Co-reporter:Mathias Schäfer, Katrin Peckelsen, Mathias Paul, Jonathan Martens, Jos Oomens, Giel Berden, Albrecht Berkessel, and Anthony J. H. M. Meijer
Journal of the American Chemical Society April 26, 2017 Volume 139(Issue 16) pp:5779-5779
Publication Date(Web):March 10, 2017
DOI:10.1021/jacs.6b10348
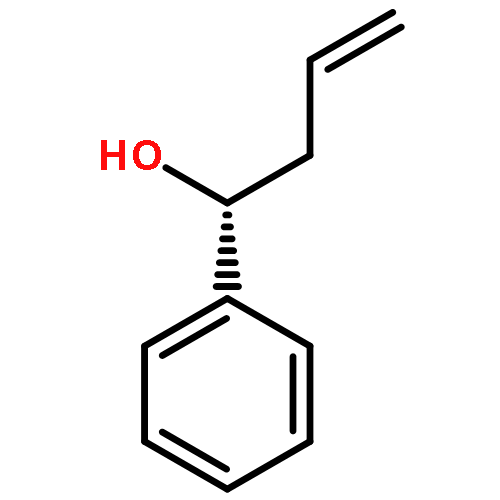
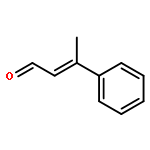
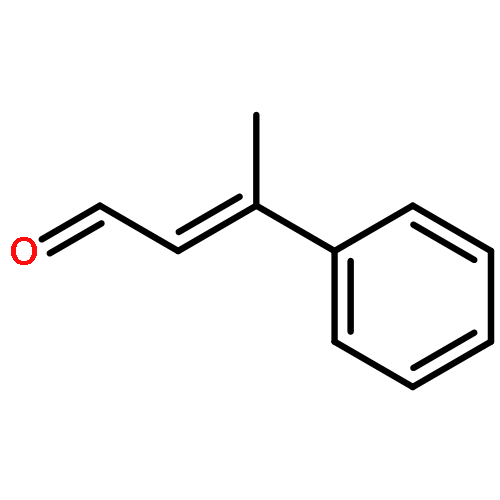
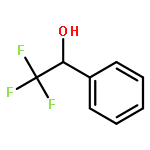
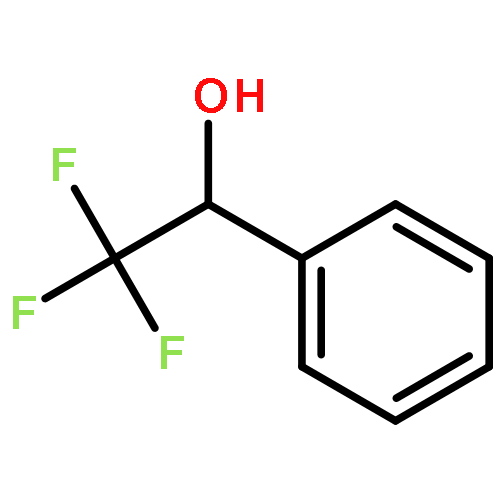
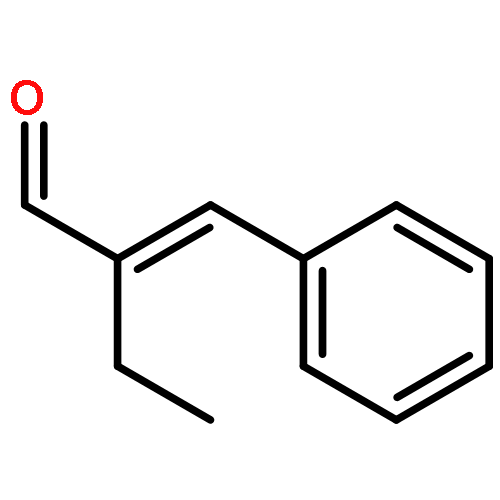
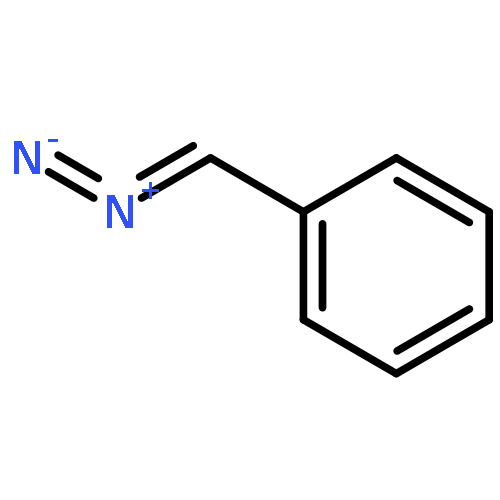
While hydrogen tunneling at elevated temperatures has, for instance, often been postulated in biochemical processes, spectroscopic proof is thus far limited to cryogenic conditions, under which thermal reactivity is negligible. We report spectroscopic evidence for H-tunneling in the gas phase at temperatures around 320–350 K observed in the isomerization reaction of a hydroxycarbene into an aldehyde. The charge-tagged carbene was generated in situ in a tandem mass spectrometer by decarboxylation of oxo[4-(trimethylammonio)phenyl]acetic acid upon collision induced dissociation. All ion structures involved are characterized by infrared ion spectroscopy and quantum chemical calculations. The charge-tagged phenylhydroxycarbene undergoes a 1,2-H-shift to the corresponding aldehyde with an half-life of about 10 s, evidenced by isomer-selective two-color (IR-IR) spectroscopy. In contrast, the deuterated (OD) carbene analogue showed much reduced 1,2-D-shift reactivity with an estimated half-life of at least 200 s under the experimental conditions, and provides clear evidence for hydrogen atom tunneling in the H-isotopologue. This is the first spectroscopic confirmation of hydrogen atom tunneling governing 1,2-H-shift reactions at noncryogenic temperatures, which is of broad significance for a range of (bio)chemical processes, including enzymatic transformations and organocatalysis.
Co-reporter:Veera Reddy Yatham; Wacharee Harnying; Darius Kootz; Jörg-M. Neudörfl; Nils E. Schlörer
Journal of the American Chemical Society 2016 Volume 138(Issue 8) pp:2670-2677
Publication Date(Web):January 21, 2016
DOI:10.1021/jacs.5b11796
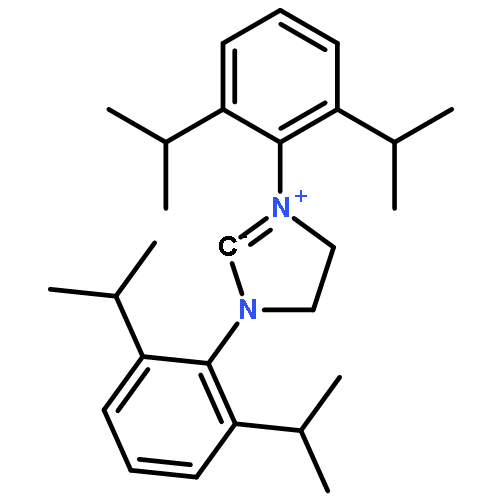
As reported by Scheidt and Bode in 2005, sterically nonencumbered α,β-enals are readily converted to saturated esters in the presence of alcohols and N-heterocyclic carbene catalysts, e.g., benzimidazolylidenes or triazolylidenes. However, substituents at the α- or β-position of the α,β-enal substrate are typically not tolerated, thus severely limiting the substrate spectrum. On the basis of our earlier mechanistic studies, a set of N-Mes- or N-Dipp-substituted 1,2,4-triazolium salts were synthesized and evaluated as (pre)catalysts in the redox esterification of various α- or β-substituted enals. In particular the 1,4-bis-Mes/Dipp-1,2,4-triazolylidenes overcome the above limitations and efficiently catalyze the redox esterification of a whole series of α/β-substituted enals hitherto not amenable to NHC-catalyzed transformations. The synthetic value of 1,4-bis-Mes/Dipp-1,2,4-triazolylidenes is further demonstrated by the one-step bicyclization of 10-oxocitral to (racemic) nepetalactone in diastereomerically pure form.
Co-reporter:Mathias Paul; Martin Breugst; Jörg-Martin Neudörfl; Raghavan B. Sunoj
Journal of the American Chemical Society 2016 Volume 138(Issue 15) pp:5044-5051
Publication Date(Web):February 14, 2016
DOI:10.1021/jacs.5b13236
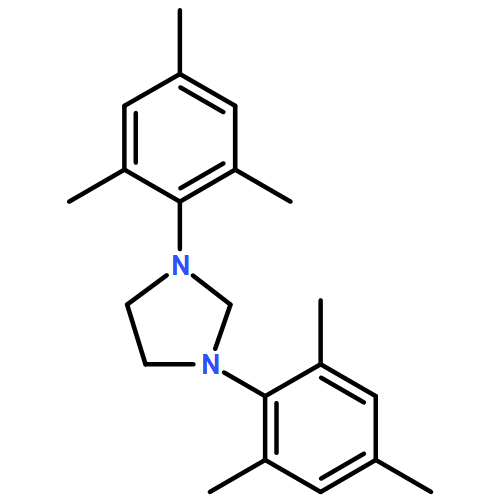
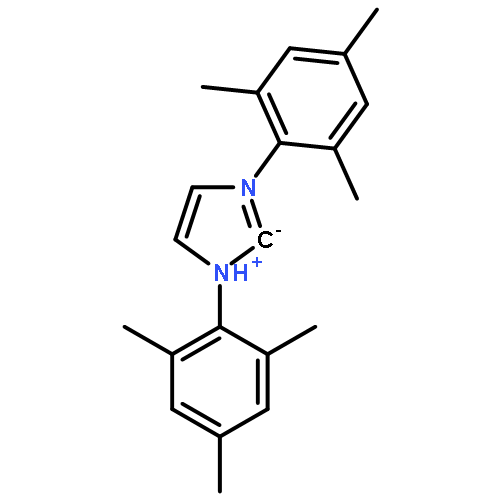
Breslow intermediates, first postulated in 1958, are pivotal intermediates in carbene-catalyzed umpolung. Attempts to isolate and characterize these fleeting amino enol species first met with success in 2012 when we found that saturated bis-Dipp/Mes imidazolidinylidenes readily form isolable, though reactive diamino enols with aldehydes and enals. In contrast, triazolylidenes, upon stoichiometric reaction with aldehydes, gave exclusively the keto tautomer, and no isolable enol. Herein, we present the synthesis of the “missing” keto tautomers of imidazolidinylidene-derived diamino enols, and computational thermodynamic data for 15 enol–ketone pairs derived from various carbenes/aldehydes. Electron-withdrawing substituents on the aldehyde favor enol formation, the same holds for N,N′-Dipp [2,6-di(2-propyl)phenyl] and N,N′-Mes [2,4,6-trimethylphenyl] substitution on the carbene component. The latter effect rests on stabilization of the diamino enol tautomer by Dipp substitution, and could be attributed to dispersive interaction of the 2-propyl groups with the enol moiety. For three enol–ketone pairs, equilibration of the thermodynamically disfavored tautomer was attempted with acids and bases but could not be effected, indicating kinetic inhibition of proton transfer.
Co-reporter:Eleonora Gianolio;Resmi Mohan
Advanced Synthesis & Catalysis 2016 Volume 358( Issue 1) pp:30-33
Publication Date(Web):
DOI:10.1002/adsc.201500820
Co-reporter:Veera Reddy Yatham, Jörg-M. Neudörfl, Nils E. Schlörer and Albrecht Berkessel
Chemical Science 2015 vol. 6(Issue 7) pp:3706-3711
Publication Date(Web):30 Apr 2015
DOI:10.1039/C5SC01027F
Since their discovery by Bode and Glorius in 2004, N-heterocyclic carbene catalyzed conjugate umpolung reactions of α,β-enals have been postulated to involve the formation of diamino dienols (“homoenolates”) and/or azolium enolates (“enolates”), typically followed by addition to electrophiles, e.g. Michael-acceptors. In this article, we provide evidence, for the first time, for the postulated individual and specific reactivity patterns of diamino dienols (γ-C–C-bond formation) vs. azolium enolates (β-C–C-bond formation). Our study is based on the pre-formation of well defined diamino dienols and azolium enolates, and the in situ NMR monitoring of their reactivities towards enone electrophiles. Additionally, reaction intermediates were isolated and characterized, inter alia by X-ray crystallography.
Co-reporter:Oue-artorn Rajachan, Mathias Paul, Veera Reddy Yatham, Jörg-M. Neudörfl, Kwanjai Kanokmedhakul, Somdej Kanokmedhakul, Albrecht Berkessel
Tetrahedron Letters 2015 Volume 56(Issue 47) pp:6537-6540
Publication Date(Web):25 November 2015
DOI:10.1016/j.tetlet.2015.09.104
The tail-to-tail oligomerization of N,N-dimethylacrylamide catalyzed by N-heterocyclic carbenes (NHCs) was investigated, giving the dimerization and trimerization products in a moderate combined yield. Reaction intermediates involved in the new oligomerization have been observed by NMR and ESI-MS. We showed the first NHC-catalyzed cross coupling of methyl methacrylate and N,N-dimethylacrylamide, and the reaction of benzaldehyde/benzoin with N,N-dimethylacrylamide.
Co-reporter:Marcel Heidlindemann, Matthias Hammel, Ulf Scheffler, Rainer Mahrwald, Werner Hummel, Albrecht Berkessel, and Harald Gröger
The Journal of Organic Chemistry 2015 Volume 80(Issue 7) pp:3387-3396
Publication Date(Web):February 24, 2015
DOI:10.1021/jo502667x
The combination of an asymmetric organocatalytic aldol reaction with a subsequent biotransformation toward a “one-pot-like” process for the synthesis of (R)-pantolactone, which to date is industrially produced by a resolution process, is demonstrated. This process consists of an initial aldol reaction catalyzed by readily available l-histidine followed by biotransformation of the aldol adduct by an alcohol dehydrogenase without the need for intermediate isolation. Employing the industrially attractive starting material isobutanal, a chemoenzymatic three-step process without intermediate purification is established allowing the synthesis of (R)-pantolactone in an overall yield of 55% (three steps) and high enantiomeric excess of 95%.
Co-reporter:Dr. Qifang Wang;Dr. Jörg-M. Neudörfl;Dr. Albrecht Berkessel
Chemistry - A European Journal 2015 Volume 21( Issue 1) pp:247-254
Publication Date(Web):
DOI:10.1002/chem.201404639
Abstract
Chiral Ti salalen complexes catalyze the asymmetric epoxidation of terminal non-conjugated olefins with hydrogen peroxide. Modular ligands based on cis-1,2-diamino-cyclohexane (cis-DACH) were developed, giving high yields and enantiomeric excesses (ee, up to 96 %) at catalyst loadings as low as 0.1–0.5 mol %, and even under solvent-free conditions.
Co-reporter:Dr. Wacharee Harnying;Dr. Albrecht Berkessel
Chemistry - A European Journal 2015 Volume 21( Issue 16) pp:6057-6061
Publication Date(Web):
DOI:10.1002/chem.201500024
Abstract
We report an efficient and practical protocol for the Cr/Ni-catalyzed vinylation of aldehydes, based on the use of Mn/Cr alloy (ca. 10 % Cr) and TMSCl. No additional Cr salts need to be added. In the presence of NiCl2 (0.3 mol %) and a bis(ketimino)-2,2′-bipyridine as N4-chelating ligand (1 mol %), the vinylations proceed smoothly at room temperature. The presence of catalytic amounts of MeOH and LiOAc as additives was found to further promote the efficiency of the catalytic system, even in the absence of the ligand. Detailed reaction monitoring revealed that LiOAc accelerates the product alcohol silylation, thus increasing the turnover rate.
Co-reporter:Somnath Das;Daniel Pekel;Dr. Jörg-M. Neudörfl;Dr. Albrecht Berkessel
Angewandte Chemie International Edition 2015 Volume 54( Issue 42) pp:12479-12483
Publication Date(Web):
DOI:10.1002/anie.201503156
Abstract
A new organocatalytic glycosylation method based on electron-deficient pyridinium salts is reported. At ambient temperature and catalyst loadings as low as 1 mol %, 2-deoxyglycosides were formed from benzyl- and silyl-protected glycals and primary or secondary glycosyl acceptors, with excellent yields and anomeric selectivity. Mechanistic investigations point to alcohol–pyridinium conjugates (1,2-addition products) as key intermediates in the catalytic cycle.
Co-reporter:Somnath Das;Daniel Pekel;Dr. Jörg-M. Neudörfl;Dr. Albrecht Berkessel
Angewandte Chemie 2015 Volume 127( Issue 42) pp:12656-12660
Publication Date(Web):
DOI:10.1002/ange.201503156
Abstract
Es wird eine neuartige organokatalytische Glycosylierung mittels elektronenarmer Pyridiniumsalze vorgestellt. 2-Deoxyglucoside konnten bei Raumtemperatur und geringen Katalysatormengen (bis zu 1 Mol-%) aus benzyl- und silylgeschützten Glycalen und primären oder sekundären Glycosylakzeptoren mit hohen Ausbeuten und Anomerenverhältnissen erhalten werden. 1,2-Alkohol-Pyridinium-Addukte fungieren vermutlich als Schlüsselintermediate des Katalysezyklus.
Co-reporter:Albrecht Berkessel;Silvia Elfert
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 2-3) pp:571-578
Publication Date(Web):
DOI:10.1002/adsc.201300801
Co-reporter:Stefan Hilken;Florian Kaletta;Angela Heinsch;Jörg-M. Neudörfl
European Journal of Organic Chemistry 2014 Volume 2014( Issue 11) pp:2231-2241
Publication Date(Web):
DOI:10.1002/ejoc.201301784
Abstract
Molybdenum cofactor (Moco) deficiency is a lethal hereditary metabolic disease. A recently developed therapy requires continuous intravenous supplementation of the biosynthetic Moco precursor cyclic pyranopterin monophosphate (cPMP). The limited stability of the latter natural product, mostly due to oxidative degradation, is problematic for oral administration. Therefore, the synthesis of more stable cPMP analogues is of great interest. In this context and for the first time, the synthesis of a cPMP analogue, in which the oxidation-labile reduced pterin unit is replaced by a pyrazine moiety, was achieved starting from the chiral pool materials D-galactose or D-arabitol. Our synthesis, 13 steps in total, includes the following key transformations: i) pyrazine lithiation, followed by acylation; ii) closure of the pyrane ring by nucleophilic aromatic substitution; and iii) introduction of phosphate.
Co-reporter:Dr. Albrecht Berkessel;Somnath Das;Daniel Pekel;Dr. Jörg-M. Neudörfl
Angewandte Chemie 2014 Volume 126( Issue 43) pp:11846-11850
