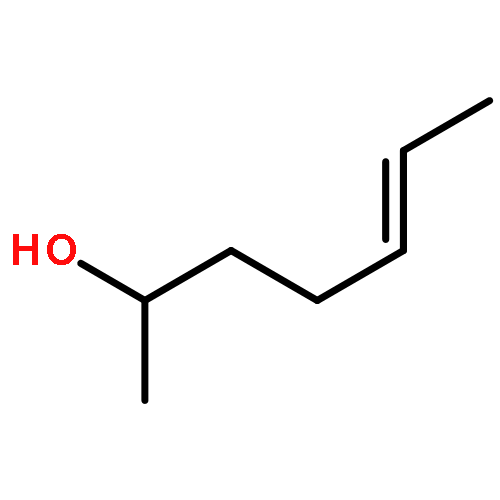
![2-Azatricyclo[3.3.1.1<sup>3,7</sup>]dec-2-yloxidanyl](http://img.cochemist.com/ccimg/57700/57625-08-8.png)
![2-Azatricyclo[3.3.1.1<sup>3,7</sup>]dec-2-yloxidanyl](http://img.cochemist.com/ccimg/57700/57625-08-8_b.png)


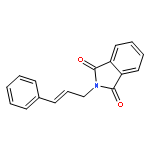
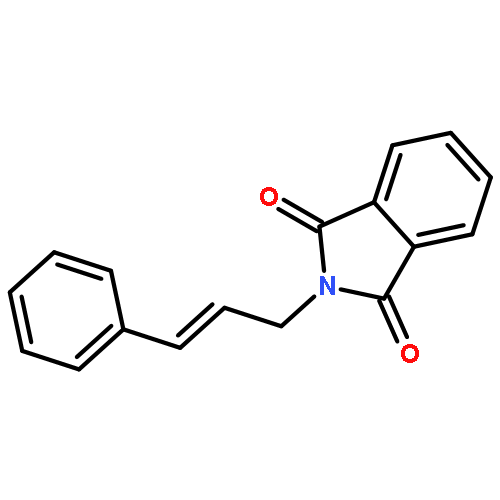


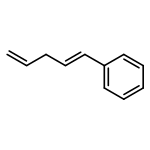

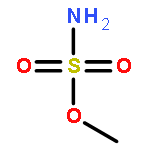
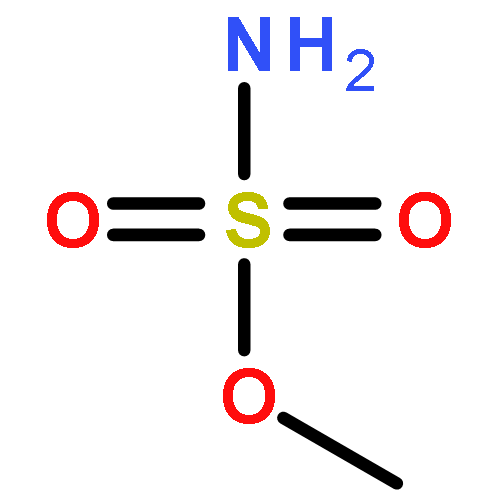
![Benzene, [1-(1-cyclohexen-1-yl)ethyl]-](/data/chemimg/3749200/54774-82-2.png)
![Benzene, [1-(1-cyclohexen-1-yl)ethyl]-](/data/chemimg/3749200/54774-82-2_b.png)
![Benzene, [(1Z)-4-methyl-1-penten-1-yl]-](/data/chemimg/1844100/54624-34-9.png)
![Benzene, [(1Z)-4-methyl-1-penten-1-yl]-](/data/chemimg/1844100/54624-34-9_b.png)







![Silane, [(1E)-1-heptenyloxy]trimethyl-](http://img.cochemist.com/ccimg/52200/52186-50-2.png)
![Silane, [(1E)-1-heptenyloxy]trimethyl-](http://img.cochemist.com/ccimg/52200/52186-50-2_b.png)


![Silane, [(1Z)-1-heptenyloxy]trimethyl-](http://img.cochemist.com/ccimg/50400/50300-19-1.png)
![Silane, [(1Z)-1-heptenyloxy]trimethyl-](http://img.cochemist.com/ccimg/50400/50300-19-1_b.png)
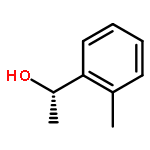
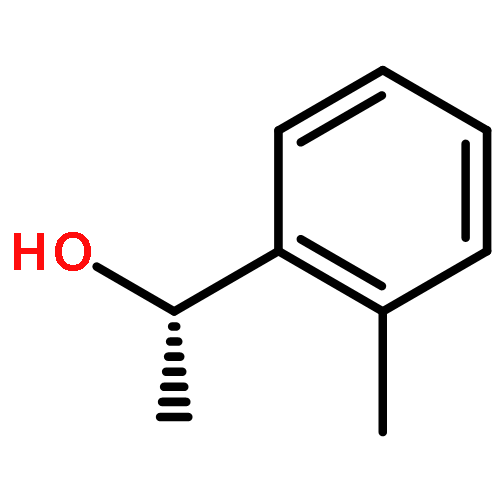


