Co-reporter:Sourabh Mishra, Ji Liu, and Aaron Aponick
Journal of the American Chemical Society March 8, 2017 Volume 139(Issue 9) pp:3352-3352
Publication Date(Web):February 16, 2017
DOI:10.1021/jacs.7b00363
By the nature of its structure, the 5-membered chiral biaryl heterocyclic scaffold represents a departure from 6-membered P,N-ligands that facilitates tuning and enables ligand evolution to address issues of selectivity and reactivity. In this vein, the Cu-catalyzed enantioselective conjugate alkynylation of Meldrum’s acid acceptors is reported using Me-StackPhos. Enabled by this new ligand, the reaction tolerates a wide range of alkynes furnishing the products in high yields and excellent enantioselectivity. The transformation provides access to highly useful chiral β-alkynyl Meldrum’s acid building blocks as demonstrated by an efficient enantioselective synthesis of the preclinical agent OPC 51803.
Co-reporter:Paulo H. S. Paioti; Khalil A. Abboud
Journal of the American Chemical Society 2016 Volume 138(Issue 7) pp:2150-2153
Publication Date(Web):February 8, 2016
DOI:10.1021/jacs.5b13387
The Cu-catalyzed synthesis of nonracemic 3-amino skipped diynes via an enantiodetermining C–C bond formation is described using StackPhos as ligand. Despite challenging issues of reactivity and stereoselectivity inherent to these chiral skipped diynes, the reaction tolerates an extremely broad substrate scope with respect to all components and provides the title compounds in excellent enantiomeric excess. The alkyne moieties are demonstrated here to be useful synthetic handles, and 3-amino skipped diynes are convenient building blocks for enantioselective synthesis.
Co-reporter:Justin A. Goodwin and Aaron Aponick
Chemical Communications 2016 vol. 52(Issue 40) pp:6731-6731
Publication Date(Web):28 Apr 2016
DOI:10.1039/C6CC90121B
Correction for ‘Regioselectivity in the Au-catalyzed hydration and hydroalkoxylation of alkynes’ by Justin A. Goodwin et al., Chem. Commun., 2015, 51, 8730–8741.
Co-reporter:Justin A. Goodwin and Aaron Aponick
Chemical Communications 2015 vol. 51(Issue 42) pp:8730-8741
Publication Date(Web):27 Mar 2015
DOI:10.1039/C5CC00120J
Over the past decade and a half, homogenous gold catalysis has emerged as a diverse and rich field of research resulting in the continuous development of new methods for organic synthesis. The activation of alkynes towards nucleophilic attack by AuI and AuIII complexes is a well-established mode of reactivity and the gold-catalyzed hydration and hydroalkoxylation of alkynes are two of the more well-explored reaction pathways. Although these classes of reactions have seen continuous development since their initial reports, achieving regioselectivity persists as one of the most challenging issues for this chemistry. This article aims to draw attention to the general problem of regioselectivity in these reactions. A select set of examples is presented to highlight the challenges and survey some of the strategies employed to address this problem.
Co-reporter:Nicholas V. Borrero, Lindsey G. DeRatt, Lais Ferreira Barbosa, Khalil A. Abboud, and Aaron Aponick
Organic Letters 2015 Volume 17(Issue 7) pp:1754-1757
Publication Date(Web):March 23, 2015
DOI:10.1021/acs.orglett.5b00528
A gold-catalyzed synthesis of cyclic 2-oxodienes from readily prepared propargyl alcohols and the subsequent Diels–Alder reaction are reported. The dehydrative cyclization reactions proceeded smoothly, and the dienes formed in situ were demonstrated to undergo cycloaddition with a variety of dienophiles. This method offers a new strategy for the synthesis of indolocarbazole alkaloids, whereby the convergent synthetic design allows for differentiation between the indole nitrogens.
Co-reporter:Barry B. Butler Jr., Jagadeesh Nagendra Manda, and Aaron Aponick
Organic Letters 2015 Volume 17(Issue 8) pp:1902-1905
Publication Date(Web):April 3, 2015
DOI:10.1021/acs.orglett.5b00711
A synthesis of the spirastrellolide A, B/C-ring monounsaturated spiroketal is reported. The key step relies on a Au-catalyzed spiroketalization of a propargyl triol employing an acetonide as a regioselectivity regulator. Through observation and analysis, a set of conditions has been developed that facilitates the use of a mixture of diastereomeric substrates, obviating the need to control the stereochemistry of the propargyl stereocenter and enabling a convenient retrosynthetic disconnection. The key reaction proceeds in 80% yield in 1 min at ambient temperature with the Me3PAuCl/AgOTf catalyst system. These conditions should be widely applicable for new synthetic endeavors as they appear to overcome all issues with the Au-catalyzed spiroketalization.
Co-reporter:Justin A. Goodwin, Carl F. Ballesteros, and Aaron Aponick
Organic Letters 2015 Volume 17(Issue 22) pp:5574-5577
Publication Date(Web):November 12, 2015
DOI:10.1021/acs.orglett.5b02725
A complementary diastereoselective gold(I) or bismuth(III) catalyzed tandem hemiacetalization/dehydrative cyclization of 1,5-monoallylic diols was developed to access 1,3-dioxolanes and dioxanes. This methodology provides rapid access to protected 1,3-diols under mild conditions with high levels of diastereoselectivity.
Co-reporter:Mukesh Pappoppula ; Aaron Aponick
Angewandte Chemie 2015 Volume 127( Issue 52) pp:16053-16056
Publication Date(Web):
DOI:10.1002/ange.201507849
Abstract
An enantioselective total synthesis of martinellic acid is described. The pyrroloquinoline alkaloid core is efficiently prepared from a quinoline, employing a method which relies on a newly developed Cu-catalyzed enantioselective alkynylation using the chiral imidazole-based biaryl P,N ligand StackPhos to establish the absolute stereochemistry. The remaining carbon atoms are then installed by means of a diastereoselective Pd-catalyzed decarboxylative allylation and the synthesis is completed after straightforward functional-group manipulation. This new synthetic method enables the most concise enantioselective synthesis of this important class of molecules to date.
Co-reporter:Mukesh Pappoppula ; Aaron Aponick
Angewandte Chemie International Edition 2015 Volume 54( Issue 52) pp:15827-15830
Publication Date(Web):
DOI:10.1002/anie.201507849
Abstract
An enantioselective total synthesis of martinellic acid is described. The pyrroloquinoline alkaloid core is efficiently prepared from a quinoline, employing a method which relies on a newly developed Cu-catalyzed enantioselective alkynylation using the chiral imidazole-based biaryl P,N ligand StackPhos to establish the absolute stereochemistry. The remaining carbon atoms are then installed by means of a diastereoselective Pd-catalyzed decarboxylative allylation and the synthesis is completed after straightforward functional-group manipulation. This new synthetic method enables the most concise enantioselective synthesis of this important class of molecules to date.
Co-reporter:Mukesh Pappoppula;Flavio S. P. Cardoso;B. Owen Garrett ; Aaron Aponick
Angewandte Chemie International Edition 2015 Volume 54( Issue 50) pp:15202-15206
Publication Date(Web):
DOI:10.1002/anie.201507848
Abstract