Publication Date(Web):
DOI:10.1002/ange.201403778
Abstract
Wir stellen ein neues Aktivierungskonzept in der Organokatalyse vor: Halogenidbindung durch Coulomb-Wechselwirkung. Dieser Katalysemodus wurde mithilfe von 3,5-Dicarbomethoxypyridiniumionen erreicht, die einen zusätzlichen elektronenziehenden Substituenten am Stickstoffatom tragen, z. B. Pentafluorbenzyl oder Cyanmethyl. Für das N-Pentafluorbenzylderivat wird die Coulomb-Wechselwirkung – im Festkörper – durch eine Anion-π-Wechselwirkung mit dem Perfluorphenylring ergänzt. Bromid und Chlorid werden in einer 1:1-Stöchiometrie durch die Kationen gebunden. Die katalytische C-C-Verknüpfung zwischen 1-Chlorisochroman (und verwandten Elektrophilen) mit Silylketenacetalen läuft bei −78 °C und mit geringen Katalysatormengen (2 Mol-%) ab.
Co-reporter:Dr. Albrecht Berkessel;Somnath Das;Daniel Pekel;Dr. Jörg-M. Neudörfl
Angewandte Chemie International Edition 2014 Volume 53( Issue 43) pp:11660-11664
Publication Date(Web):
DOI:10.1002/anie.201403778
Abstract
A new activation principle in organocatalysis is presented: halide binding through Coulombic interactions. This mode of catalysis was realized by using 3,5-di(carbomethoxy)pyridinium ions that carry an additional electron-withdrawing substituent on the nitrogen atom, for example, pentafluorobenzyl or cyanomethyl. For the N-pentafluorobenzyl derivative, Coulombic interaction with the pyridinium moiety is complemented in the solid state by anion–π interactions with the perfluorophenyl ring. Bromide and chloride are bound by these cations in a 1:1 stoichiometry. Catalysis of the CC coupling between 1-chloroisochroman (and related electrophiles) with silyl ketene acetals occurs at −78 °C and at low catalyst loading (2 mol %).
Co-reporter:Dr. Albrecht Berkessel;Jan Krämer;Dr. Florian Mummy;Dr. Jörg-M. Neudörfl;Dr. Rainer Haag
Angewandte Chemie 2013 Volume 125( Issue 2) pp:767-771
Publication Date(Web):
DOI:10.1002/ange.201206003
Co-reporter:Dr. Albrecht Berkessel;Jan Krämer;Dr. Florian Mummy;Dr. Jörg-M. Neudörfl;Dr. Rainer Haag
Angewandte Chemie 2013 Volume 125( Issue 2) pp:
Publication Date(Web):
DOI:10.1002/ange.201209130
Co-reporter:Dr. Albrecht Berkessel;Dr. Thomas Günther;Qifang Wang;Dr. Jörg-M. Neudörfl
Angewandte Chemie 2013 Volume 125( Issue 32) pp:8625-8629
Publication Date(Web):
DOI:10.1002/ange.201210198
Co-reporter:Dr. Albrecht Berkessel;Veera Reddy Yatham;Silvia Elfert ;Dr. Jörg-M. Neudörfl
Angewandte Chemie 2013 Volume 125( Issue 42) pp:11364-11369
Publication Date(Web):
DOI:10.1002/ange.201303107
Co-reporter:Dr. Albrecht Berkessel;Jan Krämer;Dr. Florian Mummy;Dr. Jörg-M. Neudörfl;Dr. Rainer Haag
Angewandte Chemie International Edition 2013 Volume 52( Issue 2) pp:739-743
Publication Date(Web):
DOI:10.1002/anie.201206003
Co-reporter:Dr. Albrecht Berkessel;Jan Krämer;Dr. Florian Mummy;Dr. Jörg-M. Neudörfl;Dr. Rainer Haag
Angewandte Chemie International Edition 2013 Volume 52( Issue 2) pp:
Publication Date(Web):
DOI:10.1002/anie.201209130
Co-reporter:Dr. Albrecht Berkessel;Dr. Thomas Günther;Qifang Wang;Dr. Jörg-M. Neudörfl
Angewandte Chemie International Edition 2013 Volume 52( Issue 32) pp:8467-8471
Publication Date(Web):
DOI:10.1002/anie.201210198
Co-reporter:Dr. Albrecht Berkessel;Veera Reddy Yatham;Silvia Elfert ;Dr. Jörg-M. Neudörfl
Angewandte Chemie International Edition 2013 Volume 52( Issue 42) pp:11158-11162
Publication Date(Web):
DOI:10.1002/anie.201303107
Co-reporter:Nongnaphat Duangdee ; Wacharee Harnying ; Giuseppe Rulli ; Jörg-M. Neudörfl ; Harald Gröger
Journal of the American Chemical Society 2012 Volume 134(Issue 27) pp:11196-11205
Publication Date(Web):May 25, 2012
DOI:10.1021/ja302511t
Aldol reactions with trifluoroacetophenones as acceptors yield chiral α-aryl, α-trifluoromethyl tertiary alcohols, valuable intermediates in organic synthesis. Of the various organocatalysts examined, Singh’s catalyst [(2S)-N-[(1S)-1-hydroxydiphenylmethyl-3-methylbutyl]-2-pyrrolidinecarboxamide] was found to efficiently promote this organocatalytic transformation in a highly enantioselective manner. Detailed reaction monitoring (19F-NMR, HPLC) showed that, up to full conversion, the catalytic transformation proceeds under kinetic control and affords up to 95% ee in a time-independent manner. At longer reaction times, the catalyst effects racemization. For the product aldols, even weak acids (such as ammonium chloride) or protic solvents, can induce racemization, too. Thus, acid-free workup, at carefully chosen reaction time, is crucial for the isolation of the aldols in high (and stable) enantiomeric purity. As evidenced by 19F-NMR, X-ray structural analysis, and independent synthesis of a stable intramolecular variant, Singh’s catalyst reversibly forms a catalytically inactive (“parasitic”) intermediate, namely a N,O-hemiacetal with trifluoroacetophenones. X-ray crystallography also allowed the determination of the product aldols’ absolute configuration (S).
Co-reporter:Jezabel Praz;Laure Guénée;Sarwar Aziz;Alexre Alexakis
Advanced Synthesis & Catalysis 2012 Volume 354( Issue 9) pp:1780-1790
Publication Date(Web):
DOI:10.1002/adsc.201101016
Abstract
Novel endo,endo-2,5-diaminonorbonane-derived tertiary C2-symmetrical diamines were synthesized via the one-pot reductive amination of enantiomerically pure norbornane-2,5-dione. These ligands were applied to various catalytic reactions such as asymmetric deprotonation, asymmetric bromine-lithium exchange, and enantioselective addition of aryl- and allkylithium reagents to aromatic aldimines.
Co-reporter:Albrecht Berkessel, Wacharee Harnying, Nongnaphat Duangdee, Jörg-M. Neudörfl, and Harald Gröger
Organic Process Research & Development 2012 Volume 16(Issue 1) pp:123-128
Publication Date(Web):December 1, 2011
DOI:10.1021/op200283q
A practical one-pot procedure for the preparation of Singh’s catalyst from either l-/d-proline or Boc-proline is described. The coupling partner, a chiral amino alcohol, can be prepared and used directly without purification from the corresponding amino acid ester. Moreover, a procedure for tert-butoxycarbonyl (Boc) group removal using concentrated HCl in MeOH–DCM was developed and utilized for the multigram-scale synthesis of Singh’s catalyst.
Co-reporter:Dr. Alexra Weyer;Dr. Dolores Díaz;Dr. Alexer Nierth;Dr. Nils E. Schlörer;Dr. Albrecht Berkessel
ChemCatChem 2012 Volume 4( Issue 3) pp:
Publication Date(Web):
DOI:10.1002/cctc.201100243
Co-reporter:Dr. Alexra Weyer;Dr. Dolores Díaz;Dr. Alexer Nierth;Dr. Nils E. Schlörer;Dr. Albrecht Berkessel
ChemCatChem 2012 Volume 4( Issue 3) pp:
Publication Date(Web):
DOI:10.1002/cctc.201290004
Co-reporter:Albrecht Berkessel, Sonja S. Vormittag, Nils E. Schlörer, and Jörg-M. Neudörfl
The Journal of Organic Chemistry 2012 Volume 77(Issue 22) pp:10145-10157
Publication Date(Web):October 2, 2012
DOI:10.1021/jo301609g
The synthesis of six enantiopure α,α,α′,α′-tetrakis(perfluoroalkyl/aryl)-2,2′-dimethyl-1,3-dioxolane-4,5-dimethanols (TEFDDOLs), by addition of perfluorinated organolithium reagents or Ruppert’s reagent (TMS-CF3) to isopropylidene tartaric dichloride, is reported. X-ray crystal structures of the TEFDDOLs alone or in complexes with H-bond acceptors such as water and DABCO revealed that this new class of highly fluorinated chiral 1,4-diols forms distinct intra- and intermolecular H-bond patterns. Intramolecular OH–OH bonding accounts for the relatively high acidity of the perfluoroalkyl TEFDDOLs (pKa in DMSO: tetrakis-CF3, 5.7; tetrakis-C2F5, 2.4). For the tetrakis(perfluorophenyl) TEFDDOL, a quite unusual “pseudo-anti” conformation of the diol, with no intramolecular (and no intermolecular) OH–OH bonds, was found both in the crystal and in solution (DOSY and NOESY NMR). The latter conformation results from a total of four intramolecular OH–Faryl hydrogen bonds overriding OH–OH bonding. Due to their H-bonding properties, the TEFDDOLs are promising new building blocks for supramolecular and potentially catalytic applications.
Co-reporter:Dr. Albrecht Berkessel;Silvia Elfert;V. Reddy Yatham;Dr. Jörg-M. Neudörfl;Dr. Nils E. Schlörer;Dr. J. Henrique Teles
Angewandte Chemie International Edition 2012 Volume 51( Issue 49) pp:12370-12374
Publication Date(Web):
DOI:10.1002/anie.201205878
Co-reporter:Dr. Albrecht Berkessel;Silvia Elfert;V. Reddy Yatham;Dr. Jörg-M. Neudörfl;Dr. Nils E. Schlörer;Dr. J. Henrique Teles
Angewandte Chemie 2012 Volume 124( Issue 49) pp:12537-12541
Publication Date(Web):
DOI:10.1002/ange.201205878
Co-reporter:Dr. Albrecht Berkessel;Silvia Elfert;V. Reddy Yatham;Dr. Jörg-M. Neudörfl;Dr. Nils E. Schlörer;Dr. J. Henrique Teles
Angewandte Chemie 2012 Volume 124( Issue 49) pp:
Publication Date(Web):
DOI:10.1002/ange.201208586
Co-reporter:Dr. Albrecht Berkessel;Silvia Elfert;V. Reddy Yatham;Dr. Jörg-M. Neudörfl;Dr. Nils E. Schlörer;Dr. J. Henrique Teles
Angewandte Chemie International Edition 2012 Volume 51( Issue 49) pp:
Publication Date(Web):
DOI:10.1002/anie.201208586
Co-reporter:Giuseppe Rulli;Nongnaphat Duangdee;Katrin Baer;Dr. Werner Hummel;Dr. Albrecht Berkessel;Dr. Harald Gröger
Angewandte Chemie 2011 Volume 123( Issue 34) pp:8092-8095
Publication Date(Web):
DOI:10.1002/ange.201008042
Co-reporter:Giuseppe Rulli;Nongnaphat Duangdee;Katrin Baer;Dr. Werner Hummel;Dr. Albrecht Berkessel;Dr. Harald Gröger
Angewandte Chemie International Edition 2011 Volume 50( Issue 34) pp:7944-7947
Publication Date(Web):
DOI:10.1002/anie.201008042
Co-reporter:Dr. Albrecht Berkessel;Dr. Ilona Jurkiewicz ;Resmi Mohan
ChemCatChem 2011 Volume 3( Issue 2) pp:319-330
Publication Date(Web):
DOI:10.1002/cctc.201000343
Abstract
In the presence of Candida antarctica lipase B, the alcoholytic ring opening of 5-substituted oxazinones proceeds as kinetic resolution (KR) and affords N-acyl β2-amino acid esters in up to 96 % ee, the remaining oxazinones were obtained in up to 99 % ee. In the presence of triethylamine as racemization catalyst, the enzyme-catalyzed alcoholytic oxazinone opening proceeds as dynamic kinetic resolution (DKR), affording quantitative yields of N-protected β2-amino acid esters in high enantiomeric purity (up to 96 % ee). N,C-double deprotection to afford the β2-amino acid can be effected without loss of enantiopurity.
Co-reporter:Adrian vonderHöh ;Dr. Albrecht Berkessel
ChemCatChem 2011 Volume 3( Issue 5) pp:861-867
Publication Date(Web):
DOI:10.1002/cctc.201000428
Abstract
A bifunctional iron-based catalyst (7), a compound related to the [Fe]- or Hmd-hydrogenase, was studied by using experimental and computational methods. Catalyst 7 bears a protic hydrogen atom at the ligand and a hydridic hydrogen atom at the iron atom, which originate from the heterolytic uptake of H2, and can be transferred to unsaturated molecules (i.e. carbonyl compounds and imines). Our DFT calculations indicate that a non-classical dihydrogen-iron complex exists, and that the hydrogen uptake is an exergonic process. The hydrogenation of carbonyl compounds proceeds through a synchronous, concerted outer-sphere mechanism, whereas the hydrogenation of the imine 18 is a two-step process. In the first step, the imine is protonated to afford an iminium ion, to which the hydride is then transferred in a second reaction step. Energy hypersurfaces were calculated by using a solvent (toluene) continuum model.
Co-reporter:Dr. Wacharee Harnying;André Kaiser;Dr. Axel Klein;Dr. Albrecht Berkessel
Chemistry - A European Journal 2011 Volume 17( Issue 17) pp:4765-4773
Publication Date(Web):
DOI:10.1002/chem.201003366
Abstract
The roles of nickel and chromium catalysts in the coupling reaction of vinyl halides and aldehydes, the so-called Nozaki–Hiyama–Kishi (NHK) reaction, have been studied by UV/Vis spectroscopy, electrochemical, and spectroelectrochemical methods. Electrochemical studies revealed that nickel plays the central role in activating the vinyl halide by reductive cleavage, to form a rapidly decomposing vinyl–Ni species. The latter can, however, be stabilized in the presence of the Cr complex. The redox behavior of the Ni complexes in the presence of vinyl halide demonstrated that the vinyl halide activation results from interaction with a one-electron reduced nickel species [formally NiI], not necessarily with a Ni0 species. It was furthermore shown by UV/Vis spectroscopy and spectroelectrochemical methods that low-valent nickel [Ni0] results from the interaction of the NiII catalyst with CrCl2.
