Co-reporter:Jessica H. Golden, John W. Facendola, Daniel Sylvinson M. R., Cecilia Quintana Baez, Peter I. Djurovich, and Mark E. Thompson
The Journal of Organic Chemistry July 21, 2017 Volume 82(Issue 14) pp:7215-7215
Publication Date(Web):July 4, 2017
DOI:10.1021/acs.joc.7b00786
Boron dipyrromethene (BODIPY) derivatives have found widespread utility as chromophores in fluorescent applications, but little is known about the photophysical properties of pyridine-based BODIPY analogues, dipyridylmethene dyes. Indeed, it has been reported that boron difluoride dipyridylmethene (DIPYR) is nonemissive, and that derivatives of DIPYR have modest, if any, luminescence. In this report, we explore this little-touched area of chemical space and investigate the photophysical properties of three simple DIPYR dyes: boron dipyridylmethene, boron diquinolylmethene, and boron diisoquinolylmethene. The three dyes strongly absorb in the blue-green part of the spectrum (λem = 450–520 nm, ε = 2.9–11 × 104 M–1 cm–1) and display green fluorescence with high quantum yields (ΦPL = 0.2, 0.8, and 0.8, respectively). Key photophysical properties in these systems were evaluated using a combination of TD-DFT and extended multiconfigurational quasidegenerate second-order perturbation theory (XMCQDPT2) methods and compared to experimental results, revealing that high quantum yields of the quinoline and isoquinoline derivatives are a result of the relative reordering of S1 and T2 state energies upon benzannulation of the parent structure. The intense absorption and high emission efficiency of the benzannulated derivatives make these compounds an intriguing class of dyes for further derivatization.
Co-reporter:Rasha Hamze;Rodolphe Jazzar;Michele Soleilhavoup;Peter I. Djurovich;Guy Bertrand
Chemical Communications 2017 vol. 53(Issue 64) pp:9008-9011
Publication Date(Web):2017/08/08
DOI:10.1039/C7CC02638B
The photophysical properties of several Cu(I) complexes coordinated with cyclic (alkyl)(amino)carbene (CAAC) ligands were examined. All the compounds were found to be phosphorescent, regardless of whether they are 2-, 3- or 4-coordinated. Aggregate and excimer emission were observed from 2-coordinate CAAC–CuCl derivatives in methylcyclohexane solution. Emission from the complex 4-coordinated with a trispyrazolylborate ligand is red-shifted with respect to both the chloro-derivative and an analogous complex with an NHC ligand.
Co-reporter:Shuyang Shi;Lee R. Collins;Mary F. Mahon;Peter I. Djurovich;Michael K. Whittlesey
Dalton Transactions 2017 vol. 46(Issue 3) pp:745-752
Publication Date(Web):2017/01/17
DOI:10.1039/C6DT04016K
The photophysical properties of four, two-coordinate, linear diamidocarbene copper(I) complexes, [(DAC)2Cu][BF4] (1), (DAC)CuOSiPh3 (2), (DAC)CuC6F5 (3) and (DAC)Cu(2,4,6-Me3C6H2) (4) (DAC = 1,3-bis(2,4,6-trimethylphenyl)-5,5-dimethyl-4,6-diketopyrimidinyl-2-ylidene) have been investigated. Complex 1 shows a high photoluminescence quantum efficiency (ΦPL) in both the solid state (ΦPL = 0.85) and in CH2Cl2 solution (ΦPL = 0.65). The emission band of 1, both as a crystalline solid and in solution, is narrow (fwhm = 2300 cm−1) relative to the emission bands of 2 (fwhm = 2900 cm−1) and 3 (fwhm = 3700 cm−1). Complexes 2 and 3 are each brightly luminescent in the solid state (ΦPL = 0.62 and 0.18, respectively), but markedly less so in CH2Cl2 solution (ΦPL = 0.03 and <0.01, respectively). Complex 4 is not emissive in either the solid state or in solution. Phosphorescence of 1 in CH2Cl2 solution shows negligible quenching by oxygen in CH2Cl2 solution. This insensitivity to quenching is attributed to the excited state redox potential being insufficient for electron transfer to oxygen.
Co-reporter:Patrick J. G. Saris and Mark E. Thompson
Organic Letters 2016 Volume 18(Issue 16) pp:3960-3963
Publication Date(Web):August 4, 2016
DOI:10.1021/acs.orglett.6b01693
The design, synthesis, and characterization of 12-phenylbenzo[f][1,7]phenanthroline, Bzp, is reported. Its use as a fluorine-free ligand for sky blue phosphorescence is demonstrated in a cyclometalated platinum complex, BzpPtDpm. BzpPtDpm phosphoresces at the same wavelength as its analogous 4,6-difluorophenylpyridine complex at both room temperature (466 nm) and 77 K (458 nm). Finally, production of a conformationally restricted derivative of BzpPtDpm with greatly increased quantum yield (46%) validates the versatility of the synthetic route.
Co-reporter:Vyacheslav V. Diev, Denise Femia, Qiwen Zhong, Peter I. Djurovich, Ralf Haiges and Mark E. Thompson
Chemical Communications 2016 vol. 52(Issue 9) pp:1949-1952
Publication Date(Web):21 Dec 2015
DOI:10.1039/C5CC09128D
A bis-phenalenyl-fused porphyrin has been synthesized by thermal dehydro-aromatization reaction regioselectively as a single syn-isomer. X-ray crystal structure revealed that both phenalenyl units of this porphyrin have close π–π contacts forming continuous network of interacting porphyrin rings. A broad and intense NIR absorption can be attributed to quinoidal character of bis-phenalenyl-fused porphyrin.
Co-reporter:Qingzhou Liu, Noppadol Aroonyadet, Yan Song, Xiaoli Wang, Xuan Cao, Yihang Liu, Sen Cong, Fanqi Wu, Mark E. Thompson, and Chongwu Zhou
ACS Nano 2016 Volume 10(Issue 11) pp:10117
Publication Date(Web):November 9, 2016
DOI:10.1021/acsnano.6b05171
We demonstrate a scalable and facile lithography-free method for fabricating highly uniform and sensitive In2O3 nanoribbon biosensor arrays. Fabrication with shadow masks as the patterning method instead of conventional lithography provides low-cost, time-efficient, and high-throughput In2O3 nanoribbon biosensors without photoresist contamination. Combined with electronic enzyme-linked immunosorbent assay for signal amplification, the In2O3 nanoribbon biosensor arrays are optimized for early, quick, and quantitative detection of cardiac biomarkers in diagnosis of acute myocardial infarction (AMI). Cardiac troponin I (cTnI), creatine kinase MB (CK-MB), and B-type natriuretic peptide (BNP) are commonly associated with heart attack and heart failure and have been selected as the target biomarkers here. Our approach can detect label-free biomarkers for concentrations down to 1 pg/mL (cTnI), 0.1 ng/mL (CK-MB), and 10 pg/mL (BNP), all of which are much lower than clinically relevant cutoff concentrations. The sample collection to result time is only 45 min, and we have further demonstrated the reusability of the sensors. With the demonstrated sensitivity, quick turnaround time, and reusability, the In2O3 nanoribbon biosensors have shown great potential toward clinical tests for early and quick diagnosis of AMI.Keywords: acute myocardial infarction diagnosis; biosensor; electronic ELISA; field-effect transistor; indium oxide semiconductor; shadow mask fabrication
Co-reporter:Andrew N. Bartynski, Stefan Grob, Theresa Linderl, Mark Gruber, Wolfgang Brütting, and Mark E. Thompson
The Journal of Physical Chemistry C 2016 Volume 120(Issue 34) pp:19027-19034
Publication Date(Web):August 9, 2016
DOI:10.1021/acs.jpcc.6b06302
Organic photovoltaic devices utilizing α-sexithiophene (6T) as a donor and tetraphenyldibenzoperiflanthene (DBP) as an acceptor were fabricated and compared to devices utilizing DBP as a donor and C60 as an acceptor. The 6T/DBP devices exhibit substantially higher open circuit voltage, 1.27 V compared to 0.86 V for DBP/C60, as a consequence of the higher energy charge transfer state formed. The 6T/DBP devices yield short-circuit current of 3.9 mA/cm2, open-circuit voltage of 1.27 V, and fill factor of 0.55, resulting in a power conversion efficiency of 2.8%. Atomic force microscopy studies show that 6T forms textured films on indium–tin oxide, and subsequent deposition of DBP infills the surface. Optical modeling provides insight into the ideal active layer and transport layer thicknesses. A power conversion efficiency of close to 3% is achieved for a fairly large process window of layer thickness combinations. The high open-circuit voltage in conjunction with absorption out to a wavelength of 650 nm make this material combination especially attractive for tandem devices.
Co-reporter:Andrew N. Bartynski; Mark Gruber; Saptaparna Das; Sylvie Rangan; Sonya Mollinger; Cong Trinh; Stephen E. Bradforth; Koen Vandewal; Alberto Salleo; Robert A. Bartynski; Wolfgang Bruetting
Journal of the American Chemical Society 2015 Volume 137(Issue 16) pp:5397-5405
Publication Date(Web):March 31, 2015
DOI:10.1021/jacs.5b00146
Low open-circuit voltages significantly limit the power conversion efficiency of organic photovoltaic devices. Typical strategies to enhance the open-circuit voltage involve tuning the HOMO and LUMO positions of the donor (D) and acceptor (A), respectively, to increase the interfacial energy gap or to tailor the donor or acceptor structure at the D/A interface. Here, we present an alternative approach to improve the open-circuit voltage through the use of a zinc chlorodipyrrin, ZCl [bis(dodecachloro-5-mesityldipyrrinato)zinc], as an acceptor, which undergoes symmetry-breaking charge transfer (CT) at the donor/acceptor interface. DBP/ZCl cells exhibit open-circuit voltages of 1.33 V compared to 0.88 V for analogous tetraphenyldibenzoperyflanthrene (DBP)/C60-based devices. Charge transfer state energies measured by Fourier-transform photocurrent spectroscopy and electroluminescence show that C60 forms a CT state of 1.45 ± 0.05 eV in a DBP/C60-based organic photovoltaic device, while ZCl as acceptor gives a CT state energy of 1.70 ± 0.05 eV in the corresponding device structure. In the ZCl device this results in an energetic loss between ECT and qVOC of 0.37 eV, substantially less than the 0.6 eV typically observed for organic systems and equal to the recombination losses seen in high-efficiency Si and GaAs devices. The substantial increase in open-circuit voltage and reduction in recombination losses for devices utilizing ZCl demonstrate the great promise of symmetry-breaking charge transfer in organic photovoltaic devices.
Co-reporter:Nadezhda V. Korovina; Saptaparna Das; Zachary Nett; Xintian Feng; Jimmy Joy; Ralf Haiges; Anna I. Krylov; Stephen E. Bradforth
Journal of the American Chemical Society 2015 Volume 138(Issue 2) pp:617-627
Publication Date(Web):December 22, 2015
DOI:10.1021/jacs.5b10550
Singlet fission is a process in which a singlet exciton converts into two triplet excitons. To investigate this phenomenon, we synthesized two covalently linked 5-ethynyl-tetracene (ET) dimers with differing degrees of intertetracene overlap: BET-X, with large, cofacial overlap of tetracene π-orbitals, and BET-B, with twisted arrangement between tetracenes exhibits less overlap between the tetracene π-orbitals. The two compounds were crystallographically characterized and studied by absorption and emission spectroscopy in solution, in PMMA and neat thin films. The results show that singlet fission occurs within 1 ps in an amorphous thin film of BET-B with high efficiency (triplet yield: 154%). In solution and the PMMA matrix the S1 of BET-B relaxes to a correlated triplet pair 1(T1T1) on a time scale of 2 ps, which decays to the ground state without forming separated triplets, suggesting that triplet energy transfer from 1(T1T1) to a nearby chromophore is essential for producing free triplets. In support of this hypothesis, selective excitation of BET-B doped into a thin film of diphenyltetracene (DPT) leads to formation of the 1(T1T1) state of BET-B, followed by generation of both DPT and BET-B triplets. For the structurally cofacial BET-X, an intermediate forms in <180 fs and returns to the ground state more rapidly than BET-B. First-principles calculations predict a 2 orders of magnitude faster rate of singlet fission to the 1(T1T1) state in BET-B relative to that of crystalline tetracene, attributing the rate increase to greater coupling between the S1 and 1(T1T1) states and favorable energetics for formation of the separated triplets.
Co-reporter:John J. Chen, Sarah M. Conron, Patrick Erwin, Michael Dimitriou, Kyle McAlahney, and Mark E. Thompson
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 1) pp:662
Publication Date(Web):December 12, 2014
DOI:10.1021/am506874k
A benzannulated boron dipyrromethene (BODIPY, bDIP) molecule exhibiting strong absorption at 640 nm was synthesized. The organic dye was used in an organic solar cell as the electron donor with C60 as the acceptor. The BODIPY dye demonstrated the best performance in lamellar architecture (indium tin oxide (ITO)/bDIP/C60/bathocuproine/Al), giving power conversion efficiency up to 4.5% with short-circuit current (JSC) of 8.7 mA/cm2 and an open-circuit voltage (VOC) of 0.81 V. Neutron reflectivity experiments were performed on the bilayer film to investigate the thickness dependence of JSC. A 13 nm mixed layer was found to be present at the donor/acceptor interface in the bilayer device, formed when the C60 was deposited onto a room temperature bDIP film. Planar-mixed heterojunction devices were fabricated to understand the extent of spontaneous mixing between the donor and acceptor materials. The native mixed region in the bilayer device was shown to most resemble 1:3 bDIP:C60 layer in the structure: (ITO/bDIP/bDIP:C60 blend/C60/bathocuproine/Al).Keywords: bilayer; BODIPY; neutron reflectivity; organic photovoltaics; planar-mixed heterojunction
Co-reporter:John W. Facendola, Martin Seifrid, Jay Siegel, Peter I. Djurovich and Mark E. Thompson
Dalton Transactions 2015 vol. 44(Issue 18) pp:8456-8466
Publication Date(Web):10 Feb 2015
DOI:10.1039/C4DT03541K
