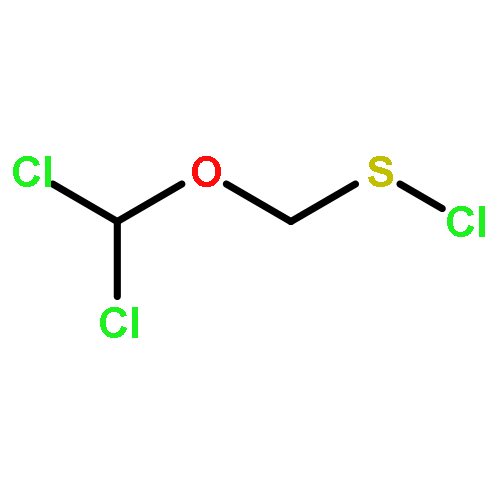
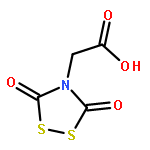
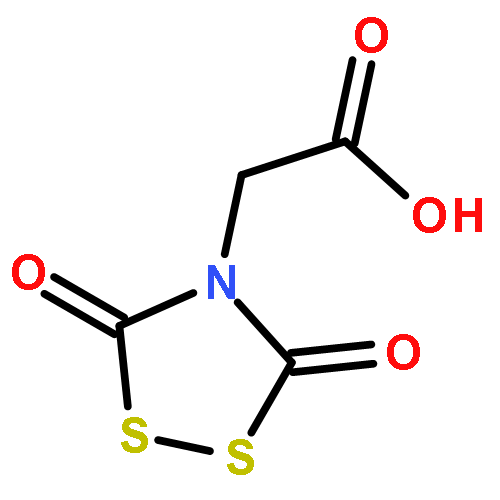
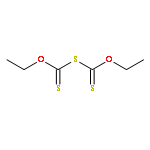
Co-reporter:George Barany, Doyle Britton, Lin Chen, Robert P. Hammer, Matthew J. Henley, Alex M. Schrader, and Victor G. Young Jr.
The Journal of Organic Chemistry 2015 Volume 80(Issue 22) pp:11313-11321
Publication Date(Web):September 29, 2015
DOI:10.1021/acs.joc.5b01826
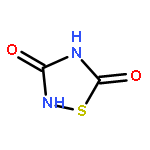
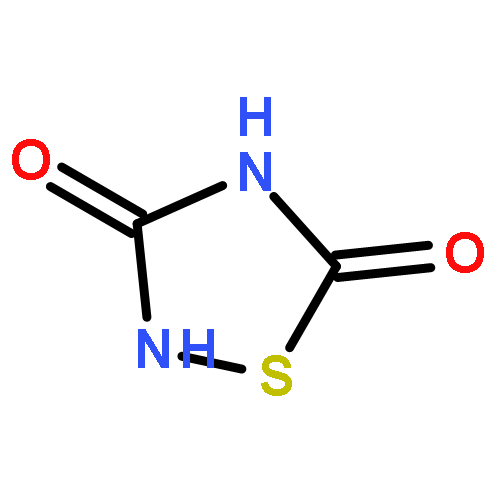
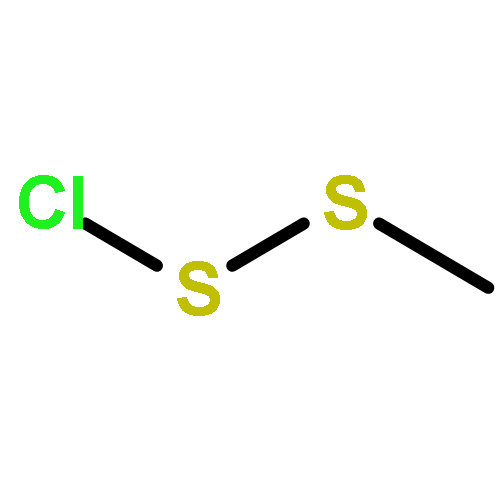
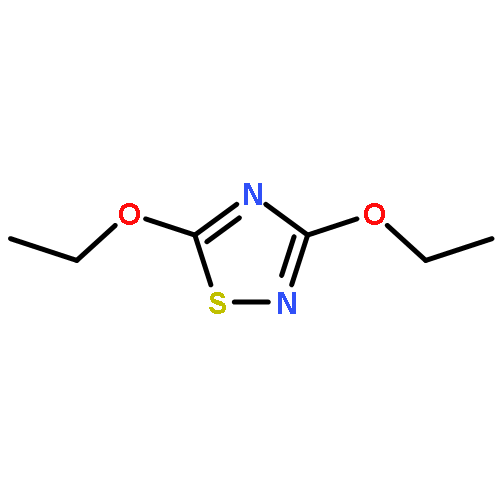
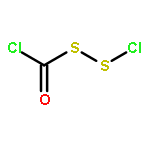
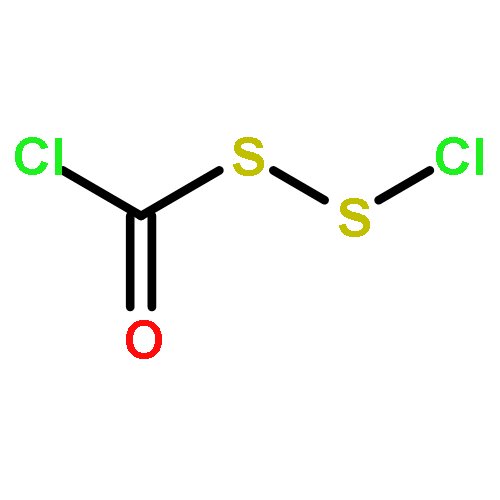
The Zumach–Weiss–Kühle (ZWK) reaction provides 1,2,4-dithiazolidine-3,5-diones [dithiasuccinoyl (Dts)-amines] by the rapid reaction of O-ethyl thiocarbamates plus (chlorocarbonyl)sulfenyl chloride, with ethyl chloride and hydrogen chloride being formed as coproducts, and carbamoyl chlorides or isocyanates generated as yield-diminishing byproducts. However, when the ZWK reaction is applied with (N-ethoxythiocarbonyl)urethane as the starting material, heterocyclization to the putative “Dts-urethane” does not occur. Instead, the reaction directly provides (chlorocarbonyl)(N-ethoxycarbonylcarbamoyl)disulfane, a reasonably stable crystalline compound; modified conditions stop at the (chlorocarbonyl)[1-ethoxy-(N-ethoxycarbonyl)formimidoyl]disulfane intermediate. The title (chlorocarbonyl)(carbamoyl)disulfane cannot be converted to the elusive Dts derivative, but rather gives (N-ethoxycarbonyl)carbamoyl chloride upon thermolysis, or (N-ethoxycarbonyl)isocyanate upon treatment with tertiary amines. Additional transformations of these compounds have been discovered, providing entries to both known and novel species. X-ray crystallographic structures are reported for the title (chlorocarbonyl)(carbamoyl)disulfane; for (methoxycarbonyl)(N-ethoxycarbonylcarbamoyl)disulfane, which is the corresponding adduct after quenching in methanol; for [1-ethoxy-(N-ethoxycarbonyl)formimidoyl](N′-methyl-N′-phenylcarbamoyl)disulfane, which is obtained by trapping the title intermediate with N-methylaniline; and for (N-ethoxycarbonylcarbamoyl)(N′-methyl-N′-phenylcarbamoyl)disulfane, which is a short-lived intermediate in the reaction of the title (chlorocarbonyl)(carbamoyl)disulfane with excess N-methylaniline. The new chemistry and structural information reported herein is expected to contribute to accurate modeling of the ZWK reaction trajectory.
Co-reporter:Alex M. Schrader, Alayne L. Schroll, and George Barany
The Journal of Organic Chemistry 2011 Volume 76(Issue 19) pp:7882-7892
Publication Date(Web):August 26, 2011
DOI:10.1021/jo201329n
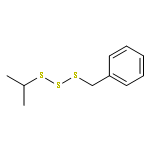
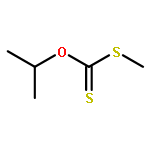
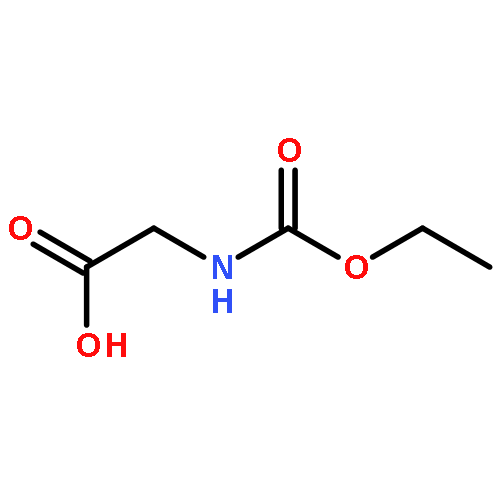
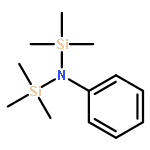
The title compound classes, (carbamoyl)sulfenyl chlorides and ((carbamoyl)dithio)carbonyl chlorides, have been implicated previously as unstable, albeit trappable, intermediates in organosulfur chemistry. The present work reports for each of these functional groups: (i) several routes to prepare it in the N-methylaniline family; (ii) its direct structural characterization by several spectroscopic techniques; (iii) its rather unexpected stability and its ultimate fate when it decomposes; (iv) a series of further chemical transformations that give highly stable derivatives, each in turn subject to thorough characterization. Relevant kinetic and mechanistic experiments were carried out, including some with p-methyl- and 2,6-dimethyl-substituted N-methylanilines. Given that the title compounds can be isolated and are relatively stable, they may find applications in the preparation of thiolyzable and/or photolabile protecting groups for the sulfhydryl function of cysteine and for the development of new protein synthesis and modification reagents.
Co-reporter:Mian Liu, Victor G. Young Jr., Sachin Lohani, David Live, George Barany
Carbohydrate Research 2005 Volume 340(Issue 7) pp:1273-1285
Publication Date(Web):23 May 2005
DOI:10.1016/j.carres.2005.02.029
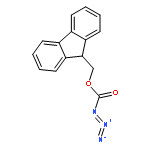
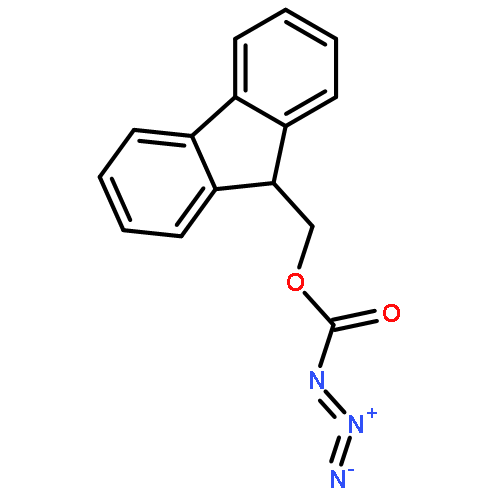
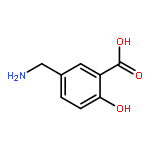
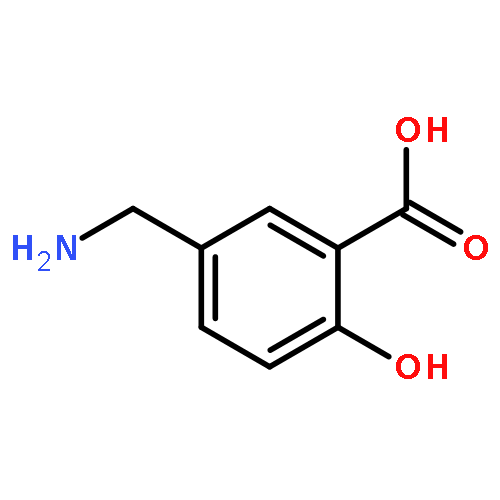
TN antigen building blocks Nα-(9-fluorenylmethoxycarbonyl)-O-(3,4,6-tri-O-acetyl-2-azido-2-deoxy-α-d-galactopyranosyl)-l-serine/l-threonine pentafluorophenyl ester [Fmoc-l-Ser/l-Thr(Ac3-α-d-GalN3)-OPfp, 13/14] have been synthesized by two different routes, which have been compared. Overall isolated yields [three or four chemical steps, and minimal intermediary purification steps] of enantiopure 13 and 14 were 5–18% and 6–10%, respectively, based on 3,4,6-tri-O-acetyl-d-galactal (1). A byproduct of the initial azidonitration reaction of the synthetic sequence, that is, N-acetyl-3,4,6-tri-O-acetyl-2-azido-2-deoxy-α-d-galactopyranosylamine (5), has been characterized by X-ray crystallography, and shown by 1H NMR spectroscopy to form complexes with lithium bromide, lithium iodide, or sodium iodide in acetonitrile-d3. Intermediates 3,4,6-tri-O-acetyl-2-azido-2-deoxy-α-d-galactopyranosyl bromide (6) and 3,4,6-tri-O-acetyl-2-azido-2-deoxy-β-d-galactopyranosyl chloride (7) were used to glycosylate Nα-(9-fluorenylmethoxycarbonyl)-l-serine/l-threonine pentafluorophenyl esters [Fmoc-l-Ser/l-Thr-OPfp, 11/12]. Previously undescribed low-level dehydration side reactions were observed at this stage; the unwanted byproducts were easily removed by column chromatography.
Co-reporter:Mian Liu, George Barany, David Live
Carbohydrate Research 2005 Volume 340(Issue 13) pp:2111-2122
Publication Date(Web):26 September 2005
DOI:10.1016/j.carres.2005.05.023
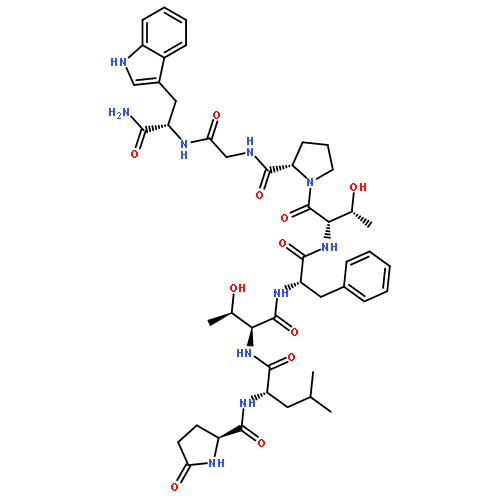
The glycopeptide, Ac-Pro-Thr(α-d-GalNAc)-Thr(α-d-GalNAc)-Thr(α-d-GalNAc)-Pro-Leu-Lys-NH2 (1), which features three consecutive O-glycosylated Thr residues and mimics a portion of mucin 2, has been prepared by solid-phase synthesis. Seven related, partially glycosylated peptides (2–8) were synthesized as well. This suite of molecules allowed a systematic analysis of synthetic protocols. Nα-(9-Fluorenylmethoxycarbonyl)-O-(3,4,6-tri-O-acetyl-2-azido-2-deoxy-α-d-galactopyranosyl)-l-threonine pentafluorophenyl ester [Fmoc-l-Thr(Ac3-α-d-GalN3)-OPfp] was used as a building block that coupled efficiently when used in a relatively low molar excess, that is, ∼1.5 equiv, with N,N-dimethylformamide (DMF) as the solvent. For conversion of the azido group to the N-acetyl function, direct treatment with thioacetic acid was preferred over a two-step procedure involving reduction with dithiothreitol (DTT) followed by N-acetylation. Effective O-deacetylation of 1–8 in solution was achieved by treatment with sodium methoxide (10–15 mM; ∼5 equiv) in methanol. On-resin deacetylation techniques were also examined, using sodium methoxide (6–10 mM) in DMF–methanol (17:3) (for 4 and 11) or hydrazine (70 mM) in methanol (for 8). The more convenient on-resin technique in DMF–methanol gave yields similar to solution conditions, and promises to be widely useful for solid-phase glycopeptide synthesis. HPLC profiles showed that free glycopeptides elute earlier than the corresponding O-acetylated derivatives, and that retention times vary systematically with the number of sugar moieties. 1H NMR studies carried out in water showed an increase in conformational organization of glycopeptides with increased density of glycosylation.
Co-reporter:Judit Tulla-Puche;Irina V. Getun;Jordi Alsina;Ferno Albericio and
European Journal of Organic Chemistry 2004 Volume 2004(Issue 22) pp:
Publication Date(Web):2 NOV 2004
DOI:10.1002/ejoc.200400507
Two cyclic analogues of the protein bovine pancreatic trypsin inhibitor (BPTI), c-[R]Smc and c-Cys5[R]Abu, have been synthesized. For the first target, a semisynthetic approach featured a cyclization that took advantage of the constraint imposed by the three disulfides of the native protein. For the second target, an entirely unstructured 58-residue thiolester, prepared by total synthesis with mild Fmoc chemistry and a side-chain anchoring strategy, was cyclized by native chemical ligation. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2004)
Co-reporter:Esther Cros;Marta Planas;Eduard Bardají
European Journal of Organic Chemistry 2004 Volume 2004(Issue 17) pp:
Publication Date(Web):17 AUG 2004
DOI:10.1002/ejoc.200400244
The N-tetrachlorophthaloyl-(TCP-)amino protecting group has been evaluated for use in solid-phase peptide synthesis. The TCP group was unaffected by exposure to either piperidine or N,N-diisopropylethylamine (DIEA), which suggests compatibility with both Fmoc and Boc solid-phase synthesis protocols. Quantitative TCP removal was achieved by treatment with hydrazine/DMF (3:17) at 35 °C for 30 min or with ethylenediamine/DMF (1:200) at 50 °C for 30 min. Several C-terminal peptide amides were synthesized successfully by following protocols that use hydrazine/DMF (3:17) at 40 °C for 1 h for repetitive deprotection. Treatment of TCP-amines with methylamine or with diamines did not give the corresponding amines (deprotected), but rather the appropriate N,N′-disubstituted tetrachlorophthalamides, which corresponds to a single ring-opening step. This observation was harnessed to prepare linear and macrocyclic peptide−arene hybrids based on the mild reaction of the parent TCP compound with 1,3-diaminopropane/DMF (1:49) at 25 °C for 5 min. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2004)
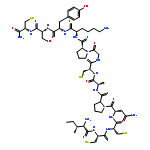
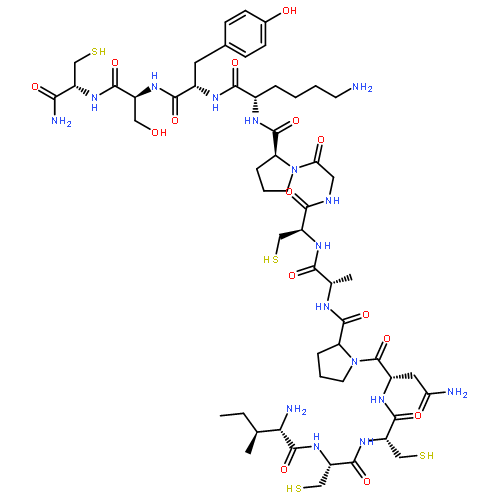
Co-reporter:Irina V. Getun, C. Kent Brown, Judit Tulla-Puche, Douglas Ohlendorf, ... George Barany
Journal of Molecular Biology (18 January 2008) Volume 375(Issue 3) pp:812-823
Publication Date(Web):18 January 2008
DOI:10.1016/j.jmb.2007.10.084
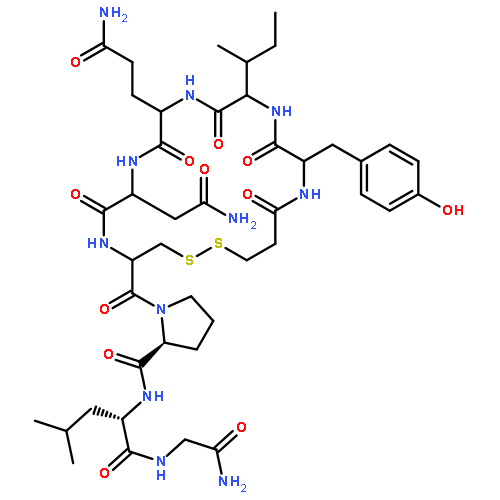
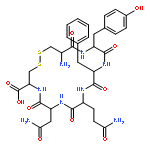
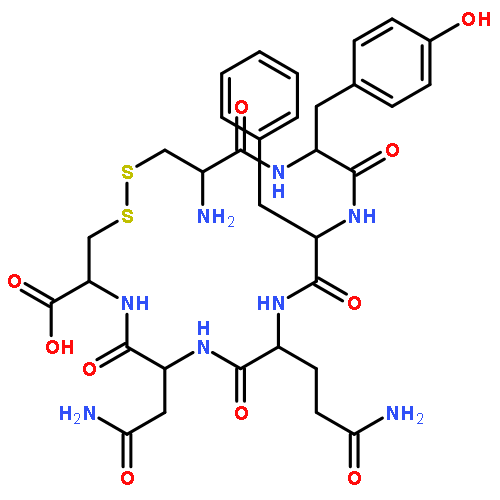
Crystal structures, at 1.7 Å resolution, were solved for complexes between each of two chemically synthesized partially folded analogues of bovine pancreatic trypsin inhibitor (BPTI) with the proteolytically inactive rat trypsin mutant S195A. The BPTI analogue termed [14–38]Abu retains only the disulfide bond between Cys14 and Cys38, while Cys5, Cys30, Cys51, and Cys55 are replaced by isosteric α-amino-n-butyric acid residues. The analogue K26P,A27D[14–38]Abu contains two further replacements, by statistically favored residues, in the type I β-turn that has been suggested to be a main site for initiation of BPTI folding. As a control, the structure of the complex between S195A trypsin and wild-type BPTI was also solved. Despite significant differences in the degree of structure detected among these three BPTIs in solution by several biophysical techniques, their tertiary folds once bound to S195A trypsin in a crystalline lattice are essentially superimposable.
.jpg)