Co-reporter:Dipl.-Chem. Philipp Christ;Dr. Anita G. Lindsay;Dipl.-Chem. Sonja S. Vormittag;Dr. Jörg-M. Neudörfl;Dr. Albrecht Berkessel;Dr. AnnMarie C. O'Donoghue
Chemistry - A European Journal 2011 Volume 17( Issue 31) pp:8524-8528
Publication Date(Web):
DOI:10.1002/chem.201101157
Co-reporter:Albrecht Berkessel, Sebastian Reichau, Adrian von der Höh, Nicolas Leconte, and Jörg-M. Neudörfl
Organometallics 2011 Volume 30(Issue 14) pp:3880-3887
Publication Date(Web):June 30, 2011
DOI:10.1021/om200459s
We herein report the first example of an asymmetric ketone hydrogenation using chirally modified derivatives of the homogeneous iron(II)–cyclopentadienone–tricarbonyl system, known as Casey’s catalyst. For the synthesis of the chirally modified catalysts, one of three carbonyl ligands was exchanged for a chiral phosphoramidite. To this end, either oxidative decarbonylation using trimethylamine-N-oxide or photolysis was applied. Photolysis was also used to convert the tricarbonyl iron precatalyst (and, analogously, the dicarbonyl phosphoramidite complexes) to the coordinatively unsaturated dicarbonyl (monocarbonyl, respectively) complexes, which are intermediates in the catalytic cycle of ketone hydrogenation. Hydrogen uptake by the latter species affords the “loaded” hydride, as evidenced by 1H NMR spectroscopy. Thus, the preparation of sensitive iron hydrides by the typically low-yielding Hieber reaction could be avoided. Instead, the catalytic cycle is accessed from air-stable carbonyl precursors. In the hydridic form of the phosphoramidite–carbonyl catalysts, the iron atom itself becomes a stereocenter. NMR spectroscopy confirmed the generation of two hydride diastereomers. With the MonoPhos iron dicarbonyl complex, moderate enantioselectivity (up to 31% ee) was achieved in the hydrogenation of acetophenone.
Co-reporter:Takafumi Yukawa ; Bianca Seelig ; Yingjie Xu ; Hiroyuki Morimoto ; Shigeki Matsunaga ; Albrecht Berkessel ;Masakatsu Shibasaki
Journal of the American Chemical Society 2010 Volume 132(Issue 34) pp:11988-11992
Publication Date(Web):August 6, 2010
DOI:10.1021/ja103294a
The catalytic asymmetric aza-Morita−Baylis−Hillman reaction using unactivated methyl acrylate is described. A simple Lewis acidic metal catalyst, such as La(OTf)3, was not suitable for the reaction, but rare earth metal alkoxide/linked-BINOL complexes possessing bifunctional Lewis acid and Brønsted base properties efficiently promoted the reaction in combination with an achiral nucleophilic organocatalyst. The combined use of a La(O-iPr)3/(S,S)-TMS-linked-BINOL complex with a catalytic amount of DABCO promoted the aza-Morita−Baylis−Hillman reaction of a broad range of N-diphenylphosphinoyl imines. Products from aryl, heteroaryl, and alkenyl imines were obtained in 67−99% yield and 81−95% ee. It is noteworthy that isomerizable alkyl imines could be employed as well, giving products in 78−89% yield and 94−98% ee. Initial rate kinetic studies as well as kinetic isotope effect experiments using α-deuterio-methyl acrylate support the importance of both the nucleophilicity of La-enolate and the Brønsted basicity of a La-catalyst for promoting the reaction.
Co-reporter:Albrecht Berkessel;Philipp Christ;Nicolas Leconte;Jörg-M. Neudörfl;Mathias Schäfer
European Journal of Organic Chemistry 2010 Volume 2010( Issue 27) pp:5165-5170
Publication Date(Web):
DOI:10.1002/ejoc.201000810
Abstract
We herein present the first synthesis of 1,1′-binaphthyl-2,2′-bis(sulfuryl)imides (JINGLEs). This new class of chiral Brønsted acids was synthesized in one step from the corresponding BINOLs and imidobis(sulfuryl chloride). A total of six enantiopure 1,1′-binaphthyl-2,2′-bis(sulfuryl)imides, carrying different 3,3′-substituents, were synthesized and characterized, inter alia, by X-ray crystallography.
Co-reporter:Albrecht Berkessel Dr.;Bianca Seelig Dr.;Silke Schwengberg Dr.;Jürgen Hescheler Dr.;Agapios Sachinidis Dr.
ChemBioChem 2010 Volume 11( Issue 2) pp:208-217
Publication Date(Web):
DOI:10.1002/cbic.200900345
Abstract
A transgenic murine embryonic stem (ES) cell lineage expressing enhanced green fluorescent protein (EGFP) under the control of α-myosine heavy chain (α-MHC) promoter (pα-MHC-EGFP) was used to investigate the effects of (thio)urea and cinchona alkaloid derivatives on cardiomyogenesis. The screening of the compounds yielded cardiomyogenesis inducing substances with good (IV-5, V-4) to very good activities (II-16, IV-8), as determined by a 50 to 80 % increase in the EGFP fluorescence compared to untreated cells. Time-dependent screening approaches in which compounds were added at different developmental stages of the ES cells appeared to be of limited suitability for the identification of potential cellular targets.
Co-reporter:Dr. A. Berkessel;M.-C. Ong;M. Nachi ;Dr. J.-M. Neudörfl
ChemCatChem 2010 Volume 2( Issue 10) pp:1215-1218
Publication Date(Web):
DOI:10.1002/cctc.201000104
Co-reporter:Dr. A. Berkessel;M.-C. Ong;M. Nachi ;Dr. J.-M. Neudörfl
ChemCatChem 2010 Volume 2( Issue 10) pp:
Publication Date(Web):
DOI:10.1002/cctc.201090039
Co-reporter:Dr. Albrecht Berkessel;Silvia Elfert;Dr. Kerstin Etzenbach-Effers;Dr. J. Henrique Teles
Angewandte Chemie 2010 Volume 122( Issue 39) pp:7275-7279
Publication Date(Web):
DOI:10.1002/ange.200907275
Co-reporter:Dr. Albrecht Berkessel;Silvia Elfert;Dr. Kerstin Etzenbach-Effers;Dr. J. Henrique Teles
Angewandte Chemie 2010 Volume 122( Issue 39) pp:
Publication Date(Web):
DOI:10.1002/ange.201001705
Co-reporter:Dr. Albrecht Berkessel;Silvia Elfert;Dr. Kerstin Etzenbach-Effers;Dr. J. Henrique Teles
Angewandte Chemie International Edition 2010 Volume 49( Issue 39) pp:7120-7124
Publication Date(Web):
DOI:10.1002/anie.200907275
Co-reporter:Dr. Albrecht Berkessel;Silvia Elfert;Dr. Kerstin Etzenbach-Effers;Dr. J. Henrique Teles
Angewandte Chemie International Edition 2010 Volume 49( Issue 39) pp:
Publication Date(Web):
DOI:10.1002/anie.201001705
Co-reporter:Albrecht Berkessel;Marc Brenburg ;Mathias Schäfer
Advanced Synthesis & Catalysis 2008 Volume 350( Issue 9) pp:1287-1294
Publication Date(Web):
DOI:10.1002/adsc.200700601
Abstract
The composition and degradation of a highly active and enantioselective titanium salalen in situ catalyst for the asymmetric epoxidation of olefins with aqueous hydrogen peroxide was investigated. Kinetic data and ESI-MS studies point to a mononuclear titanium salalen as the catalytically active species. By means of ESI-MS and selective monodeuteration of the salalen ligand, the oxidative degradation was studied. Upon exposure to aqueous hydrogen peroxide, the amine functionality of the salalen ligand is converted to the hydroxylamine, followed by loss of water and generation of the inactive titanium-salen complex. This transformation limits the activity of the catalyst in the epoxidation of less electron-rich olefins, such as 1-octene.
Co-reporter:Albrecht Berkessel Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 20) pp:3677-3679
Publication Date(Web):
DOI:10.1002/anie.200705326
Co-reporter:Albrecht Berkessel Dr.
Angewandte Chemie 2008 Volume 120( Issue 20) pp:3735-3737
Publication Date(Web):
DOI:10.1002/ange.200705326
Co-reporter:Albrecht Berkessel;Claudia Rollmann;Francoise Chamouleau;Stefanie Labs;Oliver May;Harald Gröger
Advanced Synthesis & Catalysis 2007 Volume 349(Issue 17-18) pp:
Publication Date(Web):21 NOV 2007
DOI:10.1002/adsc.200700244
A practical biocatalytic method for the synthesis of aliphatic β-halogenated (S)-alcohols as epoxide precursors by means of an enantioselective reduction of the corresponding ketones with recombinant whole cells, bearing an alcohol dehydrogenase and a glucose dehydrogenase, was developed. The biotransformations operate at high substrate concentrations of up to 208 g/L, and afford the (S)-β-halohydrins with both high conversions of >95 % and enantioselectivities of >99 % ee. Base-induced cyclization of the β-halohydrin intermediates gave the desired (S)-epoxides in high yield and enantiomeric purity (>99 % ee).
Co-reporter:Albrecht Berkessel;Marc Brenburg;Eva Leitterstorf;Julia Frey;Johann Lex;Mathias Schäfer
Advanced Synthesis & Catalysis 2007 Volume 349(Issue 14-15) pp:
Publication Date(Web):19 OCT 2007
DOI:10.1002/adsc.200700221
A modular synthesis for chiral and non-symmetrical salalen ligands (i.e., mono-reduced salens), carrying an imine and an amine functionality has been developed. The ligands can be assembled in situ by Schiff base formation of an N-(2-hydroxybenzyl)diamine with a salicylic aldehyde, thus allowing rapid and easy variation/optimization of the ligand structure. Aiming at optimal activity and enantioselectivity in the titanium-catalyzed asymmetric epoxidation of non-functionalized olefins, a positional scanning of the two ligand halves was carried out. High epoxide yields were achieved, and up to 98 % ee. The composition of the titanium complex catalysts was determined by high resolution mass spectrometry and X-ray crystallography for one selected example.
Co-reporter:Albrecht Berkessel, Erkan Ertürk, Patrick Kaiser, Axel Klein, Radoslaw M. Kowalczyk and Biprajit Sarkar
Dalton Transactions 2007 (Issue 31) pp:3427-3434
Publication Date(Web):20 Jun 2007
DOI:10.1039/B705078J
Tetraarylporphyrin ruthenium complexes [Ru(L)(aryl4Por)] (L = CO or PF3; aryl = mesityl or 10-R′-bis-methano-octahydroanthracene-9-yl with R′ = H, CF3, OCH3 or CH3) show a rich electrochemistry with at least five different stable oxidation states (including the parent state). The overall character of the redox behaviour is porphyrin-centred. However detailed spectroelectrochemical investigations using IR, UV/Vis/NIR and EPR spectroscopy (X- and K band) gave clear indication for ruthenium contributions.
Co-reporter:Albrecht Berkessel Dr.;Maria Guixà Dr.;Friederike Schmidt Dr.;Jörg M. Neudörfl Dr.;Johann Lex Dr.
Chemistry - A European Journal 2007 Volume 13(Issue 16) pp:
Publication Date(Web):9 MAR 2007
DOI:10.1002/chem.200600993
In the area of catalytic asymmetric epoxidation, the highly enantioselective transformation of cyclic enones and quinones is an extremely challenging target. With the aim to develop new and highly effective phase-transfer catalysts for this purpose, we conducted a systematic structural variation of PTCs based on quinine and quinidine. In the total of 15 new quaternary ammonium PTCs, modifications included, for example, the exchange of the quinine methoxy group for a free hydroxyl or other alkoxy substituents, and the introduction of additional elements of chirality through alkylation of the alkaloid quinuclidine nitrogen atom by chiral electrophiles. For example, the well-established 9- anthracenylmethyl group was exchanged for a “chiral” anthracene in the form of 9-chloromethyl-[(1,8-S;4,5-R)-1,2,3,4,5,6,7,8-octahydro-1,4:5,8-dimethanoanthracene. The asymmetric epoxidation of vitamin K3 was used as the test reaction for our novel PTCs. The readily available PTC 10 (derived from quinine in three convenient and high-yielding steps) proved to be the most enantioselective catalyst for this purpose known to date: At a catalyst loading of only 2.50 mol %, the quinone epoxide was obtained in 76 % yield and with 85 % ee (previously: ≤34 % ee), using commercial bleach (aqueous sodium hypochlorite) as the oxidant. To rationalize the sense of induction effected by our novel phase-transfer catalysts, a computational analysis of steric interactions in the intermediate chlorooxy enolate–PTC ion pair was conducted. Based on this analysis, the sense of induction for all 15 novel PTCs could be consistently explained.