Synthesis, structural and characterization data are provided for Pt(II) and Ir(III) complexes cyclometalated with 2-(corannulene)pyridine (corpy), (corpy)Pt(dpm) and (corpy)Ir(ppz)2 (dpm = dipivaloylmethanato, ppz = 1-phenylpyrazolyl). A third compound, (phenpy)Ir(ppz)2 (phenpy = 2-(5-phenanthryl)pyridyl), was also prepared to mimic the steric bulk of (corpy)Ir(ppz)2. X-ray analysis reveals bowl depths of 0.895 Å for (corpy)Pt(dpm) and 0.837 Å in (corpy)Ir(ppz)2. Neither complex displayed bowl-to-bowl stacking in the crystal lattice. A fluxional process for (corpy)Ir(ppz)2 attributed to bowl inversion of corrannulene is observed in solution with a barrier (ΔG‡ = 13 kcal mol−1) and rate (k = 2.5 × 103 s−1) as determined using variable temperature 1H NMR spectroscopy. All of the complexes display red phosphorescence at room temperature with quantum yields of 0.05 in solution and 0.2 in polymethyl methacrylate (PMMA).
Co-reporter:Markus J. Leitl ; Valentina A. Krylova ; Peter I. Djurovich ; Mark E. Thompson ;Hartmut Yersin
Journal of the American Chemical Society 2014 Volume 136(Issue 45) pp:16032-16038
Publication Date(Web):September 26, 2014
DOI:10.1021/ja508155x
Photophysical properties of two highly emissive three-coordinate Cu(I) complexes, (IPr)Cu(py2-BMe2) (1) and (Bzl-3,5Me)Cu(py2-BMe2) (2), with two different N-heterocyclic (NHC) ligands were investigated in detail (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene; Bzl-3,5Me = 1,3-bis(3,5-dimethylphenyl)-1H-benzo[d]imidazol-2-ylidene; py2-BMe2 = di(2-pyridyl)dimethylborate). The compounds exhibit remarkably high emission quantum yields of more than 70% in the powder phase. Despite similar chemical structures of both complexes, only compound 1 exhibits thermally activated delayed blue fluorescence (TADF), whereas compound 2 shows a pure, yellow phosphorescence. This behavior is related to the torsion angles between the two ligands. Changing this angle has a huge impact on the energy splitting between the first excited singlet state S1 and triplet state T1 and therefore on the TADF properties. In addition, it was found that, in both compounds, spin–orbit coupling (SOC) is particularly effective compared to other Cu(I) complexes. This is reflected in short emission decay times of the triplet states of only 34 μs (1) and 21 μs (2), respectively, as well as in the zero-field splittings of the triplet states amounting to 4 cm–1 (0.5 meV) for 1 and 5 cm–1 (0.6 meV) for 2. Accordingly, at ambient temperature, compound 1 exhibits two radiative decay paths which are thermally equilibrated: one via the S1 state as TADF path (62%) and one via the T1 state as phosphorescence path (38%). Thus, if this material is applied in an organic light-emitting diode, the generated excitons are harvested mainly in the singlet state, but to a significant portion also in the triplet state. This novel mechanism based on two separate radiative decay paths reduces the overall emission decay time distinctly.
Co-reporter:Zhiwei Liu, Jacky Qiu, Feng Wei, Jianqiang Wang, Xiaochen Liu, Michael G. Helander, Sarah Rodney, Zhibin Wang, Zuqiang Bian, Zhenghong Lu, Mark E. Thompson, and Chunhui Huang
Chemistry of Materials 2014 Volume 26(Issue 7) pp:2368
Publication Date(Web):March 18, 2014
DOI:10.1021/cm5006086
Phosphorescent copper(I) complexes show great promise as emitters in organic light-emitting diodes (OLEDs). However, most copper(I) complexes are neither soluble nor stable toward sublimation and, hence, not amenable to the typical methods to fabricate OLEDs. In this work, a compound 3-(carbazol-9-yl)-5-((3-carbazol-9-yl)phenyl)pyridine (CPPyC) was designed as both a good ligand and host matrix. Codeposition of CPPyC and copper iodide (CuI) gives luminescent films with photoluminescent quantum yields (PLQY) as high as 100%. A dimeric copper(I) complex Cu2I2(CPPyC)4 is formed in the thin film, characterized by X-ray absorption spectroscopy. A series of simple, highly efficient green-emitting OLEDs were demonstrated by using the codeposited film as an emissive layer. A device comprised of only CPPyC and CuI gave an external quantum efficiency (EQE) of 12.6% (42.3 cd/A) at 100 cd/m2, while a device with tailored hole and electron transporting layers gave an efficiency of 15.7% (51.6 cd/A) at the same brightness.
Co-reporter:Matthew J. Jurow, Alberto Bossi, Peter I. Djurovich, and Mark E. Thompson
Chemistry of Materials 2014 Volume 26(Issue 22) pp:6578
Publication Date(Web):October 23, 2014
DOI:10.1021/cm503336d
Solutions of facial-tris(1-phenylpyrazole)Ir(III) (fac-Ir(ppz)3), when dissolved in either tert-butyl isocyanide or in solid films of 2-naphthylisocyanide, undergo replacement of a ppz ligand by the isocyanide molecules after irradiation with UV light as demonstrated by liquid chromatograph mass spectrometer analysis. Similarly, solutions of Ir(ppz)3 and bathophenanthroline (BPhen) in CH2Cl2 or acetone-d6 form a brightly emissive species, [Ir(ppz)2(Bphen)]+ when irradiated with UV light as established by optical, mass, and 1H nuclear magnetic resonance spectroscopy. Electroluminescent data from blocked organic light-emitting diode (OLED) devices demonstrate that both mer- and fac-(Ir(ppz)3) dissociate a ligand and coordinate a neighboring BPhen molecule when the device is operated at moderate to high current levels. These experiments offer direct evidence of the dissociation of a metal–ligand bond and subsequent ligand substitution as a degradation pathway in active OLED devices during operation and provide a route to assay in situ the stability of future dopants.
Co-reporter:Francisco F. Navarro, Peter I. Djurovich, Mark E. Thompson
Organic Electronics 2014 Volume 15(Issue 11) pp:3052-3060
Publication Date(Web):November 2014
DOI:10.1016/j.orgel.2014.08.049
•We deposited Mg and Zn films using a low vacuum vapor phase deposition (VPD) system (1–10 torr).•Metal films grown in the VPD display comparable characteristics to metal films grown in the VTE.•Mg films grown in the VPD were utilized in the fabrication of optoelectronic devices.•OLEDs and OPVs exhibited analogous performance to devices fabricated in the VTE.•Complete fabrication of OLEDs/OPVs was achieved in a low vacuum VPD system.A vapor phase deposition (VPD) system has been used to deposit magnesium and zinc films and prepare optoelectronic devices under low vacuum conditions, i.e. 1 torr. An analysis of the metal films via SEM, AFM, XRD and four-point probe resistivity measurements revealed comparable characteristics to metal films deposited in a vacuum thermal evaporation (VTE) system. Magnesium cathodes were fabricated for organic light emitting diodes (OLEDs) and organic photovoltaic (OPV) devices. OLEDs were fully made in either the VPD or VTE system employing aluminum tris-(8 hydroxyquinoline) [Alq3] as the green fluorescent emitter or fac-tris(2-phenylpyridine)iridium [Ir(ppy)3] as the green emitting phosphor. Analysis of the OLED devices made in the VPD system showed external quantum efficiencies (EQE = 0.9 ± 0.1%) and (EQE = 7.6 ± 0.6%) at a luminance of 100 cd/m2 for the fluorescent and phosphorescent devices, respectively. In addition, organic photovoltaics (OPVs) were fully fabricated by both methods employing copper phthalocyanine (CuPc) and C60 as the donor/acceptor materials. Analysis of the OPV devices made in the VPD system showed a power efficiency of 0.5 ± 0.1%, an open circuit voltage of 0.45 ± 0.05% and a fill factor of 0.50 ± 0.05%.
Co-reporter:Cong Trinh ; Kent Kirlikovali ; Saptaparna Das ; Maraia E. Ener ; Harry B. Gray ; Peter Djurovich ; Stephen E. Bradforth
The Journal of Physical Chemistry C 2014 Volume 118(Issue 38) pp:21834-21845
Publication Date(Web):August 27, 2014
DOI:10.1021/jp506855t
Zinc dipyrrin complexes with two identical dipyrrin ligands absorb strongly at 450–550 nm and exhibit high fluorescence quantum yields in nonpolar solvents (e.g., 0.16–0.66 in cyclohexane) and weak to nonexistent emission in polar solvents (i.e., <10–3, in acetonitrile). The low quantum efficiencies in polar solvents are attributed to the formation of a nonemissive symmetry-breaking charge transfer (SBCT) state, which is not formed in nonpolar solvents. Analysis using ultrafast spectroscopy shows that in polar solvents the singlet excited state relaxes to the SBCT state in 1.0–5.5 ps and then decays via recombination to the triplet or ground states in 0.9–3.3 ns. In the weakly polar solvent toluene, the equilibrium between a localized excited state and the charge transfer state is established in 11–22 ps.
Co-reporter:Yifei Liu, Peter I. Djurovich, Ralf Haiges, Mark E. Thompson
Polyhedron 2014 Volume 84() pp:136-143
Publication Date(Web):14 December 2014
DOI:10.1016/j.poly.2014.06.051
The neutral bis-pincer transition metal complex Os(PCP)2 (PCP = 2,6-(CH2PPh2)2C6H3, Ph = C6H5) was prepared via two synthetic routes with up to 42% yield. The lemon-yellow complex is air stable as a neat solid and in solution, and is thermally stable as can be purified by high vacuum zone sublimation at 280-240-200 °C under 1 × 10−6 torr. The pincer ligands coordinate in a pseudo-octahedral arrangement around the metal center. X-ray crystallography reveals that the complex is in pseudo-D2 geometry symmetry due to the twist caused by the methylenes. NMR studies at varied temperatures between 223 and 343 K along with low temperature inversion recovery studies show the fluxional behavior and exchanging process between the two enantiomers (Ea = 8.6 kcal/mol). The Os(PCP)2 shows no emission in solution at room temperature, but emits at 77 K in 2-MeTHF glass (Φ = 60%). At room temperature, the neat solid of Os(PCP)2 shows yellow emission of 3% quantum yield. TDDFT predicted S0 → S1 transition shows that the lowest singlet state (S1) is metal-ligand (the Os-Xylyl fragments) to ligand’ (the PPh2 fragments) charge transfer (ML-L’CT) in character with a small contribution from the higher lying metal centered (1MC) states. The large spin–orbit coupling constant of osmium leads to effective intersystem crossing to a triplet state with increased MC character. The DFT predicted triplet spin surface of Os(PCP)2 indicates that the emission is dominated by 3MC state. Significant Os–P bonds elongation at the lowest triplet excited state also strongly supports the conclusion.A bis-pincer osmium complex Os(PCP)2 (PCP = 2,6-(CH2PPh2)2C6H3, Ph = C6H5) has been prepared in up to 42% yield. The complex is composed of a racemic mixture and shows metal centered emission with a 3% quantum yield in the solid state (298 K).
Co-reporter:Andrew N. Bartynski, Cong Trinh, Anurag Panda, Kevin Bergemann, Brian E. Lassiter, Jeramy D. Zimmerman, Stephen R. Forrest, and Mark E. Thompson
Nano Letters 2013 Volume 13(Issue 7) pp:3315-3320
Publication Date(Web):June 10, 2013
DOI:10.1021/nl401531t
We demonstrate the concentration dependence of C60 absorption in solid solutions of C60 and bathocuprione (BCP), revealing a nonlinear decrease of the C60 charge transfer (CT) state absorption. These blends are utilized to study the photocurrent contribution of the CT in bilayer organic photovoltaics (OPVs); 1:1 blends produce 40% less photocurrent. As exciton blocking electron transporting layers, the blends achieve power conversion efficiencies of 5.3%, an increase of 10% compared to conventional buffers.
Co-reporter:Cong Trinh ; Kent O. Kirlikovali ; Andrew N. Bartynski ; Christopher J. Tassone ; Michael F. Toney ; George F. Burkhard ; Michael D. McGehee ; Peter I. Djurovich
Journal of the American Chemical Society 2013 Volume 135(Issue 32) pp:11920-11928
Publication Date(Web):July 17, 2013
DOI:10.1021/ja4043356
Fullerenes are currently the most popular electron-acceptor material used in organic photovoltaics (OPVs) due to their superior properties, such as good electron conductivity and efficient charge separation at the donor/acceptor interface. However, low absorptivity in the visible spectral region is a significant drawback of fullerenes. In this study, we have designed a zinc chlorodipyrrin derivative (ZCl) that absorbs strongly in the visible region (450–600 nm) with an optical density 7-fold higher than a C60 film. ZCl efficiently transfers absorbed photoenergy to C60 in mixed films. Application of ZCl as an energy sensitizer in OPV devices leads to an increase in the photocurrent from the acceptor layer, without changing the other device characteristics, i.e., open circuit voltage and fill factor. For example, C60-based OPVs with and without the sensitizer give 4.03 and 3.05 mA/cm2, respectively, while both have VOC = 0.88 V and FF = 0.44. Our ZCl sensitization approach improves the absorbance of the electron-acceptor layer while still utilizing the beneficial characteristics of C60 in OPVs.
Co-reporter:Alberto Bossi ; Andreas F. Rausch ; Markus J. Leitl ; Rafał Czerwieniec ; Matthew T. Whited ; Peter I. Djurovich ; Hartmut Yersin
Inorganic Chemistry 2013 Volume 52(Issue 21) pp:12403-12415
Publication Date(Web):October 11, 2013
DOI:10.1021/ic4011532
A detailed examination was performed on photophysical properties of phosphorescent cyclometalated (C∧N)Pt(O∧O) complexes (ppy)Pt(dpm) (1), (ppy)Pt(acac) (1′), and (bzq)Pt(dpm) (2) and newly synthesized (dbq)Pt(dpm) (3) (C∧N = 2-phenylpyridine (ppy), benzo[h]quinoline (bzq), dibenzo[f,h]quinoline (dbq); O∧O = dipivolylmethanoate (dpm), acetylacetonate (acac)). Compounds 1, 1′, 2, and 3 were further characterized by single crystal X-ray diffraction. Structural changes brought about by cyclometalation were determined by comparison with X-ray data from model C∧N ligand precursors. The compounds emit from metal-perturbed, ligand-centered triplet states (E0–0 = 479 nm, 1; E0–0 = 495 nm, 2; E0–0 = 470 nm, 3) with disparate radiative rate constants (kr = 1.4 × 105 s–1, 1; kr = 0.10 × 105 s–1, 2; kr = 2.6 × 105 s–1, 3). Zero-field splittings of the triplet states (ΔEIII–I = 11.5 cm–1, 1′; ΔEIII–I < 2 cm–1, 2; ΔEIII–I = 46.5 cm–1, 3) were determined using high resolution spectra recorded in Shpol’skii matrices. The fact that the E0–0 energies do not correspond to the extent of π-conjugation in the aromatic C∧N ligand is rationalized on the basis of structural distortions that occur upon cyclometalation using data from single crystal X-ray analyses of the complexes and ligand precursors along with the triplet state properties evaluated using theoretical calculations. The wide variation in the radiative rate constants and zero-field splittings is also explained on the basis of how changes in the electronic spin density in the C∧N ligands in the triplet state alter the spin–orbit coupling in the complexes.