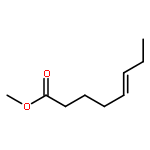
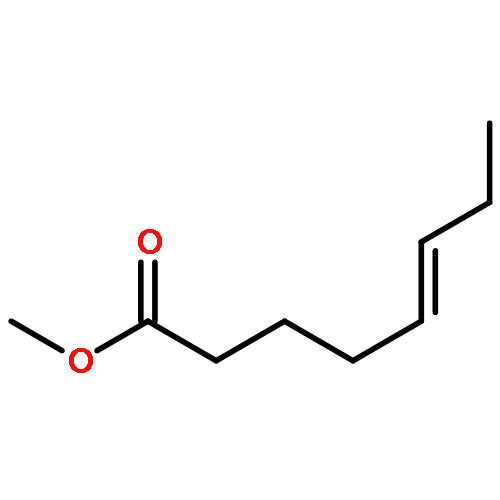















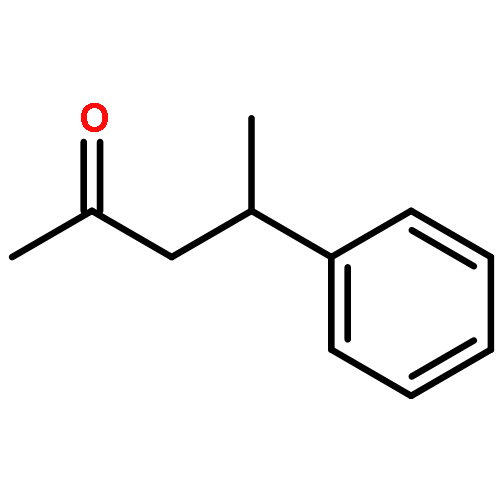
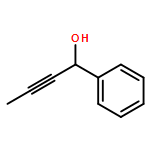
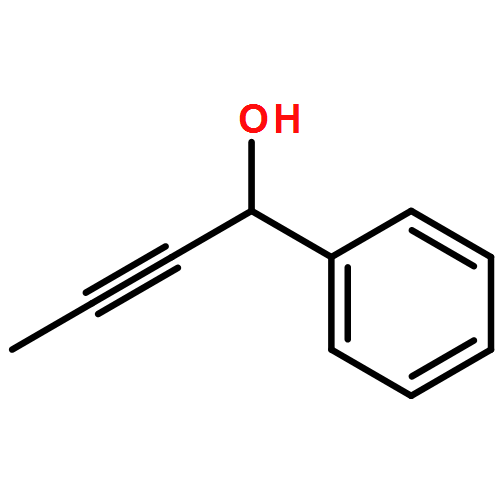
![9-Azabicyclo[3.3.1]non-9-yloxy](/data/chemimg/1187500/31785-68-9.png)
![9-Azabicyclo[3.3.1]non-9-yloxy](/data/chemimg/1187500/31785-68-9_b.png)



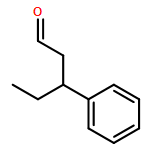
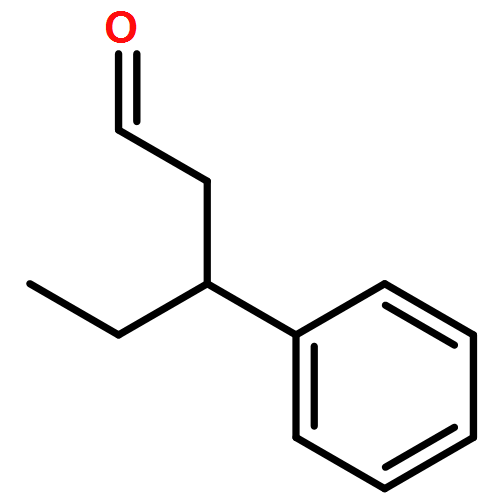
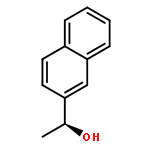

![Benzene, 1,1'-[(1E)-1-butene-1,4-diyl]bis-](/data/chemimg/1171700/27066-35-9.png)
![Benzene, 1,1'-[(1E)-1-butene-1,4-diyl]bis-](/data/chemimg/1171700/27066-35-9_b.png)
](http://img.cochemist.com/ccimg/27000/26913-06-4.png)
](http://img.cochemist.com/ccimg/27000/26913-06-4_b.png)
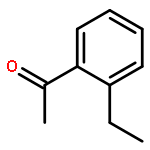
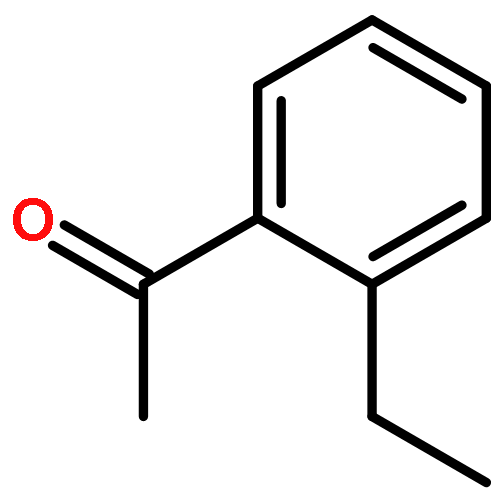




