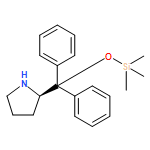
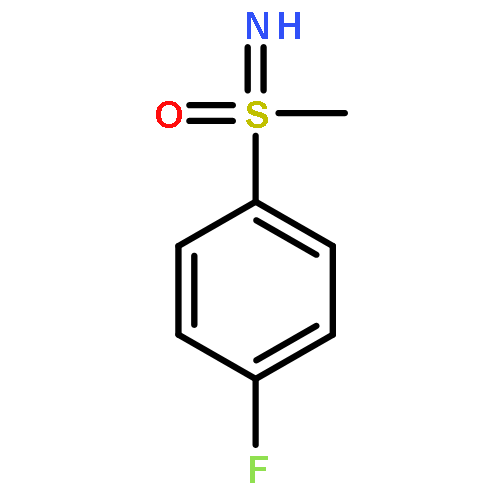
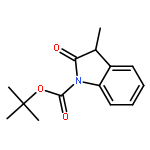
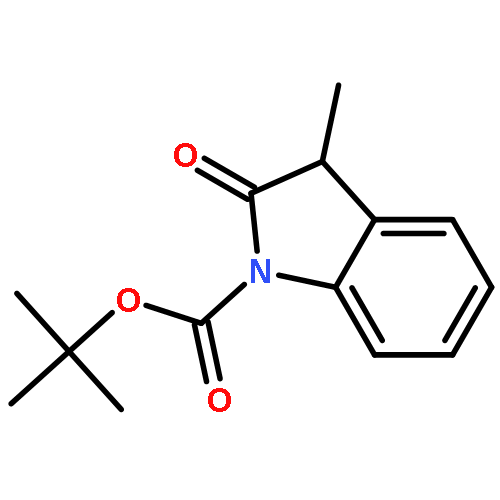
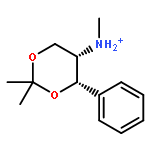
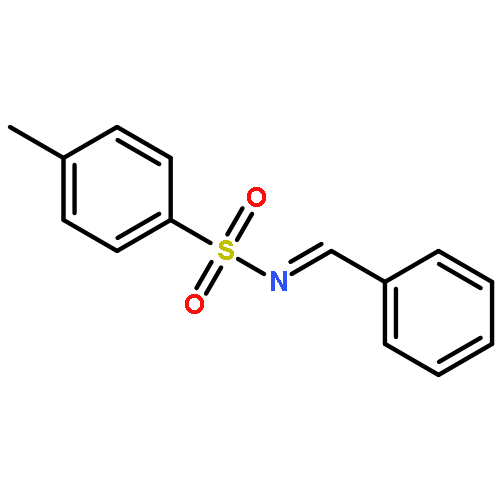
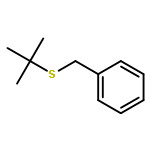
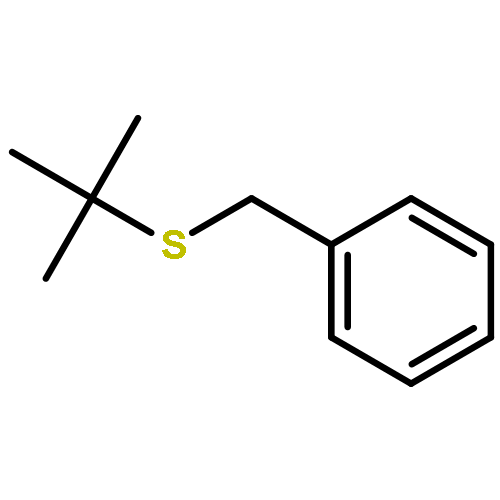
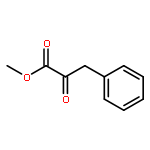
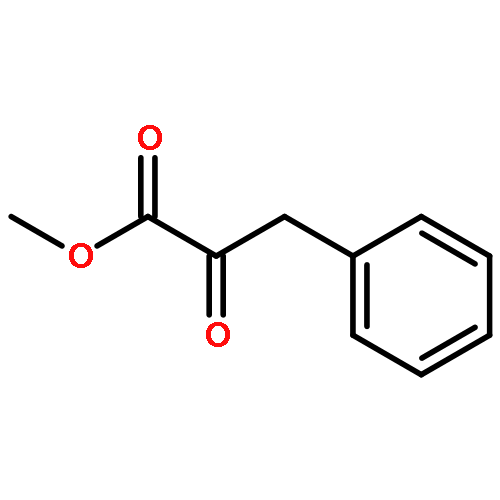
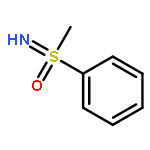
Enantiomerically pure triflones R1CH(R2)SO2CF3 have been synthesized starting from the corresponding chiral alcohols via thiols and trifluoromethylsulfanes. Key steps of the syntheses of the sulfanes are the photochemical trifluoromethylation of the thiols with CF3Hal (Hal=halide) or substitution of alkoxyphosphinediamines with CF3SSCF3. The deprotonation of RCH(Me)SO2CF3 (R=CH2Ph, iHex) with nBuLi with the formation of salts [RC(Me)SO2CF3]Li and their electrophilic capture both occurred with high enantioselectivities. Displacement of the SO2CF3 group of (S)-MeOCH2C(Me)(CH2Ph)SO2CF3 (95 % ee) by an ethyl group through the reaction with AlEt3 gave alkane MeOCH2C(Me)(CH2Ph)Et of 96 % ee. Racemization of salts [R1C(R2)SO2CF3]Li follows first-order kinetics and is mainly an enthalpic process with small negative activation entropy as revealed by polarimetry and dynamic NMR (DNMR) spectroscopy. This is in accordance with a CαS bond rotation as the rate-determining step. Lithium α-(S)-trifluoromethyl- and α-(S)-nonafluorobutylsulfonyl carbanion salts have a much higher racemization barrier than the corresponding α-(S)-tert-butylsulfonyl carbanion salts. Whereas [PhCH2C(Me)SO2tBu]Li/DMPU (DMPU = dimethylpropylurea) has a half-life of racemization at −105 °C of 2.4 h, that of [PhCH2C(Me)SO2CF3]Li at −78 °C is 30 d. DNMR spectroscopy of amides (PhCH2)2NSO2CF3 and (PhCH2)N(Ph)SO2CF3 gave NS rotational barriers that seem to be distinctly higher than those of nonfluorinated sulfonamides. NMR spectroscopy of [PhCH2C(Ph)SO2R]M (M=Li, K, NBu4; R=CF3, tBu) shows for both salts a confinement of the negative charge mainly to the Cα atom and a significant benzylic stabilization that is weaker in the trifluoromethylsulfonyl carbanion. According to crystal structure analyses, the carbanions of salts {[PhCH2C(Ph)SO2CF3]Li⋅L}2 (L=2 THF, tetramethylethylenediamine (TMEDA)) and [PhCH2C(Ph)SO2CF3]NBu4 have the typical chiral CαS conformation of α-sulfonyl carbanions, planar Cα atoms, and short CαS bonds. Ab initio calculations of [MeC(Ph)SO2tBu]− and [MeC(Ph)SO2CF3]− showed for the fluorinated carbanion stronger nCσ* and nOσ* interactions and a weaker benzylic stabilization. According to natural bond orbital (NBO) calculations of [R1C(R2)SO2R]− (R=tBu, CF3) the nCσ*SR interaction is much stronger for R=CF3. Ab initio calculations gave for [MeC(Ph)SO2tBu]Li⋅2 Me2O an O,Li,Cα contact ion pair (CIP) and for [MeC(Ph)SO2CF3]Li⋅2 Me2O an O,Li,O CIP. According to cryoscopy, [PhCH2C(Ph)SO2CF3]Li, [iHexC(Me)SO2CF3]Li, and [PhCH2C(Ph)SO2CF3]NBu4 predominantly form monomers in tetrahydrofuran (THF) at −108 °C. The NMR spectroscopic data of salts [R1(R2)SO2R3]Li (R3=tBu, CF3) indicate that the dominating monomeric CIPs are devoid of CαLi bonds.
The CD spectra of the pyrrole-imidazole alkaloids, (−)-dibromophakellin and (−)-dibromophakellstatin, have been calculated employing the quantum-chemical time-dependent density functional theory. Comparison of calculated and measured spectra showed that this well-established method is also a useful tool to elucidate the absolute stereochemistry of this class of compounds. The computational results have further been used to analyze the spectra measured in methanol and to explain the remarkable red shift of one CD band when trifluoroethanol is used as a solvent instead of methanol. Chirality, 2007. © 2007 Wiley-Liss, Inc.
![Borate(1-),[1,2:5,6-bis-O-(1-methylethylidene)-a-D-glucofuranosato-kO3]-1,5-cyclooctanediylhydro-, potassium, (T-4)- (9CI)](http://img.cochemist.com/ccimg/101700/101696-41-7.png)
![Borate(1-),[1,2:5,6-bis-O-(1-methylethylidene)-a-D-glucofuranosato-kO3]-1,5-cyclooctanediylhydro-, potassium, (T-4)- (9CI)](http://img.cochemist.com/ccimg/101700/101696-41-7_b.png)