Co-reporter:Qiwen Zhong, Vyacheslav V. Diev, Sean T. Roberts, Priscilla D. Antunez, Richard L. Brutchey, Stephen E. Bradforth, and Mark E. Thompson
ACS Nano 2013 Volume 7(Issue 4) pp:3466
Publication Date(Web):March 10, 2013
DOI:10.1021/nn400362e
A systematic study of the interaction between π-extended porphyrins and single-walled carbon nanotubes (SWNTs) is reported here. Zinc porphyrins with 1-pyrenyl groups in the 5,15-meso positions, 1, as well as compounds where one or both of the pyrene groups have been fused at the meso and β positions of the porphyrin core, 2 and 3, respectively, have been examined. The strongest binding to SWNTs is observed for porphyrin 3, leading to debundling of the nanotubes and formation of stable suspensions of 3–SWNT hybrids in a range of common organic solvents. Absorption spectra of 3–SWNT suspensions are broad and continuous (λ = 400–1400 nm), and the Q-band of 3 displays a significant bathochromic shift of 33 nm. The surface coverage of the SWNTs in the nanohybrids was estimated by spectroscopic and analytical methods and found to reach 64% for (7,6) nanotubes. The size and shape of π-conjugated porphyrins were found to be important factors in determining the strength of the π–π interactions, as the linear anti-3 isomer displays more than 90% binding selectivity compared to the bent syn-3 isomer. Steady-state photoluminescence measurements show quenching of porphyrin emission from the nanohybrids. Femtosecond transient absorption spectroscopy reveals that this quenching results from ultrafast electron transfer from the photoexcited porphyrin to the SWNT (1/kCT = 260 fs) followed by rapid charge recombination on a picosecond time scale. Overall, our data demonstrate that direct π–π interaction between fused porphyrins and SWNTs leads to electronically coupled stable nanohybrids.Keywords: femtosecond transient absorption; isomer selectivity; nanohybrids; noncovalent π−π interaction; single-walled carbon nanotubes; ultrafast electron transfer; π-extended porphyrins
Co-reporter:Sean T. Roberts ; R. Eric McAnally ; Joseph N. Mastron ; David H. Webber ; Matthew T. Whited ; Richard L. Brutchey ; Mark E. Thompson ;Stephen E. Bradforth
Journal of the American Chemical Society 2012 Volume 134(Issue 14) pp:6388-6400
Publication Date(Web):March 20, 2012
DOI:10.1021/ja300504t
Singlet exciton fission is a process that occurs in select organic semiconductors and entails the splitting of a singlet excited state into two lower triplet excitons located on adjacent chromophores. Research examining this phenomenon has recently seen a renaissance due to the potential to exploit singlet fission within the context of organic photovoltaics to prepare devices with the ability to circumvent the Shockley–Queisser limit. To date, high singlet fission yields have only been reported for crystalline or polycrystalline materials, suggesting that molecular disorder inhibits singlet fission. Here, we report the results of ultrafast transient absorption and time-resolved emission experiments performed on 5,12-diphenyl tetracene (DPT). Unlike tetracene, which tends to form polycrystalline films when vapor deposited, DPT’s pendant phenyl groups frustrate crystal growth, yielding amorphous films. Despite the high level of disorder in these films, we find that DPT exhibits a surprisingly high singlet fission yield, with 1.22 triplets being created per excited singlet. This triplet production occurs over two principal time scales, with ∼50% of the triplets appearing within 1 ps after photoexcitation followed by a slower phase of triplet growth over a few hundred picoseconds. To fit these kinetics, we have developed a model that assumes that due to molecular disorder, only a subset of DPT dimer pairs adopt configurations that promote fission. Singlet excitons directly excited at these sites can undergo fission rapidly, while singlet excitons created elsewhere in the film must diffuse to these sites to fission.
Co-reporter:Cong Trinh, Matthew T. Whited, Andrew Steiner, Christopher J. Tassone, Michael F. Toney, and Mark E. Thompson
Chemistry of Materials 2012 Volume 24(Issue 13) pp:2583
Publication Date(Web):June 14, 2012
DOI:10.1021/cm3012777
We present a chemical annealing process for organic thin films. In this process, a thin film of a molecular material, such as zinc tetraphenylporphyrin (ZnTPP), is exposed to a vapor of nitrogen-based ligand (e.g., pyrazine, pz, and triazine, tz), forming a film composed of the metal–ligand complex. Fast and quantitative formation of the complex leads to marked changes in the morphology and optical properties of the film. X-ray diffraction studies show that the chemical annealing process converts amorphous ZnTPP films to crystalline ZnTPP·ligand films, whose porphryin planes lie nearly parallel to the substrate (average deviation is 8° for the ZnTPP·pz film). Organic solar cells were prepared with ZnTPP donor and C60 acceptor layers. Devices were prepared with and without chemical annealing of the ZnTPP layer with a pyrazine ligand. The devices with chemically annealed ZnTPP donor layer show an increase in short-circuit current (JSC) and fill factor (FF) relative to analogous unannealed devices, presumably because of enhanced exciton diffusion length and improved charge conductivity. The open circuit voltages (VOC) of the chemically annealed devices are lower than their unannealed counterpart because of enhanced polaron pair recombination at the donor/acceptor heterojunction. A net improvement of 5–20% in efficiency has been achieved, after chemical annealing of ZnTPP films with pyrazine.Keywords: chemical annealing; crystalline organic film; organic solar cell; porphyrin; structural reorganization;
Co-reporter:Hsiao-Fan Chen, Chao Wu, Ming-Cheng Kuo, Mark E. Thompson and Ken-Tsung Wong
Journal of Materials Chemistry A 2012 vol. 22(Issue 19) pp:9556-9561
Publication Date(Web):09 Mar 2012
DOI:10.1039/C2JM30443K
Two anionic iridium complexes Na[Ir(2-(p-tolyl)pyridine)2(CN)2] (A1) and Na[Ir(2-phenylquinoline)2(CN)2] (A2) were synthesized and characterized for use in light-emitting electrochemical cells (LECs). The photophysical and electrochemical studies show that the emission wavelengths and the LUMO energy levels of these complexes are governed by the conjugation length of the cyclometalated ligands. Single-layer LEC devices incorporating anionic complexes were fabricated and tested. Suffering from poor solubility of the anionic complexes, the device performances were rather limited due to the lack of film uniformity. The solubility issue was circumvented by using 18-crown-6 as an additive, efficiently eliminating the cluster structure formed by ion-dipole (sodium ion–lone pair of the nitrile ligand) interaction. The maximum brightness and peak external quantum efficiency achieved by crown-mediated LEC were up to 69 cd m−2 and 1.38%, respectively. The device efficiencies exhibit a great dependence with the LUMO level of the complex, suggesting substantial importance of the electron injection process.
Co-reporter:Matthew T. Whited, Niral M. Patel, Sean T. Roberts, Kathryn Allen, Peter I. Djurovich, Stephen E. Bradforth and Mark E. Thompson
Chemical Communications 2012 vol. 48(Issue 2) pp:284-286
Publication Date(Web):21 Nov 2011
DOI:10.1039/C1CC12260F
We report the synthesis and characterization of symmetric BODIPY dyads where the chromophores are attached at the meso position, using either a phenylene bridge or direct linkage. Both molecules undergo symmetry-breaking intramolecular charge transfer in the excited state, and the directly linked dyad serves as a visible-light-absorbing analogue of 9,9′-bianthryl.
Co-reporter:Zhiwei Liu, Peter I. Djurovich, Matthew T. Whited, and Mark E. Thompson
Inorganic Chemistry 2012 Volume 51(Issue 1) pp:230-236
Publication Date(Web):December 1, 2011
DOI:10.1021/ic2015226
A series of Cu4I4 clusters (1–5) supported by two P∧N-type ligands 2-[(diRphosphino)methyl]pyridine (1, R = phenyl; 2, R = cyclohexyl; 3, R = tert-butyl; 4, R = iso-propyl; 5, R = ethyl) have been synthesized. Single crystal X-ray analyses show that all five clusters adopt a rare “octahedral” geometry. The central core of the cluster consists of the copper atoms arranged in a parallelogram with μ4-iodides above and below the copper plane. The copper atoms on the two short edges of the parallelogram (Cu–Cu = 2.525(2)–2.630(1) Å) are bridged with μ2-iodides, whereas the long edges (Cu–Cu = 2.839(3)–3.035(2) Å) are bridged in an antiparallel fashion by the P∧N ligands. This Cu4I4 geometry differs significantly from the “cubane” and “stairstep” geometries reported for other Cu4I4L4 clusters. Luminescence spectra of clusters 3 and 4 display a single emission around 460 nm at room temperature that is assigned to emission from a triplet halide-to-ligand charge-transfer (3XLCT) excited state, whereas clusters 1, 2, and 5 also have a second band around 570 nm that is assigned to a Cu4I4 cluster-centered (3CC) excited state. The structural and photophysical properties of a dinuclear Cu2I2(P∧N)2 complex obtained during the sublimation of cluster 3 is also provided.
Co-reporter:Kenneth Hanson, Luke Roskop, Niral Patel, Laurent Griffe, Peter I. Djurovich, Mark S. Gordon and Mark E. Thompson
Dalton Transactions 2012 vol. 41(Issue 28) pp:8648-8659
Publication Date(Web):07 Jun 2012
DOI:10.1039/C2DT30354J
A series of twelve platinum(II) complexes of the form (N^N^N)PtX have been synthesized and characterized where N^N^N is 1,3-bis(2-pyridylimino)isoindolate ligands (BPI) or BPI ligands whose aryl moieties are substituted with tert-butyl, nitro, alkoxy, iodo or chloro groups, and X is a chloride, fluoride, cyano, acetate, phenyl or 4-(dimethylamino)phenyl ligand. All complexes display at least one irreversible oxidation and two reversible reduction waves at potentials dependent on the position and the electron donating or withdrawing nature of both X and the substituted N^N^N ligand. Broad room temperature phosphorescence ranging in energy from 594 to 680 nm was observed from the complexes, with quantum efficiencies ranging from 0.01 to 0.05. The efficiency of emission is dictated largely by nonradiative processes since the rate constants for nonradiative deactivation [(1.1–100) × 105 s−1] show greater variation than those for radiative decay [(0.57–4.0) × 04 s−1]. Nonradiative deactivation for compounds with X = Cl follow the energy gap law, i.e. the nonradiative rate constants increase exponentially with decreasing emission energy. Deactivation of the excited state appears to be strongly influenced by a non-planar distortion of the BPI ligand.
Co-reporter:Valentina A. Krylova, Peter I. Djurovich, Jacob W. Aronson, Ralf Haiges, Matthew T. Whited, and Mark E. Thompson
Organometallics 2012 Volume 31(Issue 22) pp:7983-7993
Publication Date(Web):October 19, 2012
DOI:10.1021/om300656v
A series of four neutral luminescent three-coordinate Cu(I) complexes (IPr)Cu(N∧N), where IPr is a monodentate N-heterocyclic carbene (NHC) ligand (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) and N∧N denotes monoanionic pyridyl-azolate ligands, have been synthesized and characterized. A monomeric, three-coordinate geometry, best described as distorted trigonal planar, has been established by single-crystal X-ray analyses for three of the derivatives. In contrast to the previously reported (IPr)Cu(N∧N) complexes, the compounds described here display a perpendicular orientation between the chelating N∧N ligands and the imidazolylidene ring of the carbene ligand. The geometrical preferences revealed by X-ray crystallography correlate well with the NMR data. The conformational behavior of the complexes, investigated by variable-temperature 1H NMR spectroscopy, indicate free rotation about the CNHC–Cu bond in solution. The complexes display broad, featureless luminescence at both room temperature and 77 K, with emission maxima that vary between 555 and 632 nm depending on sample conditions. Luminescence quantum efficiencies of the complexes in solution (Φ ≤ 17%) increase markedly in the solid state (Φ ≤ 62%). On the basis of time-dependent density functional theory (TD-DFT) calculations and the experimental data, luminescence is assigned to phosphorescence from a metal-to-ligand charge-transfer (MLCT) triplet state admixed with ligand-centered (LC) character.
Co-reporter:Vyacheslav V. Diev, Cody W. Schlenker, Kenneth Hanson, Qiwen Zhong, Jeramy D. Zimmerman, Stephen R. Forrest, and Mark E. Thompson
The Journal of Organic Chemistry 2012 Volume 77(Issue 1) pp:143-159
Publication Date(Web):November 13, 2011
DOI:10.1021/jo201490y
A systematic study of the preparation of porphyrins with extended conjugation by meso,β-fusion with polycyclic aromatic hydrocarbons (PAHs) is reported. The meso-positions of 5,15-unsubstituted porphyrins were readily functionalized with PAHs. Ring fusion using standard Scholl reaction conditions (FeCl3, dichloromethane) occurs for perylene-substituted porphyrins to give a porphyrin β,meso annulated with perylene rings (0.7:1 ratio of syn and anti isomers). The naphthalene, pyrene, and coronene derivatives do not react under Scholl conditions but are fused using thermal cyclodehydrogenation at high temperatures, giving mixtures of syn and anti isomers of the meso,β-fused porphyrins. For pyrenyl-substituted porphyrins, a thermal method gives synthetically acceptable yields (>30%). Absorption spectra of the fused porphyrins undergo a progressive bathochromic shift in a series of naphthyl (λmax = 730 nm), coronenyl (λmax = 780 nm), pyrenyl (λmax = 815 nm), and perylenyl (λmax = 900 nm) annulated porphyrins. Despite being conjugated with unsubstituted fused PAHs, the β,meso-fused porphyrins are more soluble and processable than the parent nonfused precursors. Pyrenyl-fused porphyrins exhibit strong fluorescence in the near-infrared (NIR) spectral region, with a progressive improvement in luminescent efficiency (up to 13% with λmax = 829 nm) with increasing degree of fusion. Fused pyrenyl-porphyrins have been used as broadband absorption donor materials in photovoltaic cells, leading to devices that show comparatively high photovoltaic efficiencies.