Co-reporter:Albrecht Berkessel;Erkan Ertürk
Advanced Synthesis & Catalysis 2006 Volume 348(Issue 18) pp:
Publication Date(Web):15 DEC 2006
DOI:10.1002/adsc.200606181
The hydrolytic kinetic resolution (HKR) of terminal epoxides, using chiral chromium(III)-salen catalysts based on DIANANE (endo,endo-2,5-diaminonorbornane), was studied. A broad substrate scope was found for the chromium(III)-DIANANE catalysts, and very low loadings (down to 0.05 mol %) were needed to achieve high enantiomeric purities of both the remaining epoxides and the product diols (up to >99 % ee). Besides monosubstituted epoxides, 2-methyl-2-n-pentyloxirane, which is an example for 2,2-disubstituted epoxides, could be ring-opened in an asymmetric fashion with water in the presence of an electronically tuned chromium(III)-DIANANE complex.
Co-reporter:Albrecht Berkessel;Erkan Ertürk;Cécile Laporte
Advanced Synthesis & Catalysis 2006 Volume 348(Issue 1-2) pp:
Publication Date(Web):19 JAN 2006
DOI:10.1002/adsc.200505249
Starting from enantiomerically pure 5,10,15,20-tetrakis[(1S,4R,5R,8S)-1,2,3,4,5,6,7,8-octahydro-1,4 : 5,8-dimethanoanthracene-9-yl]porphyrin, treatment with CrCl2 and subsequent air oxidation afforded the corresponding Cr(III) complex, with chloride as counterion, in 96% yield. Anion exchange with AgBF4 gave the corresponding tetrafluoroborate. These hitherto unknown chiral chromium porphyrins are efficient and highly enantioselective catalysts for the hetero-Diels–Alder reaction of aliphatic, aromatic, and heteroaromatic aldehydes with dienes of varying electron density. In the case of 1-methoxy-3-(trimethylsilyloxy)butadiene (“Danishefsky's diene”), enantiomeric excesses >90% were achieved in a number of cases, with furfural affording the highest ee (97%). Metal-coordinating aldehydes such as pyridine-2-carbaldehyde do not inactivate the Cr(III) porphyrin catalyst. The cycloaddition of less electron-rich dienes (such as 1-methoxybutadiene) is effected as well, affording diastereoselectivities up to >99 : 1, and enantiomeric excesses >80%.
Co-reporter:Albrecht Berkessel, Santanu Mukherjee, Thomas N. Müller, Felix Cleemann, Katrin Roland, Marc Brandenburg, Jörg-M. Neudörfl and Johann Lex
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 23) pp:4319-4330
Publication Date(Web):01 Nov 2006
DOI:10.1039/B607574F
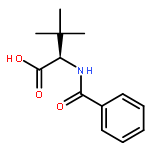
This article describes the synthesis of a library of structurally diverse bifunctional organocatalysts bearing both a quasi-Lewis acidic (thio)urea moiety and a Brønsted basic tertiary amine group. Sequential modification of the modular catalyst structure and subsequent screening of the compounds in the alcoholytic dynamic kinetic resolution (DKR) of azlactones revealed valuable structure–activity relationships. In particular, a “hit-structure” was identified which provides e.g.N-benzoyl-tert-leucine allyl ester in an excellent enantiomeric excess of 95%.
Co-reporter:Albrecht Berkessel;Nadine Vogl
European Journal of Organic Chemistry 2006 Volume 2006(Issue 22) pp:
Publication Date(Web):11 SEP 2006
DOI:10.1002/ejoc.200600359
A new type of chromium-salen complex bearing DIANANE (endo,endo-2,5-diaminonorbornane) as chiral backbone has been synthesized and applied to the hetero-Diels–Alder reaction of Danishefsky’s diene with various aldehydes. The reactions afford the corresponding 2-substituted 2,3-dihydro-4H-pyran-4-ones in high yields and enantioselectivities (up to 96 % ee). The effect of different counteranions of the complex on reactivity as well as enantioselectivity has been investigated. Besides Danishefsky’s diene, reaction of the less reactive 1-methoxybutadiene with benzaldehyde and ethyl glyoxylate can be effected by the chromium catalysts as well.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Co-reporter:Albrecht Berkessel Dr.;M. Luisa Sebastian-Ibarz;Thomas N. Müller Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 39) pp:
Publication Date(Web):4 SEP 2006
DOI:10.1002/anie.200600379
Racemization wanted: The dynamic kinetic resolution of secondary alcohols can be achieved by a simple and readily available catalyst system. Substrate racemization is effected at room temperature by a combination of (racemic) 1,1′-bi-2-naphthol (binol) or 2,2′-biphenol with AlMe3, and a lipase performs enantiospecific acylation (see scheme).
Co-reporter:Thomas Wielpütz Dipl.-Chem.;Thomas Sottmann Dr.;Reinhard Strey Dr.;Friederike Schmidt Dr. Dr.
Chemistry - A European Journal 2006 Volume 12(Issue 29) pp:
Publication Date(Web):28 SEP 2006
DOI:10.1002/chem.200690092
Co-reporter:Thomas Wielpütz Dipl.-Chem.;Thomas Sottmann Dr.;Reinhard Strey Dr.;Friederike Schmidt Dr. Dr.
Chemistry - A European Journal 2006 Volume 12(Issue 29) pp:
Publication Date(Web):6 SEP 2006
DOI:10.1002/chem.200600550
The ability of microemulsions to dissolve polar and non-polar components with a huge internal interface can overcome the reagent incompatibilities frequently encountered in organic reactions. We investigated model epoxidation reactions of α,β-unsaturated enones and alkaline hydrogen peroxide in different nonionic microemulsions, both in the presence and absence of a phase-transfer agent (PTA). The obtained reaction profiles were compared with those for the corresponding surfactant-free two-phase systems. In addition, we defined a time constant τ as a measure for the rate of turnover. The epoxidation of trans-chalcone using an n-alkyl-polyoxyethylene surfactant based microemulsion was fastest in the system with the PTA (τ=66 min) and slightly slower without the PTA (τ=77 min). It was still slower in the two-phase system with a PTA (τ=114 min) and extremely sluggish without a phase-transfer agent. With n-alkyl β-D-glucopyranoside as the surfactant the conversion was twice as fast than in the former microemulsion systems, but the PTA did not accelerate the reaction further (τ=35 and 33 min). The epoxidation of vitamin K3, the second model system, was extremely accelerated. It proceeded a factor of approximately 35 faster in the microemulsion (τ=1.44 min) than in the corresponding two-phase system (τ=57 min).
Co-reporter:Albrecht Berkessel Dr.;M. Luisa Sebastian-Ibarz;Thomas N. Müller Dr.
Angewandte Chemie 2006 Volume 118(Issue 39) pp:
Publication Date(Web):4 SEP 2006
DOI:10.1002/ange.200600379
Racemisierung erwünscht: Die dynamische kinetische Racematspaltung sekundärer Alkohole gelang mit einem einfachen und sehr leicht zugänglichen Katalysatorsystem: In situ wird aus (racemischem) 1,1′-Bi-2-naphthol (Binol) oder 2,2′-Biphenol und AlMe3 ein Aluminiumkatalysator erzeugt, der das Substrat bei Raumtemperatur racemisiert, und eine Lipase gewährleistet die enantiospezifische Acylierung (siehe Schema).
Co-reporter:Albrecht Berkessel, Santanu Mukherjee, Felix Cleemann, Thomas N. Müller and Johann Lex
Chemical Communications 2005 (Issue 14) pp:1898-1900
Publication Date(Web):15 Feb 2005
DOI:10.1039/B418666D
Bifunctional organocatalysts of the thiourea-tert-amine type, carrying two “matched” elements of chirality, effect the alcoholytic dynamic kinetic resolution of a variety of azlactones with up to 95% ee.
Co-reporter:Albrecht Berkessel, Felix Cleemann,Santanu Mukherjee
Angewandte Chemie International Edition 2005 44(45) pp:7466-7469
Publication Date(Web):
DOI:10.1002/anie.200502003
Co-reporter:Albrecht Berkessel Dr.;Felix Cleemann Dipl.-Chem.;Santanu Mukherjee M. Sc.
Angewandte Chemie 2005 Volume 117(Issue 45) pp:
Publication Date(Web):24 OCT 2005
DOI:10.1002/ange.200502003
Eine profitable Scheidung: Ein organokatalytisches Verfahren überführt die leicht zugänglichen racemischen Oxazinone 1 durch kinetische Racematspaltung unter katalytischer Ringöffnung in die wertvollen enantiomerenreinen β-Aminosäurederivate 2 (bei 53 % Umsatz >99 % ee des zurückbleibenden Oxazinons 1, 88 % ee des Esters). Der Organokatalysator 3, ein Thioharnstoff, ist modular aufgebaut, leicht zugänglich und kann in Mengen von nur 1 Mol-% eingesetzt werden.
Co-reporter:Albrecht Berkessel Dr.;Felix Cleemann Dipl.-Chem.;Santanu Mukherjee;Thomas N. Müller Dipl.-Chem.;Johann Lex Dr.
Angewandte Chemie 2005 Volume 117(Issue 5) pp:
Publication Date(Web):18 JAN 2005
DOI:10.1002/ange.200590014
Co-reporter:Albrecht Berkessel Dr.;Felix Cleemann Dipl.-Chem.;Santanu Mukherjee;Thomas N. Müller Dipl.-Chem.;Johann Lex Dr.
Angewandte Chemie 2005 Volume 117(Issue 5) pp:
Publication Date(Web):18 JAN 2005
DOI:10.1002/ange.200461442
Neue Aufgaben für ehrwürdige Moleküle: Bifunktionale Organokatalysatoren mit einer quasi-Lewis-sauren Harnstoff- oder Thioharnstoff-Einheit und einer Brønsted-basischen Aminofunktion bewirken die dynamische kinetische Racemattrennung von Azlactonen mit hoher Enantioselektivität: N-Benzoylaminosäureester mit bis zu 91 % ee konnten in hohen Ausbeuten aus racemischen Azlactonen gewonnen werden.
Co-reporter:Albrecht Berkessel Dr.;Felix Cleemann Dipl.-Chem.;Santanu Mukherjee;Thomas N. Müller Dipl.-Chem.;Johann Lex Dr.
Angewandte Chemie International Edition 2005 Volume 44(Issue 5) pp:
Publication Date(Web):18 JAN 2005
DOI:10.1002/anie.200590014
Co-reporter:Albrecht Berkessel Dr.;Felix Cleemann Dipl.-Chem.;Santanu Mukherjee;Thomas N. Müller Dipl.-Chem.;Johann Lex Dr.
Angewandte Chemie International Edition 2005 Volume 44(Issue 5) pp:
Publication Date(Web):18 JAN 2005
DOI:10.1002/anie.200461442
Novel tasks for a venerable molecule: Bifunctional organocatalysts of the type shown, with a quasi Lewis acidic urea or thiourea unit and a Brønsted basic amine functionality, effect the dynamic kinetic resolution of azlactones with high enantioselectivity. N-Benzoyl amino acid esters were obtained with up to 91 % ee in high yields from racemic azlactones.
Co-reporter:Albrecht Berkessel;Jens A. Adrio
Advanced Synthesis & Catalysis 2004 Volume 346(Issue 2-3) pp:
Publication Date(Web):29 MAR 2004
DOI:10.1002/adsc.200303222
The epoxidation of cyclooctene and 1-octene with hydrogen peroxide was studied kinetically in HFIP (1,1,1,3,3,3-hexafluoro-2-propanol)/1,4-dioxane mixtures. In the case of cyclooctene, no additional catalyst was applied, whereas the epoxidation of 1-octene was run in the presence of phenylarsonic acid as catalyst. For both reaction types, two kinetic regimes can be distinguished: at low HFIP concentration (nHFIP/ntotal≤0.15), both the catalyzed and the uncatalyzed reaction show first order dependence on the HFIP concentration. This result is interpreted by the HFIP “charge template” for the uncatalyzed epoxidation (as postulated by Shaik and Neumann) and by the formation of an arsonic acid mono-HFIP ester for the catalyzed reaction. At high HFIP concentration (nHFIP/ntotal≥0.5), both reaction types are up to ca. 5 orders of magnitude faster and show ca. 12th order dependence on the HFIP concentration. We propose that the dramatic accelerations observed at high HFIP concentrations are brought about by HFIP clusters. Based on literature data (Fioroni, Roccatano, Hong, Suhm), it is concluded that the epoxidation reactions studied here take place in a HFIP coordination sphere comprising ca. 12 HFIP molecules, and that this coordination sphere effects the enormous increases in epoxidation rates. This mechanistic model bears resemblance to enzymatic reactions taking place in and being catalyzed by a protein matrix.
Co-reporter:Albrecht Berkessel;Burkhard Koch;Johann Lex
Advanced Synthesis & Catalysis 2004 Volume 346(Issue 9-10) pp:
Publication Date(Web):21 SEP 2004
DOI:10.1002/adsc.200404126