![3H-Pyrazol-3-one, 2,4-dihydro-4-[(4-methoxyphenyl)methylene]-5-methyl-2-phenyl-, (4Z)-](/data/chemimg/1823800/39143-07-2.png)
![3H-Pyrazol-3-one, 2,4-dihydro-4-[(4-methoxyphenyl)methylene]-5-methyl-2-phenyl-, (4Z)-](/data/chemimg/1823800/39143-07-2_b.png)
![4-[(4-carboxyphenyl)methylidene]-2-methyl-1,2,3,4-tetrahydroacridine-9-carboxylic acid](http://img.cochemist.com/ccimg/5700/5628-16-0.png)
![4-[(4-carboxyphenyl)methylidene]-2-methyl-1,2,3,4-tetrahydroacridine-9-carboxylic acid](http://img.cochemist.com/ccimg/5700/5628-16-0_b.png)




![Sulfoximine,S-methyl-S-phenyl-N-[2-[[[2,4,6-tris(1-methylethyl)phenyl]methyl]amino]phenyl]-,[S(S)]- (9CI)](http://img.cochemist.com/ccimg/825700/825612-43-9.png)
![Sulfoximine,S-methyl-S-phenyl-N-[2-[[[2,4,6-tris(1-methylethyl)phenyl]methyl]amino]phenyl]-,[S(S)]- (9CI)](http://img.cochemist.com/ccimg/825700/825612-43-9_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2_b.png)
![Quinoline, 8-[(pentafluorophenyl)methoxy]-](http://img.cochemist.com/ccimg/122700/122687-47-2.png)
![Quinoline, 8-[(pentafluorophenyl)methoxy]-](http://img.cochemist.com/ccimg/122700/122687-47-2_b.png)