Co-reporter:Zhiwei Liu ; Munzarin F. Qayyum ; Chao Wu ; Matthew T. Whited ; Peter I. Djurovich ; Keith O. Hodgson ; Britt Hedman ; Edward I. Solomon
Journal of the American Chemical Society 2011 Volume 133(Issue 11) pp:3700-3703
Publication Date(Web):March 2, 2011
DOI:10.1021/ja1065653
We demonstrate a new approach for utilizing CuI coordination complexes as emissive layers in organic light-emitting diodes that involves in situ codeposition of CuI and 3,5-bis(carbazol-9-yl)pyridine (mCPy). With a simple three-layer device structure, pure green electroluminescence at 530 nm from a Cu(I) complex was observed. A maximum luminance and external quantum efficiency (EQE) of 9700 cd/m2 and 4.4%, respectively, were achieved. The luminescent species was identified as [CuI(mCPy)2]2 on the basis of photophysical studies of model complexes and X-ray absorption spectroscopy.
Co-reporter:Siyi Wang, Lincoln Hall, Vyacheslav V. Diev, Ralf Haiges, Guodan Wei, Xin Xiao, Peter I. Djurovich, Stephen R. Forrest, and Mark E. Thompson
Chemistry of Materials 2011 Volume 23(Issue 21) pp:4789
Publication Date(Web):October 18, 2011
DOI:10.1021/cm2020803
We report new derivatives of symmetric squaraine dyes with N,N-diarylanilino substituents that have high solubility and high absorptivity (ε = 0.71–4.1 × 105 M–1 cm–1) in the red solar spectral region (λmax = 645–694 nm) making them promising candidates for application in organic photovoltaics (OPVs). Unsymmetrical N,N-diisobutylanilino- and N,N-diphenylanilino(diphenylamino)squaraines have also been prepared that give blue-shifted absorption spectra (λmax = 529–535 nm) relative to their symmetric counterparts. Compared to bis(N,N-diisobutylanilino)squaraine, both symmetrical and unsymmetrical N,N-diarylanilino squaraines show markedly broader absorption bands in solution than their N,N-dialkylanilino squaraine counterparts: the full width at half-maximum (fwhm) for N,N-diarylanilino squaraines range from 1280–1980 cm–1, while the fwhm value for the N,N-diisobutylanilino squarine is only 630 cm–1. The absorption bands for thin films of N,N-diarylanilino squaraines broaden further to 2500–3300 cm–1. N,N-Diarylanilino squaraines are fluorescent, albeit with lower quantum yields than bis(N,N-diisobutylanilino)squaraine (ϕPL = 0.02–0.66 and 0.80, respectively). OPVs were prepared with solution processed squaraine layers using the following structure: ITO/squaraine (66–85 Å)/C60 (400 Å)/BCP (100 Å)/Al (1000 Å), BCP = bathocuproine. Devices using thin films of the bis(N,N-diarylanilino)squaraines as donor layers show improved performance relative to OPVs prepared with bis(N,N-dialkylanilino)squaraines, i.e. bis(N,N-diisobutylanilino)squaraine: open-circuit voltage Voc = 0.59 ± 0.05 V, short-circuit current Jsc = 5.58 ± 0.16 mA/cm2, fill factor FF = 0.56 ± 0.03, and power conversion efficiency η = 1.8 ± 0.2% under 1 sun, AM1.5G simulated illumination, compared with bis(N,N-diphenylanilino)squaraine: Voc = 0.82 ± 0.02 V, Jsc = 6.71 ± 0.10 mA/cm2, FF = 0.59 ± 0.01, and η = 3.2 ± 0.1%. Morphological studies of thin films suggest that the solubility of bis(N,N-diarylanilino)squaraines plays an important role in controlling the optoelectronic properties of the OPVs.Keywords: ; organic photovoltaic; solar cell; squaraine dyes;
Co-reporter:Rui Zhang, Marco Curreli, and Mark E. Thompson
ACS Applied Materials & Interfaces 2011 Volume 3(Issue 12) pp:4765
Publication Date(Web):November 1, 2011
DOI:10.1021/am2012454
Selective electrochemically activated biofunctionalization of In2O3 nanowires (NWs) has been achieved, using monolayer coatings of p-dimethoxybenzene derivatives. Monolayer coatings of 4-(2,5-dimethoxyphenyl)butyl-phosphonic acid (DMP-PA) were deposited on planar indium–tin oxide (ITO) electrodes and In2O3 NWs. The electrochemical behavior of the monolayer coating was first studied using ITO electrodes, as a model system for In2O3 nanowires. When a potential of 950 mV vs a Ag/AgCl reference electrode is applied to an ITO electrode coated with DMP-PA in PBS buffer, the p-dimethoxyphenyl groups are converted to p-benzoquinone (BQ). The electrochemically formed benzoquinone groups react readily with alkyl thiol groups via a Michael addition. The reaction strategy optimized on ITO was applied to an In2O3 NW mat sample coated with DMP-PA. Applying a potential of 950 mV to metal electrodes deposited on NWs converts the DMP-PA NW coating to BQ-PA, which reacts with a thiol-terminated 20-base oligonucleotide. These NWs showed strong fluorescence response after paring with the dye labeled compliment, demonstrating that the probe was bound to the NW surface and that it remained active toward hybridization with its compliment. The unactivated DMP-PA coated NWs showed no response, demonstrating the selective electrochemical functionalization of NWs and the potential of using them in multiplex sensing. We also compared the p-dimethoxybenzene derivative to the conventional hydroquinone analog. The results show that the former can largely enhance the selectivity during the functionalization of both ITO and In2O3 NWs.Keywords: ; benzoquinone; electrochemical activation; Indium oxide nanowire; Indium−tin oxide; p-dimethoxybenzene; selective biofunctionalization;
Co-reporter:Vincent S. Barlier, Cody W. Schlenker, Stephanie W. Chin and Mark E. Thompson
Chemical Communications 2011 vol. 47(Issue 13) pp:3754-3756
Publication Date(Web):04 Feb 2011
DOI:10.1039/C0CC05164K
Alkyne linked tetracene dimers were synthesized from naphthacenequinone and terminal acetylenes. The silylethynyl tetracene dimers exhibit good solubility, high photostability, and broad absorbance leading to photocurrent generation in an organic photovoltaic device.
Co-reporter:Chao Wu ; Hsiao-Fan Chen ; Ken-Tsung Wong
Journal of the American Chemical Society 2010 Volume 132(Issue 9) pp:3133-3139
Publication Date(Web):February 10, 2010
DOI:10.1021/ja9097725
Three Ir-based materials were synthesized through metathesis reaction between halide and alkali metal salts of two cationic and three anionic Ir complexes, respectively. The resulting “soft salt” complexes are composed of an organometallic cation and an organometallic anion. The electrochemical and photophysical characterization of these compounds is reported. The redox potentials of the soft salts are shown to be determined by the lowest energy potentials of the two ions. Energy transfer between the ions in solution is observed, and found to take place at diffusion controlled rates. Organic LEDs were prepared with each of the three soft salts, using the simple structure of anode/PVK/soft salt/BCP/cathode. The soft salts yielded maximal external quantum efficiencies (EQE) ranging from 0.2% to 4.7%. The study suggests that the internal energy alignment between two ions in the soft salts is responsible for the widely disparate results. To achieve a high EQE, it is critical to have the HOMO and LUMO values of one of the ions fall between those of the other ion, that is, one ion has both the lowest oxidation potential and the least negative reduction potential.
Co-reporter:Kenneth Hanson ; Luke Roskop ; Peter I. Djurovich ; Federico Zahariev ; Mark S. Gordon
Journal of the American Chemical Society 2010 Volume 132(Issue 45) pp:16247-16255
Publication Date(Web):October 21, 2010
DOI:10.1021/ja1075162
Benzannulation of aromatic molecules is often used to red-shift absorption and emission bands of organic and inorganic, molecular, and polymeric materials; however, in some cases, either red or blue shifts are observed, depending on the site of benzannulation. A series of five platinum(II) complexes of the form (N∧N∧N)PtCl are reported here that illustrate this phenomenon, where N∧N∧N represents the tridentate monoanionic ligands 2,5-bis(2-pyridylimino)3,4-diethylpyrrolate (1), 1,3-bis(2-pyridylimino)isoindolate (2), 1,3-bis(2-pyridylimino)benz(f)isoindolate (3), 1,3-bis(2-pyridylimino)benz(e)isoindolate (4), and 1,3-bis(1-isoquinolylimino) isoindolate (5). For this series of molecules, either a blue shift (2 and 3) or a red shift (4 and 5) in absorption and emission maxima, relative to their respective nonbenzannulated compounds, was observed that depends on the site of benzannulation. Experimental data and first principles calculations suggest that a similar HOMO energy level and a destabilized or stabilized LUMO with benzannulation is responsible for the observed trends. A rationale for LUMO stabilization/destabilization is presented using simple molecular orbital theory. This explanation is expanded to describe other molecules with this unusual behavior.
Co-reporter:Matthew T. Whited ; Peter I. Djurovich ; Sean T. Roberts ; Alec C. Durrell ; Cody W. Schlenker ; Stephen E. Bradforth
Journal of the American Chemical Society 2010 Volume 133(Issue 1) pp:88-96
Publication Date(Web):December 10, 2010
DOI:10.1021/ja108493b
Multichromophoric arrays provide one strategy for assembling molecules with intense absorptions across the visible spectrum but are generally focused on systems that efficiently produce and manipulate singlet excitations and therefore are burdened by the restrictions of (a) unidirectional energy transfer and (b) limited tunability of the lowest molecular excited state. In contrast, we present here a multichromophoric array based on four boron dipyrrins (BODIPY) bound to a platinum benzoporphyrin scaffold that exhibits intense panchromatic absorption and efficiently generates triplets. The spectral complementarity of the BODIPY and porphryin units allows the direct observation of fast bidirectional singlet and triplet energy transfer processes (kST(1BDP→1Por) = 7.8 × 1011 s−1, kTT(3Por→3BDP) = 1.0 × 1010 s−1, kTT(3BDP→3Por) = 1.6 × 1010 s−1), leading to a long-lived equilibrated [3BDP][Por]⇌[BDP][3Por] state. This equilibrated state contains approximately isoenergetic porphyrin and BODIPY triplets and exhibits efficient near-infrared phosphorescence (λem = 772 nm, Φ = 0.26). Taken together, these studies show that appropriately designed triplet-utilizing arrays may overcome fundamental limitations typically associated with core−shell chromophores by tunable redistribution of energy from the core back onto the antennae.
Co-reporter:Wei Wei, Peter I. Djurovich and Mark E. Thompson
Chemistry of Materials 2010 Volume 22(Issue 5) pp:1724
Publication Date(Web):January 12, 2010
DOI:10.1021/cm903146x
Multifluorenyl silanes have been studied as potential hosts for organic light emitting diodes. Four molecules, (9,9′-dimethylfluoren-2-yl)nSi(phenyl)4-n (SiFln, n = 1, 2, 3, and 4), with an increasing number of fluorene units have been synthesized and investigated. These compounds possess high triplet energies (2.9 eV), large HOMO−LUMO gaps (∼5.2 eV), and high glass transition temperatures. Their glass transition and sublimation temperatures increase linearly as the fluorene ratio increases, but there are only small changes in their electrochemical or photophysical properties. These studies suggest that the Si center helps maintain the high singlet and triplet energy levels of these molecules. These materials exhibit ambipolar transport characteristics in undoped OLED devices, and the charge conductivity of the devices was enhanced by increasing the fluorene ratios in the host molecules. Compared with phenylsilanes, the fluorenylsilanes show better hole injecting and charge transporting abilities. SiFl4 was investigated as a host material for red, green, and blue phosphorescent devices, giving peak efficiencies of 8, 8, and 3%, respectively.
Co-reporter:Valentina A. Krylova, Peter I. Djurovich, Matthew T. Whited and Mark E. Thompson
Chemical Communications 2010 vol. 46(Issue 36) pp:6696-6698
Publication Date(Web):17 Aug 2010
DOI:10.1039/C0CC01864C
Cationic and neutral monomeric three-coordinate phosphorescent Cu(I) complexes were synthesized and characterized by XRD analysis, electrochemistry and photophysical studies in different environments. DFT calculations have aided the assignment of the electronic structure and excited state behavior of these complexes.
Co-reporter:Kenneth Hanson ; Arnold Tamayo ; Vyacheslav V. Diev ; Matthew T. Whited ; Peter I. Djurovich
Inorganic Chemistry 2010 Volume 49(Issue 13) pp:6077-6084
Publication Date(Web):June 8, 2010
DOI:10.1021/ic100633w
A series of seven dipyrrin-based bis-cyclometalated Ir(III) complexes have been synthesized and characterized. All complexes display a single, irreversible oxidation wave and at least one reversible reduction wave. The electrochemical properties were found to be dominated by dipyrrin centered processes. The complexes were found to display room temperature luminescence with emission maxima ranging from 658 to 685 nm. Through systematic variation of the cyclometalating ligand and the meso substituent of the dipyrrin moiety, it was found that the observed room temperature emission was due to phosphorescence from a dipyrrin-centered triplet state with quantum efficiencies up to 11.5%. Bis-cyclometalated Ir(III) dipyrrin based organic light emitting diodes (OLEDs) display emission at 682 nm with maximum external quantum efficiencies up to 1.0%.
Co-reporter:VyacheslavV. Diev Dr.;Kenneth Hanson Dr.;JeramyD. Zimmerman Dr.;StephenR. Forrest ;MarkE. Thompson
Angewandte Chemie 2010 Volume 122( Issue 32) pp:5655-5658
Publication Date(Web):
DOI:10.1002/ange.201002669
Co-reporter:VyacheslavV. Diev Dr.;Kenneth Hanson Dr.;JeramyD. Zimmerman Dr.;StephenR. Forrest ;MarkE. Thompson
Angewandte Chemie International Edition 2010 Volume 49( Issue 32) pp:5523-5526
Publication Date(Web):
DOI:10.1002/anie.201002669
Co-reporter:M. Dolores Perez ; Carsten Borek ; Stephen R. Forrest
Journal of the American Chemical Society 2009 Volume 131(Issue 26) pp:9281-9286