![2-[[(2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-3-phenylpropanoyl]amino]acetic Acid](http://img.cochemist.com/ccimg/25700/25616-33-5.png)
![2-[[(2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-3-phenylpropanoyl]amino]acetic Acid](http://img.cochemist.com/ccimg/25700/25616-33-5_b.png)
![Diphosphoric acid,P-[(2E,6E,10E)-3,7,11,15-tetramethyl-2,6,10,14-hexadecatetraen-1-yl] ester](http://img.cochemist.com/ccimg/6700/6699-20-3.png)
![Diphosphoric acid,P-[(2E,6E,10E)-3,7,11,15-tetramethyl-2,6,10,14-hexadecatetraen-1-yl] ester](http://img.cochemist.com/ccimg/6700/6699-20-3_b.png)
![13H-4,6:21,24-Dietheno-8,12-metheno-1H-pyrido[3',2':14,15][1,11]dioxacycloeicosino[2,3,4-ij]isoquinolinium,2,3,13a,14,15,16,25,25a-octahydro-9,18,19,29-tetramethoxy-1,1,14,14-tetramethyl-,(13aR,25aS)-](http://img.cochemist.com/ccimg/5200/5152-30-7.png)
![13H-4,6:21,24-Dietheno-8,12-metheno-1H-pyrido[3',2':14,15][1,11]dioxacycloeicosino[2,3,4-ij]isoquinolinium,2,3,13a,14,15,16,25,25a-octahydro-9,18,19,29-tetramethoxy-1,1,14,14-tetramethyl-,(13aR,25aS)-](http://img.cochemist.com/ccimg/5200/5152-30-7_b.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4_b.png)
![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1.png)
![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1_b.png)
![Pentanoic acid,5-[[9-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-9H-xanthen-2-yl]oxy]-](http://img.cochemist.com/ccimg/137100/137052-29-0.png)
![Pentanoic acid,5-[[9-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-9H-xanthen-2-yl]oxy]-](http://img.cochemist.com/ccimg/137100/137052-29-0_b.png)
![Pentanoic acid,5-[4-[[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]methyl]-3,5-dimethoxyphenoxy]-](http://img.cochemist.com/ccimg/115200/115109-65-4.png)
![Pentanoic acid,5-[4-[[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]methyl]-3,5-dimethoxyphenoxy]-](http://img.cochemist.com/ccimg/115200/115109-65-4_b.png)

