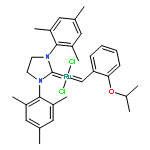
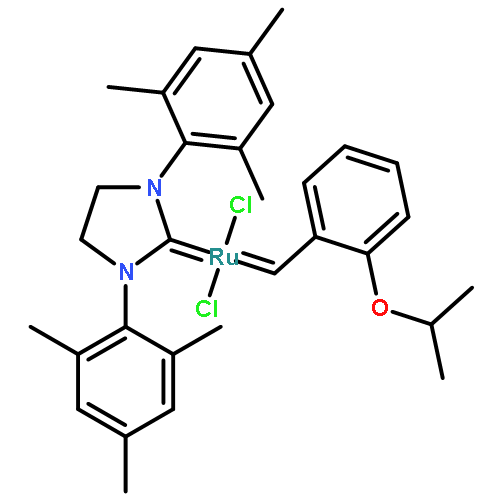
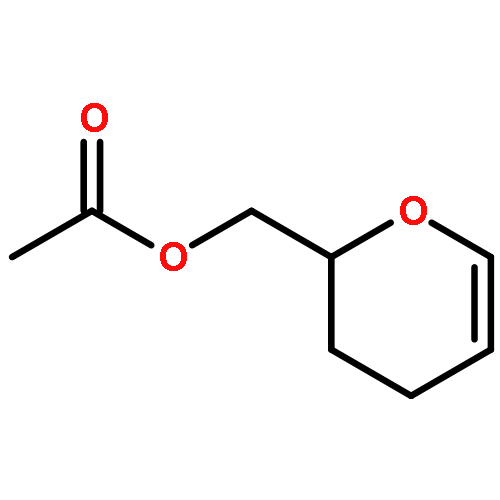
Co-reporter:Dr. George Karageorgis;Dr. Mark Dow;Dr. Anthony Aimon;Dr. Stuart Warriner; Adam Nelson
Angewandte Chemie International Edition 2015 Volume 54( Issue 46) pp:
Publication Date(Web):
DOI:10.1002/anie.201584661
Co-reporter:Dr. George Karageorgis;Dr. Mark Dow;Dr. Anthony Aimon;Dr. Stuart Warriner; Adam Nelson
Angewandte Chemie 2015 Volume 127( Issue 46) pp:13742-13748
Publication Date(Web):
DOI:10.1002/ange.201506944
Abstract
Activity-directed synthesis (ADS), a novel discovery approach in which bioactive molecules emerge in parallel with associated syntheses, was exploited to develop a weakly binding fragment into novel androgen receptor agonists. Harnessing promiscuous intermolecular reactions of carbenoid compounds enabled highly efficient exploration of chemical space. Four substrates were prepared, yet exploited in 326 reactions to explore diverse chemical space; guided by bioactivity alone, the products of just nine of the reactions were purified to reveal diverse novel agonists with up to 125-fold improved activity. Remarkably, one agonist stemmed from a novel enantioselective transformation; this is the first time that an asymmetric reaction has been discovered solely on the basis of the biological activity of the product. It was shown that ADS is a significant addition to the lead generation toolkit, enabling the efficient and rapid discovery of novel, yet synthetically accessible, bioactive chemotypes.
Co-reporter:Dr. George Karageorgis;Dr. Mark Dow;Dr. Anthony Aimon;Dr. Stuart Warriner; Adam Nelson
Angewandte Chemie International Edition 2015 Volume 54( Issue 46) pp:13538-13544
Publication Date(Web):
DOI:10.1002/anie.201506944
Abstract
Activity-directed synthesis (ADS), a novel discovery approach in which bioactive molecules emerge in parallel with associated syntheses, was exploited to develop a weakly binding fragment into novel androgen receptor agonists. Harnessing promiscuous intermolecular reactions of carbenoid compounds enabled highly efficient exploration of chemical space. Four substrates were prepared, yet exploited in 326 reactions to explore diverse chemical space; guided by bioactivity alone, the products of just nine of the reactions were purified to reveal diverse novel agonists with up to 125-fold improved activity. Remarkably, one agonist stemmed from a novel enantioselective transformation; this is the first time that an asymmetric reaction has been discovered solely on the basis of the biological activity of the product. It was shown that ADS is a significant addition to the lead generation toolkit, enabling the efficient and rapid discovery of novel, yet synthetically accessible, bioactive chemotypes.
Co-reporter:Dr. George Karageorgis;Dr. Mark Dow;Dr. Anthony Aimon;Dr. Stuart Warriner; Adam Nelson
Angewandte Chemie 2015 Volume 127( Issue 46) pp:
Publication Date(Web):
DOI:10.1002/ange.201584661
Co-reporter:Dr. Nathalie M. Valette; Sheena E. Radford;Dr. Sarah A. Harris;Dr. Stuart L. Warriner
ChemBioChem 2012 Volume 13( Issue 2) pp:271-281
Publication Date(Web):
DOI:10.1002/cbic.201100607
Abstract
Despite the importance of post-translational modifications in controlling the solubility and conformational properties of proteins and peptides, precisely how the aggregation propensity of different peptide sequences is modulated by chemical modification remains unclear. Here we have investigated the effect of phosphorylation on the aggregation propensity of a 13-residue synthetic peptide incorporating one or more phosphate groups at seven different sites at various pH values. Fibril formation was shown to be inhibited when a single phosphate group was introduced at all seven locations in the peptide sequence at pH 7.5, when the phosphate group is fully charged. By contrast, when the same peptides were analysed at pH 1.1, when the phosphate is fully protonated, fibrils from all seven peptide sequences form rapidly. At intermediate pH values (pH 3.6) when the phosphate group is mono-anionic, the aggregation propensity of the peptides was found to be highly dependent on the position of the phosphate group in the peptide sequence. Using this information, combined with molecular dynamics (MD) simulations of the peptide sequence, we provide evidence consistent with the peptide forming amyloid fibrils with a class 7 architecture. The results highlight the potential utility of phosphorylation as a method of reversibly controlling the aggregation kinetics of peptide sequences both during and after synthesis. Moreover, by exploiting the ability of the phosphate group to adopt different charge states as a function of pH, and combining experimental insights with atomistic information calculated from MD simulations as pH is varied, we show how the resulting information can be used to predict fibril structures consistent with both datasets, and use these to rationalise their sensitivity of fibrillation kinetics both to the location of the phosphate group and its charge state.