![Acetic acid, [(4-methoxyphenyl)imino]-, ethyl ester](http://img.cochemist.com/ccimg/115300/115276-75-0.png)
![Acetic acid, [(4-methoxyphenyl)imino]-, ethyl ester](http://img.cochemist.com/ccimg/115300/115276-75-0_b.png)
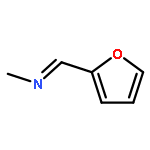
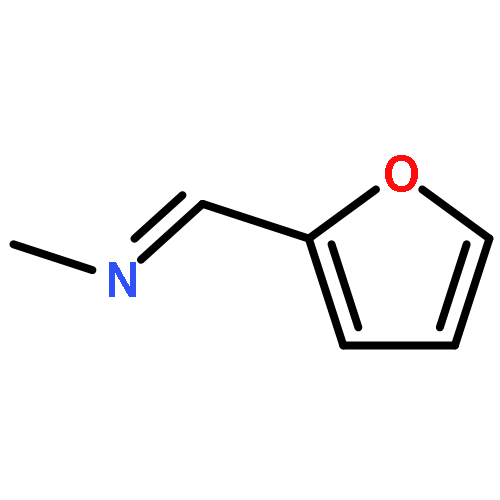
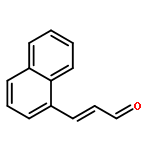
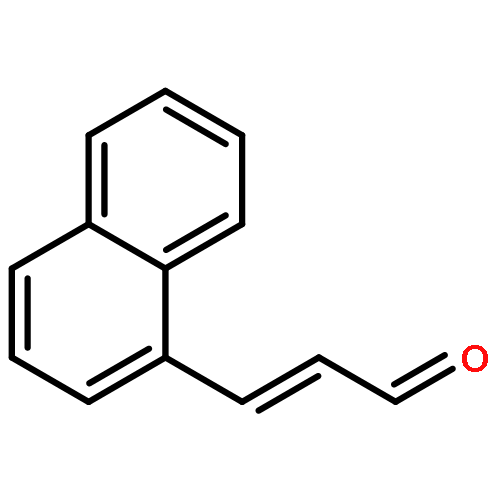




![Ethanone,1-[3-(acetyloxy)-6-chloro-1H-indol-1-yl]-](http://img.cochemist.com/ccimg/108800/108761-33-7.png)
![Ethanone,1-[3-(acetyloxy)-6-chloro-1H-indol-1-yl]-](http://img.cochemist.com/ccimg/108800/108761-33-7_b.png)






![Propanedioic acid, [(1R,2E)-1,3-diphenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/96500/96482-67-6.png)
![Propanedioic acid, [(1R,2E)-1,3-diphenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/96500/96482-67-6_b.png)
![Propanedioic acid, [(1S,2E)-1,3-diphenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/96500/96482-64-3.png)
![Propanedioic acid, [(1S,2E)-1,3-diphenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/96500/96482-64-3_b.png)
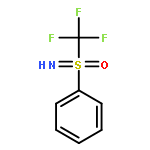
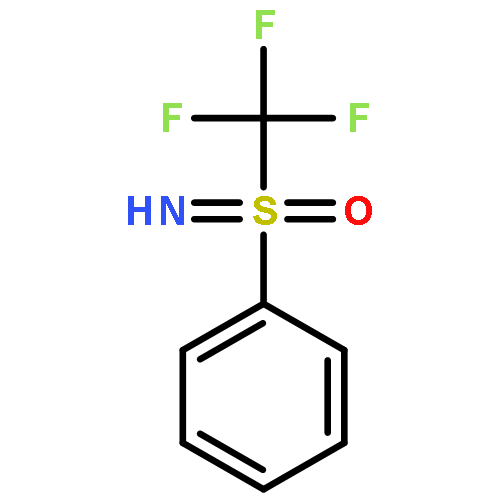


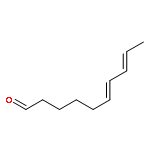
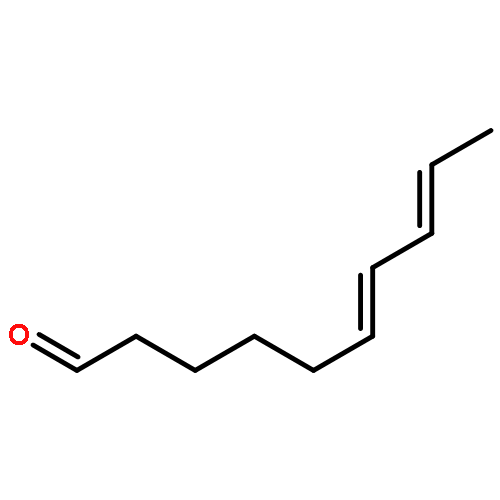