Publication Date(Web):June 11, 2009
DOI:10.1021/ja9007722
We explore the dependence of the dark current of C60-based organic photovoltaic (OPV) cells on molecular composition and the degree of intermolecular interaction of several molecular donor materials. The saturation dark current density, JS, is an important factor in determining the open circuit voltage, Voc. The Voc values of OPVs show a strong inverse correlation with JS. Donor materials that show evidence for aggregation in their thin-film absorption spectra and polycrystallinity in thin film X-ray diffraction result in a high dark current, and thus a low Voc. In contrast, donor materials with structures that hinder intermolecular π-interaction give amorphous thin films and reduced values of JS, relative to donors with strong intermolecular π-interactions, leading to a high Voc. This work provides guidance for the design of materials and device architectures that maximize OPV cell power conversion efficiency.
Co-reporter:Chao Wu;Peter I. Djurovich
Advanced Functional Materials 2009 Volume 19( Issue 19) pp:3157-3164
Publication Date(Web):
DOI:10.1002/adfm.200900357
Abstract
A device structure is used in which the hole-transporting layer (HTL) of an OLED is doped with either fluorescent or phosphorescent emitters, that is, anode/HTL-host/hole blocker/electron-transporting layer/cathode. The HTL hosts have higher HOMO energy allowing holes to be transported without being trapped by dopant molecules, avoiding direct recombination on the dopant. The unconventional mismatch of HOMO energies between host and dopant allow for the study of energy transfer in these host/guest systems and triplet exciton diffusion in the HTL-host layers of OLED devices, without the complication of charge trapping at dopants. The host materials examined here are tetraaryl-p-phenylenediamines. Data shows that Förster energy transfer between these hosts and emissive dopant in devices is inefficient. Triplet exciton diffusion in these host materials is closely related to molecular structure and the degree of intermolecular interaction. Host materials that contain naphthyl groups demonstrate longer triplet exciton diffusion lengths than those with phenyl substituents, consistent with DFT calculations and photophysical measurements.
Co-reporter:M. Dolores Perez, Peter I. Djurovich, Azad Hassan, Grace Y. Cheng, Tim J. Stewart, Kristen Aznavour, Robert Bau and Mark E. Thompson
Chemical Communications 2009 (Issue 28) pp:4215-4217
Publication Date(Web):08 Jun 2009
DOI:10.1039/B901510H
An intensely phosphorescent Pt complex in cyclohexane is efficiently quenched by exciplex formation with extremely weak Lewis bases such as toluene and other aromatic compounds.
Co-reporter:Paulin N. Wahjudi;Jin H. Oh;Salam O. Salman;Jason A. Seabold;Damien C. Rodger;Yu-Chong Tai
Journal of Biomedical Materials Research Part A 2009 Volume 89A( Issue 1) pp:206-214
Publication Date(Web):
DOI:10.1002/jbm.a.31929
Abstract
A general method for chemical surface functionalization of parylene C [PC, (para-CH2-C6H3Cl-CH2-)n] films is reported. Friedel-Crafts acylation is used to activate the surface of the PC film, and the resulting carbonyl groups are then used to form a range of different organic functional groups to the surface of the parylene film, including alcohol, imine, thiol, phthalimide, amine, and maleimide. The presence of these functional groups on the parylene surface was confirmed by Fourier transform infrared spectroscopy. Static water drop contact angle measurements were also used to demonstrate the changes in hydrophilicity of the PC film surface, consistent with each of the surface modifications. Enhanced metal (gold) adhesion was achieved by anchoring a thiol group onto the acylated surface of PC film. Acylation of parylene with 2-chloropropionyl chloride gave a surface bound chloropropionyl group. Grafting of poly-N-isopropylacrylamide (pNIPAM) onto the chloropropionyl substituted PC film via atom transfer radical polymerization (ATRP) was carried out. The grafted pNIPAM on the parylene surface leads to temperature-dependent cellular tissue adhesion on the PC film. © 2008 Wiley Periodicals, Inc. J Biomed Mater Res, 2009
Co-reporter:Fumiaki N. Ishikawa, Hsiao-Kang Chang, Marco Curreli, Hsiang-I Liao, C. Anders Olson, Po-Chiang Chen, Rui Zhang, Richard W. Roberts, Ren Sun, Richard J. Cote, Mark E. Thompson and Chongwu Zhou
ACS Nano 2009 Volume 3(Issue 5) pp:1219
Publication Date(Web):May 7, 2009
DOI:10.1021/nn900086c
Antibody mimic proteins (AMPs) are polypeptides that bind to their target analytes with high affinity and specificity, just like conventional antibodies, but are much smaller in size (2−5 nm, less than 10 kDa). In this report, we describe the first application of AMP in the field of nanobiosensors. In2O3 nanowire based biosensors have been configured with an AMP (Fibronectin, Fn) to detect nucleocapsid (N) protein, a biomarker for severe acute respiratory syndrome (SARS). Using these devices, N protein was detected at subnanomolar concentration in the presence of 44 μM bovine serum albumin as a background. Furthermore, the binding constant of the AMP to Fn was determined from the concentration dependence of the response of our biosensors.Keywords: antibody mimetic protein; biosensor; nanowire; nucleocapsid (N) protein
Co-reporter:Carsten Borek;Kenneth Hanson;Peter I. Djurovich Dr.;Mark E. Thompson ;Kristen Aznavour;Robert Bau ;Yiru Sun;Stephen R. Forrest ;Jason Brooks Dr.;Lech Michalski;Julie Brown Dr.
Angewandte Chemie 2007 Volume 119(Issue 7) pp:
Publication Date(Web):9 JAN 2007
DOI:10.1002/ange.200604240
Rotverschiebung über Rot hinaus: Der nichtplanare Porphyrinkomplex [Pt(tpbp)] (tpbp=Tetraphenyltetrabenzoporphyrin) wurde als phosphoreszierendes Dotierungsmittel in hocheffizienten elektrophosphoreszierenden Bauteilen eingesetzt, die im Nah-IR-Bereich emittieren (siehe normiertes Emissionsspektrum). Die hohen Effizienzen lassen diese NIR-Funktionseinheiten für Nachtsichtanzeigen und Sensoren geeignet erscheinen.
Co-reporter:Carsten Borek;Kenneth Hanson;Peter I. Djurovich Dr.;Mark E. Thompson ;Kristen Aznavour;Robert Bau ;Yiru Sun;Stephen R. Forrest ;Jason Brooks Dr.;Lech Michalski;Julie Brown Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 7) pp:
Publication Date(Web):9 JAN 2007
DOI:10.1002/anie.200604240
Red-shifting beyond red: The nonplanar porphyrin complex [Pt(tpbp)] (tpbp=tetraphenyltetrabenzoporphyrin) has been used as a phosphorescent dopant in highly efficient electrophosphorescent devices that emit in the near-infrared region (see normalized emission spectrum). The high efficiencies of these NIR devices make them amenable to many night-vision display and sensing applications.
Co-reporter:B. Ma;P. I. Djurovich;S. Garon;B. Alleyne;M. E. Thompson
Advanced Functional Materials 2006 Volume 16(Issue 18) pp:
Publication Date(Web):3 NOV 2006
DOI:10.1002/adfm.200600614
Efficient blue-, green-, and red-light-emitting organic diodes are fabricated using binuclear platinum complexes as phosphorescent dopants. The series of complexes used here have pyrazolate bridging ligands and the general formula C∧NPt(μ-pz)2PtC∧N (where C∧N = 2-(4′,6′-difluorophenyl)pyridinato-N,C2′, pz = pyrazole (1), 3-methyl-5-tert-butylpyrazole (2), and 3,5-bis(tert-butyl)pyrazole (3)). The Pt–Pt distance in the complexes, which decreases in the order 1 > 2 > 3, solely determines the electroluminescence color of the organic light-emitting diodes (OLEDs). Blue OLEDs fabricated using 8 % 1 doped into a 3,5-bis(N-carbazolyl)benzene (mCP) host have a quantum efficiency of 4.3 % at 120 Cd m–2, a brightness of 3900 Cd m–2 at 12 V, and Commission Internationale de L'Eclairage (CIE) coordinates of (0.11, 0.24). Green and red OLEDs fabricated with 2 and 3, respectively, also give high quantum efficiencies (∼ 6.7 %), with CIE coordinates of (0.31, 0.63) and (0.59, 0.46), respectively. The current-density–voltage characteristics of devices made using dopants 2 and 3 indicate that hole trapping is enhanced by short Pt–Pt distances (< 3.1 Å). Blue electrophosphorescence is achieved by taking advantage of the binuclear molecular geometry in order to suppress dopant intermolecular interactions. No evidence of low-energy emission from aggregate states is observed in OLEDs made with 50 % 1 doped into mCP. OLEDs made using 100 % 1 as an emissive layer display red luminescence, which is believed to originate from distorted complexes with compressed Pt–Pt separations located in defect sites within the neat film. White OLEDs are fabricated using 1 and 3 in three different device architectures, either with one or two dopants in dual emissive layers or both dopants in a single emissive layer. All the white OLEDs have high quantum efficiency (∼ 5 %) and brightness (∼ 600 Cd m–2 at 10 V).
Co-reporter:Biwu Ma, Peter I. Djurovich, Mark E. Thompson
Coordination Chemistry Reviews 2005 Volume 249(13–14) pp:1501-1510
Publication Date(Web):July 2005
DOI:10.1016/j.ccr.2005.02.004
Luminescence quenching studies of a cyclometalated complex, platinum(II) (2-(4′,6′-difluorophenyl)pyridinato-N,C2′)(2,4-pentanedionato-O,O) (FPt), in solution at room temperature are reported. The FPt complex undergoes efficient self-quenching in solution at room temperature that can be successfully modeled by a monomer/excimer phosphorescence mechanism with a diffusion limited rate constant (4.2 ± 0.3 × 109 M−1 s−1). The emission lifetimes for FPt monomer and excimer in 2-methyltetrahydrofuran at room temperature are 330 ns (±15 ns) and 135 ns (±10 ns), respectively. The excited state properties of FPt were also investigated. A triplet energy ET of 2.8 eV and excited-state reduction potential E(FPt*/−) of 0.81 V versus SCE were determined from quenching studies in agreement with values estimated from emission spectra and a thermochemical cycle. The excited-state oxidation potential E(FPt*/+) cannot be determined from electrochemical data since FPt undergoes irreversible oxidation. However, a value of E(FPt*/+) = −1.41 V versus SCE was established from an electron transfer quenching study and thus, a ground state oxidation potential for FPt can be estimated to be 1.30 V versus SCE.
Co-reporter:Vadim I. Adamovich, Steven R. Cordero, Peter I. Djurovich, Arnold Tamayo, Mark E. Thompson, Brian W. D’Andrade, Stephen R. Forrest
Organic Electronics 2003 Volume 4(2–3) pp:77-87
Publication Date(Web):September 2003
DOI:10.1016/j.orgel.2003.08.003
Three strategies for preparing high efficiency OLEDs are demonstrated, which involve the use of hole and electron blocking layers. The first of these strategies involves the use of a cyclometallated iridium compound (bis(2-(4,6-difluorophenyl)pyridyl-N,C2′)iridium(III) picolinate, FIrpic) as a hole-blocking material for green and blue emissive OLEDs. Devices which utilized FIrpic as a combined hole blocking and electron transporting layer gave external quantum efficiencies > 14% (device structure: anode/HTL/EL/FIrpic/cathode, HTL=hole transport layer, EL=emissive layer). When the FIrpic layer of this device was replaced with bathocuproine (BCP), the device efficiency dropped to 12%. A host-guest approach to the formation of a hole blocking layer (HBL) has also been demonstrated. FIrpic was doped into two different wide energy band-gap organic matrix materials (i.e. octaphenyl–cyclooctatetraene, OPCOT, and 1,3,5-tris-phenyl-2-(4-biphneyl)benzene, SC5) forming a mixed HBL. Devices with doped OPCOT gave quantum efficiencies comparable to those with a BCP HBL, while the SC5 based devices gave higher efficiency than their BCP blocked counterparts. When blue electrophosphorescent devices are prepared in a conventional OLED structure (i.e. anode/HTL/EL/HBL/ETL/cathode), excessive HTL emission is often observed, resulting from electron leakage from the doped CBP layer into the HTL. This electron leakage can be eliminated by inserting an electron blocking layer (EBL) between the HTL and luminescent layers. Both fac-tris(1-phenylpyrazolato,N,C2′)iridium(III) (Irppz) and Iridium(III) bis(1-phenylpyrazolato,N,C2′)(2,2,6,6-tetramethyl-3,5-heptanedionato-O,O) have been used as efficient EBLs. The insertion of an EBL leads to both improved color purity and quantum efficiency, relative to devices without EBLs. For example, a white emitting device with the structure ITO/HTL/EL/HBL/ETL/LiF/Al gave an external efficiency of 1.9% and nearly exclusively HTL emission. Addition of a 100 Å Irppz layer between the HTL and EL gave a device with an external quantum efficiency of 3.3% and electroluminescence from only the EL.
Co-reporter:D.E. Loy;B.E. Koene;M.E. Thompson
Advanced Functional Materials 2002 Volume 12(Issue 4) pp:
Publication Date(Web):18 APR 2002
DOI:10.1002/1616-3028(20020418)12:4<245::AID-ADFM245>3.0.CO;2-5
We report a new class of diamine hole-transporting materials (HTMs) based upon a fluorene core. Using a fluorene core, rather than a biphenyl group, leads to enhanced thermal stability, as evidenced by glass-transition (Tg) temperatures as high as 161 °C for N,N′-iminostilbenyl-4,4′-fluorene (ISF). The fluorene-based HTMs have lower ionization potentials (Ip) than their biphenyl analogs, which leads to more efficient injection of holes from the indium tin oxide (ITO) anode, and higher quantum efficiencies. Devices prepared with fluorene-based HTMs were operated under thermal stress. The failure of an organic light-emitting diode (OLED) under thermal stress has a direct correlation with the thermal stability of the HTM that is in contact with the ITO anode. OLEDs based on ISF are stable to over 140 °C.
Co-reporter:Vadim Adamovich, Jason Brooks, Arnold Tamayo, Alex M. Alexander, Peter I. Djurovich, Brian W. D'Andrade, Chihaya Adachi, Stephen R. Forrest and Mark E. Thompson
New Journal of Chemistry 2002 vol. 26(Issue 9) pp:1171-1178
Publication Date(Web):12 Aug 2002
DOI:10.1039/B204301G