Co-reporter:Frederick Campbell, Jeffrey P. Plante, Thomas A. Edwards, Stuart L. Warriner and Andrew J. Wilson
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 10) pp:2344-2351
Publication Date(Web):18 Mar 2010
DOI:10.1039/C001164A
Generic approaches for the design and synthesis of small molecule inhibitors of protein–protein interactions (PPIs) represent a key objective in modern chemical biology. Within this context, the α-helix mediated PPIs have received considerable attention as targets for inhibition using small molecules, foldamers and proteomimetics. This manuscript describes a novel N-alkylated aromatic oligoamide proteomimetic scaffold and its solid-phase synthesis—the first time such an approach has been used for proteomimetics. The utility of these scaffolds as proteomimetics is exemplified through the identification of potent μM inhibitors of the p53–hDM2 helix mediated PPI—a key oncogenic target.
Co-reporter:Sarah Murrison;David Glowacki Dr.;Christian Einzinger Dr.;James Titchmarsh;Stephen Bartlett Dr.;Ben McKeever-Abbas Dr. Dr.;Adam Nelson
Chemistry - A European Journal 2009 Volume 15( Issue 9) pp:2185-2189
Publication Date(Web):
DOI:10.1002/chem.200802127
Co-reporter:Nicola S. Skoulding, Gopal Chowdhary, Mara J. Deus, Alison Baker, ... Stuart L. Warriner
Journal of Molecular Biology (13 March 2015) Volume 427(Issue 5) pp:1085-1101
Publication Date(Web):13 March 2015
DOI:10.1016/j.jmb.2014.12.003
•Peroxisome targeting predictions for mutagenized PTS1 domains were tested in vivo.•Quantitative binding data between PEX5 and PTS1 cargo were determined and compared.•A combination of methods better defines the threshold that predicts functional PTS1s.•More accurate in silico prediction of plant peroxisomal proteomes can be achieved.Most peroxisomal matrix proteins possess a C-terminal targeting signal type 1 (PTS1). Accurate prediction of functional PTS1 sequences and their relative strength by computational methods is essential for determination of peroxisomal proteomes in silico but has proved challenging due to high levels of sequence variability of non-canonical targeting signals, particularly in higher plants, and low levels of availability of experimentally validated non-canonical examples. In this study, in silico predictions were compared with in vivo targeting analyses and in vitro thermodynamic binding of mutated variants within the context of one model targeting sequence. There was broad agreement between the methods for entire PTS1 domains and position-specific single amino acid residues, including residues upstream of the PTS1 tripeptide. The hierarchy Leu>Met>Ile>Val at the C-terminal position was determined for all methods but both experimental approaches suggest that Tyr is underweighted in the prediction algorithm due to the absence of this residue in the positive training dataset. A combination of methods better defines the score range that discriminates a functional PTS1. In vitro binding to the PEX5 receptor could discriminate among strong targeting signals while in vivo targeting assays were more sensitive, allowing detection of weak functional import signals that were below the limit of detection in the binding assay. Together, the data provide a comprehensive assessment of the factors driving PTS1 efficacy and provide a framework for the more quantitative assessment of the protein import pathway in higher plants.Download high-res image (81KB)Download full-size image
Co-reporter:Frederick Campbell, Jeffrey P. Plante, Thomas A. Edwards, Stuart L. Warriner and Andrew J. Wilson
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 10) pp:NaN2351-2351
Publication Date(Web):2010/03/18
DOI:10.1039/C001164A
Generic approaches for the design and synthesis of small molecule inhibitors of protein–protein interactions (PPIs) represent a key objective in modern chemical biology. Within this context, the α-helix mediated PPIs have received considerable attention as targets for inhibition using small molecules, foldamers and proteomimetics. This manuscript describes a novel N-alkylated aromatic oligoamide proteomimetic scaffold and its solid-phase synthesis—the first time such an approach has been used for proteomimetics. The utility of these scaffolds as proteomimetics is exemplified through the identification of potent μM inhibitors of the p53–hDM2 helix mediated PPI—a key oncogenic target.