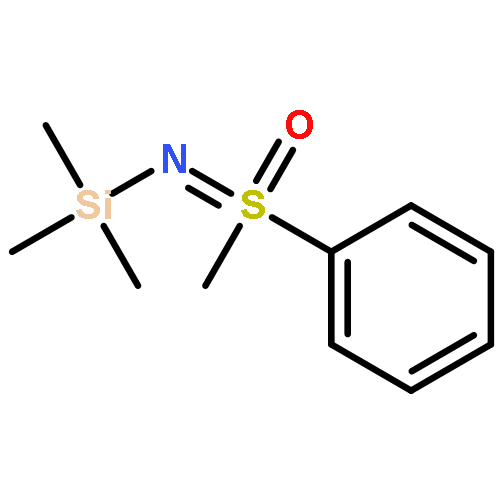
![1H-Isoindole-1,3(2H)-dione, 2-[(4-nitrophenyl)thio]-](http://img.cochemist.com/ccimg/88700/88683-47-0.png)
![1H-Isoindole-1,3(2H)-dione, 2-[(4-nitrophenyl)thio]-](http://img.cochemist.com/ccimg/88700/88683-47-0_b.png)
![1H-ISOINDOLE-1,3(2H)-DIONE, 2-[(3-NITROPHENYL)THIO]-](http://img.cochemist.com/ccimg/88700/88683-46-9.png)
![1H-ISOINDOLE-1,3(2H)-DIONE, 2-[(3-NITROPHENYL)THIO]-](http://img.cochemist.com/ccimg/88700/88683-46-9_b.png)

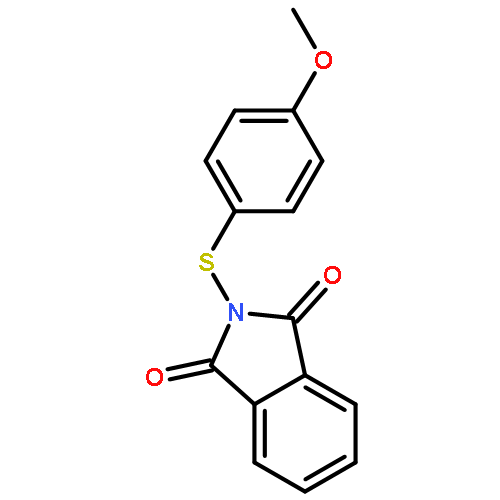
![1H-PYRROLE, 1-METHYL-2-[(TRIMETHYLSILYL)OXY]-](http://img.cochemist.com/ccimg/87900/87884-52-4.png)
![1H-PYRROLE, 1-METHYL-2-[(TRIMETHYLSILYL)OXY]-](http://img.cochemist.com/ccimg/87900/87884-52-4_b.png)

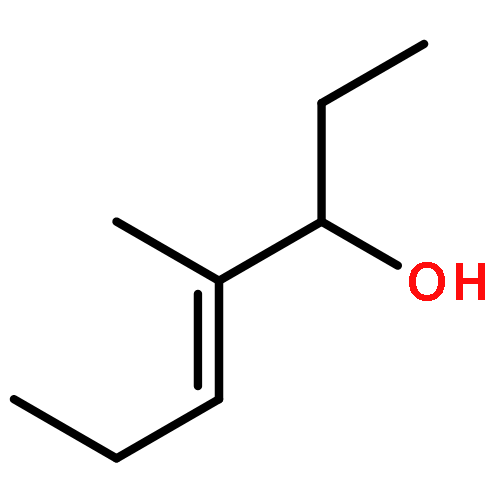

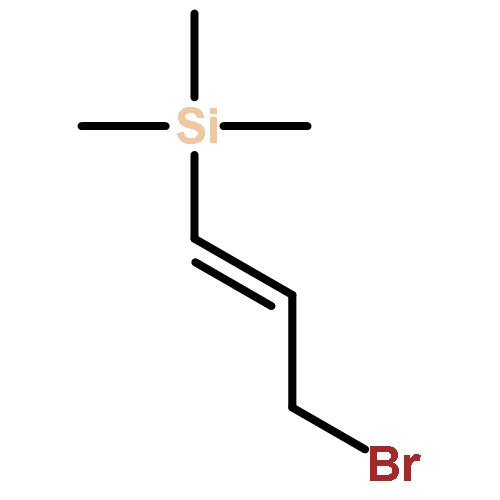
























![Benzenesulfonamide, 4-methyl-N-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/73000/72984-27-1.png)
![Benzenesulfonamide, 4-methyl-N-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/73000/72984-27-1_b.png)
![Benzene, [(1,1-dimethyl-2-propenyl)sulfonyl]-](http://img.cochemist.com/ccimg/72900/72863-20-8.png)
![Benzene, [(1,1-dimethyl-2-propenyl)sulfonyl]-](http://img.cochemist.com/ccimg/72900/72863-20-8_b.png)


![Propane, 2-methyl-2-[(1-methylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/69500/69489-93-6.png)
![Propane, 2-methyl-2-[(1-methylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/69500/69489-93-6_b.png)
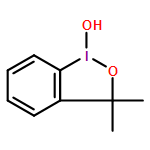