A highly enantioselective copper-catalyzed alkynylation of quinolinium salts is reported. The reaction employs StackPhos, a newly developed imidazole-based chiral biaryl P,N ligand, and copper bromide to effect a three-component reaction between a quinoline, a terminal alkyne, and ethyl chloroformate. Under the reaction conditions, the desired products are delivered in high yields with ee values of up to 98 %. The transformation tolerates a wide range of functional groups with respect to both the alkyne and the quinoline starting materials and the products are easily transformed into useful synthons. Efficient, enantioselective syntheses of the tetrahydroquinoline alkaloids (+)-galipinine, (+)-angustureine, and (−)-cuspareine are reported.
Co-reporter:Mukesh Pappoppula;Flavio S. P. Cardoso;B. Owen Garrett ; Aaron Aponick
Angewandte Chemie 2015 Volume 127( Issue 50) pp:15417-15421
Publication Date(Web):
DOI:10.1002/ange.201507848
Abstract
A highly enantioselective copper-catalyzed alkynylation of quinolinium salts is reported. The reaction employs StackPhos, a newly developed imidazole-based chiral biaryl P,N ligand, and copper bromide to effect a three-component reaction between a quinoline, a terminal alkyne, and ethyl chloroformate. Under the reaction conditions, the desired products are delivered in high yields with ee values of up to 98 %. The transformation tolerates a wide range of functional groups with respect to both the alkyne and the quinoline starting materials and the products are easily transformed into useful synthons. Efficient, enantioselective syntheses of the tetrahydroquinoline alkaloids (+)-galipinine, (+)-angustureine, and (−)-cuspareine are reported.
Co-reporter:Paulo H. S. Paioti, John M. Ketcham, and Aaron Aponick
Organic Letters 2014 Volume 16(Issue 20) pp:5320-5323
Publication Date(Web):October 1, 2014
DOI:10.1021/ol5024954
A novel gold-catalyzed synthesis of unsaturated spiroketals that addresses regioselectivity issues commonly reported in metal-catalyzed spiroketalization of alkynes is reported. The reaction sequence is regulated by an acetonide protecting group which undergoes extrusion of acetone to deliver the desired spiroketals in good yields and diastereoselectivities. The reaction, which is carried out under very mild conditions employing AuCl as the catalyst, should be widely applicable in the synthesis of a broad range of spiroketals.
Co-reporter:Flavio S. P. Cardoso ; Khalil A. Abboud
Journal of the American Chemical Society 2013 Volume 135(Issue 39) pp:14548-14551
Publication Date(Web):September 17, 2013
DOI:10.1021/ja407689a
A new strategy for increasing the barrier to rotation in biaryls has been developed that allows for the incorporation of 5-membered aromatic heterocycles into chiral atropisomers. Using this concept, an imidazole-based biaryl P,N-ligand has been designed and prepared as a single enantiomer. This ligand performs exceptionally well in the enantioselective A3-coupling, demonstrating the potential of this new design element.
Co-reporter:John M. Ketcham, Berenger Biannic and Aaron Aponick
Chemical Communications 2013 vol. 49(Issue 39) pp:4157-4159
Publication Date(Web):06 Nov 2012
DOI:10.1039/C2CC37166A
The Au(I)-catalyzed intermolecular hydroalkoxylation of alkynes with allylic alcohols to provide allyl vinyl ethers that subsequently undergo Claisen rearrangement is reported. This new cascade reaction strategy facilitates the direct formation of γ,δ-unsaturated ketones from simple starting materials in a single step.
Co-reporter:John M. Ketcham;Flavio S. P. Cardoso;Berenger Biannic;Henri Piras
Israel Journal of Chemistry 2013 Volume 53( Issue 11-12) pp:923-931
Publication Date(Web):
DOI:10.1002/ijch.201300041
Abstract
Mild conditions for the gold-catalyzed dehydrative cyclization of carbamate-protected azaallylic alcohols to form saturated nitrogen heterocycles are reported. The cyclization reactions are high-yielding, operationally easy to perform, and provide heterocycles with a synthetically useful vinyl group, strategically located on the ring system, which can facilitate further transformations for target oriented synthesis. It is also demonstrated through chirality transfer experiments that the mechanism can be either cationic in nature or a Au-catalyzed addition/elimination sequence. The diverging mechanistic scenario is dependent on the nature of the substituents on the allylic alcohol and necessitates judicious substrate design.
Co-reporter:Thomas Ghebreghiorgis, Brian H. Kirk, Aaron Aponick, and Daniel H. Ess
The Journal of Organic Chemistry 2013 Volume 78(Issue 15) pp:7664-7673
Publication Date(Web):July 17, 2013
DOI:10.1021/jo4012283
Density functional calculations and experiments were used to examine mechanisms of Pd(II) catalyzed intramolecular cyclization and dehydration in acyclic and bicyclic monoallylic diols, a formal SN2′ reaction. In contrast to the previously proposed syn-oxypalladation mechanism for acyclic monoallylic diols, calculations and experiments strongly suggest that hydrogen bonding templates a hydroxyl group and Pd addition across the alkene and provides a low energy pathway via anti-addition (anti-oxypalladation) followed by intramolecular proton transfer and anti-elimination of water. This anti-addition, anti-elimination pathway also provides a simple rationale for the observed stereospecificity. For bicyclic monoallylic diol compounds, Pd(II) is capable of promoting either anti- or syn-addition. In addition, palladium chloride ligands can mediate proton transfer to promote dehydration when direct intramolecular proton transfer between diol groups is impossible.
Co-reporter:Dr. Jean A. Palmes;Paulo H. S. Paioti;Leonardo Perez deSouza ;Dr. Aaron Aponick
Chemistry - A European Journal 2013 Volume 19( Issue 35) pp:11613-11621
Publication Date(Web):
DOI:10.1002/chem.201301723
Abstract
A high-yielding stereoselective method for forming spiroketals from simple ketoallylic diols is reported. Employing catalytic [PdCl2(MeCN)2] in THF at 0 °C, these dehydrative cyclization reactions require only mild conditions to produce vinyl-substituted spiroketals in high yields after brief reaction times with water as the only byproduct. Using this method, the stereochemical information embedded at the nucleophile is transmitted “down-the-chain” and efficiently sets the stereochemistry at both the anomeric carbon atom and the newly formed allylic stereocenter.
Co-reporter:Thomas Ghebreghiorgis ; Berenger Biannic ; Brian H. Kirk ; Daniel H. Ess
Journal of the American Chemical Society 2012 Volume 134(Issue 39) pp:16307-16318
Publication Date(Web):September 4, 2012
DOI:10.1021/ja306333a