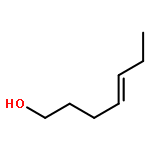
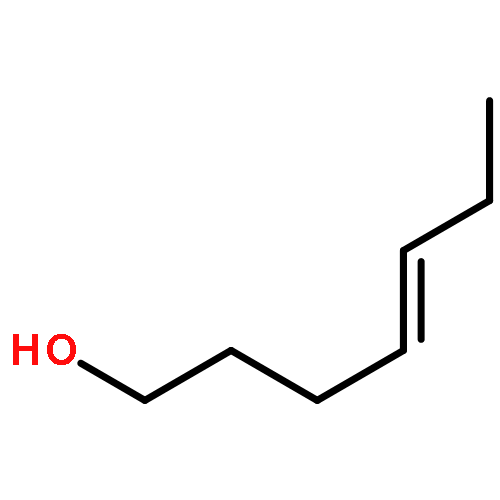
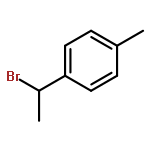


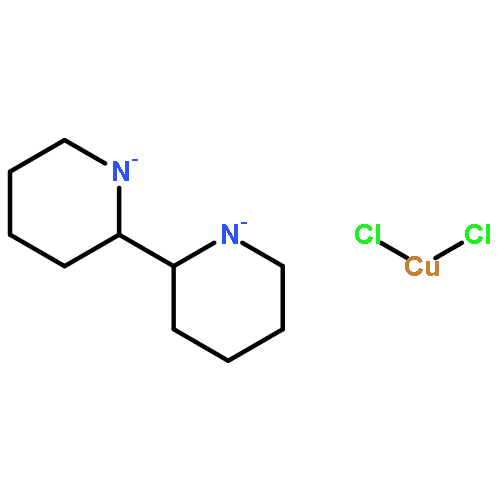
![Benzene, 1-methoxy-4-[(1E,3E)-4-phenyl-1,3-butadien-1-yl]-](/data/chemimg/1794300/22145-08-0.png)
![Benzene, 1-methoxy-4-[(1E,3E)-4-phenyl-1,3-butadien-1-yl]-](/data/chemimg/1794300/22145-08-0_b.png)
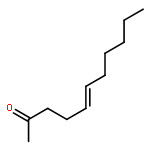
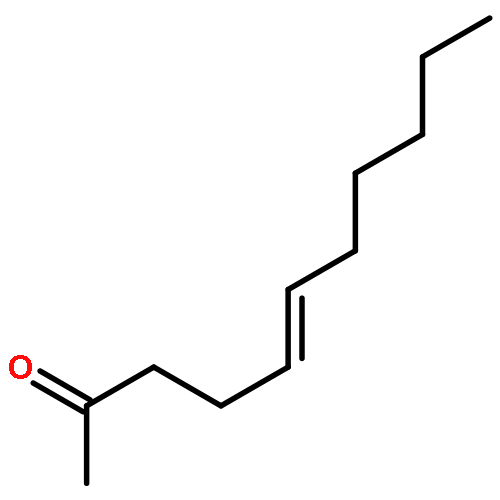
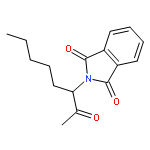









![Benzene, 1-chloro-4-[(1E)-2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/18900/18878-89-2.png)
![Benzene, 1-chloro-4-[(1E)-2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/18900/18878-89-2_b.png)


![Butanoic acid, 3-oxo-2-[(trimethylsilyl)methyl]-, ethyl ester](/data/chemimg/2152500/17906-77-3.png)
![Butanoic acid, 3-oxo-2-[(trimethylsilyl)methyl]-, ethyl ester](/data/chemimg/2152500/17906-77-3_b.png)




![6-chlorodibenzo[d,f][1,3,2]dioxaphosphepine](http://img.cochemist.com/ccimg/16700/16611-68-0.png)
![6-chlorodibenzo[d,f][1,3,2]dioxaphosphepine](http://img.cochemist.com/ccimg/16700/16611-68-0_b.png)