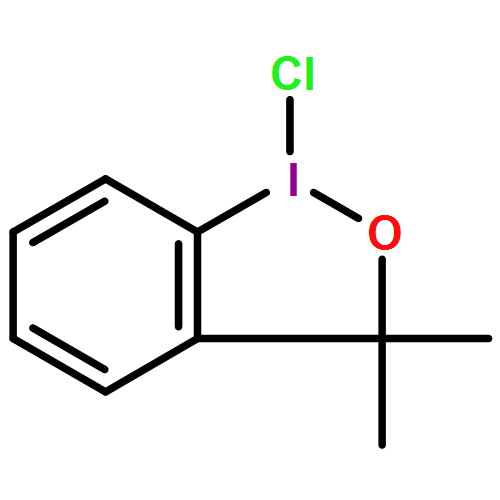
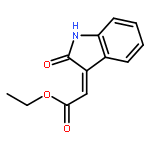
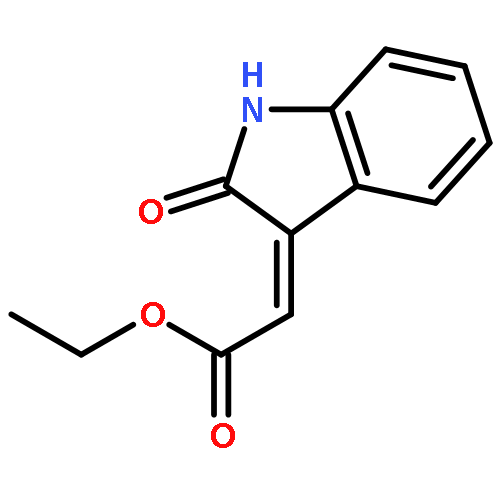
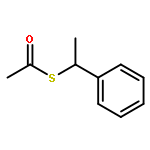
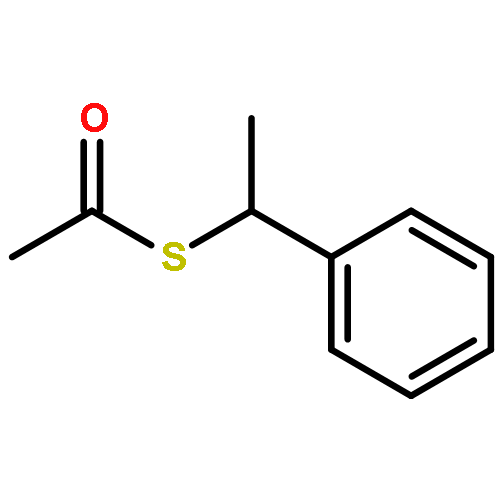


![BENZENESULFONAMIDE, 4-METHYL-N-[(1S)-1-PHENYLETHYL]-](http://img.cochemist.com/ccimg/66600/66558-04-1.png)
![BENZENESULFONAMIDE, 4-METHYL-N-[(1S)-1-PHENYLETHYL]-](http://img.cochemist.com/ccimg/66600/66558-04-1_b.png)






![9-Borabicyclo[3.3.1]nonane,9-[(1R,2S,3R,5R)-2,6,6-trimethylbicyclo[3.1.1]hept-3-yl]-](http://img.cochemist.com/ccimg/64200/64106-79-2.png)
![9-Borabicyclo[3.3.1]nonane,9-[(1R,2S,3R,5R)-2,6,6-trimethylbicyclo[3.1.1]hept-3-yl]-](http://img.cochemist.com/ccimg/64200/64106-79-2_b.png)










![Benzene, [1-[(1,1-dimethylethyl)thio]ethyl]-](http://img.cochemist.com/ccimg/62300/62252-48-6.png)
![Benzene, [1-[(1,1-dimethylethyl)thio]ethyl]-](http://img.cochemist.com/ccimg/62300/62252-48-6_b.png)
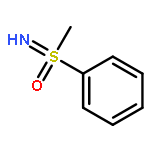
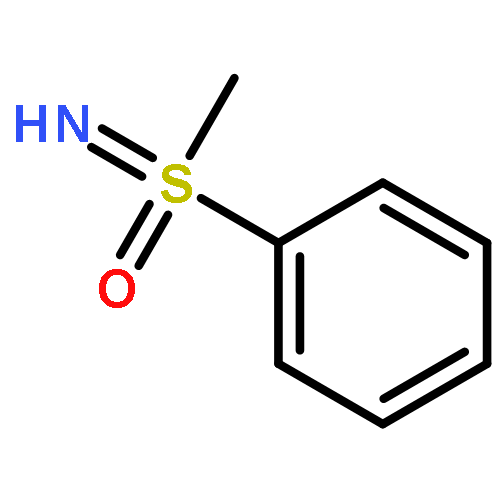


![Propanedioic acid, [(4-methylphenyl)methylene]-, dimethyl ester](http://img.cochemist.com/ccimg/59900/59832-45-0.png)
![Propanedioic acid, [(4-methylphenyl)methylene]-, dimethyl ester](http://img.cochemist.com/ccimg/59900/59832-45-0_b.png)











![METHANAMINE, N-[(2-METHYLPHENYL)METHYLENE]-, (E)-](http://img.cochemist.com/ccimg/53700/53699-35-7.png)
![METHANAMINE, N-[(2-METHYLPHENYL)METHYLENE]-, (E)-](http://img.cochemist.com/ccimg/53700/53699-35-7_b.png)
![METHANAMINE, N-[(4-METHYLPHENYL)METHYLENE]-, (E)-](http://img.cochemist.com/ccimg/53700/53699-34-6.png)
![METHANAMINE, N-[(4-METHYLPHENYL)METHYLENE]-, (E)-](http://img.cochemist.com/ccimg/53700/53699-34-6_b.png)


![Benzene, 1,1'-[1-[(trifluoromethyl)sulfonyl]-1,2-ethanediyl]bis-](http://img.cochemist.com/ccimg/52300/52208-98-7.png)
![Benzene, 1,1'-[1-[(trifluoromethyl)sulfonyl]-1,2-ethanediyl]bis-](http://img.cochemist.com/ccimg/52300/52208-98-7_b.png)
![Benzene, [2-[(trifluoromethyl)sulfonyl]ethyl]-](http://img.cochemist.com/ccimg/52300/52208-95-4.png)
![Benzene, [2-[(trifluoromethyl)sulfonyl]ethyl]-](http://img.cochemist.com/ccimg/52300/52208-95-4_b.png)
![Isoxazole, 5-[(1E)-2-(4-methoxyphenyl)ethenyl]-3-methyl-4-nitro-](http://img.cochemist.com/ccimg/52000/51978-98-4.png)
![Isoxazole, 5-[(1E)-2-(4-methoxyphenyl)ethenyl]-3-methyl-4-nitro-](http://img.cochemist.com/ccimg/52000/51978-98-4_b.png)