![1,3,2-Dioxaborolane, 2-[3,4-bis(hexyloxy)phenyl]-4,4,5,5-tetramethyl-](http://img.cochemist.com/ccimg/881300/881209-78-5.png)
![1,3,2-Dioxaborolane, 2-[3,4-bis(hexyloxy)phenyl]-4,4,5,5-tetramethyl-](http://img.cochemist.com/ccimg/881300/881209-78-5_b.png)

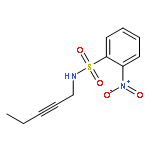
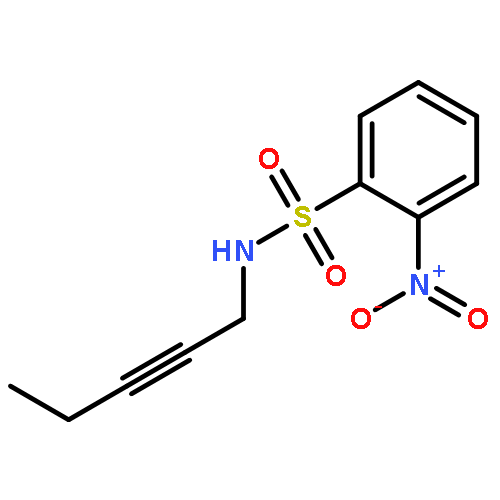
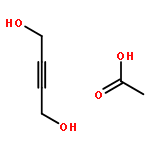