Efficient white electrophosphorescence has been achieved with a single emissive dopant. The dopant in these white organic light emitting diodes (WOLEDs) emits simultaneously from monomer and aggregate states, leading to a broad spectrum and high quality white emission. The dopant molecules are based on a series of platinum(II) [2-(4,6-difluorophenyl)pyridinato-N,C2′] β-diketonates. All of the dopant complexes described herein have identical photophysics in dilute solution with structured blue monomer emission (λmax=468, 500, 540 nm). A broad orange aggregate emission (λmax≈580 nm) is also observed, when doped into OLED host materials. The intensity of the orange band increases relative to the blue monomer emission, as the doping level is increased. The ratio of monomer to aggregate emission can be controlled by the doping concentration, the degree of steric bulk on the dopant and by the choice of the host material. A doping concentration for which the monomer and excimer bands are approximately equal gives an emission spectrum closest to standard white illumination sources. WOLEDs have been fabricated with doped CBP and mCP luminescent layers (CBP=N,N′-dicarbazolyl-4,4′-biphenyl, mCP=N,N′-dicarbazolyl-3,5-benzene). The best efficiencies and color stabilities were achieved when an electron/exciton blocking layer (EBL) is inserted into the structure, between the hole transporting layer and doped CBP or mCP layer. The material used for an EBL in these devices was fac-tris(1-phenylpyrazolato-N,C2′)iridium(III). The EBL material effectively prevents electrons and excitons from passing through the emissive layer into the hole transporting NPD layer. CBP based devices gave a peak external quantum efficiency of 3.3±0.3%
(7.3±0.7 lm W−1) at 1 cd m−2, and 2.3±0.2%
(5.2±0.3 lm W−1) at 500 cd m−2. mCP based devices gave a peak external quantum efficiency of 6.4%
(12.2 lm W−1, 17.0 cd A−1), CIE coordinates of 0.36, 0.44 and a CRI of 67 at 1 cd m−2
(CIE=Commission Internationale de l'Eclairage, CRI=color rendering index). The efficiency of the mCP based device drops to 4.3±0.5%
(8.1±0.6 lm W−1, 11.3 cd A−1) at 500 cd m−2, however, the CIE coordinates and CRI remain unchanged.
Co-reporter:Y. You;Y. He;P. E. Burrows;S. R. Forrest;N. A. Petasis;M. E. Thompson
Advanced Materials 2000 Volume 12(Issue 22) pp:
Publication Date(Web):1 DEC 2000
DOI:10.1002/1521-4095(200011)12:22<1678::AID-ADMA1678>3.0.CO;2-H
Co-reporter:Mark E. Thompson, Paul E. Burrows, Stephen R. Forrest
Current Opinion in Solid State and Materials Science 1999 Volume 4(Issue 4) pp:369-372
Publication Date(Web):August 1999
DOI:10.1016/S1359-0286(99)00037-6
Co-reporter:B.K. Thilini Batagoda, Peter I. Djurovich, Stefan Bräse, Mark E. Thompson
Polyhedron (25 September 2016) Volume 116() pp:
Publication Date(Web):25 September 2016
DOI:10.1016/j.poly.2016.04.039
The synthesis and characterization of a series of neutral cyclometalated Pt(II) and Ir(III) heteroleptic complexes containing cyclophane substituted chelates is reported. The complexes have the general formula of (C^N)Pt(O^O) or (C^N)2Ir(O^O) where C^N is a monoanionic cyclophane ligand 2-([2.2]-paracyclophane-4-yl)pyridyl (pCpy) or 1-([2.2]-paracyclophane-4-yl)pyrazolyl (pCpz) and (O^O) is an ancillary ligand; acetylacetonato (acac) or dipivolylmethanato (dpm). The pCpyH and pCpzH ligand precursors were obtained as racemates due to the planar chirality of the cyclophane ring system. Two diastereomers of (pCpz)2Ir(acac), one with C1 symmetry (ΛRS, ΔSR) and the other with C2 symmetry (ΛRR, ΔSS) were isolated, whereas only one C1 diastereomer was obtained for (pCpy)2Ir(acac). Inherent strain in the cyclophane core and transannular interaction between the bridged phenyl rings leads to a 250–400 mV cathodic shift in the oxidation potentials relative to analogous complexes with ppy or ppz ligands. The destabilization of the HOMO leads to a corresponding red shift in the absorption and emission spectra from the complexes with pCpy and pCpz ligands. The Pt and Ir complexes with pCpy ligands are strongly emissive at room temperature, whereas complexes with pCpz ligands are only weakly emissive as they still suffer from temperature-dependent nonradiative deactivation to metal-centered ligand field states.The synthesis and characterization of a series of neutral cyclometalated Pt(II) and Ir(III) heteroleptic complexes containing cyclophane substituted chelates is reported. The complexes have the general formula of (C^N)Pt(O^O) or (C^N)2Ir(O^O) where C^N is a monoanionic cyclophane ligand 2-([2.2]-paracyclophane-4-yl)pyridyl (pCpy) or 1-([2.2]-paracyclophane-4-yl)pyrazolyl (pCpz) and (O^O) is an ancillary ligand; acetylacetonato (acac) or dipivolylmethanato (dpm).
Co-reporter:Hsiao-Fan Chen, Chao Wu, Ming-Cheng Kuo, Mark E. Thompson and Ken-Tsung Wong
Journal of Materials Chemistry A 2012 - vol. 22(Issue 19) pp:
Publication Date(Web):
DOI:10.1039/C2JM30443K
Co-reporter:Shuyang Shi, Lee R. Collins, Mary F. Mahon, Peter I. Djurovich, Mark E. Thompson and Michael K. Whittlesey
Dalton Transactions 2017 - vol. 46(Issue 3) pp:NaN752-752
Publication Date(Web):2016/12/12
DOI:10.1039/C6DT04016K
The photophysical properties of four, two-coordinate, linear diamidocarbene copper(I) complexes, [(DAC)2Cu][BF4] (1), (DAC)CuOSiPh3 (2), (DAC)CuC6F5 (3) and (DAC)Cu(2,4,6-Me3C6H2) (4) (DAC = 1,3-bis(2,4,6-trimethylphenyl)-5,5-dimethyl-4,6-diketopyrimidinyl-2-ylidene) have been investigated. Complex 1 shows a high photoluminescence quantum efficiency (ΦPL) in both the solid state (ΦPL = 0.85) and in CH2Cl2 solution (ΦPL = 0.65). The emission band of 1, both as a crystalline solid and in solution, is narrow (fwhm = 2300 cm−1) relative to the emission bands of 2 (fwhm = 2900 cm−1) and 3 (fwhm = 3700 cm−1). Complexes 2 and 3 are each brightly luminescent in the solid state (ΦPL = 0.62 and 0.18, respectively), but markedly less so in CH2Cl2 solution (ΦPL = 0.03 and <0.01, respectively). Complex 4 is not emissive in either the solid state or in solution. Phosphorescence of 1 in CH2Cl2 solution shows negligible quenching by oxygen in CH2Cl2 solution. This insensitivity to quenching is attributed to the excited state redox potential being insufficient for electron transfer to oxygen.
Co-reporter:Matthew T. Whited, Niral M. Patel, Sean T. Roberts, Kathryn Allen, Peter I. Djurovich, Stephen E. Bradforth and Mark E. Thompson
Chemical Communications 2012 - vol. 48(Issue 2) pp:NaN286-286
Publication Date(Web):2011/11/21
DOI:10.1039/C1CC12260F
We report the synthesis and characterization of symmetric BODIPY dyads where the chromophores are attached at the meso position, using either a phenylene bridge or direct linkage. Both molecules undergo symmetry-breaking intramolecular charge transfer in the excited state, and the directly linked dyad serves as a visible-light-absorbing analogue of 9,9′-bianthryl.
Co-reporter:Vincent S. Barlier, Cody W. Schlenker, Stephanie W. Chin and Mark E. Thompson
Chemical Communications 2011 - vol. 47(Issue 13) pp:NaN3756-3756
Publication Date(Web):2011/02/04
DOI:10.1039/C0CC05164K
Alkyne linked tetracene dimers were synthesized from naphthacenequinone and terminal acetylenes. The silylethynyl tetracene dimers exhibit good solubility, high photostability, and broad absorbance leading to photocurrent generation in an organic photovoltaic device.
Co-reporter:Valentina A. Krylova, Peter I. Djurovich, Matthew T. Whited and Mark E. Thompson
Chemical Communications 2010 - vol. 46(Issue 36) pp:NaN6698-6698
Publication Date(Web):2010/08/17
DOI:10.1039/C0CC01864C
Cationic and neutral monomeric three-coordinate phosphorescent Cu(I) complexes were synthesized and characterized by XRD analysis, electrochemistry and photophysical studies in different environments. DFT calculations have aided the assignment of the electronic structure and excited state behavior of these complexes.
Co-reporter:M. Dolores Perez, Peter I. Djurovich, Azad Hassan, Grace Y. Cheng, Tim J. Stewart, Kristen Aznavour, Robert Bau and Mark E. Thompson
Chemical Communications 2009(Issue 28) pp:NaN4217-4217
Publication Date(Web):2009/06/08
DOI:10.1039/B901510H
An intensely phosphorescent Pt complex in cyclohexane is efficiently quenched by exciplex formation with extremely weak Lewis bases such as toluene and other aromatic compounds.
Co-reporter:Kenneth Hanson, Luke Roskop, Niral Patel, Laurent Griffe, Peter I. Djurovich, Mark S. Gordon and Mark E. Thompson
Dalton Transactions 2012 - vol. 41(Issue 28) pp:NaN8659-8659
Publication Date(Web):2012/06/07
DOI:10.1039/C2DT30354J
A series of twelve platinum(II) complexes of the form (N^N^N)PtX have been synthesized and characterized where N^N^N is 1,3-bis(2-pyridylimino)isoindolate ligands (BPI) or BPI ligands whose aryl moieties are substituted with tert-butyl, nitro, alkoxy, iodo or chloro groups, and X is a chloride, fluoride, cyano, acetate, phenyl or 4-(dimethylamino)phenyl ligand. All complexes display at least one irreversible oxidation and two reversible reduction waves at potentials dependent on the position and the electron donating or withdrawing nature of both X and the substituted N^N^N ligand. Broad room temperature phosphorescence ranging in energy from 594 to 680 nm was observed from the complexes, with quantum efficiencies ranging from 0.01 to 0.05. The efficiency of emission is dictated largely by nonradiative processes since the rate constants for nonradiative deactivation [(1.1–100) × 105 s−1] show greater variation than those for radiative decay [(0.57–4.0) × 04 s−1]. Nonradiative deactivation for compounds with X = Cl follow the energy gap law, i.e. the nonradiative rate constants increase exponentially with decreasing emission energy. Deactivation of the excited state appears to be strongly influenced by a non-planar distortion of the BPI ligand.
Co-reporter:Valentina A. Krylova, Peter I. Djurovich, Brian L. Conley, Ralf Haiges, Matthew T. Whited, Travis J. Williams and Mark E. Thompson
Chemical Communications 2014 - vol. 50(Issue 54) pp:NaN7179-7179
Publication Date(Web):2014/05/23
DOI:10.1039/C4CC02037E
A series of three phosphorescent mononuclear (NHC)–Cu(I) complexes were prepared and characterized. Photophysical properties were found to be largely controlled by the NHC ligand chromophore. Variation of the NHC ligand leads to emission colour tuning over 200 nm range from blue to red, and emission efficiencies of 0.16–0.80 in the solid state.
Co-reporter:Vyacheslav V. Diev, Denise Femia, Qiwen Zhong, Peter I. Djurovich, Ralf Haiges and Mark E. Thompson
Chemical Communications 2016 - vol. 52(Issue 9) pp:NaN1952-1952
Publication Date(Web):2015/12/21
DOI:10.1039/C5CC09128D
A bis-phenalenyl-fused porphyrin has been synthesized by thermal dehydro-aromatization reaction regioselectively as a single syn-isomer. X-ray crystal structure revealed that both phenalenyl units of this porphyrin have close π–π contacts forming continuous network of interacting porphyrin rings. A broad and intense NIR absorption can be attributed to quinoidal character of bis-phenalenyl-fused porphyrin.
Co-reporter:John W. Facendola, Martin Seifrid, Jay Siegel, Peter I. Djurovich and Mark E. Thompson
Dalton Transactions 2015 - vol. 44(Issue 18) pp:NaN8466-8466
Publication Date(Web):2015/02/10
DOI:10.1039/C4DT03541K
Synthesis, structural and characterization data are provided for Pt(II) and Ir(III) complexes cyclometalated with 2-(corannulene)pyridine (corpy), (corpy)Pt(dpm) and (corpy)Ir(ppz)2 (dpm = dipivaloylmethanato, ppz = 1-phenylpyrazolyl). A third compound, (phenpy)Ir(ppz)2 (phenpy = 2-(5-phenanthryl)pyridyl), was also prepared to mimic the steric bulk of (corpy)Ir(ppz)2. X-ray analysis reveals bowl depths of 0.895 Å for (corpy)Pt(dpm) and 0.837 Å in (corpy)Ir(ppz)2. Neither complex displayed bowl-to-bowl stacking in the crystal lattice. A fluxional process for (corpy)Ir(ppz)2 attributed to bowl inversion of corrannulene is observed in solution with a barrier (ΔG‡ = 13 kcal mol−1) and rate (k = 2.5 × 103 s−1) as determined using variable temperature 1H NMR spectroscopy. All of the complexes display red phosphorescence at room temperature with quantum yields of 0.05 in solution and 0.2 in polymethyl methacrylate (PMMA).
Co-reporter:Biwu Ma ; Peter I. Djurovich ; Muhammed Yousufuddin ; Robert Bau
The Journal of Physical Chemistry C () pp:
Publication Date(Web):
DOI:10.1021/jp800594j
The synthesis, characterization, and photophysical properties are reported for a series of (C^N)Pt dyad complexes with cyclometalated ligands (C^N: F = 2-(4′,6′-difluorophenyl)pyridyl, tpy = 2-(4′-methylphenyl)pyridyl, thpy = 2-(2-thienyl)pyridyl, and btp = 2-(2-benzothienyl)pyridyl). The dyads are connected with a bridging ligand, sym-tetraacetylethane (tae), that consists of two 2,4-pentadionate units covalently linked at the 3-position. Stepwise synthesis was applied to obtain both homoleptic and heteroleptic dyads that have been characterized by 1H NMR and elemental analysis. X-ray crystallographic analysis shows a near orthogonal orientation of the two diketonate moieties in the di-tpyPt (84°) and FPt-thpyPt (89°). An investigation of the photophysical properties of the dyads has been carried out. For homodyads, the emission characteristics are governed by the nature of the cyclometalating ligand, allowing the emission to be tuned throughout the visible spectrum. The di-FPt and di-tpyPt dyads show a modest decrease in luminescent lifetimes compared to their mononuclear analogs. The di-FPt complex undergoes efficient self-quenching by an excimer that is presumed to have only π−π interactions. The heterodyads exhibit efficient intramolecular triplet energy transfer leading to the luminescence almost exclusively from the lower-energy moiety. The energy transfer is presumed to be mediated through the bridging tae ligand by a Dexter-like mechanism involving a combination of hopping and superexchange processes.
.jpg)
![Palladium, [6,13,20,27-tetraphenyl-29H,31H-tetrabenzo[b,g,l,q]porphinato(2-)-κN29,κN30,κN31,κN32]-, (SP-4-1)-](/data/chemimg/134800/119654-64-7.png)
![Palladium, [6,13,20,27-tetraphenyl-29H,31H-tetrabenzo[b,g,l,q]porphinato(2-)-κN29,κN30,κN31,κN32]-, (SP-4-1)-](/data/chemimg/134800/119654-64-7_b.png)