![ETHANONE, 1-[1,1'-BIPHENYL]-4-YL-2-(PHENYLSULFONYL)-](http://img.cochemist.com/ccimg/41100/41024-57-1.png)
![ETHANONE, 1-[1,1'-BIPHENYL]-4-YL-2-(PHENYLSULFONYL)-](http://img.cochemist.com/ccimg/41100/41024-57-1_b.png)




![Acetamide, N-[4-(2-bromoethyl)phenyl]-](http://img.cochemist.com/ccimg/39300/39232-06-9.png)
![Acetamide, N-[4-(2-bromoethyl)phenyl]-](http://img.cochemist.com/ccimg/39300/39232-06-9_b.png)
![Naphthalene, 1-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/37700/37630-26-5.png)
![Naphthalene, 1-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/37700/37630-26-5_b.png)
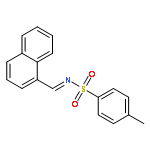
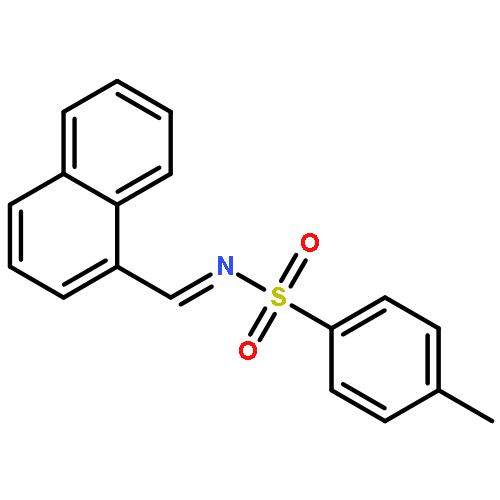




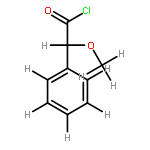
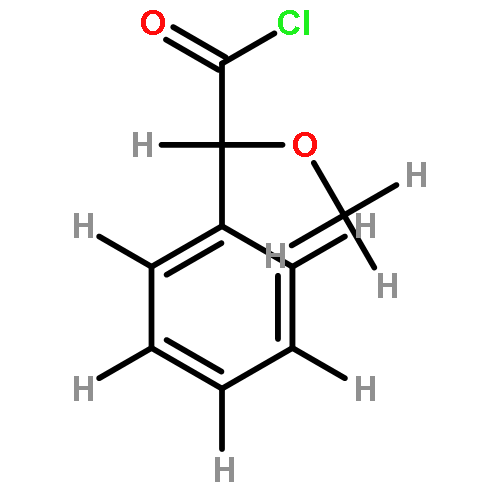























![Benzene, 1,2-dimethoxy-4-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/22600/22568-47-4.png)
![Benzene, 1,2-dimethoxy-4-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/22600/22568-47-4_b.png)


![Benzene, 1-methoxy-2-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/21300/21298-69-1.png)
![Benzene, 1-methoxy-2-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/21300/21298-69-1_b.png)
![Sulfoximine,S-methyl-S-(4-methylphenyl)-, [S(R)]-](http://img.cochemist.com/ccimg/20500/20414-85-1.png)
![Sulfoximine,S-methyl-S-(4-methylphenyl)-, [S(R)]-](http://img.cochemist.com/ccimg/20500/20414-85-1_b.png)







![Silane, [(1-ethoxyethenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/18300/18295-66-4.png)
![Silane, [(1-ethoxyethenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/18300/18295-66-4_b.png)










![1H-Indene-1,3(2H)-dione, 2-[(2-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/15900/15875-56-6.png)
![1H-Indene-1,3(2H)-dione, 2-[(2-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/15900/15875-56-6_b.png)
![1H-Indene-1,3(2H)-dione, 2-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/15900/15875-51-1.png)
![1H-Indene-1,3(2H)-dione, 2-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/15900/15875-51-1_b.png)
![1H-Indene-1,3(2H)-dione, 2-[(3-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/15900/15875-45-3.png)
![1H-Indene-1,3(2H)-dione, 2-[(3-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/15900/15875-45-3_b.png)
![2-(Benzo[d]oxazol-2-yl)acetonitrile](http://img.cochemist.com/ccimg/15400/15344-56-6.png)
![2-(Benzo[d]oxazol-2-yl)acetonitrile](http://img.cochemist.com/ccimg/15400/15344-56-6_b.png)
![1H-Isoindole-1,3(2H)-dione,2-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/15200/15199-26-5.png)
![1H-Isoindole-1,3(2H)-dione,2-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/15200/15199-26-5_b.png)
![6H-1,3-Dioxolo[4,5-g][1]benzopyran-6-one, 8-hydroxy-](http://img.cochemist.com/ccimg/15000/14991-61-8.png)
![6H-1,3-Dioxolo[4,5-g][1]benzopyran-6-one, 8-hydroxy-](http://img.cochemist.com/ccimg/15000/14991-61-8_b.png)
![Benzene,[(1-methylpropyl)thio]-](http://img.cochemist.com/ccimg/15000/14905-79-4.png)
![Benzene,[(1-methylpropyl)thio]-](http://img.cochemist.com/ccimg/15000/14905-79-4_b.png)
![Benzenesulfonamide, 4-methyl-N-[(3-nitrophenyl)methylene]-](http://img.cochemist.com/ccimg/14700/14674-36-3.png)
![Benzenesulfonamide, 4-methyl-N-[(3-nitrophenyl)methylene]-](http://img.cochemist.com/ccimg/14700/14674-36-3_b.png)