Density functional calculations and experiment were used to examine the mechanism, reactivity, and origin of chirality transfer in monophosphine Au-catalyzed monoallylic diol cyclization reactions. The lowest energy pathway for cyclization involves a two-step sequence that begins with intramolecular C–O bond formation by anti-addition of the non-allylic hydroxyl group to the Au-coordinated alkene followed by concerted hydrogen transfer/anti-elimination to liberate water. Concerted SN2′-type transition states were found to be significantly higher in energy. The two-step cyclization pathway is extremely facile due to hydrogen bonding between diol groups that induces nucleophilic attack on the alkene and then proton transfer between diol groups after C–O bond formation. Importantly, intramolecular proton transfer and elimination provides an extremely efficient avenue for catalyst regeneration from the Au–C σ-bond intermediate, in contrast to other Au-catalyzed cyclization reactions where this intermediate severely restricts catalyst turnover. The origin of chirality transfer and the ensuing alkene stereochemistry is also the result of strong hydrogen-bonding interactions between diol groups. In the C–O bond-forming step, requisite hydrogen bonding biases the tethered nucleophilic moiety to adopt a chair-like conformation with substituents in either axial or equatorial positions, dictating the stereochemical outcome of the reaction. Since this hydrogen bonding is maintained throughout the course of the reaction, establishment of the resultant olefin geometry is also attributed to this templating effect. These computational conclusions are supported by experimental evidence employing bicyclic systems to probe the facial selectivity.
Co-reporter:Nicholas V. Borrero and Aaron Aponick
The Journal of Organic Chemistry 2012 Volume 77(Issue 19) pp:8410-8416
Publication Date(Web):September 21, 2012
DOI:10.1021/jo301835e
The total synthesis of acortatarin A relying on a Pd(II)-catalyzed spiroketalization is reported. This strategy allows a single stereocenter in the spiroketalization substrate to produce the target efficiently under mild conditions, installing the necessary oxygenation in the backbone through an allylic transposition. The synthesis also verifies that pollenopyrroside B and acortatarin A are the same compound, and electrochemical studies suggest that the reported bioactivity is not due to simple antioxidant properties.
Co-reporter:Kinga Chojnacka, Stefano Santoro, Radi Awartani, Nigel G. J. Richards, Fahmi Himo and Aaron Aponick
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 15) pp:5350-5353
Publication Date(Web):08 Jun 2011
DOI:10.1039/C1OB05751K
We report a new method for constructing the ABC ring system of strigolactones, in a single step from a simple linear precursor by acid-catalyzed double cyclization. The reaction proceeds with a high degree of stereochemical control, which can be qualitatively rationalized using DFT calculations. Our concise synthetic approach offers a new model for thinking about the (as yet) unknown chemistry that is employed in the biosynthetic pathways leading to this class of plant hormones.
Co-reporter:Berenger Biannic
European Journal of Organic Chemistry 2011 Volume 2011( Issue 33) pp:6605-6617
Publication Date(Web):
DOI:10.1002/ejoc.201100858
Abstract
The activation of allylic and propargylic alcohols for use as electrophiles represents a burgeoning new area in homogeneous gold catalysis. This review covers Au-catalyzed substitution reactions of unsaturated alcohols in which water serves as leaving group. Attention is given to different substrate classes and the mechanisms by which the reactions are believed to occur. Recent advances in chirality transfer and enantioselective reactions are also reviewed.
Co-reporter:Aaron Aponick, Berenger Biannic and Michael R. Jong
Chemical Communications 2010 vol. 46(Issue 36) pp:6849-6851
Publication Date(Web):20 Aug 2010
DOI:10.1039/C0CC01961E
The gold(I)-catalyzed endo-cyclization of o-(1-hydroxyallyl)phenols to form chromenes is reported. The title compounds are prepared in high yield from readily available substrates. The system tolerates both electron rich and deficient aryl rings and a high degree of substitution on the allyl moiety.
Co-reporter:Aaron Aponick, Chuan-Ying Li, Jeremy Malinge and Emerson Finco Marques
Organic Letters 2009 Volume 11(Issue 20) pp:4624-4627
Publication Date(Web):September 22, 2009
DOI:10.1021/ol901901m
Furans, pyrroles, and thiophenes are efficiently prepared by gold-catalyzed dehydrative cyclizations of readily available, heteroatom-substituted propargylic alcohols. The reactions are rapid, high-yielding, and procedurally simple, giving essentially pure aromatic heterocycles in minutes under open-flask conditions with catalyst loadings as low as 0.05 mol %.
Co-reporter:Justin A. Goodwin and Aaron Aponick
Chemical Communications 2016 - vol. 52(Issue 40) pp:NaN6731-6731
Publication Date(Web):2016/04/28
DOI:10.1039/C6CC90121B
Correction for ‘Regioselectivity in the Au-catalyzed hydration and hydroalkoxylation of alkynes’ by Justin A. Goodwin et al., Chem. Commun., 2015, 51, 8730–8741.
Co-reporter:Aaron Aponick, Berenger Biannic and Michael R. Jong
Chemical Communications 2010 - vol. 46(Issue 36) pp:NaN6851-6851
Publication Date(Web):2010/08/20
DOI:10.1039/C0CC01961E
The gold(I)-catalyzed endo-cyclization of o-(1-hydroxyallyl)phenols to form chromenes is reported. The title compounds are prepared in high yield from readily available substrates. The system tolerates both electron rich and deficient aryl rings and a high degree of substitution on the allyl moiety.
Co-reporter:Kinga Chojnacka, Stefano Santoro, Radi Awartani, Nigel G. J. Richards, Fahmi Himo and Aaron Aponick
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 15) pp:NaN5353-5353
Publication Date(Web):2011/06/08
DOI:10.1039/C1OB05751K
We report a new method for constructing the ABC ring system of strigolactones, in a single step from a simple linear precursor by acid-catalyzed double cyclization. The reaction proceeds with a high degree of stereochemical control, which can be qualitatively rationalized using DFT calculations. Our concise synthetic approach offers a new model for thinking about the (as yet) unknown chemistry that is employed in the biosynthetic pathways leading to this class of plant hormones.
Co-reporter:John M. Ketcham, Berenger Biannic and Aaron Aponick
Chemical Communications 2013 - vol. 49(Issue 39) pp:NaN4159-4159
Publication Date(Web):2012/11/06
DOI:10.1039/C2CC37166A
The Au(I)-catalyzed intermolecular hydroalkoxylation of alkynes with allylic alcohols to provide allyl vinyl ethers that subsequently undergo Claisen rearrangement is reported. This new cascade reaction strategy facilitates the direct formation of γ,δ-unsaturated ketones from simple starting materials in a single step.
Co-reporter:Justin A. Goodwin and Aaron Aponick
Chemical Communications 2015 - vol. 51(Issue 42) pp:NaN8741-8741
Publication Date(Web):2015/03/27
DOI:10.1039/C5CC00120J
Over the past decade and a half, homogenous gold catalysis has emerged as a diverse and rich field of research resulting in the continuous development of new methods for organic synthesis. The activation of alkynes towards nucleophilic attack by AuI and AuIII complexes is a well-established mode of reactivity and the gold-catalyzed hydration and hydroalkoxylation of alkynes are two of the more well-explored reaction pathways. Although these classes of reactions have seen continuous development since their initial reports, achieving regioselectivity persists as one of the most challenging issues for this chemistry. This article aims to draw attention to the general problem of regioselectivity in these reactions. A select set of examples is presented to highlight the challenges and survey some of the strategies employed to address this problem.