![PROPANOIC ACID, 3-[[(2,4,6-TRIMETHYLPHENYL)METHYL]THIO]-](http://img.cochemist.com/ccimg/78300/78221-81-5.png)
![PROPANOIC ACID, 3-[[(2,4,6-TRIMETHYLPHENYL)METHYL]THIO]-](http://img.cochemist.com/ccimg/78300/78221-81-5_b.png)




![2H-Pyran, tetrahydro-2-[[(2E,6E)-3,7,11-trimethyl-2,6,10-dodecatrien-1-yl]oxy]-](/data/chemimg/1918800/67858-93-9.png)
![2H-Pyran, tetrahydro-2-[[(2E,6E)-3,7,11-trimethyl-2,6,10-dodecatrien-1-yl]oxy]-](/data/chemimg/1918800/67858-93-9_b.png)


![Glycine, N-[(1,1-dimethylethoxy)carbonyl]-L-tyrosyl-](http://img.cochemist.com/ccimg/64000/63909-62-6.png)
![Glycine, N-[(1,1-dimethylethoxy)carbonyl]-L-tyrosyl-](http://img.cochemist.com/ccimg/64000/63909-62-6_b.png)

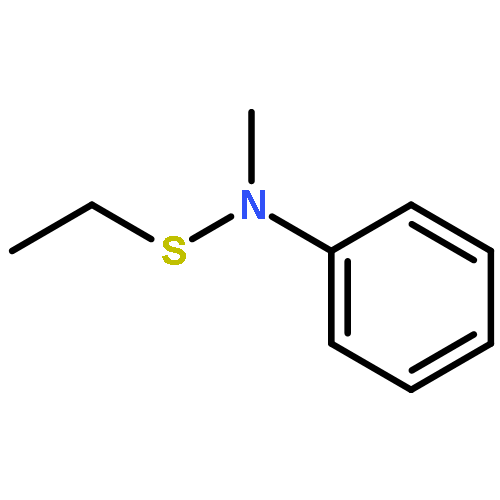

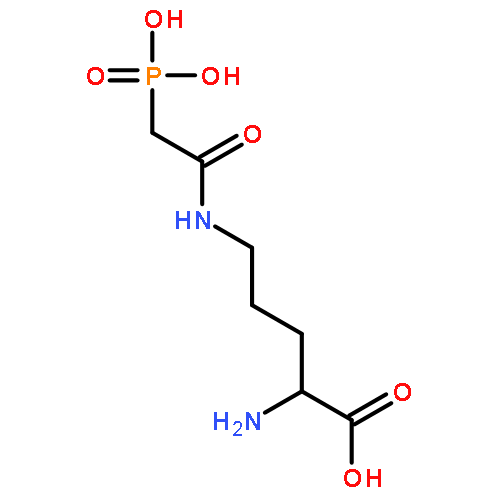
![N-[2-(diethylamino)ethyl]acetamide](http://img.cochemist.com/ccimg/63300/63224-19-1.png)
![N-[2-(diethylamino)ethyl]acetamide](http://img.cochemist.com/ccimg/63300/63224-19-1_b.png)
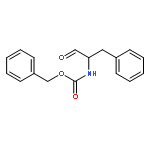
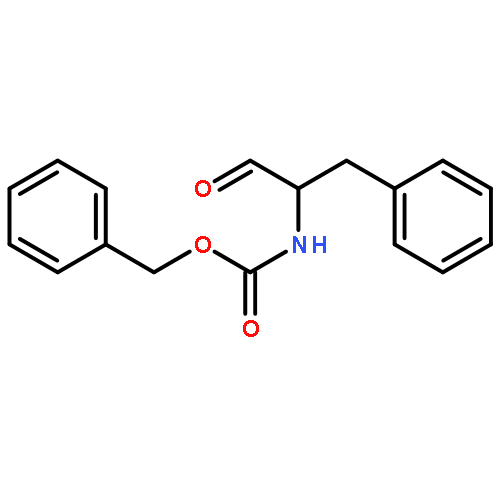
![Glycine,N-[N-[N-[N-[(1,1-dimethylethoxy)carbonyl]-L-alanyl]-L-alanyl]-L-alanyl]-](http://img.cochemist.com/ccimg/62600/62570-97-2.png)
![Glycine,N-[N-[N-[N-[(1,1-dimethylethoxy)carbonyl]-L-alanyl]-L-alanyl]-L-alanyl]-](http://img.cochemist.com/ccimg/62600/62570-97-2_b.png)





![2H-Pyran, 2-[(3,7-dimethyl-2,6-octadienyl)oxy]tetrahydro-, (E)-](http://img.cochemist.com/ccimg/59700/59632-99-4.png)
![2H-Pyran, 2-[(3,7-dimethyl-2,6-octadienyl)oxy]tetrahydro-, (E)-](http://img.cochemist.com/ccimg/59700/59632-99-4_b.png)
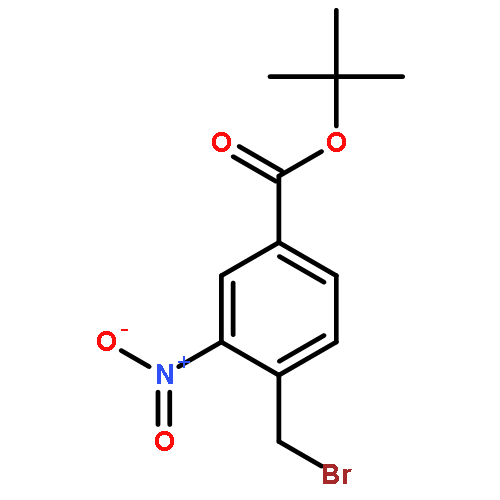
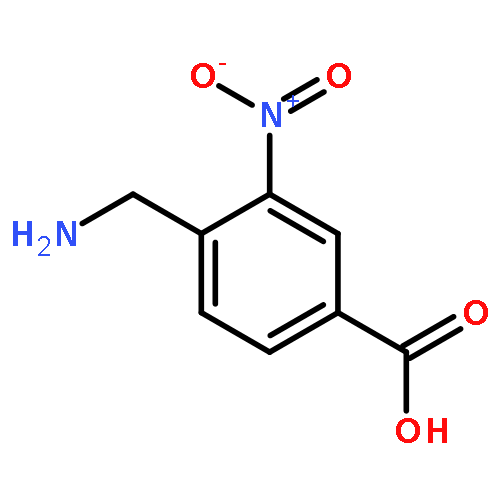
![Benzoic acid, 4-[[[(1,1-dimethylethoxy)carbonyl]amino]methyl]-3-nitro-](http://img.cochemist.com/ccimg/56100/56047-52-0.png)
![Benzoic acid, 4-[[[(1,1-dimethylethoxy)carbonyl]amino]methyl]-3-nitro-](http://img.cochemist.com/ccimg/56100/56047-52-0_b.png)


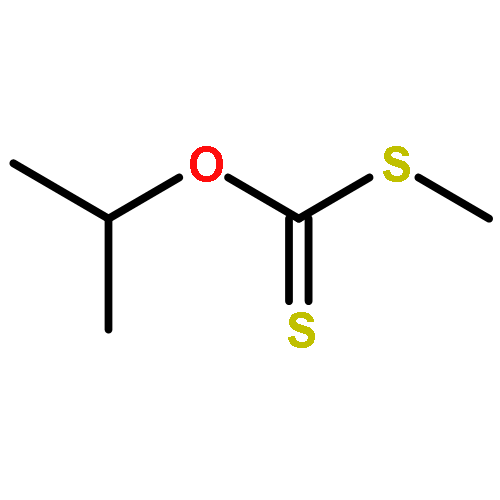


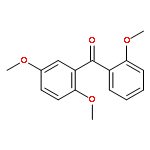
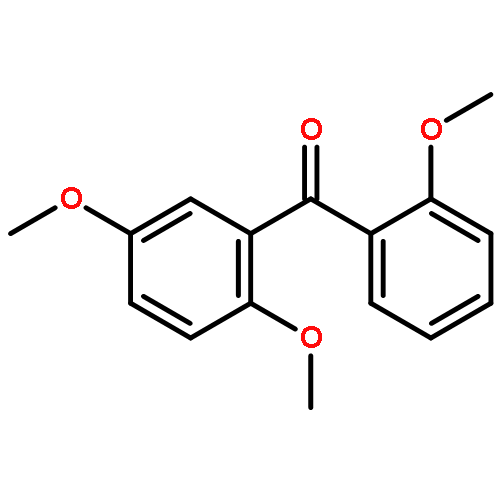
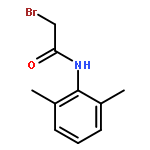
![Ethanaminium,2-[[(dodecyloxy)hydroxyphosphinyl]oxy]-N,N,N-trimethyl-, inner salt](http://img.cochemist.com/ccimg/29600/29557-51-5.png)
![Ethanaminium,2-[[(dodecyloxy)hydroxyphosphinyl]oxy]-N,N,N-trimethyl-, inner salt](http://img.cochemist.com/ccimg/29600/29557-51-5_b.png)
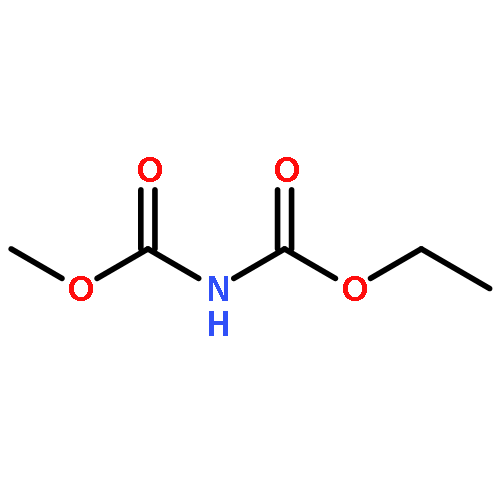
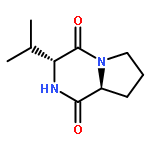
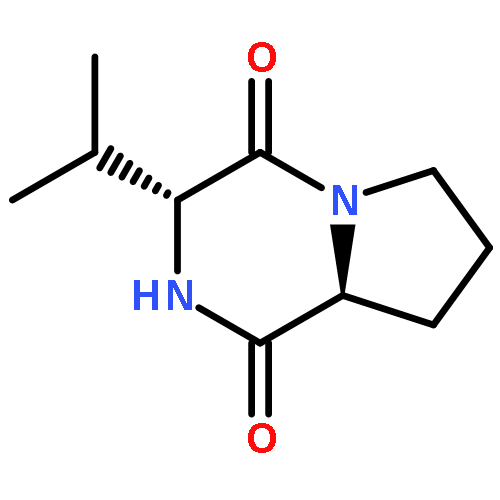