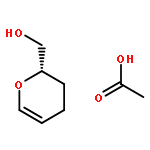
![2-Propanol, 1-[[(4-methoxyphenyl)methyl]amino]-, (2R)-](http://img.cochemist.com/ccimg/570400/570398-15-1.png)
![2-Propanol, 1-[[(4-methoxyphenyl)methyl]amino]-, (2R)-](http://img.cochemist.com/ccimg/570400/570398-15-1_b.png)


![[1,1'-Biphenyl]-2,2'-dimethanamine](http://img.cochemist.com/ccimg/70900/70898-14-5.png)
![[1,1'-Biphenyl]-2,2'-dimethanamine](http://img.cochemist.com/ccimg/70900/70898-14-5_b.png)

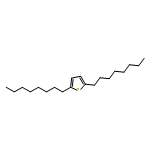


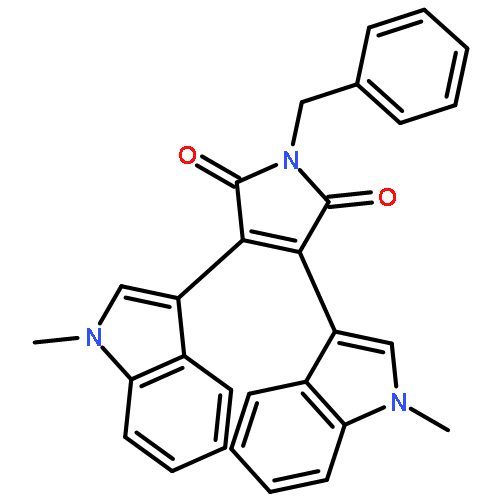
![Glycine, N-[4-(aminosulfonyl)benzoyl]glycylglycylglycylglycyl-](http://img.cochemist.com/ccimg/754200/754159-45-0.png)
![Glycine, N-[4-(aminosulfonyl)benzoyl]glycylglycylglycylglycyl-](http://img.cochemist.com/ccimg/754200/754159-45-0_b.png)
![Benzenesulfonamide, N-[2-(hydroxymethyl)phenyl]-4-nitro-](http://img.cochemist.com/ccimg/90400/90312-01-9.png)
![Benzenesulfonamide, N-[2-(hydroxymethyl)phenyl]-4-nitro-](http://img.cochemist.com/ccimg/90400/90312-01-9_b.png)
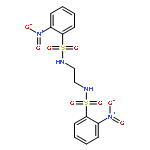

![GLYCINE, N-[4-(AMINOSULFONYL)BENZOYL]GLYCYL-](http://img.cochemist.com/ccimg/165700/165682-39-3.png)
![GLYCINE, N-[4-(AMINOSULFONYL)BENZOYL]GLYCYL-](http://img.cochemist.com/ccimg/165700/165682-39-3_b.png)
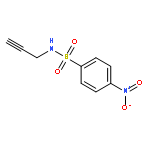
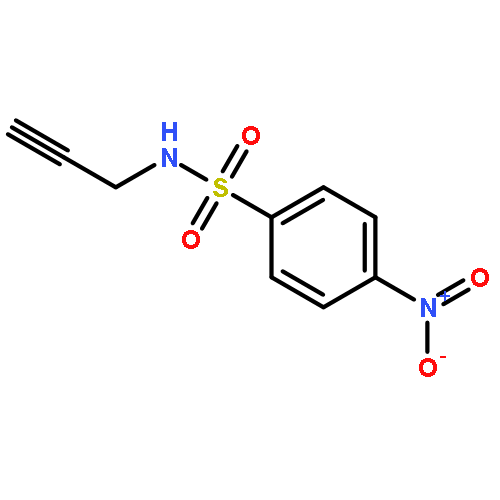
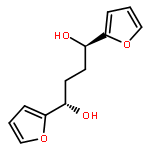
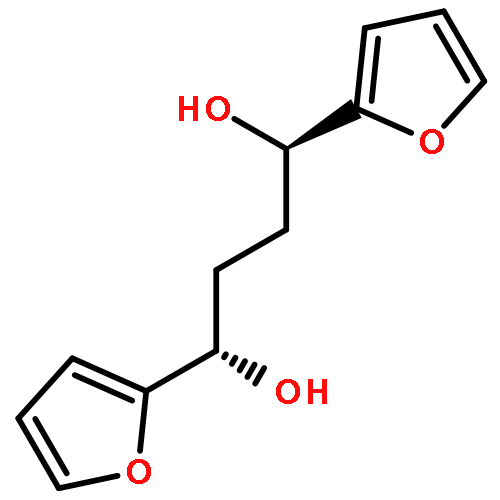
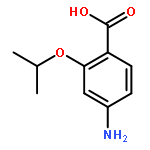