![2-[1-[2-Chloro-4-(1-pyrrolidinyl)benzoyl]-2,3,4,5-tetrahydro-1H-benzazepin-5(R)-yl]-N-isopropylacetamide](http://img.cochemist.com/ccimg/192600/192514-54-8.png)
![2-[1-[2-Chloro-4-(1-pyrrolidinyl)benzoyl]-2,3,4,5-tetrahydro-1H-benzazepin-5(R)-yl]-N-isopropylacetamide](http://img.cochemist.com/ccimg/192600/192514-54-8_b.png)
![1,3-Dioxane-4,6-dione, 5-[(1S)-1,3-diphenyl-2-propynyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/862200/862123-01-1.png)
![1,3-Dioxane-4,6-dione, 5-[(1S)-1,3-diphenyl-2-propynyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/862200/862123-01-1_b.png)
![1,3-Dioxane-4,6-dione,5-[(1R)-1-ethyl-3-phenyl-2-propynyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/862200/862123-00-0.png)
![1,3-Dioxane-4,6-dione,5-[(1R)-1-ethyl-3-phenyl-2-propynyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/862200/862123-00-0_b.png)
![Benzenesulfonamide, 4-methyl-N-[2-(3-oxo-1-propynyl)phenyl]-](http://img.cochemist.com/ccimg/852300/852292-42-3.png)
![Benzenesulfonamide, 4-methyl-N-[2-(3-oxo-1-propynyl)phenyl]-](http://img.cochemist.com/ccimg/852300/852292-42-3_b.png)
![BENZENESULFONAMIDE, N-[2-(3-HYDROXY-1-PROPYNYL)PHENYL]-4-METHYL-](http://img.cochemist.com/ccimg/665100/665033-49-8.png)
![BENZENESULFONAMIDE, N-[2-(3-HYDROXY-1-PROPYNYL)PHENYL]-4-METHYL-](http://img.cochemist.com/ccimg/665100/665033-49-8_b.png)


