Proline catalysis of the asymmetric direct aldol reaction involves both the secondary amine function and the carboxyl group of the amino acid. N-Sulfonylcarboxamides are known to be of similar acidity as carboxylic acids, and three N-arylsulfonyl derivatives of L-proline amide were synthesized as functionalized and versatile derivatives of L-Pro. Their catalytic performance was evaluated in the direct aldol addition of acetone to 4-nitrobenzaldehyde. Significantly improved reactivities and enantioselectivities were achieved in various solvents at low catalyst loadings (5–10 mol %) and at room temperature, with ees ranging up to 98%, whereas L-proline itself afforded a maximum ee of 80% (in DMSO). Thus, N-arylsulfonyl derivatives of proline amide represent a novel class of highly enantioselective catalysts for direct aldol reactions. Furthermore, the N-arylsulfonyl substituent suggests possibilities for incorporation into larger catalyst assemblies (including immobilization) without affecting the catalytically active functional groups.
Co-reporter:Albrecht Berkessel ;Patrick Kaiser Dr.;Johann Lex Dr.
Chemistry - A European Journal 2003 Volume 9(Issue 19) pp:
Publication Date(Web):26 SEP 2003
DOI:10.1002/chem.200305045
We report the use of three enantiomerically pure and electronically tuned ruthenium carbonyl porphyrin catalysts for the asymmetric cyclopropanation and epoxidation of a variety of olefinic substrates. The D4-symmetric ligands carry a methoxy, a methyl or a trifluoromethyl group at the 10-position of each of the 9-[anti-(1,2,3,4,5,6,7,8-octahydro-1,4:5,8-dimethanoanthracene)]-substituents at the meso-positions of the porphyrin. Introduction of a CF3-substituent in this remote position resulted in greatly improved catalyst stability, and turnover numbers of up to 7 500 were achieved for cyclopropanation, and up to 14 200 for epoxidation, with ee values typically >90 % and ≈80 %, respectively. In one example, the axial CO ligand at the ruthenium was exchanged for PF3, resulting in the first chiral ruthenium porphyrin with a PF3 ligand reported to date. In cyclopropanations with ethyl diazoacetate, the latter catalyst performed exceedingly well, and gave a 95 % ee in the case of 1,1-diphenylethylene as substrate.
Co-reporter:Albrecht Berkessel Dr.;Dirk Menche Dr.;Christoph A. Sklorz Dr.;Michael Schröder Dipl.-Chem.;Ian Paterson Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 9) pp:
Publication Date(Web):26 FEB 2003
DOI:10.1002/anie.200390265
Catalytic asymmetric addition of vinylic halides and triflates to aldehydes, with useful levels of stereoinduction, has been achieved for the first time using the salen-type ligand (S,S)-5 (see scheme; PMB=para-methoxybenzyl), which contains the novel endo,endo-2,5-diamino norbornane building block. This asymmetric Nozaki–Hiyama–Kishi reaction leads to the coupling of various halides and aldehydes with high yields and enantioselectivities (up to 92 % ee).
Co-reporter:Albrecht Berkessel;Katja Glaubitz;Johann Lex
European Journal of Organic Chemistry 2002 Volume 2002(Issue 17) pp:
Publication Date(Web):12 AUG 2002
DOI:10.1002/1099-0690(200209)2002:17<2948::AID-EJOC2948>3.0.CO;2-E
Enantiomerically pure trans-2-aminocyclohexanecarboxylic acid is an important building block for helical β-peptides. We report here that this amino acid can be obtained from trans-cyclohexane-1,2-dicarboxylic acid in good yield by a simple one-pot procedure comprising cyclization to the anhydride, amide formation with ammonia, and a subsequent Hofmann-type degradation with phenyliodine(III) bis(trifluoroacetate) (PIFA) as the oxidant. The N-Fmoc- and N-BOC-protected derivatives were obtained by treatment of the amino acid with Fmoc-OSu and BOC2O, respectively. The N-BOC derivative could be prepared in even better overall yield by a one-pot procedure leading directly from trans-cyclohexane-1,2-dicarboxylic acid to the N-BOC-protected amino acid. Both enantiomers of the starting trans-1,2-cyclohexanedicarboxylic acid can be obtained easily and in large quantities by separating commercially available racemic trans-1,2-cyclohexanedicarboxylic acid using either (R)- or (S)-1-phenethylamine. X-ray crystallography of the diastereomerically pure salt obtained from (R)-1-phenethylamine revealed that the configuration of the diacid component is (1R,2R), and not (1S,2S) as reported in the literature. (© Wiley-VCH Verlag GmbH, 69451 Weinheim, Germany, 2002)
Co-reporter:Albrecht Berkessel;Matthias R. Vennemann;Johann Lex
European Journal of Organic Chemistry 2002 Volume 2002(Issue 16) pp:
Publication Date(Web):24 JUL 2002
DOI:10.1002/1099-0690(200208)2002:16<2800::AID-EJOC2800>3.0.CO;2-4
We report the synthesis of three novel and chiral salicylic aldehyde building blocks 6−8, each in both enantiomerically pure forms. Two of these salicylic aldehydes were prepared from (+)-camphene and each bear a [2.2.1]bicycloheptyl substituent ortho to the salicylic hydroxy group. In the third case, the chiral element at the 6-position is a (1-phenylethyl) group. The synthetic sequences consisted of ortho-alkylation of para-cresol with either camphene or styrene and subsequent ortho-formylation of the product phenols. The chromatographic separation of enantiomers was accomplished through the diastereomeric imines obtained from condensation of the racemic salicylic aldehydes with (R)-phenylglycinol. Finally, the absolute configurations of two of the salicylic aldehydes were established by X-ray crystallography. For this purpose, the (1-phenylethyl)-substituted salicylic aldehyde was condensed with L-valinamide, and the relative configuration of the resulting Schiff base diastereomer 12 was determined. In the second case, the racemic intermediate phenol rac-15 was separated by HPLC on a chiral stationary phase, ortho-brominated, and analyzed by anomalous X-ray scattering. (© Wiley-VCH Verlag GmbH, 69451 Weinheim, Germany, 2002)
Co-reporter:Albrecht Berkessel ;Marc R. M. Andreae;Hans Schmickler Dr.;Johann Lex Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 23) pp:
Publication Date(Web):27 NOV 2002
DOI:10.1002/1521-3773(20021202)41:23<4481::AID-ANIE4481>3.0.CO;2-7
What a difference the solvent makes! Unlike in conventional solvents, nonstrained ketones such as cyclohexanone react smoothly with hydrogen peroxide in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) to give lactones. The reaction proceeds via an isolatable spiro-bisperoxide, which undergoes a highly exothermic acid-catalyzed rearrangement to two equivalents of lactone (see Equation and IR thermogram).
Co-reporter:Albrecht Berkessel ;Marc R. M. Andreae;Hans Schmickler Dr.;Johann Lex Dr.
Angewandte Chemie 2002 Volume 114(Issue 23) pp:
Publication Date(Web):27 NOV 2002
DOI:10.1002/1521-3757(20021202)114:23<4661::AID-ANGE4661>3.0.CO;2-H
Das Solvens macht den Unterschied: Anders als in konventionellen Lösungsmitteln reagieren ungespannte Ketone wie Cyclohexanon mit Wasserstoffperoxid in 1,1,1,3,3,3-Hexafluor-2-propanol (HFIP) glatt zu Lactonen. Die Reaktion verläuft über ein isolierbares Spiro-Bisperoxid, das in einer stark exothermen Reaktion säurekatalysiert zu zwei Äquivalenten des Lactons umlagert (siehe Gleichung und IR-Thermogramm).
Co-reporter:Albrecht Berkessel;David A. Hérault
Angewandte Chemie 1999 Volume 111(Issue 1‐2) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19990115)111:1/2<99::AID-ANGE99>3.0.CO;2-Q
Das kombinatorische Auffinden katalytischer Aktivität erfordert das Zusammenspiel von Bibliothekssynthese und geeigneten Assays. Mit dem Testsubstrat 1 wurden in einer Bibliothek Undecapeptide gefunden (z. B. A), die in Gegenwart von Zr4+ die Phosphathydrolyse beschleunigen.
Co-reporter:Albrecht Berkessel;David A. Hérault
Angewandte Chemie International Edition 1999 Volume 38(Issue 1‐2) pp:
Publication Date(Web):18 JAN 1999
DOI:10.1002/(SICI)1521-3773(19990115)38:1/2<102::AID-ANIE102>3.0.CO;2-H
The combinatorial discovery of novel catalytic activity requires both library synthesis and suitable assays. By means of the test substrate 1, undecapeptides such as A were identified within a library that accelerate phosphate hydrolysis in the presence of Zr4+.
Co-reporter:Dipl.-Chem. Martin Picard;Dr. Jürgen Gross; Dr. Albrecht Berkessel;Dipl.-Chem. Ellen Lübbert;Sabine Tölzer; Dr. Karl-Heinz van Pée;Susanne Krauss
Angewandte Chemie 1997 Volume 109(Issue 11) pp:
Publication Date(Web):31 JAN 2006
DOI:10.1002/ange.19971091118
Co-reporter:Giuseppe Rulli, Marcel Heidlindemann, Albrecht Berkessel, Werner Hummel, Harald Gröger
Journal of Biotechnology (November 2013) Volume 168(Issue 3) pp:271-276
Publication Date(Web):1 November 2013
DOI:10.1016/j.jbiotec.2013.08.031
•ADHs from Lactobacillus kefir and Rhodococcus sp., respectively, were immobilized with their cofactors on a superabsorber based on a literature-known method.•Applying the immobilized ADH from L. kefir in the reduction of acetophenone as a model substrate led to high conversion (>95%).•Immobilized ADHs are suitable catalysts for the diastereoselective reduction of an organocatalytically prepared enantiomerically enriched aldol adduct.•At a lower catalyst and cofactor amount being still sufficient for biotransformations with “free” enzymes the immobilized ADH only showed high conversion and >99% ee for the first reaction cycle.The alcohol dehydrogenases (ADHs) from Lactobacillus kefir and Rhodococcus sp., which earlier turned out to be suitable for a chemoenzymatic one-pot synthesis with organocatalysts, were immobilized with their cofactors on a commercially available superabsorber based on a literature known protocol. The use of the immobilized ADH from L. kefir in the reduction of acetophenone as a model substrate led to high conversion (>95%) in the first reaction cycle, followed by a slight decrease of conversion in the second reaction cycle. A comparable result was obtained when no cofactor was added although a water rich reaction media was used. The immobilized ADHs also turned out to be suitable catalysts for the diastereoselective reduction of an organocatalytically prepared enantiomerically enriched aldol adduct, leading to high conversion, diastereomeric ratio and enantioselectivity for the resulting 1,3-diols. However, at a lower catalyst and cofactor amount being still sufficient for biotransformations with “free” enzymes the immobilized ADH only showed high conversion and >99% ee for the first reaction cycle whereas a strong decrease of conversion was observed already in the second reaction cycle, thus indicating a significant leaching effect of catalyst and/or cofactor.Download full-size image
Co-reporter:Albrecht Berkessel, Erkan Ertürk, Patrick Kaiser, Axel Klein, Radoslaw M. Kowalczyk and Biprajit Sarkar
Dalton Transactions 2007(Issue 31) pp:NaN3434-3434
Publication Date(Web):2007/06/20
DOI:10.1039/B705078J
Tetraarylporphyrin ruthenium complexes [Ru(L)(aryl4Por)] (L = CO or PF3; aryl = mesityl or 10-R′-bis-methano-octahydroanthracene-9-yl with R′ = H, CF3, OCH3 or CH3) show a rich electrochemistry with at least five different stable oxidation states (including the parent state). The overall character of the redox behaviour is porphyrin-centred. However detailed spectroelectrochemical investigations using IR, UV/Vis/NIR and EPR spectroscopy (X- and K band) gave clear indication for ruthenium contributions.
Co-reporter:Veera Reddy Yatham, Jörg-M. Neudörfl, Nils E. Schlörer and Albrecht Berkessel
Chemical Science (2010-Present) 2015 - vol. 6(Issue 7) pp:NaN3711-3711
Publication Date(Web):2015/04/30
DOI:10.1039/C5SC01027F
Since their discovery by Bode and Glorius in 2004, N-heterocyclic carbene catalyzed conjugate umpolung reactions of α,β-enals have been postulated to involve the formation of diamino dienols (“homoenolates”) and/or azolium enolates (“enolates”), typically followed by addition to electrophiles, e.g. Michael-acceptors. In this article, we provide evidence, for the first time, for the postulated individual and specific reactivity patterns of diamino dienols (γ-C–C-bond formation) vs. azolium enolates (β-C–C-bond formation). Our study is based on the pre-formation of well defined diamino dienols and azolium enolates, and the in situ NMR monitoring of their reactivities towards enone electrophiles. Additionally, reaction intermediates were isolated and characterized, inter alia by X-ray crystallography.



