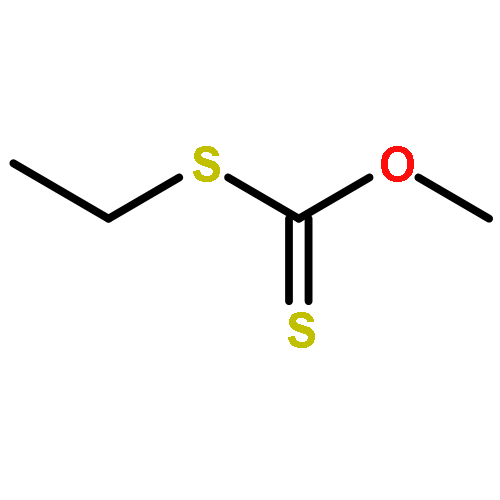
![Glycine, N-[(1,1-dimethylethoxy)carbonyl]-L-tryptophyl-](http://img.cochemist.com/ccimg/25700/25691-58-1.png)
![Glycine, N-[(1,1-dimethylethoxy)carbonyl]-L-tryptophyl-](http://img.cochemist.com/ccimg/25700/25691-58-1_b.png)
![Acetic acid,2-[(ethoxythioxomethyl)thio]-](http://img.cochemist.com/ccimg/25600/25554-84-1.png)
![Acetic acid,2-[(ethoxythioxomethyl)thio]-](http://img.cochemist.com/ccimg/25600/25554-84-1_b.png)








![Benzoic acid, 4-[(formylamino)methyl]-](http://img.cochemist.com/ccimg/17900/17847-25-5.png)
![Benzoic acid, 4-[(formylamino)methyl]-](http://img.cochemist.com/ccimg/17900/17847-25-5_b.png)
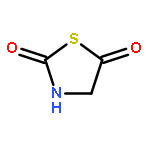
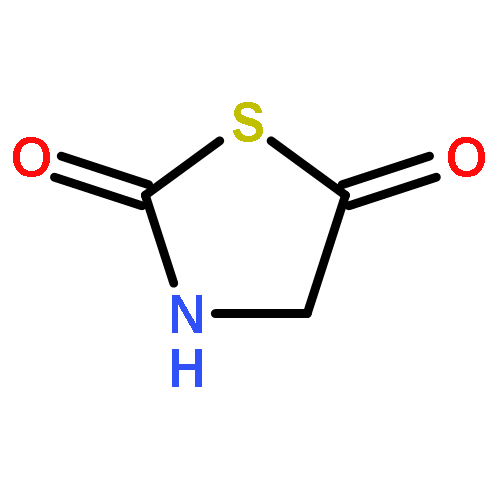



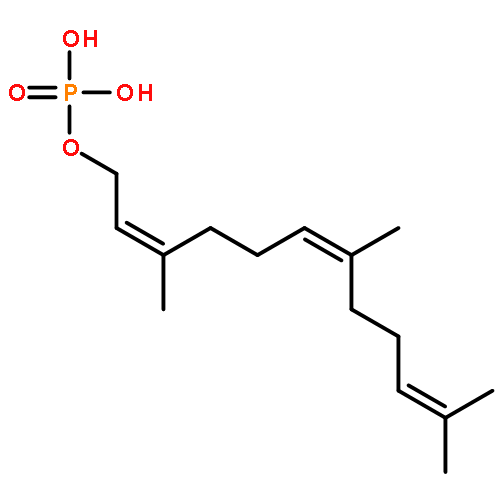
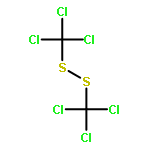
![Carbamothioic acid, methyl-, O-[2-(dimethylamino)ethyl] ester](http://img.cochemist.com/ccimg/14200/14128-41-7.png)
![Carbamothioic acid, methyl-, O-[2-(dimethylamino)ethyl] ester](http://img.cochemist.com/ccimg/14200/14128-41-7_b.png)


















![2,6-di-tert-butyl-4-[(2,5-dimethoxyphenyl)imino]cyclohexa-2,5-dien-1-one](http://img.cochemist.com/ccimg/5900/5813-75-2.png)
![2,6-di-tert-butyl-4-[(2,5-dimethoxyphenyl)imino]cyclohexa-2,5-dien-1-one](http://img.cochemist.com/ccimg/5900/5813-75-2_b.png)
![N-[2-(1-piperidinyl)ethyl]-Acetamide](http://img.cochemist.com/ccimg/5700/5661-20-1.png)
![N-[2-(1-piperidinyl)ethyl]-Acetamide](http://img.cochemist.com/ccimg/5700/5661-20-1_b.png)
![N-(3,4-dimethoxybenzyl)thieno[2,3-d]pyrimidin-4-amine](http://img.cochemist.com/ccimg/5600/5585-23-9.png)
![N-(3,4-dimethoxybenzyl)thieno[2,3-d]pyrimidin-4-amine](http://img.cochemist.com/ccimg/5600/5585-23-9_b.png)

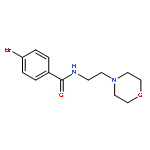
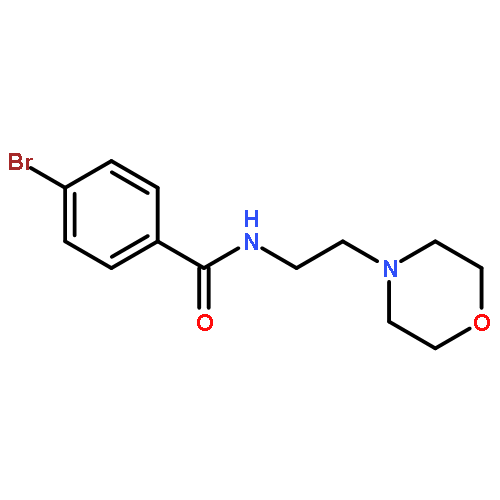
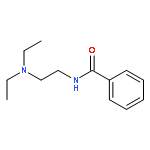
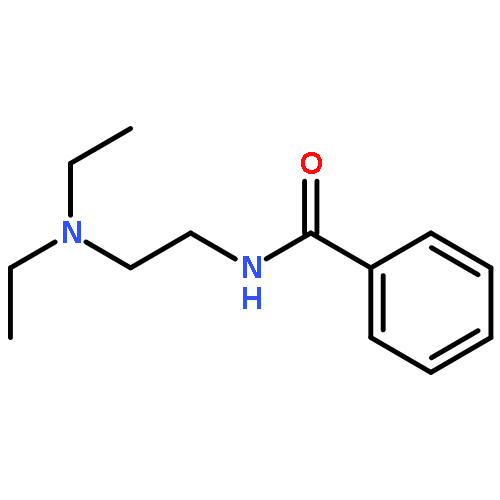
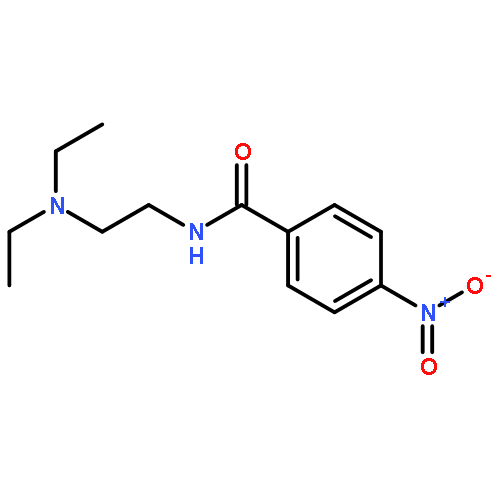
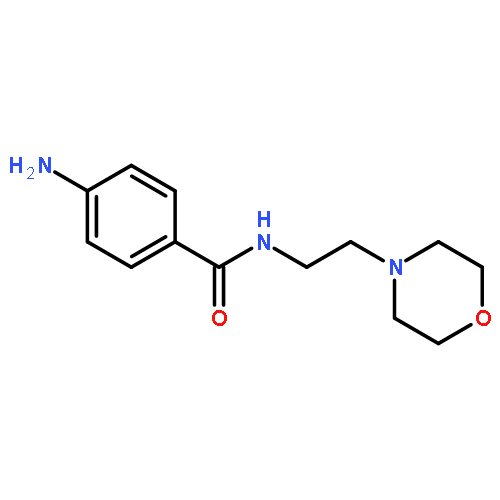
![Benzamide, N-[2-(4-morpholinyl)ethyl]-](http://img.cochemist.com/ccimg/4500/4476-13-5.png)
![Benzamide, N-[2-(4-morpholinyl)ethyl]-](http://img.cochemist.com/ccimg/4500/4476-13-5_b.png)
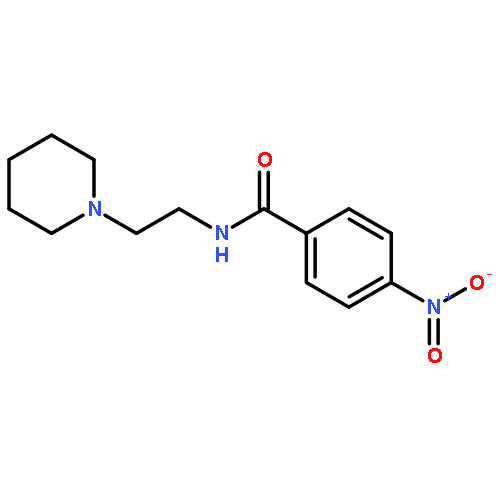
![BENZAMIDE, N-[2-(1-PIPERIDINYL)ETHYL]-](http://img.cochemist.com/ccimg/4400/4380-82-9.png)
![BENZAMIDE, N-[2-(1-PIPERIDINYL)ETHYL]-](http://img.cochemist.com/ccimg/4400/4380-82-9_b.png)


![Benzamide, N-[2-(4-morpholinyl)ethyl]-4-nitro-](http://img.cochemist.com/ccimg/4000/3948-64-9.png)
![Benzamide, N-[2-(4-morpholinyl)ethyl]-4-nitro-](http://img.cochemist.com/ccimg/4000/3948-64-9_b.png)


![2-[(2-amino-4-methylpentanoyl)amino]-3-phenylpropanoic Acid](http://img.cochemist.com/ccimg/3100/3063-05-6.png)
![2-[(2-amino-4-methylpentanoyl)amino]-3-phenylpropanoic Acid](http://img.cochemist.com/ccimg/3100/3063-05-6_b.png)