![1,3-Dioxane-4,6-dione, 5-[(2-chlorophenyl)methylene]-2,2-dimethyl-](http://img.cochemist.com/ccimg/15900/15851-88-4.png)
![1,3-Dioxane-4,6-dione, 5-[(2-chlorophenyl)methylene]-2,2-dimethyl-](http://img.cochemist.com/ccimg/15900/15851-88-4_b.png)
![1,3-Dioxane-4,6-dione, 5-[(4-chlorophenyl)methylene]-2,2-dimethyl-](http://img.cochemist.com/ccimg/15900/15851-87-3.png)
![1,3-Dioxane-4,6-dione, 5-[(4-chlorophenyl)methylene]-2,2-dimethyl-](http://img.cochemist.com/ccimg/15900/15851-87-3_b.png)
![1,3-Dioxane-4,6-dione, 5-[(4-methoxyphenyl)methylene]-2,2-dimethyl-](http://img.cochemist.com/ccimg/15800/15795-54-7.png)
![1,3-Dioxane-4,6-dione, 5-[(4-methoxyphenyl)methylene]-2,2-dimethyl-](http://img.cochemist.com/ccimg/15800/15795-54-7_b.png)















![Silane, (1,1-dimethylethyl)dimethyl[[(1R)-1-methyl-2-propen-1-yl]oxy]-](/data/chemimg/183000/1375015-06-7.png)
![Silane, (1,1-dimethylethyl)dimethyl[[(1R)-1-methyl-2-propen-1-yl]oxy]-](/data/chemimg/183000/1375015-06-7_b.png)
![(2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)-1',4,4',5-tetrahydro-3H-spiro[furan-2,3'-pyrrolo-[2,1-c][1,4]oxazine]-6'-carbaldehyde](http://img.cochemist.com/ccimg/1324000/1323902-17-5.png)
![(2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)-1',4,4',5-tetrahydro-3H-spiro[furan-2,3'-pyrrolo-[2,1-c][1,4]oxazine]-6'-carbaldehyde](http://img.cochemist.com/ccimg/1324000/1323902-17-5_b.png)
![Silane, (1,1-dimethylethyl)dimethyl[[(1S)-1-methyl-2-propen-1-yl]oxy]-](/data/chemimg/182900/1287305-44-5.png)
![Silane, (1,1-dimethylethyl)dimethyl[[(1S)-1-methyl-2-propen-1-yl]oxy]-](/data/chemimg/182900/1287305-44-5_b.png)
![6-AMINO-6,8-DIHYDRO-5H-IMIDAZO[1,2-A]PYRIMIDIN-7-ONE](http://img.cochemist.com/ccimg/1159900/1159825-32-7.png)
![6-AMINO-6,8-DIHYDRO-5H-IMIDAZO[1,2-A]PYRIMIDIN-7-ONE](http://img.cochemist.com/ccimg/1159900/1159825-32-7_b.png)