![1H-Pyrrole, 2,5-dihydro-1-[(2-nitrophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/219600/219583-73-0.png)
![1H-Pyrrole, 2,5-dihydro-1-[(2-nitrophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/219600/219583-73-0_b.png)






![BENZONITRILE, 4-[(2-PHENYLETHYL)AMINO]-2-(TRIFLUOROMETHYL)-](http://img.cochemist.com/ccimg/884900/884855-03-2.png)
![BENZONITRILE, 4-[(2-PHENYLETHYL)AMINO]-2-(TRIFLUOROMETHYL)-](http://img.cochemist.com/ccimg/884900/884855-03-2_b.png)


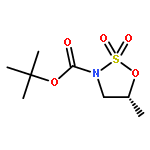
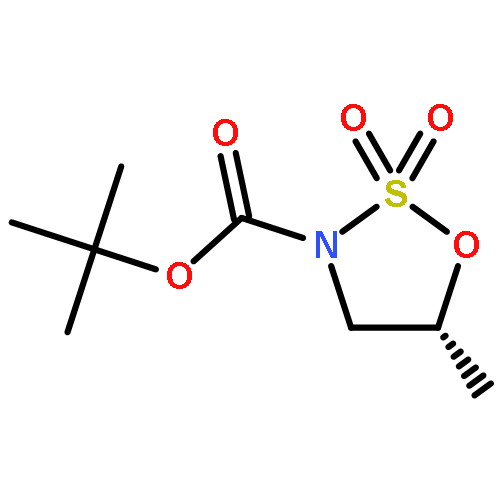
![1,6-DIOXASPIRO[4.5]DEC-3-ENE-2,10-DIOL, 7-(2-FURANYL)-, (5S,7R,10S)-](http://img.cochemist.com/ccimg/637800/637759-42-3.png)
![1,6-DIOXASPIRO[4.5]DEC-3-ENE-2,10-DIOL, 7-(2-FURANYL)-, (5S,7R,10S)-](http://img.cochemist.com/ccimg/637800/637759-42-3_b.png)
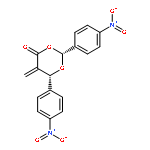
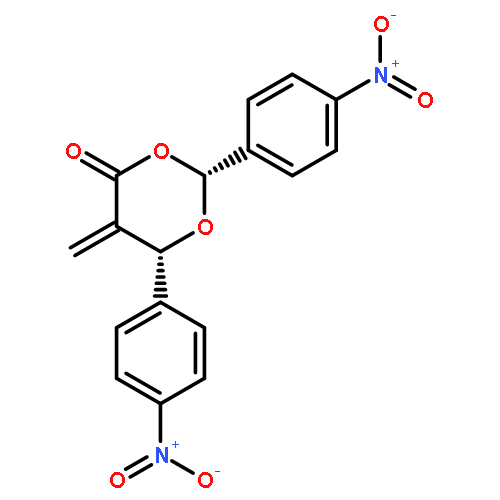
![Glycine, N-[4-(aminosulfonyl)benzoyl]glycylglycyl-](http://img.cochemist.com/ccimg/165700/165682-40-6.png)
![Glycine, N-[4-(aminosulfonyl)benzoyl]glycylglycyl-](http://img.cochemist.com/ccimg/165700/165682-40-6_b.png)
![2-PROPANOL, 1-[[(4-METHOXYPHENYL)METHYL]AMINO]-, (2S)-](http://img.cochemist.com/ccimg/570400/570398-16-2.png)
![2-PROPANOL, 1-[[(4-METHOXYPHENYL)METHYL]AMINO]-, (2S)-](http://img.cochemist.com/ccimg/570400/570398-16-2_b.png)