![Bis(2-methyldibenzo[f,h]quinoxaline) (acetylacetonate) iridium (III)](/data/chemimg/108400/536755-34-7.png)
![Bis(2-methyldibenzo[f,h]quinoxaline) (acetylacetonate) iridium (III)](/data/chemimg/108400/536755-34-7_b.png)






![6H-[2]Benzopyrano[4,3-b]pyridin-6-one](http://img.cochemist.com/ccimg/39900/39883-80-2.png)
![6H-[2]Benzopyrano[4,3-b]pyridin-6-one](http://img.cochemist.com/ccimg/39900/39883-80-2_b.png)

![Bisbenz[5,6]indeno[1,2,3-cd:1',2',3'-lm]perylene, 5,10,15,20-tetraphenyl-](/data/chemimg/116100/175606-05-0.png)
![Bisbenz[5,6]indeno[1,2,3-cd:1',2',3'-lm]perylene, 5,10,15,20-tetraphenyl-](/data/chemimg/116100/175606-05-0_b.png)

















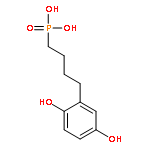
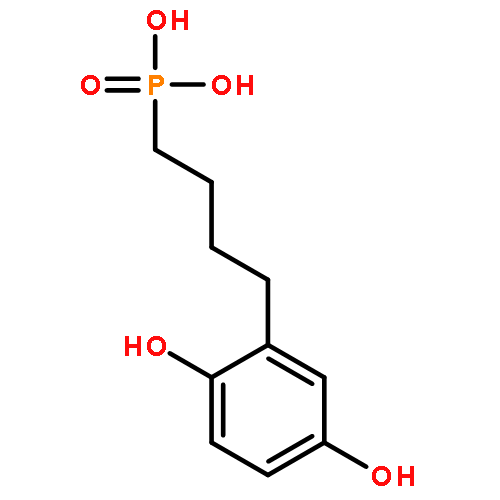