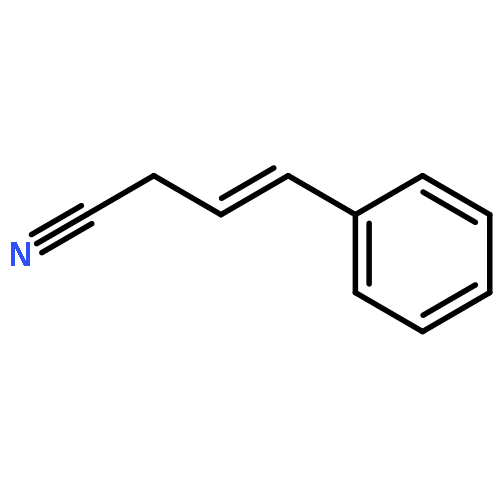


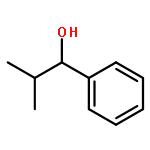
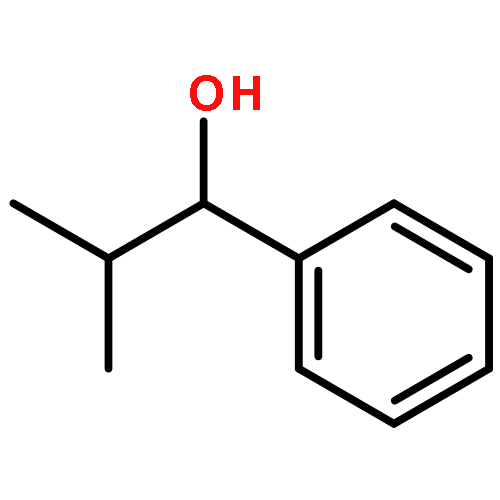
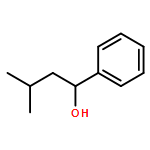

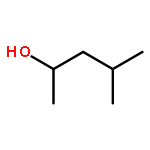

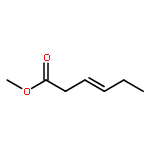
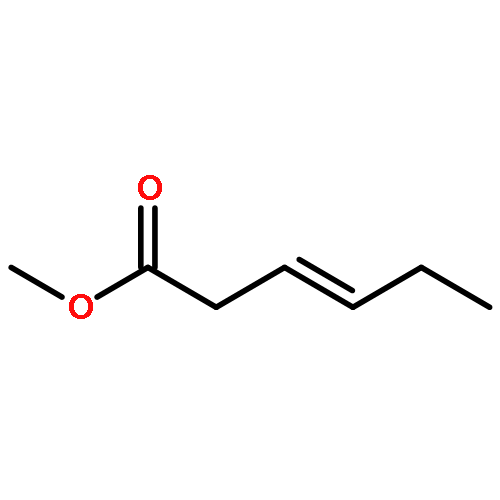








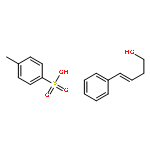
![Benzene, [(1E)-4-bromo-1-butenyl]-](http://img.cochemist.com/ccimg/7600/7515-41-5.png)
![Benzene, [(1E)-4-bromo-1-butenyl]-](http://img.cochemist.com/ccimg/7600/7515-41-5_b.png)












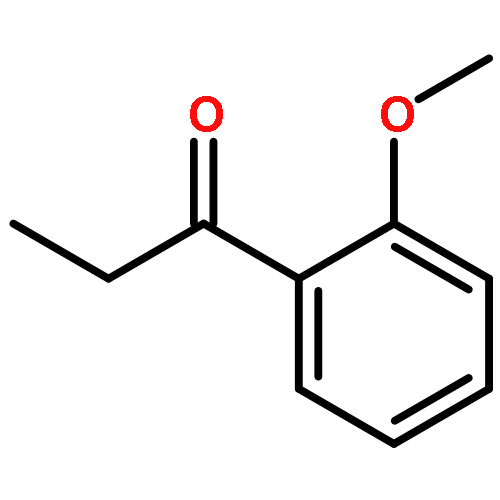


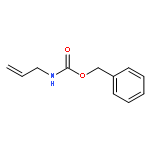



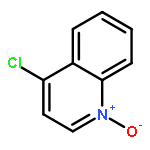
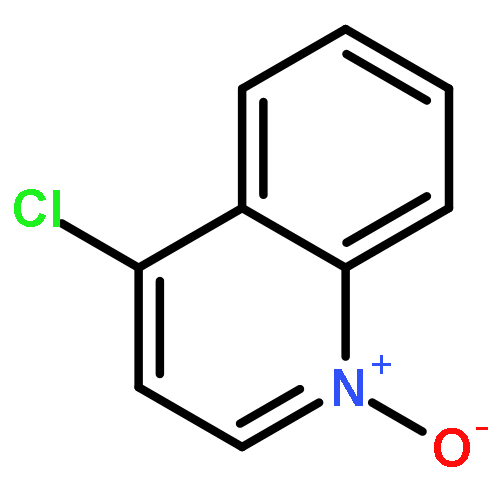
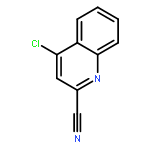
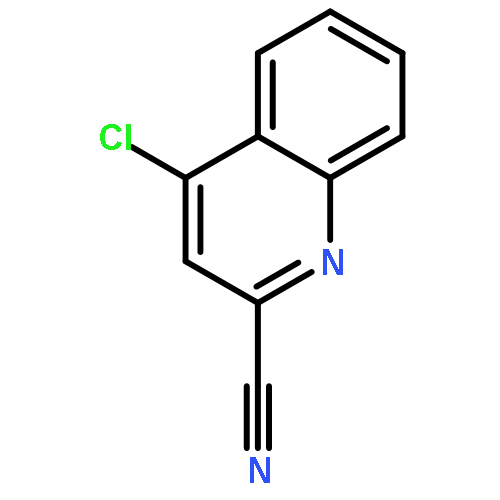