![Carbamic acid, [1-(aminocarbonyl)-3-methylbutyl]-,9H-fluoren-9-ylmethyl ester, (S)-](http://img.cochemist.com/ccimg/139600/139551-97-6.png)
![Carbamic acid, [1-(aminocarbonyl)-3-methylbutyl]-,9H-fluoren-9-ylmethyl ester, (S)-](http://img.cochemist.com/ccimg/139600/139551-97-6_b.png)







![Benzoic acid,4-[(2-methylpropyl)amino]-](http://img.cochemist.com/ccimg/372000/371958-90-6.png)
![Benzoic acid,4-[(2-methylpropyl)amino]-](http://img.cochemist.com/ccimg/372000/371958-90-6_b.png)
![Bicyclo[5.1.0]octane, 8,8-dibromo-, cis-](http://img.cochemist.com/ccimg/52800/52750-35-3.png)
![Bicyclo[5.1.0]octane, 8,8-dibromo-, cis-](http://img.cochemist.com/ccimg/52800/52750-35-3_b.png)


![Benzaldehyde,4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/160800/160744-60-5.png)
![Benzaldehyde,4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/160800/160744-60-5_b.png)
![6-Nitrobenzo[d]thiazole-2-carbonitrile](http://img.cochemist.com/ccimg/188700/188672-83-5.png)
![6-Nitrobenzo[d]thiazole-2-carbonitrile](http://img.cochemist.com/ccimg/188700/188672-83-5_b.png)














![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)




![Benzoic acid, 4-[(phenylmethyl)amino]-](http://img.cochemist.com/ccimg/61500/61439-54-1.png)
![Benzoic acid, 4-[(phenylmethyl)amino]-](http://img.cochemist.com/ccimg/61500/61439-54-1_b.png)
![Benzoic acid, 4-[(2-phenylethyl)amino]-](http://img.cochemist.com/ccimg/61500/61439-45-0.png)
![Benzoic acid, 4-[(2-phenylethyl)amino]-](http://img.cochemist.com/ccimg/61500/61439-45-0_b.png)














![Glycine, N-[4-(aminosulfonyl)benzoyl]-](http://img.cochemist.com/ccimg/143300/143288-21-5.png)
![Glycine, N-[4-(aminosulfonyl)benzoyl]-](http://img.cochemist.com/ccimg/143300/143288-21-5_b.png)










![9-((3AR,4R,6R,6AR)-2,2-DIMETHYL-6-((METHYLAMINO)METHYL)-TETRAHYDROFURO[3,4-D][1,3]DIOXOL-4-YL)-9H-PURIN-6-AMINE](http://img.cochemist.com/ccimg/34300/34245-49-3.png)
![9-((3AR,4R,6R,6AR)-2,2-DIMETHYL-6-((METHYLAMINO)METHYL)-TETRAHYDROFURO[3,4-D][1,3]DIOXOL-4-YL)-9H-PURIN-6-AMINE](http://img.cochemist.com/ccimg/34300/34245-49-3_b.png)