![1H-Isoindole-1,3(2H)-dione, 2-[(phenylmethyl)thio]-](http://img.cochemist.com/ccimg/14300/14204-26-3.png)
![1H-Isoindole-1,3(2H)-dione, 2-[(phenylmethyl)thio]-](http://img.cochemist.com/ccimg/14300/14204-26-3_b.png)


![tricyclo[8.2.2.2~4,7~]hexadeca-1(12),4,6,10,13,15-hexaen-5-amine](http://img.cochemist.com/ccimg/10200/10122-95-9.png)
![tricyclo[8.2.2.2~4,7~]hexadeca-1(12),4,6,10,13,15-hexaen-5-amine](http://img.cochemist.com/ccimg/10200/10122-95-9_b.png)




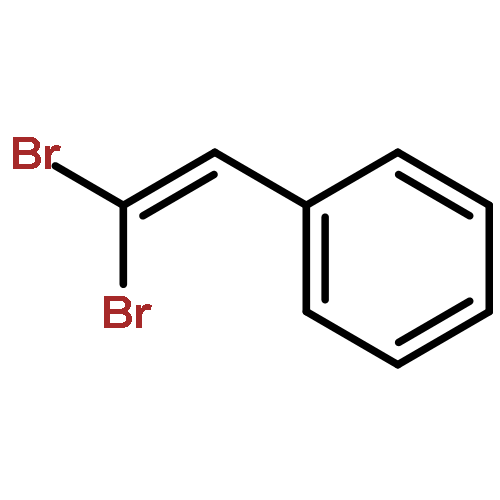






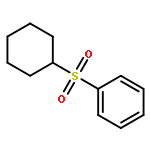

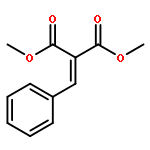
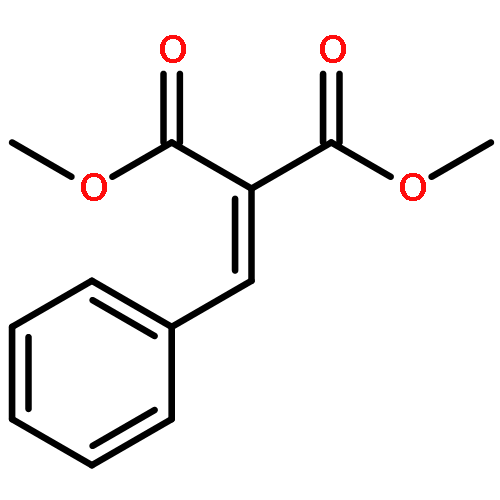












![BENZENE, [[(TRIFLUOROMETHYL)SULFONYL]METHYL]-](http://img.cochemist.com/ccimg/4900/4855-02-1.png)
![BENZENE, [[(TRIFLUOROMETHYL)SULFONYL]METHYL]-](http://img.cochemist.com/ccimg/4900/4855-02-1_b.png)














![Propanedinitrile,2-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/2900/2826-26-8.png)
![Propanedinitrile,2-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/2900/2826-26-8_b.png)
![2-[(4-methylphenyl)methylidene]propanedinitrile](http://img.cochemist.com/ccimg/2900/2826-25-7.png)
![2-[(4-methylphenyl)methylidene]propanedinitrile](http://img.cochemist.com/ccimg/2900/2826-25-7_b.png)
![Propanedinitrile, [(4-fluorophenyl)methylene]-](http://img.cochemist.com/ccimg/2900/2826-22-4.png)
![Propanedinitrile, [(4-fluorophenyl)methylene]-](http://img.cochemist.com/ccimg/2900/2826-22-4_b.png)




![Propanedinitrile,2-[(4-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/1900/1867-38-5.png)
![Propanedinitrile,2-[(4-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/1900/1867-38-5_b.png)



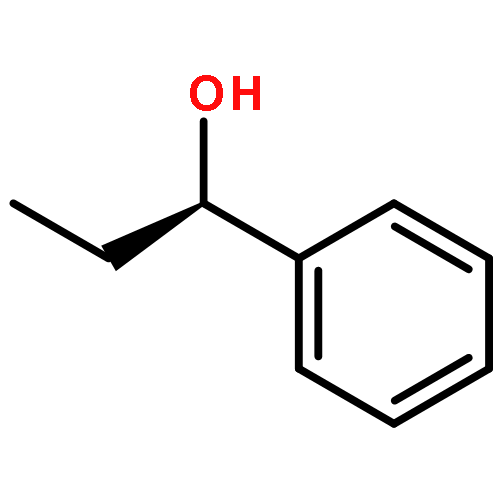
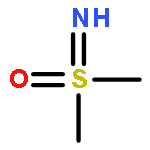
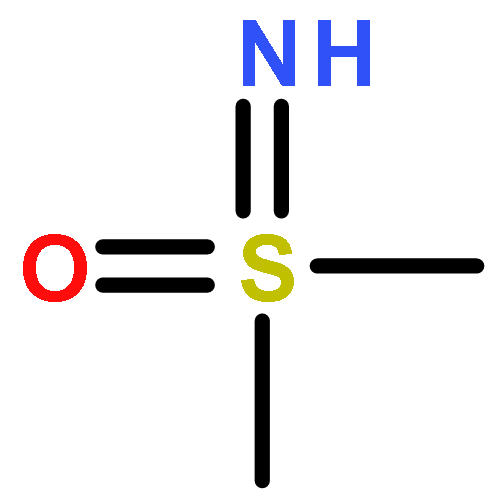