![Piperidine, 1-[(tetrahydro-2-furanyl)methyl]-](http://img.cochemist.com/ccimg/1200/1199-81-1.png)
![Piperidine, 1-[(tetrahydro-2-furanyl)methyl]-](http://img.cochemist.com/ccimg/1200/1199-81-1_b.png)
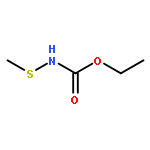
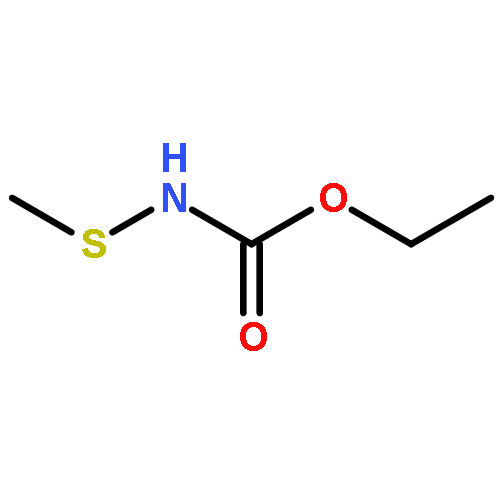






















![2H-Pyran, 2-[[(2E)-6-azido-3,7-dimethyl-2,7-octadienyl]oxy]tetrahydro-](http://img.cochemist.com/ccimg/328200/328122-52-7.png)
![2H-Pyran, 2-[[(2E)-6-azido-3,7-dimethyl-2,7-octadienyl]oxy]tetrahydro-](http://img.cochemist.com/ccimg/328200/328122-52-7_b.png)
![2-Methyl-3-oxo-3,4-dihydro-2H-benzo[b][1,4]thiazine-6-carboxylic acid](http://img.cochemist.com/ccimg/272500/272437-85-1.png)
![2-Methyl-3-oxo-3,4-dihydro-2H-benzo[b][1,4]thiazine-6-carboxylic acid](http://img.cochemist.com/ccimg/272500/272437-85-1_b.png)

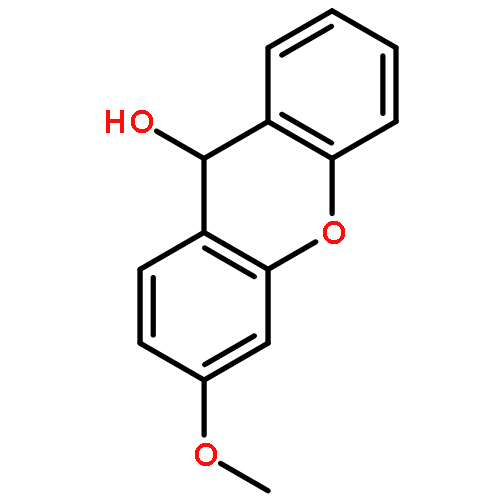

![Pentanoic acid, 5-[(9-amino-9H-xanthen-2-yl)oxy]-](http://img.cochemist.com/ccimg/143700/143677-66-1.png)
![Pentanoic acid, 5-[(9-amino-9H-xanthen-2-yl)oxy]-](http://img.cochemist.com/ccimg/143700/143677-66-1_b.png)
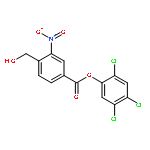
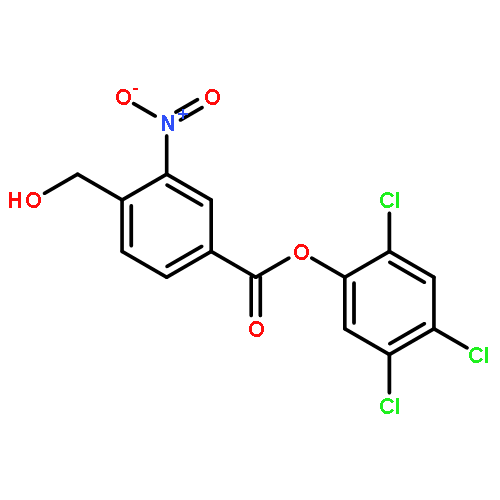
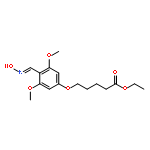
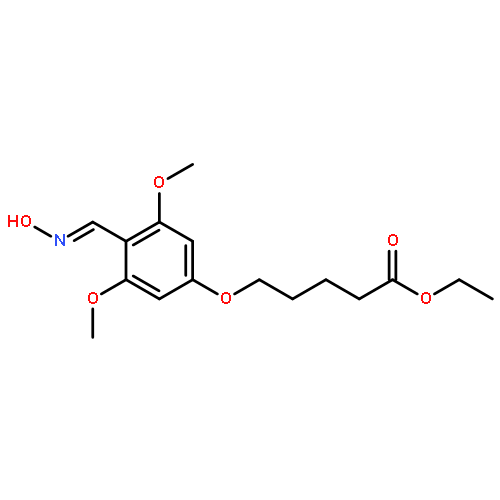
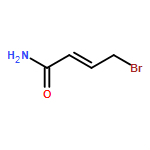
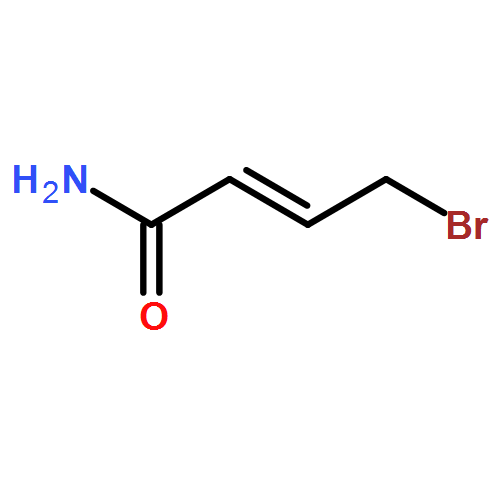
![Pentanoic acid, 5-[2-[(hydroxyimino)methyl]-3,5-dimethoxyphenoxy]-](http://img.cochemist.com/ccimg/115200/115109-62-1.png)
![Pentanoic acid, 5-[2-[(hydroxyimino)methyl]-3,5-dimethoxyphenoxy]-](http://img.cochemist.com/ccimg/115200/115109-62-1_b.png)

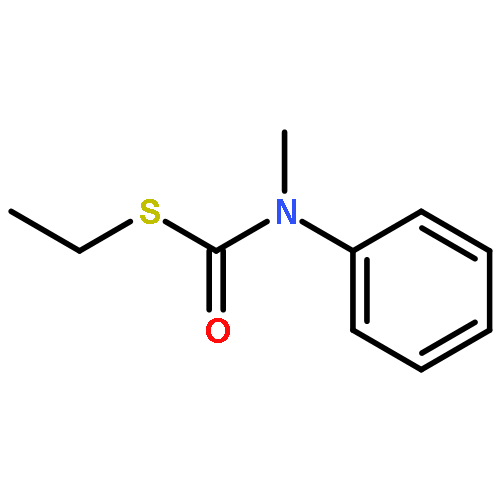
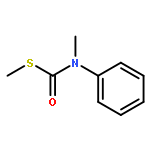
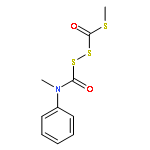
![Benzenamine, N-methyl-N-[[(methylthio)carbonyl]thio]-](http://img.cochemist.com/ccimg/95300/95271-87-7.png)
![Benzenamine, N-methyl-N-[[(methylthio)carbonyl]thio]-](http://img.cochemist.com/ccimg/95300/95271-87-7_b.png)






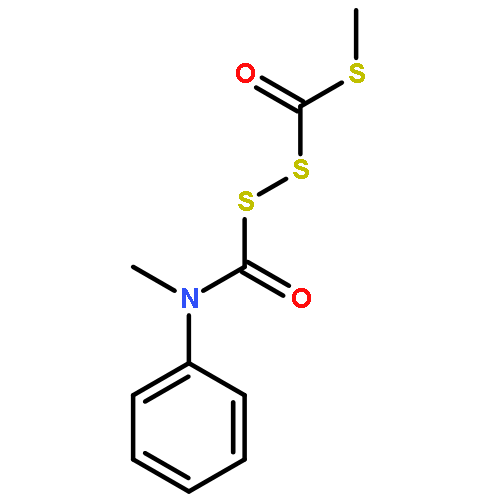
![BENZENAMINE, N-METHYL-N-[[(METHYLPHENYLAMINO)CARBONYL]DITHIO]-](http://img.cochemist.com/ccimg/88800/88766-58-9.png)
![BENZENAMINE, N-METHYL-N-[[(METHYLPHENYLAMINO)CARBONYL]DITHIO]-](http://img.cochemist.com/ccimg/88800/88766-58-9_b.png)



![BENZENAMINE, N-[(METHOXYCARBONYL)DITHIO]-N-METHYL-](http://img.cochemist.com/ccimg/88800/88766-30-7.png)
![BENZENAMINE, N-[(METHOXYCARBONYL)DITHIO]-N-METHYL-](http://img.cochemist.com/ccimg/88800/88766-30-7_b.png)
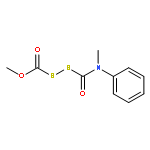
![BENZENAMINE, N-[(METHOXYCARBONYL)THIO]-N-METHYL-](http://img.cochemist.com/ccimg/87500/87463-14-7.png)
![BENZENAMINE, N-[(METHOXYCARBONYL)THIO]-N-METHYL-](http://img.cochemist.com/ccimg/87500/87463-14-7_b.png)

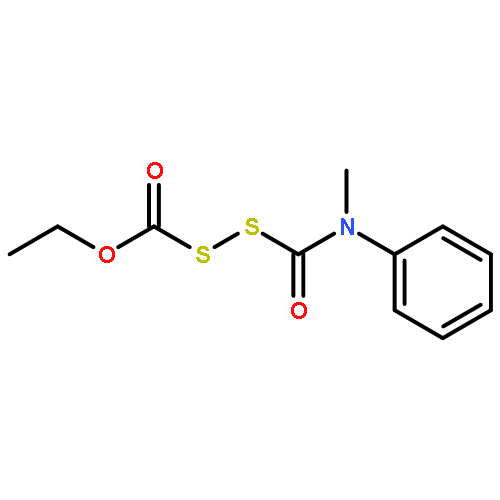


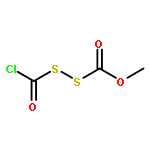
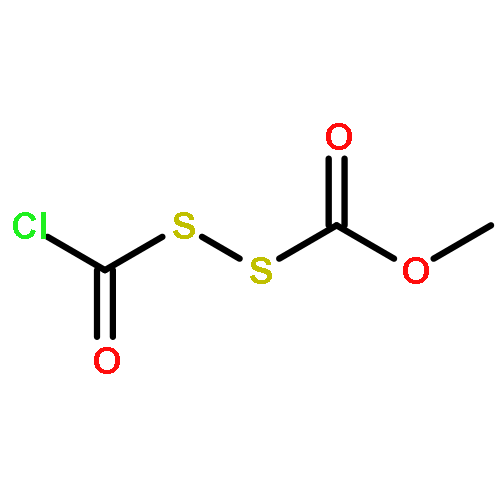
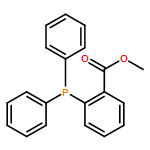