![1,3,2-Dioxaphosphorino[4',5':5,6]pyrano[3,2-g]pteridin-10(4H)-one,8-amino-4a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-, 2-oxide,(4aR,5aR,11aR,12aS)-](http://img.cochemist.com/ccimg/150900/150829-29-1.png)
![1,3,2-Dioxaphosphorino[4',5':5,6]pyrano[3,2-g]pteridin-10(4H)-one,8-amino-4a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-, 2-oxide,(4aR,5aR,11aR,12aS)-](http://img.cochemist.com/ccimg/150900/150829-29-1_b.png)




![Benzene, 1-bromo-2-[(S)-methylsulfinyl]-](http://img.cochemist.com/ccimg/146300/146235-11-2.png)
![Benzene, 1-bromo-2-[(S)-methylsulfinyl]-](http://img.cochemist.com/ccimg/146300/146235-11-2_b.png)




![Bicyclo[2.2.1]heptane-2,5-dione, (1S)-](http://img.cochemist.com/ccimg/145100/145092-68-8.png)
![Bicyclo[2.2.1]heptane-2,5-dione, (1S)-](http://img.cochemist.com/ccimg/145100/145092-68-8_b.png)






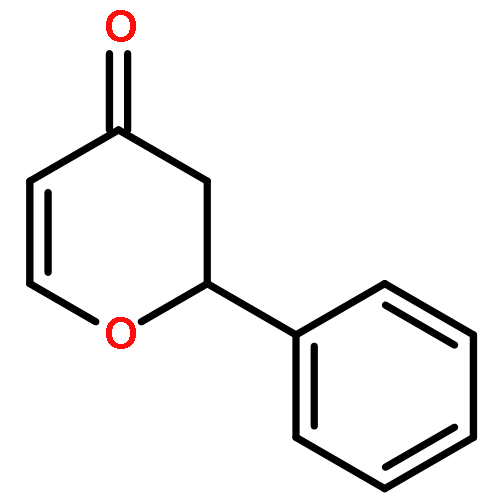
![4H-Pyran-4-one, 2,3-dihydro-2-[(1E)-2-phenylethenyl]-, (2R)-](http://img.cochemist.com/ccimg/139700/139627-54-6.png)
![4H-Pyran-4-one, 2,3-dihydro-2-[(1E)-2-phenylethenyl]-, (2R)-](http://img.cochemist.com/ccimg/139700/139627-54-6_b.png)