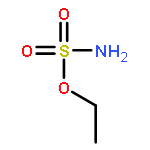
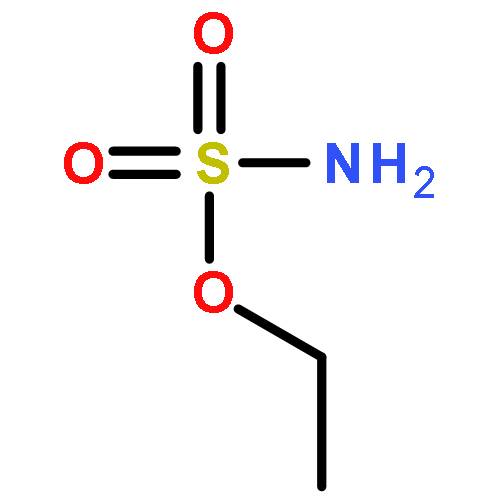





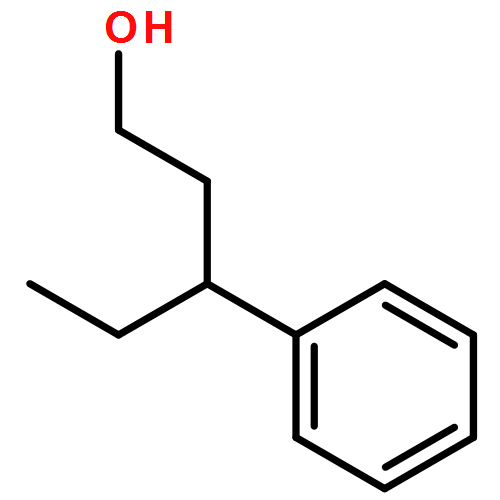
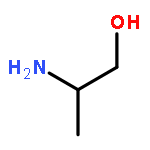
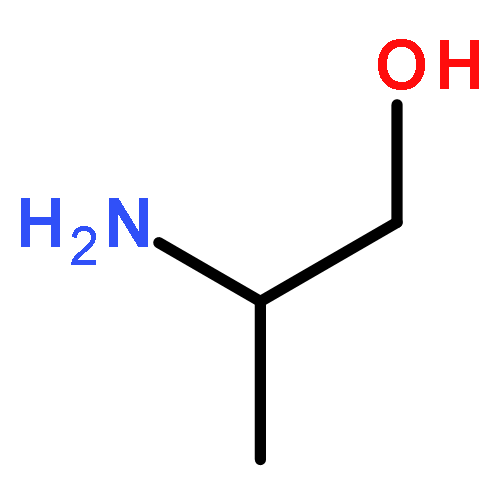






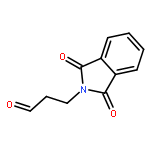
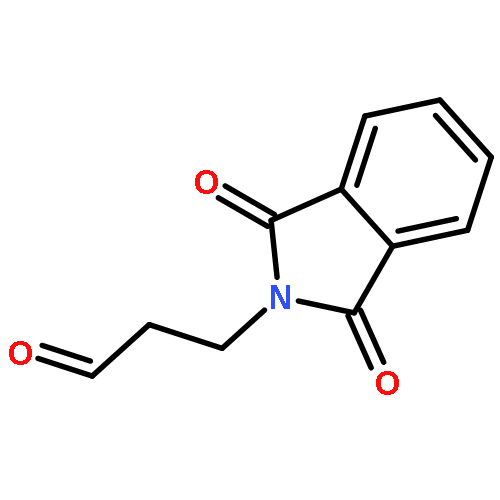
![1-[2-(propan-2-yl)phenyl]ethanone](http://img.cochemist.com/ccimg/2200/2142-65-6.png)
![1-[2-(propan-2-yl)phenyl]ethanone](http://img.cochemist.com/ccimg/2200/2142-65-6_b.png)




![1-methoxy-2-[(1E)-prop-1-en-1-yl]benzene](http://img.cochemist.com/ccimg/2100/2077-36-3.png)
![1-methoxy-2-[(1E)-prop-1-en-1-yl]benzene](http://img.cochemist.com/ccimg/2100/2077-36-3_b.png)


![4-[1-(4-hydroxyphenyl)propyl]phenol](http://img.cochemist.com/ccimg/1600/1576-13-2.png)
![4-[1-(4-hydroxyphenyl)propyl]phenol](http://img.cochemist.com/ccimg/1600/1576-13-2_b.png)
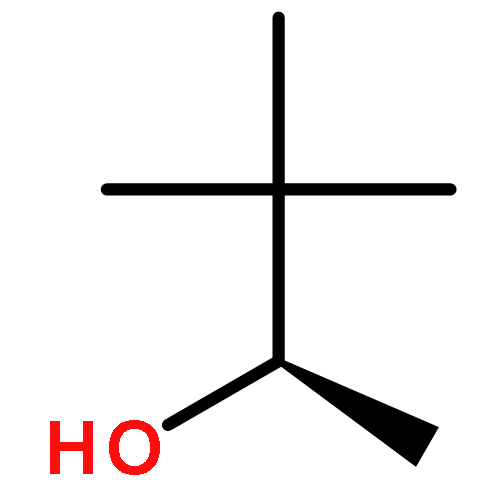