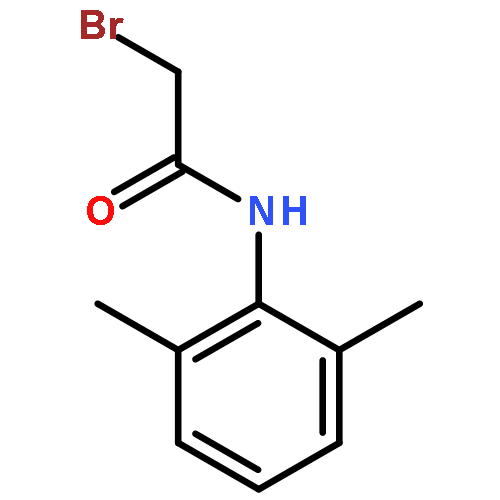
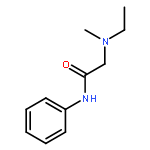
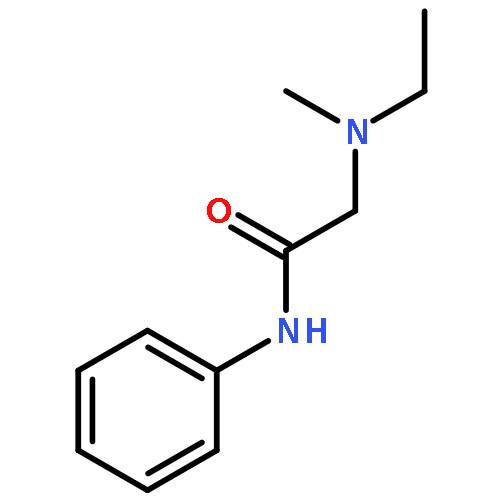
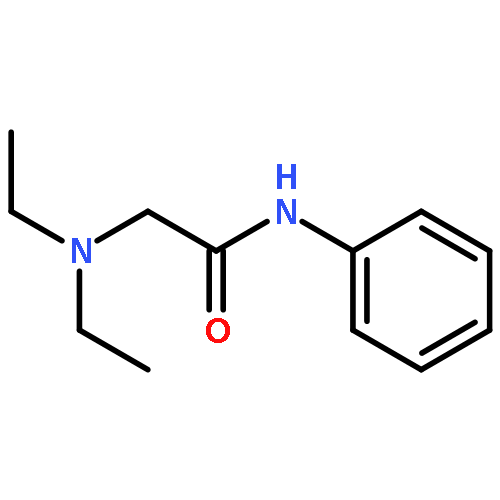
![N-(2,6-DIMETHYLPHENYL)-2-[ETHYL(METHYL)AMINO]ACETAMIDE](http://img.cochemist.com/ccimg/74700/74634-66-5.png)
![N-(2,6-DIMETHYLPHENYL)-2-[ETHYL(METHYL)AMINO]ACETAMIDE](http://img.cochemist.com/ccimg/74700/74634-66-5_b.png)

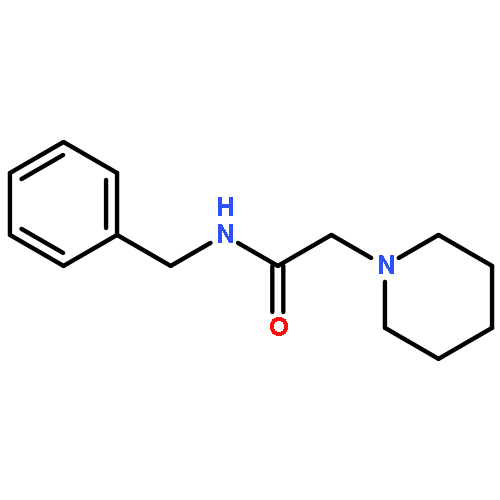
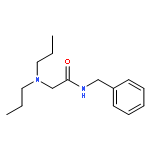
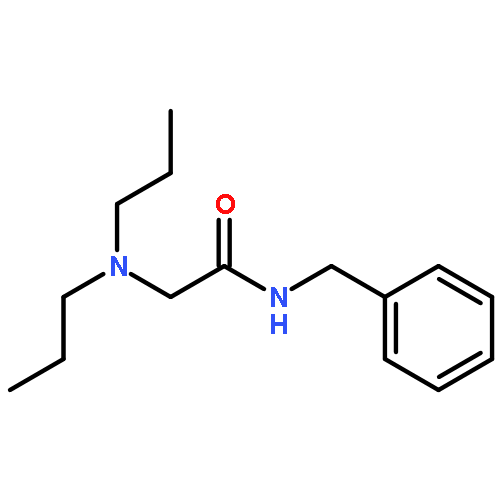






![Acetic acid, [4-(hydroxymethyl)phenoxy]-, 2,4,5-trichlorophenyl ester](http://img.cochemist.com/ccimg/68900/68858-22-0.png)
![Acetic acid, [4-(hydroxymethyl)phenoxy]-, 2,4,5-trichlorophenyl ester](http://img.cochemist.com/ccimg/68900/68858-22-0_b.png)
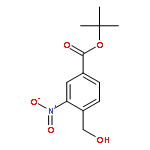



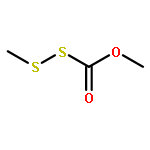
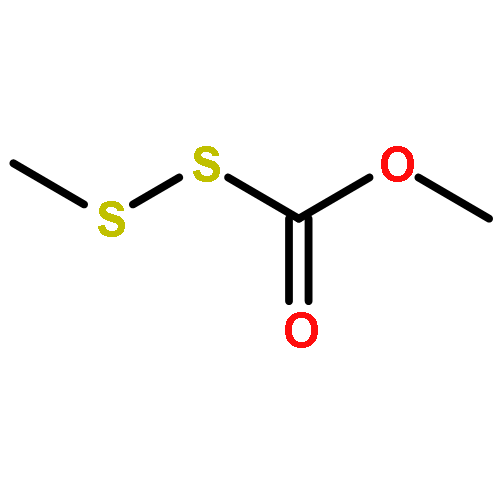


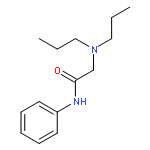
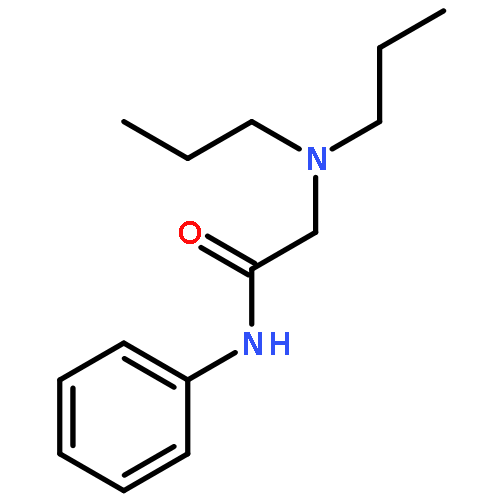




![2,6-Octadien-1-ol, 2,6-dimethyl-8-[(tetrahydro-2H-pyran-2-yl)oxy]-, (2E,6E)-](/data/chemimg/1815800/38290-53-8.png)
![2,6-Octadien-1-ol, 2,6-dimethyl-8-[(tetrahydro-2H-pyran-2-yl)oxy]-, (2E,6E)-](/data/chemimg/1815800/38290-53-8_b.png)
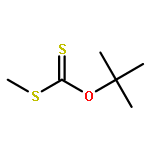







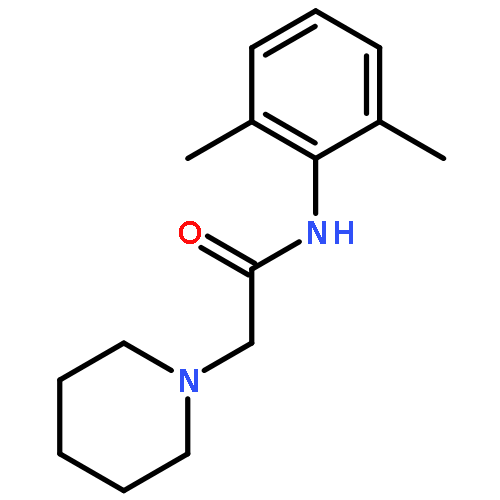




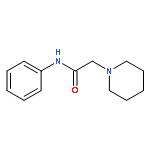
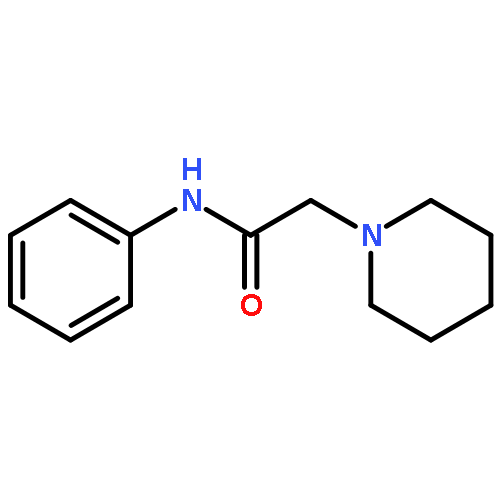


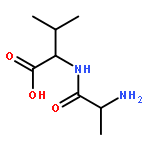
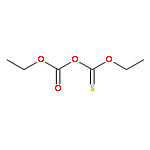
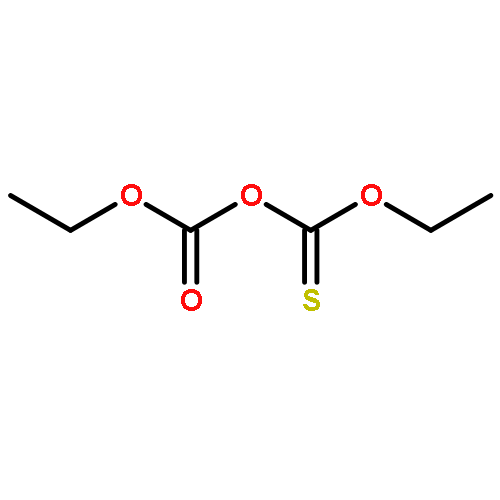

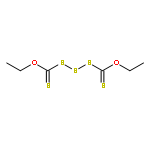
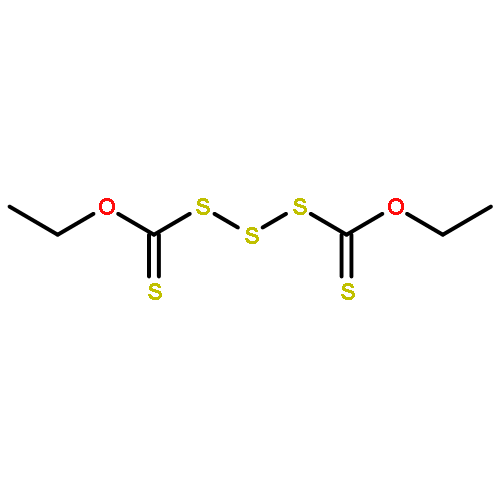
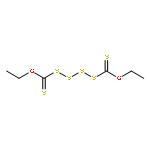
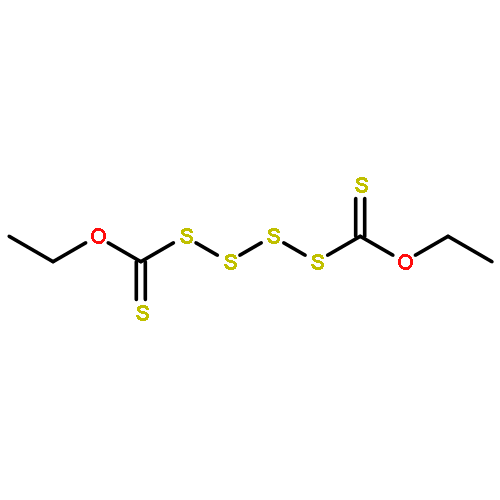
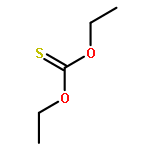
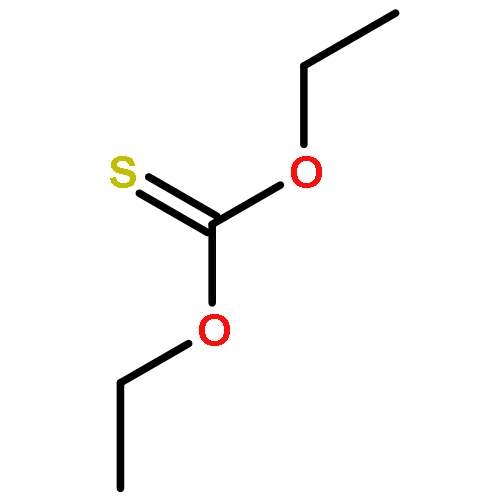