![(Acetonitrile)[(2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate](http://img.cochemist.com/ccimg/866700/866641-66-9.png)
![(Acetonitrile)[(2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate](http://img.cochemist.com/ccimg/866700/866641-66-9_b.png)
![Silane, [[3-(chloromethyl)-3-butenyl]oxy](1,1-dimethylethyl)dimethyl-](http://img.cochemist.com/ccimg/605700/605664-46-8.png)
![Silane, [[3-(chloromethyl)-3-butenyl]oxy](1,1-dimethylethyl)dimethyl-](http://img.cochemist.com/ccimg/605700/605664-46-8_b.png)
![4-Piperidinecarboxaldehyde, 1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/553700/553666-09-4.png)
![4-Piperidinecarboxaldehyde, 1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/553700/553666-09-4_b.png)
![Carbamic acid, N-[(4-methylphenyl)sulfonyl]-N-4-pentyn-1-yl-, 1,1-dimethylethyl ester](/data/chemimg/110200/309947-63-5.png)
![Carbamic acid, N-[(4-methylphenyl)sulfonyl]-N-4-pentyn-1-yl-, 1,1-dimethylethyl ester](/data/chemimg/110200/309947-63-5_b.png)
![Indium, tris[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/250700/250643-77-7.png)
![Indium, tris[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/250700/250643-77-7_b.png)