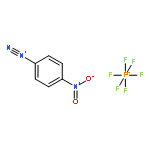
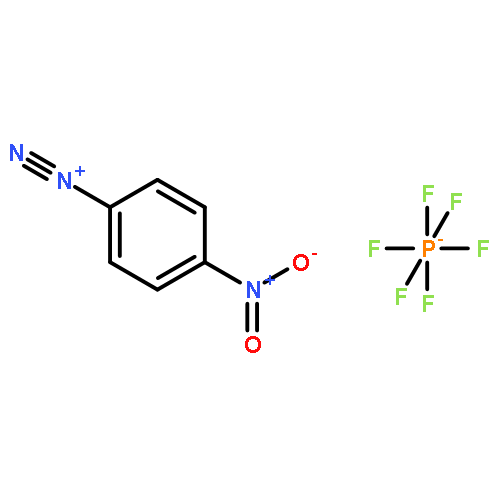
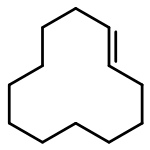








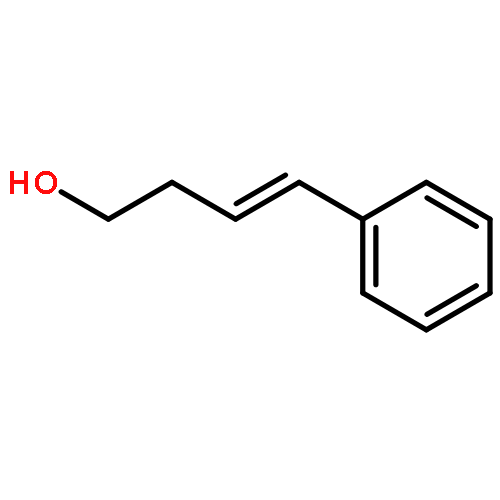
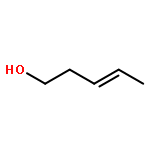
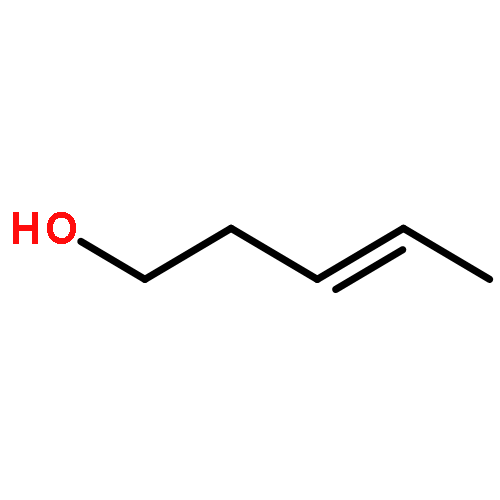

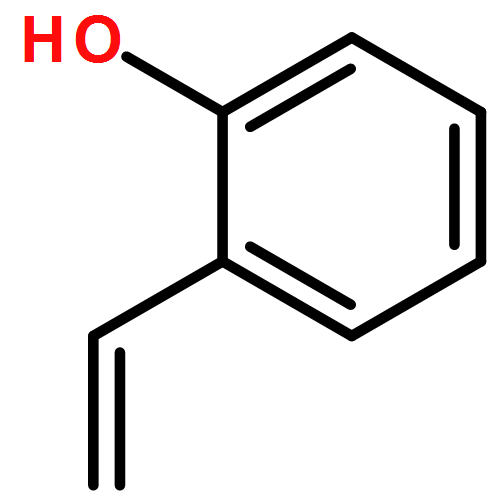





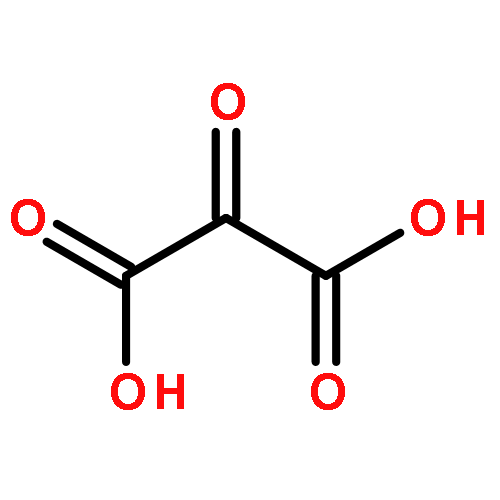






![Pyridine, 2-[(4S)-4-(1,1-dimethylethyl)-4,5-dihydro-2-oxazolyl]-4-(trifluoromethyl)-](/data/chemimg/3721600/1257527-14-2.png)
![Pyridine, 2-[(4S)-4-(1,1-dimethylethyl)-4,5-dihydro-2-oxazolyl]-4-(trifluoromethyl)-](/data/chemimg/3721600/1257527-14-2_b.png)
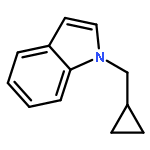