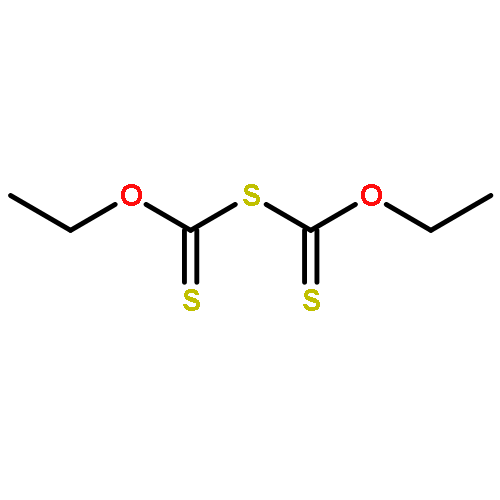
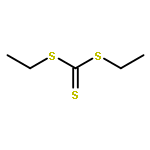
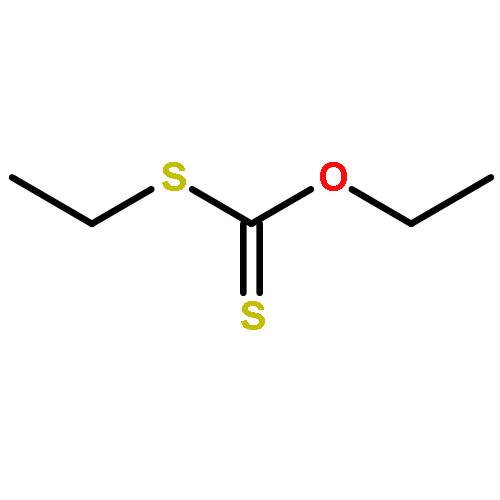
![Thioperoxydicarbonic acid ([(HO)C(S)] S ), OC,OC'-diethyl ester](/data/chemimg/1504100/502-55-6.png)
![Thioperoxydicarbonic acid ([(HO)C(S)] S ), OC,OC'-diethyl ester](/data/chemimg/1504100/502-55-6_b.png)






![Pentanoic acid, 5-[(9-hydroxy-9H-xanthen-2-yl)oxy]-](http://img.cochemist.com/ccimg/141800/141701-91-9.png)
![Pentanoic acid, 5-[(9-hydroxy-9H-xanthen-2-yl)oxy]-](http://img.cochemist.com/ccimg/141800/141701-91-9_b.png)
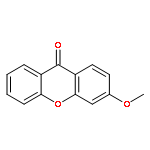
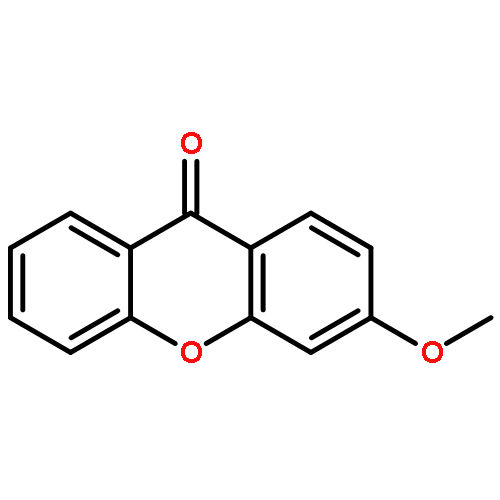
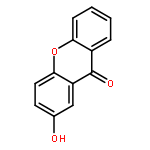
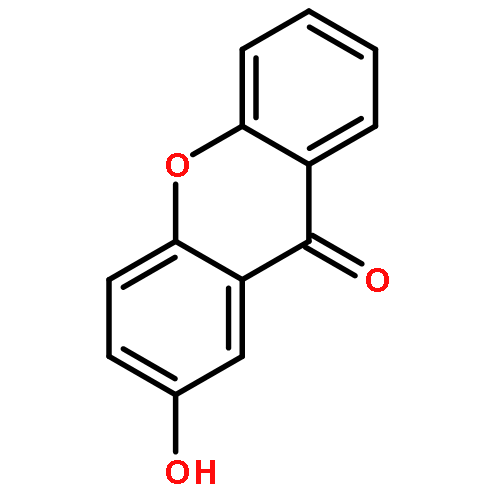
![Pentanoic acid, 5-[(9-oxo-9H-xanthen-2-yl)oxy]-](http://img.cochemist.com/ccimg/141800/141701-90-8.png)
![Pentanoic acid, 5-[(9-oxo-9H-xanthen-2-yl)oxy]-](http://img.cochemist.com/ccimg/141800/141701-90-8_b.png)



![BENZENAMINE, N-[(METHOXYTHIOXOMETHYL)THIO]-N-METHYL-](http://img.cochemist.com/ccimg/89300/89264-59-5.png)
![BENZENAMINE, N-[(METHOXYTHIOXOMETHYL)THIO]-N-METHYL-](http://img.cochemist.com/ccimg/89300/89264-59-5_b.png)



![L-Serine, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-, pentafluorophenyl ester](http://img.cochemist.com/ccimg/86100/86061-05-4.png)
![L-Serine, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-, pentafluorophenyl ester](http://img.cochemist.com/ccimg/86100/86061-05-4_b.png)
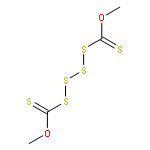
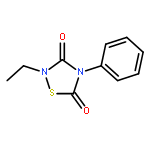
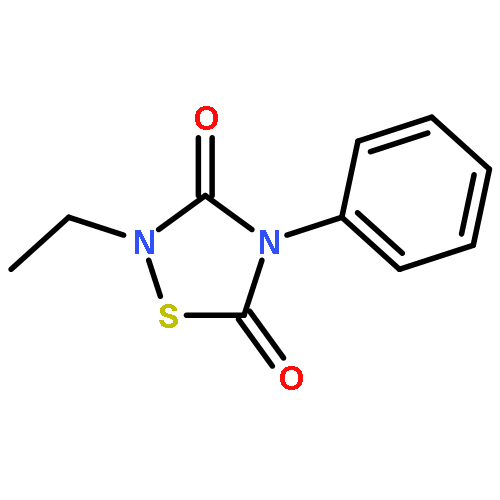
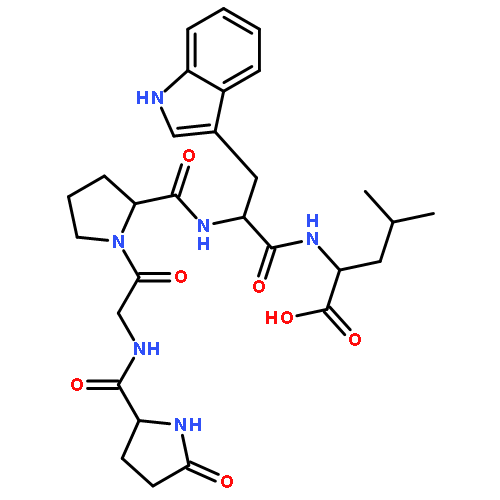
![L-Valine, N-[(3,5-dioxo-1,2,4-dithiazolidin-4-yl)acetyl]-](http://img.cochemist.com/ccimg/70800/70711-37-4.png)
![L-Valine, N-[(3,5-dioxo-1,2,4-dithiazolidin-4-yl)acetyl]-](http://img.cochemist.com/ccimg/70800/70711-37-4_b.png)
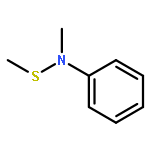









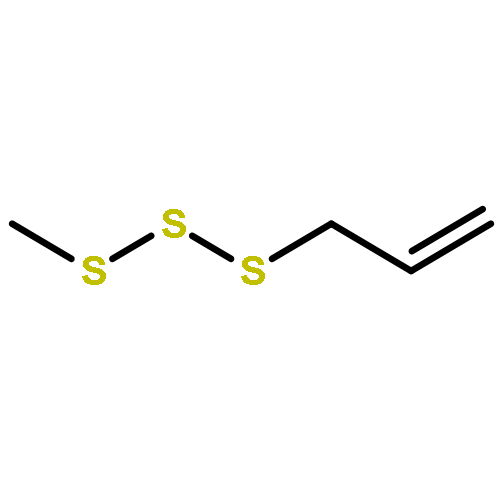
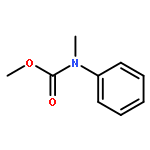


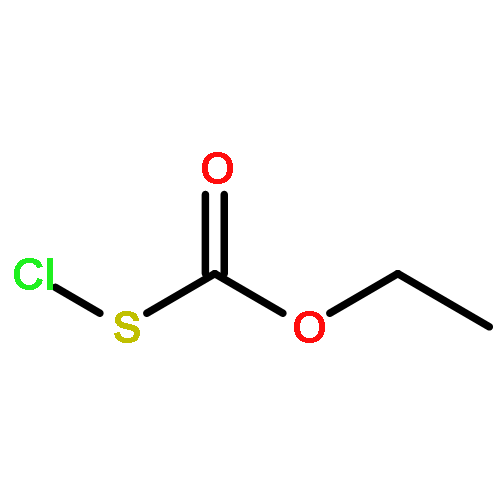

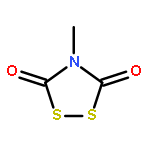
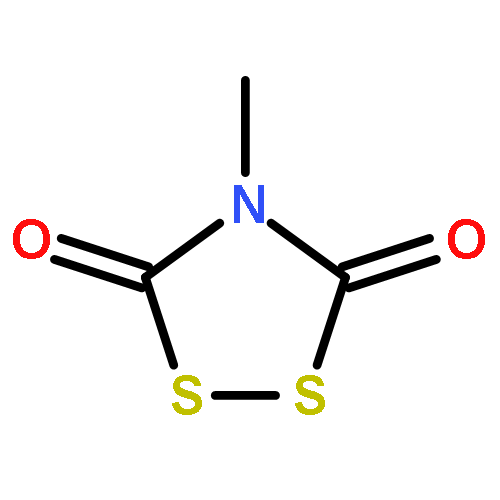
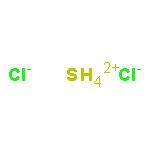