![3,11a-Epidithio-11aH-pyrazino[1',2':1,5]pyrrolo[2,3-b]indole-1,4-dione,2,3,5a,6,10b,11-hexahydro-3-(hydroxymethyl)-10b-[(1S,4S)-3-[[4-(hydroxymethyl)-5,7-dimethyl-6,8-dioxo-2,3-dithia-5,7-diazabicyclo[2.2.2]oct-1-yl]methyl]-1H-indol-1-yl]-2-methyl-,(3S,5aR,10bS,11aS)-](http://img.cochemist.com/ccimg/1500/1403-36-7.png)
![3,11a-Epidithio-11aH-pyrazino[1',2':1,5]pyrrolo[2,3-b]indole-1,4-dione,2,3,5a,6,10b,11-hexahydro-3-(hydroxymethyl)-10b-[(1S,4S)-3-[[4-(hydroxymethyl)-5,7-dimethyl-6,8-dioxo-2,3-dithia-5,7-diazabicyclo[2.2.2]oct-1-yl]methyl]-1H-indol-1-yl]-2-methyl-,(3S,5aR,10bS,11aS)-](http://img.cochemist.com/ccimg/1500/1403-36-7_b.png)
![Ethanol,2-[(triphenylmethyl)thio]-](http://img.cochemist.com/ccimg/29200/29167-28-0.png)
![Ethanol,2-[(triphenylmethyl)thio]-](http://img.cochemist.com/ccimg/29200/29167-28-0_b.png)
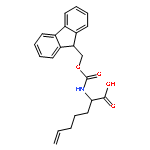





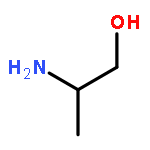
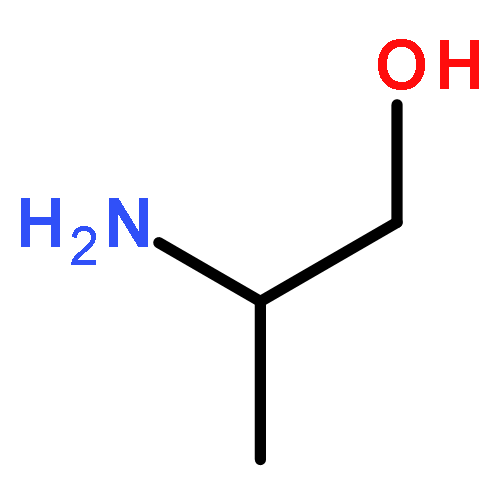
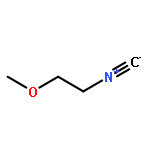
















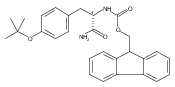
















![N-[4-(1H,1H,2H,2H-PERFLUORODECYL)BENZYLOXYCARBONYLOXY]SUCCINIMIDE](http://img.cochemist.com/ccimg/356100/356056-15-0.png)
![N-[4-(1H,1H,2H,2H-PERFLUORODECYL)BENZYLOXYCARBONYLOXY]SUCCINIMIDE](http://img.cochemist.com/ccimg/356100/356056-15-0_b.png)


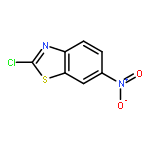
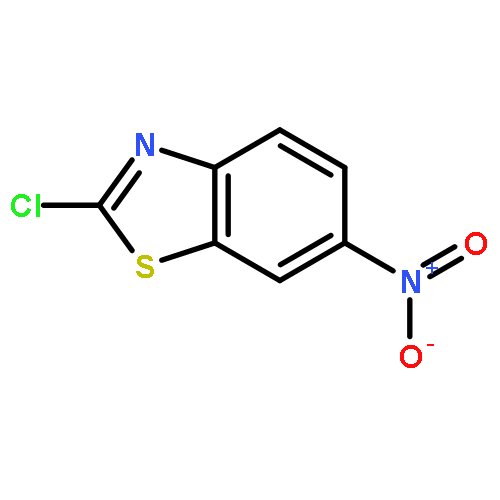
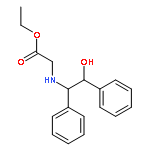


![Adenosine,5'-[[(3S)-3-amino-3-carboxypropyl]methylamino]-5'-deoxy- (9CI)](http://img.cochemist.com/ccimg/111800/111770-79-7.png)
![Adenosine,5'-[[(3S)-3-amino-3-carboxypropyl]methylamino]-5'-deoxy- (9CI)](http://img.cochemist.com/ccimg/111800/111770-79-7_b.png)