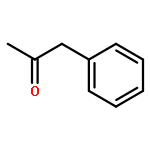
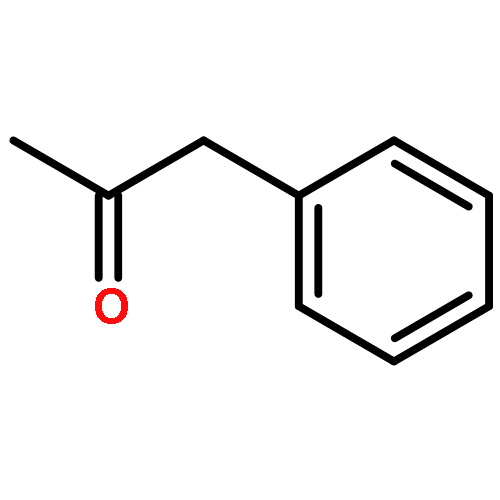
![1-Nonen-5-one, 9-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/198700/198637-26-2.png)
![1-Nonen-5-one, 9-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/198700/198637-26-2_b.png)

![5-Hepten-1-ol, 7-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, (5E)-](http://img.cochemist.com/ccimg/197300/197219-29-7.png)
![5-Hepten-1-ol, 7-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, (5E)-](http://img.cochemist.com/ccimg/197300/197219-29-7_b.png)















![Propanal, 3-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/128500/128461-65-4.png)
![Propanal, 3-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/128500/128461-65-4_b.png)