![Silane, [[(1Z,2E)-1-ethylidene-2-butenyl]oxy]trimethyl-](http://img.cochemist.com/ccimg/138100/138080-92-9.png)
![Silane, [[(1Z,2E)-1-ethylidene-2-butenyl]oxy]trimethyl-](http://img.cochemist.com/ccimg/138100/138080-92-9_b.png)











![Propanal, 3-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/128500/128461-65-4.png)
![Propanal, 3-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/128500/128461-65-4_b.png)
![Oxirane,2-[(4-nitrophenoxy)methyl]-, (2S)-](http://img.cochemist.com/ccimg/125300/125279-82-5.png)
![Oxirane,2-[(4-nitrophenoxy)methyl]-, (2S)-](http://img.cochemist.com/ccimg/125300/125279-82-5_b.png)


![(3R)-1-Azabicyclo[2.2.2]octan-3-amine](http://img.cochemist.com/ccimg/123600/123536-15-2.png)
![(3R)-1-Azabicyclo[2.2.2]octan-3-amine](http://img.cochemist.com/ccimg/123600/123536-15-2_b.png)
![2-[2-(TRIFLUOROMETHYL)PHENYL]PHENOL](http://img.cochemist.com/ccimg/122900/122801-60-9.png)
![2-[2-(TRIFLUOROMETHYL)PHENYL]PHENOL](http://img.cochemist.com/ccimg/122900/122801-60-9_b.png)











![Benzamide, N-[2-(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/93100/93007-80-8.png)
![Benzamide, N-[2-(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/93100/93007-80-8_b.png)
![Ruthenium, carbonyl[5,10,15,20-tetrakis(2,4,6-trimethylphenyl)-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-31)-](/data/chemimg/2246300/92669-43-7.png)
![Ruthenium, carbonyl[5,10,15,20-tetrakis(2,4,6-trimethylphenyl)-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-31)-](/data/chemimg/2246300/92669-43-7_b.png)




![6H-Indeno[1,2-b]oxirene,1a,6a-dihydro-, (1aR,6aS)-](http://img.cochemist.com/ccimg/85400/85354-35-4.png)
![6H-Indeno[1,2-b]oxirene,1a,6a-dihydro-, (1aR,6aS)-](http://img.cochemist.com/ccimg/85400/85354-35-4_b.png)













































![2,4-ditert-butyl-6-[(3,5-ditert-butyl-2-hydroxyphenyl)disulfanyl]phenol](http://img.cochemist.com/ccimg/65000/64953-47-5.png)
![2,4-ditert-butyl-6-[(3,5-ditert-butyl-2-hydroxyphenyl)disulfanyl]phenol](http://img.cochemist.com/ccimg/65000/64953-47-5_b.png)
![Phenol, 4-methyl-2-[(1R,2S,4R,6S)-5,5,6-trimethylbicyclo[2.2.1]hept-2-yl]-, rel-](/data/chemimg/1287400/64374-17-0.png)
![Phenol, 4-methyl-2-[(1R,2S,4R,6S)-5,5,6-trimethylbicyclo[2.2.1]hept-2-yl]-, rel-](/data/chemimg/1287400/64374-17-0_b.png)




![Magnesium, [2,6-bis(1-methylethyl)phenyl]bromo-](http://img.cochemist.com/ccimg/63500/63488-11-9.png)
![Magnesium, [2,6-bis(1-methylethyl)phenyl]bromo-](http://img.cochemist.com/ccimg/63500/63488-11-9_b.png)






![Methanone, phenyl[(2S,3R)-3-phenyloxiranyl]-](http://img.cochemist.com/ccimg/61900/61840-93-5.png)
![Methanone, phenyl[(2S,3R)-3-phenyloxiranyl]-](http://img.cochemist.com/ccimg/61900/61840-93-5_b.png)
![[1,1'-Biphenyl]-2-ylhydrazine](http://img.cochemist.com/ccimg/60000/59964-94-2.png)
![[1,1'-Biphenyl]-2-ylhydrazine](http://img.cochemist.com/ccimg/60000/59964-94-2_b.png)







![(6R)-6-METHYL-7-OXABICYCLO[4.1.0]HEPTANE](http://img.cochemist.com/ccimg/56300/56246-60-7.png)
![(6R)-6-METHYL-7-OXABICYCLO[4.1.0]HEPTANE](http://img.cochemist.com/ccimg/56300/56246-60-7_b.png)

![[1,1'-Binaphthalene]-2,2'-diol, 3,3'-dimethyl-, (1S)-](/data/chemimg/1847300/55515-99-6.png)
![[1,1'-Binaphthalene]-2,2'-diol, 3,3'-dimethyl-, (1S)-](/data/chemimg/1847300/55515-99-6_b.png)


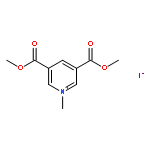
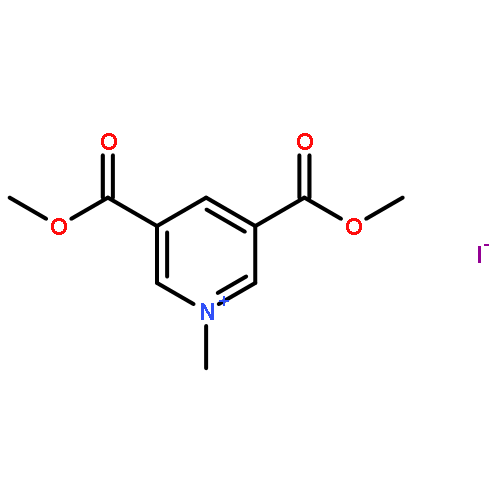
















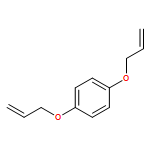
![2-{[4-(prop-2-en-1-yloxy)phenoxy]methyl}oxirane](http://img.cochemist.com/ccimg/52300/52210-93-2.png)
![2-{[4-(prop-2-en-1-yloxy)phenoxy]methyl}oxirane](http://img.cochemist.com/ccimg/52300/52210-93-2_b.png)

![1-[6-(6-ACETYLPYRIDIN-2-YL)PYRIDIN-2-YL]ETHANONE](http://img.cochemist.com/ccimg/49700/49669-27-4.png)
![1-[6-(6-ACETYLPYRIDIN-2-YL)PYRIDIN-2-YL]ETHANONE](http://img.cochemist.com/ccimg/49700/49669-27-4_b.png)























![Silane, [(1-methoxyethenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/36900/36850-80-3.png)
![Silane, [(1-methoxyethenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/36900/36850-80-3_b.png)








![Benzamide, N-[2,6-bis(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/35400/35336-02-8.png)
![Benzamide, N-[2,6-bis(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/35400/35336-02-8_b.png)


















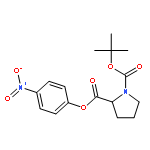
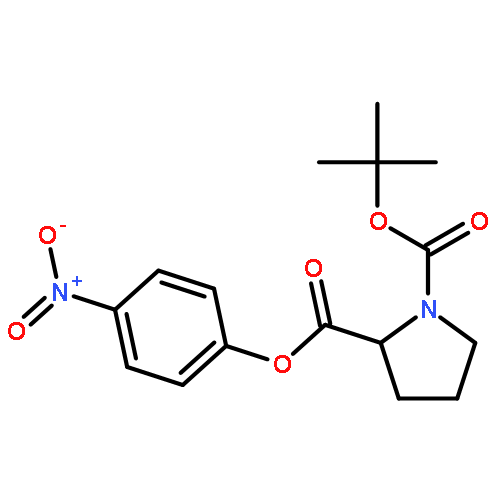
![Bicyclo[2.2.1]heptane-2,5-dione](http://img.cochemist.com/ccimg/28000/27943-47-1.png)
![Bicyclo[2.2.1]heptane-2,5-dione](http://img.cochemist.com/ccimg/28000/27943-47-1_b.png)
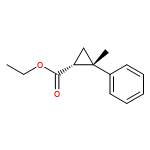
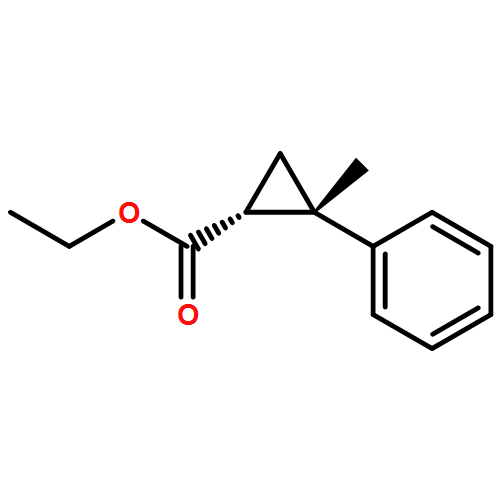









![(3aS,6aR)-3,3a,6,6a-Tetrahydro-2H-cyclopenta[b]furan-2-one](http://img.cochemist.com/ccimg/26100/26054-46-6.png)
![(3aS,6aR)-3,3a,6,6a-Tetrahydro-2H-cyclopenta[b]furan-2-one](http://img.cochemist.com/ccimg/26100/26054-46-6_b.png)



















![Acetic acid, [4-[(1E)-2-phenylethenyl]phenoxy]-](http://img.cochemist.com/ccimg/19900/19882-38-3.png)
![Acetic acid, [4-[(1E)-2-phenylethenyl]phenoxy]-](http://img.cochemist.com/ccimg/19900/19882-38-3_b.png)






![Benzo[a]phenoxazin-7-ium,5-amino-9-(dimethylamino)-10-methyl-, chloride (1:1)](http://img.cochemist.com/ccimg/18500/18472-89-4.png)
![Benzo[a]phenoxazin-7-ium,5-amino-9-(dimethylamino)-10-methyl-, chloride (1:1)](http://img.cochemist.com/ccimg/18500/18472-89-4_b.png)
![Benzene, [(S)-methylsulfinyl]-](http://img.cochemist.com/ccimg/18500/18453-46-8.png)
![Benzene, [(S)-methylsulfinyl]-](http://img.cochemist.com/ccimg/18500/18453-46-8_b.png)









![Naphth[2,3-b]oxirene-2,7-dione,1a,7a-dihydro-1a-methyl-](http://img.cochemist.com/ccimg/15500/15448-59-6.png)
![Naphth[2,3-b]oxirene-2,7-dione,1a,7a-dihydro-1a-methyl-](http://img.cochemist.com/ccimg/15500/15448-59-6_b.png)