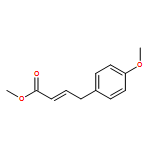
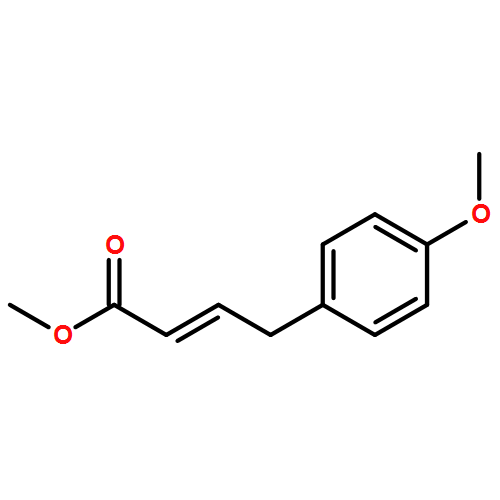
![Pyridine, 4-chloro-2-[(4S)-4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]-](/data/chemimg/3721600/235742-70-8.png)
![Pyridine, 4-chloro-2-[(4S)-4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]-](/data/chemimg/3721600/235742-70-8_b.png)
![Benzene, 1-methoxy-4-[(2E)-1-methyl-3-phenyl-2-propen-1-yl]-](/data/chemimg/138000/223510-20-1.png)
![Benzene, 1-methoxy-4-[(2E)-1-methyl-3-phenyl-2-propen-1-yl]-](/data/chemimg/138000/223510-20-1_b.png)


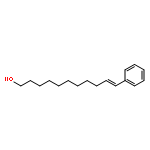
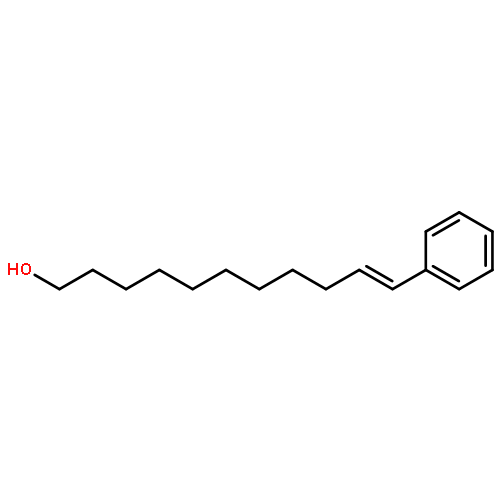
![Propanal, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/144700/144681-67-4.png)
![Propanal, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/144700/144681-67-4_b.png)


![Pyridine, 2-[(4S)-4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]-](/data/chemimg/108000/108915-04-4.png)
![Pyridine, 2-[(4S)-4,5-dihydro-4-(1-methylethyl)-2-oxazolyl]-](/data/chemimg/108000/108915-04-4_b.png)
![Benzene, 1,1'-[(1E)-1-pentene-1,5-diyl]bis-](http://img.cochemist.com/ccimg/97500/97455-11-3.png)
![Benzene, 1,1'-[(1E)-1-pentene-1,5-diyl]bis-](http://img.cochemist.com/ccimg/97500/97455-11-3_b.png)
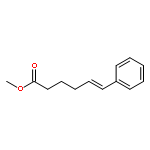

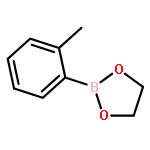
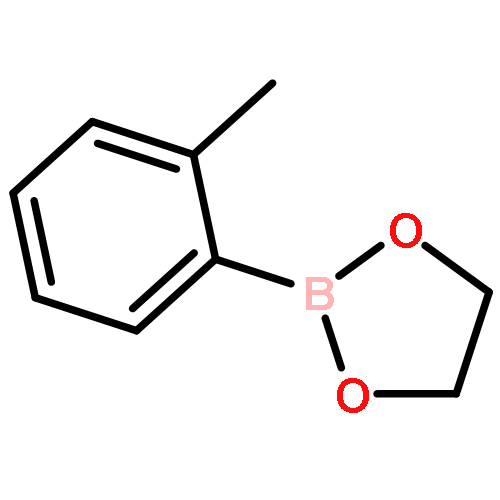
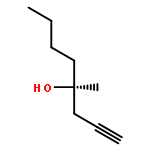
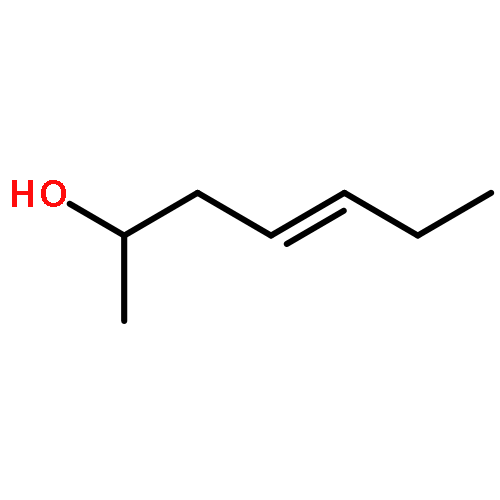