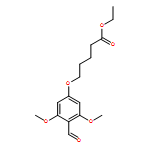
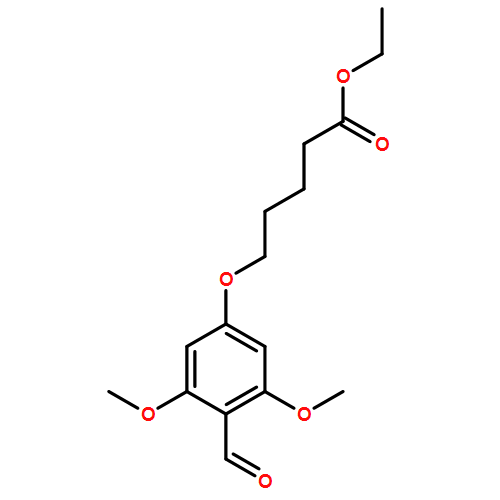
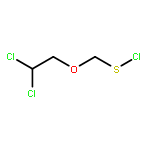
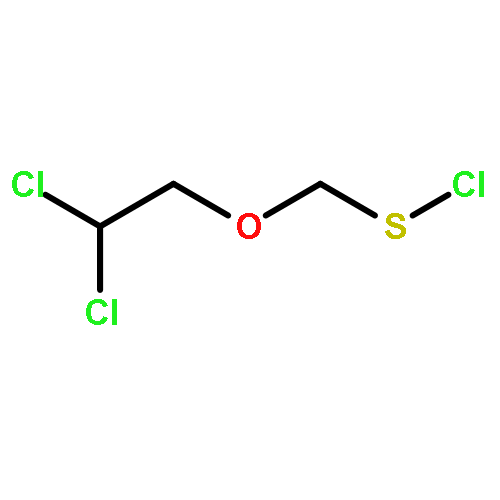
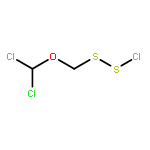

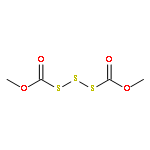
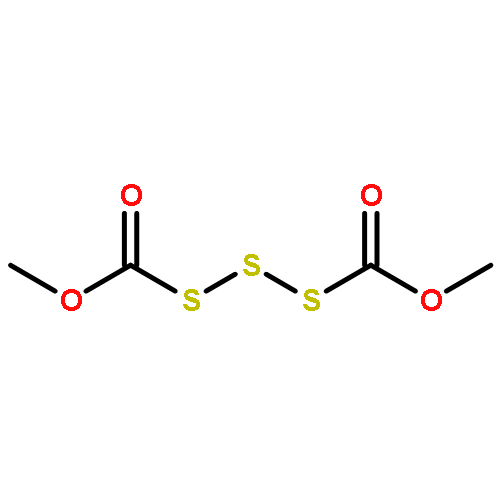

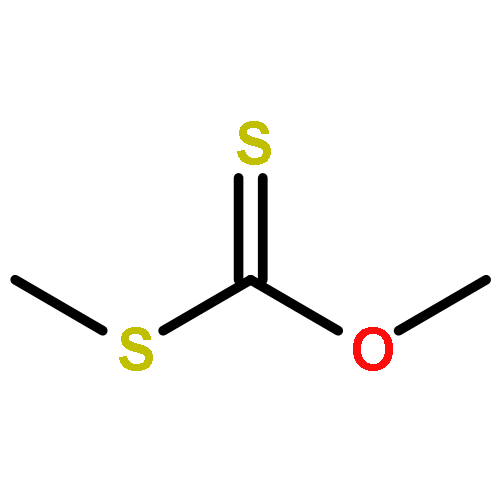
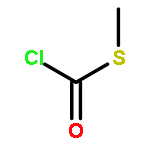
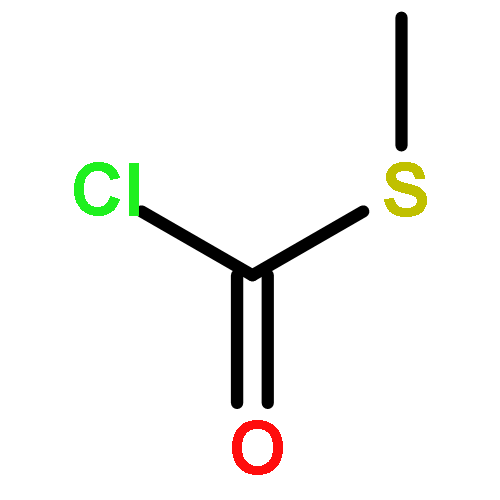
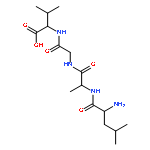
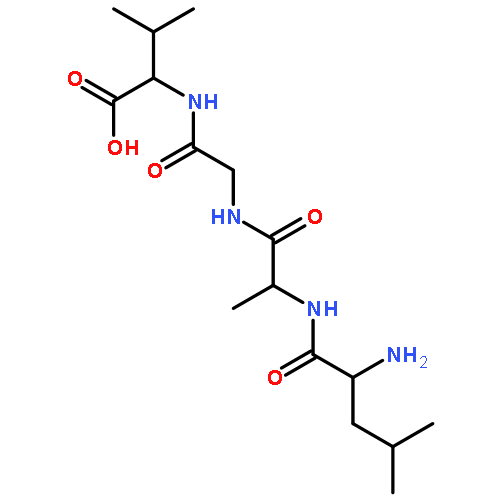
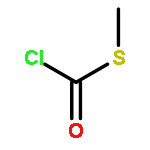
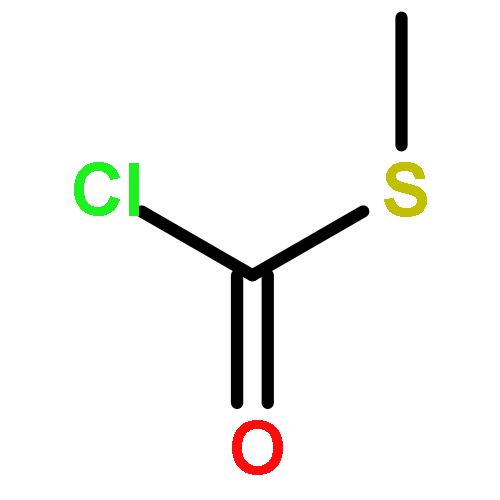


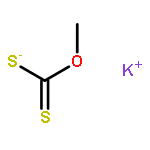
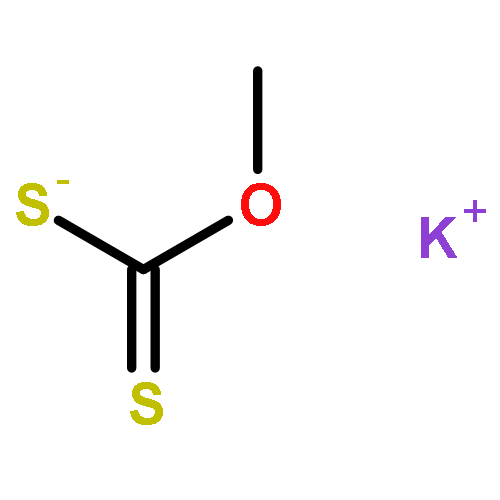
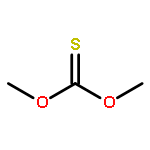
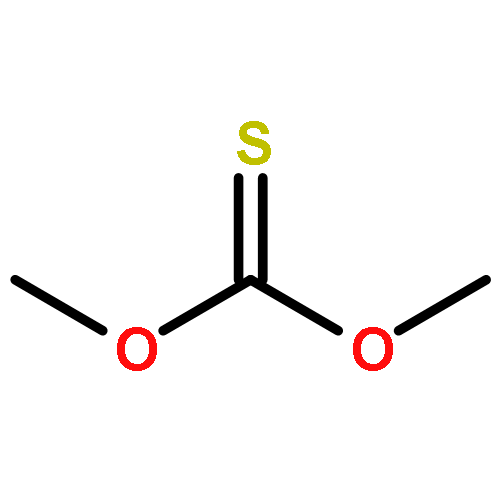
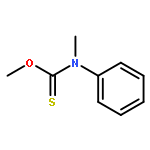
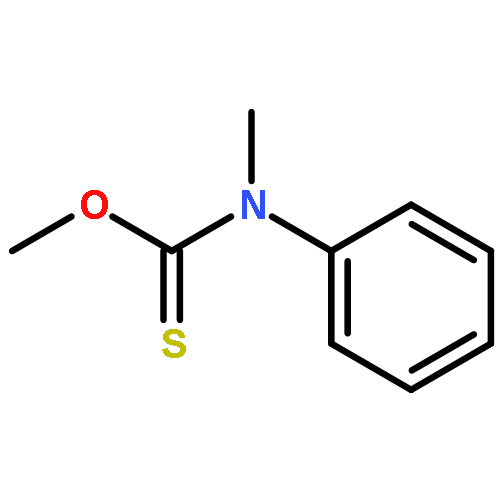



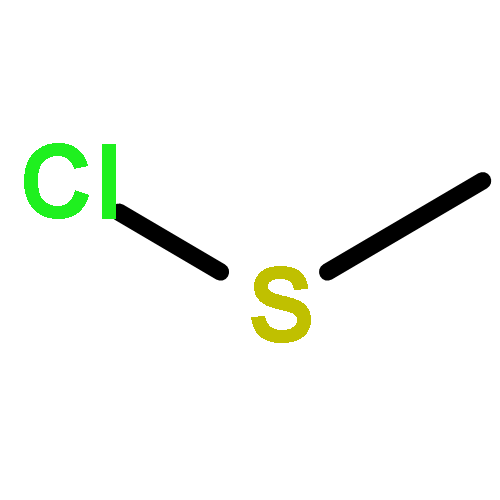
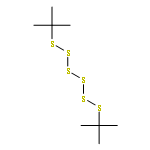
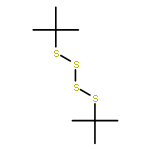
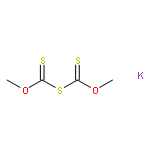
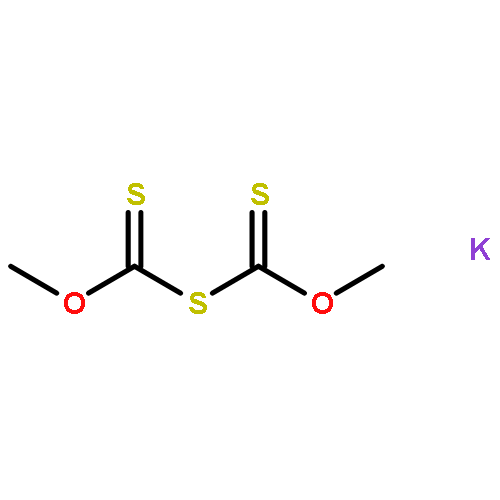
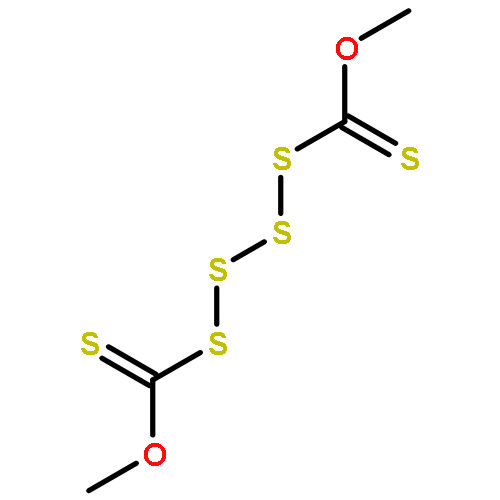
![Thioperoxydicarbonicacid ([(HO)C(S)]2S2), O,O'-dimethyl ester](http://img.cochemist.com/ccimg/1500/1468-37-7.png)
![Thioperoxydicarbonicacid ([(HO)C(S)]2S2), O,O'-dimethyl ester](http://img.cochemist.com/ccimg/1500/1468-37-7_b.png)
![Benzenamine, N-methyl-N-[[(methylphenylamino)carbonyl]thio]-](http://img.cochemist.com/ccimg/87500/87463-10-3.png)
![Benzenamine, N-methyl-N-[[(methylphenylamino)carbonyl]thio]-](http://img.cochemist.com/ccimg/87500/87463-10-3_b.png)