![Benzaldehyde, 2-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/126700/126680-71-5.png)
![Benzaldehyde, 2-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/126700/126680-71-5_b.png)
![1-Propanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-](/data/chemimg/110800/125244-91-9.png)
![1-Propanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-](/data/chemimg/110800/125244-91-9_b.png)
![7-[TERT-BUTYL(DIMETHYL)SILYL]OXYHEPT-2-EN-1-OL](http://img.cochemist.com/ccimg/121400/121372-78-9.png)
![7-[TERT-BUTYL(DIMETHYL)SILYL]OXYHEPT-2-EN-1-OL](http://img.cochemist.com/ccimg/121400/121372-78-9_b.png)




![PROPANAL, 3-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-2-METHYL-](http://img.cochemist.com/ccimg/91800/91751-25-6.png)
![PROPANAL, 3-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-2-METHYL-](http://img.cochemist.com/ccimg/91800/91751-25-6_b.png)



![2-Buten-1-ol, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, (Z)-](http://img.cochemist.com/ccimg/87800/87770-83-0.png)
![2-Buten-1-ol, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, (Z)-](http://img.cochemist.com/ccimg/87800/87770-83-0_b.png)


![[2-[[tert-butyl(dimethyl)silyl]oxymethyl]phenyl]methanol](http://img.cochemist.com/ccimg/81200/81168-17-4.png)
![[2-[[tert-butyl(dimethyl)silyl]oxymethyl]phenyl]methanol](http://img.cochemist.com/ccimg/81200/81168-17-4_b.png)






















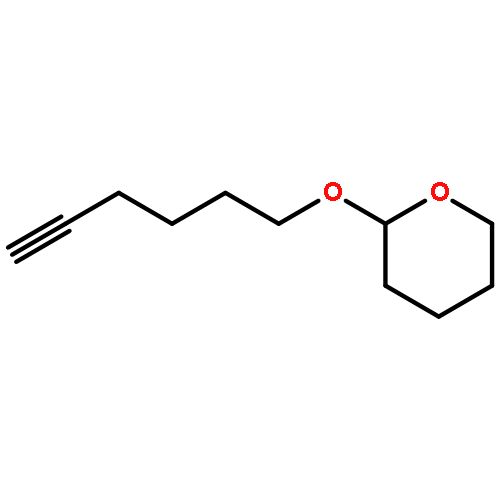
![Benzene, [(3-butynyloxy)methyl]-](http://img.cochemist.com/ccimg/22300/22273-77-4.png)
![Benzene, [(3-butynyloxy)methyl]-](http://img.cochemist.com/ccimg/22300/22273-77-4_b.png)
![3,3-dimethyl-3H-benzo[f]chromene](http://img.cochemist.com/ccimg/19900/19836-62-5.png)
![3,3-dimethyl-3H-benzo[f]chromene](http://img.cochemist.com/ccimg/19900/19836-62-5_b.png)











![6H-1,3-Dioxolo[4,5-g][1]benzopyran](http://img.cochemist.com/ccimg/300/269-55-6.png)
![6H-1,3-Dioxolo[4,5-g][1]benzopyran](http://img.cochemist.com/ccimg/300/269-55-6_b.png)


![3H-Naphtho[2,1-b]pyran](http://img.cochemist.com/ccimg/300/229-80-1.png)
![3H-Naphtho[2,1-b]pyran](http://img.cochemist.com/ccimg/300/229-80-1_b.png)


![1H-Imidazole, 2-[2-(diphenylphosphino)-1-naphthalenyl]-1-[(2,3,4,5,6-pentafluorophenyl)methyl]-4,5-diphenyl-](/data/chemimg/3726300/1454371-42-6.png)
![1H-Imidazole, 2-[2-(diphenylphosphino)-1-naphthalenyl]-1-[(2,3,4,5,6-pentafluorophenyl)methyl]-4,5-diphenyl-](/data/chemimg/3726300/1454371-42-6_b.png)


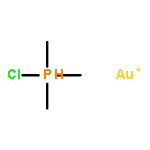




![Gold,[[1,1'-biphenyl]-2-ylbis(1,1-dimethylethyl)phosphine]chloro-](http://img.cochemist.com/ccimg/854100/854045-93-5.png)
![Gold,[[1,1'-biphenyl]-2-ylbis(1,1-dimethylethyl)phosphine]chloro-](http://img.cochemist.com/ccimg/854100/854045-93-5_b.png)