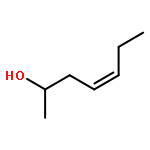
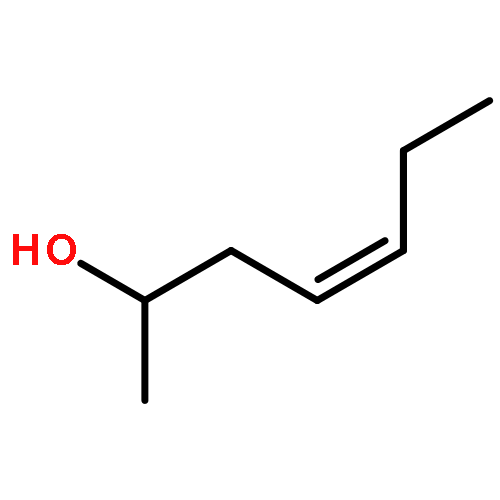


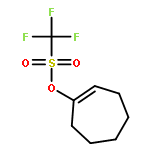
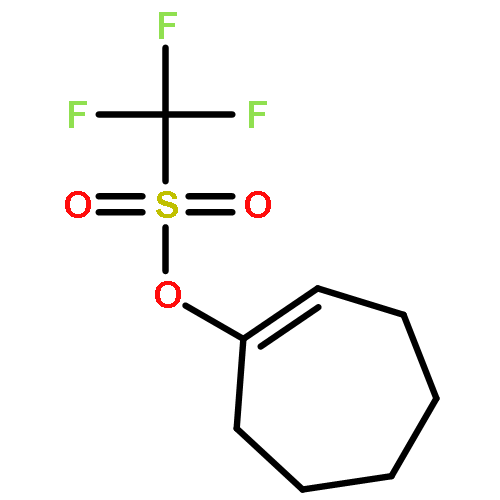




![Pyridine, 2-[(4S)-4-(1,1-dimethylethyl)-4,5-dihydro-2-oxazolyl]-4-methyl-](/data/chemimg/3721700/1257527-18-6.png)
![Pyridine, 2-[(4S)-4-(1,1-dimethylethyl)-4,5-dihydro-2-oxazolyl]-4-methyl-](/data/chemimg/3721700/1257527-18-6_b.png)
![Pyridine, 4-chloro-2-[(4S)-4-(1,1-dimethylethyl)-4,5-dihydro-2-oxazolyl]-](/data/chemimg/3721700/1257527-16-4.png)
![Pyridine, 4-chloro-2-[(4S)-4-(1,1-dimethylethyl)-4,5-dihydro-2-oxazolyl]-](/data/chemimg/3721700/1257527-16-4_b.png)




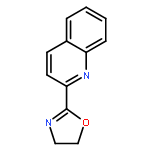
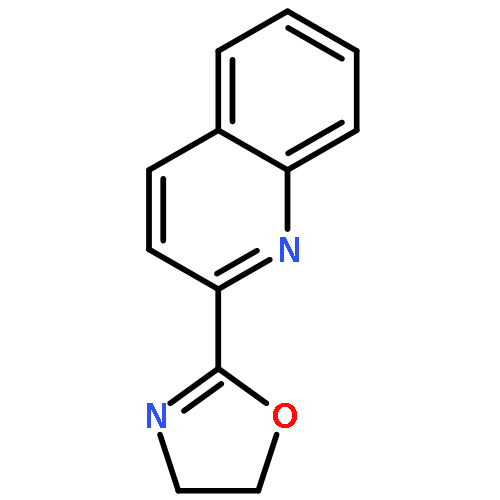

![Benzene, 1,1'-[(1E)-3-methyl-1-propene-1,3-diyl]bis-](http://img.cochemist.com/ccimg/7400/7302-01-4.png)
![Benzene, 1,1'-[(1E)-3-methyl-1-propene-1,3-diyl]bis-](http://img.cochemist.com/ccimg/7400/7302-01-4_b.png)
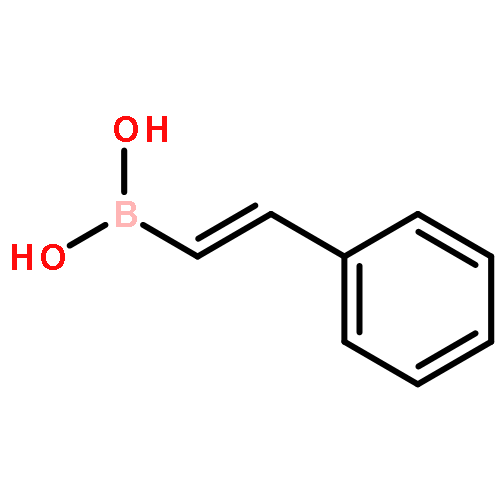





![Dichloro[2-(4,5-dihydro-2-oxazolyl)quinoline]palladium(II)](http://img.cochemist.com/ccimg/1150100/1150097-98-5.png)
![Dichloro[2-(4,5-dihydro-2-oxazolyl)quinoline]palladium(II)](http://img.cochemist.com/ccimg/1150100/1150097-98-5_b.png)


![2-[(4s)-4-(2-methyl-2-propanyl)-4,5-dihydro-1,3-oxazol-2-yl]-5-(t Rifluoromethyl)pyridine](/data/chemimg/482100/1416819-91-4.png)
![2-[(4s)-4-(2-methyl-2-propanyl)-4,5-dihydro-1,3-oxazol-2-yl]-5-(t Rifluoromethyl)pyridine](/data/chemimg/482100/1416819-91-4_b.png)