![1H-Indene-1,3(2H)-dione, 2-[(4-bromophenyl)methylene]-](http://img.cochemist.com/ccimg/1000/961-20-6.png)
![1H-Indene-1,3(2H)-dione, 2-[(4-bromophenyl)methylene]-](http://img.cochemist.com/ccimg/1000/961-20-6_b.png)






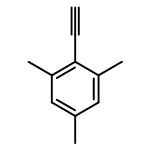
![Benzenemethanol, a-[(1S)-1-aminoethyl]-, (aS)-](http://img.cochemist.com/ccimg/500/492-39-7.png)
![Benzenemethanol, a-[(1S)-1-aminoethyl]-, (aS)-](http://img.cochemist.com/ccimg/500/492-39-7_b.png)









![Benzene, [(1-ethylpropyl)sulfonyl]-](http://img.cochemist.com/ccimg/813500/813436-63-4.png)
![Benzene, [(1-ethylpropyl)sulfonyl]-](http://img.cochemist.com/ccimg/813500/813436-63-4_b.png)









![Phenol, 4-methyl-2-[(1E)-2-nitroethenyl]-](/data/chemimg/103700/264224-82-0.png)
![Phenol, 4-methyl-2-[(1E)-2-nitroethenyl]-](/data/chemimg/103700/264224-82-0_b.png)




![2-Propynal, 3-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/183900/183801-13-0.png)
![2-Propynal, 3-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/183900/183801-13-0_b.png)






















![Benzene, [1-[(1,1-dimethylethyl)sulfonyl]ethyl]-](http://img.cochemist.com/ccimg/63900/63801-21-8.png)
![Benzene, [1-[(1,1-dimethylethyl)sulfonyl]ethyl]-](http://img.cochemist.com/ccimg/63900/63801-21-8_b.png)




![Benzene, [2-[(1,1-dimethylethyl)thio]ethyl]-](http://img.cochemist.com/ccimg/62300/62252-51-1.png)
![Benzene, [2-[(1,1-dimethylethyl)thio]ethyl]-](http://img.cochemist.com/ccimg/62300/62252-51-1_b.png)




![Methanamine,N-[(S)-methyloxidophenyl-l4-sulfanylidene]-](http://img.cochemist.com/ccimg/34000/33993-53-2.png)
![Methanamine,N-[(S)-methyloxidophenyl-l4-sulfanylidene]-](http://img.cochemist.com/ccimg/34000/33993-53-2_b.png)






![2-Propenal, 3-[4-(dimethylamino)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/20500/20432-35-3.png)
![2-Propenal, 3-[4-(dimethylamino)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/20500/20432-35-3_b.png)
![[(tert-butylsulfonyl)methyl]benzene](http://img.cochemist.com/ccimg/20300/20282-89-7.png)
![[(tert-butylsulfonyl)methyl]benzene](http://img.cochemist.com/ccimg/20300/20282-89-7_b.png)


![Benzene, [(bromomethoxy)methyl]-](http://img.cochemist.com/ccimg/17700/17690-16-3.png)
![Benzene, [(bromomethoxy)methyl]-](http://img.cochemist.com/ccimg/17700/17690-16-3_b.png)






















![Phenol, 4-chloro-2-[(1E)-2-nitroethenyl]-](/data/chemimg/144800/1220194-30-8.png)
![Phenol, 4-chloro-2-[(1E)-2-nitroethenyl]-](/data/chemimg/144800/1220194-30-8_b.png)
![N-[3,5-Bis(trifluoromethyl)phenyl]-N-[(8a,9S)-6-methoxy-9-cinchonanyl]thiourea](/data/chemimg/103500/852913-16-7.png)
![N-[3,5-Bis(trifluoromethyl)phenyl]-N-[(8a,9S)-6-methoxy-9-cinchonanyl]thiourea](/data/chemimg/103500/852913-16-7_b.png)




![Benzene, 1-methoxy-3-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/55500/55446-68-9.png)
![Benzene, 1-methoxy-3-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/55500/55446-68-9_b.png)





![Tricyclo[8.2.2.24,7]hexadeca-4,6,10,12,13,15-hexaene,5-bromo-](http://img.cochemist.com/ccimg/2000/1908-61-8.png)
![Tricyclo[8.2.2.24,7]hexadeca-4,6,10,12,13,15-hexaene,5-bromo-](http://img.cochemist.com/ccimg/2000/1908-61-8_b.png)


![Cyclohexane, [(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/132900/132839-80-6.png)
![Cyclohexane, [(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/132900/132839-80-6_b.png)




![1-chloro-2-[(e)-2-nitrovinyl]benzene](http://img.cochemist.com/ccimg/22600/22568-07-6.png)
![1-chloro-2-[(e)-2-nitrovinyl]benzene](http://img.cochemist.com/ccimg/22600/22568-07-6_b.png)
![1,3-Benzodioxole,5-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/22600/22568-48-5.png)
![1,3-Benzodioxole,5-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/22600/22568-48-5_b.png)