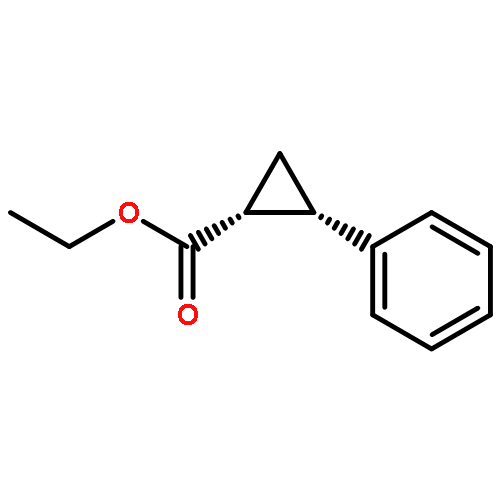
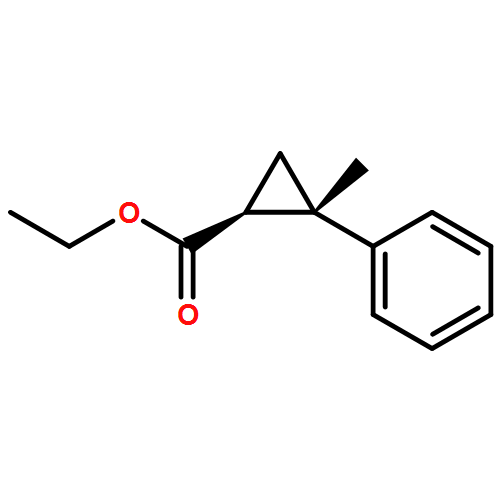
![Benzene,1,1'-[(1R,2S)-1-methyl-1,2-cyclopropanediyl]bis-, rel- (9CI)](http://img.cochemist.com/ccimg/14200/14161-73-0.png)
![Benzene,1,1'-[(1R,2S)-1-methyl-1,2-cyclopropanediyl]bis-, rel- (9CI)](http://img.cochemist.com/ccimg/14200/14161-73-0_b.png)
![Benzene,1,1'-[(1R,2R)-1-methyl-1,2-cyclopropanediyl]bis-, rel- (9CI)](http://img.cochemist.com/ccimg/14200/14161-72-9.png)
![Benzene,1,1'-[(1R,2R)-1-methyl-1,2-cyclopropanediyl]bis-, rel- (9CI)](http://img.cochemist.com/ccimg/14200/14161-72-9_b.png)






















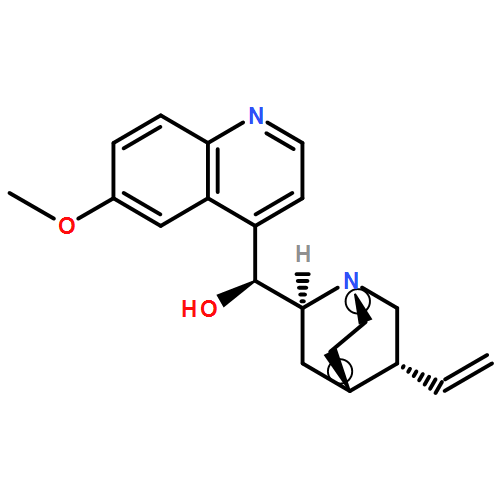
![4-[(5-ethyl-1-azabicyclo[2.2.2]octan-2-yl)-hydroxymethyl]quinolin-6-ol](http://img.cochemist.com/ccimg/6000/5962-19-6.png)
![4-[(5-ethyl-1-azabicyclo[2.2.2]octan-2-yl)-hydroxymethyl]quinolin-6-ol](http://img.cochemist.com/ccimg/6000/5962-19-6_b.png)














![6-phenyl-7-oxabicyclo[4.1.0]heptane](http://img.cochemist.com/ccimg/4900/4829-01-0.png)
![6-phenyl-7-oxabicyclo[4.1.0]heptane](http://img.cochemist.com/ccimg/4900/4829-01-0_b.png)







































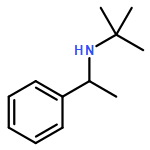
![Benzenemethanamine, N-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/700/622-72-0.png)
![Benzenemethanamine, N-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/700/622-72-0_b.png)



![[4-(2-HYDROXY-2-METHYLPROPYL)PHENYL]ACETIC ACID](/data/chemimg/99000/572-59-8.png)
![[4-(2-HYDROXY-2-METHYLPROPYL)PHENYL]ACETIC ACID](/data/chemimg/99000/572-59-8_b.png)


![6,7,13,14-TETRAOXADISPIRO[4.2.4<SUP>8</SUP>.2<SUP>5</SUP>]TETRADECANE](http://img.cochemist.com/ccimg/400/311-38-6.png)
![6,7,13,14-TETRAOXADISPIRO[4.2.4<SUP>8</SUP>.2<SUP>5</SUP>]TETRADECANE](http://img.cochemist.com/ccimg/400/311-38-6_b.png)











![THIOUREA, N,N'-BIS[(1R,2R)-2-(DIMETHYLAMINO)CYCLOHEXYL]-](/data/chemimg/2717900/852202-60-9.png)
![THIOUREA, N,N'-BIS[(1R,2R)-2-(DIMETHYLAMINO)CYCLOHEXYL]-](/data/chemimg/2717900/852202-60-9_b.png)
![Urea, N-[(1R,2R)-2-(dimethylamino)cyclohexyl]-N'-[(1S)-1-phenylethyl]-](http://img.cochemist.com/ccimg/852300/852202-54-1.png)
![Urea, N-[(1R,2R)-2-(dimethylamino)cyclohexyl]-N'-[(1S)-1-phenylethyl]-](http://img.cochemist.com/ccimg/852300/852202-54-1_b.png)
![Urea, N-[(1R,2R)-2-(dimethylamino)cyclohexyl]-N'-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/852300/852202-53-0.png)
![Urea, N-[(1R,2R)-2-(dimethylamino)cyclohexyl]-N'-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/852300/852202-53-0_b.png)
![Thiourea, N-[(1R,2R)-2-(dimethylamino)cyclohexyl]-N'-methyl-](http://img.cochemist.com/ccimg/852300/852202-51-8.png)
![Thiourea, N-[(1R,2R)-2-(dimethylamino)cyclohexyl]-N'-methyl-](http://img.cochemist.com/ccimg/852300/852202-51-8_b.png)





![Bicyclo[2.2.1]heptane-2,5-diamine, (1S,2S,4S,5S)-](http://img.cochemist.com/ccimg/698400/698353-42-3.png)
![Bicyclo[2.2.1]heptane-2,5-diamine, (1S,2S,4S,5S)-](http://img.cochemist.com/ccimg/698400/698353-42-3_b.png)




![1-[(2S,4S,5R)-5-ethenyl-1-azabicyclo[2.2.2]oct-2-yl]methanamine](http://img.cochemist.com/ccimg/290900/290817-84-4.png)
![1-[(2S,4S,5R)-5-ethenyl-1-azabicyclo[2.2.2]oct-2-yl]methanamine](http://img.cochemist.com/ccimg/290900/290817-84-4_b.png)


![Chromium,chloro[[2,2'-[(1R,2R)-1,2-cyclohexanediylbis[(nitrilo-kN)methylidyne]]bis[4,6-bis(1,1-dimethylethyl)phenolato-kO]](2-)]-, (SP-5-13)-](http://img.cochemist.com/ccimg/165000/164931-83-3.png)
![Chromium,chloro[[2,2'-[(1R,2R)-1,2-cyclohexanediylbis[(nitrilo-kN)methylidyne]]bis[4,6-bis(1,1-dimethylethyl)phenolato-kO]](2-)]-, (SP-5-13)-](http://img.cochemist.com/ccimg/165000/164931-83-3_b.png)













![Methanone, phenyl[(2R,3S)-3-phenyloxiranyl]-](http://img.cochemist.com/ccimg/61900/61840-92-4.png)
![Methanone, phenyl[(2R,3S)-3-phenyloxiranyl]-](http://img.cochemist.com/ccimg/61900/61840-92-4_b.png)









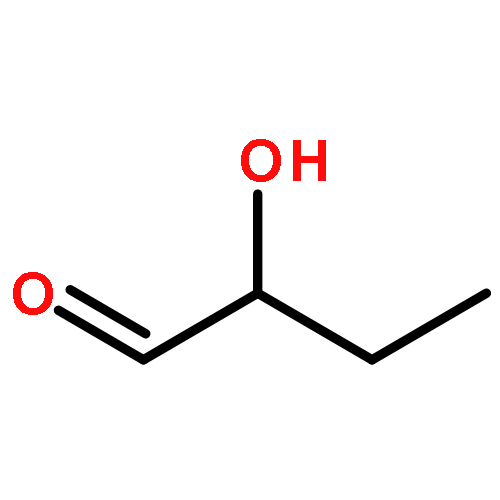
![(2S)-N-[(1S)-1-(Hydroxydiphenylmethyl)-3-methylbutyl]-2-pyrrolidinecarboxam ide](http://img.cochemist.com/ccimg/910200/910110-45-1.png)
![(2S)-N-[(1S)-1-(Hydroxydiphenylmethyl)-3-methylbutyl]-2-pyrrolidinecarboxam ide](http://img.cochemist.com/ccimg/910200/910110-45-1_b.png)
![UREA, N-[(1R,2R)-2-(DIMETHYLAMINO)CYCLOHEXYL]-N'-(3,5-DINITROPHENYL)-](http://img.cochemist.com/ccimg/849100/849024-93-7.png)
![UREA, N-[(1R,2R)-2-(DIMETHYLAMINO)CYCLOHEXYL]-N'-(3,5-DINITROPHENYL)-](http://img.cochemist.com/ccimg/849100/849024-93-7_b.png)






![Benzene, [(5-hexenyloxy)methyl]-](http://img.cochemist.com/ccimg/59200/59137-50-7.png)
![Benzene, [(5-hexenyloxy)methyl]-](http://img.cochemist.com/ccimg/59200/59137-50-7_b.png)
![Naphth[1,2-b]oxirene, 1a,2,3,7b-tetrahydro-, (1aS,7bR)-](http://img.cochemist.com/ccimg/58900/58800-12-7.png)
![Naphth[1,2-b]oxirene, 1a,2,3,7b-tetrahydro-, (1aS,7bR)-](http://img.cochemist.com/ccimg/58900/58800-12-7_b.png)








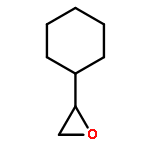
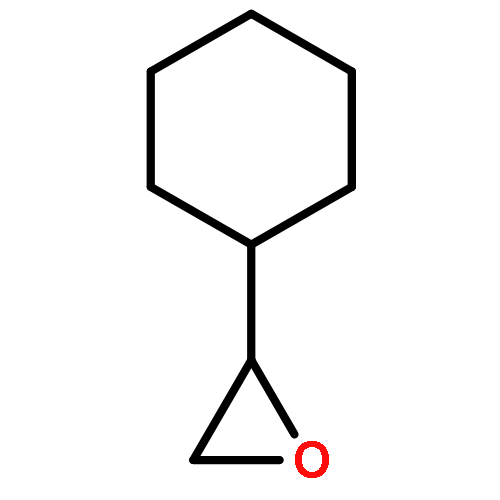




![Thiourea, N-cyclohexyl-N'-[(1R,2R)-2-(dimethylamino)cyclohexyl]-](http://img.cochemist.com/ccimg/852300/852202-52-9.png)
![Thiourea, N-cyclohexyl-N'-[(1R,2R)-2-(dimethylamino)cyclohexyl]-](http://img.cochemist.com/ccimg/852300/852202-52-9_b.png)






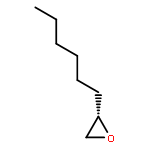
![2-Hydroxy-[1,1'-biphenyl]-3-carbaldehyde](http://img.cochemist.com/ccimg/14600/14562-10-8.png)
![2-Hydroxy-[1,1'-biphenyl]-3-carbaldehyde](http://img.cochemist.com/ccimg/14600/14562-10-8_b.png)