![1,3-Benzenediol, 5-[bis(2-methylpropyl)amino]-](/data/chemimg/68500/481634-76-8.png)
![1,3-Benzenediol, 5-[bis(2-methylpropyl)amino]-](/data/chemimg/68500/481634-76-8_b.png)
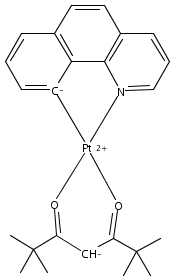






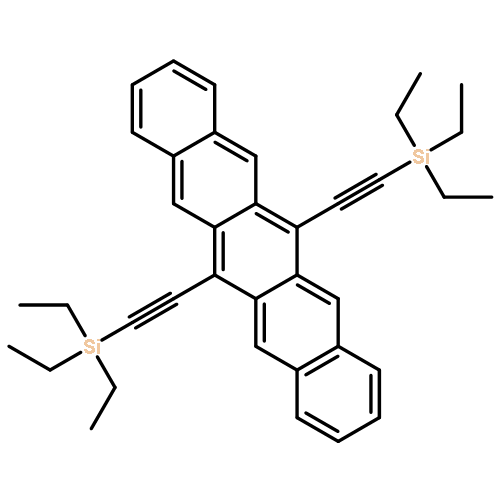


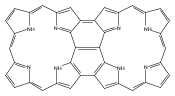
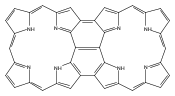


![2-[5-(trifluoromethyl)-1h-1,2,4-triazol-3-yl]pyridine](http://img.cochemist.com/ccimg/219600/219508-27-7.png)
![2-[5-(trifluoromethyl)-1h-1,2,4-triazol-3-yl]pyridine](http://img.cochemist.com/ccimg/219600/219508-27-7_b.png)

![Poly[[2-[(3,7-dimethyloctyl)oxy]-5-methoxy-1,4-phenylene]-1,2-ethenediyl]](http://img.cochemist.com/ccimg/177800/177716-59-5.png)
![Poly[[2-[(3,7-dimethyloctyl)oxy]-5-methoxy-1,4-phenylene]-1,2-ethenediyl]](http://img.cochemist.com/ccimg/177800/177716-59-5_b.png)
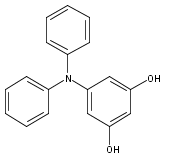


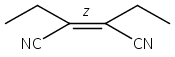


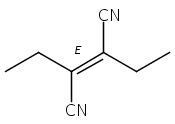
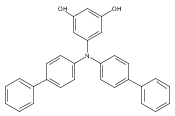









![Dichlorotetrakis[2-(2-Pyridyl)Phenyl]Diiridium(Iii)](http://img.cochemist.com/ccimg/92300/92220-65-0.png)
![Dichlorotetrakis[2-(2-Pyridyl)Phenyl]Diiridium(Iii)](http://img.cochemist.com/ccimg/92300/92220-65-0_b.png)
![[1,1'-Biphenyl]-2-amine, N-[1,1'-biphenyl]-2-yl-](http://img.cochemist.com/ccimg/92000/91923-32-9.png)
![[1,1'-Biphenyl]-2-amine, N-[1,1'-biphenyl]-2-yl-](http://img.cochemist.com/ccimg/92000/91923-32-9_b.png)

![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(2,6-dimethylphenyl)-](/data/chemimg/673600/76372-76-4.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(2,6-dimethylphenyl)-](/data/chemimg/673600/76372-76-4_b.png)

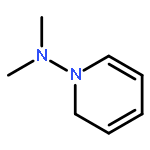






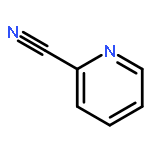
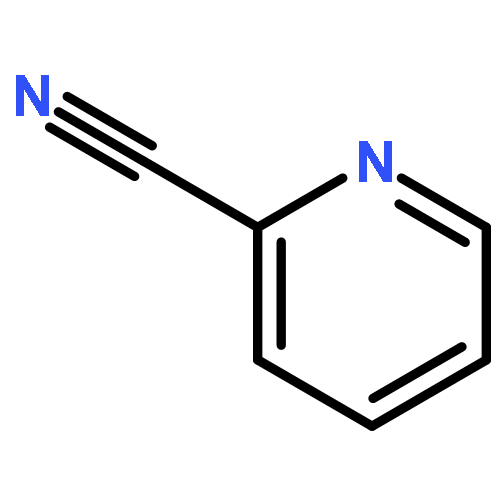


![Zinc, [5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/478800/14074-80-7.png)
![Zinc, [5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/478800/14074-80-7_b.png)




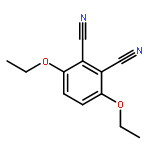
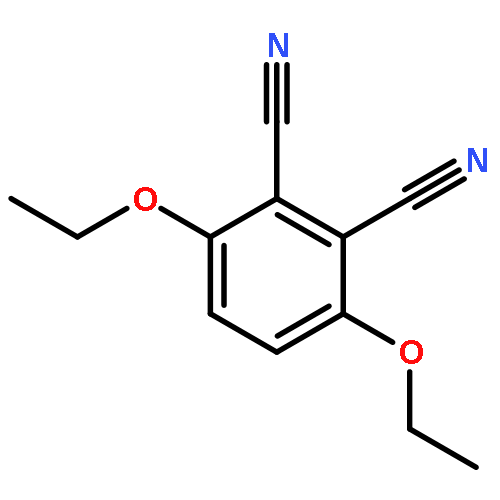


![2-[5-(TRIFLUOROMETHYL)-1H-PYRAZOL-3-YL]PYRIDINE](http://img.cochemist.com/ccimg/4000/3974-71-8.png)
![2-[5-(TRIFLUOROMETHYL)-1H-PYRAZOL-3-YL]PYRIDINE](http://img.cochemist.com/ccimg/4000/3974-71-8_b.png)
![Dibenzo[f,h]quinoline](http://img.cochemist.com/ccimg/300/217-65-2.png)
![Dibenzo[f,h]quinoline](http://img.cochemist.com/ccimg/300/217-65-2_b.png)










![Platinum, [6,13,20,27-tetraphenyl-29H,31H-tetrabenzo[b,g,l,q]porphinato(2-)-κN29,κN30,κN31,κN32]-, (SP-4-1)-](/data/chemimg/107100/166174-05-6.png)
![Platinum, [6,13,20,27-tetraphenyl-29H,31H-tetrabenzo[b,g,l,q]porphinato(2-)-κN29,κN30,κN31,κN32]-, (SP-4-1)-](/data/chemimg/107100/166174-05-6_b.png)




![Poly[(chloro-1,4-phenylene)-1,2-ethanediyl]](http://img.cochemist.com/ccimg/9100/9052-19-1.png)
![Poly[(chloro-1,4-phenylene)-1,2-ethanediyl]](http://img.cochemist.com/ccimg/9100/9052-19-1_b.png)